
Abstract
Enantioselective dicarbofunctionalization of alkenes is a powerful strategy for constructing functionalized sp³-rich molecules, yet it remains challenging for unactivated substrates lacking directing groups. While asymmetric multicomponent reactions catalyzed by d¹⁰ transition metals have advanced for activated alkenes, enantioselective multicomponent cross-coupling of unactivated alkenes, particularly enabling remote functionalization to stereoselectively generate the nonadjacent stereocenters, is still underdeveloped. Herein, we report a palladium-catalyzed asymmetric migratory dicarbofunctionalization of directing-group-free, trisubstituted unactivated alkenes. This method forges remote stereogenic centers, enabling both 1,3-diarylation and 1,4-diarylation with high enantioselectivity and diastereoselectivity. Mechanistic studies indicate a chain-walking process involving irreversible Pd-H migration, rationalizing the observed regiocontrol.
Similar content being viewed by others
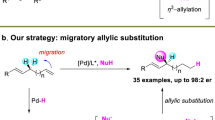
Introduction
Olefins are indispensable feedstocks for bulk chemicals, bioactive molecules, and functional materials, making the development of highly selective alkene transformations a central pursuit in organic synthesis. Among these, the stereoselective conversion of planar alkenes into diverse, highly functionalized three-dimensional sp³-hybridized molecules is particularly valuable. This is underscored by the well-established benefit of increased sp³-character in organic molecules for the success of clinical drug candidates1. Since Sigman’s foundational work2,3,4, asymmetric multicomponent cross-coupling reactions of alkenes catalyzed by d¹⁰ transition metals–specifically, dicarbofunctionalization–have emerged as a powerful synthetic strategy5,6,7,8,9,10,11,12,13,14,15,16. This approach enables the simultaneous incorporation of two distinct carbon fragments across the π-bond, facilitating rapid assembly of molecular complexity (Fig. 1). Prior efforts focused predominantly on reactive alkenes, such as acrylates, enamides, or styrenes2,17,18,19,20,21,22,23,24,25,26,27,28,29,30. In this realm, these functional groups regulate regioselectivity, also enhance reactivity, thus enabling facile migratory insertion of carbon-metal bonds or carbon-radical addition. However, enantioselective migratory dicarbofunctionalization of activated alkenes still remains scarce; only two cases include Sigman’s 1,1-diarylation of acrylates and our 1,3-diarylation of enamides17,31,32 (Fig. 1a, top). Despite these impressive advances, stereoselective intermolecular dicarbofunctionalization of unactivated alkenes remains largely underdeveloped, yet is highly desirable in organic synthesis13,14,15,16.

a Enantioselective Dicarbofunctionalization (DCF) of Acyclic Alkenes. b Enantioselective 1,3 and 1,4-Diarylation of Unactivated Trisubstituted Acyclic Alkenes (this work).
To accelerate the reactivity of unactivated alkenes, chelation-assisted strategies have been elegantly developed for chiral nickel-catalyzed 1,2-dicarbofunctionalization of unbiased alkenes33,34,35,36,37,38 (Fig. 1a, bottom). Representative examples include the Chu group’s enantioselective reductive 1,2-alkyl-alkenylation via five-membered nickellacycle formation33,34, the Engle group’s redox-neutral 1,2-diarylation, as well as Chen group’s reductive 1,2-diarylation through stereoselective migratory insertion35,36,37. However, enantioselective dicarbofunctionalization of the simple unactivated alkenes persisted as a formidable unmet challenge–with only one isolated example that the collaborative work between Koh and Shi presented the asymmetric 1,2-dicarbofunctionalization of light alkenes enabled by bulky chiral N-heterocyclic carbene-ligated nickel catalysis38. Beyond these advances, remote functionalization of unactivated alkenes is a distinct challenge, where successful examples could enable unconventional C–C bond formation at distant sites39,40,41,42,43,44,45,46,47,48,49,50,51. Notably, while several racemic 1,3-dicarbofunctionalization via nickel catalysis have been independently reported by Giri52,53, Zhao54, Martin55, and Shu group56, the asymmetric versions remained elusive, representing a critical synthetic gap.
Based on our continued research interest in d¹⁰ transition metal-catalyzed asymmetric dicarbofunctionalization17,18,57,58,59,60,61,62,63,64, we herein address these limitations by achieving the Pd-catalyzed asymmetric migratory dicarbofunctionalization of unactivated internal alkenes (Fig. 1b). This method accomplishes the enantioselective 1,3- and 1,4-dicarbofunctionalization of unactivated alkenes, forging remote stereogenic centers with high enantioselectivity; moreover, the broad reactivity without pre-installed directing group can overcome fundamental substrate constraints in alkene difunctionalization. This strategy enables expedient construction of 1,3- and 1,4-nonadjacent stereocenters with high enantioselectivity and diastereoselectivity, addressing a long-standing synthetic challenge in organic chemistry65,66,67,68,69,70. Mechanistic studies confirm a chain-walking process involving irreversible Pd-H migration, rationalizing the observed regiocontrol.
Results and discussion
Reaction optimization for diarylation of alkenes
Initial optimization of asymmetric remote dicarbofunctionalization employed non-activated trisubstituted alkene 1a, aryl diazonium salt 2a, and arylboronic acid 3a as model substrates with 5.0 mol% Pd(OAc)₂, 10 mol% (S)-Cy-Box (L1) as chiral ligand, and 16 mol% diethyl fumarate as achiral ligand in presence of 1.0 equivalent Ag2CO3 (Table 1). Pleasingly, this ligand-swap strategy proved effective for palladium-catalyzed 1,3-diarylation, affording the desired product 4a in 48% yield with 88% ee in DME (Table 1, entry 1). It should be noted that the addition of Ag₂CO₃ facilitates the transmetalation step18. Reactions in 1,4-dioxane showed comparable yield and marginally improved enantioselectivity (Table 1, entry 2), prompting further investigation of mixed DME/1,4-dioxane solvent ratios. Notably, a 1:3 DME/1,4-dioxane mixture delivered 4a in 57% yield and 92% ee (Table 1, entries 3–5). Subsequent screening of various substituted chiral BiOx ligands (Table 1, entries 6–10) revealed minimal impact on yield and enantioselectivity, with the isopropyl-substituted BiOx ligand L3 proving optimal, delivering 4a in 62% yield and 93% ee (Table 1, entry 7). Given the importance of additives in promoting efficient cross-coupling by accelerating reductive elimination, several fumarate diesters with different substituents (isopropyl A2, isobutyl A3, tert-butyl A4, phenyl A5, methyl A6) were evaluated (Table 1, entries 11-15). Diphenyl fumarate (A5) successfully facilitated the reaction, maintaining a slightly higher yield and similar enantioselectivity. Further optimization of the palladium catalyst identified [Pd(allyl)Cl]2 as optimal, furnishing 4a in 79% isolated yield with 93% ee (Table 1, entry 16). The current three-component cross-coupling protocol was not suitable for aryl halides as electrophiles.
Substrate scope of unactivated trisubstituted alkenes
With optimized conditions established, we evaluated the scope of unactivated trisubstituted alkenes (Fig. 2). Symmetric alkenes bearing a benzyl substituent were systematically investigated. Both electron-withdrawing and electron-donating para-substituted phenyl groups at the terminal site afforded products 4b–4e in moderate to good yields (61–72%) with excellent enantioselectivities (91–94% ee). The reaction tolerated a meta-fluoro-substituted phenyl counterpart, providing 4f in 40% yield and 91% ee. ortho-Substituted alkenes proved particularly effective, yielding 4g in 74% yield and 97% ee. Notably, an alkene bearing an acetal group was efficiently converted to the ketone-containing product 4h (55%, 94% ee), demonstrating direct ketone formation and expanding synthetic utility. Intriguingly, an unactivated alkene bearing methyl and ethyl substituents delivered the 1,3-diarylation product 4i bearing nonadjacent stereocenters in 44% isolated yield, 91% ee and >20:1 dr under minimally modified solvent conditions, highlighting the stereoselective Pd-H migration pathway. A trifluoromethyl-substituted alkene at the terminal position of a linear alkyl chain afforded 4j (43%, 91% ee). Diverse aromatic substituents, including methoxy (4k) and benzodioxole (4l) provided products in moderate yields with good stereoselectivity. It should be noted that all 1,3-diarylation proceeded with high stereoselectivity, with no diastereomers observed in the reaction mixture. However, the migratory diarylation of a linear 1,2-disubstituted alkene analog afforded the desired product only in very low yield, and the reaction failed entirely with fully substituted alkenes under the standard conditions. Further efforts will be devoted to addressing this limitation.

Reaction conditions: alkene 1 (1.0 equiv, 0.20 mmol), aryl diazonium salt 2a (2.0 equiv, 0.40 mmol), arylboronic acid 3a (3.0 equiv, 0.60 mmol), [Pd(allyl)Cl]2 (2.5 mol %, 0.005 mmol), L3 (10 mol %, 0.02 mmol), A5 (16 mol %, 0.032 mmol), and Ag2CO3 (1.0 equiv, 0.20 mmol) in DME/1,4-Dioxane (0.4 mL (v/v = 1/3), 0.5 M) at 25 °C. Isolated yields were reported, and the ee values were determined by chiral HPLC. aDCM/1,4-Dioxane (0.4 mL (v/v = 1/3), 0.5 M) was used.
Substrate scope of aryldiazonium salts and arylboronic acids
Subsequently, we examined aryl diazonium salts and arylboronic acids, evaluating electronic effects and substituent positions (Fig. 3). para-Functionalized aryl diazonium salts afforded products 4m–4o in 45–62% yield with 90–94% ee. Naphthalene-derived (4p) and benzodioxole-derived (4q) diazonium salts gave good yields and enantioselectivities. Diazonium salts bearing meta-substituents (–Cl, –OBn, –OEt; 4r–4t) delivered products with excellent enantiocontrol (up to 94% ee). Arylboronic acids with diverse para-substituents (–tBu 4u, –OMe 4v, –TMS 4w, –CF3 4x) provided target products in high yields (up to 85%) with 94% ee. meta-Substituted arylboronic acids (4y–4ab) exhibited excellent enantioselectivities (up to 95% ee). Sterically encumbered biphenyl-2-ylboronic acid afforded 4ac in 63% yield and 96% ee. Electron-withdrawing substituents on the boronic acid (4ad) did not diminish stereoselectivity. The configuration of 4ae was unambiguously confirmed by X-ray crystallography, demonstrating stereoselective Pd-H chain walking process.

Reaction conditions: alkene 1 (1.0 equiv, 0.20 mmol), aryl diazonium salt 2 (2.0 equiv, 0.40 mmol), arylboronic acid 3 (3.0 equiv, 0.60 mmol), [Pd(allyl)Cl]2 (2.5 mol %, 0.005 mmol), L3 (10 mol %, 0.02 mmol), A5 (16 mol %, 0.032 mmol), and Ag2CO3 (1.0 equiv, 0.20 mmol) in DME/1,4-Dioxane (0.4 mL (v/v = 1/3), 0.5 M) at 25 oC. Isolated yields were reported and the ee values were determined by chiral HPLC. aDCM/1,4-Dioxane (0.4 mL (v/v = 1/3), 0.5 M) was used.
Beyond controlling stereoselectivity at the C-3 position, we investigated whether this migratory 1,n-diarylation strategy could extend to longer distances. Sigman and coworkers demonstrated that Pd–H species undergo stereoselective reinsertion into alkene intermediates in asymmetric remote Heck reactions4, a similar reactivity pattern also observed in our prior Pd-catalyzed diastereoselective 1,1-diarylation of 1,1-arylethylenes62. Applying standard conditions to unactivated trisubstituted alkene bearing a phenethyl substituent 5 smoothly delivered the 1,4-diarylation product 6a in 52% yield (Fig. 4). Notably, enantioselectivity decreased relative to the 1,3-diarylation (83% ee vs. 93% ee for 4a). Further optimization of chiral ligands, solvents, and palladium catalysts failed to improve enantiocontrol. This result suggests partial dissociation of the Pd–H species from the alkene intermediate occurs during extended migration. The reaction tolerated diverse aryl boronic acids, affording products 6b–6i in 44–62% yield while establishing a new stereocenter at the C-4 position with 78–86% ee. It was noteworthy that this 1,4-diarylation also proceeds smoothly for the unsymmetric trisubstituted alkenes, delivering the desired product 6j with 89% ee and more than 20:1 dr. The presence of an aromatic ring at the alkene terminus is therefore critical for the observed 1,3- and 1,4-diarylation, an effect we attribute to the stabilization of a key benzylic palladium intermediate.

Reaction conditions: alkene 5 (1.0 equiv, 0.20 mmol), aryl diazonium salt 2a (2.0 equiv, 0.40 mmol), arylboronic acid 3 (3.0 equiv, 0.60 mmol), [Pd(allyl)Cl]2 (2.5 mol %, 0.005 mmol), L3 (10 mol %, 0.02 mmol), A5 (16 mol %, 0.032 mmol), and Ag2CO3 (1.0 equiv, 0.20 mmol) in DME/1,4-Dioxane (0.4 mL (v/v = 1/3), 0.5 M) at 25 °C. Isolated yields were reported, and the ee values were determined by chiral HPLC.
Mechanistic investigation
To further elucidate the impact of alkyl chain length on the stereoselective chain-walking process and the reversibility of Pd-H migration, preliminary mechanistic investigations were conducted (Fig. 5). While remote 1,n-diarylation of unactivated alkenes yielded products 8, 10, 12 in moderate isolated yields (45–57%), enantioselectivity declined significantly: 1,5-diarylation afforded product 8 in 26% ee, 1,6-diarylation yielded product 10 in 8% ee, and near-racemization occurred in 1,7-diarylation with only 4% ee (Fig. 5a). This erosion in enantioselectivity suggests insufficient weak interactions, such as potential coordination between phenyl ring at the terminal site of the unfunctionalized alkene intermediate with the palladium center during the long distance migration process such as 1,6- and 1,7-diarylations71, leading to undesired Pd-H dissociation. While the undesired dissociation rate maybe slower than the re-insertion of Pd-H intermediate in the short-distance migration, such as 1,3-diarylation and 1,4-diarylation processes. Another possibility is a geometrically defined alkene intermediate cannot be formed when the stereogenic center is far from to the carbon-palladium site that undergoing the β-H elimination.

a Effect of the alkyl chain length. b Deuterium-labeling experiment. c Cross-over experiment. d Proposed mechanism.
Deuterium-labeling experiments then probed the Pd-H chain-walking pathway: reaction of 5a-D (dideuterated at the benzylic position) gave 6a-D with >99% deuterium at C-3, and reaction of 7-D (dideuterated at C-4) resulted in regioselective deuterium migration (>99%) from C-4 to C-3, with no labeling at C-1, C-2, or C-5 (Fig. 5b). These results collectively indicate that the chain-walking migration proceeds exclusively in a net forward direction toward the reaction site (eg. from C-4 to C-5 position), without detectable reversal. This regioselective, irreversible progression supports the proposed Pd–H insertion into the transient alkene intermediate, thereby preventing deuterium scrambling, while a behavior that stands in sharp contrast to the reversible chain-walking often observed in NiH-catalyzed cross-coupling chemistry42. Furthermore, a crossover experiment between deuterated 5a-D and non-deuterated 5b in the 1,4-diarylation yielded only 6a-D and 6k, with no crossover product, demonstrating that the Pd-H species remains tightly bound to the alkene chain during 1,4-migration, thereby preserving high enantioselectivity (Fig. 5c). Based on these findings and previous results17,18, a plausible mechanism for the 1,3-diarylation is proposed (Fig. 5d). The reaction is initiated with oxidative addition of Pd(0) catalyst with the aryl diazonium salts, forming intermediate Int-A. Stereoselective migratory insertion of Int-A across the alkene C=C bond yields Int-B. Syn β-hydride elimination from Int-B then produces Int-C42. The resulting Pd–H species undergoes chain-walking migration followed by stereoselective re-insertion into the alkene, affording the stabilized benzyl palladium species Int-D72,73. Subsequent transmetalation of Int-D with an arylboronic acid assisted by ligand exchange18, followed by reductive elimination promoted by the electronically deficient diphenyl fumarate, delivers the desired migratory product 4 and regenerates the Pd(0) catalyst.
In conclusion, we have developed a palladium-catalyzed enantioselective migratory dicarbofunctionalization of unactivated internal alkenes. This method enables facile 1,3- and 1,4-diarylation by integrating aryl diazonium salts and arylboronic acids as coupling partners. Notably, 1,3- and 1,4-non-contiguous stereocenters are formed with high enantio- and diastereoselectivity. Mechanistic studies indicate the migration proceeds via irreversible Pd–H insertion. Current efforts focus on tackling the unactivated fully substituted alkenes asymmetric cross-coupling for accessing non-contiguous quaternary stereocenters.
Methods
General procedure for diarylation of alkenes
To a dried 8-mL vial was charged with [Pd(allyl)Cl]2 (2.5 mol %), L3 (10 mol %), A5 (16 mol %), Ag2CO3 (1.0 equiv), aryl diazonium salts (2.0 equiv), arylboronic acids (3.0 equiv) and alkene (1.0 equiv). After evacuated and backfilled nitrogen three times, DME/1,4-Dioxane (v/v = 1/3, 0.5 M) or DCM/1,4-Dioxane (v/v = 1/3, 0.5 M) was added. The reaction was allowed to stirred at 25 °C for 36 ~ 48 h, and then the reaction was filtered with celite and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography to yield the diarylation product.
Data availability
All data to support the conclusions are available in the main text or the supplementary materials. The X-ray Crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC) under deposition numbers CCDC 2474573 (4ae) and 2475482 (6a-D). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Data supporting the findings of this manuscript are also available from the corresponding author upon request.
References
Lovering, F., Bikker, J. & Humblet, C. Escape from flatland: in creasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756 (2009).
Stokes, B. J., Liao, L., de Andrade, A. M., Wang, Q. & Sigman, M. S. A palladium-catalyzed three-component-coupling strategy for the differential vicinal diarylation of terminal 1,3-dienes. Org. Lett. 16, 4666–4669 (2014).
Werner, E. W., Mei, T.-S., Burckle, A. J. & Sigman, M. S. Enantioselective Heck arylations of acyclic alkenyl alcohols using a redox-relay strategy. Science 338, 1455–1458 (2012).
Mei, T.-S., Patel, H. H. & Sigman, M. S. Enantioselective construction of remote quaternary stereocenters. Nature 508, 340–344 (2014).
McDonald, R. I., Liu, G. & Stahl, S. S. Palladium(II)-catalyzed alkene functionalization via nucleopalladation: stereochemical pathways and enantioselective catalytic applications. Chem. Rev. 111, 2981–3019 (2011).
Giri, R. & Kc, S. Strategies toward dicarbofunctionalization of unactivated olefins by combined Heck carbometalation and cross-coupling. J. Org. Chem. 83, 3013–3022 (2018).
Kang, T., Apolinar, O. & Engle, K. M. Ni- and Pd-catalyzed enantioselective 1,2-dicarbofunctionalization of alkenes. Synthesis 56, 1–15 (2024).
Qi, X. & Diao, T. Nickel-catalyzed dicarbofunctionalization of alkenes. ACS Catal. 10, 8542–8556 (2020).
Luo, Y.-C., Xu, C. & Zhang, X. Nickel-catalyzed dicarbofunctionalization of alkenes. Chin. J. Chem. 38, 1371–1394 (2020).
Zhu, S., Zhao, X., Li, H. & Chu, L. Catalytic three-component dicarbofunctionalization reactions involving radical capture by nickel. Chem. Soc. Rev. 50, 10836–10856 (2021).
Li, Z.-L., Fang, G.-C., Gu, Q.-S. & Liu, X.-Y. Recent advances in copper-catalysed radical-involved asymmetric 1,2-difunctionalization of alkenes. Chem. Soc. Rev. 49, 32–48 (2020).
Xu, B., Wang, Q., Fang, C., Zhang, Z.-M. & Zhang, J. Recent advances in Pd-catalyzed asymmetric cyclization reactions. Chem. Soc. Rev. 53, 883–971 (2024).
Coombs, J. R. & Morken, J. P. Catalytic enantioselective functionalization of unactivated terminal alkenes. Angew. Chem. Int. Ed. 55, 2636–2649 (2016).
Dhungana, R. K., KC, S., Basnet, P. & Giri, R. Transition metal-catalyzed dicarbofunctionalization of unactivated olefins. Chem. Rec. 18, 1314–1340 (2018).
Wang, H. & Koh, M. J. Directing group-free approaches for three-component catalytic dicarbofunctionalization of unactivated alkenes. Cell Rep. Phys. Sci. 3, 100901 (2022).
Han, J., He, R. & Wang, C. Transition metal-catalyzed asymmetric three-component dicarbofunctionalization of unactivated alkenes. Chem. Catal. 3, 100690 (2023).
Xi, Y. et al. Catalytic asymmetric diarylation of internal acyclic styrenes and enamides. J. Am. Chem. Soc. 144, 8389–8398 (2022).
Huang, W. et al. Palladium-catalyzed enantioselective multi-component cross-coupling of trisubstituted olefins. J. Am. Chem. Soc. 146, 16892–16901 (2024).
Gu, J. W., Min, Q.-Q., Yu, L.-C. & Zhang, X. Tandem difluoro-alkylation-arylation of enamides catalyzed by nickel. Angew. Chem. Int. Ed. 55, 12270–12274 (2016).
Chierchia, M., Xu, P., Lovinger, G. J. & Morken, J. P. Enantioselective radical addition/cross-coupling of organozinc reagents, alkyl iodides, and alkenyl boron reagents. Angew. Chem. Int. Ed. 58, 14245–14249 (2019).
Anthony, D., Lin, Q., Baudet, J. & Diao, T. Nickel-catalyzed asymmetric reductive diarylation of vinylarenes. Angew. Chem. Int. Ed. 58, 3198–3202 (2019).
Wei, X., Shu, W., García-Domínguez, A., Merino, E. & Nevado, C. Asymmetric Ni-catalyzed radical relayed reductive coupling. J. Am. Chem. Soc. 142, 13515–13522 (2020).
Guo, L. et al. General method for enantioselective three-component carboarylation of alkenes enabled by visible-light dual photoredox/nickel catalysis. J. Am. Chem. Soc. 142, 20390–20399 (2020).
Liu, C.-F. et al. Synthesis of tri- and tetrasubstituted stereocenters by nickel-catalyzed enantioselective olefin cross-couplings. Nat. Catal. 5, 934–942 (2022).
Li, X. et al. Three-component enantioselective alkenylation of organophosphonates via nickel metallaphotoredox catalysis. Chem 9, 154–169 (2023).
Wang, Y.-Z. et al. Enantioselective reductive cross-couplings of olefins by merging electrochemistry with nickel catalysis. J. Am. Chem. Soc. 145, 23910–23917 (2023).
Hu, X., Cheng-Sánchez, l., Kong, W., Molander, G. A. & Nevado, C. C. et al. Nickel-catalysed enantioselective alkene dicarbofunctionalization enabled by photochemical aliphatic C–H bond activation. Nat. Catal. 7, 655–665 (2024).
Li, X. et al. Metallaphotoredox-catalyzed three-component asymmetric cross electrophile coupling for chiral boronate synthesis. ACS. Catal. 14, 15790–15798 (2024).
Ye, F., Xu, Y., Zheng, S., Huang, G. & Yuan, W. Enantioselective synthesis of chiral γ-amino acid esters via photoredox/nickel-catalyzed aryl-aminoalkylation of alkenes. Chin. J. Chem. 43, 1862–1868 (2025).
Gao, X. et al. Nickel/photoredox-catalyzed asymmetric three-component cross coupling to access enantioenriched 1,1-diaryl(heteroaryl)alkanes. Org. Lett. 26, 8792–8797 (2024).
Yamamoto, E. et al. Development and analysis of a Pd(0)-catalyzed enantioselective 1,1-diarylation of acrylates enabled by chiral anion phase transfer. J. Am. Chem. Soc. 138, 15877–15880 (2016).
Orlandi, M., Hilton, M. J., Yamamoto, E., Toste, F. D. & Sigman, M. S. Mechanistic investigations of the Pd(0)-catalyzed enantioselective 1,1-diarylation of benzyl acrylates. J. Am. Chem. Soc. 139, 12688–12695 (2017).
Tu, H.-Y. et al. Enantioselective three-component fluoroalkylarylation of unactivated olefins through nickel-catalyzed cross-electrophile coupling. J. Am. Chem. Soc. 142, 9604–9611 (2020).
Wang, F., Pan, S., Zhu, S. & Chu, L. Selective three-component reductive alkylalkenylation of unbiased alkenes via carbonyl-directed nickel catalysis. ACS Catal. 12, 9779–9789 (2022).
Apolinar, O. et al. Three-Component asymmetric Ni-catalyzed 1,2-dicarbofunctionalization of unactivated alkenes via stereoselective migratory insertion. J. Am. Chem. Soc. 144, 19337–19343 (2022).
Dong, Z. et al. Directed asymmetric nickel-catalyzed reductive 1,2-diarylation of electronically unactivated alkenes. Angew. Chem. Int. Ed. 135, e202218286 (2023).
Dong, Z. et al. Enantioselective directed nickel-catalyzed three-component reductive arylalkylation of alkenes via the carbometalation/radical cross-coupling Sequence. ACS Catal. 14, 4395–4406 (2024).
Wang, Z.-C. et al. Enantioselective C–C cross-coupling of unactivated alkene. Nat. Catal. 6, 1087–1097 (2023).
Vasseur, A., Bruffaerts, J. & Marek, I. Remote functionalization through alkene isomerization. Nat. Chem. 8, 209–219 (2016).
Li, Y., Wu, D., Cheng, H.-G. & Yin, G. Difunctionalization of alkenes involving metal migration. Angew. Chem. Int. Ed. 59, 7990–8003 (2020).
Dhungana, R. K., Sapkota, R. R., Niroula, D. & Giri, R. Walking metals: catalytic difunctionalization of alkenes at nonclassical sites. Chem. Sci. 11, 9757–9774 (2020).
Wang, Y., He, Y. & Zhu, S. NiH-catalyzed functionalization of remote and proximal olefins: new reactions and innovative strategies. Acc. Chem. Res. 55, 3519–3536 (2022).
Rodrigalvarez, J., Haut, F.-L. & Martin, R. Regiodivergent sp3 C−H functionalization via Ni-catalyzed chain-walking reactions. JACS Au 3, 3270–3282 (2023).
Han, C. Palladium-catalyzed remote 1,n-arylamination of unactivated terminal alkenes. ACS Catal. 9, 4196–4202 (2019).
Hamasaki, T., Aoyama, Y., Kawasaki, J., Kakiuchi, F. & Kochi, T. Chain walking as a strategy for carbon−carbon bond formation at unreactive sites in organic synthesis: catalytic cycloisomerization of various 1,n-dienes. J. Am. Chem. Soc. 137, 16163–16171 (2015).
Lin, L., Romano, C. & Mazet, C. Palladium-catalyzed long-range deconjugative isomerization of highly substituted α,β-unsaturated carbonyl compounds. J. Am. Chem. Soc. 138, 10344–10350 (2016).
Kohler, D. G., Gockel, S. N., Kennemur, J. L., Waller, P. J. & Hull, K. L. Palladium-catalysed anti-markovnikov selective oxidative amination. Nat. Chem. 10, 333–340 (2018).
Li, X. et al. Regio- and enantioselective remote dioxygenation of internal alkenes. Nat. Chem. 15, 862–871 (2023).
Chen, X.-X., Luo, H., Chen, Y.-W., Liu, Y. & He, Z.-T. Enantioselective palladium-catalyzed directed migratory allylation of remote dienes. Angew. Chem. Int. Ed. 62, e20230726 (2023).
Wang, Z., Zhu, J., Wang, M. & Lu, P. Palladium-catalyzed divergent enantioselective functionalization of cyclobutenes. J. Am. Chem. Soc. 146, 12691–12701 (2024).
Wang, Y.-C., Liu, J.-B. & He, Z.-T. Palladium-catalyzed asymmetric hydrofunctionalizations of conjugated dienes. Chin. J. Org. Chem. 43, 2614–2627 (2023).
Basnet, P. et al. Ni-catalyzed regioselective β,δ-diarylation of unactivated olefins in ketimines via ligand-enabled contraction of transient nickellacycles: rapid access to remotely diarylated ketones. J. Am. Chem. Soc. 140, 7782–7786 (2018).
Dhungana, R. et al. Ni(I)-catalyzed β,δ-vinylarylation of γ,δ-alkenyl α-cyanocarboxylic esters via contraction of transient nickellacycles. ACS Catal. 9, 10887–10893 (2019).
Li, W., Boon, J. & Zhao, Y. Nickel-catalyzed difunctionalization of allyl moieties using organoboronic acids and halides with divergent regioselectivities. Chem. Sci. 9, 600–607 (2018).
Liu, Z., D’Amico, F. & Martin, R. Regiodivergent radical-relay alkene dicarbofunctionalization. J. Am. Chem. Soc. 146, 28624–28629 (2024).
Meng, H. et al. Ni-catalyzed regioselective and site-divergent reductive arylalkylations of allylic amines. Chem. Sci. 16, 4442–4449 (2025).
Wu, X., Qu, J. & Chen, Y. Quinim: A new ligand scaffold enables nickel-catalyzed enantioselective synthesis of α-alkylated γ-lactam. J. Am. Chem. Soc. 142, 15654–15660 (2020).
Wu, X. et al. Catalytic desymmetric dicarbofunctionalization of unactivated alkenes. Angew. Chem. Int. Ed. 61, e202111598 (2022).
Wu, X. et al. Nickel-catalyzed enantioselective reductive alkyl-carbamoylation of internal alkenes. Angew. Chem. Int. Ed. 61, e202207536 (2022).
Wu, X., Li, H., He, F., Qu, J. & Chen, Y. Nickel/Quinim enabled asymmetric carbamoyl-acylation of unactivated alkenes. Chin. J. Chem. 41, 1673–1678 (2023).
Xi, Y., Wang, C., Zhang, Q., Qu, J. & Chen, Y. Palladium-catalyzed regio-, diastereo-, and enantioselective 1,2-arylfluorination of internal enamides. Angew. Chem. Int. Ed. 60, 2699–2703 (2021).
Zhang, C., Xi, Y., Qu, J. & Chen, Y. Pd-catalyzed diastereoselective 1,1-diarylation of 1,1-disubstituted alkenes enabling the modular synthesis of 1,1,2,2-tetraarylethanes. Sci. China Chem. 66, 3539–3545 (2023).
Wang, C., Xi, Y., Xia, T., Qu, J. & Chen, Y. Pd(0)-catalyzed diastereoselective and enantioselective intermolecular Heck–Miyaura borylation of internal enamides for the β-aminoboronate ester synthesis. ACS Catal. 14, 418–425 (2024).
Xi, Y. et al. Palladium-catalyzed intermolecular asymmetric dearomatizative arylation of indoles and benzofurans. Sci. Adv. 11, eadw4471 (2025).
Ji, C.-L., Zou, X.-Z. & Gao, D.-W. Catalytic asymmetric construction of nonadjacent stereoelements. Angew. Chem. Int. Ed. 64, e202504224 (2025).
Kong, M. M. et al. Catalytic reductive cross coupling and enantioselective protonation of olefins to construct remote stereocenters for azaarenes. J. Am. Chem. Soc. 143, 4024–4031 (2021).
Deng, Y. et al. One-step asymmetric construction of 1,4-stereocenters via tandem Mannich-isomerization reactions mediated by a dual-functional betaine catalyst. JACS Au 2, 2678–2685 (2022).
Chang, X. et al. Stereodivergent construction of 1,4-nonadjacent stereocenters via hydroalkylation of racemic allylic alcohols enabled by copper/ruthenium relay catalysis. Angew. Chem. Int. Ed. 61, e202206517 (2022).
Zhang, J. et al. Synergistic Pd/Cu-catalyzed 1,5-double chiral inductions. J. Am. Chem. Soc. 146, 9241–9251 (2024).
Zhao, W., Lin, E.-Z., Chen, K.-Z., Sun, Y.-W. & Li, B.-J. Diastereo- and enantioselective 1,3-hydrofunctionalization of trisubstituted alkenes by a directing relay. Nat. Chem. https://doi.org/10.1038/s41557-025-01936-3 (2025).
Catellani, M. et al. Palladium−Arene interactions in catalytic intermediates: an experimental and theoretical investigation of the soft rearrangement between η1 and η2 coordination modes. J. Am. Chem. Soc. 124, 4336–4346 (2002).
Muto, K., Hatanaka, M., Kakiuchi, F. & Kochi, T. Conformational isomerization as a process to determine selectivity over reaction pathways: effect of alkene rotation in chain walking via cis alkene intermediates. J. Org. Chem. 89, 4712–4721 (2024).
Zhang, S., Yamamoto, Y. & Bao, M. Benzyl palladium intermediates: unique and versatile reactive intermediates for aromatic functionalization. Adv. Synth. Catal. 363, 587–601 (2021).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22371071, 22571081, 92356301, Y.C.), Science and Technology Commission of Shanghai Municipality (grant No.24DX1400200, Y.C.), the Program of Introducing Talents of Discipline to Universities (B16017, Y.C.), the China Postdoctoral Science Foundation (2024M750901, 2023TQ0118, Y.X.), Postdoctoral Fellowship Program of CPSF (GZB20230212, Y.X.), and the Fundamental Research Funds for the Central Universities. We thank the Analysis and Testing Center of East China University of Science and Technology for help with NMR and HRMS analysis.
Author information
Authors and Affiliations
Contributions
Y.C. conceived the projects. L.F., Y.X., H.G., and W.H. performed the experiments under the supervision of W.-H. Z., J.Q., and Y.C. Y.X. and Y.C. wrote the manuscript with the feedback of all other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jiuzhong Huang and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fan, L., Xi, Y., Gu, H. et al. Palladium-catalyzed asymmetric migratory diarylation of unactivated directing-group-free internal alkenes. Nat Commun 17, 1247 (2026). https://doi.org/10.1038/s41467-025-68006-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68006-7
