
Abstract
Chemoselective functionalization of hetero-gem-dimetalloid represents an attractive strategy in terms of diversity-oriented synthesis. In particular, desilylative functionalization of gem-silylboronate esters remains a challenging task and existing solutions heavily relied on ionic reactions. Herein, we report a desilylative functionalization of allylic gem-silylboronate esters with aldehydes under synergistic photoredox and chromium(II) catalysis. With different substrates, both α- and γ-functionalization are realized with exclusive regioselectivity and diastereoselectivity, which is dictated by the chair-like transition state of predominant isomer of CrIII allyl intermediate. Moreover, γ-functionalization products bearing CF2 unit are acquired when gem-difluoroalkene-containing substrates are employed. In the presence of chiral ligand, enantioselective allylation of aldehydes is successfully accomplished, affording alkenylated 1,2-diols after oxidative workup with excellent regio-, diastereo- and enantioselectivity. The current protocol displays wide substrate generality and broad functional group compatibility. In addition, diverse post-transformations converted obtained products into a variety of valuable structures.
Similar content being viewed by others
Introduction
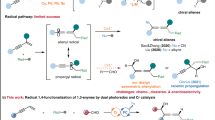
In recent decades, sp3-geminated dimetallic/metalloidal compounds have been widely established as bis-nucleophilic reagents, being capable of undergoing a wide array of organic transformations to enable facile assembly of structures with diversity and complexity1,2,3. In particular, reagents bearing geminated hetero-metallic/metalloid pairs have garnered considerable attention, as chemoselective sequential transformations of these reagents allow for their function as linchpins for modular assembly of structures with densely substituted sp3 carbon centers, which align with the prevalence of “escape from the flatland” endeavor in medical chemistry4. Therefore, new geminated dimetallic/metalloids with preparation/operation simplicity and tunable reactivity are always sought after (Fig. 1A). Among the various gem-dimetallic/metalloids developed thus far, gem-silylboronate esters, thanks to the rich reaction patterns of silyl and boronate groups engaged in, have been demonstrated to be versatile bis-nucleophilic agents to participate in a broad range of reactions with different electrophiles. Notwithstanding the lower electronegativity of Si than B in the Pauling electronegativity scale (χB = 2.04, χSi = 1.90), the boronate ester is more Lewis acidic than the silyl group and hence is predisposed to be activated by Lewis basic reagents towards electrophiles or transmetalation to transition metals (nucleophilic activation). For instance, Wang et al. have reported that in the presence of Pd catalyst and K2CO3 as base, benzylic gem-silylboronate ester underwent smooth cross-coupling with aryliodides with selective cleavage of C‒B bond5,6. In another case, the boronate group in allylic gem-silylboronate ester was able to be activated by oxygen atom in aldehyde, thereby furnishing deboronative γ-allylation of aldehydes in a highly regio- and diastereoselective manner under catalyst-free conditions, through a chair-like six-membered cyclic transition state7,8,9,10,11(Fig. 1B). On the other hand, the HOMO energy of C‒Si σ bond is higher than that of C‒B σ bond, which endows better interaction between C‒Si σ bond and adjacent carbocation through hyperconjugation, also known as “β-silicon effect”12,13. In other words, upon generation of an α-carbocation of gem-silylboronate, a desilylation process would be favored over cleavage of the C–B bond. Taking advantage of the above unique property of the silyl group, a series of selective desilylative γ-functionalization of allylic gem-silylboronate ester was successfully disclosed using an electrophilic activation strategy. For example, in the presence of Lewis/Brønsted acid, reactions of α-silyl-substituted allylboronates with aldehydes proceeded via a Sakurai-type allylation pathway14,15 through cleavage of the C‒Si bond (Fig. 1B)16,17,18. In addition to the activated carbonyl group, Selectfluor19 and transition metal complex20 were also competent electrophiles for the SE2’ reaction of allylic gem-silylboronate ester. Despite these impressive prior arts, desilylative transformation of allylic gem-silylboronate ester with α-selectivity remains however, virtually undeveloped to the best of our knowledge. Against this backdrop and with our continuous interest in radical chemistry21,22,23,24,25,26, we envisage that a radical-involved manifold might offer a mechanistically distinct solution to this unsolved issue. In our design, if α-silyl-substituted allylboronate is single electron oxidized by a highly oxidizing species, for example excited photocatalyst, the thus produced radical cation intermediate might induce spontaneous C‒Si bond cleavage to produce the boron-substituted allylic radical intermediate. By tailoring of substrate structure and judicious choice of reaction conditions, highly chemo- and regioselective transformation of allylic gem-silylboronate ester with tunable regioselectivity might be viable (Fig. 1C).

A Chemoselective functionalization of sp3-geminated dimetallic/metalloids. B γ-Selective divergent functionalization of allylic gem-silylboronate. C Our design: SET-driven desilylative functionalization of allylic gem-silylboronate esters. D Synergistic photoredox and Cr-catalyzed desilylative allylation of aldehydes with allylic gem-silylboronate esters. (This work) SET, single electron transfer.
Herein, we report that by diverting the previously reported ionic mode to a radical-type pathway under synergistic photoredox and Cr catalysis27,28,29,30,31,32,33,34, a highly efficient desilylative addition of gem-silylboronate ester with aldehyde with exclusive α-regioselectivity is successfully accomplished. The reactions proceed efficiently to give the corresponding silyl-protected and boron-containing homoallylic alcohols in moderate to good yields with high chemo-, regio- and diastereoselectivity. Remarkably, a highly enantioselective version (up to 99% ee) is also realized by employing chiral bisoxazoline (BOX) as ligand. In addition, with γ-substituted allylic gem-silylboranes as substrate, a completely reversed γ-selectivity is observed under slightly modified conditions, leading to the desired alkenylboron-containing homoallylic alcohols with excellent control of regioselectivity, diastereoselectivity, and double bond geometry (Fig. 1D).
Result and discussion
Reaction development
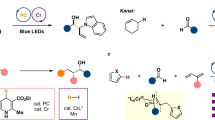
We initiated our investigation with 4-phenylbenzaldehyde (1a) and α-trimethylsilyl allylboronate ester 2a as model substrates (Table 1). After examining a range of reaction parameters, we identified that the allylation of aldehyde proceeded effectively in the presence of 2 mol% acridinium photocatalyst PC-1 and 10 mol% CrCl2 in MeCN (0.1 M) under irradiation of blue LEDs, enabling facile generation of the desired allylboration product 3aa in excellent yield with exclusive α-regioselectivity and diastereoselectivity (entry 1). 3aa was further oxidized to diol 4aa in nearly quantitative yield for the simplicity of purification. Control experiments indicated that continuous irradiation, chromium salt, and photocatalyst are all indispensable for the smooth generation of 3aa. Under conditions of these control experiments, a side deboronative allylation of 2a took place to afford 3aa’. The exclusive chemoselectivity and preference for the Z-isomer was in accordance with literature reports (entries 2–4)17. The complete suppression of the background reaction under standard conditions underscored the prominent chemoselectivity of the current protocol. In contrast, the use of other photocatalysts (entries 5–7) or solvents (entries 8–14) either failed to trigger the titled allylation reaction or resulted in attenuated yields. The distinct performance in MeCN is possibly caused by the weak nucleophilicity of the solvent, which was able to promote the cleavage of the C‒Si bond35. In addition, the use of MeCN in combination with other solvents as reaction media at varied temperatures resulted in eroded reaction performance (entries 15–18). Product 3aa was prone to elimination under basic conditions36 and no desired product was obtained in the presence of a stoichiometric amount of K2CO3 as base (entry 19). When pyridine was employed as an alternative organic base, the reaction was completely shut down, possibly due to the coordination effect of pyridine, which attenuate the reactivity of chromium (entry 20). Subsequent survey of other reaction parameters, including decreasing the dosage of 2a (entries 21-22) or reversing the equivalent of 1a and 2a at 50 °C (entry 23), all had detrimental effect on the yield. Finally, using CrCl3 instead of CrCl2 as a catalyst also afforded 3aa in moderate yield (entry 24).
Substrate scope
With the successful establishment of the optimal reaction conditions, we proceeded to examine the substrate scope of the α-regioselective allylation reactions between α-trimethylsilyl allylboronate ester 2a and various aldehydes. As shown in Fig. 2, a wide variety of aldehydes 1 efficiently underwent the allylations with 2a via C‒Si bond cleavage with exclusive α-regioselectivity and excellent diastereoselectivity, affording the corresponding diols 4 in moderate to good yields after oxidative workup. Aryl aldehydes 1b-q decorating with electron-withdrawing groups on the phenyl ring, such as cyano, trifluoromethyl, trifluoromethoxy, methoxycarbonyl, fluoro, chloro, bromo, iodo, Bpin, and ethynyl, all effectively reacted with 2a to give the diols 4ba-qa in 38–95% yields. Similarly, electron-rich aryl aldehydes 1s-aa bearing electron-donating substituents, including methyl, ethyl, tert-butyl, methoxy, acetoxyl, methylthio, and acetamido, were also proven to be competent reaction partners for the allylation reactions, affording the desired products 4sa-aaa in 47-82% yields. Notably, aldehydes 1j and 1t possessing ortho-substituted fluoro and ethyl groups in the benzene ring, worked equally well, irrespective of the steric congestion. In addition, naphthyl-substituted aldehyde 1ab and heteroaryl aldehydes 1ac-af derived from furan, thiophene, benzofuran, and pyridine were amenable to the present allylation, and the target products 4aba-aafa were delivered in reasonable yields. Moreover, α, β-unsaturated aldehydes 1ag-ai with alkenyl or alkynyl substituents were good reactants as well under the optimized conditions, furnishing 4aga-aia in 53–79% yields without detection of product derived from Michael addition. Furthermore, a series of diversely substituted aliphatic aldehydes were examined. It was found that the allylations could also be applied to alkyl aldehydes 1aj-ar under the standard conditions, producing the anticipated products 4aja-ara in moderate to good yields. Remarkably, a broad range of functional groups or substituents, including CN, CF3, OCF3, ester, halogen, Bpin, thioether, amide, alkene, alkyne, cyclopropyl and carbonyl, successfully survived the reaction conditions, providing a platform for downstream synthetic manipulations.

Reaction conditions: 1 (0.1 mmol, 1.0 equiv.), 2a (0.15 mmol, 1.5 equiv.), PC-1 (2 mol%), CrCl2 (10 mol%), MeCN (1 mL), blue LEDs, N2, rt, 12 - 48 h; oxidation of C‒B bond: H2O2 (0.6 mmol, 6.0 equiv.), NaOH (0.3 mmol, 3.0 equiv.), THF (1 mL), 0 °C - rt, 3 h; dr was determined by crude 1H NMR analysis; isolated yield of diol 4, NMR yield of 3 in parentheses; acorresponding 4,4,6-trimethyl-1,3,2-dioxaborinane as substrate; b4-OAcC6H4CHO was used as substrate; cNaH2PO4 was used instead of NaOH for the oxidation.
The gem-silylboronate ester substrate generality was then investigated with a plethora of structurally varied α-silyl-substituted allylboronate esters 2. As summarized in Fig. 3, α-trimethylsilyl allylboronate esters 2b-j tethered with different R1/R2 substituents, including methyl, n-butyl, chloropropyl, bromoethyl, phenyl, methyl/methyl, and ethyl/ethyl, all participated in the allylations efficiently with a reversed γ-selectivity to give the corresponding OTMS-substituted alkenyl boronates 5ib-ij, which furnished the desired hydroxylated alkenylboronates 6ib-ij in modest to excellent yields upon acidic hydrolysis. In addition to aromatic aldehyde, aliphatic aldehydes were also viable reaction partners as demonstrated by 6aji-ami. It is noteworthy that the reaction exhibited excellent stereocontrol as products were obtained in both excellent diastereoselectivity (> 95:5 dr) and C‒C double bond geometry (> 99:1 E/Z). When α-TMS-substituted allylboronate esters 2k-l derived from cyclopentane and cyclohexane, which possessed R3 substituent, were employed as substrates, the reactions could proceeded well, albeit with compromised yields (4ik-al). Interestingly, when allylic gem-silylboronate ester 2m with gem-difluoroalkene moiety was introduced to the allylation37, its desilylallylation also occurred with exclusive γ-selectivity under the optimized conditions to give the corresponding alkenylborons 6am-vm bearing CF2 unit in moderate yields.

Reaction conditions for 6: 1 (0.1 mmol, 1.0 equiv.), 2 (0.15 mmol, 1.5 equiv.), PC-1 (2 mol%), CrCl2 (10 mol%), MeCN/DCM (V/V = 1/1, 1 mL), blue LEDs, N2, rt, 12–18 h, then HCl (2 M, 2.0 equiv.), 30 min; a3 mol% PC-1; b20 mol% CrCl2; cMeOH (2.0 equiv.) was added without a deprotection step; dbenzyldimethylsilyl substrate was used.
Next, the enantioselective version of the present desilylallylation was also investigated (Fig. 4). After many trials with the use of various chiral ligands (see Supplementary Table S1 in Supplemental Information for details), it was gratifying to find that the asymmetric allylation of aldehyde 1a with α-trimethylsilyl allylboronate ester 2a proceeded efficiently by employing a chiral bisoxazoline (BOX) as ligand and TMSCl as additive under otherwise standard conditions, leading to the corresponding diol 4aa* in 82% yield with exclusive α-regioselectivity, excellent diastereoselectivity (> 95:5 dr), and enantioselectivity (89% ee) after oxidative workup. Subsequent evaluation of other aldehydes 1 revealed that the enantioselective allylations could also be applied to electron-poor or electron-rich benzaldehydes 1c-aa, naphthyl/heteroaryl aldehydes 1ab and 1af, as well as aliphatic aldehyde 1ao-au, all affording the desired alkenyl-substituted diols 4* in moderate to good yields with good to excellent enantiocontrol (83–99% ee). The absolute configuration of product 4* was unambiguously determined by single crystal X-ray diffraction analysis of product 4aaa* (CCDC 2451142). In addition, the absolute configuration of 4 was further confirmed by comparison of the optical rotation of 4ra* with the reported value of an identical compound38.

Reaction conditions for 4*: 1 (0.1 mmol, 1.0 equiv.), 2a (0.30 mmol, 3.0 equiv.), PC-1 (2 mol%), CrCl2 (10 mol%), L* (12 mol%), TMSCl (0.5 equiv.), MeCN (1 mL), blue LEDs, N2, rt, 18 h, then H2O2 (1.2 mmol, 12.0 equiv.), NaOH (0.6 mmol, 6.0 equiv.), THF (2 mL), 0 °C - rt, 4 h; dr and ee were determined by HPLC analysis; isolated yields of 4*; aH2O2 (2.4 mmol, 24.0 equiv.) and NaH2PO4 (0.25 mmol, 2.5 equiv.) were used for oxidation; b0.15 equiv. TMSCl; c1.5 equiv. 2a.
Subsequently, we proceeded to investigate the utility of the present desilylative allylation in the late-stage functionalization of complex molecules. As shown in Fig. 5A, aldehyde 1as derived from biologically active dehydrocholic acid was employed in the allylation with 2a to furnish the desired product 4asa in 41% yield. Analogously, the allylation protocol involving 1as and allylating agent 2i worked equally well to afford the target product 6asi in 51% yield. Next, synthetic transformations of the product 3, 4, 6 were explored (Fig. 5B, C). For example, allylboronate ester 3aa could be readily transformed into 1,5-diols 7 and 7’ in 59% yield (2 steps) through a second allylation of aldehyde 1at followed by desilylation with TBAF39. In addition, 3aa underwent smooth one-carbon homologation in the presence of CH2BrCl/nBuLi to yield CH2Bpin-tethered homoallylic alcohol 8 after acidic workup40. Moreover, following Sharpless dihydroxylation with AD-mix-α41, 4aa was facilely converted to 1,2,3,4-tetraol 9 in 67% yield. Additionally, olefin metathesis of 4aa with methyl acrylate in the presence of Hoveyda-Grubbs catalyst (2nd generation) delivered unsaturated ester 10 in 37% yield with elusive alkene geometry (> 99:1 E/Z). Finally, a hydroboration/oxidation sequence of 4aa afforded TBS-protected triol 11 in 57% yield after in-situ silyl protection (Fig. 5B). A series of functional group transformations of the vinyl boronate ester were then performed. Suzuki coupling of 3-bromopyridine with 6ii yielded the coupling product 12 smoothly in high yield42. Deboronative bromination with CuBr converted 6ai to 13 in 62% yield43. In addition, after protection of the hydroxyl group, 6ai was oxidized to aldehyde 14 in high yield by NaBO3‧4H2O44. Interestingly, when 6ai was directly treated with NaBO3‧4H2O, a cyclic lactol 15 was produced in almost quantitative yield. Further transformation of 15, either by oxidation with PCC41 or reduction with TFA/Et3SiH45, afforded lactone 16 and cyclic ether 17 in 92% and 99% yields, respectively (Fig. 5C).

A Late-stage functionalization of complex molecules. B Synthetic transformations of 3 and 4. C Synthetic transformations of 6.
Mechanistic studies
To gain insight into the reaction mechanism, several mechanistic experiments were performed (Fig. 6). We envisioned that if the current reaction occurs via a radical pathway as we designed, the use of vinyl-Bpin allylsilane 18 instead of 2a would gave the same product because an identical allylic radical intermediate was produced. As expected, when 18 was subjected to standard conditions with 1i, the corresponding diol 4ia was obtained in 4% yield after oxidative workup (Fig. 6a). The extremely low yield could be rationalized by the higher oxidative potential of 18 than 2a, caused by the electron-withdrawing Bpin substituent on the alkene, which resulted in reduced single-electron oxidation efficiency. In addition, the existence of TEMPO as a radical scavenger under standard conditions completely inhibited the model reaction, and 19 arising from the coupling of allylic radical with TEMPO was isolated in 28% yield (Fig. 6b). Moreover, radical clock experiment with cyclopropane-tethered allylic gem-silylboronate ester 2n provided the corresponding ring-opening dienyl alcohol 20 in 36% yield (Fig. 6c). Taken together, all these reaction outcomes strongly suggested a radical-involved reaction pathway. In addition, cyclic voltammetry (CV) analysis showed that the oxidation potential of 2a, 2g, 2i and 2l was measured to fall between + 1.4 to + 1.8 V (vs. SCE in MeCN), which all located within the capacity of the excited photocatalyst (E01/2 = + 2.08 V vs. SCE in MeCN) (Fig. 6d). To further verify which species is responsible for quenching the excited photocatalyst, a Stern-Volmer experiment was conducted (Fig. 6e). It was revealed that both the Cr(II) and allylic gem-silylboronate ester 2a exhibited efficient luminescence quenching of the excited photocatalyst. Considering the fact that the oxidation potential of 2 is significantly beyond the oxidative power of Cr(III)42, we believe that single electron oxidation of Cr(II) by the excited photocatalyst should be a reversible and unproductive process, and SET between the excited photocatalyst and allylic gem-silylboronate ester 2 is the working pathway. Finally, a light “on-off” experiment was conducted, and the result confirmed the necessity of constant irradiation for the reaction to proceed, which indicated that the reaction is not likely to proceed through a highly efficient chain propagation process (Fig. 6f).

a Control experiment with vinyl-Bpin allylsilane 18. b Radical quenching experiment. c Radical clock experiment. d Cyclic voltammetry experiment. e Stern-Volmer experiment. f Light “on-off” experiment. g Proposed reaction mechanism.
Based on the above experimental results and previous reports27,28,29,30,31,32,33,34, a plausible reaction mechanism is tentatively proposed (Fig. 6g). Initially, visible light emission stimulated the acridinium photocatalyst to its excited-state. Then, reductive quenching of the excited photocatalyst by α-TMS allylboronate ester 2 generates Bpin-substituted allylic radical I, along with dissociation of TMS cation. Next, allylic radical I was trapped by Cr(II) to give an allylchromium(III) species II, which is in equilibrium with its regioisomer III through π-allyl chromium(III) IV. For substrate 2a (R1 = R2 = H), due to steric factors, follow-up reactions predominantly proceeded via isomer II. While for γ-substituted 2 (R1, R2 ≠ H), isomer III was more favored than II as Bpin might provide extra stabilization for adjacent C‒Cr species, possibly through electronic interaction between C‒Cr σ bond and the p orbital of boron46,47. The nucleophilic species II then added to aldehyde 1 in a highly diastereoselective manner, which was secured by a six-membered Zimmerman-Traxler transition state, to give intermediate V. The allylation product 3 was then produced through the replacement of Cr(III) in V by the TMS cation. Finally, single electron transfer between the reduced photocatalyst and Cr(III) species afforded the ground state photocatalyst and Cr(II), thereby closing both the photocatalysis cycle and chromium catalysis cycle. Similarly, the γ-allylation product 5 was produced from allylchromium(III) species III and aldehyde 1, also through a chair-like six-membered transition state (see Supplementary Information for details). Moreover, our proposed rationale for the elusive regioselectivity in forming 3 is supported by DFT calculation (see Supplementary Information and Supplementary Data 1 for details).
In conclusion, by taking advantage of a radical-type reaction pathway under synergistic photoredox and Cr catalysis, we have successfully developed the first highly efficient desilylative allylation of allylic gem-silylboronate esters with aldehydes with exclusively α-selectivity, enabling facile entry to homoallylic alcohols bearing allylic Bpin group in modest to excellent yields with excellent chemo-, regio- and diastereoselectivity. In addition, when γ-substituted allylic gem-silylboronate esters were employed as substrates, their reactions with aldehydes proceeded with a reversed γ-selectivity with excellent stereocontrol. Moreover, enantioselective transformation was also realized with a bisoxazoline-type chiral ligand. Mechanistic experiments provided concrete evidence for a radical-involved mechanism. The synthetic potential of the current protocol was demonstrated by late-stage modification of complex molecules as well as a wide range of downstream transformations.
Methods
General procedure for allylation with 2a
To an oven-dried 10 mL Schlenk tube was added PC-1 (1.2 mg, 2 µmol, 2 mol%), the tube was transferred to a nitrogen-filled glovebox, and CrCl2 (1.2 mg, 10 µmol, 10 mol%) was added. After sealing and removing the tube from the glovebox, aldehyde 1 (0.1 mmol, 1.0 equiv.) in MeCN (1.0 mL, 0.1 M) and 2a (45 μL, 0.15 mmol, 1.5 equiv.) were added quickly under a nitrogen atmosphere. The reaction mixture was irradiated with blue LEDs (5 W, λmax = 456 nm) at room temperature for 12–48 h. After completion of the reaction, the volatiles were removed in vacuo before THF (1 mL), aqueous NaOH (150 μL, 0.3 mmol, 3.0 equiv., 2 M in water), and 30% H2O2 (60 μL, 0.6 mmol, 6.0 equiv.) were sequentially added at 0 °C (ice-water bath), followed by further stirring at room temperature for 3 h. The reaction mixture was quenched with H2O (5 mL) and EtOAc (5 mL), then the organic phase was separated. The aqueous phase was further extracted with EtOAc. The combined organic phases were filtered over a short silica gel plug and concentrated under reduced pressure. Further purification by silica gel column chromatography afforded the desired product 4.
General procedure for allylation with 2b-2n
To an oven-dried 10 mL Schlenk tube was added PC-1 (1.2 mg, 2 µmol, 2 mol%), the tube was transferred to a nitrogen-filled glovebox and CrCl2 (1.2 mg, 10 µmol, 10 mol%) was added. After sealing and removing the tube from the glovebox, aldehyde 1a or 1i (0.1 mmol, 1.0 equiv.) in MeCN/DCM (1:1, 1.0 mL, 0.1 M) and α-silyl-substituted allylboronate reagent 2 (0.15 mmol, 1.5 equiv.) were added quickly under a nitrogen atmosphere. The reaction mixture was irradiated with blue LEDs (5 W, λmax = 456 nm) at room temperature for 12–18 h. After completion of the reaction, the volatiles were removed in vacuo before THF (1 mL) and aqueous HCl (100 μL, 0.2 mmol, 2.0 equiv., 2 M in water) were added, and the reaction mixture was stirred at room temperature for 30 min before quenching with H2O (5 mL) and EtOAc (5 mL). Then the organic phase was separated, and the aqueous phase was further extracted with EtOAc. The combined organic phases were filtered over a short silica gel plug and concentrated under reduced pressure. Further purification by silica gel column chromatography afforded the desired product 6.
Data availability
Experimental procedures, characterizations of new compounds, results of DFT calculation, details of XRD analysis, NMR and HPLC spectra of new compounds are included in the Supplementary Information (PDF). CCDC 2451142 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Center (www.ccdc.cam.ac.uk/data_request/cif). All Data supporting the findings of this manuscript are also available from the corresponding author upon request. Details of the DFT calculation have been provided in Supplementary Data 1 accompanying this paper.
References
Marek, I. & Normant, J.-F. Synthesis and reactivity of sp3-geminated organodimetallics. Chem. Rev. 96, 3241–3268 (1996).
Marek, I. Synthesis and reactivity of sp2 geminated organobismetallic derivatives. Chem. Rev. 100, 2887–2900 (2000).
Banchini, F. et al. Stereoselective double functionalization of geminated C(sp3)-organodimetallic linchpins. ChemCatChem 16, e202301495 (2024).
Lovering, F., Bikker, J. & Humblet, C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756 (2009).
Wu, C., Bao, Z., Xu, X. & Wang, J. Metal-free synthesis of gem-silylboronate esters and their Pd(0)-catalyzed cross-coupling with aryliodides. Org. Biomol. Chem. 17, 5714–5724 (2019).
Chen, W. W. et al. Exploring benzylic gem-C(sp3)–boron–silicon and boron–tin centers as a synthetic platform. Chem. Sci. 12, 10514–10521 (2021).
Yamamoto, Y., Yatagai, H. & Maruyama, K. -Silyl- or -stannyl-substituted crotyl-9-borabicyclo[3.3.1]nonane as a new reagent for the stereoregulated synthesis of acrylic systems. J. Am. Chem. Soc. 103, 3229–3231 (1981).
Chen, M. & Roush, W. R. Enantioselective synthesis of (E)-δ-Silyl-anti-homoallylic alcohols via an enantiodivergent hydroboration-crotylboration reaction of a racemic allenylsilane. Org. Lett. 15, 1662–1665 (2013).
Tong, R. et al. 3-Silyl-3-Borylhex-4-enoate: a chiral reagent for asymmetric crotylboration of aldehydes. Org. Lett. 24, 7822–7827 (2022).
Huang, M. Y., Zhao, Y. T., Zhang, C. D. & Zhu, S. F. Highly regio-, stereo-, and enantioselective copper-catalyzed B–H bond insertion of α-silylcarbenes: efficient access to chiral allylic gem-silylboranes. Angew. Chem. Int. Ed. 61, e202203343 (2022).
Huang, M.-Y. et al. Enantioselective α-boryl carbene transformations. J. Am. Chem. Soc. 146, 9871–9879 (2024).
Lambert, J. B. et al. The β effect of silicon and related manifestations of σ conjugation. Acc. Chem. Res. 32, 183–190 (1999).
Roberts, D. D. & McLaughlin, M. G. Strategic applications of the β-silicon effect. Adv. Synth. Catal. 364, 2307–2332 (2022).
Masse, C. E. & Panek, J. S. Diastereoselective reactions of chiral allyl and allenyl silanes with activated C:X.pi.-bonds. Chem. Rev. 95, 1293–1316 (1995).
Lee, J. H. Use of the Hosomi-Sakurai allylation in natural product total synthesis. Tetrahedron 76, 131351 (2020).
Shimizu, M., Kitagawa, H., Kurahashi, T. & Hiyama, T. 1-Silyl-1-boryl-2-alkenes: Reagents for stereodivergent allylation leading to 4-Oxy-(E)-1-alkenylboronates and Oxy-(Z)-1-alkenylsilanes. Angew. Chem. Int. Ed. 40, 4283–4286 (2001).
Carosi, L., Lachance, H. & Hall, D. G. Additions of functionalized α-substituted allylboronates to aldehydes under the novel Lewis and brønsted acid catalyzed manifolds. Tetrahedron Lett. 46, 8981–8985 (2005).
Chen, J., Gao, S. & Chen, M. Stereoselective syntheses of γ,δ-bifunctionalized homoallylic alcohols and ethers via chemoselective allyl addition to aldehydes. Org. Lett. 21, 9893–9897 (2019).
Jung, Y. & Cho, S. H. Chemo-, regio-, and stereoselective access to (E)-Boryl-substituted allyl fluorides via electrophilic fluorodesilylation. Synlett 34, 2165–2168 (2023).
Jung, Y. et al. Iridium-catalyzed chemo-, diastereo-, and enantioselective allyl-allyl coupling: accessing all four stereoisomers of (E)-1-boryl-substituted 1,5-dienes by chirality pairing. Angew. Chem. Int. Ed. 62, e202218794 (2023).
Liu, H. et al. Photocatalytic multisite functionalization of unactivated terminal alkenes by merging polar cycloaddition and radical ring-opening process. Angew. Chem. Int. Ed. 63, e202407928 (2024).
Xu, Y. et al. Stereoselective photoredox catalyzed (3+3) dipolar cycloaddition of nitrone with aryl cyclopropane. Angew. Chem. Int. Ed. 62, e202310671 (2023).
Ge, L. et al. Photoredox-catalyzed C–C bond cleavage of cyclopropanes for the formation of C(sp3)–heteroatom bonds. Nat. Commun. 13, 5938 (2022).
Li, X. et al. A novel type of radical-addition-induced β-fragmentation and ensuing remote functionalization. Chem 8, 2245–2259 (2022).
Ge, L. et al. Photoredox-catalyzed oxo-amination of aryl cyclopropanes. Nat. Commun. 10, 4367 (2019).
Zhang, Y. et al. Intermolecular carboamination of unactivated alkenes. J. Am. Chem. Soc. 140, 10695–10699 (2018).
Xiong, Y. & Zhang, G. Enantioselective 1,2-difunctionalization of 1,3-butadiene by sequential alkylation and carbonyl allylation. J. Am. Chem. Soc. 140, 2735–2738 (2018).
Schwarz, J. L., Schäfers, F., Tlahuext-Aca, A., Lückemeier, L. & Glorius, F. Diastereoselective allylation of aldehydes by dual photoredox and chromium catalysis. J. Am. Chem. Soc. 140, 12705–12709 (2018).
Mitsunuma, H., Tanabe, S., Fuse, H., Ohkubo, K. & Kanai, M. Catalytic asymmetric allylation of aldehydes with alkenes through allylic C(sp3)–H functionalization mediated by organophotoredox and chiral chromium hybrid catalysis. Chem. Sci. 10, 3459–3465 (2019).
Huang, H.-M., Bellotti, P. & Glorius, F. Merging carbonyl addition with photocatalysis. Acc. Chem. Res. 55, 1135–1147 (2022).
Tanabe, S., Mitsunuma, H. & Kanai, M. Catalytic allylation of aldehydes using unactivated alkenes. J. Am. Chem. Soc. 142, 12374–12381 (2020).
Tang, S.-Y., Wang, Z.-J., Ao, Y., Wang, N. & Huang, H.-M. Photoredox/Cr- catalyzed enantioselective radical-polar crossover transformation via C-H functionalization. Nat. Commun. 16, 1354 (2025).
Zhang, F.-H., Guo, X., Zeng, X. & Wang, Z. Asymmetric 1,4-functionalization of 1,3-enynes via dual photoredox and chromium catalysis. Nat. Commun. 13, 5036 (2022).
Shen, H., Zhang, Z., Shi, Z., Gao, K. & Wang, Z. A modular approach to stereoselective homoaldol reaction via photoredox/Cr/Co triple catalysis. Chem 10, 998–1014 (2024).
Yoon, U. C. & Mariano, P. S. Mechanistic and synthetic aspects of amine-enone single electron transfer photochemistry. Acc. Chem. Res. 25, 233–240 (1992).
LaPorte, A. J., Shi, Y., Hein, J. E. & Burke, M. D. Stereospecific Csp3 suzuki–miyaura cross-coupling that evades β-oxygen elimination. ACS Catal. 12, 10905–10912 (2022).
Oyama, N., Akiyama, S., Kubota, K., Imamoto, T. & Ito, H. Cu(I)-Catalyzed enantioselective γ-boryl substitution of trifluoromethyl- and silyl-substituted alkenes. Eur. J. Org. Chem. 2022, e202200664 (2022).
Han, S. B., Han, H. & Krische, M. J. Diastereo- and enantioselective anti-alkoxyallylation employing allylic gem-dicarboxylates as allyl donors via iridium-catalyzed transfer hydrogenation. J. Am. Chem. Soc. 132, 1760–1761 (2010).
McGhie, L. et al. Photogeneration of α-bimetalloid radicals via selective activation of multifunctional C1 units. J. Am. Chem. Soc. 146, 15850–15859 (2024).
Yuan, X. et al. Asymmetric radical oxyboration of β-substituted styrenes via late-stage stereomutation. Angew. Chem. Int. Ed. 62, e202313770 (2023).
Ondrusek, B. A. & McQuade, D. T. Synthesis of methyl axenoside and methyl 3-epi-axenoside via ate-mediated allylic substitution (AMAS). Synlett 25, 1547–1549 (2014).
Wheatley, E., Zanghi, J. M. & Meek, S. J. Diastereo-, enantio-, and anti-selective formation of secondary alcohol and quaternary carbon stereocenters by cu-catalyzed additions of B-substituted allyl nucleophiles to carbonyls. Org. Lett. 22, 9269–9275 (2020).
Schäfers, F., Dutta, S., Kleinmans, R., Mück-Lichtenfeld, C. & Glorius, F. Asymmetric addition of allylsilanes to aldehydes: a Cr/photoredox dual catalytic approach complementing the Hosomi–Sakurai reaction. ACS Catal. 12, 12281–12290 (2022).
Chen, J., Gao, S., Gorden, J. D. & Chen, M. Stereoselective syntheses of γ-boryl substituted syn-β-Alkoxy- and syn-β-amino-homoallylic alcohols via a regio- and stereoselective allene diboration and aldehyde allylboration reaction sequence. Org. Lett. 21, 4638–4641 (2019).
Kraus, G. A., Molina, M. T. & Walling, J. A. Reduction of cyclic hemiacetals. The synthesis of demethoxyeleutherin and nanaomycin A. J. Chem. Soc. Chem. Commun. 1568−1569, https://doi.org/10.1039/C39860001568 (1986).
Lin, E. E. et al. Regio- and stereoselective synthesis of tetra- and triarylethenes by N-methylimidodiacetyl borondirected palladium-catalysed three-component coupling. Commun. Chem. 2, 34 (2019).
Sun, Y. et al. Copper−hydride-catalyzed enantioselective processes with allenyl boronates. mechanistic nuances, scope, and utility in target-oriented synthesis. J. Am. Chem. Soc. 141, 12087–12099 (2019).
Acknowledgements
We gratefully acknowledge the financial support of the National Natural Science Foundation of China (grant no. 22271151(C.F.), 22301133(C.Z.)) and the Natural Science Foundation of Jiangsu Province (grant no. BK20220327(C.Z.)).
Author information
Authors and Affiliations
Contributions
C.F. conceived the project and directed the investigations. L.L. performed the reaction development and condition optimization. L.L. performed the synthetic experiments, mechanistic study and analyzed the experimental data with contributions from H.L., M.Z., and R.D. L.T. performed the DFT calculation. Z.S., C.Z., Y.Z., and C.F. wrote and revised the manuscript with input from all authors. K.G. and C.F. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Qingmin Wang, and the other anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, L., Li, H., Zhang, M. et al. Desilylative allylation of 3,3-borylsilylpropene under metallaphotoredox catalysis. Nat Commun 17, 1547 (2026). https://doi.org/10.1038/s41467-025-68269-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68269-0
