
Abstract
The central circadian clock of the suprachiasmatic nucleus (SCN) comprises a network of diverse neuronal and glial cell types, yet its operating mechanism remains elusive. Here, by monitoring cellular calcium rhythms in vivo using dual-color fiber photometry in mice lacking vasoactive intestinal polypeptide (VIP), we demonstrate that arginine vasopressin (AVP) neurons oscillate intrinsically with a short period and reduced amplitude. This indicates that VIP normally amplifies and phase-delays the AVP neuronal rhythm each day, thereby lengthening its period to approximately 24 h in constant darkness. Consistently, the behavioral circadian period is shortened by AVP neuron-specific VIP receptor dysfunction and lengthened by AVP neuron-specific blockade of neurotransmitter release. VIP neurons and other SCN cell types occasionally exhibit weak, unstable, long-period calcium rhythms only when the AVP neuronal oscillation is attenuated due to VIP deficiency or AVP neuron-specific Bmal1 deletion. Given that AVP neurons serve as the primary pacesetter cells of the SCN ensemble rhythm, these results indicate that an SCN neuronal feedback loop (SNFL), composed of the AVP cellular oscillator and VIPergic signaling, is essential for generating robust circadian rhythms.
Similar content being viewed by others


Introduction
The suprachiasmatic nucleus (SCN) acts as the central circadian pacemaker in mammals, orchestrating multiple circadian rhythms of behavior and physiological function according to the environmental light/dark (LD) cues conveyed from the eye1,2,3,4. The SCN contains ~20,000 cells, most of which can generate circadian oscillations. In individual cells, the circadian rhythm is driven by the autoregulatory transcription-translation feedback loop (TTFL) consisting of clock genes such as Per1/2/3, Cry1/2, Bmal1, and Clock1,2. Notably, the TTFL is not unique to SCN cells and is common to almost all peripheral cells2,5. Rather, the SCN constructs a network of many TTFL-driven cellular clocks to generate a highly robust and coherent circadian rhythm as the central clock, which entrains a large number of peripheral clocks throughout the body1,2,3,4. Thus, intercellular communication among SCN clock cells through the neuronal and diffusible network distinguishes the SCN from the peripheral clocks. However, the network mechanism underlying the SCN central clock remains unclear.
The SCN is a heterogeneous structure composed of multiple types of neurons and glial cells1,2,3,4. Most SCN neurons are GABAergic, and several populations of these GABAergic neurons also co-express neuropeptides. These include arginine vasopressin (AVP)-producing neurons located predominantly in the dorsomedial part (SCN shell) and vasoactive intestinal polypeptide (VIP)-producing neurons in the ventrolateral part (SCN core). VIP is the most important contributor to the maintenance and synchrony of SCN cellular clocks with varying periods6,7. Therefore, mice deficient in VIP (Vip−/−) or its receptor VPAC2 (Vipr2−/−) demonstrate a drastic attenuation of the free-running rhythm in constant darkness (DD) with a shortened period or unstable multiple periods. In the latter, the free-running period changes suddenly or two periods emerge simultaneously6,8,9,10. VIP neurons receive direct projections from the retina, are involved in the photoentrainment, and send dense projections dorsally to the SCN shell1,9,11. However, the precise mechanisms by which VIP regulates SCN circuit remain incompletely understood.
On the other hand, our studies and those of others have suggested that the TTFL-driven cellular clocks in AVP neurons may play a dominant role than those of other SCN neurons in the generation of circadian oscillation by the SCN network, regulating the circadian oscillation of other cells and setting the SCN ensemble period3. Thus, AVP neuron-specific disruption of the TTFL by deleting Bmal1 (Avp-Bmal1−/− mice) results in an unstable, attenuated free-running rhythm with a lengthened period and the activity time (the interval between the onset and offset of locomotor activity)12,13,14,15,16. Furthermore, the cellular period length of AVP neurons appears to be the primary determinant of the SCN ensemble period16,17. In contrast, similar genetic manipulations of disrupting the TTFL or altering the cellular period in VIP neurons have little effect on the amplitude and period of the free-running behavior rhythm13,14,15,16,18, suggesting that VIP peptide is critical but the TTFL in VIP neurons may be dispensable for the circadian timekeeping. Besides, our previous studies indicated the importance of in vivo analysis of SCN network dynamics16,17.
Thus, there is a functional differentiation between the shell and the core within the SCN network. However, how the two integrate to generate the circadian rhythm of the SCN ensemble is still unclear. In the current study, we investigated the regulation of AVP neuronal circadian oscillation by VIP in vivo. To this end, we applied dual-color fiber photometry recording to VIP-deficient mice, allowing us to simultaneously monitor the cellular circadian rhythms from two types of SCN neurons. We then analyzed the circadian behavior rhythms of multiple mouse strains that had various impairments in the communication between VIP and AVP neurons. Our results reveal a feedback loop neuronal circuit of these two neuron types that may be the essential network mechanism of the SCN central circadian pacemaker.
Results
AVP neurons maintain a short-period Ca2+ rhythm in VIP-deficient mice
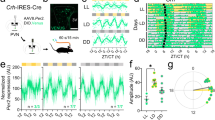
To elucidate the mechanism of how VIP regulates the cellular clocks of AVP neurons, we first aimed to compare the circadian rhythm of intracellular Ca2+ in SCN AVP neurons between control and VIP-deficient mice. Intracellular Ca2+ rhythms are a nice measure of cellular circadian rhythms in vivo that can be recorded in a neuron type-specific manner11,16,19,20,21. We used the homozygous Vip-tTA knock-in mice (ViptTA/tTA), which are equivalent to Vip knock-out mice and exhibit a spectrum of phenotypes such as simple short-period or complicated multiple-period behavioral rhythms10. In addition, their specific expression of tTA in VIP neurons allowed us to record Ca2+ rhythms in SCN AVP and VIP neurons (AVP- and VIP-Ca2+) separately and simultaneously when combined with hemizygous Avp-Cre BAC transgenic mice12. Thus, we generated VIP-deficient Avp-Cre; ViptTA/tTA mice and heterozygous control Avp-Cre; Vipwt/tTA mice and then targeted the expression of green (jGCaMP7s)22 and red (jRGECO1a)23 fluorescent Ca2+ sensor proteins in AVP and VIP neurons, respectively, by simultaneous injection of Cre- and tTA-dependent AAV vectors (AAV-CAG-FLEX-jGCaMP7s and AAV-TRE-jRGECO1a) (Fig. 1a, b).

a Schematic diagram of viral vector (AAV-CAG-FLEX-jGCaMP7s and AAV-TRE-jRGECO1a) injection and optical fiber implantation at the SCN in control (Avp-Cre; Vipwt/tTA) or VIP-deficient (Avp-Cre; ViptTA/tTA) mice for fiber photometry recording. b A representative coronal SCN section of mice with jGCaMP7s expression in AVP neurons and jRGECO1a in VIP neurons. A white dotted square shows the estimated position of the implanted optical fiber. Green, jGCaMP7s; magenta, jRGECO1a. Scale bar, 200 μm. Similar images were consistently observed across all mice used in this experiment. c Representative plots of the in vivo jGCaMP7s signal of AVP neurons (AVP-Ca2+, green) and jRGECO1a signal of VIP neurons (VIP-Ca2+, magenta) overlaid with the locomotor activity (home-cage activity) (black) in actograms. Detrended Ca2+ signals are shown with normalization in each row. Control and VIP-deficient mice were initially housed in 12:12-h LD and then in DD. Gray shading indicates the time when the lights were off. Time courses of AVP-Ca2+ (d) and VIP-Ca2+ (e) rhythms for 26 days (6 days in LD, 20 days in DD) (left) and their daily amplitude (right, peak - trough). Detrended and smoothened data from control (lavender) and VIP-deficient (orange) mice are shown. f Plots of locomotor activity onset (black), AVP-Ca2+ onset (green), AVP-Ca2+ offset (cyan), AVP-Ca2+ midpoint (blue), VIP-Ca2+ onset (magenta), VIP-Ca2+ offset (light red), and VIP-Ca2+ midpoint (yellow) in control and VIP-deficient mice. Behavior parameters were determined by actograms, while Ca2+ parameters were calculated from the wave forms in (d, e), as described in Methods. VIP-Ca2+ values are unmeasurable in VIP-deficient mice after DD9 due to their attenuated rhythmicity. g Day-by-day changes in the periods of AVP-Ca2+ (green), VIP-Ca2+ (magenta) (the intervals between two adjacent midpoints), and the locomotor activity (the intervals between two adjacent locomotor activity onsets, black) rhythms in LD and DD. *p < 0.05, ***p < 0.001 by two-way repeated measures ANOVA. ns, not significant. Values are mean ± SEM. n = 5 for control; n = 8 for VIP-deficient mice (d–g). The exact p-values are as follows: LD, 0.0006; DD, 0.0008 (d), LD, 0.4957; DD, 0.0132 (e), Control LD, 0.6686; DD, 0.9920; VIP-deficient LD, 0.0530; DD, 0.0314 (g). Source data are provided as a Source data file.
In vivo dual-color fiber photometry with simultaneous monitoring of spontaneous locomotor activity revealed robust, temporally coordinated AVP- and VIP-Ca2+ rhythms in control mice in both light/dark (LD) and dark/dark (DD), higher during the (subjective) day (Fig. 1c–e and Supplementary Fig. 1a–c). In VIP-deficient mice, the VIP-Ca2+ rhythm was comparable to that of controls in LD but rapidly attenuated in DD, even when the locomotor activity still showed rhythmicity (Fig. 1c, e and Supplementary Fig. 2). This made it difficult to determine the daily onset and offset of the VIP-Ca2+ rhythm from its waveforms after a week in DD (Fig. 1e–g). The number of jRGECO1a-expressing cells was comparable between control and VIP-deficient mice (Supplementary Fig. 1e), suggesting that the lack of VIP did not decline the VIP neuronal health. In contrast, the amplitude of the AVP-Ca2+ rhythm was substantially reduced in both LD and DD, but it remained stable in DD (Fig. 1c, d and Supplementary Fig. 2). Consistent with the changes in the behavior rhythm, the AVP-Ca2+ rhythm showed a phase advance in LD (midpoint phase: control, ZT 4.11 ± 0.38; VIP-deficient, ZT 21.62 ± 0.80, p < 0.001) and a shortened period in DD (control, ~23.7 h; VIP-deficient, ~22.5 h, p < 0.05) (Fig. 1f, g and Supplementary Fig. 1b–d). Notably, the latter was almost identical to the short free-running period of the behavior rhythm (determined based on the locomotor activity onsets), even when behavior changed to a long-period rhythm later in DD (Fig. 1c, g and Supplementary Fig. 2).
These results suggest that AVP neurons intrinsically oscillate with a short period and that VIP enhances and delays the oscillation of AVP neurons to make their period closer to 24 h in DD. In turn, the enhanced AVP neuronal rhythm may confer a robust circadian oscillation of VIP neurons, regulating the timing of VIP release. In LD, light may be unable to appropriately entrain and boost the AVP-Ca2+ rhythm in the absence of VIP.
In this study, we used mice of both sexes and analyzed them collectively. However, we confirmed that the results remain fundamentally unchanged when analyzing males and females separately (e.g., Supplementary Fig. 3; also see “Animals” in the “Methods”). The sex of each mouse is disclosed in the Supplementary Figs. and the Source Data file.
VIP-Ca2+ changes to a weak, long-period rhythm in VIP-deficient mice
In VIP-deficient mice, the period length of AVP-Ca2+ rhythm was comparable with the short-period behavior rhythm in DD. As a result, in more than half of VIP-deficient mice, the AVP-Ca2+ rhythm dissociated from the long-period behavior rhythm in the later stage of DD. The VIP-Ca2+ rhythm in actograms initially followed the short-period AVP-Ca2+ and behavior rhythms but gradually disappeared and became very weak and noisy in DD (Fig. 1c–g and Supplementary Figs. 1 and 2). Considering the multiple periodicities of the behavior rhythm in VIP-deficient mice6,8,9,10, we performed periodogram analyses of Ca2+ rhythms throughout three consecutive weeks in DD to detect weak rhythms with different periods objectively and quantitatively. The VIP-Ca2+ rhythm demonstrated two period peaks in most VIP-deficient mice (Supplementary Fig. 2). Averaging periodograms of all the recorded VIP-deficient mice clarified two peaks in the VIP-Ca2+ rhythm, a primary short period (22.67 h) and a secondary long period (24.33 h), whereas AVP-Ca2+ rhythm showed only one short period (22.50 h) (Fig. 2a; period values are multiple of 1/6 because of 10-min bins of Ca2+ recordings). In contrast, both AVP- and VIP-Ca2+ rhythms of control mice presented a robust single period within the normal range (23.67 h), which matched that of the behavior rhythm (Fig. 2a and Supplementary Fig. 1a). We obtained a similar result by first detecting two period peaks for each mouse and then calculating the mean values for the short and long periods of each mouse (AVP-Ca2+: control, 23.60 ± 0.09 h; VIP-deficient, 22.61 ± 0.07 h; VIP-Ca2+: control, 23.60 ± 0.09 h; VIP-deficient primary, 22.67 ± 0.08 h; secondary, 24.23 ± 0.13 h) (Fig. 2b, c).

a Averaged periodograms of detrended AVP-Ca2+ (green), VIP-Ca2+ (magenta) signals, and the locomotor activity (black) of control and VIP-deficient mice for days 1–20 in DD (10-min bins). Black dotted line, 24 h. b, c Mean period and amplitude (Qp values) of Ca2+ rhythms in DD. Here, peak periods are first calculated from individual mice’s periodograms and then averaged. Time courses of periods (d) and amplitudes (Qp) (e) of AVP- Ca2+ (green), VIP-Ca2+ (magenta), and the locomotor activity (black) rhythms in DD by sequential periodogram analyses with 5-day shifting windows. Values are mean ± SEM. n = 5 for control; n = 8 for VIP-deficient mice. *p < 0.05, **p < 0.01, ***p < 0.001 by two-tailed Student’s or Welch’s t test (b, c), one-way (b, c) or two-way repeated measures (d) ANOVA; ns not significant. The exact p-values are as follows: AVP-Ca2+, <0.0001; VIP-Ca2+, Control vs. VIP-deficient (Primary), <0.0001, Control vs. VIP-deficient (Secondary), 0.0037, VIP-deficient (Primary) vs. VIP-deficient (Secondary), <0.0001 (b), AVP-Ca2+, 0.0009; VIP-Ca2+, Control vs. VIP-deficient (Primary), <0.0001, Control vs. VIP-deficient (Secondary), <0.0001, VIP-deficient (Primary) vs. VIP-deficient (Secondary), 0.1153 (c), Control, 0.9178; VIP-deficient, AVP-Ca2+ vs. VIP-Ca2+, 0.0033, AVP-Ca2+ vs. Locomotor activity, 0.0006, VIP-Ca2+ vs. Locomotor activity, >0.9999 (d). Gray lines indicate the 99.9% confidence levels in χ2 periodograms (a, e). Source data are provided as a Source Data file.
To determine the stage of DD at which the secondary long period occurred, we next calculated the time courses of amplitudes (Qp) and periods of AVP-Ca2+, VIP-Ca2+, and behavior rhythms by sequential periodogram analyses with 5-day shifting windows. Although 5 days may be too short to precisely calculate the period length, it may be sufficient to briefly track the time course of changing periods. In VIP-deficient mice, the peak Qp values of the VIP-Ca2+ rhythm decreased continuously during the first 7–8 days (early stage) and then sustained at a relatively low value until the end of the recording (late stage) (Fig. 2e, right). At the early stage, there was no significant difference in the period length between AVP-Ca2+, VIP-Ca2+, and behavior rhythms (Fig. 2d, right). At the late stage, however, the period of the VIP-Ca2+ rhythm was lengthened close to the long period of the behavior rhythm (VIP-Ca2+ vs. Locomotor activity, p > 0.99), whereas the AVP-Ca2+ period remained short (AVP-Ca2+ vs. VIP-Ca2+, p < 0.01; AVP-Ca2+ vs. Locomotor activity, p < 0.001, Fig. 2d, right and Supplementary Fig. 2). In control mice, all periods of Ca2+ and behavior rhythms were stable and consistent (Fig. 2d, e, right and Supplementary Fig. 1a). These results suggest that VIP neurons may be a subordinate slow oscillator and exhibit a weak, long-period rhythm that correlates with the behavior rhythm period in vivo when the AVP cellular rhythm is attenuated by VIP deficiency. However, the causal relationship between these weak VIP-Ca2+ and behavior rhythms remains unknown.
AVP neurons are responsible for the short-period behavior rhythm in VIP-deficiency
As described above, we observed a correlation between the AVP-Ca2+ and the short-period behavior rhythms in VIP-deficient mice (Figs. 1g and 2d). To test for the causal relationship, we measured the behavior rhythm of VIP-deficient mice with an artificially lengthened TTFL period in AVP neurons due to the specific deletion of the casein kinase 1 delta (CK1δ) gene (Avp-Cre; CK1δflox/flox; ViptTA/tTA = Avp-CK1δ−/−; ViptTA/tTA mice).
As reported previously17, control mice (Avp-CK1δ−/−; Vipwt/tTA) exhibited a robust free-running rhythm in DD with a lengthened period (24.70 ± 0.05 h) (Fig. 3 and Supplementary Fig. 4a). In contrast, Avp-CK1δ−/−; ViptTA/tTA mice showed an attenuated behavior rhythm with unstable periods, the mean of which was significantly shorter than that of control Avp-CK1δ−/− mice (24.23 ± 0.11 h) (Fig. 3 and Supplementary Fig. 4b). However, unlike CK1δ-intact VIP-deficient mice (Fig. 1g and Supplementary Fig. 2), they never initiated the free-running rhythm with a short period around 22.6 h (Fig. 3a, b, e and Supplementary Fig. 4b). These results indicate that the AVP cellular period regulates the short-period component of the behavior rhythm in VIP-deficient mice.

a Representative locomotor activity actograms of Avp-CK1δ−/−; Vipwt/tTA (control) and Avp-CK1δ−/−; ViptTA/tTA mice. Gray shading indicates the time when the light was off. b Day-by-day changes in the periods of locomotor activity rhythms (the intervals between two adjacent locomotor activity onsets) in LD and DD. c Averaged periodograms of the locomotor activity for days 1–20 in DD (1-min bins). d Mean free-running period and amplitude (Qp values) of the locomotor activity rhythm in DD. e Time courses of amplitudes (Qp) and periods of the locomotor activity rhythms in DD by sequential periodogram analyses with 5-day shifting windows. Values are mean ± SEM; n = 6 for control, n = 5 for Avp-CK1δ−/−; ViptTA/tTA mice. *p < 0.05, **p < 0.01 by two-tailed Student’s t test (d) or two-way repeated measures ANOVA (b, e); ns, not significant. The exact p-values are as follows: LD, 0.3960; DD, 0.0133 (b), period, 0.0030; amplitude, 0.0087 (d), amplitude, 0.0274; period, 0.2071 (e). Gray lines indicate the 99.9% confidence levels in χ2 periodograms (c, e). Source data are provided as a Source Data file.
Ca2+ rhythm of CCK neurons in VIP-deficient mice rapidly attenuates in DD
Next, we aimed to know how unique the consistently short-period AVP-Ca2+ and the late-onset, weak, long-period VIP-Ca2+ rhythms are. In particular, CCK neurons enriched in the anterior shell region of the SCN were of interest as another candidate for a long-period oscillator, because they reportedly play an important role in the behavior under a long-day photoperiod, and their ablation slightly shortened the behavioral free-running period in DD19.
To test this possibility, we introduced a Cck-ires-Cre allele24 to VIP-deficient (Cckwt/ires-Cre; ViptTA/tTA) and control (Cckwt/ires-Cre; Vipwt/tTA) mice and labeled CCK and VIP neurons with jGCaMP7s and jRGECO1a, respectively, by injecting AAV vectors into the SCN (Fig. 4a, b). Our fiber photometry recording successfully recapitulated the reported CCK-Ca2+ rhythm in control mice, with its peak in the second half of the (subjective) day (Fig. 4c, d and Supplementary Fig. 5a)19. In VIP-deficient mice, the CCK-Ca2+ rhythm was robust in LD but rapidly attenuated in DD, as did VIP-Ca2+ (Fig. 4c, d and Supplementary Fig. 5b). However, in contrast to the VIP-Ca2+, the averaged periodogram of the CCK-Ca2+ rhythm for 3-week recordings in DD showed only one period peak (22.00 h), which was even slightly shorter than that of AVP-Ca2+ (22.50 h) but with a broad distribution of significant period lengths on the long-period side up to ~24.3 h, i.e., the secondary peak of the VIP-Ca2+ period (Fig. 4f and Supplementary Fig. 5b). Sequential periodogram analysis revealed that the CCK-Ca2+ rhythm gradually reduced its amplitude (Qp) and maintained a short period on average, but with a large variability (Fig. 4g, h and Supplementary Fig. 5b). In contrast, in control mice, CCK-Ca2+, VIP-Ca2+, and behavioral periods were stable and consistent (23.50 h) (Fig. 4f–h and Supplementary Fig. 5a). These results suggest that the CCK-Ca2+ rhythm in VIP deficiency is weak and includes more heterogeneous periods in DD. Furthermore, the late-onset, weak, long-period rhythm is unlikely to be unique to VIP neurons. Rather, it may be a more general feature of the multiple types of SCN cells that appear when AVP neuronal rhythm is attenuated due to VIP deficiency.

a Schematic diagram of viral vector (AAV-CAG-FLEX-jGCaMP7s and AAV-TRE-jRGECO1a) injection and optical fiber implantation at the SCN in control (Cckwt/ires-Cre; Vipwt/tTA) or VIP-deficient mice (Cckwt/ires-Cre; ViptTA/tTA) for fiber photometry recording. b A representative coronal SCN section of mice with jGCaMP7s expression in CCK neurons and jRGECO1a in VIP neurons. A white dotted square shows the estimated position of the implanted optical fiber. Green, jGCaMP7s; magenta, jRGECO1a. Scale bar, 200 μm. Similar images were consistently observed across all mice used in this experiment. c Representative plots of the in vivo jGCaMP7s signal of CCK neurons (CCK-Ca2+, green) and jRGECO1a signal of VIP neurons (VIP-Ca2+, magenta) overlaid with the locomotor activity (black) in actograms. Detrended Ca2+ signals are shown with normalization in each row. Control and VIP-deficient mice were initially housed in 12:12-h LD and then in DD. Gray shading indicates the time when the light was off. Time courses of CCK-Ca2+ (d) and VIP-Ca2+ (e) rhythms for 26 days (6 days in LD, 20 days in DD). Detrended and smoothened data from control (lavender) or VIP-deficient (orange) mice are shown. f Averaged periodogram of detrended CCK-Ca2+ (green), VIP-Ca2+ (magenta) signals and the locomotor activity (black) of control and VIP-deficient mice for days 1–20 in DD (10-min bins). Gray line, significance level; black dotted line, 24 h. Time courses of periods (g) and amplitudes (Qp) (h) of CCK-Ca2+ (green), VIP-Ca2+ (magenta), and the locomotor activity (black) rhythms in DD by sequential periodogram analyses with shifting 5-day windows. *p < 0.05, **p < 0.01 by two-way repeated measures ANOVA; ns, not significant. Values are mean ± SEM. n = 3 for control; n = 6 for VIP-deficient mice (d–h). The exact p-values are as follows: LD, 0.1034; DD, 0.0018 (d), LD, 0.1242; DD, 0.0057 (e), Control, 0.6993; VIP-deficient, CCK-Ca2+ vs. VIP-Ca2+, 0.2802, CCK-Ca2+ vs. Locomotor activity, 0.0825, VIP-Ca2+ vs. Locomotor activity, >0.9999 (g). Gray lines indicate the 99.9% confidence levels in χ2 periodograms (f, h). Source data are provided as a Source data file.
Such heterogeneity may partly reflect that CCK neurons contain at least two subpopulations, one overlapping with a small subpopulation of AVP neurons25,26. To estimate the overlap of AVP and CCK neurons, we mated Cck-ires-Cre mice with Rosa26-LSL-tdTomato (Ai14) reporter mice to label CCK neurons with tdTomato27. Then, we stained their SCN sections with an anti-AVP antibody. Approximately 30% of tdTomato+ cells were also AVP+, whereas ~20% of AVP+ cells were tdTomato+ (Supplementary Fig. 6). These AVP-positive and -negative CCK neurons may correspond to Cck+/C1ql3+ and Cck+/Bdnf1+ cells, respectively, reported by Wen et al.25. Thus, the CCK-Ca2+ rhythm recorded here by fiber photometry likely derived from a mixture of these two CCK neuron subpopulations.
Ablating VIP neurons in adulthood causes a simple short-period rhythm
VIP-deficient ViptTA/tTA mice free-ran initially with a short period in DD, but about 60% of the mice changed the free-running rhythm to a long period after 1–2 weeks (Supplementary Figs. 2 and 5). This observation was consistent with the previous reports6,8,9. On the other hand, mice in which VIP neurons were ablated in adulthood were reported to retain the wheel running rhythm but with a shortened period (~22.7 h)28. We also performed a similar experiment to measure the locomotor activity (home cage activity) rhythm by injecting an AAV vector expressing activated Caspase 3 in a Cre-dependent manner (AAV-CAG-FLEX-taCasp3-TEVp) bilaterally into the SCN of Vipwt/ires-Cre mice24. We obtained a similar result, i.e., a shortened free-running period and reduced amplitude of the rhythm (22.77 ± 0.09 h vs. 23.67 ± 0.04 h, p < 0.001) (Supplementary Fig. 7). We observed an apparent sex difference in the amplitude reduction but not in the shortening of the period in VIP neuron-ablated mice (Supplementary Fig. 7e, f). These results suggest that VIP neurons contribute to circadian timekeeping by lengthening the circadian period and promoting the rhythmicity in adults. Some compensatory mechanisms may cause multiple periods in VIP-deficient mice during development28.
VPAC2 dysfunction in AVP neurons acutely shortens the free-running period
We next investigated the importance of VIP/VPAC2 signaling in AVP neurons for the pacesetting of the SCN network. We used in vivo genome editing to selectively disrupt the Vipr2 gene in AVP neurons. First, we crossed Avp-Cre mice with Rosa26-LSL-SpCas9 mice, which express SpCas9 Cre-dependently29. We then injected AAV vectors expressing gRNAs targeting the Vipr2 (AAV-U6-gVipr2-EF1α-DIO-mCherry, Avp-Vipr2−/− mice) or control gRNAs (AAV-U6-gControl-EF1α-DIO-mCherry) bilaterally into the SCN of Avp-Cre; Rosa26-LSL-SpCas9 mice (Fig. 5a, b). Substantial reduction of VPAC2 expression specific to the SCN shell was confirmed in Avp-Vipr2−/− mice by immunohistochemistry (Fig. 5b, c).

a Schematic diagram showing the injection of AAV-U6-gVipr2-EF1α-DIO-mCherry (Avp-Vipr2−/−) or AAV-U6-gControl-EF1α-DIO-mCherry (control) into the SCN of Avp-Cre; Rosa26-LSL-SpCas9-2A-EGFP mice. b Representative coronal SCN sections prepared from control and Avp-Vipr2−/−mice stained with an anti-VPAC2 antibody. Scale bars, 300 (low magnification) or 50 (high magnification) μm; green, SpCas9-2A-EGFP; magenta, mCherry; light blue, VPAC2 immunostaining). c Quantification of VPAC2 immunostaining in the SCN shell and core regions indicated in (b) by dotted circles. d Representative locomotor activity actograms of control and Avp-Vipr2−/− mice. Recordings were started immediately after the AAV injection to track the course of behavior change. Mice were initially housed in LD for 3 days and then in DD. Gray shading indicates the time when the light was off. e Averaged daily profiles of the locomotor activity of 1st and 2nd stages in DD. f Mean free-running period and amplitude (Qp values) of the locomotor activity rhythm in DD. Because Avp-Vipr2−/− obviously altered the free-running period in the middle of DD, periodogram analysis was performed separately before (1st stage) and after (2nd stage) the transition point for 10 days when the periods were stable. Values are mean ± SEM; n = 6 for control, n = 8 for Avp-Vipr2−/− mice. ***p < 0.001 by two-way repeated measures ANOVA; ns, not significant. The exact p-values are as follows: shell, <0.0001; core >0.9999 (c); period 1st stage, <0.0001; 2nd stage, >0.2723; amplitude 1st stage, 0.5122; 2nd stage, >0.9999 (f). Source data are provided as a Source data file.
Avp-Vipr2−/− mice free-ran with a period significantly shorter than controls when released into DD (23.21 ± 0.05 h vs. 23.73 ± 0.05 h, p < 0.001) (Fig. 5d, f and Supplementary Fig. 8b). Surprisingly, however, after a while, Avp-Vipr2−/− mice suddenly changed their free-running period to the one comparable to controls (23.85 ± 0.03 h vs. 23.71 ± 0.03 h, p = 0.27). This observation led us to start recording locomotor activity immediately after the AAV injection in most of the mice examined, with recordings in LD only for 3 days before DD (Supplementary Fig. 8b). Strikingly, all Avp-Vipr2−/− mice exhibited changes in the free-running period around 21 days after AAV injection. Control mice never showed such a sudden change in the free-running period (Fig. 5d, f and Supplementary Fig. 8a). These results suggest that disruption of VIP/VPAC2 signaling in AVP neurons in adulthood acutely shortens the free-running period due to the reduced phase-delaying effect of VIP on the intrinsic short period of AVP neuronal clocks. On the other hand, the period shortening in Avp-Vipr2−/− mice was less than in VIP-deficient and VIP neuron-ablated mice (23.21 ± 0.05 h vs. 22.67 ± 0.08 h and 22.77 ± 0.09 h). Moreover, the shortening was abolished ~3 weeks after the blockade of the VIP → AVP neuron axis. These observations may be due to an incomplete elimination of VPAC2 from AVP neurons. In addition, we speculate that the indirect pathways may exist that mediate the regulation of AVP neurons by VIP neurons and are involved in the compensatory recovery of the standard period in Avp-Vipr2−/− mice.
Disrupting TTFLs in AVP neurons leaves an attenuated, long-period VIP-Ca2+ rhythm
We previously reported that Bmal1 deletion specific to AVP neurons (Avp-Cre; Bmal1flox/flox = Avp-Bmal1−/− mice) leaves an attenuated circadian behavior rhythm with a lengthening of the free-running period and the activity time12. In contrast, mice become completely arrhythmic when Bmal1 is deleted in all SCN neurons using forebrain-specific CamKII-Cre driver mice16,30. These results may be explained by the weak circadian oscillators of SCN non-AVP cells with a long period as a population in vivo, including VIP and CCK neurons as described above. To test this possibility, we recorded AVP-Ca2+ and VIP-Ca2+ rhythms in Avp-Bmal1−/− mice. To do so, we generated Avp-Bmal1−/−; Vipwt/tTA mice and injected AAV vectors into the SCN (Fig. 6a, b). Similar fiber photometry recordings from Avp-Cre (Bmal1wt/wt); Vipwt/tTA mice (Figs. 1 and 2) were regarded as controls. As expected, the AVP-Ca2+ exhibited little circadian rhythmicity (Fig. 6c, d, f and Supplementary Fig. 9b, c) in Avp-Bmal1−/−; Vipwt/tTA mice. Notably, the VIP-Ca2+ retained a rhythm with a long period (24.54 ± 0.16 h) that is consistent with the period of the behavior rhythm (24.47 ± 0.16 h) in DD (Fig. 6e–g and Supplementary Fig. 9b, c). The duration of high VIP-Ca2+ in the subjective day was compressed according to the prolongation of the activity time of the behavior rhythm (Fig. 6c and Supplementary Fig. 9b). Consistent with the previous report12, one out of six Avp-Bmal1−/−; Vipwt/tTA mice demonstrated the multiple period phenotype. In this mouse, the VIP-Ca2+ rhythm was significant but largely attenuated (Supplementary Fig. 9b, mouse #6). These results support the idea that an attenuated oscillation of non-AVP cells with a long period on average manifests in the circadian behavior rhythm when the AVP cellular clock is disrupted.

a Schematic diagram of viral vector (AAV-CAG-FLEX-jGCaMP7s and AAV-TRE-jRGECO1a) injection and optical fiber implantation at the SCN in Avp-Bmal1−/−; Vipwt/tTA mice for fiber photometry recording. b A representative coronal SCN section of mice with jGCaMP7s expression in AVP neurons and jRGECO1a in VIP neurons. A white dotted square shows the estimated position of the implanted optical fiber. Green, jGCaMP7s; magenta, jRGECO1a. Scale bar, 200 μm. Similar images were consistently observed across all mice used in this experiment. c Representative plots of the in vivo jGCaMP7s signal of AVP neurons (AVP-Ca2+, green) and jRGECO1a signal of VIP neurons (VIP-Ca2+, magenta) overlaid with the locomotor activity (black) in actograms. For comparison, representative actograms of an Avp-Cre; (Bmal1wt/wt); Vipwt/tTA analyzed in Figs. 1 and 2, but different from the one shown in Fig. 1c, are also presented. Detrended Ca2+ signals are shown with normalization in each row. Mice were initially housed in LD and then in DD. Gray shading indicates the time when the lights were off. Time courses of AVP-Ca2+ (d) and VIP-Ca2+ (e) rhythms for 26 days (6 days in LD, 20 days in DD). Detrended and smoothened data from control (lavender) or Avp-Bmal1−/−; Vipwt/tTA (orange) mice are shown. f Averaged periodogram of detrended AVP-Ca2+ (green), VIP-Ca2+ (magenta) signals, and the locomotor activity (black) for the last 7 days in DD (10-min bins). g Mean period and amplitude (Qp values) of Ca2+ and behavior rhythms in DD. Here, peak periods are first calculated from individual mice’s periodograms and then averaged. Values are mean ± SEM. n = 5 for control (recordings same as those in Figs. 1 and 2); n = 5 for Avp-Bmal1−/−; Vipwt/tTA mice. *p < 0.05, **p < 0.01, ***p < 0.01 by two-way repeated measures ANOVA; ns, not significant. The exact p-values are as follows: LD, 0.0697; DD, 0.0337 (e); period AVP-Ca2+, 0.0031; VIP-Ca2+, 0.0082; locomotor activity, 0.0205); amplitude AVP-Ca2+, < 0.0001; VIP-Ca2+, 0.0045; locomotor activity, 0.2281 (g). Gray lines indicate the 99.9% confidence levels in χ2 periodograms (f). Source data are provided as a Source data file.
Blocking neurotransmitter release from AVP neurons lengthens the free-running period
To further explore the evidence that AVP neurons act as the principal oscillator with an intrinsically short period, but its attenuation uncovers a weak, long-period circadian oscillation of non-AVP cells, we next examined the behavioral changes in mice in which the neurotransmitter release from AVP neurons was blocked by AAV-mediated expression of tetanus toxin light chain (Avp-TeNT mice, AAV-EF1α-DIO-GFP::TeNT injected bilaterally into the SCN of Avp-Cre mice) (Fig. 7a, b). Avp-TeNT mice showed a longer free-running period (24.48 ± 0.06 h) in DD than control EGFP-expressing mice (23.28 ± 0.06 h, p < 0.001, Fig. 7c–e and Supplementary Fig. 10). These data further confirmed that AVP neurons intrinsically encode a short-period circadian rhythm.

a Schematic diagram showing the injection of AAV-EF1α-DIO-GFP::TeNT or AAV-CAG-FLEX-EGFP (control) into the SCN of Avp-Cre mice. b Representative coronal SCN sections of mice with GFP::TeNT (immunostained with anti-GFP antibody) or EGFP expression. Scale bar, 200 μm. Similar images were consistently observed across all mice used in this experiment. c Representative locomotor activity actograms of Avp-EGFP (control) or Avp-TeNT mice. Animals were initially housed in LD and then in DD. Gray shading indicates the time when the light was off. d Averaged periodogram of locomotor activity for days 1–20 in DD (1-min bins). e Mean free-running period and amplitude (Qp values) of the locomotor activity rhythm in DD. Values are mean ± SEM; n = 7 for Avp-EGFP, n = 15 for Avp-TeNT mice. ***p < 0.001 by two-tailed Student’s t test; ns not significant. The exact p-values are as follows: period, <0.0001; amplitude, 0.8577 (e). Gray lines indicate the 99.9% confidence levels in χ2 periodograms (d). Source data are provided as a Source data file.
The amplitude of the locomotor activity rhythm (periodogram Qp) was not significantly different between groups (p = 0.86, Fig. 7d, e). However, individual Avp-TeNT mice exhibited diverse daily patterns of locomotor activity that deviated significantly from the normal nocturnal and bimodal pattern with clear evening (E) and morning (M) locomotor activities (Fig. 7c and Supplementary Fig. 10b). To our surprise, many Avp-TeNT mice showed varying degrees of diurnal locomotor activity rhythms in both LD and DD. To understand the underlying cause, we followed the time course of the emergence of apparent diurnal rhythms in LD after the onset of TeNT expression. Interestingly, TeNT expression in AVP neurons rapidly altered the coupling of E and M locomotor activities (Supplementary Fig. 10c). Namely, Avp-TeNT mice quickly exhibited a prolonged activity time, specifically manifested as a slight advancement in locomotor activity onset (E activity) and a significant delay in locomotor activity offset (M activity). A new steady state of the M-E (M is closer to the following E than to the preceding E) phase relation, rather than the normal E-M phase relation, generated an apparent diurnal behavior rhythm in some mice. Such features can be regarded as a more severe phenotype of the lengthened activity time observed in Avp-Bmal1−/− and Avp-Vgat−/−mice12,20. It should be noted that the free-running period was consistently lengthened in Avp-TeNT mice regardless of their diverse daily patterns of locomotor activity. Thus, these data suggest that blocking neurotransmission of AVP neurons impairs both the pacesetting of the ensemble period and the phase-setting of both E and M locomotor activities.
Neuronal feedback loop model of the SCN explains experimental results
Taken together with previous findings that AVP neurons act as the primary circadian pacesetter in vivo3,12,16,17, the above results suggest a feedback loop neural circuit of AVP and VIP neurons in the SCN (SNFL: SCN neuronal feedback loop) as a critical network machinery of the mammalian central clock (Fig. 8a), which generates robust circadian rhythms with a period of ~24 h in DD. In this context, AVP neurons are the principal oscillatory part of the SCN and regulate the circadian rhythms of VIP and other SCN cells16. AVP neurons constitute a fast oscillator with an intrinsically short period. VIP peptide, released rhythmically, acts on AVP neurons, delaying the AVP neuronal rhythm every day to lengthen its period closer to 24 h and increase its amplitude (Fig. 8a, right). In addition to this primary direct pathway, there may also be minor indirect routes through which VIP regulates AVP neurons via other SCN cells (Fig. 5). Consequently, in the absence of VIP, the AVP neuronal rhythm expresses an intrinsic short period in the attenuated behavior rhythm (Fig. 8a, middle). However, when AVP neuronal oscillation reduces its amplitude too much to control the various clock cells in the SCN, which have different cellular period lengths but an average long period, a weak long-period behavior rhythm emerges (background circadian oscillation or default clock state, discussed below in the Discussion) (Fig. 8a, left). Such rhythms also appear when the pacesetting ability of AVP neurons is impaired by specifically disrupting TTFL or blocking transmitter release. They also correlate with weak, slow Ca2+ rhythms in non-AVP cells, such as VIP neurons.

a Schematic diagram of the SCN Neuronal Feedback Loop (SNFL). As a default state or background oscillation, the SCN is an assembly of weak oscillators with varying periods but slightly longer than 24 h on average in vivo (left). On top of that, the SNFL of AVP and VIP neurons enables the SCN to autonomously generate a robust circadian rhythm with a period of around 24 h (right). Here, AVP neurons are the principal oscillatory part and regulate the circadian rhythm of VIP and other cells. However, they have an intrinsically short period. VIP, in turn, acts on the AVP neurons, delaying their rhythm to lengthen their period closer to 24 h and also amplifying their rhythm daily in DD. In the absence of VIP, the AVP neuronal rhythm is attenuated but still expresses its intrinsic short period in the behavior rhythm (middle). However, when these neurons further reduce rhythmicity and lose control over the default oscillators in the SCN, background oscillation emerges (left). b Schematic diagram (top) and a modified Kuramoto model (middle) illustrating the VIP effect in lengthening the period of AVP neurons. The model consists of two coupled oscillators (A: AVP neurons, nA: non-AVP cells including VIP neurons) with an inhibitory term mediated by VIP (KV). Parameters: θA, θnA (phase of oscillator A and nA); ωA, ωnA (intrinsic angular velocities, ωA = 2π / 22.7 h, ωnA = 2π / 24.3 h); KnA→A (coupling strength from nA to A); KA→nA (coupling strength from A to nA); KV (VIP-mediated inhibitory modulation, active only when sin(θnA) > 0 and sin(θA - π/2) > 0. (bottom) Heatmaps of simulated AVP, non-AVP, and ensemble periods. The ratios between KA→nA and KnA→A (α) is set to 0, 0.2, 1, 5, and 25. In normal mice, α is postulated to be 5 or 25, considering the dominant role of the AVP cellular oscillator. Black regions indicate the final ensemble periods between oscillators A and nA differed by > 0.2 h. A 25-h period and those exceeding it are displayed in the same color.
We next built a simplified mathematical model consisting of two oscillators to test if the free-running period of normal mice (i.e., the synchronized, SCN ensemble period) could be explained by the periods of AVP- and VIP-Ca2+ rhythms measured in this study (Fig. 8b and Supplementary Fig. 11). Here, the VIP-Ca2+ period represented non-AVP cells. The oscillations were obtained using a phase oscillator model with coupling31. Based on the Ca2+ rhythms in VIP-deficient mice in DD (Fig. 2a), we estimated the in vivo intrinsic period lengths of the dominant fast oscillator (i.e., AVP neurons: τA) and the presumed weak slow oscillator (non-AVP cells: τnA) to be ~22.7 h and ~24.3 h, respectively. The free-running periods of Ca2+ and the locomotor activity rhythm in control mice were ~23.7 h (Figs. 1g and 2a) and regarded as the normal SCN ensemble period.
Since AVP neurons are dominant, the coupling strength from AVP to non-AVP cells (KA→nA) should be greater than the coupling strength from non-AVP to AVP cells (KnA→A). We set KA→nA = 5x KnA→A (α = 5) based on an explant study13, or 25× KnA→A (α = 25) considering the reports that AVP neurons may control other SCN cells more effectively in vivo than ex vivo16,17. As in the SNFL model, we paid particular attention to the phase-delaying effect of VIP (KV) by introducing a separate term to the AVP neuronal oscillator equation. We roughly estimated that VIP is released during subjective day (CT0 ~ 12) when VIP-Ca2+ is high (Fig. 1) (red underline in Fig. 8b). Additionally, based on the phase response curves of the SCN explant PER2::LUC rhythm to VIP application32 and the behavior rhythm to in vivo optogenetic stimulation of VIP neurons33, we assumed that AVP neurons respond to VIP around CT6–CT18 (green underline in Fig. 8b) primarily by delaying their phases. Using these equations, we identified parameter ranges that produce a synchronized ensemble period of ~23.7 h (Fig. 8b, bottom). In contrast, without the additional special term (Fig. 8b, KV = 0), two oscillators synchronized; however, the resultant period remained close to the short period of the AVP neuronal oscillator and never approached the measured value of ~23.7 h when the AVP cellular oscillator dominated the non-AVP cellular oscillator (α = 5 or 25).
In VIP-deficient mice, VIP’s phase-delaying effect (KV) was reduced to zero, and both AVP (KA→nA) and non-AVP (KnA→A) cellular rhythms were weakened. Nevertheless, as far as the AVP neuronal oscillator dominated the non-AVP cellular oscillator (α = 5 or 25; KnA→A is not too close to zero), the two oscillators synchronized, expressing a short period close to that of the AVP cellular oscillator. However, when the AVP cellular oscillator was attenuated to the point of losing control over non-AVP cells (α and KnA→A approaching zero), AVP and non-AVP cells oscillated with different short and long periods, respectively. This is similar to what was often observed in the later stage of DD. In Avp-Vipr2−/− mice, the synchronized period was shortened due to reduced KV. In contrast, in Avp-Bmal1−/− and Avp-TeNT mice, the periods lengthened due to reduced KA→nA (i.e., α = 0 or 0.2). Furthermore, this model could similarly explain the period length of Avp-CK1δ−/−; Vipwt/tTA and Avp-CK1δ−/−; ViptTA/tTA mice by just lengthening the AVP cellular intrinsic period by 1–1.5 h (Supplementary Fig. 11d).
Thus, the SNFL model may appropriately represent the logic of SCN pacesetting, both qualitatively and quantitatively.
Discussion
First, this study used fiber photometry to record Ca2+ rhythms in neuronal populations defined by the expression of specific neuropeptides (i.e., AVP, VIP, or CCK)11,16,19,20,21. Therefore, the estimated period, phase, and amplitude values may reflect a mixture of various circadian oscillations from multiple single neurons within each population. In addition, each population still contains molecularly heterogeneous subpopulations25,26,34. Thus, we could not determine whether the attenuation of Ca2+ rhythms in VIP-deficient mice resulted from the heterogeneity of recorded neuronal populations, a reduction in the synchrony between individual recorded neurons, or diminished cellular oscillations within each recorded neuron. On the other hand, the detection of significant Ca2+ rhythms in VIP-deficient mice indicates that there is some degree of circadian synchrony between the recorded cells.
Intriguingly, Stowie et al. recorded single-cell Ca2+ rhythms in SCN AVP neurons of normal mice in vivo and concluded that most individual AVP neurons do not exhibit significant circadian Ca2+ rhythms in either LD or DD35. However, these cells showed significant rhythmicity at the population level when the equal contributions of cells from each recorded mouse to the population measure were ensured (Bootstrapping analysis in Stowie et al). In contrast, individual AVP neurons in SCN explants were reported to demonstrate clear circadian Ca2+ rhythms36, and AVP neurons reportedly exhibited a circadian rhythm of Fos expression in vivo19. It might just be possible that AVP neurons show a Ca2+ rhythm at the population level without rhythmicity at the single-cell level in vivo. At the same time, this apparent discrepancy might derive from differences in experimental conditions, such as the number of cells recorded, signal-to-noise ratio that depends on GCaMP expression levels, sampling frequency, and the recording duration.
Thus, while individual AVP neurons may be sloppy circadian oscillators with significantly diverse periods and phases, the AVP neuronal population may show a significant circadian Ca2+ rhythm, further underscoring the importance of VIP signaling as the feedback amplifier and synchronizer of their oscillation. As discussed above, the fiber photometry recordings in this study had some technical limitations, which may have led to oversimplification. Nevertheless, this method is, to date, the only one that allows for long-term (more than a week) recordings of SCN dynamics sufficient to measure the free-running periods in genetically defined neuronal populations in vivo. Furthermore, the dual-color fiber photometry approach established in this study enabled simultaneous in vivo monitoring of Ca2+ rhythms in two types of neurons, together with locomotor activity. As discussed below, the results provided essential information for proposing the SNFL model (Fig. 8) as the network mechanism underlying the SCN central clock.
By explicitly considering the effect of VIP signaling to phase-delay the AVP neuronal rhythm, the SNFL model successfully explains seemingly contradictory experimental results: the integrated ensemble period of the SCN network determined by the coupling of stronger fast (AVP neurons) and weaker slow (other unidentified cell populations, including VIP neurons) oscillators is closer to that of the weak oscillator. We previously proposed that the TTFL period of AVP neurons is the primary determinant (i.e., pacesetter) of the SCN ensemble period in vivo16. This view remained unchanged in this study but was further updated to account for the ensemble period length being ~1 h longer than the intrinsic AVP cellular period due to the phase-delaying effect of VIP signaling. In this sense, AVP neurons are not fully autonomous pacesetters. Instead, they are equipped with feedback neural circuits that perfect their role as the pacesetter. Such a feedback loop circuit may also explain the observation that the VIP-Ca2+ rhythm quickly attenuated in DD in the absence of VIP even though VIP neurons express little VPAC226,34.
The scheme of the SNFL model fits well with the reported significant phase-delaying effects of VIP and VIP neuronal activation on the SCN and behavior rhythms32,33,37,38, which has been regarded as the cellular/molecular mechanism of phase-delay by the external light pulse. Here, we propose that a similar mechanism operates daily on AVP neurons for endogenous SCN timekeeping in DD, where no external zeitgeber is present. The exact intracellular events downstream of VPAC2 in AVP neurons to delay their TTFLs remain elusive. However, they may resemble those of SCN explants responding to VIP peptide application, such as activation of cAMP, phospholipase C, and MAPK/ERK pathways that lead to PER2 induction32,39. Because AVP and VIP neurons are reciprocally connected directly and indirectly16,40, the intra-SCN network may be sufficient for SNFL function. However, this model does not exclude the involvement of extra-SCN neurons, because the SCN also receives inputs from its surroundings41.
Besides, the SNFL scheme is highly flexible in setting the ensemble period by adjusting the phase-delaying power of VIP signaling (KV), even longer than that of the long-period weak oscillator. Therefore, it may be suitable for light entrainment by transiently modifying the VIP signaling intensity and subsequently the ensemble period to cause phase shifts in both directions. It should be noted that the SNFL model does not exclude the functions of AVP and VIP neurons as the output arms of the SCN, such as those regulating timed drinking behavior42, neurosecretion via the SCN-OVLT portal system43, and corticosterone release44,45. This view may be analogous to the intracellular timekeeping in which TTFL ticks the cellular clock and also drives output clock-controlled genes in individual cells.
Approximately 40 % of Vip−/− or Vipr2−/− mice demonstrate an attenuated circadian behavior rhythm with a single short period in DD6,8,9,10. Even in Vip−/− or Vipr2−/− mice showing multiple periodicities, the dominant periods are often short6. Indeed, ablation of VIP neurons in adult mice results in a single short period (Supplementary Fig. 7)28, suggesting that the multiple periodicities may result from a chronic lack of VIP signaling throughout development. Furthermore, the absence of VPAC2 in AVP neurons significantly shortened the free-running period (Fig. 5), underscoring the importance of the VIP neuron → AVP neuron peptidergic axis38. Intriguingly, however, the period shortening caused by Vipr2 mutation in AVP neurons was less than that caused by VIP deficiency and disappeared after ~3 weeks. Therefore, it is tempting to speculate that the indirect, redundant, and compensatory pathways play a role in regulating the AVP neuronal period by VIP to tightly control the SCN ensemble period (Fig. 8a).
It remains unknown whether such a short-period rhythm of AVP neurons is inherent in individual neurons or arises from the in vivo condition. Several studies have reported that AVP-rich regions of the SCN show shorter periods in organotypic slices13,46,47,48,49,50. Alternatively, in vivo conditions of the SCN, such as the network among AVP neurons and the interaction between AVP and other neurons, may set the short period of the AVP neuronal population. Indeed, we previously reported that the ability of AVP neurons to set the SCN ensemble period in vivo is lost in slices16.
Because AVP neurons were responsible for the short-period behavior rhythm, we next questioned the identity of the slow oscillators relevant to the long-period behavior rhythm in VIP-deficient mice. CCK neurons were a good candidate because they have been reported to track the onset of circadian behavioral activities and to coordinate the circadian activities under long photoperiods. Moreover, ablation of CCK neurons shortened the free-running period in DD by 10 ~ 20 min, although their TeNT expression did not alter the period19. We expected the presumable slow oscillator to continue consistently and independently of the fast oscillator in DD. However, the CCK-Ca2+ rhythm behaved similarly to VIP neurons in VIP-deficient mice, initially following the fast oscillator and rapidly reducing its amplitude. These observations may rule out the possibility that CCK neurons act as the discrete slow oscillator in DD. On the other hand, the slight shortening of the free-running period in DD by ablating CCK neurons may reflect their contribution to the indirect pathway controlling the AVP neurons’ short-period oscillation by VIP, which was discussed above. This idea is supported by the fact that CCK neurons respond to the VIP neuronal activation and activate AVP neurons19.
Instead, VIP neurons often recovered weak Ca2+ rhythms with a lengthened period comparable to the behavioral period at the late stage after the initial attenuation. However, the causal relation between these weak VIP-Ca2+ and behavior rhythms remains unknown. Because these VIP neurons lack VIP peptide and thus have substantially attenuated effects on the other cells, their weak, slow Ca2+ rhythm may be a passive, merely correlated oscillation. The late-stage periods of the CCK-Ca2+ rhythm were more variable, ranging from a shorter period than AVP-Ca2+ to a longer period similar to VIP-Ca2+. Therefore, the late-onset, slow, weak Ca2+ rhythms may be a general feature of multiple types of SCN cells, potentially reflecting something like a background circadian oscillation or a default clock state of the SCN, which may play a marginal role in the pacesetting of the intact SCN network (Fig. 8). Indeed, these long-period Ca2+ rhythms appear only when the functions of AVP neurons are attenuated (e.g., VIP-deficient mice, Avp-Bmal1−/− mice, and Avp-TeNT mice).
What makes such a presumable background oscillation? In mammals, almost every cell throughout the body has a TTFL, as do all SCN cells, with widely varying period lengths1,2. Therefore, it is not surprising that weak circadian rhythms with various periods emerge when the control of primary oscillators (AVP neurons) over the rest of the SCN cellular oscillators is attenuated. Indeed, TTFL disruption in all SCN neurons resulted in a complete arrhythmicity (CaMKII2α-Bmal1−/− mice)16,30. Why is the background oscillation period longer than 24 h? It is challenging to know the accurate intrinsic cellular periods of individual SCN cells in vitro, as they seem to depend largely on culture conditions (e.g., slice or dispersal, density of dispersal) and measurement methods (e.g., clock gene reporters, Ca2+, firing rate). Nevertheless, not all, but many mouse studies using Per1-Luc, Per2::Luc, or GCaMP reporters suggested that the average of highly heterogeneous cellular periods is slightly longer than 24 h51,52,53,54,55. Such a feature is common to peripheral tissues and even to fibroblasts53,56,57,58. Because the regulation of the SCN network by AVP neurons attenuates ex vivo16, the SCN slices may tend to show circadian periods longer than 24 h. The firing rate rhythms of individual SCN neurons also demonstrate diverse period lengths with an average of around 23.5 h in neonatal dispersals or slices of normal mice6,59,60,61,62,63. Notably, VIP- or VPAC2-deficiency further broadens their distribution, which is consistent with the emergence of multiple periodicities in the behavior rhythms6,63.
TeNT expression in AVP neurons not only lengthened the free-running period but also altered the phase relationship of E and M locomotor activities, resulting in an apparent split or diurnal behavior rhythm in most mice examined. This phenotype was consistent with the lengthened activity time in Avp-Bmal1−/− and Avp-Vgat−/− mice12,20, suggesting that AVP neurons have two parallel functions: pacesetting of the ensemble period and phase-setting of both E and M locomotor activities. Previous reports have shown that GABAergic transmission from AVP neurons may play little role in the pacesetting but regulates the timing of SCN neuronal firing, temporally restricting circadian locomotor activity to appropriate time windows in the AVP cellular clock20. Blocking the entire neurotransmission of AVP neurons by TeNT might impair both pacesetting and phase-setting functions of these neurons, resulting in a long-period behavior rhythm with a split phenotype more severe than that of Avp-Vgat−/− mice. At the same time, the existence of both E and M locomotor activities in many Avp-TeNT mice, albeit split, suggests that AVP neurons are unlikely to contain neurons (or E and M oscillators) that send the timed output signals to the locomotion center. Rather, AVP neurons may transmit the timing information to output neurons, indicating when locomotor activity should occur. Importantly, this view is compatible with the encoding of photoperiod by the phase distribution of clock neurons in the SCN64. A comparison to the standard dual oscillator model, especially that of the fly’s central clock network, is discussed further in the Supplementary Discussion.
In most species, the behavioral free-running periods differ significantly from 24 h and are adjusted to 24 h by the external LD cycle. This mechanism has been considered advantageous for stabilizing the phase relationship between the LD cycle and the behavioral rhythm65. A similar strategy may be used in the central clock network to stabilize the ensemble period of the free-running rhythm, in which the VIP signal entrains AVP neuronal oscillators daily. Such a network architecture may also be adaptive to the photic entrainment.
Methods
Animals
Avp-Cre (C57BL/6J-Tg(Avp-icre)#Meid/Rbrc, RBRC12048) and Vip-tTA (B6(Cg)-Vipem1(tTA2)Miem/Rbrc, RBRC12109) mice were reported previously10,12. Cck-ires-Cre (Ccktm1.1(cre)Zjh/J, JAX:012706)24, Rosa26-LSL-tdTomato (Ai14) (B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J, JAX:007914)27, Rosa26-LSL-SpCas9-2A-EGFP (B6J.129(B6N)-Gt(ROSA)26Sortm1(CAG-cas9*,-EGFP)Fezh/J, JAX:026175)29, Vip-ires-Cre (Viptm1(cre)Zjh/J, JAX:010908)24, CK1δ flox (B6.129S4-Csnk1dtm1Drw/J, JAX:010487)66, and Bmal1 flox (B6.129S4(Cg)-Bmal1tm1Weit/J, JAX:007668)67 mice were obtained from Jackson Laboratory. All lines were congenic on C57BL/6J. We compared the mutant mice with controls whose genetic backgrounds were comparable. Avp-Cre, Cck-ires-Cre, Vip-ires-Cre, Rosa26-LSL-tdTomato, and Rosa26-LSL-SpCas9-2A-EGFP mice were used in hemizygous or heterozygous conditions. We used both male and female mice in our experiments. Whether we pooled data from both sexes or analyzed them separately, the conclusions we reached remained almost the same. Previous studies reported slight but statistically significant sex differences in the behavior rhythms of VIP knockout mice (e.g., 0.05 h longer free-running period in females)68,69. However, the differences observed between genotypes in this study were large enough not to be affected by sex differences. The sex of individual mice is described in Supplementary Figs. and the Source Data file. Mice were maintained under a strict 12-h light/12-h dark cycle in a temperature- and humidity-controlled room and fed ad libitum. All experimental procedures involving animals were approved by the appropriate institutional animal care and use committees of Kanazawa University.
Viral vector and surgery
The AAV-2 ITR containing plasmid pGP-AAV-CAG-FLEX-jGCaMP7s-WPRE (Addgene plasmid #104495, a gift from Dr. Douglas Kim and GENIE Project)22, was obtained from Addgene. pAAV-TRE-ChR2-YFP (Addgene #171622, a gift from Hyungbae Kwon)70, was modified to construct pAAV-TRE-jRGECO1a by replacing ChR2-YFP cDNA with a jRGECO1a cDNA fragment amplified by PCR from the plasmid pAAV.CAG.Flex.NES-jRGECO1a.WPRE.SV40 (Addgene #100852, a gift from Douglas Kim & GENIE Project)23, using the following primers: 5′-CTAGGATCCGCCACCATGCTGCAGAACGAGCTTGC-3’ and 5’-CTGAAGCTTTCACTTCGCTGTCATCATTTGTAC-3′. Then, an EcoRI-HindIII fragment of the resultant plasmid containing jRGECO1a sequence was used to replace an EcoRI-HindIII fragment containing ChrimsonR-mCherry from pAAV-TRE-ChrimsonR-mCherry (Addgene #92207, a gift from Alice Ting)71. pAAV-CAG-FLEX-taCasp3-TEVp was made by replacing a BamHI-EcoRV fragment containing EGFP of pAAV-CAG-FLEX-EGFP20 with a BamHI-EcoRV fragment containing taCasp3-TEVp from pAAV-flex-taCasp3-TEVp (Addgene #45580, a gift from Nirao Shah & Jim Wells)72. pAAV-U6-gVipr2-EF1α-DIO-mCherry, a plasmid for CRISPR-Cas9-mediated Vipr2 gene disruption, was generated as follows. The target sites for CRISPR-Cas9 were designed by CRISPOR73. Two sequences targeting Vipr2 gene were selected: 5′-TGCTGGCGCCCGGCAGACGT-3′ and 5′-CCAGATTTCATAGATGCGTG-3′. Oligonucleotides encoding the guide sequences were cloned into the BbsI and BsaI sites of pX333 (Addgene #64073, a gift from Dr. Andrea Ventura)74. Then, a fragment containing two tandem units of U6-gRNA was amplified by PCR, using the following primers: 5′-agtacgcgTCGAGCATGCTCGAGAATGG-3′ and 5′-agtacgcgtCGGGTACCCCATTTGTCTGC-3′, and cloned into the MluI site of pAAV-EF1a-DIO-mCherry (a gift from Dr. Bryan Roth) as described previously75. pAAV-U6-gControl-EF1α-DIO-mCherry contains spacer sequences from pX333 instead of gRNA sequences for Vipr2. pAAV-EF1α-DIO-GFP::TeNT was kindly provided by Dr. Richard D. Palmiter76.
Recombinant AAV vectors (AAV2-rh10) were produced using a triple-transfection, helper-free method and purified as described previously12. Briefly, 293A cells (Thermo Fisher Scientific) were transfected with pHelper (Stratagene), pAAV2-rh10 (provided by Penn Vector Core), and one of AAV-2 ITR containing plasmids using a standard calcium phosphate method. Three days later, the transfected cells were collected and suspended in PBS containing 1 mM MgCl2. After 2 freeze-thaw cycles, the cell lysate was treated with benzonase nuclease (Merck) at 37 °C for 30 min, and centrifuged 2 times at 16,000 × g for 10 min. The supernatant was used as the virus-containing solution. The titers of recombinant AAV vectors were determined by quantitative PCR: AAV-CAG-DIO-jGCaMP7s, 3.4 × 1013; AAV-TRE-jRGECO1a, 8.6 × 1012; AAV-CAG-FLEX-taCasp3-TEVp, 5.7 × 1012; AAV-CAG-FLEX-nls-mCherry, 6.1 × 1012; AAV-U6-gVipr2-EF1α-DIO-mCherry, 6.8 × 1012; AAV-U6-gControl-EF1α-DIO-mCherry, 2.6 × 1012; AAV-EF1α-DIO-GFP::TeNT, 1.8 × 1012 and AAV-CAG-FLEX-EGFP, 1.0 × 1013 genome copies/ml. Stereotaxic injection of AAV vectors was performed as described previously12. Two weeks or immediately after surgery, we began monitoring the mice for their locomotor activity.
In vivo fiber photometry
We used 8 Avp-Cre; ViptTA/tTA, 6 Cckwt/ires-Cre; ViptTA/tTA, 8 control (5 Avp-Cre; Vipwt/tTA and 3 Cckwt/ires-Cre; Vipwt/tTA), and 6 Avp-Bmal1−/− × Vip-tTA (Avp-Cre; Bmal1flox/flox; Vipwt/tTA) mice. Mice were anesthetized by administering a cocktail of medetomidine (0.3 mg/kg), midazolam (4 mg/kg), and butorphanol (5 mg/kg) and were secured to the stereotaxic apparatus (Muromachi Kikai). Lidocaine (1%) was applied for local anesthesia before making the surgical incision. We drilled a small hole in the exposed region of the skull using a dental drill. We injected 1.0 μL of the mixture of AAV-CAG-FLEX-jGCaMP7s and AAV-TRE-jRGECO1a (ratio 1:1, flow rate = 0.1 μL/min) into the right SCN (posterior: 0.5 mm, lateral: 0.25 mm, depth: 5.7 mm from the bregma) with a 33 G Hamilton syringe (1701RN Neuros Syringe, Hamilton) to label AVP and VIP neurons. We then placed an implantable optical fiber (400 μm core, N.A. 0.39, 6 mm, ferrule 2.5 mm, Thorlabs or RWD) above the SCN (posterior: 0.2 mm, lateral: 0.2 mm, depth: 5.3 mm from the bregma) with dental cement (Super-bond C&B, Sun Medical). The dental cement was painted black. Atipamezole (0.3 mg/kg) was administered postoperatively to shorten the anesthetized period. Mice were used for experiments 2–7 weeks after the virus injection and optical fiber implantation. Their ages ranged from 3 to 10 months, including both males and females.
A fiber photometry system (FP3002, Neurophotometrics) was used to record the calcium signal of SCN neurons in freely moving mice77,78. Excitation light sources were a 470-nm LED for detecting Ca2+-dependent jGCaMP7s fluorescence signal (F470), a 560-nm LED for detecting Ca2+-dependent jRGECO1a fluorescence signal (F560), and a 415-nm LED for Ca2+-independent isosbestic fluorescence signal (F415). The duration of the excitation lights was 50 ms, and the onsets of the excitation timing of the LEDs were interleaved. The lights passed through excitation bandpass filters, dichroic mirrors, and then to the animal via fiber-optic patch cords (BBP(4)_400/440/900-0.37_1m_FCM-4xFCM_LAF, MFP_400/440/LWMJ-0.37_1m_FCM-ZF2.5_LAF, Doric Lenses) and the implanted optical fiber. Subsequently, all signals were detected using a CMOS camera through the optical fibers, dichroic mirrors, and emission bandpass filters. The green emission signal and the red emission signal were recorded separately and simultaneously. Ca2+ signals were recorded every 10 min for 30 s using Bonsai software at a sampling rate of 6.6 Hz for each color. The excitation intensities of the 470-nm, 560-nm, and 415-nm LEDs at the tip of the patch cord on the animal side ranged from 60 to 120 μw. During recording, the mouse was housed in a 12-h light–dark cycle for more than 7 days (LD condition) and then transferred to continuous darkness for more than 21 days (DD condition) in a custom-made acrylic cage surrounded by a sound-attenuating chamber.
Ratio (R) was defined as the ratio between F470 and F415 in green channel (F470/F415 green) or the ratio between F560 and F415 in red channel (F560/F415 red) for calibration and reducing motion artifacts. R was averaged within a 30-s session20. To detrend the gradual decrease of the signal during the recording days, ±12 h average from the time (145 points) was calculated as baseline (R0). The data were subsequently detrended by the subtraction of R0 (ΔR). Then, the ΔR/R0 value was calculated. To determine the daily peak phase of jGCaMP7s or jRGECO1a Ca2+ signal, ΔR/R0 were smoothened with a 21-point moving average, then the middle of the time points crossing value 0 upward (Ca2+ onset) and downward (Ca2+ offset) were defined as the midpoint that reflects the peak phases. Additionally, the intervals between two adjacent midpoints were defined as the daily periods (Fig. 1f,g)12. Because VIP-Ca2+ rhythm in VIP deficient mice attenuated drastically in DD, we failed to determine its Ca2+ onset, offset, and daily period at the late stage. A double-plotted actogram of the jGCaMP7s or jRGECO1a signal was prepared by converting all ΔR/R0 to positive values by subtracting the minimum value of ΔR/R0. Subsequently, these values were multiplied by 100 or 1000 and rounded off. The plots were made via ClockLab (Actimetrics) with normalization in each row to emphasize weak VIP-Ca2+ and CCK-Ca2+ rhythmicity in VIP-deficient mice at the late stage in DD. To quantitively analyze the weak rhythms and their changes, periods and amplitudes of Ca2+ signals (ΔR/R0) were calculated by periodogram with 10-min bins for all three weeks and shifting five-day time windows in DD, respectively (Figs. 2, 4, and 6; Supplementary Figs. 1, 2, 5, and 9).
During the fiber photometry recordings, the animal’s locomotor activity was monitored using an infrared sensor (AMN31112 and Arduino) in 10-min bins. A double-plotted actogram of locomotor activity was also prepared and overlaid on that of the jGCaMP7s and jRGECO1a signal. The onset of locomotor activity was determined using the actograms. Initially, we attempted to automatically detect the onset; however, it was followed by a manual visual inspection and modifications by the experimenter. The intervals between two adjacent locomotor onsets were defined as the daily periods (Fig. 1f, g). To calculate the CT of the midpoint phases of jGCaMP7s and jRGECO1a signals, we have defined the regression line of locomotor activity onsets as CT12. Period and amplitude of locomotor activity were also calculated by χ2 periodogram analyses for all days or 5 consecutive days in DD to analyze weak rhythmicity of VIP-deficient mice (Figs. 2, 4, and 6; Supplementary Figs. 1, 2, and 5).
We confirmed the jGCaMP7s and jRGECO1a expressions and the position of the optical fiber by slicing the brains into 30 μm coronal sections using a cryostat (Leica). The sections were mounted on glass slides with a mounting medium (VECTASHIELD HardSet with DAPI, H-1500, Vector Laboratories; Dako Fluorescence Mounting Medium, Agilent Technologies) and observed via epifluorescence microscope (BZ-9000, Keyence).
Behavioral analyses
Male and female Avp-CK1δ−/−; ViptTA/tTA (Avp-Cre; CK1δflox/flox; ViptTA/tTA) or control (Avp-Cre; CK1δflox/flox; Vipwt/tTA) (Fig. 2), Vip-Casp3 (Vipwt/ires-Cre mice injected bilaterally with AAV-CAG-FLEX-taCasp3-TEVp) or control (Vipwt/ires-Cre mice injected with AAV-CAG-FLEX-nls-mCherry and wildtype C57BL/6J mice injected with AAV-CAG-FLEX-taCasp3-TEVp) (Suppelementary Fig. 7), Avp-Vipr2−/− (Avp-Cre; Rosa26-LSL-Cas9-2A-EGFP mice injected bilaterally with AAV-U6-gVipr2-EF1α-DIO-mCherry) or control (Avp-Cre; Rosa26-LSL-Cas9-2A-EGFP mice injected with AAV-U6-gControl-EF1a-DIO-mCherry) (Fig. 5), and Avp-TeNT (Avp-Cre mice injected bilaterally with AAV-EF1α-DIO-GFP::TeNT) or control (Avp-Cre mice injected with AAV-CAG-FLEX-EGFP) (Fig. 7) mice, aged 8 to 30 weeks, were housed individually in a cage placed in a light-tight chamber (light intensity was approximately 100 lux). Spontaneous locomotor activity (home-cage activity) was monitored by infrared motion sensors (O’Hara) in 1-min bins with a custom-made program as described previously12. Actogram, activity profile, and χ2 periodogram analyses were performed via ClockLab (Actimetrics). The free-running period and amplitude were measured by periodogram for all days in DD (Avp-CK1δ−/−; ViptTA/tTA and Avp-TeNT) or the last 14 days in DD (Vip-Casp3). In the case of Avp-Vipr2−/− and control mice, their periods and amplitudes were measured by periodogram for 10 days in 1st or 2nd stage in DD (Fig. 5).
Immunohistochemistry
Immunostaining was performed as described previously10,12,75. Mice were killed approximately at ZT2 by transcardial perfusion of PBS followed by 4% paraformaldehyde (PFA) in PBS. For estimating co-expression of CCK and AVP, Cck-ires-Cre; Rosa26-LSL-tdTomato mice were pretreated with intracerebroventricular injections of colchicine (40 µg in 1 µl saline) 48 h before perfusion around ZT6. Then, the mice brains were postfixed in the 4% PFA at 4 ˚ C overnight, followed by immersion in 30% sucrose solution at 4 °C for 2 days. Serial coronal brain sections (30 μm thickness) were made with a cryostat (CM1860, Leica) and collected in 4 series—one of which was further immunostained. For immunofluorescence staining, sections were washed with PBS containing 0.3% Triton X-100 (PBST) and blocked with PBST plus 3% BSA (blocking solution). Then, slices were incubated overnight with the designated primary antibodies in the blocking solution at 4 °C. Antibodies used were rabbit anti-GFP antibody (1:1000; A11122, Thermo Fisher Scientific), rabbit anti-VIP antibody (1:1,000; #20077, Immunostar), rabbit anti-AVP antibody (Millipore, 1:2000), and rabbit anti-VPAC2 antibody (Abcam, 1:1000). Then, slices were washed with PBST, followed by incubation with the designated secondary antibodies in blocking solution for 4 h. Secondary antibodies used in this study were Alexa Fluor 488–conjugated donkey anti-rabbit IgG antibody (1:2000, A-21206; Thermo Fisher Scientific), Alexa Fluor 594–conjugated donkey anti-rabbit IgG antibody (1:2000, A-21207; Thermo Fisher Scientific), and Alexa Fluor 647–conjugated goat anti-Rabbit IgG antibody (1:600, 111-605-003; Jackson Immuno Research Laboratories). After incubation with secondary antibody, slices were washed with PBS, mounted on slide glasses, air dried, and coverslipped using Mounting Medium (H-1500, Vector Laboratories; Dako Fluorescence Mounting Medium, Agilent Technologies). Images were taken using an epifluorescence microscope (BZ-9000, Keyence) or a confocal microscope (Fluoview Fv10i, Olympus). Fluorescent signal of Alexa Fluor 647 was pseudo-colored in light blue (Fig. 5 and Supplementary Fig. 7). The VPAC2 expression levels were quantified by ImageJ as follows (Fig. 5b). First, the mean intensities of pixels within the ROIs in the SCN shell and core were calculated. Then, the values of the region lateral to the SCN were regarded as background and were subtracted from those of the SCN.
Mathematical model construction and simulation
The two-oscillator model with coupling was modified by introducing a term in which VIP neurons delay the phase of AVP neuron oscillator31, reflecting the phase-dependent receptivity of AVP to VIP’s delaying effect.
where θA, θnA denote the phases of oscillator A (representing AVP neurons) and oscillator nA (non-AVP neurons, including VIP neurons), respectively. ωA, ωnA are the intrinsic angular velocities (ωA = 2π/τA, ωnA = 2π/τnA), and τA, τnA are the intrinsic periods (τA = 22.7 h, except for Supplementary Fig. 11d where τA = 23.7 h; τnA = 24.3 h), based on the AVP-Ca2+ and VIP-Ca2+ fiber photometry results presented in this study. KnA→A denotes the coupling strength from oscillator nA to A, KA→nA denotes the coupling strength from oscillator A to nA, and KV represents inhibitory modulation (e.g., VIP-mediated inhibition), which is active only when sin(θnA) > 0 and sin(θA − δ) > 0. δ represents the phase difference between oscillator A and its receptivity, and was set to π/2 (Fig. 8b), 0 (Supplementary Fig. 11a), π (Supplementary Fig. 11b), or 3π/2 (Supplementary Fig. 11c). The ratio between KA→nA and KnA→A (α) was set to 0, 0.2, 1, 5, and 25. In the heatmaps, gray regions indicate simulations in which a stable period could not be determined, whereas black regions indicate the final periods between oscillators A and nA are different (>0.2 h). All simulations were performed in MATLAB.
Statistical analysis
All results are expressed as mean ± SEM. Statistical analyses were performed using Prism 8.0 software (GraphPad). For comparisons of 2 groups, two-tailed unpaired Student’s or Welch’s t tests were performed. For comparisons of multiple groups with no difference of variance by Bartlett test, two-way repeated measures ANOVA followed by Bonferroni, Tukey or Holm-Sidak post hoc test were performed. For circular data, Rayleigh test, Watson–Williams test, and Harrison–Kanji test were performed with Circstat MATLAB Toolbox for Circular Statistics79. All p values less than 0.05 were considered as statistically significant. Only relevant information from the statistical analysis was indicated in the text and figures. Detailed results of the statistical analyses are provided in Supplementary Data 1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the article and its supplementary files. Source Data are provided with this paper. All datasets required to reproduce the figures are included. Any further requests can be addressed to the corresponding author. Source data are provided with this paper.
Code availability
The custom codes for the mathematical model (Fig. 8 and Supplementary Fig. 11) can be accessed at: https://github.com/MiedaLaboratory/Wang2026NatureCommunications on GitHub (https://doi.org/10.5281/zenodo.17861530).
References
Welsh, D. K., Takahashi, J. S. & Kay, S. A. Suprachiasmatic nucleus: cell autonomy and network properties. Annu. Rev. Physiol. 72, 551–577 (2010).
Herzog, E. D., Hermanstyne, T., Smyllie, N. J. & Hastings, M. H. Regulating the suprachiasmatic nucleus (SCN) circadian clockwork: interplay between cell-autonomous and circuit-level mechanisms. Cold Spring Harb. Perspect. Biol. 9, a027706 (2017).
Mieda, M. The central circadian clock of the suprachiasmatic nucleus as an ensemble of multiple oscillatory neurons. Neurosci. Res. 156, 24–31 (2020).
Ono, D. et al. The suprachiasmatic nucleus at 50: looking back, then looking forward. J. Biol. Rhythms 39, 135–165 (2024).
Balsalobre, A., Damiola, F. & Schibler, U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 93, 929–937 (1998).
Aton, S. J., Colwell, C. S., Harmar, A. J., Waschek, J. & Herzog, E. D. Vasoactive intestinal polypeptide mediates circadian rhythmicity and synchrony in mammalian clock neurons. Nat. Neurosci. 8, 476–483 (2005).
Maywood, E. S. et al. Synchronization and maintenance of timekeeping in suprachiasmatic circadian clock cells by neuropeptidergic signaling. Curr. Biol. 16, 599–605 (2006).
Harmar, A. J. et al. The VPAC(2) receptor is essential for circadian function in the mouse suprachiasmatic nuclei. Cell 109, 497–508 (2002).
Colwell, C. S. et al. Disrupted circadian rhythms in VIP- and PHI-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 285, R939–R949 (2003).
Peng, Y. et al. Cell type-specific genetic manipulation and impaired circadian rhythms in ViptTA knock-in mice. Front. Physiol. 13, 895633 (2022).
Jones, J. R., Simon, T., Lones, L. & Herzog, E. D. SCN VIP neurons are essential for normal light-mediated resetting of the circadian system. J. Neurosci. 38, 7986–7995 (2018).
Mieda, M. et al. Cellular clocks in AVP neurons of the SCN are critical for interneuronal coupling regulating circadian behavior rhythm. Neuron 85, 1103–1116 (2015).
Shan, Y. et al. Dual-color single-cell imaging of the suprachiasmatic nucleus reveals a circadian role in network synchrony. Neuron 108, 164–179.e7 (2020).
Todd, W. D. et al. Suprachiasmatic VIP neurons are required for normal circadian rhythmicity and comprised of molecularly distinct subpopulations. Nat. Commun. 11, 4410 (2020).
Tonsfeldt, K. J. et al. Female fertility does not require Bmal1 in suprachiasmatic nucleus neurons expressing arginine vasopressin, vasoactive intestinal peptide, or neuromedin-S. Front. Endocrinol. 13, 956169 (2022).
Tsuno, Y. et al. In vivo recording of suprachiasmatic nucleus dynamics reveals a dominant role of arginine vasopressin neurons in circadian pacesetting. PLoS Biol. 21, e3002281 (2023).
Mieda, M., Okamoto, H. & Sakurai, T. Manipulating the cellular circadian period of arginine vasopressin neurons alters the behavioral circadian period. Curr. Biol. 26, 2535–2542 (2016).
Lee, I. T. et al. Neuromedin s-producing neurons act as essential pacemakers in the suprachiasmatic nucleus to couple clock neurons and dictate circadian rhythms. Neuron 85, 1086–1102 (2015).
Xie, L. et al. Cholecystokinin neurons in mouse suprachiasmatic nucleus regulate the robustness of circadian clock. Neuron 111, 2201–2217.e4 (2023).
Maejima, T. et al. GABA from vasopressin neurons regulates the time at which suprachiasmatic nucleus molecular clocks enable circadian behavior. Proc. Natl. Acad. Sci. USA 118, e2010168118 (2021).
Mei, L. et al. Long-term in vivo recording of circadian rhythms in brains of freely moving mice. Proc. Natl. Acad. Sci. USA 115, 4276–4281 (2018).
Dana, H. et al. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods 16, 649–657 (2019).
Dana, H. et al. Sensitive red protein calcium indicators for imaging neural activity. Elife 5, e12727 (2016).
Taniguchi, H. et al. A Resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71, 995–1013 (2011).
Wen, S. et al. Spatiotemporal single-cell analysis of gene expression in the mouse suprachiasmatic nucleus. Nat. Neurosci. 23, 456–467 (2020).
Xu, P. et al. NPAS4 regulates the transcriptional response of the suprachiasmatic nucleus to light and circadian behavior. Neuron 109, 3268–3282.e6 (2021).
Madisen, L. et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010).
Mazuski, C., Chen, S. P. & Herzog, E. D. Different roles for VIP neurons in the neonatal and adult suprachiasmatic nucleus. J. Biol. Rhythms 35, 465–475 (2020).
Platt, R. J. et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455 (2014).
Izumo, M. et al. Differential effects of light and feeding on circadian organization of peripheral clocks in a forebrain Bmal1 mutant. Elife 3, e04617 (2014).
Kuramoto, Y. Chemical Oscillations, Waves, and Turbulence. Vol. 19 (Springer, 1984).
An, S., Irwin, R. P., Allen, C. N., Tsai, C. & Herzog, E. D. Vasoactive intestinal polypeptide requires parallel changes in adenylate cyclase and phospholipase C to entrain circadian rhythms to a predictable phase. J. Neurophysiol. 105, 2289–2296 (2011).
Mazuski, C. et al. Entrainment of circadian rhythms depends on firing rates and neuropeptide release of VIP SCN neurons. Neuron 99, 555–563.e5 (2018).
Morris, E. L. et al. Single-cell transcriptomics of suprachiasmatic nuclei reveal a Prokineticin-driven circadian network. EMBO J 40, e108614 (2021).
Stowie, A. et al. Arginine–vasopressin-expressing neurons in the murine suprachiasmatic nucleus exhibit a circadian rhythm in network coherence in vivo. Proc. Natl. Acad. Sci. USA 120, e2209329120 (2023).
Enoki, R. et al. Synchronous circadian voltage rhythms with asynchronous calcium rhythms in the suprachiasmatic nucleus. Proc. Natl. Acad. Sci. USA 114, E2476–E2485 (2017).
Jones, J. R., Tackenberg, M. C. & McMahon, D. G. Manipulating circadian clock neuron firing rate resets molecular circadian rhythms and behavior. Nat. Neurosci. 18, 373–375 (2015).
Patton, A. P. et al. The VIP-VPAC2 neuropeptidergic axis is a cellular pacemaking hub of the suprachiasmatic nucleus circadian circuit. Nat. Commun. 11, 3394 (2020).
Hamnett, R., Crosby, P., Chesham, J. E. & Hastings, M. H. Vasoactive intestinal peptide controls the suprachiasmatic circadian clock network via ERK1/2 and DUSP4 signalling. Nat. Commun. 10, 542 (2019).
Varadarajan, S. et al. Connectome of the suprachiasmatic nucleus: new evidence of the core-shell relationship. eNeuro 5, ENEURO.0205-18.2018 (2018).
Moga, M. M. & Moore, R. Y. Organization of neural inputs to the suprachiasmatic nucleus in the rat. J. Comp. Neurol. 389, 508–534 (1997).
Gizowski, C., Zaelzer, C. & Bourque, C. W. Clock-driven vasopressin neurotransmission mediates anticipatory thirst prior to sleep. Nature 537, 685–688 (2016).
Yao, Y., Taub, A. B., LeSauter, J. & Silver, R. Identification of the suprachiasmatic nucleus venous portal system in the mammalian brain. Nat. Commun. 12, 5643 (2021).
Paul, S. et al. Output from VIP cells of the mammalian central clock regulates daily physiological rhythms. Nat. Commun. 11, 1453 (2020).
Jones, J. R., Chaturvedi, S., Granados-Fuentes, D. & Herzog, E. D. Circadian neurons in the paraventricular nucleus entrain and sustain daily rhythms in glucocorticoids. Nat. Commun. 12, 5763 (2021).
Noguchi, T., Watanabe, K., Ogura, A. & Yamaoka, S. The clock in the dorsal suprachiasmatic nucleus runs faster than that in the ventral. Eur. J. Neurosci. 20, 3199–3202 (2004).
Noguchi, T. & Watanabe, K. Regional differences in circadian period within the suprachiasmatic nucleus. Brain Res. 1239, 119–126 (2008).
Shinohara, K., Honma, S., Katsuno, Y., Abe, H. & Honma, K. Two distinct oscillators in the rat suprachiasmatic nucleus in vitro. Proc. Natl. Acad. Sci. USA 92, 7396–7400 (1995).
Myung, J. et al. Period coding of Bmal1 oscillators in the suprachiasmatic nucleus. J. Neurosci. 32, 8900–8918 (2012).
Morimoto, T., Yoshikawa, T., Nagano, M. & Shigeyoshi, Y. Regionality of short and long period oscillators in the suprachiasmatic nucleus and their manner of synchronization. PLoS ONE 17, e0276372 (2022).
Webb, A. B., Angelo, N., Huettner, J. E. & Herzog, E. D. Intrinsic, nondeterministic circadian rhythm generation in identified mammalian neurons. Proc. Natl. Acad. Sci. USA 106, 16493–16498 (2009).
Hirata, Y. et al. Circadian rhythms in Per1, PER2 and Ca2+ of a solitary SCN neuron cultured on a microisland. Sci. Rep. 9, 18271 (2019).
Liu, A. C. et al. Intercellular coupling confers robustness against mutations in the SCN circadian clock network. Cell 129, 605–616 (2007).
Yamaguchi, S. et al. Synchronization of cellular clocks in the suprachiasmatic nucleus. Science 302, 1408–1412 (2003).
Enoki, R., Ono, D., Kuroda, S., Honma, S. & Honma, K.-I. Dual origins of the intracellular circadian calcium rhythm in the suprachiasmatic nucleus. Sci. Rep. 7, 41733 (2017).
Nagoshi, E. et al. Circadian gene expression in individual fibroblasts: cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell 119, 693–705 (2004).
Welsh, D. K., Yoo, S.-H., Liu, A. C., Takahashi, J. S. & Kay, S. A. Bioluminescence imaging of individual fibroblasts reveals persistent, independently phased circadian rhythms of clock gene expression. Curr. Biol. 14, 2289–2295 (2004).
Li, Y. et al. Noise-driven cellular heterogeneity in circadian periodicity. Proc. Natl. Acad. Sci. USA 117, 10350–10356 (2020).
Herzog, E. D., Takahashi, J. S. & Block, G. D. Clock controls circadian period in isolated suprachiasmatic nucleus neurons. Nat. Neurosci. 1, 708–713 (1998).
Nakamura, W., Honma, S., Shirakawa, T. & Honma, K. Clock mutation lengthens the circadian period without damping rhythms in individual SCN neurons. Nat. Neurosci. 5, 399–400 (2002).
Ono, D., Honma, S. & Honma, K. Cryptochromes are critical for the development of coherent circadian rhythms in the mouse suprachiasmatic nucleus. Nat. Commun. 4, 1666 (2013).
Herzog, E. D., Aton, S. J., Numano, R., Sakaki, Y. & Tei, H. Temporal precision in the mammalian circadian system: a reliable clock from less reliable neurons. J. Biol. Rhythms 19, 35–46 (2004).
Brown, T. M., Colwell, C. S., Waschek, J. A. & Piggins, H. D. Disrupted neuronal activity rhythms in the suprachiasmatic nuclei of vasoactive intestinal polypeptide-deficient mice. J. Neurophysiol. 97, 2553–2558 (2007).
VanderLeest, H. T. et al. Seasonal encoding by the circadian pacemaker of the SCN. Curr. Biol. 17, 468–473 (2007).
Pittendrigh, C. S. & Daan, S. A functional analysis of circadian pacemakers in nocturnal rodents. J. Comp. Physiol. A 106, 291–331 (1976).
Etchegaray, J.-P. et al. Casein kinase 1 delta regulates the pace of the mammalian circadian clock. Mol. Cell Biol. 29, 3853–3866 (2009).
Storch, K.-F. et al. Intrinsic circadian clock of the mammalian retina: importance for retinal processing of visual information. Cell 130, 730–741 (2007).
Loh, D. H. et al. Disrupted reproduction, estrous cycle, and circadian rhythms in female mice deficient in vasoactive intestinal peptide. J. Biol. Rhythms 29, 355–369 (2014).
Wahba, L. R., Perez, B., Nikhil, K., Herzog, E. D. & Jones, J. R. Circadian rhythms in multiple behaviors depend on sex, neuropeptide signaling, and ambient light. Preprint at https://doi.org/10.1101/2022.08.18.504454 (2022).
Hyun, J. H. et al. Tagging active neurons by soma-targeted Cal-Light. Nat. Commun. 13, 7692 (2022).
Wang, W. et al. A light- and calcium-gated transcription factor for imaging and manipulating activated neurons. Nat. Biotechnol. 35, 864–871 (2017).
Yang, C. F. et al. Sexually dimorphic neurons in the ventromedial hypothalamus govern mating in both sexes and aggression in males. Cell 153, 896–909 (2013).
Concordet, J.-P. & Haeussler, M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245 (2018).
Maddalo, D. et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 516, 423–427 (2014).
Islam, M. T. et al. Vasopressin neurons in the paraventricular hypothalamus promote wakefulness via lateral hypothalamic orexin neurons. Curr. Biol. 32, 3871–3885.e4 (2022).
Padilla, S. L. et al. Kisspeptin neurons in the arcuate nucleus of the hypothalamus orchestrate circadian rhythms and metabolism. Curr. Biol. 29, 592–604.e4 (2019).
Kim, C. K. et al. Simultaneous fast measurement of circuit dynamics at multiple sites across the mammalian brain. Nat. Methods 13, 325–328 (2016).
Martianova, E., Aronson, S. & Proulx, C. D. Multi-fiber photometry to record neural activity in freely-moving animals. J. Vis. Exp. https://doi.org/10.3791/60278 (2019).
Berens, P. CircStat: a MATLAB toolbox for circular statistics. J. Stat. Softw. 31, 1–21 (2009).
Acknowledgements
We thank I. Tokuda for valuable discussion; H. Okamoto for the Avp-Cre mouse; S. Horike and T. Daikoku for the Vip-tTA mouse; Z. J. Huang and I. Fukunaga for the Cck-ires-Cre mouse; Z. J. Huang for the Vip-ires-Cre mouse; H. Zeng for the Ai14 mouse; F. Zhang for the Rosa26-LSL-SpCas9-2A-EGFP mouse; D. R. Weaver for the CK1δflox mouse; C.J. Weitz for the Bmal1flox mouse; Penn Vector Core for pAAV2-rh10; D. Kim & GENIE Project for pGP-AAV-CAG-FLEX-jGCaMP7s-WPRE and pAAV.CAG.Flex.NES-jRGECO1a.WPRE.SV40; H. Kwon for pAAV-TRE-EGFP and pAAV-TRE-ChR2-YFP; pAAV-TRE-ChrimsonR-mCherry for A. Ting; pAAV-flex-taCasp3-TEVp for N. Shah and J. Wells; pX333 for A. Ventura; pAAV-EF1a-DIO-mCherry for B. Roth; and pAAV-EF1α-DIO-GFP::TeNT for R.D. Palmiter; We thank all lab members, including M. Kawabata and Y. Nishiwaki. This work was supported in part by JSPS KAKENHI Grant Numbers JP24KJ1189 (Y.P.); JP23K06345 (Y.T.); JP22K20738 (A.M.); JP24K02137 (T.M.); JP23K24064; 25K02440; 25K22589; the Takeda Science Foundation; the Terumo Life Science Foundation; the Research Foundation for Opto-Science and Technology; the Koyanagi Foundation; the NOVARTIS Foundation for the Promotion of Science (M.M.); JST SPRING Grant Number JPMJSP2135 (M.W., Y.P.); and Japanese Government Scholarship for International Students (M.T.I.).
Author information
Authors and Affiliations
Contributions
M.W. and M.M. designed research; M.W., Y.T., Y.P., M.T.I., A.M., T.M., and M.M. performed research; M.W., Y.T., and M.M. analyzed data; Y.T. conducted simulations; M.W. and M.M. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Anat Kahan and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, M., Tsuno, Y., Peng, Y. et al. Neuronal feedback loop of the suprachiasmatic nucleus generates robust circadian rhythms. Nat Commun 17, 1505 (2026). https://doi.org/10.1038/s41467-025-68218-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68218-x
