
Abstract
Generalized pustular psoriasis (GPP) is a severe subtype of psoriasis characterized by epidermal neutrophil infiltration, often presenting as acute, potentially life-threatening flares. However, the characterization of the immune micro-environment in GPP lesions remains largely unknown. Here, we use single-cell RNA profiling to interrogate the transcriptomes of 60,000 single cells from GPP lesional skin (n = 13) and healthy adult skin (n = 4), combined with spatial transcriptomics. We identify a neutrophil subset lacking CASP8 expression but exhibiting elevated levels of inflammatory pathway genes, including RIPK1, NFKB1, IL1B, CXCL1, and CXCL8 in GPP flares, illustrating neutrophil transition from pre-inflammatory to a pro-inflammatory state, and activation of a communication network between IL36G+ keratinocytes and neutrophils in GPP lesions, with TNFSF15 (TL1A) released from neutrophils exaggerating the inflammatory crosstalk. We further demonstrate that fibroblasts and capillary endothelial cells function as central communication hubs in GPP, through dynamic receptor-ligand interactions with several spatially proximate immune cells, including T cells, neutrophils, and macrophages. In this work, we provide an in-depth view of immune cell participation and highlight the role of neutrophil-keratinocyte crosstalk in GPP pathogenesis.
Similar content being viewed by others

Introduction
Generalized pustular psoriasis (GPP) is a rare, severe subtype of psoriasis characterized by potentially life-threatening autoinflammatory episodes involving the widespread formation of sterile neutrophil-rich pustules in the epidermis1. GPP pathogenesis involves the interactions of inborn errors of the innate immune system with environmental triggers, leading to uncontrolled activation of inflammatory pathways and intercellular networks, which are only partially understood to date, and remain a topic of debate2. Deregulated innate immune skin and systemic responses driven by hyperactivation of neutrophils have long been postulated to be a canonical feature of GPP1,2,3. On the other hand, the involvement of CD4+ T cells and the IL-23/IL-17 axis, which play a crucial role in plaque psoriasis, has also been implicated in GPP pathogenesis4,5,6. To date, the above-mentioned understanding of GPP pathogenesis largely depends on immunohistochemistry and global gene analyses7,8, primarily focusing on a limited number of cellular contributors, including neutrophils, keratinocytes, and IL-17-producing CD4+ (Th17) cells9. In contrast, the potential pathogenic contributions of other cell populations have received limited attention, largely due to the lack of detailed genetic and molecular profiling of individual cells to delineate their roles in pathogenesis.
Research conducted over the past decade revealed that aberrant activation of IL-36 signaling, promoting proinflammatory responses in keratinocytes as a central driver of GPP pathogenesis10,11, which laid the groundwork for the development of targeted therapies, such as the anti-IL-36 receptor blocker, spesolimab12,13. Intriguingly, IL-36 cytokines (IL-36α, IL-36β, and IL-36γ) are initially expressed as inactive precursors that require activation by neutrophil granule-derived proteases14. After activation, IL-36 signaling proceeds via a complex of IL-36 receptor and IL-1R accessory protein, propagating inflammatory responses in keratinocytes15. The signaling cascade subsequently induces the production of numerous cytokines, such as IL-1β, IL-17A, IL-23, and TNF-α, as well as chemokines like CXCL1 and CXCL8, which collectively establish a chemokine gradient that drives a substantial influx of dermal neutrophils into the epidermis10. Of note, we previously demonstrated that IL-36-dependent amplification of inflammatory responses occurs primarily within the supraspinous layer of the psoriatic epidermis16, aligning with the observed phenotype of neutrophil accumulation manifesting as pustules, predominantly in the corneum of GPP epidermis17. Yet, the detailed mechanisms driving cellular interactions at this specific disease site remain to be elucidated.
Recent clinical and preclinical studies have implicated the contribution of pro-inflammatory fibroblasts in psoriasis lesions16,18. Thus, we questioned whether fibroblasts also play a crucial role in GPP pathogenesis. Notably, while the well-established feedback loop between keratinocytes and neutrophil recruitment explains much of the inflammatory process, it does not fully clarify how neutrophils initially accumulate in GPP lesions, especially given the absence of microvasculature within the epidermis. Here, we provide a comprehensive view of GPP immunopathogenesis by employing single-cell RNA-sequencing (scRNA-seq) on biopsy samples from GPP and healthy control skin, combined with spatial transcriptomics to provide robust validation of cell–cell interactions. Our findings implicate how neutrophil-keratinocyte interactions, mediated through TNFSF15 (TL1A) signaling, contribute to disease progression and suggest this pathway as a potential target for future therapeutic interventions. Furthermore, we identify several cell subtypes, including CASP8- neutrophils and SFRP2+ fibroblasts, which are implicated in the initial recruitment of neutrophils to GPP lesions.
Results
Census of scRNA-seq and spatial-seq reveals diverse cell types and their spatial locations in GPP skin
To elucidate the cellular heterogeneity and cell state of healthy and GPP, we performed scRNA-seq on archived FFPE biopsies of 4 healthy and 13 clinically and histologically confirmed GPP patients (Supplementary Fig. 1A). After quality control, we collected a total of 59,886 cells and identified 18 clusters, which we annotated into 11 distinct primary cell types: keratinocytes (basal, differentiated, keratinized), eccrine cells, endothelial cells, fibroblasts, mast cells, melanocytes, myeloid cells, pericytes, and T cells, across the two groups (Fig. 1A and Supplementary Fig. 1B). Cluster annotation was corroborated by canonical lineage-specific genes published in previous skin disease scRNA-seq studies16,19 (Fig. 1B and Supplementary Fig. 1C). Within myeloid and T cell clusters, cells from GPP versus healthy samples had distinct distributions (Fig. 1C), suggesting dynamic cellular changes in the disease state. Among major cell types, myeloid cells were predominantly derived from GPP lesions (Fig. 1D and Supplementary Fig. 1D). Furthermore, analysis of absolute abundance per sample revealed increased numbers of myeloid cells rather than T cells in GPP lesions (Fig. 1E), consistent with a shift towards innate immune mechanisms in GPP10. Strikingly, ligand-receptor analyses undertaken with CellChat20 demonstrated a widespread up-regulation in the number of cell–cell interactions originating from fibroblasts to several immune cell types (Fig. 1F), underscoring an important role of fibroblasts in recruiting immune cells into GPP lesions.

A UMAP plot showing 59,886 cells colored by cell types. B Dot plot showing representative marker genes for each cell type. The color scale represents the scaled expression average of each gene. The size of the dot represents the percentage of cells expressing each gene. C UMAP plot showing all cells colored by disease conditions. GPP generalized pustular psoriasis. D Bar charts showing proportion of main cell types in different disease conditions. E Cell number of myeloid cells (left) and T cells (right) in disease conditions. Every dot represents a sample; Control (n = 4) and GPP (n = 13). Box plots show the median (center line), interquartile range (box), and 1.5× interquartile range (whiskers). F Heatmap depicting the number of ligand-receptor interactions between cell types. Red represents an increase in the GPP group. Blue represents a decrease in the GPP group. G Representative spatial transcriptomics plots of GPP lesional skin, with corresponding cluster annotation. The statistical significance was determined by two-tailed unpaired Student’s t test (E).
To explore the spatial organization of the identified cell types in GPP lesions, we performed spatial-seq using the Xenium platform on five skin samples (two healthy donors and three GPP patients). By deconvoluting RNA expression in each spot with the scRNA-seq gene expression profiles of the major cell types, we identified the cell type composition in each capture spot (Fig. 1G and Supplementary Fig. 2). Intriguingly, consistent with our previous findings in psoriasis16, we observed that IL-36 signaling was abundantly activated in the upper layers of epidermis.
Single-cell RNA sequencing defines unique myeloid subtypes in GPP skin
Myeloid cells were the most prominent cell type in GPP lesions as defined by our scRNA-seq data. To address myeloid contribution to the disease process in GPP, we initially homed in on myeloid subtypes. We subclustered the myeloid cells into eleven sub-clusters (Fig. 2A) and identified nine subpopulations: MRC1+CXCL12+ Macrophage (Mac), MRC1+CCL18+ Mac, IL1B+ Mac, Neutrophil, plasmacytoid Dendritic cells, Langerhans cells, classical type 1 Dendritic cells, classical type 2 Dendritic cell subset A (cDC2A) and classical type 2 Dendritic cell subset B (Fig. 2B, C). Analysis of disease composition revealed a stark contrast, with MRC1+CXCL12+ macrophages being less prevalent in GPP, while MRC1+CCL18+ macrophages and IL1B+ macrophages were increased (Fig. 2D–F), pointing to a marked shift among these three macrophage subtypes in GPP. To corroborate this, we found that IL1B and TLR2 were predominantly expressed by IL1B+ macrophages, distinguishing them as M1-like macrophages from the other M2-like macrophage subtypes (Supplementary Fig. 3B). We also performed Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the identified signature genes in the IL1B+ Mac and found enrichment for pathways associated with lysosomes and bacterial killing (Supplementary Fig. 3C), consistent with macrophage activation via Toll-like receptors21. Given the well-established observation that the elevated ratio of classically activated inflammatory macrophages (M1) to anti-inflammatory macrophages (M2) is a hallmark of inflammatory skin disease22, we sought to evaluate the relationship between M1/M2 polarization and the underlying pathophysiology of GPP. Indeed, increased gene expression signatures of M1-differentiation and reduced module score of M2-differentiation distinguished GPP macrophages from healthy skin (Fig. 2G). To further explore the differentiation process of macrophages, we performed pseudotime analysis on the three sub-clusters using Monocle23 with the pseudotime assigned from MRC1+CXCL12+ Mac (Control, early state) to IL1B+ Mac (GPP, late state) (Fig. 2H–J). Previously reported principal M1-differentiation markers, including LYZ, FOS, TLR2, and IL1B21,24, were highly enriched along the M1-like polarization path (Fig. 2K) and displayed a prominent and positive correlation with the development from control to GPP state (Fig. 2L).

A UMAP plot showing myeloid cells colored by sub-clusters. B UMAP plot showing myeloid cells colored by subtypes. C Dot plot showing the top marker genes for each myeloid cell subtype. The color scale represents the scaled expression of each gene. The size of the dot represents the percentage of cells expressing the gene of interest. D UMAP plot showing myeloid cells colored by disease conditions. E Bar charts showing proportion of myeloid cell subtypes in different disease conditions. F Cell percentage of IL1B+ Mac (left), MRC1+CCL18+ Mac (middle) and MRC1+CXCL12+ Mac (right) in disease conditions. Every dot represents a sample; Control (n = 4) and GPP (n = 13). Box plots show the median (center line), interquartile range (box), and 1.5× interquartile range (whiskers). G Box plot showing the M1 and M2 differentiation module scores in the macrophages (containing IL1B+, MRC1+CCL18+, and MRC1+CXCL12+ Mac), split by the disease conditions. Each dot represents the module score in a single cell. In the box plot, the centerlines denote median values (50th percentile); the bounds of the boxes represent the first and third quartiles (25th and 75th percentiles); the whiskers indicate the smallest and largest values within 1.5× the interquartile range. H Pseudotime trajectory colored by the pseudotime of three macrophage subtypes. I Pseudotime trajectory colored by the disease condition of three macrophage subtypes. J Pseudotime trajectory colored by the subtype identity of three macrophage subtypes. Expression of transcriptional signature plotted over pseudotime, colored by subtypes (K) or disease conditions (L). The statistical significance was determined by two-tailed unpaired Student’s t test (F) and Wilcoxon signed-rank test (G).
Strikingly, among classical dendritic cell subsets, only cDC2A was significantly enriched in GPP compared to healthy control (Supplementary Fig. 4A). We also found that co-stimulatory molecules CD40, CD80 and CD83 were primarily expressed by cDC2A (Supplementary Fig. 4B), indicating that cDC2A may be responsible for T cell activation and expansion in GPP lesions. Furthermore, cDC2A expressed a high level of interferon and JAK-STAT signaling (Supplementary Fig. 4C)25,26.
Functional heterogeneity of neutrophils highlights the involvement of CASP8 - subset in GPP inflammation
A distinct pattern of pathway enrichment was observed for each myeloid sub-cluster, with significant enrichment of inflammatory pathways in neutrophils (Supplementary Fig. 3A), aligning with the hyperactivation of neutrophils as a key driver of GPP pathogenesis1. While neutrophil infiltration is a hallmark of pathology, as demonstrated by immunofluorescent staining (Fig. 3L), the understanding of dynamic changes in cell states remains limited10, inspiring us to examine neutrophil heterogeneity in GPP.

A UMAP plot showing neutrophils colored by sub-clusters. B UMAP plot showing neutrophils colored by subtypes. C UMAP plot showing neutrophils colored by disease conditions. D Dot plot showing the top ten Gene Ontology pathways enriched for differentially expressed genes in each neutrophil subtype. Gene Ontology pathway enrichment was performed using a hypergeometric test, and p-values were adjusted for multiple testing using the Benjamini–Hochberg method. E Violin plot showing the expression of genes split by neutrophil subtypes. F Individual cell AUC score overlay for TNFA_SIGNALING_VIA_NFKB and INFLAMMATORY_RESPONSE activity in neutrophil subtypes. G Heatmap showing the expression of genes split by subtype. H Pseudotime trajectory colored by the sub-clusters of neutrophils. I Boxplots showing neutrophil sub-clusters ordered by pseudotime; data are median (interquartile range, minimum-maximum). Each dot represents the pseudotime in a single cell. J Bar plot showing the top ten Gene Ontology pathways enriched for the up-regulated genes in neutrophil sub-cluster 0 and 5. Gene Ontology pathway enrichment was performed using a hypergeometric test. K Heatmap showing the five expression patterns of top twenty variable genes along the pseudotime of the neutrophils. L Immunofluorescence showing the colocalization of CASPASE-8 and MPO in healthy control and GPP skin tissues. Images shown are representative of n = 3.
KEGG analysis and Gene set enrichment analysis (GSEA) of differentially expressed genes (DEGs) in GPP versus Control samples showed dramatic up-regulation of several inflammation signaling pathways associated with neutrophil activation (Supplementary Fig. 5A, B). The top distinguishing genes represented in these enrichment terms included interleukin-1 signaling (IL1B and IL1RN), neutrophil chemotaxis (CXCL8), and transcriptional factor (FOS and NFKBIA) (Supplementary Fig. 5C, D). These findings, along with a higher enrichment score of “TNFA_SIGNALING_VIA_NFKB”, “MTORC1_SIGNALING” and “OXIDATIVE_PHOSPHORYLATION” in GPP lesions (Supplementary Fig. 5E), suggest that neutrophils broadly underlie inflammatory responses in GPP pathology.
To probe the molecular shifts of neutrophils in GPP lesions, we sub-clustered all neutrophils and annotated the sub-clusters as CXCL8 + , S100A12+ and CASP8+ neutrophils (Fig. 3A, B) based on their characteristic marker gene expression (Fig. 3E). Strikingly, CXCL8+ neutrophils, exclusively found in GPP lesions, were distinct from CASP8+ neutrophils found in healthy samples (Fig. 3C), with immunofluorescence images further validating the absence of CASP8+ neutrophils in GPP samples (Fig. 3L). To investigate the mechanisms driving this shift, we analyzed enriched Gene Ontology (GO) biological processes and calculated the hallmark score of canonical pathways across individual sub-clusters, highlighting the regulation of cell death and inflammation as the most prominent program specifically exhibited in CXCL8+ neutrophils (Fig. 3D, F). Previous studies have established a pathogenic role of neutrophils in psoriasis via releasing pro-inflammatory mediators and neutrophil extracellular traps (NETs)27. In addition, NET formation involves a programmed cell death of neutrophils28,29, consistent with the phenotype of CXCL8+ neutrophils in our data. In contrast, CASP8+ neutrophils dominantly originated from healthy samples (Fig. 3E) and demonstrated an inactive inflammatory response (Fig. 3F).
Our results thus far raised the hypothesis that the transition of neutrophils in GPP pathology from a pre-inflammation to a pro-inflammatory state depends on turning off CASP8. To corroborate this, we performed pseudotime analysis to clarify the development of CXCL8+ neutrophil phenotypes. Neutrophil clusters followed a branching trajectory from CASP8+ to CXCL8+ neutrophils (Supplementary Fig. 6A–C), culminating in two distinct endpoints, sub-cluster 0 and 5, both belonging to the CXCL8+ neutrophil subset (Fig. 3H, I). Sub-cluster 5 was predominantly positioned in the middle-late stage rather than the terminal stage (Fig. 3I). To identify the potential cytokines that drive this differentiation, we split the variable genes along the pseudotime into five expression patterns, characterized by upregulated expression of CXCL8, IL1RN, IL1B, and PTGS2 in the middle-to-late stage of pseudotime (Fig. 3K), where sub-cluster five was positioned. GO analysis of the top-listed genes in cluster 5 implicated dramatic alterations in immune processes linked to the activation status of the neutrophils (Fig. 3J). Intriguingly, key molecules involved in necroptosis signaling and necroptosis-related inflammation30,31, including IL1B, MLKL, NFKBIZ, FOS, and RIPK1, were all expressed in a higher percentage of cells in sub-cluster 5 compared to sub-clusters 0 (Supplementary Fig. 6D), indicating the role of sub-cluster 5 in supporting CXCL8+ neutrophil phenotype. In addition, CytoTRACE analysis32 suggested that the CASP8+ sub-cluster represented a less differentiated state, and CXCL8+ neutrophils represented the most differentiated state (Supplementary Fig. 6E, F).
T cells exhibit both cytotoxicity and inflammatory responses
Pathogenic T helper type 17 cells (Th17) have long been regarded as an important player in orchestrating GPP pathology6. However, a recent scRNA-seq study on PBMCs proposed cytotoxic T as a potential contributor to GPP pathogenesis33. To define the exact role of T cell subtypes in GPP lesions, we performed sub-clustering of T cells and annotated the sub-clusters using canonical T cell lineage markers (Fig. 4A). We identified six lymphocyte subtypes: ETS1+CD4+ cells, Th17, cytotoxic T cells (Tc), regulatory T cells (Treg), CD4+ effector memory T cells (CD4+ Tem) and natural killer cells (NK) (Fig. 4B). Analysis of sub-cluster composition between the two groups revealed a significantly increased proportion of Th17 cells, along with a notable rise in Tc cells, in GPP (Fig. 4C, D, I and Supplementary Fig. 7A), indicating dual contribution of T cells to GPP inflammation. As expected, GSEA further showed a significant upregulation of leukocyte differentiation signaling in GPP Th17 cells compared to the control groups (Fig. 4E). Consistently, type 17 immune genes such as IL17RB, IL22, CCR6, and IL26 were highly ranked among the upregulated genes in GPP (Fig. 4F), although we were not able to detect increased expression of IL17A or IL17F. Furthermore, expression of key transcriptional factors during Th17 differentiation34 and module score on Th17 differentiation pathway were both elevated in Th17 cells from GPP lesions (Fig. 4G, H).

A UMAP plot showing T cells colored by subtypes. B Heatmap showing the scaled mean expression of genes associated with T cell markers. C Bar charts showing proportion of T cell subtypes in different disease conditions. D Cell percentage of Th17 (left), and Tc (right) in disease conditions. Every dot represents a sample. Control (n = 4) and GPP (n = 13). Box plots show the median (center line), interquartile range (box), and 1.5× interquartile range (whiskers). E GSEA showing the enrichment of the pathway from GPP Th17 compared with healthy control Th17. F Dot plot showing the 15 differentially expressed genes comparing GPP to control in Th17 cells. G Box plot showing the Th17 differentiation module scores in the Th17, split by the disease conditions. Each dot represents the module score in a single cell. In the box plot, the centerlines denote median values (50th percentile); the bounds of the boxes represent the first and third quartiles (25th and 75th percentiles); the whiskers indicate the smallest and largest values within 1.5× the interquartile range. H Violin plot showing the expression of genes in Th17 cells split by disease condition. I Spatial transcriptome image of lesional sections showing the distribution of Th17 cells (identified by expression of CD4, IL22, and IL26). The size bar represents 500 µm. The statistical significance was determined by two-tailed unpaired Student’s t test (D) and Wilcoxon signed-rank test (G).
As another potential function of T cells in GPP pathogenesis, we observed higher expression of GZMB, CCL5, NKG7, and PRF1 in cytotoxic T cells from GPP compared to healthy controls (Supplementary Fig. 7B). The module score aggregating the expression of cytotoxic genes was significantly higher in GPP lesions (Supplementary Fig. 7C).
SFRP2+ fibroblasts promote myeloid cell chemotaxis into GPP lesions
Fibroblasts (FBs) are one of the most frequent cell types in GPP lesions, as defined by our scRNA-seq data. To characterize the difference between GPP and healthy FBs. We totally identified 10 clusters (Fig. 5A), which were annotated into five main FB subtypes based on previously published marker genes16, including SFRP2+, TYMP+, S100A9+, LSP1+, and SFRP4+FBs (Fig. 5B, D). Strikingly, GPP FBs were mostly distinct from control FBs by dimensionality reduction using UMAP (Fig. 5C), representing a marked transcriptomic shift in GPP lesions. To characterize the potential functions of FB subtypes, we conducted gene set variation analysis (GSVA) for each FB cluster based on each cluster’s gene expression pattern. We observed significant enrichment of inflammatory pathways in S100A9+ and SFRP2+FBs (Supplementary Fig. 8B). Additionally, spatial mapping and immunofluorescence confirmed the presence of S100A9+ and SFRP2 + FBs in GPP lesions (Supplementary Fig. 8D, F). Intriguingly, S100A9 + FBs drove intercellular interaction with keratinocytes and myeloid cells in GPP lesions (Supplementary Fig. 9A, B). Although S100A9 + FBs were derived exclusively from GPP lesions (Fig. 5E), differential expression analysis (GPP vs control) showed that the majority of the changes were observed in SFRP2 + FBs (Supplementary Fig. 8C), identifying SFRP2 + FBs as primary responders to GPP pathogenesis.

A UMAP plot showing fibroblasts colored by sub-clusters. B UMAP plot showing fibroblasts colored by subtypes. C UMAP plot showing fibroblasts colored by disease conditions. D Violin plot showing the expression of genes split by fibroblast subtypes. E Bar charts showing the proportion of fibroblast subtypes in different disease conditions. F Volcano plot showing the differential expressed genes comparing GPP to control in SFRP2+ fibroblasts. P-value was calculated by two-sided Wilcoxon rank-sum test with Bonferroni correction. G Dot plot showing the top ten canonical pathways enriched using the differentially expressed genes comparing GPP to control in SFRP2+ fibroblasts. KEGG pathway enrichment was performed using a hypergeometric test. H Heatmap depicting the number of ligand-receptor interactions between myeloid cell subtypes and SFRP2+ fibroblasts. Red represents an increase in the GPP group. Blue represents a decrease in the GPP group. I Cellular interactions of CXCL1 signaling pathway in control and GPP group.
To further characterize the functions of GPP-associated SFRP2 + FBs, we performed analysis of DEGs and upregulated canonical pathways. These upregulated genes included chemokines (CCL2, CXCL1, and CXCL2), pro-inflammatory cytokines (IL6) and key transcriptional factors of inflammation signaling (CEBPB, FOS, JUNB, STAT1, NFKB1, and IRF1) (Fig. 5F and Supplementary Fig. 8E). As expected, SFRP2 + FBs showed functions associated with IL-17 signaling and TNF signaling (Fig. 5G). The overexpression of specific chemokines in SFRP2 + FBs raised the possibility that SFRP2 + FBs may be a key orchestrator in recruiting myeloid cells35,36. We, therefore, conducted receptor-ligand analyses using CellChat to infer communication between SFRP2 + FBs and myeloid cell subtypes. Indeed, the ligand-receptor analyses showed an enhanced CXCL signal interaction from SFRP2 + FBs to neutrophils in the GPP group (Fig. 5H, I). Furthermore, we confirmed this potential chemotaxis effect of SFRP2+ fibroblasts on neutrophils through spatial transcriptomics analysis, in which co-localization of SFRP2 + CXCL1 + FBs and MPO+ neutrophils was robustly detected in the upper dermis of GPP lesion (Supplementary Fig. 8F). Similarly, NicheNet analysis predicted an interaction between the ligand (originating from SFRP2 + FBs) and respective receptor on MRC1+ Macrophage in GPP, which showed an increased CSF1 level in GPP SFRP2 + FBs (Supplementary Fig. 8G, H). Combined with the well-established role of CSF-1 in promoting macrophage maturation37,38, these observations suggest that SFRP2 + FBs facilitate neutrophil infiltration and macrophage activation in GPP lesions.
To address endothelial contribution to inflammatory chemotaxis into GPP lesions, we assessed the contribution of endothelial cells in immune cell infiltration. We subclustered endothelial cells and identified five subsets based on published scRNA-seq studies on vasculature39,40, including Venules-1 (SELE+/ACKR1+), Capillaries-1 (SPARC+/PLVAP+), Venules-2 (VWF+/CLU+), Capillaries-2 (STAB2+/SNCG+), and Arterioles (GJA4+/SEMA3G) (Supplementary Fig. 10A–D). While both Venules-1 and Capillaries-1 were nearly completely derived from GPP lesional skin (Supplementary Fig. 10E), upregulation of leukocyte chemotaxis (CCL21) was observed in GPP Capillaries-2 cells (Supplementary Fig. 10F), facilitating the migration of T cells towards sites of skin inflammation. To validate the spatial specificity of these responses, we demonstrated that infiltrating T cells were surrounded by CCL21+STAB+ capillary cells in GPP lesions, whereas healthy skin lacked co-expression of CCL21 and STAB in the dermis (Supplementary Fig. 10G). Collectively, the heterogeneity observed in GPP SFRP2+ fibroblasts and Capillaries-2 cells, particularly their involvement in chemotaxis and macrophage activation, likely contributes to the aforementioned inflammatory responses driven by myeloid cells and T cells.
IL-36 amplifies inflammation response in the supraspinous epidermis of GPP lesions
Keratinocytes (KCs) constituted the majority of cells sequenced in GPP lesions (Fig. 1A). To delineate molecular changes of epidermal pathology, we subclustered KCs into 12 clusters and annotated cycling, basal, spinous, and supraspinous KCs based on marker gene expression (Fig. 6A–D). GPP-dominated subclusters corresponded to sub-populations within cycling and supraspinous KCs (Fig. 6E). Intriguingly, several risk genes of GPP, including IL1RL2, IL1RN, IL36RN, IL36A, IL36G, and CARD147,17, were constitutively expressed in supraspinous KCs (Supplementary Fig. 11B, C), pointing to a role of supraspinous KCs in driving GPP pathology. Consistent with this, spatial mapping of two representative genes of chemotaxis, CXCL1 and CXCL2, illustrated their up-regulation in GPP and localized distribution in the supraspinous layer of the epidermis (Supplementary Fig. 8F).

A UMAP plot showing keratinocytes colored by sub-clusters. B UMAP plot showing keratinocytes colored by subtypes. C UMAP plot showing keratinocytes colored by disease conditions. D Heatmap showing marker genes with the highest fold change for each keratinocyte subtype. The color scale represents the scaled expression of each gene. E Bar charts showing proportion of keratinocyte subtypes in different disease conditions. F Violin plot showing the cytokine module scores in keratinocyte subtypes, and each subtype is split by the disease conditions. G Dot plot showing the top differentially expressed genes comparing GPP to control in the spinous (left) and supraspinous (right) keratinocytes. H UMAP plot showing keratinocytes of interest, colored by original subtypes. I Feature plots showing expression of IL36G in keratinocytes of interest. J UMAP plot showing keratinocytes of interest, colored by reannotated subtypes. K UMAP plot showing keratinocytes of interest, colored by disease conditions. L Individual cell AUC score overlay for INFLAMMATORY_RESPONSE activity in keratinocyte reannotated subtypes. The statistical significance was determined by Wilcoxon signed-rank test (L).
To investigate the key upstream regulators of activated KCs, we applied cytokine response signatures previously conducted by our group, containing genes induced by the stimulation of primary human KCs with a panel of cytokines, such as IL-36γ, IL-17A, and IL-1β, to calculate the module score for each cytokine. GPP lesions showed increased scores for IL-36γ, IL-17A, and IL-1β responses in spinous and supraspinous subtypes compared with healthy skin, with the highest responses observed in supraspinous KCs (Fig. 6F).
Next, we performed differential gene expression analysis between GPP and healthy control within spinous and supraspinous KCs to identify top distinguishing transcripts (Fig. 6G). Expression of genes associated with keratinocyte activation (S100A7, S100A8, S100A9, KRT6A, KRT6B, and KRT6C) was markedly elevated in spinous and supraspinous layers of GPP. Moreover, IL-36-induced genes, including IL36G, IL1RN, and IL36RN, were likewise upregulated in both KC subsets, in line with previous GPP studies7,10. In addition, we assessed enriched pathways between two groups within each epidermal layer and identified the IL-17 signaling pathway as the top mediator in cycling, spinous, and supraspinous KCs (Supplementary Fig. 11D–G).
IL-36 is a member of the IL-1 family of cytokines, which plays important roles in the pathogenesis of GPP10. Given the prominence of spinous and supraspinous KCs in response to IL-36, we sought to define heterogeneity within these subsets. We re-clustered spinous and supraspinous KCs and annotated four KC subpopulations: IL36G- spinous, IL36G+ spinous, IL36G- supraspinous, and IL36G+ supraspinous (Fig. 6H–J). Strikingly, the majority of IL36G+ spinous and supraspinous keratinocytes were derived from GPP samples (Fig. 6K). Furthermore, a striking increase from spinous to supraspinous KCs was also observed for “INFLAMMATORY_RESPONSE,” with IL36G+ supraspinous KCs exhibiting the highest levels on average (Fig. 6L), confirming that IL36G+ supraspinous KCs may broadly underlie the epidermal pathology in GPP-associated inflammation.
Considering that active IL-36 isoforms bind to the IL-36 receptor on keratinocytes (KCs) and drive the expression of proinflammatory mediators, including IL-36 itself41, we hypothesized that IL36G- KCs represent a transient state rather than an exact cell subtype. These cells may be further induced to differentiate into IL36G + KCs upon exposure to inflammatory stimuli such as TNF and IL-1716. To explore the upstream transcriptional regulation driving the emergence of IL36G⁺ spinous KCs, we performed Ingenuity Pathway Analysis (IPA) based on the transcriptomic differences between IL36G- and IL36G+ spinous subsets. Our analysis demonstrated an enrichment for SP1, MYC, KLF4, STAT4, and RELA as top-ranked transcriptional factor genes in IL36G+ spinous (Supplementary Fig. 12A).
In addition, we conducted pseudotime trajectory analysis to reconstruct the developmental path of spinous KCs (Supplementary Fig. 12B–D). Interestingly, during the transition from healthy to GPP-associated spinous KCs, a distinct population of IL36G- spinous cells (State 3) persisted, even within the inflammatory milieu of GPP lesions (Supplementary Fig. 12C, D). In contrast, IL36G+ spinous cells were largely confined to a separate trajectory branch (State 1), suggesting that these two subpopulations (State 1 and 3) from GPP lesions may follow divergent developmental paths despite being exposed to the same proinflammatory environment. Intriguingly, state 1 exhibited markedly elevated expression of both differentiation markers (FLG and IVL) and pro-inflammatory markers (S100A7A and IL36G) compared to other states, underscoring the potential role of IL36G+ spinous cells in epidermal maturation and inflammatory responses (Supplementary Fig. 12E, F). These findings suggest that State 1, rather than State 3, plays an indispensable role in the pathogenesis of GPP.
Ligand–receptor interaction analysis between the IL-36γ-expressing keratinocytes and neutrophils in GPP skin
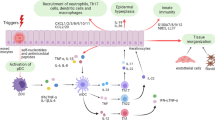
While the synergy of IL-36γ-expressing keratinocytes with neutrophils is considered a hallmark of GPP pathophysiology, the nature of the cellular interactions remains largely unknown. To address this gap, we analyzed cell–cell communication between putative pathogenic keratinocytes and neutrophils in GPP skin. Generally, CellChat network analysis revealed a dominant communication between IL36G+ spinous, IL36G+ supraspinous, and neutrophils in GPP (Fig. 7A, B). In addition, several signal pathways were markedly upregulated in GPP lesions, including IFN-I, IGF, CSF, VEGI, and CXCL signaling (Fig. 7C). Intriguingly, network analysis of GPP lesions revealed an unreported VEGI signaling between neutrophils to IL36G+ spinous KCs (Fig. 7D). To corroborate this further, we detected an enhanced TL1A (encoded by TNFSF15)–DR3 (encoded by TNFRSF25) interaction from neutrophils to IL36G+ spinous KCs (Fig. 7E).

A Cell–cell interaction number analysis among IL36G- Spinous, IL36G+ Spinous, IL36G- Supraspinous, IL36G+ Supraspinous, and neutrophils in control and GPP group. B The incoming and outgoing signaling strength of different subtypes in control and GPP group. C The significant signaling pathways were ranked based on their differences in overall information flow within the inferred networks between control and GPP group. D Cellular interactions of VEGI signaling pathway in GPP group. E Signal pair interactions from neutrophils to keratinocyte subtypes identified with CellChat analysis in GPP group. F Feature plots showing expression of TNFSF15 in myeloid cells. G Spatial transcriptome image of lesional sections showing the co-localization of IL36G + TNFRSF25+ differentiated keratinocytes and TNFSF15 + MPO+ neutrophils. The pink frame represents differentiated keratinocyte. The green frame represents myeloid cell. The size bar represents 500 µm (left, zoom out) or 100 µm (right, zoom in). H Schematic of the experimental design for the in vitro coculture assay. Created in BioRender. Jiang, R. (2025) https://BioRender.com/lcyuhhl<https://urldefense.com/v3/__https://BioRender.com/lcyuhhl__;!!NnSTv5QBqPjS9UMk!JEO9XaAu3G1endKkUtPBlL_enDqU_76Al307YLCYnUt2lDpHO_kWRIx-Yn_M0xHteCCZv_1plokWETudQJQZeRZYkw$>. I qRT-PCR of inflammatory genes in primary keratinocytes after co-culture with neutrophils (n = 3 biologically independent samples; one-way ANOVA; mean ± SEM). J qRT-PCR of inflammatory genes in WT/IL36G KO N/TERTs after co-culture with neutrophils (n = 3 biologically independent samples; one-way ANOVA; mean ± SEM).
TNFSF15, and its protein ligand TL1A, is a member of the tumor necrosis factor family. TL1A competitively binds to DR3, providing a stimulatory signal for downstream inflammation response implicated in several autoimmune diseases, including rheumatoid arthritis, inflammatory bowel diseases, and psoriasis42. In contrast to previous studies demonstrating macrophages’ ability to express TL1A43, we found that TNFSF15 was primarily derived from neutrophils rather than other myeloid cell subsets in GPP lesions (Fig. 7F). Furthermore, the close proximity of TNFSF15 + MPO+ neutrophils to IL36G + TNFRSF25+ keratinocytes was identified in the spinous layer of the epidermis, rather than in the supraspinous layer, as simultaneously revealed by both spatial-seq mapping (Fig. 7G) and immunohistochemistry staining (Supplementary Fig. 13A). As expected, immunofluorescent staining on TL1A + MPO+ cells complemented the expression of TL1A in neutrophils during GPP pathogenesis (Supplementary Fig. 13B). Notably, serum levels of TL1A protein were also specifically elevated in patients with GPP, rather than psoriasis vulgaris patients (Supplementary Fig. 13D), suggesting that TL1A signal might be a specific pathogenic signal in GPP. Collectively, these results indicate that neutrophils may contribute to keratinocyte inflammatory response through TL1A signal in GPP skin.
To determine whether TL1A signals act directly on keratinocytes to potentiate the inflammatory response, we conducted an in vitro assay using sorted neutrophils from healthy donors. These neutrophils were first stimulated with phorbol 12-myristate 13-acetate (PMA) to activate them for processing full-length IL-36γ protein10 and then co-cultured with human primary keratinocytes or N/TERT cells, an immortalized keratinocyte line44 (Fig. 7H). Firstly, full-length IL-36γ protein (inactive form) was added to the culture system for primary keratinocytes to reflect the participation of neutrophil proteases in cleaving pro-IL-36γ into the mature form in this model system. Intriguingly, in our preliminary experiments, untreated neutrophils remained quiescent and failed to induce a robust inflammatory response in keratinocytes, whereas PMA-stimulated neutrophils elicited pronounced inflammatory activation (Supplementary Fig. 13C). Based on these findings, we employed PMA-treated neutrophils in subsequent assays to assess the efficacy of TL1A blockade in vitro and observed that high-dose inhibition of TL1A effectively attenuated the keratinocyte inflammatory response (Fig. 7I). Next, to elucidate the crucial role of IL36G+KCs, we used CRISPR-Cas9 to knock out IL36G in N/TERT cells, which were primed with IL-36γ (aa 18-169) and IL-1β for 8 h to induce the new production of pro-IL-36γ. After the medium was replaced, N/TERT cells were cocultured with PMA-preactivated neutrophils. Consistent with the primary keratinocyte phenotype, we observed notably lower expression of antimicrobial and proinflammatory genes in the WT group after TL1A blockade. In contrast, when keratinocytes lost the expression of IL36G, no additional inhibition on proinflammation genes was observed (Fig. 7J), underscoring that IL-36γ expressing keratinocyte–neutrophil communication relies on TL1A signaling.
Discussion
Collectively, through a combination of scRNA-Seq, spatial transcriptomics, and function experiment validation, our data describe the cellular composition and architecture of GPP pathogenesis at unprecedented resolution. While previous studies utilized scRNA-seq analysis of PBMCs from whole blood of GPP patients33, they included only two GPP patients and lacked insights into the spatial compartments where cell–cell interactions occur at the disease site. Our data bridge the gap in understanding the neutrophil-keratinocyte signal interaction network in GPP pathogenesis, demonstrating an amplified communication between IL36G+ keratinocytes and neutrophils in GPP lesions, with TNFSF15 (TL1A) released from neutrophils intensifying the inflammatory crosstalk. We further identify that stromal subtype enriched in GPP lesional skin, SFRP2+ fibroblasts, and capillary endothelial cells impact other spatially proximate cell types, including neutrophils, macrophage, and T cells, via the production of CXCL1, CXCL2, CCL2, CSF1, and CCL21 (Fig. 5F and Supplementary Figs. 8F, 10F), underscoring the underappreciated contribution of stromal cells to shape and perpetuate the immune cells infiltration.
While the contribution of neutrophils to GPP has been appreciated for a long time, the precise cellular heterogeneity in neutrophils is unclear. Our studies pinpoint the precise clarification of the transition process from CASP8+ to CASP8- neutrophils, highlighting another key pathologic characteristic in GPP. Caspase-8 has historically been considered to promote cell apoptosis45, but recent observations have also provided abundant evidence for its role in preventing necroptosis, an inflammatory form of cell death, particularly through enzymatically degrading crucial necroptosis mediators, including RIPK1 and RIPK33046, therefore restraining the downstream inflammation pathway. Here, our single cell and spatial transcriptomics data suggested a substantial heterogeneity across neutrophil subtypes, characterized by a gradual loss of CASP8 and interferon receptor (IFNAR1), combined with upregulation of necroptosis mediators (RIPK1), transcription factors (NFKB1 and FOS), and proinflammatory cytokines (IL1B and CXCL1) as CASP8+ neutrophils transition into CXCL8+ neutrophils (Fig. 3G), in line with previously report that Caspase-8 inhibition induced chemokine production and neutrophil recruitment47.
Neutrophils have long been recognized as enzymatic machines that process precursor IL-36, predominantly secreted by keratinocytes, through the action of granule-derived proteases such as elastase, cathepsin G, and proteinase 314,48, underlying basic crosstalk between keratinocytes and neutrophils. Of note, our results further identify neutrophils as an underestimated source of pro-inflammatory cytokines in GPP, particularly through the release of TL1A signals. Unexpectedly, contrary to the prior understanding that TL1A is primarily produced by macrophages and dendritic cells upon Toll-like receptor activation43,49, our findings indicate that in GPP lesions, TL1A is predominantly derived from neutrophils, rather than other myeloid cell subsets (Fig. 7F), underscoring a unique role of neutrophils in driving the inflammatory response in GPP.
Recent clinical trials have demonstrated the efficacy of monoclonal antibodies targeting TL1A as a potential therapeutic approach for treating patients with autoimmune diseases50. Mechanistically, TL1A binds to its receptor DR3, activating downstream pathways such as MAPK, NF-κB, and caspase-8, thereby modulating innate and adaptive immune responses51,52. To date, DR3 is reported to be expressed on lymphoid lineage, especially activated lymphocytes, in which TL1A acts as a co-stimulator that increases IL-2 responsiveness and secretion of proinflammatory cytokines53. In contrast, our data demonstrated constitutive cutaneous expression of TL1A and DR3 in GPP epidermis, especially within the spinous layer (Supplementary Fig. 13A), consistent with similar findings in psoriatic skin lesions54. Aligned with this, we observe obvious TL1A to DR3 ligand-receptor interactions between IL36G+ spinous KCs and neutrophils in our single-cell and spatial-seq data. Considering that TL1A triggers the recruitment of downstream adapter proteins such as TRAF and RIPK1 to the membrane complex, subsequently activating the transcription factor NF-κB55, the TL1A–DR3 axis indirectly amplifies this autocrine inflammatory loop between keratinocytes and neutrophils. Thus, it is tempting to speculate that targeting TL1A could serve as a potential strategy to regulate the neutrophil-keratinocyte crosstalk and mitigate the manifestations of GPP.
The data presented here also implicate keratinocyte populations in GPP pathogenesis. Particularly, the contribution of keratinocytes to psoriasis has been appreciated for a long time56. Likewise, in accordance with our previous finding in psoriasis16, we also demonstrate that the strongest downstream responses to IL-17A, IL-1β, and IL-36 in GPP KCs occur in their supraspinous compartment. Interestingly, the seemingly paradoxical observation of a strong inflammatory response phenotype in supraspinous KCs alongside TL1A-DR3 interactions within spinous KCs suggests the intriguing possibility that interactions between IL36G+ spinous KCs and neutrophils may serve as the preliminary and foundational step in pro-inflammatory keratinocyte differentiation during GPP pathogenesis. This hypothesis offers a dynamic perspective on the interaction between keratinocytes and neutrophils in GPP, suggesting that spinous rather than supraspinous keratinocytes act as the initial responders activated by neutrophils, ultimately acquiring the capacity to recruit neutrophils following terminal differentiation. Indeed, additional research is required to validate this process in greater detail.
In sum, the data presented here identify a cell–cell interaction network—particularly between keratinocytes and neutrophils—in the pathogenesis of GPP. Our study has several limitations. First, while we focused primarily on GPP, other neutrophilic dermatoses, including Sweet’s Syndrome57, were not included in our analysis. Incorporating these conditions in future investigations may provide broader insight into the shared and distinct mechanisms underlying neutrophil-driven skin inflammation. Second, although GPP is well known for its association with IL36RN mutations in keratinocytes58, the current study primarily focused on the shared pathogenic mechanisms of GPP, irrespective of IL36RN mutation status. Future investigations may help delineate distinct and precise molecular pathways in patients with and without IL36RN mutations. Third, the interaction between IL36G+ spinous KCs and neutrophils via the TL1A–DR3 signaling axis, as observed at the scRNA-seq level, was validated by comparing the inflammatory responses between IL36G knockout and wild-type keratinocytes (Fig. 7J). However, this approach may not fully recapitulate the specific behavior of IL36G+ spinous keratinocytes. Further validation using targeted methods such as IL36G-specific siRNA knockdown should be considered to strengthen this hypothesis. Finally, although we validated the crosstalk between neutrophils and keratinocytes through co-culture experiments in this study, other predicted cell–cell interactions, such as those between SFRP2+ fibroblasts and neutrophils/macrophages, remain to be experimentally confirmed. Future co-culture studies using fibroblasts and neutrophils/macrophages derived from GPP lesions and healthy controls may help to further elucidate these GPP-specific pathogenic mechanisms.
Methods
Human sample acquisition
Thirteen patients with GPP (Table S1) and 4 healthy controls were recruited for this study. The study was approved by the University of Michigan Institutional Review Board (IRB), and all patients provided written informed consent. Skin biopsies (6-mm punch) were collected from lesional or healthy areas. The study was conducted according to the Declaration of Helsinki Principles.
Single-cell RNA-seq library preparation, sequencing, and alignment
Tissue dissociation and library construction were prepared by the University of Michigan Advanced Genomics Core on the 10X Chromium system per the manufacturer’s instructions. Briefly, three 50 µm FFPE tissue scrolls were placed into a GentleMACS C-tube (Miltenyi Biotech) and processed using the GentleMACS Octo Dissociator. The scrolls were deparaffinized, washed, and then enzymatically dissociated into a single-cell suspension using Liberase TH.
Library preparation was performed on the 10X Chromium platform using Next GEM chemistry. Sequencing was conducted on the Illumina NovaSeq X to generate 150 bp paired-end reads.
Cell clustering and cell type annotation
The Seurat R package (v5.0.1) was employed for cell clustering on the combined matrix59. Cells containing fewer than 200 features or more than 10% mitochondrial gene expression were filtered as low-quality cells. The NormalizeData function, with default parameters, was utilized to normalize expression levels across individual cells. Highly variable genes were subsequently identified using the FindVariableFeatures function with default settings. The dataset was scaled and centered using the ScaleData function. Principal component analysis was conducted on the highly variable genes, using the first 20 principal components for clustering and UMAP dimensionality reduction. Cell clusters were determined using the FindNeighbors and FindClusters functions, with a resolution of 0.5. Marker genes for each cluster were identified through the FindAllMarkers function. Cell type annotations were assigned by cross-referencing the cluster marker genes with known canonical cell type signature genes.
Cell type sub-clustering
Sub-clustering was conducted for the most abundant cell types using the same functions described previously. Sub-clusters that were defined dominantly by mitochondrial gene expression, indicating low quality, were removed from further analysis. Subtypes were annotated by cross-referencing the marker genes of sub-clusters with canonical subtype signature genes. To investigate the characteristic differences among cell clusters, the “FindAllMarkers” function in the Seurat package was applied to identify DEGs in individual clusters using the Wilcoxon rank-sum test. Genes with p_val < 0.05 and avg_log2FC > 1 were considered upregulated, while genes with p_val < 0.05 and avg_log2FC < –1 were classified as downregulated. GO and KEGG pathway enrichment analyses were conducted using the clusterProfiler package (v4.8.3). GSEA for DEGs was performed using the fgsea package (v1.26.0). IPA was applied to the DEGs to determine the potential upstream regulators. The upstream regulators with an activation z-score ≥2 were considered significant.
Calculation of enrichment score
The “AddModuleScore” function was used to calculate module scores for different functions. Specifically, the cytotoxicity score was defined as the average expression of GZMA, GZMB, GZMH, NKG7, CCL5, GNLY, CST7, TCF7, IFNG, and RPF1. The Th17 differentiation score was defined as the average expression of IL17A, IL17F, IL22, IL26, RORA, RORC, CCR6, IL23R, and STAT3. The M1 differentiation score was defined as the average expression of IL1B, CCL1, CXCL9, CXCL10, LYZ, TLR2, TLR4, ROS1, NOS2, NFKB1, NFKB2, RELA, FOS, and STAT1. The M2 differentiation score was defined as the average expression of IL10, CXCL12, CCL18, CD163, MRC1, CD209, IGF1, EGF, FCGR3A, ARG1, CCL17, and CCL2260.
To describe the function features of cell subtypes, we gained the gene sets of 50 hallmarks from the MSigDB database61. The score of above-mentioned pathways was calculated using GSVA package (V1.48.3)62, and AUCell package (V1.22.0)63. Wilcoxon rank test was used to perform significance tests using the ggpubr package (V0.6.0).
Integration with KC cytokine signatures
We used RNA-seq-based KC cytokine response signatures for the following cytokines: IL-17A (10 ng/mL), IL-1β (10 ng/mL), or IL-36 (10 ng/mL)64. Briefly, primary human KCs from 50 donors were treated with a panel of cytokines as above for 8 h and harvested for RNA isolation. Bulk RNA-seq was performed on the Illumina NovaSeq 6000 sequencer with the assistance of the University of Michigan Advanced Genomics Core. For each stimulation condition versus unstimulated controls, differential expression analysis was performed using DESeq265. DEGs (two-fold increase; false discovery rate, <0.05) were used to construct response signatures for each cytokine.
Cell–cell interaction inference
The ligand-receptor (LR) interaction analysis was first performed through CellChat (v1.6.1)66. The primary analysis focused on interactions between myeloid cell subtypes and fibroblast subtypes, as well as between neutrophils and keratinocyte subtypes. A separate run was performed for each sample group, and the number of significant interactions was calculated for each cell type pair. In addition, the cellular interaction was further confirmed by NicheNet (v2.0.6) package to analyze the different LR pairs between GPP and healthy control group67. For example, in the Supplementary Fig. 8G, fibroblast subtypes were defined as “sender” cells, and MRC1+ macrophages were defined as “receiver” cells. Results were generated using the “nichenet_seruatobj_aggregate” function.
Pseudotime trajectory construction
Pseudotime trajectory analysis was performed using the Monocle package (v2.28.0)23. Raw cell counts from the Seurat analysis were normalized using the estimateSizeFactors and estimateDispersions functions with default settings. Genes with an average expression above 0.5 and present in at least 10 cells were retained for further analysis. Variable genes were identified using the FindVariableFeatures function. Cell ordering was determined using the orderCells function, and the trajectory was constructed with the reduceDimension function, both applied with default parameters.
Spatial sequencing library preparation
Spatial transcriptomics was performed by embedding tissues into a single block, followed by a 5 µm section mounted onto a Xenium slide (10X Genomics). Xenium spatial library generation and transcriptomics were completed by the University of Michigan Advanced Genomics Core, where tissue sections were hybridized with fluorescently labeled oligonucleotide probes targeting specific mRNAs, using a custom-made 480 gene panel following the manufacturer’s instructions.
Spatial sequencing data analysis
The output images and expression profiles were evaluated by Xenium Explorer (version 3.1.1, 10x Genomics). After the Xenium run, H&E staining was performed on the Xenium slide. For quality control of Xenium analysis, we checked the output summary HTML file and confirmed that the number of detected transcripts is compatible with that in the literature reported by 10x Genomics68. We visualized the cell annotation and gene expression using the Xenium Explorer software. Gene expression k-means clustering (k = 10) was applied to partition the cells into 10 distinct populations, using the UMAP projection as a basis. These 10 clusters, defined by their top marker genes, were annotated according to the scRNA-seq results and overlaid on the spatial-seq data. The Xenium Explorer was used to perform the gene expression analysis and define the co-localization of specific cell types.
Immunohistochemical and immunofluorescence staining
Paraffin-embedded tissue sections from punch biopsies of GPP patients and healthy controls were incubated at 60 °C for 30 min, followed by deparaffinization and rehydration. Slides were placed in antigen retrieval buffer and heated at 125 °C for 30 s in a pressure cooker water bath. After cooling, slides were treated with 3% H2O2 for 5 min (only for immunohistochemical staining) and blocked using 10% goat serum for 30 min. Overnight incubation was then performed using anti-MPO (Thermo Fisher Scientific, cat. PA5-16672), anti-CASPASE8 (Abcam, cat. ab220171), anti-TL1A (Abcam, cat. ab234300), anti-KRT5 (Thermo Fisher Scientific, cat. MA5-12596), and anti-DR3 (R&D Systems, cat. MAB943) at 4 °C. Images presented are representative of at least three biological replicates.
Neutrophil isolation from whole human blood
Peripheral blood samples from healthy donors were collected at the University of Michigan Hospital. The study was approved by the University of Michigan IRB, and all donors provided written informed consent. Peripheral blood polymorphonuclear leukocytes were isolated by centrifugation using Polymorphprep (COSMO BIO USA, cat. AXS-1114683) according to the manufacturer’s recommendations. The polymorphonuclear cell layer was collected, and red blood cells were removed using RBC Lysing Buffer (Biolegend, cat. 420302). Freshly isolated neutrophils were suspended at 1 × 107/mL in PBS for a later co-culture experiment.
Cell culture and in vitro co-culture model
Primary keratinocytes and N/TERTs, an immortalized keratinocyte line44, were cultured in Keratinocyte-SFM medium (Thermo Fisher Scientific, cat. 17005-042) supplemented with 30 µg/mL bovine pituitary extract, 0.2 ng/mL epidermal growth factor, and 0.3 mM calcium chloride. Knock-out cell lines (IL36G KO) via CRISPR/Cas9 had been generated in our previous work16.
Neutrophils were isolated as described above and seeded at a concentration of 2 × 106/mL in a 12-well plate. Neutrophils were cultured in RPMI 1640 medium (Gibco, cat. 11875093) supplemented with 500 nM Phorbol 12-myristate 13-acetate (Sigma, cat. 524400) to induce NET formation before co-culture with keratinocytes. After 4 h, the supernatant was carefully removed, and NETs and neutrophils were collected by washing the plate with cold PBS, followed by a centrifugation step (500 × g, 10 min).
Prior to co-culture with N/TERTs and neutrophils, N/TERTs were stimulated with activated IL-36γ (R&D Systems, cat. 6835-IL-010, 100 ng/mL) and IL-1β (Sino, cat. 10139-HNAE, 50 ng/mL) for 8 h to induce an intracellular inflammatory response. After stimulation, the supernatant was carefully removed and replaced with fresh Keratinocyte-SFM medium without additional cytokines, followed by culturing with PMA-treated neutrophils (2 × 106 per well) for the next 20 h. This allowed us to evaluate the effect of IL36G KO in the co-culture system, as only wild-type cell lines were capable of producing abundant IL-36γ precursors. Concurrently, anti-TL1A (R&D Systems, cat. 1319-TL-010, 0.4 μg/mL) was added to the culture medium to inhibit TL1A signaling.
For the co-culture with primary keratinocytes and neutrophils, full-length IL-36γ (Sino, cat. 10124-HNAE, 100 ng/mL) was consistently included in the culture medium to ensure an adequate supply of precursors for neutrophils to cleave. Additionally, anti-TL1A (0.04 μg/mL or 0.4 μg/mL) was added immediately after initiating co-culture with neutrophils (2 × 106 per well). This co-culture system was also sustained for 20 h.
RNA extraction and qRT-PCR
After co-culture for 20 h, keratinocytes (both primary keratinocytes and N/TERTs) were then harvested for RNA extraction. RNAs were isolated from cell cultures using the RNeasy Plus kit (Qiagen, cat. 74136). Reversed transcription was performed using a High-Capacity cDNA Transcription kit (Thermo Fisher Scientific, cat. 4368813). qPCR was performed on a 7900HT Fast Real-time PCR system (Thermo Fisher Scientific) with TaqMan Universal PCR Master Mix (Thermo Fisher Scientific, cat. 4304437) using TaqMan primers (Thermo Fisher Scientific, CXCL2: Hs00601975_m1; CXCL8: Hs00174103_m1; IL36G: Hs00219742_m1; S100A8: Hs00374264_g1). RPLP0 (Thermo Fisher Scientific, Hs99999902_m1) was used as an endogenous control.
Statistical data analysis
The Wilcoxon rank-sum test (two-sided) was employed for scRNA-seq marker gene analyses and differential expression analysis. Enrichment analysis was performed using the hypergeometric test. Group comparisons were conducted using Prism software (GraphPad) with a two-tailed unpaired Student’s t test for two-group comparisons and ordinary one-way analysis of variance for comparisons across three or more groups. Data are presented as individual data points with means ± SEM. P value of <0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The scRNA-seq data generated are available in GEO under accession number GSE309097 (GEO Accession viewer). The spatial transcriptomics data generated for GPP samples are available in Zenodo (GPP Xenium). The remaining data are available within the Supplementary Information or Source data file from the corresponding author on request. Source data are provided with this paper.
References
Marrakchi, S. & Puig, L. Pathophysiology of generalized pustular psoriasis. Am. J. Clin. Dermatol. 23, 13–19 (2022).
Rivera-Díaz, R., Daudén, E., Carrascosa, J. M., Cueva, P. & Puig, L. Generalized pustular psoriasis: a review on clinical characteristics, diagnosis, and treatment. Dermatol. Ther. 13, 673–688 (2023).
Onoufriadis, A. et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am. J. Hum. Genet. 89, 432–437 (2011).
Arakawa, A., Ruzicka, T. & Prinz, J. C. Therapeutic efficacy of interleukin 12/interleukin 23 blockade in generalized pustular psoriasis regardless of IL36RN mutation status. JAMA Dermatol. 152, 825–828 (2016).
Prinz, J. et al. Chimaeric CD4 monoclonal antibody in treatment of generalised pustular psoriasis. Lancet 338, 320–321 (1991).
Arakawa, A. et al. Unopposed IL-36 activity promotes clonal CD4(+) T-cell responses with IL-17A production in generalized pustular psoriasis. J. Investig. Dermatol. 138, 1338–1347 (2018).
Swindell, W. R. et al. RNA-Seq analysis of IL-1B and IL-36 responses in epidermal keratinocytes identifies a shared MyD88-dependent gene signature. Front. Immunol. 9, 80 (2018).
Catapano, M. et al. IL-36 promotes systemic IFN-I responses in severe forms of psoriasis. J. Investig. Dermatol. 140, 816–826.e813 (2020).
Twelves, S. et al. Clinical and genetic differences between pustular psoriasis subtypes. J. Allergy Clin. Immunol. 143, 1021–1026 (2019).
Johnston, A. et al. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J. Allergy Clin. Immunol. 140, 109–120 (2017).
Marrakchi, S. et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 365, 620–628 (2011).
Bachelez, H. et al. Trial of spesolimab for generalized pustular psoriasis. N. Engl. J. Med. 385, 2431–2440 (2021).
Morita, A. et al. Efficacy and safety of subcutaneous spesolimab for the prevention of generalised pustular psoriasis flares (Effisayil 2): an international, multicentre, randomised, placebo-controlled trial. Lancet 402, 1541–1551 (2023).
Henry, C. M. et al. Neutrophil-derived proteases escalate inflammation through activation of IL-36 family cytokines. Cell Rep. 14, 708–722 (2016).
Iznardo, H. & Puig, L. Exploring the role of IL-36 cytokines as a new target in psoriatic disease. Int. J. Mol. Sci. 22, https://doi.org/10.3390/ijms22094344 (2021).
Ma, F. et al. Single-cell and spatial sequencing define processes by which keratinocytes and fibroblasts amplify inflammatory responses in psoriasis. Nat. Commun. 14, 3455 (2023).
Hoegler, K. M., John, A. M., Handler, M. Z. & Schwartz, R. A. Generalized pustular psoriasis: a review and update on treatment. J. Eur. Acad. Dermatol. Venereol. 32, 1645–1651 (2018).
Francis, L. et al. Single-cell analysis of psoriasis resolution demonstrates an inflammatory fibroblast state targeted by IL-23 blockade. Nat. Commun. 15, 913 (2024).
Ma, F. et al. Systems-based identification of the Hippo pathway for promoting fibrotic mesenchymal differentiation in systemic sclerosis. Nat. Commun. 15, 210 (2024).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821 (2019).
West, A. P. et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–480 (2011).
Yang, L. et al. Hsa_circ_0004287 inhibits macrophage-mediated inflammation in an N(6)-methyladenosine-dependent manner in atopic dermatitis and psoriasis. J. Allergy Clin. Immunol. 149, 2021–2033 (2022).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014).
Sarode, P. et al. Reprogramming of tumor-associated macrophages by targeting β-catenin/FOSL2/ARID5A signaling: a potential treatment of lung cancer. Sci. Adv. 6, eaaz6105 (2020).
Duong, E. et al. Type I interferon activates MHC class I-dressed CD11b(+) conventional dendritic cells to promote protective anti-tumor CD8(+) T cell immunity. Immunity 55, 308–323.e309 (2022).
Walker, J. G. et al. Characterisation of a dendritic cell subset in synovial tissue which strongly expresses Jak/STAT transcription factors from patients with rheumatoid arthritis. Ann. Rheum. Dis. 66, 992–999 (2007).
Chen, J. et al. CREB1-driven CXCR4(hi) neutrophils promote skin inflammation in mouse models and human patients. Nat. Commun. 14, 5894 (2023).
Wigerblad, G. & Kaplan, M. J. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat. Rev. Immunol. 23, 274–288 (2023).
Brinkmann, V. et al. Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535 (2004).
Oberst, A. et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 (2011).
Weinlich, R., Oberst, A., Beere, H. M. & Green, D. R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 18, 127–136 (2017).
Gulati, G. S. et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367, 405–411 (2020).
Haskamp, S. et al. Transcriptomes of MPO-deficient patients with generalized pustular psoriasis reveal expansion of CD4(+) Cytotoxic T cells and an involvement of the complement system. J. Investig. Dermatol. 142, 2149–2158.e2110 (2022).
Hall, J. A. et al. Transcription factor RORα enforces stability of the Th17 cell effector program by binding to a Rorc cis-regulatory element. Immunity 55, 2027–2043.e2029 (2022).
Drummond, R. A. et al. CARD9(+) microglia promote antifungal immunity via IL-1β- and CXCL1-mediated neutrophil recruitment. Nat. Immunol. 20, 559–570 (2019).
Yu, K. et al. Bacterial indole-3-lactic acid affects epithelium-macrophage crosstalk to regulate intestinal homeostasis. Proc. Natl. Acad. Sci. USA 120, e2309032120 (2023).
Hume, D. A. & MacDonald, K. P. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood 119, 1810–1820 (2012).
Guilliams, M., Thierry, G. R., Bonnardel, J. & Bajenoff, M. Establishment and maintenance of the macrophage niche. Immunity 52, 434–451 (2020).
Li, X. et al. The dynamically evolving cell states and ecosystem from benign nevi to melanoma. Clin. Cancer Res. https://doi.org/10.1158/1078-0432.ccr-24-2971 (2025).
Pan, X. et al. Tumour vasculature at single-cell resolution. Nature 632, 429–436 (2024).
Ahmad, F. et al. Emerging role of the IL-36/IL-36R axis in multiple inflammatory skin diseases. J. Investig. Dermatol. 144, 206–224 (2024).
Xu, W. D., Li, R. & Huang, A. F. Role of TL1A in inflammatory autoimmune diseases: a comprehensive review. Front. Immunol. 13, 891328 (2022).
Bamias, G. et al. Expression, localization, and functional activity of TL1A, a novel Th1-polarizing cytokine in inflammatory bowel disease. J. Immunol. 171, 4868–4874 (2003).
Dickson, M. A. et al. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell. Biol. 20, 1436–1447 (2000).
Frisch, S. M. Caspase-8: fly or die. Cancer Res. 68, 4491–4493 (2008).
Newton, K. et al. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 574, 428–431 (2019).
Lentini, G. et al. Caspase-8 inhibition improves the outcome of bacterial infections in mice by promoting neutrophil activation. Cell Rep. Med. 4, 101098 (2023).
Towne, J. E. et al. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J. Biol. Chem. 286, 42594–42602 (2011).
Bayry, J. Immunology: TL1A in the inflammatory network in autoimmune diseases. Nat. Rev. Rheumatol. 6, 67–68 (2010).
Danese, S. et al. Anti-TL1A antibody PF-06480605 safety and efficacy for ulcerative colitis: a phase 2a single-arm Study. Clin. Gastroenterol. Hepatol. 19, 2324–2332.e2326 (2021).
Chinnaiyan, A. M. et al. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science274, 990–992 (1996).
Pobezinskaya, Y. L., Choksi, S., Morgan, M. J., Cao, X. & Liu, Z. G. The adaptor protein TRADD is essential for TNF-like ligand 1A/death receptor 3 signaling. J. Immunol. 186, 5212–5216 (2011).
Migone, T. S. et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity 16, 479–492 (2002).
Bamias, G. et al. Upregulation and nuclear localization of TNF-like cytokine 1A (TL1A) and its receptors DR3 and DcR3 in psoriatic skin lesions. Exp. Dermatol. 20, 725–731 (2011).
Bamias, G., Menghini, P., Pizarro, T. T. & Cominelli, F. Targeting TL1A and DR3: the new frontier of anti-cytokine therapy in IBD. Gut 74, 652–668 (2025).
McGuire, J. & Arnesen, S. Control of keratinocyte division in vitro. J. Investig. Dermatol. 59, 84–90 (1972).
Pulido-Pérez, A. & Bergon-Sendin, M. Sweet’s syndrome. N. Engl. J. Med. 382, 1543 (2020).
Sugiura, K. The genetic background of generalized pustular psoriasis: IL36RN mutations and CARD14 gain-of-function variants. J. Dermatol. Sci. 74, 187–192 (2014).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Sica, A. & Mantovani, A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Investig. 122, 787–795 (2012).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Corridoni, D. et al. Single-cell atlas of colonic CD8(+) T cells in ulcerative colitis. Nat. Med. 26, 1480–1490 (2020).
Billi, A. C. et al. Nonlesional lupus skin contributes to inflammatory education of myeloid cells and primes for cutaneous inflammation. Sci. Transl. Med. 14, eabn2263 (2022).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genom. Biol. 15, 550 (2014).
Jin, S. et al. Inference and analysis of cell–cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020).
Janesick, A. et al. High-resolution mapping of the tumor microenvironment using integrated single-cell, spatial and in situ analysis. Nat. Commun. 14, 8353 (2023).
Acknowledgements
This work was supported by a non-restricted research grant from Boehringer Ingelheim (J.E.G.), the National Institutes of Health (K01AR072129 and R01AR080662 to L.C.T.; 1P30AR075043 to L.C.T., M.T.P., and J.E.G.; UC2 AR081033 to L.C.T. and J.E.G.). Sinocare Diabetes Foundation Scholarship for the Michigan-Xiangya MD/PhD Dual Degree Program (awarded to R.J.).
Author information
Authors and Affiliations
Contributions
Conceptualization: R.J. and J.E.G.; Methodology: R.J., J.K., J.F., J.W., R.B., M.K.S., S.S., and J.E.G.; Imaging: X.X. and R.J.; Investigation: R.J., J.K., J.F., X.X., J.W., M.K.S., R.B., T.D., A.C., C.C., O.P., J.E.R., H.Z., J.M.K., P.W.H., H.B., L.C.T., S.S., and A.C.B.; Writing: R.J. and J.E.G.; Funding Acquisition: L.C.T. and J.E.G. Supervision: H.B., X.C., and J.E.G.
Corresponding authors
Ethics declarations
Competing interests
This was an independent, investigator-initiated study supported by Boehringer Ingelheim Pharmaceuticals, Inc. (BIPI). BIPI had no role in the design, analysis or interpretation of the results in this study. BIPI was given the opportunity to review the manuscript for medical and scientific accuracy as it relates to BIPI substances, as well as intellectual property considerations. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mayumi Komine, Shigetoshi Sano and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jiang, R., Kirma, J., Fox, J. et al. Dynamic neutrophil-keratinocyte communication network centered on IL-36/TNFSF15 responses characterizes inflammatory responses in generalized pustular psoriasis. Nat Commun 17, 1156 (2026). https://doi.org/10.1038/s41467-025-67917-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67917-9
