
Abstract
Biogas upgrading to pipeline-grade methane (>95% CH4) represents a critical pathway for enhancing the utilization and value of biogas. Direct methanation of biogas is promising, but due to the performance limitations of conventional nickel-based methanation catalysts, reactant circulating or multi-stage process is required as engineering compensation. Here, through process simulation and economic analysis, we demonstrate substantial economic benefits of the single-stage methanation process over conventional routes in terms of both capital investment and operation costs. A structured inverse catalyst with CeZrOx/Ni inverse configuration grown on porous Ni-foam featuring specific surface area and water removal enhancements over powder inverse catalyst is developed and realizes scale-up. The remarkable low-temperature activity of the structured inverse catalysts achieves a near-equilibrium conversion of 97.8% CO2 in biogas, allowing the production of pipeline-grade methane through the single-stage biogas methanation process. With the innovation of catalysts and process, the price of low-carbon methane produced from biogas with industrial by-product hydrogen is competitive with natural gas.
Similar content being viewed by others

Introduction
Biogas, an anaerobic fermentation product of organic matter, is generally considered as an efficient localized renewable energy source supporting community need of cooking and electricity1,2,3. It is one of the potential solutions dealing with two critical challenges of modern society, reducing the increasing organic wastes and greenhouse gas (GHG) emission4,5. According to the International Energy Agency (IEA)6, the biogas potential reached 560 million tons of oil equivalent (Mtoe) in 2018, which equals to 15% of natural gas consumption annually used by human beings7. The efficient utilization of biogas can reduce over 1 billion tons of GHG emission in 2040 under optimistically scenario8,9. However, biogas contains only 50-70% of methane. Upgrading it to pipeline-grade natural gas by removing the impurities (mainly CO2), is necessary for the biogas to raise the quality and gain access to transport via the national grid or international natural gas networks (Fig. 1a)10,11.

a Schematic illustration of biogas upgrading methods for the production of Pipeline-grade methane; b The process diagrams illustrating the single-stage biogas methanation process (iii. SS) and conventional processes with single-stage biogas circulation (i. SSC) and multiple-stage with intermediate water removal (ii. MS); c Economic analysis of three typical processes on fixed capital investment (FCI) and operation & maintenance costs (excluding the cost of raw materials).
Currently, the methane production demonstration from biogas typically involves a physical separation (PSA/membrane) step or chemical absorption processes to remove CO2 from biogas12,13. However, this approach often results in a waste of concentrated CO2 (over 30 vol%) unless an additional utilization process is implemented14. An alternative strategy, direct methanation of biogas15,16,17, integrates biogas upgrading with methanation process to maximize the utilization of CO2 and nearly double the methane production (Fig. 1b)18,19,20. Pipeline-grade natural gas which has high standard on the purity, generally requires the concentration of CH4 and H2 higher than 95% and lower than 5%21,22, respectively. Therefore, the direct methanation process of biogas has to be performed with a near equilibrium CO2 conversion higher than 97.3%, 97.8% and 98.2% for 40%CO2, 50%CO2 and 60% CO2 in biogas respectively in order to reach the pipeline-grade (stoichiometric CO2/H2 = 1/4 as an example composition). Considering the reducing thermodynamic conversion of the methanation reaction with elevated temperature, single-stage methanation reactor must operate temperature below 242, 278 and 337 °C at 1 bar, 5 bar and 15 bar (Supplementary Fig. 1). A major challenge in single-stage direct biogas methanation is the poor low-temperature activity of conventional Sabatier catalysts, often leading to limited CO2 conversion (<90%) and consequently a substandard CH4 purity of 70-85%, which is below pipeline-grade specifications23,24,25,26. Due to the limitation of catalysts, current commercial demonstrations and process optimization studies of the direct biogas methanation usually conducted at high temperature of 350-500 °C, and thus employ engineering approaches such as single-stage circulation process (SSC, Fig. 1b, pathway-i)27 or multi-stage CO2 methanation reactors (MS, Fig. 1b, pathway-ii)28 with intermediate water removal step for compensation. Although effective, these complex designs increase the capital and operating costs of methane production, which introduce high economic barrier to widespread adoption in the underdeveloped regions with abundant biogas resources, like rural communities, pastures and organic waste landfills etc.27,29. Therefore, addressing these limitations through advanced catalyst engineering represents the pivotal research frontier for unlocking the full potential of single-stage methanation technology.
Inverse catalysts, where oxide nanoparticles are dispersed on metallic substrates30,31,32,33 rather than adopting the conventional metal-on-oxide configuration, have recently emerged as an efficient strategy in CO2 methanation34,35. Notably, the CeZrOx/Ni35 and ZrO2/Ni (Co)34,36 inverse catalyst shows a CO2 conversion of over 90% at a low-temperature of 200 °C, which exceeds that of some active Ru-based catalyst, showing pronounced application potential for the single-stage biogas methanation process. However, the metal atoms utilization of powder inverse catalyst is not sufficient, especially after molding into pellets37,38,39. The limited heat and mass transfer efficiency will also inhibit the removal of reaction heat and product water, which is harmful for the hydrothermal stability of applied methanation catalyst. Foam-structured metal substrates have been known for its excellent heat and mass transport efficiency in industrial applications40,41,42. Therefore, constructing active species of Ni-based inverse sites on highly porous Ni foam skeleton can benefits the apparent performance and life-time of inverse catalyst in single-stage biogas upgrade process.
Here, we demonstrate that the CeZrOx/Ni/Ni-foam inverse structured catalyst enables the single-stage direct methanation process for the first time to produce pipeline-grade methane from biogas. Specifically, this innovative approach reduces fixed asset investment by 38% and 11% compared to conventional SSC and MS methanation processes, respectively. Furthermore, it delivers substantial operational cost savings of 33% and 15% relative to these established technologies, presenting a compelling case for its implementation in sustainable energy systems. The specific activity of dispersed CeZrOx/Ni inverse composites anchored on Ni-Foam skeleton is identified 18.3% higher than powdered inverse catalysts. The structured inverse catalyst also improves H2O removal compared powdered catalysts and modulates CO2/H2 coverage. By unlocking the single-stage biogas catalyst technology, the price of methane produced via biogas upgrading becomes less sensitive to the price of hydrogen, making the current direct biogas methanation process profitable using fossil fuel derived or byproduct H2 as hydrogen source. From the perspective of green hydrogen and biogas development in China in 2050, the inverse catalyst driven single-stage methanation process is able to produce zero-CO2 emission biomethane competitive with local pipeline natural gas in 14.7% area of China.
Results
Biogas upgrading and comparative economic analysis
To quantitatively assess the advantages of single-stage biogas methanation process (SS) compared with conventional methods (i) single-stage with circulation (SSC) and (ii) multi-stage reactors with intermediate water removal (MS) (Supplementary Fig. 2)26,43, an economic analysis of three biogas methanation processes (e.g., CO2:CH4 = 4:6) was conducted at a scale of 1000 Nm3/h methane plant (Fig. 1b, can be used to consume the biogas production of a full-size biogas fermentation plant). The CO2 methanation activity of commercial Ni-based catalyst (Catalyst designation: SCST-241-5Q produced by Shutai Corporation, China) was applied for the assess of SSC and MS processes (Supplementary Fig. 3, Supplementary Tables 1, 2), and the simulation for SS process was based on the ideal catalyst which can achieve desired CO2 conversion of 97.8% in single-stage process (Supplementary Table 3). The energy and process economy were simulated using the Aspen Plus and AEA44. Notably, process simulation and economy analysis results indicate that the SS process incurs a fixed capital investment (FCI) of $4.8 million/plant, $2.9 million and $0.6 million lower than that of processes SSC and MS, respectively. Furthermore, the simplified operation of the single-stage system reduces operation cost (OC) by 46.7% and 17.3% compared to SSC and MS (Fig. 1c, Supplementary Table 4) due to the compact process. The economic analysis demonstrates single-stage methanation process will emerge as a promising advantageous design in biogas upgrading, as long as the low-temperature CO2 methanation catalyst can be developed.
Catalytic performance of the structured catalysts
Constructing Ni-based inverse configurations on Ni foam skeletons is an effective method to synergistically integrate the excellent low-temperature methanation activity and enhanced heat and mass transport efficiency, which offers an opportunity to meet the restrict kinetic and engineering challenges of direct biogas methanation35,42. To achieve this goal, a pretreatment strategy of simultaneously etching and precipitating Ni2+ on the Ni foam skeleton was adopted to form a vertically aligned, interconnected Ni(OH)2 nanosheets arrays on the surface of Ni foam as a high surface area intermediate layer (Supplementary Fig. 4 and Supplementary Table 5). Then, the high-density of oxide nanoparticles are loaded on the Ni(OH)2 nanosheets by a hydrothermal method. After the calcination and subsequent reduction process, the MOx/Ni/Ni-foam structured inverse catalyst with abundant oxide/Ni inverse composites on the Ni foam skeletons were synthesized (Fig. 2a, b and Supplementary Figs. 6, 7). It was further confirmed that the inverse composition was firmly anchored on the foam, as the weight loss observed by ultrasonic treatment in methanol for 5 h was less than 3 wt% (Supplementary Fig. 8). The Ni metal surface area of the CeZrOx/Ni configuration grown on the Ni-foam substrate reached 58.5 mNi2/gactive phase, which is 3.0-fold higher than that of CeZrOx/Ni powdered catalysts synthesized in the same method (Fig. 2c and Supplementary Table 5), underscoring the improved synthesis procedure successfully increases the exposure of inverse active sites.

a Schematic illustration of MOx/NiO/Ni-foam structured inverse catalyst; b Scanning electron microscope (SEM) images of CeZrOx/NiO/Ni-foam (urea) catalyst; c Comparison of Ni metal surface area among Ni/CeZrOx, CeZrOx/Ni (urea), and CeZrOx/Ni/Ni-foam catalysts. Comparison of water reaction orders (d) and reaction orders with respect to H2 and CO2 (e) among Ni/CeZrOx, CeZrOx/Ni (oxalic acid), and CeZrOx/Ni/Ni-foam catalysts; f Comparative catalytic performance of various MOx/Ni/Ni-foam catalysts in direct biogas methanation (Reaction conditions: GHSV = 10000 h−1, 240 °C, CO2:CH4 = 50%:50%, CO2:H2 = 1:4, P = 1 bar).
Kinetic evaluations demonstrate the advantages of porous Ni foam in handling reactants and products under CO2 methanation conditions. The apparent reaction order of product H2O on CeZrOx/Ni/Ni-foam is determined as -0.06, while the values of CeZrOx/Ni inverse catalyst and Ni/CeZrOx catalyst are -0.18 and -0.22, respectively (Fig. 2d and Supplementary Table 6). Based on the Langmuir-Hinshelwood mechanism, the negative reaction order of product indicates that the catalytic activity would be inhibited by the product due to the partial occupation of active sites45. The much smaller negative value of water order (-0.06) demonstrates the porous structure of Ni foam substrate accelerates water removal from catalyst surface (Supplementary Fig. 9), facilitating the release of active sites by preventing strong water absorption and inbibitation oxidative deactivation. Moreover, the apparent reaction orders of H2 and CO2 on the CeZrOx/Ni/Ni-foam structured inverse catalyst was 0.56 and 0.48, respectively, comparing with 0.60 and 0.38 on the CeZrOx/Ni inverse catalyst, 0.91 and 0.04 on the conventional Ni/CeZrOx catalyst (Fig. 2e, Supplementary Fig. 10 and Supplementary Tables 7, 8). The similar H2 and CO2 reaction orders for structured and powdered inverse catalyst further indicate that the synthetic structured inverse catalyst has a similar inverse interfacial structure with the active powdered inverse catalyst. The balanced C/H coverages on inverse catalyst compared to the conventional methanation catalysts is the most important kinetic advantage for CO2 methanation35,46, suggesting the kinetic self-poisoning problems are restrained. The catalytic performance of the structured MOx/Ni/Ni-foam (M = Zr, Al, Ce, Mg) in the methanation biogas (e.g., 50% CO2-50% CH4) to produce pipeline-grade methane was evaluated at atmospheric pressure and up to 240 °C (the maximum temperature for single-stage biogas methanation) (Fig. 2f and Supplementary Fig. 11). Supported oxide particles play a vital role in enhancing the activity of CO2 methanation. Among the tested oxide particles, the Ce-Zr mixed oxide showed the best biogas direct methanation performance of 97.8% CO2 conversion at 240 °C, meeting the requirement for single-stage process. After optimization, Ce/Zr = 2/1 was identified as the most active composition (Supplementary Fig. 12).
Structural characterization of catalysts
High angle dark field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray (EDX) elemental mapping of the CeZrOx/NiO species scraped from structured Ni foam indicates that the NiO nanoparticles are in a continuous phase to support isolated CeZrOx nano particles of ~2 nm (the lattice fringes of 0.24 nm and 0.31 nm are identical to the NiO (111) and CeZrOx (110)) (Fig. 3a–c). As depicted in Supplementary Fig. 13, element mapping validates that the 30 mol% Ni/CeZrOx catalyst has an oxide-supported on NiO configuration. In-situ X-ray photoelectron spectroscopy (XPS, Fig. 3d) demonstrates the percentage of Ce3+ species on reduced CeZrOx/Ni/Ni-foam reached as high as 60%, indicating the reducible properties of oxide are promoted significantly by nano-sizes and inverse configuration47. The simultaneous positive shift of Zr 3d binding energy with the reduction of Ce is due to the oxygen vacancy formation within the Ce-O-Zr linkage, which indicates the formation of Ce-Zr mixed oxide species48. Raman spectroscopy reveals that the deposition of CeZrOx species can induce a red-shift of the Ni-O bonding of NiO (Supplementary Fig. 14), suggesting the strong interaction of the Ce or Zr species with the NiO lattice49. The synthesis of the structured inverse catalyst was even scaled up to the dimension from 6 mm*6 mm to 20 mm*20 mm and 40 mm*40 mm. The catalytic performance of the D = 6 mm structured inverse catalysts disks scissored randomly from the large sheets reproduced that of the original synthesized ones (Fig. 3e and Supplementary Fig. 15), demonstrating the excellent scalability of structured inverse catalysts for the biogas methanation process.

a Aberration-corrected HAADF-STEM image of scraped CeZrOx/NiO catalyst; b EDS elemental mappings of scraped CeZrOx/NiO catalyst, showing the distribution of Ni, Ce and Zr; c High resolution HAADF-STEM image of the 1# area in (a) and the analysis of lattice fringes; d In-situ XPS of Ce 3d, Ni 2p, O 1s and Zr 3d of calcined and reduced CeZrOx/Ni/Ni-foam catalyst; e Large area integral CeZrOx/Ni/Ni-foam catalyst preparation (6 mm, 20 mm, 40 mm).
Direct biogas methanation performance
When tested in simulated biogas atmosphere (CO2/CH4 = 50%/50%, 10000 h−1), the CeZrOx/Ni/Ni-foam catalyst (Ce:Zr = 2:1, optimized) structured inverse catalyst displayed outstanding low-temperature CO2 methanation performance at 1 bar, 5 bar and 15 bar respectively (Fig. 4a). It achieves CO2 conversion of 97.9% at 240 °C under atmospheric pressure, ensuring the methane concentration in outlet exceed 95%, which meets the threshold of natural gas pipeline grid. On the contrary, the CO2 conversion was less than 10% at 240 °C at 1 bar on the most active 30 mol% Ni/CeZrOx methanation catalyst with conventional configuration50. Elevating the working pressure to 5 bar, CeZrOx/Ni/Ni-foam catalyst could fulfill the biogas upgrading requirement from 220 to 260 °C. At 15 bar, the normal inlet pressure of natural gas grid, the appropriate reaction temperature range can be further expanded to 200-280 °C. The operating window temperature can be expanded from a single point of 240 °C at 1 bar to 40 °C at 5 bar and 60 °C at 15 bar, significantly improving the feasibility and convenience of single-stage methanation process. More importantly, under the same condition, the conventional supported Ni/CeZrOx catalyst has never succeeded in producing qualified high-purity methane in a single-stage reaction. At 15 bar and 340 °C, the CO2 conversion of Ni/CeZrOx catalyst almost reaches the equilibrium limit, but still lower than the purity threshold (95%). To demonstrate the excellent adaptability of CeZrOx/Ni/Ni-foam inverse catalyst for treating different biogas sources with different compositions, the CH4/CO2 ratio was tuned and tested (Fig. 4b). Remarkably, even at elevated CO2 concentrations up to 60%, the inverse catalyst is still capable to convert the simulated biogas into relative pure methane which fits for transport via natural gas networks. And, the kinetic orders for varied biogas composition remain similar, indicating the concentration of CH4 has little influence to methanation process (Supplementary Fig. 16). Stability evaluation demonstrates the inverse catalyst remained stable after 350 h on stream without any deactivation regardless the composition variation in the gas feed (Fig. 4c). The O2-TPO and HAADF STEM characterization of spent catalyst also shows no apparent carbon deposition or sintering (Supplementary Figs. 17, 18). This structured CeZrOx/Ni/Ni-foam inverse catalyst has confirmed its applicability in single-stage biogas methanation processes (Fig. 4d). The structured inverse catalyst wss also evaluated in CO2 methanation under the same reaction conditon as most of commercial and advanced Ni-based methanation catalysts (Fig. 4d● and Supplementary Fig. 19). The CeZrOx/Ni/Ni-foam was attaining 97.8% CO2 conversion with >99.9% CH4 selectivity at 240-260 °C (◯ in the color-filled region). And, it is the first catalyst residing within the “sweet zone” (color-filled region) defined by thermodynamic equilibrium calculations for direct biogas methanation (biogas: 30 ~ 50% CO2/CH4) (Fig. 4 and Supplementary Table 9).
and Supplementary Table 9).

a Temperature-dependent activities of Ni-based catalysts in simulated biogas atmosphere under different pressure (Reaction conditions: GHSV = 10,000 h−1, 180-380 °C, CO2:CH4 = 50%:50%, CO2:H2 = 1:4, P = 1-15 bar), methane concentration in dehumidified post-reaction outlet gas at CO₂ conversion rates of 92%–98% (upper panel); b Influence of gas composition of simulated biogas on the catalytic performance of inverse and conventional catalysts; c The long-term stability of CeZrOx/Ni/Ni-foam catalyst under varied operating conditions (P = 15 bar). d thermodynamic equilibrium of biogas direct methanation of 50% CO2/CH4 and CO2 methanation performance of state-of-the-art Ni-based catalysts. The gray dot line refers to the minimum CO2 conversion (97.8%) corresponding to the purity of outlet methane of 95% in the biogas direct methanation process (50% CO2-50% CH4). The color-filled region is defined by thermodynamic equilibrium and the minimum CO2 conversion is a “sweet area” for qualified single-stage biogas methanation catalysts. ● with number inside indicates pure CO₂ methanation performance of reported Ni-based catalysts; open ◯ represents pure CO2 methanation performance for CeZrOx/Ni/Ni-foam catalyst;  presents the direct biogas methanation performance of CeZrOx/Ni/Ni-foam catalyst (50% CO2/CH4). The ratio of CO2:H2 fixed at 1:4 for catalytic evaluation.
presents the direct biogas methanation performance of CeZrOx/Ni/Ni-foam catalyst (50% CO2/CH4). The ratio of CO2:H2 fixed at 1:4 for catalytic evaluation.
Economic analysis of biogas methanation processes
The evaluation of catalytic performance and kinetic studies demonstrates that the structured CeZrOx/Ni/Ni-foam inverse catalyst is a promising solution for biogas upgrading in a single-stage CO2 methanation reactor (SS), offering remarkable activity and stability simultaneously. The success in the development of catalysts will benefit the application of the economy-benign process and further mitigate the dependence of the retail price of methane from biogas on the cost of hydrogen. The economic analysis was updated using based on the performance of inverse catalyst. In fact, based on current hydrogen prices in China, hydrogen costs account for 55.54-62.25% of the production costs across the three production routes. This highlights the reliance of biomethane production via methanation on hydrogen costs, underscoring the necessity of conducting sensitivity analyses on hydrogen prices. The sensitivity analysis of hydrogen price on methane from biogas cost (Fig. 5a, Supplementary Methods-Economic analysis of biogas-to-methane process) shows that the single-stage process allows for higher tolerances in hydrogen price than the conventional pathways, which are about $0.77/kgH2 and $0.24/kgH2 compared with SSR and MS processes at the same price of methane, thereby lowering the technical readiness level required for hydrogen production. The advanced single-stage biogas upgrading technology powered by inverse catalyst is able to produce price-competitive pipeline-grade methane by using the “gray hydrogen” and “blue hydrogen”51 produced from fossil fuels including coals and by-product hydrogen etc. (Supplementary Table 10). For example, according to the price of coal-based hydrogen provided by IEA52, using coal produced hydrogen as substrate, the pipeline-grade methane obtained can be reduced to $0.6-0.8/kgmethane, close to the commercial natural gas (Fig. 5b and Supplementary Table 12). To further assess the application potential of the single-stage process in producing the zero-CO2 emission biomethane in the near future, according to the techno-economic evaluation model of green hydrogen proposed in the literatures53,54, assessments of the renewable hydrogen cost in 2050 (Fig. 5c) and the distribution of biogas resources of China were performed (Supplementary Table 12). Since the economic collection radius of biomass is about 50 km55, t the screening conditions for feasible areas calculation are limited by green hydrogen price and sufficient local biogas resources within the economic range of the upgrade plant (Fig. 5d). It can be seen that the area which can produce biomethane cheaper than the local pipeline natural gas reaches 1.42 million km2 for SS methanation technology, covering most part of Shandong, Henan, Zhejiang and part of Guangdong, Shanxi, Sichuan and Tibet (about 14.7% of China’s land territory, Fig. 5d), in which 146 billion Nm3/year biogas resources are hosted, ~27.4% of total biogas potential of China (Supplementary Table 12). Compared with the conventional MS process, the feasible area and influenced biogas resources to produce biomethane with the same price increase by 26.1% and 41.8% (Supplementary Fig. 20), respectively, highlighting the extraordinary advantages of the structured inverse catalyst powered single-stage methanation technique for biogas upgrading. Considering the green premium (GP) of the zero-CO2 emission biomethane, the feasible regions were further screened with the GP up to 25%. From the results, the area can expand to 4.66 million km2, near half of China, suggesting most of eastern and southern part of China is suitable to develop the sustainable biomethane industry to consume the biogas with the efficient single-stage process. The fully development of the biogas methanation processes can consume over 79.4% of the biogas potential in China, leading to a sustainable future by reducing the organic wastes and producing more green synthetic fuels.

a Biomethane price sensitivity with the price of hydrogen reactants. The yellow lines represent the reduction rate in unit cost of the single-stage process compared to those of two conventional processes; b The anticipated price of biomethane synthesized with different types of gray and blue hydrogen; c Anticipated green hydrogen cost in different regions of China in 2050; d Regions feasible to produce economically appealing biomethane (SS) from biogas using green hydrogen in China in 2050. Levels 1-6 corresponds to the green premium level of the biomethane over the price of industrial pipeline natural gas (Level 1-6 corresponds to 0, 5, 10, 15, 20, 25%). The prices of industrial PNG in the capital cities of each province are used as a reference. Areas where the local biomethane price exceeds the reference value are considered unavailable area.
Discussion
In summary, the processes of direct biogas methanation were compared and the single stage process which requires efficient low-temperature CO2 methanation catalyst, was determined as the simplest and economic feasible one to upgrade biogas to biomethane. The CeZrOx/Ni/Ni-foam inverse structured catalyst was developed and demonstrated to meet the critical activity and thermodynamic requirement of the single-stage direct CO2 methanation of biogas (composition of 40% CO2/CH4) process, achieving a CH4 concentration of >95% in the outlet gas at 240 °C and atmospheric pressure. When the operation pressure was raised to the normal inlet pressure of natural gas grid (15 bar), the operating temperature range of the inverse catalyst that meets the biomethane standard can be expanded from 200 to 300 °C. Additionally, the structured Ni foam substrate enhanced the water removal and inhibited the accumulation of undesirable carbon deposition, improving the coke-resistance of CeZrOx/Ni/Ni-foam catalyst in the strong exothermic reaction. Kinetics and mechanism studies have confirmed that the inverse catalyst overcomes the kinetic poisoning disadvantages of conventional catalysts, and exhibits a high methanation activity at low temperature region, thereby fully utilizing the advantage of high CO2 equilibrium conversion and greatly simplifying the separation module. The superior performance originates from the inverse oxide-metal interface regulating the H/C species coverage and the porous foam structure enhancing water removal ability, which together ensuring high activity and stability in CO2 methanation. With the advancement of renewable energy collection and green hydrogen production technologies, biogas upgrading techniques powered by low-temperature structured inverse catalysts will be able to improve the efficiency and economic feasibility of the biogas methanation processes, and boosts the exploit and utilization of green bioenergy for the sustainable development.
Methods
Materials
Analytical grade chemicals including the anhydrous oxalic acid (H2C2O4, 99 wt% purity), sodium carbonate (Na2CO3, 99 wt% purity), sodium hydroxide (NaOH, 99 wt% purity), Urea (CH4N2O, 99 wt% purity), nickelous nitrate hexahydrate (Ni(NO3)2·6H2O, 98 wt% purity), cerium nitrate hexahydrate (Ce(NO3)2·6H2O, 99 wt% purity), Zirconium nitrate pentahydrate (Zr(NO3)4·5H2O, 99 wt% purity), aluminum nitrate nonahydrate (Al(NO3)3·9H2O, 99 wt% purity) and Magnesium nitrate hexahydrate (Mg(NO3)4·5H2O, 99 wt% purity) was purchased from Sinopharm Chemical Reagent Co., Ltd. The Ni-foam felt was purchased from Suzhou Taili Material Co. All chemicals were used as received without any further purification.
Catalyst synthesis
Preparation of CeZrOx/Ni/Ni-foam catalyst
The Ni(OH)2/Ni foam substrate was prepared as follows: Circular Ni foam slices (6 mm in diameter, 1.0 mm thick, 110 PPI porosity) were cut from Ni foam plates and sonicated in acetone for 20 min to remove residual organic impurities. The slices were then immersed in 0.1 M HCl solution and sonicated at room temperature for an additional 20 min to eliminate surface nickel oxide, followed by thorough rinsing with deionized water. The cleaned Ni foam slices were transferred into a stainless-steel autoclave lined with a 50 mL polytetrafluoroethylene (PTFE) container containing 35 mL of a solution composed of urea (6.3 mmol) and Ni(NO3)2·6H2O (0.875 mmol). Hydrothermal treatment was performed at 160 °C for 12 h, after which the Ni foam, now coated with deep green Ni(OH)2 crystals, was rinsed with deionized water and dried under vacuum at 80 °C for 12 h. Next, a solution containing Zr(NO3)2·5H2O (1.6 mmol), Ce(NO3)3·6H2O (3.2 mmol), and urea (6.0 mmol) in 35 mL deionized water was prepared and stirred for 60 min. The solution and the Ni(OH)2/Ni foam slices (0.4 g) were then transferred into a 100 mL Teflon-lined autoclave and subjected to hydrothermal treatment at 180 °C for 12 h. After cooling to room temperature, the sample was washed with ethanol and deionized water, dried under vacuum at 60 °C for 12 h, and finally calcined at 400 °C for 3 h to yield the CeZrOx/NiO/Ni foam catalyst. The Ni/Ni-foam and MOx/Ni/Ni-foam (M = Mg, Al, Ce, Zr, CeAlOx, ZrAlOx) catalysts were fabricated using the identical synthetic procedure described above.
Catalytic evaluation
Performance evaluation of structured Ni based inverse Catalyst
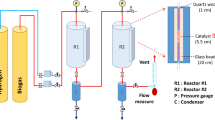
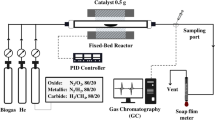
The performance evaluation for CO2 hydrogenation to CH4 was performed in an ordinary pressure fixed-bed flow stainless steel reactor (Purchased from Quzhou Vodo Instrument Co., LTD). The prepared catalyst sheets (0.4 g, diameter 6 mm) are loaded into a quartz tube (inner diameter = 6 mm and length = 60 cm) and put into the reactor. The catalyst is preprocessed in 20% H2 at 450 °C for 3 h, cooled to the reaction temperature (130–300 °C). Next, model biogas (45% CH4, 45% CO2, and 10% N2 as an internal standard) and H2 were fed into the reactor at the desired gas hourly space velocity (GHSV) with a H2/CO2 molar ratio of 4. The effect of the reaction temperature (130–360 °C), pressure (1, 5, and 15 bar), and GHSV (10,000–50,000 h−1) were studied by univariate analysis. The actual temperature of the catalyst bed is measured using a thermocouple located at the middle of the catalyst bed. Gas-phase products are analyzed using a gas chromatograph (GC-8860, Agilent) equipped with a thermal conductivity detector, Porapak Q and 5A molecular sieve columns.
where F denotes the gas flow into the reactor, mL/min; C denotes the concentration, %; A denotes the gas chromatographic peak area, Vin (CO2) denotes the CO2 flow rate before the reaction, mL/min; X (CO2) denotes the CO2 conversion, %; S (CH4) denotes the CH4 selectivity, %; Vfoam denotes the volume of the catalyst, mL; mfoam denotes the mass of the catalyst, g; macticve phase denotes the loading mass of the active phase on the nickel foam substrate, g.
The Arrhenius plots are depicted at a high GHSV of 15,000–40,000 h−1 to keep the CO2 conversions below 15%, where the heat and mass transfer effect is negligible. Also, the differential mass-normalized reaction rates are obtained in the kinetic regime.
Catalyst characterization
Thermodynamic equilibrium calculation for biogas methanation
The thermodynamic equilibrium reported in Supplementary Fig. 1 was calculated using Aspen Plus V11 with the RGibbs reactor module based on Gibbs free energy minimization. The biogas feed was modeled according to Eq. (8), where the CH4 fraction was varied within 40–60 vol% to represent typical compositions.
Inductively coupled plasma-optical emission spectrometer (ICP-OES)
The ICP-OES analysis was conducted using a Varian ICP-OES 720 instrument.
Sample preparation: a specific amount of the sample was weighed into a PTFE container and mixed with 5 mL of concentrated nitric acid, 3 mL of hydrochloric acid, 1 mL of hydrofluoric acid, and 2 mL of hydrogen peroxide. The container was sealed in a microwave digestion furnace, where the sample was heated at 1200 W to 130 °C for 20 min, held at this temperature for 5 min, then further heated to 180 °C for 20 min, and maintained at this temperature for 40 min. The sample was subsequently cooled to room temperature.
Testing procedure: the cooled solution was transferred into a 25 mL plastic volumetric flask and diluted to the mark with deionized water. The resulting solution was analyzed sequentially. Samples exceeding the calibration curve range were further diluted and re-analyzed.
Standard solution preparation: the standard solutions used for calibration were prepared using certified reference materials with concentration points of 0, 0.5, 1.0, 2.0, and 5.0 mg/L.
X-ray diffraction (XRD)
X-ray diffraction (XRD) patterns were recorded with a PANalytical X’Pert PRO powder diffractometer using Cu Kα radiation (λ = 0.1541 nm). The working voltage was 40 kV and the working current was 40 mA. The patterns were collected with a 2θ range from 10° to 80°at a step of 0.0167°. The average crystallite sizes of the samples were calculated with the Scherrer equation based on the strongest h k l (111) diffraction peak of Ni.
Surface area measurement
To obtain the textural parameters, N₂ adsorption–desorption analysis was conducted at –196 °C using a BSD-PS2 system after sample degassing (200 °C, 4 h, vacuum). The Brunauer–Emmett–Teller (BET) surface area was derived from the adsorption data, while the pore size distribution was calculated by applying the Barrett–Joyner–Halenda (BJH) model to the desorption branch.
H2 temperature programmed reduction (H2-TPR)
H2-TPR profiles were obtained using a BELCAT-B instrument. Prior to measurement, a 50 mg sample was pretreated in flowing He (30 mL min−1, 130 °C, 1 h) and cooled. Following the introduction of a 10% H2/Ar flow (30 mL min−1), the temperature was ramped from 50 to 700 °C at 10 °C min−1. The corresponding hydrogen consumption was recorded via a thermal conductivity detector (TCD).
H2 titration experiment
The active metal dispersion was determined by H2 pulse chemisorption. Prior to measurement, the catalyst (0.1 g) was reduced in situ (20% H2/Ar, 30 mL min−1, 450 °C, 3 h) and purged with Ar at 50 °C for 30 min. Controlled pulses of 10% H2/Ar were then introduced at 50 °C using a precision injection loop. The consumption was monitored by a thermal conductivity detector, and the titration was considered complete after three successive pulses yielded consistent peak areas.
CO2 temperature programmed desorption (CO2-TPD)
CO2-TPD profiles were acquired using a Microtrac BEL Cat II system. Following in-situ reduction (20% H2/Ar, 450 °C, 3 h, 5 °C min−1) and He purge at 50 °C, the catalyst (50 mg) was saturated with 10% CO2/He for 1 h. After removing physisorbed CO2 via a 40-min He purge, temperature-programmed desorption was carried out from 50 to 800 °C at 10 °C min⁻¹ under He flow.
Scanning electronic microscopy (SEM)
FE-SEM imaging and elemental mapping were performed on a HITACHI Regulus 8100 microscope at an acceleration voltage of 20 kV, combined with an Oxford Ultim Max 65 energy-dispersive X-ray (EDX) detector for compositional analysis.
Transmission electron microscope (TEM)
TEM images were acquired on a Tecnai G2 F30 S-Twin FEG-TEM at 300 kV. Following dispersion in ethanol via sonication, the sample was drop-cast onto carbon-coated copper grids and dried under ambient conditions for analysis.
Scanning Transmission Electron Microscope (STEM)
STEM and EDX experiments are conducted on a Thermo Scientific Spectra 300 Double-Corrected Transmission Electron Microscope equipped with a Gatan Imaging Filter. The STEM-EDX elemental mapping is carried out using a scanning point of 150 × 150, and an acceleration voltage of 300 kV was applied.
X-ray photoelectron spectroscopy (XPS)
X-ray photoelectron spectroscopy analysis is performed on a ThermoFischer ESCALAB 250Xi equipped with an in-situ reactor. The specific parameters are as follows: excitation source using Al Kalpha radiation (hv = 1486.6 eV); analysis chamber vacuum level of 8 × 10−10 mbar; working voltage of 12.5 kV; filament current of 16 mA; and signal accumulation for ~10 cycles. The Passing Energy is set to 30 eV with a step size of 0.1 eV. The specific operational procedure is as follows: the catalyst sample, in the form of a disc, is placed inside the reactor chamber. It is pre-treated for 1 h at a set temperature in a H2/N2 atmosphere (20 vol% H2) with a flow rate of 20 mL/min. After cooling to room temperature, the sample is transferred to the measurement chamber without exposure to air. The measurement chamber is evacuated to a vacuum level below 8 × 10−10 mbar before conducting the analysis. Charging correction of the binding energy is performed using C1s (284.6 eV) as a reference42.
Temperature-programmed oxidation (TPO) of spent catalysts
To quantify coke deposition, spent catalysts were subjected to TPO analysis. Following a 30-min purge with 20% O2/Ar (50 mL/min) at room temperature, the temperature was ramped to 700 °C (10 °C/min) and held for 10 min. The effluent gases were analyzed using a DECRA mass spectrometer, primarily detecting CO2.
Raman spectroscopy analysis
Raman spectra are obtained using the Renishaw In Via Reflex spectrometer with a 532 nm laser excitation source. The scanning range is set from 100 to 2000 cm−1 with an accuracy of 2 cm−1. The scan test is considered complete when consistent results are obtained from at least three positions on each sample.
Temperature programmed surface reaction-mass spectrum (TPSR)
The test procedure for CH4 dissociation: 100 mg of sample, pretreat it at 450 °C for 3 h under 40 mL∙min−1 20% H2/Ar purge. Then cool down to room temperature (~25 °C), and switch the 20% H2/Ar to 25 mL∙min−1 10% CH4/Ar to record mass baseline. After the baseline is stable, the temperature is increased to 750 °C with a heating rate of 10 °C∙min−1, while the mass spectrum is recorded at the same time42.
Data availability
The data that support the plots within this paper and other finding of this study are available from the corresponding author upon request. Source data is provided as a Source Data file. Source data are provided with this paper.
References
Ahmed, S. F. et al. Biogas upgrading, economy and utilization: a review. Environ. Chem. Lett. 19, 4137–4164 (2021).
Fernández-González, J. M. et al. Biomethane injection into natural gas network vs composting and biogas production for electricity in Spain: an analysis of key decision factors. Sustain. Cities Soc. 60, 102242 (2020).
Clark, M. A. et al. Global food system emissions could preclude achieving the 1.5° and 2 °C climate change targets. Science 370, 705–708 (2020).
Zhou, H. et al. Oxygen-vacancy-mediated catalytic methanation of lignocellulose at temperatures below 200 °C. Joule 5, 3031–3044 (2021).
Si, X. et al. Catalytic production of low-carbon footprint sustainable natural gas. Nat. Commun. 13, 258 (2022).
IEA. Outlook for biogas and biomethane: prospects for organic growth, IEA, Paris https://www.iea.org/reports/outlook-for-biogas-and-biomethane-prospects-for-organic-growth (2020).
Rosa, L. et al. Assessment of carbon dioxide removal potential via BECCS in a carbon-neutral Europe. Energy Environ. Sci. 14, 3086–3097 (2021).
Verbeeck, K. et al. Upgrading the value of anaerobic digestion via chemical production from grid injected biomethane. Energy Environ. Sci. 11, 1788–1802 (2018).
Springmann, M. et al. Options for keeping the food system within environmental limits. Nature 562, 519–525 (2018).
Bidart, C. et al. Biogas catalytic methanation for biomethane production as fuel in freight transport - a carbon footprint assessment. Renew. Sustain. Energy Rev. 168, 112802 (2022).
Glenk, G. et al. Reversible Power-to-Gas systems for energy conversion and storage. Nat. Commun. 13, 2010 (2022).
Chaemchuen, S. et al. Metal–organic frameworks for upgrading biogas via CO2 adsorption to biogas green energy. Chem. Soc. Rev. 42, 9304–9332 (2013).
Khan, M. U. et al. Current status of biogas upgrading for direct biomethane use: a review. Renew. Sustain. Energy Rev. 149, 111343 (2021).
Jepleting, A. et al. Potential of low-cost materials for biogas purification, a review of recent developments. Renew. Sustain. Energy Rev. 210, 115152 (2025).
Song, S. et al. Production of hydrogen from offshore wind in China and cost-competitive supply to Japan. Nat. Commun. 12, 6953 (2021).
Chen, X. et al. Pathway toward carbon-neutral electrical systems in China by mid-century with negative CO2 abatement costs informed by high-resolution modeling. Joule 5, 2715–2741 (2021).
Bakkaloglu, S. et al. A comparative study of biogas and biomethane with natural gas and hydrogen alternatives. Energy Environ. Sci. 17, 1482–1496 (2024).
Witte, J. et al. Direct catalytic methanation of biogas – part I: new insights into biomethane production using rate-based modelling and detailed process analysis. Energy Convers. Manage. 171, 750–768 (2018).
Glenk, G. et al. Advances in power-to-gas technologies: cost and conversion efficiency. Energy Environ. Sci. 16, 6058–6070 (2023).
Mallapragada, D. S. et al. Decarbonization of the chemical industry through electrification: barriers and opportunities. Joule 7, 23–41 (2023).
Kim, Y. et al. Renewable natural gas value chain based on cryogenic carbon capture, utilization and storage, and power-to-gas for a net-zero CO2 economy. Renew. Sustain. Energy Rev. 212, 115425 (2025).
Erdener, B. C. et al. A review of technical and regulatory limits for hydrogen blending in natural gas pipelines. Int. J. Hydrogen Energy 48, 5595–5617 (2023).
Giarnieri, I. et al. Direct biogas methanation at moderate pressure: mechanism investigation over Ni-based catalysts. J. CO2 Util. 93, 103045 (2025).
Aieamsam-Aung, P. et al. Upgradation of methane in the biogas by hydrogenation of CO2 in a prototype reactor with double pass operation over optimized Ni-Ce/Al-MCM-41 catalyst. Sci. Rep. 13, 9342 (2023).
Stangeland, K. et al. Active and stable Ni based catalysts and processes for biogas upgrading: The effect of temperature and initial methane concentration on CO2 methanation. Appl. Energy 227, 206–212 (2018).
González-Arias, J. et al. Optimizing biogas methanation over nickel supported on ceria-alumina catalyst: towards CO2-rich biomass utilization for a negative emissions society. Environ. Res. 242, 117735 (2024).
Witte, J. et al. Direct catalytic methanation of biogas – part II: techno-economic process assessment and feasibility reflections. Energy Convers. Manage. 178, 26–43 (2018).
Calbry-Muzyka, A. S., et al. Direct methanation of biogas—technical challenges and recent progress. Front. Energy Res. 8, 570887 (2020).
Giglio, E. et al. Direct biogas methanation via renewable-based Power-to-Gas: Techno-economic assessment based on real industrial data. Energy Convers. Manage. 332, 119775 (2025).
Li, Y. et al. Metal–support interactions in metal/oxide catalysts and oxide–metal interactions in oxide/metal inverse catalysts. ACS Catal. 12, 1268–1287 (2022).
Kattel, S. et al. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 355, 1296–1299 (2017).
Kattel, S. et al. Tuning selectivity of CO2 hydrogenation reactions at the metal/oxide interface. J. Am. Chem. Soc. 139, 9739–9754 (2017).
Rodriguez, J. A. et al. Inverse oxide/metal catalysts in fundamental studies and practical applications: a perspective of recent developments. J. Phys. Chem. Lett. 7, 2627–2639 (2016).
Tang, X. et al. Spontaneous nano-ZrO2 exsolution from Ni-Zr-O mixed oxides enables facile fabrication of ZrO2/Ni inverse catalysts for efficient COx methanation. Angew. Chem. Int. Ed. 64, e202511453 (2025).
Song, C. et al. Engineering MOx/Ni inverse catalysts for low-temperature CO2 activation with high methane yields. Nat. Chem. Eng. 1, 638–649 (2024).
Zhou, H. et al. The inverse configuration confers high activity and stability to ZrO2/Ni catalysts for CO2 methanation. Adva. Func. Mater. 35, e25888 (2025).
Xu, Y. et al. Cu-supported nano-ZrZnOx as a highly active inverse catalyst for low temperature methanol synthesis from CO2 hydrogenation. Appl. Catal. B Environ. 344, 123656 (2024).
Xu, Y. et al. Insights into the interfacial structure of Cu/ZrO2 catalysts for methanol synthesis from CO2 hydrogenation: effects of Cu-supported nano-ZrO2 inverse interface. Chem. Eng. J. 470, 144006 (2023).
Wu, C. et al. Inverse ZrO2/Cu as a highly efficient methanol synthesis catalyst from CO2 hydrogenation. Nat. Commun. 11, 5767 (2020).
Zhu, J. et al. Superior FeNi3-FeOx/Ni-foam catalyst for gas-phase hydrogenation of dimethyl oxalate to ethanol. Appl. Catal. B Environ. 270, 118873 (2020).
Zhu, J. et al. Superb Ni-foam-structured nano-intermetallic InNi3C0.5 catalyst for hydrogenation of dimethyl oxalate to ethylene glycol. Chem. Eng. J. 426, 130857 (2021).
Tang, X. et al. Thermally stable Ni foam-supported inverse CeAlOx/Ni ensemble as an active structured catalyst for CO2 hydrogenation to methane. Nat. Commun. 15, 3115 (2024).
Cañada-Barcala, A. et al. Synthetic natural gas production through biogas methanation using a sorption-enhanced reaction process. Sep. Purif. Technol. 331, 125714 (2024).
Silla, H. Chemical Process Engineering: Design and Economics (CRC Press, 2003).
Zou, T. et al. Descriptors of InZrOx vs ZnZrOx catalysts for CO2 hydrogenation to methanol. Adv. Energy Mater. 15, 2404967 (2025).
Lee, K. et al. Engineering nanoscale H supply chain to accelerate methanol synthesis on ZnZrOx. Nat. Commun. 14, 819 (2023).
Hu, B. et al. Distinct crystal-facet-dependent behaviors for single-atom palladium-on-ceria catalysts: enhanced stabilization and catalytic properties. Adv. Mater. 34, 2107721 (2022).
Feng, W.-H. et al. Insights into bimetallic oxide synergy during carbon dioxide hydrogenation to methanol and dimethyl ether over GaZrOx oxide catalysts. ACS Catal 11, 4704–4711 (2021).
Zhao, Z. et al. Inverse ceria-nickel catalyst for enhanced C–O bond hydrogenolysis of biomass and polyether. Nat. Commun. 15, 8444 (2024).
Ashok, J. et al. Enhanced activity of CO2 methanation over Ni/CeO2-ZrO2 catalysts: influence of preparation methods. Catal. Today 281, 304–311 (2017).
Saha, P., et al. Grey, blue, and green hydrogen: a comprehensive review of production methods and prospects for zero-emission energy. Int. J. Green Energy 21, 1383–1397 (2024).
IEA, The Future of Hydrogen, IEA, Paris https://www.iea.org/reports/the-future-of-hydrogen (2019).
Pan, G. et al. Cost and low-carbon competitiveness of electrolytic hydrogen in China. Energy Environ. Sci. 14, 4868–4881 (2021).
Zhang, X. et al. High-resolution maps highlighting the cost competitiveness of China’s green hydrogen production. Energy Convers. Manage. 322, 119112 (2024).
Shanbhag, N. C., Parmar, H., Salve, V. K. & Cherian, I. Case study: radiological evidence of intracisternal bleed spread into brain perivascular spaces. Acad. Med. 2, https://doi.org/10.20935/acadmed7616 (2025).
Acknowledgements
This work is financially supported by the National Key R&D Program of China (2024YFB4105300), Zhejiang Provincial Natural Science Foundation of China (LR22B030003 and LQ24B030016), Natural Science Foundation of China (22278367, U22A20415, 22308314), Fundamental Research Funds for the Provincial Universities of Zhejiang (RF-C2025003), Young Elite Scientists Sponsorship Program by Zhejiang Association for Science and Technology. We acknowledge the Electron Microscopy Center of Zhejiang University of Technology for the AC-TEM test. AP-XPS experiments were conducted at the Analytical Instrumentation Center of Peking University. We acknowledge the participation of Mr. J.W. from Zhentai Energy Technology Co., Ltd. in discussions.
Author information
Authors and Affiliations
Contributions
L.L., D.M., and S.Y. designed the study. X.T., Z.S., and W.L. performed most of the reactions. X.Z. and Z.L. did the energy and economy analysis. X.T. and C.S. did the most data analysis. W.L., X.Y., and Y.W. carried out the stability experiments. Y.X. did the Quasi-situ XPS characterization and analysis. X.T., X.Z., L.L., and S.Y. wrote the paper. X.T., X.Z. Z.S., C.S., Y.X., X.Y., Y.W., H.L., S.Y., Z.L., X.L., D.M. and L.L. performed certain experiments, discussed and revised the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tang, X., Zhang, X., Song, Z. et al. Economical biogas direct methanation to pipeline grade natural gas via structured Ni based inverse catalyst. Nat Commun 17, 1371 (2026). https://doi.org/10.1038/s41467-025-68119-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68119-z
