
Abstract
Zygotes inherit parental genomes with distinct chromatin structures. In eutherian mammals, this asymmetry is considered crucial for embryonic development, notably because it enables genomic imprinting. Besides the well-established role of DNA methylation in this process, a transient form of imprinting in mice has been shown to rely instead on H3K27me3. Here, we show that maternal deletion of Ezhip, encoding a negative regulator of PRC2, initially increases the asymmetric distribution of H3K27me3 among the parental genomes at the zygotic stage but subsequently impairs H3K27me3-dependent imprinting and mitigates X-chromosome inactivation in pre-implantation embryos. We show that EZHIP protein, translated from the maternal mRNA pool, is present during the first cell divisions post-fertilization and limits PRC2 enzymatic activity. In its absence, the H3K27me3 landscape is both expanded and flattened, and the asymmetry between the two parental genomes is lost. Our study reveals the deleterious consequences on early embryonic development of unleashing PRC2 activity.
Similar content being viewed by others

Introduction
In many sexually reproducing organisms, the zygote inherits genomes from the male and female gametes that are folded within chromatin with distinct properties. The case of the asymmetric distribution of DNA methylation between parental genomes has received a great deal of attention in mammals, in particular in mice. It has been shown that this mark is largely erased post-fertilization, enabling the establishment of the embryonic methylation pattern to ensure accurate transcription patterns for proper development of the newly formed embryo1. However, specific genomic regions bypass this remodeling process and can drive parental-allele-specific gene expression, a phenomenon known as genomic imprinting. Imprinted genes often rely on differential DNA methylation to silence one copy (or prevent its activation), and these modified parental alleles can be inherited in specific cell types from the embryo to adulthood2.
Recently, a second type of imprinting mechanism was described that relies on chromatin regulation by the Polycomb machinery3. H3K27me3, a repressive mark catalyzed by the Polycomb PRC2 complex, was shown to be asymmetrically enriched at thousands of regions from the zygote to the blastocyst stage, when resetting of this mark is reported to take place4,5. Erasing H3K27me3 from the oocyte was shown to trigger the deregulation of a small subset of genes (76 genes) that are normally kept silent by H3K27me3 on their maternal copy during mouse preimplantation, and that are solely expressed from the paternal allele3. This asymmetric parental expression is mostly transient except for a few genes (fewer than 10) that maintain a parentally biased expression in the extraembryonic tissues after implantation. Of note, while inhibiting the activity of either Polycomb complex, PRC1 or PRC2, during oocyte maturation impairs H3K27me3-dependent imprinting, erasure just before fertilization of H3K27me3 but not H2AK119ub leads to a loss of this imprinting5,6,7. This suggests that while both marks contribute to the establishment of the imprinting, only H3K27me3 is required to transmit the imprinting information from the oocyte to the zygote.
One of the transiently imprinted genes is Xist8, which encodes the long non-coding RNA responsible for X-chromosome inactivation (XCI) in XX embryos. This process ensures gene-dosage compensation between XY and XX individuals9. While loss of autosomal H3K27me3-dependent imprinting is associated with placental enlargement at term (E18.5), the reduced viability of male embryos with a maternal deletion of Eed (Eed-matKO) at birth is proposed to be due to ectopic maternal Xist expression triggering the aberrant inactivation of their single X chromosome6,7,8,10,11.
Recently, a new cofactor of the PRC2 complex named EZHIP (for Enhancer of Zeste Homologs Inhibitory Protein) has been identified12,13,14,15,16. EZHIP limits the enzymatic activity of PRC2 by direct interaction with EZH2, the enzymatic subunit of PRC2, through a mechanism proposed to be similar to the one described for the “oncohistone” H3K27M15,16,17. In normal physiological conditions, Ezhip is primarily expressed in adult gonads. Its RNA is very abundant in oocytes and in zygotes but quickly drops at the 2-cell stage in mice14. Constitutive knockout of Ezhip results in a strong accumulation of H3K27me3 in spermatogonia and mature oocytes, and an age-related exhaustion of ovarian reserve, as well as a diminished progeny number14.
Here, we set out to study the consequences of the loss of maternal Ezhip, an opportunity to explore how gain of H3K27me3 in the oocyte impacts H3K27me3-dependent imprinting. We confirm that maternal Ezhip knockout leads to a global increase of H3K27me3 in the oocyte. Surprisingly, maternal deletion of Ezhip also affects H3K27me3 deposition post-fertilization including on the paternally inherited genome. We show that this is due to the presence of maternal EZHIP protein during cleavage stages. When Ezhip is maternally deleted, H3K27me3 deposition is markedly increased on both genomes, resulting in the loss of the asymmetric distribution of the mark and its flattening throughout the genome. This reshaping of the H3K27me3 landscape is accompanied by a loss of H3K27me3-dependent imprinting. Our study reveals the crucial role of EZHIP-mediated PRC2 inhibition during the first cell divisions post-fertilization.
Results
Ezhip loss alters H3K27me3 but barely the RNA pool of oocytes
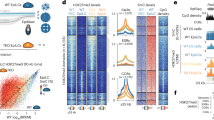
We previously showed by immunofluorescence that global levels of H3K27me3 increase in Ezhip-KO oocytes at the germinal vesicle (GV) and metaphase of meiosis II (MII) stages as compared to wild-type counterparts14. Here, we performed low-input CUT&RUN with spike-in (see Methods & ref. 18) to map the genomic location of H3K27me3 in Ezhip-deficient GVs (Ezhip-KO) compared to wild type control GVs (Ctr). Broad views of H3K27me3 enrichment suggest an overall increase in H3K27me3 deposition in Ezhip-KO versus Ctr (Fig. 1a, top) with a tendency to flatten around the main peaks (Fig. 1a, bottom). To make a more quantitative evaluation, we split the genome into 10 kb bins and analyzed H3K27me3 fold-change enrichment as a function of the initial abundance of the mark (Fig. 1b). We confirmed a global increase in H3K27me3, as indicated by the fact that most peaks have a positive a Log2(FC). These regions are generally poorly enriched for this mark in control GVs (Fig. 1b, c) and include non-coding regions (Fig. 1e). We also observed loss of H3K27me3 in regions initially enriched for H3K27me3 (Fig. 1d), mostly found at promoters and exonic regions (Fig. 1e). Of note, adjacent bins with a similar trend were merged resulting in intervals that are larger for the gains than for the losses (See scale in Fig. 1c, d).

a Genome browser views of H3K27me3 CUT&RUN for Ctr or Ezhip-KO mature GVs. It is centered on chromosome 6 including the Hoxa gene cluster at two different resolutions. b Density scatter plot showing the Fold Change (Log2) in H3K27me3 CUT&RUN signal distribution between Ctr and Ezhip-KO GVs. The genome is split in 10 kb bins across the genome (see also methods). c, d Heatmaps showing gain (UP) or loss (DOWN) of H3K27me3 signal enrichment (rpkm) in KO versus Ctr GVs, signals corresponding to the squares defined in b. The H3K27me3 regions is centered and a flanking region of 0.2 Mb is considered in c and, of 20 kb in (d). Rows are ordered by region length. e Genomic distribution of up and down regions in KO GVs. f Average H3K27me3 CUT&RUN signal for Ctr (light red) or KO (dark red) GVs at Polycomb associated domains defined in ref. 19. g Similar as f for HiC clusters defined in ref. 20. Left panel corresponds to clusters 1 and 2, while the right panel represents analysis over cluster 4 to 9. CUT&RUN signal represents the pooled from independent experiments performed on n = 125 and n = 198 oocytes for Ctr and Ezhip-KO respectively.
This indication that the losses are rather focal contrasts with the unusual distribution of H3K27me3 in oocytes characterized notably by broad domains (up to 1–2 Mb in size) correlating with densely compacted chromatin and referred to as Polycomb associated domains (PADs; see ref. 19). We therefore evaluated H3K27me3 enrichment in Ezhip-KO oocytes at the PADs and observed a clear enrichment despite the focal losses reported previously (Fig. 1f). We obtained similar results when focusing on HiC-informed chromatin architecture domains of the early embryos enriched in H3K27me3 (Fig. 1g; see ref. 20).
We then investigated whether Ezhip ablation affects the composition of the maternal pool of RNA in mature preovulatory oocytes. We performed single-oocyte RNA sequencing (RNA-seq) and analyzed differential gene expression after pooling control and Ezhip-KO germinal vesicle stage (GV) oocytes (Supplementary Fig. 1a). This revealed minimal changes in RNA abundance, and no correlation between the H3K27me3 changes in the mutant oocytes and the expression levels of corresponding genes (Supplementary Fig. 1b). These results are consistent with previous observations in MII oocytes14.
We conclude that the absence of EZHIP at the GV stage in oogenesis leads to an increase in H3K27me3 deposition, both within and outside Polycomb domains yet these alterations do not have substantial consequences on the RNA content of mutant oocytes.
Ezhip maternal deletion alters H3K27me3 post-fertilization
We then evaluated whether the maternal deletion of Ezhip (constitutive deletion of Ezhip in mother14, hereafter designated as Ezhip-matKO) could impact the early stages of embryonic development. Importantly, Ezhip RNA is very abundant in the maternal pool of RNAs transmitted by the oocyte but barely expressed post-fertilization in the embryos (see ref. 14 and Supplementary Fig. 2a). Ezhip-matKO embryos are therefore Ezhip wt/- for females and -/Y for males, yet Ezhip mRNA is mostly absent in both cases. We first performed immunofluorescence to detect any global modification of H3K27me3 deposition focusing on 2-cells (2 C) and blastocyst stages. This revealed a pronounced gain in H3K27me3 signal for Ezhip-matKO embryos as compared to control embryos (Ctr) at 2-cell stage (Fig. 2a). Interestingly, this difference in H3K27me3 signal detected by immunofluorescence remains at blastocyst stage. This prompted us to determine what part of the genome is affected by those gains and whether they equally affect the maternally and paternally inherited genomes. Toward this end, we generated highly polymorphic hybrid embryos enabling us to distinguish the maternal (in a C57B6/J background) from the paternal genome (CAST Ei/J strain) by CUT&RUN analysis of preimplantation embryos (2-Cell and blastocyst stages). The distribution of H3K27me3 appears globally altered in Ezhip-matKO blastocysts (Fig. 2b). Similarly to the oocytes, we observed a global increase of H3K27me3 deposition with a flattening of the profile at a high plateau resulting in much less defined Polycomb domains (Supplementary Fig. 2b). Another important change is that, whereas the control group (Ctr) displayed the expected asymmetric distribution of H3K27me3 between parental genomes4, the profiles of this mark on the maternally and paternally inherited genomes in mutant blastocysts are very similar (Fig. 2b and Supplementary Fig. 2b). This loss of asymmetry is further underscored by our hierarchical clustering analysis of the H3K27me3 distribution, which shows that the maternal and paternal genomes cluster together in Ezhip-matKO embryos at the blastocyst stage, in contrast to the control condition in which they cluster separately as expected (Fig. 2c). Overall, the impact of Ezhip deletion on H3K27me3 pattern of deposition appears more pronounced on embryos at the blastocyst stage than on oocytes and 2-Cell stage embryos (Fig. 2bversus Fig. 1a). We investigated whether this alteration of the epigenome correlates with transcriptional alterations by performing RNA-seq on single hybrid embryos (2-Cell embryos and blastocysts). A comparative analysis showed that more genes are affected at 2-Cell stage than in oocytes (Fig. 2d versus Supplementary Fig. 1a), and this difference is even more pronounced at the blastocyst stage (Fig. 2e), with a similar proportion of genes being up or down regulated. Of note, these changes concerned both genomes (Supplementary Fig. 2c, d) yet changes in H3K27me3 enrichment and transcriptomic changes were not clearly correlated suggesting that transcriptomic changes are often indirect (Supplementary Fig. 2e). Importantly, we compared these transcriptomic alterations with those reported upon loss of maternal H3K27me3 (overexpression of Kdm6b, see refs. 3,5) but did not observe any correlation, indicating that preventing H3K27me3 inheritance produces a different transcriptional outcome in embryos compared to maternal deletion of Ezhip (Fig. 2f).

a Representative image of H3K27me3 immunofluorescence signal for Ctr and Ezhip-matKO embryos at 2 C stage (Top) and blastocyst stages (Bottom). Counterstaining with Phalloidin (2 C) and DAPI (Blastocyst). Representative equatorial slice for 2 C stage and representative Z projection for blastocyst. Numbers of embryos analyzed for each stage are respectively for Ctr n = 12 and n = 5 and Ezhip-matKO n = 9 and n = 6. Scale bar is 10 μm. Right panel represents the mean nuclear to cytoplasmic (N/C) ratio of H3K27me3 staining for individual embryos (averaged over both cells in 2 C embryos, and over 10 cells in blastocysts). Mean and single values. Statistics: Welch’s t test two-tailed, P-value on graph. Raw data as a Source Data file. b Genome browser views for H3K27me3 CUT&RUN for Ctr or Ezhip-matKO at blastocyst stage. It is centered on chromosome 6 including the Hoxa gene cluster at two different resolutions. n⩾7 per sex and per genotype for CUT&RUN. c Hierarchical clustering analysis of allelic H3K27me3 distribution (10 kb windows) at different stages (oocyte (GVs) to blastocyst) for Ctr or Ezhip-matKO. d, e MA plot representing Log2 ratio of Ezhip-matKO/Ctr rpkm depending on the average rpkm in Ctr for embryos at the 2 C and blastocyst stages respectively. n⩾5 per sex, per genotype and per stage for RNAseq. f Correlation plot representing Log2(FoldChange) Ezhip-matKO/Ctr RNAseq at 2-cell stage in function of Log2(FoldChange) Kdm6b OverExpression/Ctr RNAseq at 2-cell stage (GSE153531). Linear fitting and corresponding equation are represented on the graph.
Our results demonstrate that the absence of the maternal pool of Ezhip deeply affects the epigenome and transcriptome during early development.
EZHIP is present in the embryo post-fertilization
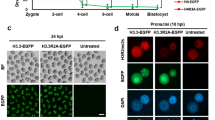
Given that the effects of maternal deletion of Ezhip on embryos post-fertilization are so pronounced, and that they notably include a strong gain of H3K27me3 on the paternally inherited genome, we reasoned that they are unlikely to result only from the inheritance of an altered maternal epigenome. We therefore investigated whether EZHIP protein remains present post-fertilization despite the sharp drop in Ezhip mRNA at the 2-Cell stage14. To address this question, we first needed to raise an antibody suitable for immunofluorescence. Our previous attempt to immunize rabbits against small peptides of mEZHIP did not produce sufficiently specific antibodies14, we therefore tried again using a larger fragment of mEZHIP (amino acids 1-415). After affinity purification, the antibody specificity was checked by western blot using nuclear extracts of HeLa cells transiently over-expressing mouse or human EZHIP (Supplementary Fig. 3a, b). The antibody solely recognized the murine version of EZHIP, in contrast to a commercially available anti-human EZHIP antibody, which only detects the human ortholog. Immunofluorescence analysis against EZHIP revealed a diffuse signal in the cytoplasm from control zygote to morula stage embryos, signal which was completely absent in Ezhip-matKO embryos (Fig. 3a). Higher magnification examination of embryos nonetheless showed that EZHIP also localizes within the nucleus at the 2-Cell and morula stages, still with no signal detected in Ezhip-matKO embryos (Fig. 3b, c).

a Representative z- stack confocal images of immunofluorescence detecting EZHIP (in green) for Ctr and Ezhip-matKO embryos at zygote, 2 C or morula stages (respective numbers for Ctr are n = 14, 15 and 9; for Ezhip-matKO n = 10; 7 and 4). Scale bar is 10 μm and DNA is counterstained with DAPI (grey). Control and mutant samples were processed in parallel and acquired using similar settings. b, c Nucleus views of single plane images of embryos shown in a for Ezhip Ctr (b) or Ezhip-matKO (c). Mat and PN stand respectively for maternal and paternal pronuclei. Scale bar is 10 μm and DNA is counterstained with DAPI (grey). Representative images from n = 14/15/9 (WT) and n = 10/7/2 (matKO) embryos at zygote/2 C/morula respectively. d Histone Methyl Transferase assays monitoring recombinant PRC2 enzymatic activity in presence of different cofactors (PRC2/J2/A = PRC2 in presence of JARID2 and AEBP2) or of H3K27me3 peptide with or without EZHIP. HMTs were analyzed on SDS-PAGE. Top, radiography detecting the 3H, bottom corresponding coomassie staining. Representative result of n = 3. Raw data provided as a Source Data file. e Same as in d but in presence of either octamer or nucleosome. Representative result of n = 2. Raw data provided as a Source Data file. f Fractions 1 to 12 of 10–25% sucrose gradient analyzed for either DNA (agarose gel) or protein (Dot Blot detecting Strep-EZHIP). Main elution fractions are underlined in red. Representative result of n = 2. Raw data provided as a Source Data file.
The results obtained so far suggest that the presence of EZHIP in early embryos globally restricts PRC2 enzymatic activity but allows a residual activity on the maternally inherited and H3K27me3-enriched genome, and that it exerts its activity without being tightly associated to chromatin. This prompted us to investigate how these observations could be reconciled with the protein’s biochemical properties. First, we compared the inhibitory potential of EZHIP on PRC2 in presence or absence of peptides carrying the H3K27me3 modification and/or regulatory cofactors by performing in vitro enzymatic assays monitoring the incorporation of 3H-SAM (Fig. 3d). The degree of inhibition of PRC2 by EZHIP is proportionally similar in all conditions tested (Supplementary Fig. 3c). Its residual activity therefore remains higher when the starting activity is higher (i.e., when stimulated by its cofactors and/or by allosteric regulation). We speculate that this limited activity might be sufficient to maintain H3K27me3 on the maternally inherited genome but not on the paternally inherited genome which requires de novo establishment of the mark. We then tested whether EZHIP inhibits PRC2 exclusively in the context of chromatin by comparing its inhibitory activity on nucleosomes versus histone octamers (Fig. 3e). We found that EZHIP inhibits PRC2-mediated H3K27 methylation equally well on both substrates (Supplementary Fig. 3d). To further probe the relationship between EZHIP and chromatin, we investigated whether EZHIP interacts with either DNA or chromatin by analyzing its capacity to form a complex as detected by a shift toward the densest fractions on sucrose gradient. The experiment revealed that, when incubated together, a small fraction of EZHIP and of DNA are shifted on sucrose gradient, indicative of direct interaction (Fig. 3f, underlined with a red lane). Yet, this shift is not observed when EZHIP is put in presence of chromatin, supporting the hypothesis that they do not interact directly. Of note, in the same assay, PRC2 is shifted of about one fraction in presence of chromatin and, even in the presence of PRC2, EZHIP does not appear to bind chromatin (Supplementary Fig. 3e).
Altogether, these results suggest a model whereby EZHIP, present from the zygote at least until the morula stage, globally inhibits PRC2, nonetheless allowing a limited enzymatic activity compatible with the maintenance of maternally inherited H3K27me3.
EZHIP is required for H3K27me3-dependent imprinting
The results presented so far show that the maternal deletion of Ezhip does not noticeably alter the asymmetric distribution of H3K27me3 between the gametes (since H3K27me3 is increased in the Ezhip-KO oocytes and the paternal contribution not affected) but rather impairs the maintenance of this asymmetry post-fertilization. This led us to wonder whether Ezhip deletion had any impact on H3K27me3-dependent imprinting. We examined the genes that are subject to this imprinting3 and analyzed their enrichment for H3K27me3 at different developmental stages. In mature oocytes, these genes display a strong density of H3K27me3, which is even more pronounced in mutant condition (Fig. 4a). At 2-cell stage, the H3K27me3 density is substantially reduced but remains higher on the maternally inherited alleles than on the paternally inherited ones, even in mutant embryos. At the blastocyst stage, however, wild-type and Ezhip-matKO embryos diverge substantially. In Ezhip-matKO blastocysts, H3K27me3 density globally increases on both genomes and, the asymmetric distribution of the mark is lost (Fig. 4a). The same trend is observed when focusing on the DNA-methylation dependent imprinted genes (Supplementary Fig. 4a).

a Enrichment of H3K27me3 over the gene body of H3K27me3 imprinted genes in Ctr and Ezhip-matKO from GVs to blastocyst stages. b Heatmaps representing the allelic expression of H3K27me3 dependent imprinted genes in presence (Ctr) or absence (Ezhip-matKO) of EZHIP at the indicated developmental stages. Cluster 1 includes imprinted gene classified as “persistent” and cluster 2 genes imprinted only at blastocyst stage. The coloured key indicates the ratio of Mat/ Pat+Mat reads for genes with > 10 SNP-containing reads. Each column represents a single embryo, and each line corresponds to a gene. c Relative gene expression levels in Ezhip-matKO versus Ctr represented in Log 2 (Fold Change) for H3K27me3 dependent imprinted genes as in b. Statistics: Student t test two-tailed, P-value on graph. d Gene expression level (rpkm) for factors involved in trophoblast lineage in Ctr versus Ezhip matKO. Data are extracted from 6.5 ExE RNAseq (b). Mean and single values (n = 10 Ctr & n = 8 matKO embryos). Statistics: Welch’s t test two-tailed, P-value on graph. Raw data as a Source Data file. e Violin plots indicating the relative weight ratio embryo over placenta. Related to Supplementary Fig. 4c. Single values. Statistics: Welch’s t test two-tailed, P-value on graph. Raw data as a Source Data file. f Bar plot representing the average embryo numbers at indicated gestation days post coitum. Number of embryos are indicated in brackets. Genotype indicated in the legend. Mean and single values. Statistics: Welch’s t test two-tailed, P-value on graph. Raw data as a Source Data file.
We investigated the transcriptional consequences of this altered H3K27me3 deposition by analyzing RNAseq from hybrid embryos either at the blastocyst stage or later in extra-embryonic annexes from 6.5 dpc (E6.5 ExE). This later stage is used for the few genes whose H3K27me3-dependent imprinting persists beyond implantation3. We assessed 16 genes with sufficient allelic coverage (Fig. 4b) that we further subdivided into two groups based on the persistence of the imprinting at E6.5 ExE (cluster 1) or its disappearance (cluster 2) (Fig. 4b). Most of the genes, which are normally predominantly expressed from the paternally inherited genome, are expressed from both genomes in Ezhip-matKO embryos with nonetheless a strong heterogeneity in the extent of allelic changes between genes (e.g. Gab1 and Smoc1), (Fig. 4b). Besides, with the exception of Mbnl2, the bi-allelic expression of those gene correlate with either no change or an increase of their total RNA levels, indicating that it is no due to silencing on both alleles but that it occurs while both alleles tend to be transcriptionally active (Fig. 4c). None of the DNA methylation-dependent imprinted genes were affected in their parental expression biases by the absence of EZHIP (Supplementary Fig. 4b). In E6.5 ExE, most of the cluster 1 genes still display allelic ratio alterations upon loss of maternal EZHIP.
We then investigated the functional consequences of this impaired H3K27me3-dependent imprinting and showed that it correlates with transcriptional alterations of some genes important for development including trophoblast lineage development, implantation and placentation (Fig. 4d and Supplementary Fig. 4c)21,22,23,24,25. Besides, at E13.5, the weight of mutant embryos was significantly lower than that of the control embryos, while the placenta weights were comparable in the two conditions (Supplementary Fig. 4d). This decrease results in an aberrant weight ratio between the embryos and their annexes, as also reported in Eed-MatKO embryos11, which could contribute to the phenotype (Fig. 4e). We previously reported that Ezhip-knockout impairs female fertility14; here we confirmed this result, as well the lack of sex bias in the progeny at weaning (Supplementary Fig. 4e). Looking during development at the number of embryos, we observed a large part of the lethality resulting from Ezhip maternal deletion had already occurred at E13.5 (Fig. 4f), and we speculate that it takes place around E6.5, this could explain the high variability in the number of embryos at that stage (Fig. 4f). The potential precocity of the embryonic lethality resulting from the maternal deletion of Ezhip suggests that factors other than impaired placental function might underlie this phenotype.
Altogether, our results show that the aberrant deposition of H3K27me3 resulting from Ezhip maternal deletion impairs H3K27me3-dependent imprinting and is associated with long-lasting developmental defects, including in extraembryonic tissues.
Aberrant Xist allelic expression in Ezhip mutant blastocysts
Xist is among the H3K27me3-imprinted genes that can contribute to early embryonic lethality when biallelically expressed7,26,27. During mouse preimplantation development, the long noncoding RNA Xist is exclusively expressed from the paternal allele in female embryos, due to H3K27me3-dependent imprinting8. Of note, the CUT&RUN for H3K27me3 did not reveal any modification of H3K27me3 deposition at the Xist locus in Ezhip-KO GVs (Supplementary Fig. 5a), while both alleles seem to gain H3K27me3 at blastocyst stage (Supplementary Fig. 5b). Analysis of our single-embryo RNA-seq performed on C57B6/J-CAST hybrid blastocysts revealed that Xist is aberrantly expressed on the maternal allele in Ezhip-matKO female embryos resulting in Xist biallelic expression in contrast to the expected strict paternal expression observed in control embryos (Fig. 5a). Importantly, Xist is also robustly upregulated in male mutant embryos (Fig. 5a). We then implemented smRNA-FISH to evaluate Xist expression in blastocyst at single cell level (Fig. 5b). As expected, we can detect one Xist “cloud” in most cells of control female blastocysts. In contrast, in Ezhip-matKO female blastocysts, a fraction of the cells (26%) displays two clouds indicative of aberrant X inactivation on both alleles (Fig. 5c). Regarding the males, no expression of Xist was detected in control blastocyst whereas while in about half the cells of Ezhip-matKO blastocysts, a cloud was observable.

a Heatmaps representing Xist expression levels (top row; scale in Z score, green to purple legend) and allelic ratios (bottom row, blue to red legend) for female and male embryos. Each column represents a single blastocyst. b smRNA-FISH probing Xist in blastocysts either Ctr or Ezhip-matKO. Blastocysts were counterstained with DAPI. A representative Z projection is shown. Scale bar is 10 μm. Higher magnification of one cell of the blastocyst is provided on the right panel. c Manual quantification of Xist cloud (0, 1 or 2), % of cell with each number of clouds. The number of blastocysts analyzed for each genotype and sexe is indicated on top. Exact values provided as a Source Data file.
Impaired XCI in early mouse embryos upon Maternal EZHIP depletion
To understand whether this abnormal Xist expression leads to aberrant XCI, we compared the relative X-linked gene expression levels in control and Ezhip-matKO embryos for both sexes. At the blastocyst stage, the global expression level of X-linked genes, but not autosomal genes, is lower in mutant embryos of both sexes (Fig. 6a). When looking at each conceptus individually, there are no apparent differences in X-linked gene levels at the 2 C and E6.5 ExE stages between control and mutant, while approximately half of the blastocyst stage embryos of either sex exhibit a decrease in X-linked gene expression (Supplementary Fig. 6). We then focused on females and analyzed the allelic ratio changes at different development stages on the X chromosome. We confirmed the lack of noticeable differences at the 2-Cell stage while at the blastocyst stage Ezhip-matKO embryos display mostly biallelic expression when control embryos show mostly maternal expression (Fig. 5b). The alterations in allelic expression on the X chromosome remain detectable at E6.5 in ExE tissues. When we monitored gene expression instead of allelic ratio, differences between wild-type and Ezhip-matKO embryos were less consistent (Fig. 5d and Supplementary Fig. 6). Half of the embryos at the blastocyst stage (#4 to #7) display a global reduction of X-linked gene expression while the other three seem to have a rather normal X-linked gene expression pattern. At E6.5 in ExE tissues, expression of X-linked genes appears very similar between control and Ezhip-matKO. Of note, because of the potential timing of embryonic lethality near E6.5, we cannot exclude the possibility that the embryos from which we were able to recover extra-embryonic tissues are biased toward milder phenotypes. Nonetheless, this suggests that about half of the embryos find a mechanism to restore near-normal X-linked gene expression despite aberrant biallelic silencing. Finally, to further confirm that the aberrant biallelic expression of Xist in Ezhip-matKO is able to trigger functional X chromosome gene inactivation, we crossed male carrying a Xist deletion (female embryo cannot survive to the lack of Xist26,28) with female Ezhip-KO and analyzed the sex of the embryo. The double mutant embryos had a normal sex ratio strongly suggesting that the aberrant Xist expression from the maternally inherited X partially compensates for the absence of Xist expression from the paternally inherited X (Fig. 6d, bottom). However, the size of the litter did not substantially increase in the double mutant suggesting that the aberrant regulation of Xist in Ezhip-matKO embryos is not the only element contributing to embryonic lethality (Fig. 6d, top).

a Box plots showing the relative gene expression between Ctr and Ezhip-matKO blastocysts (both sexes) per individual chromosomes. The middle line in the box represents the median, box edges indicate 25/75th percentile, while the whiskers and the box encompass 90 percent of the data. Female Ctr n = 6 & matKO n = 7, Male Ctr n = 8 & matKO n = 7 embryos. b Heat maps representing the mean allele specific expression ratios per embryonic stage and genotype for each informative and expressed X linked genes (ordered as basis of their genomic position, from centromere to telomere orientation). Colour gradients are used in between two values (one for maternal, zero for paternal) as shown in the key. Xist position is indicated on the left. c Heatmaps representing gene expression for X linked genes (one line per gene, one row per embryo at the indicated stage for each genotype). d Top, mean weaned pup number +/− SD depending on the genotype of the parents. Statistics: Welch’s t test two-tailed, P-value on graph. The number of litter and total number of animals considered is indicated below between brackets. Graph data as a Source Data file. Bottom, bar plot for sex ratio. Controls same data set as in Supplementary Fig. 4d.
Discussion
Our study reveals the critical role of EZHIP as a negative regulator of PRC2 in pre-implantation embryos. Hence, while Ezhip is highly transcribed in oocyte and while its mRNA level drops sharply post-fertilization, the protein remains present at least until the morula stage. The absence of EZHIP unleashes PRC2 enzymatic activity leading to a global increase of H3K27me3 deposition and the loss of asymmetrical, parental-specific H3K27me3 enrichment. Immunofluorescence reveals a rather cytoplasmic subcellular localization of EZHIP from zygote to morula, a subcellular localization also reported upon overexpression in HEK-29315, raising the question of its mode of action on PRC2. Previous studies have shown that EZHIP colocalizes with PRC2 at chromatin and proposed that EZHIP preferentially inhibits the allosterically activated form of EZH217. Yet, our experiments indicate that EZHIP does not interact with chromatin in vitro and inhibits PRC2 to the same extent regardless of the cofactors or substrates that we added in the assays. Putting together these biochemical properties and subcellular localization data, this led us to favor a model wherein EZHIP-mediated PRC2 inhibition can persist even after EZHIP dissociates from PRC2, as was previously proposed for the oncohistone H3K27M29, and does not require continuous colocalization with PRC2 for inhibition.
The aberrant deposition of H3K27me3 has major transcriptomic consequences, including the loss of the H3K27me3-dependent imprinting. Consequently, in mice, the lncRNA Xist, which is normally expressed only from the paternally inherited genome in female, becomes bi-allelically expressed in mutant embryos. The most straight-forward explanation for the loss of the H3K27me3-dependent imprinting in Ezhip-matKO embryos would be that the lack of asymmetry in H3K27me3 enrichment between maternal and paternal genomes results in bi-allelic expression. However, this hypothesis is difficult to reconcile with the aberrant expression of Xist in Ezhip-matKO male embryos. Indeed, in that case the asymmetry is not part of the equation since males have only one X. This result instead suggests that when H3K27me3 is too abundant, the repressive function of the mark is thwarted. This would explain that male embryos that have not inherited the maternal Ezhip are unable to maintain the silencing of Xist on the unique X chromosome even though H3K27me3 density is equal to or higher than its level in wild type embryos. This hypothesis would also explain the seemingly paradoxical observation that, in the case of H3K27me3-dependent imprinted genes other than Xist, both alleles are expressed despite a high H3K27me3 density at those genes.
If H3K27me3 does not properly prevent gene expression in the Ezhip-matKO context, one could argue that the aberrant activation of PRC2 may ultimately translate into a PRC2 loss of function. Yet, the distinct transcriptomic responses to H3K27me3 erasure through kdm6b overexpression in the zygote and to maternal deletion of Ezhip indicates that the excess deposition of H3K27me3 is not equivalent to a PRC2 loss of function. Interestingly, broadening of H3K27me3 deposition with an overall flattening of the peaks is reminiscent of what we and others have reported in cells expressing the gain of function mutation of EZH2 found in a subset of follicular and diffuse large B-cell lymphomas30,31. In that case as well, the mutation which creates a hyperactive form of PRC2 leads to transcriptomic changes (both up and downregulations) that do not overlap with genes deregulated upon PRC2 loss of function31. We speculate that the broadening of the H3K27me3 distribution across the genome might have effects on chromatin regulation that are not straightforward but eventually alter transcription regulation. For instance, H3K27me3 is known to stabilize the recruitment of a subset of PRC1 complexes (PRC1.2 and PRC1.4; for review32), and it will be interesting to determine what happens to their recruitment when H3K27me3 is more broadly distributed and whether their function is altered as previously reported with specific mutation of histone (H3K36M33). The same question applies to other proteins reported to bind H3K27me3 such as CDYL, BAHD1 or BAHCC134,35,36. We also envision that the aberrant deposition of H3K27me3 might indirectly modulate the deposition of other chromatin modifications. Hence, interplays between H3K27me3 deposition and either H3K27ac or H3K36me3 were previously described37,38 and would also deserve to be further investigated in the context of maternal deletion of Ezhip.
Another intriguing result is that male and female pups from Ezhip KO mothers are born at a similar ratio, albeit with a significantly lower number compared to pups from control mothers. This contrasts with the maternal deletion of Eed, which displays similar loss of Xist imprinting and a high resorption rate but with a preferential survival of females7. Of note, the sex bias is related to Xist expression since it is restored by the combined deletion of Eed and Xist11. Around implantation, about half of the embryos succeed in escaping the aberrant X chromosome inactivation by a yet unknown mechanism as also observed upon maternal deletion of Eed7. We speculate that lack of H3K27me3 asymmetry in the context of low levels of this mark (maternal deletion of Eed) or in a context of excess of the mark (maternal deletion of Ezhip) has different transcriptional outcome. Understanding how a subset of embryos manages to correct X chromosome inactivation might shed light on this question.
Altogether, this study shows the crucial role that EZHIP plays in limiting PRC2 enzymatic activity in the pre-implantation embryos. In rodent, this affects H3K27me3-dependent imprinting however it is unclear that this process exists in non-rodent eutherian39,40. Yet, orthologs are found among most eutherian mammals, their capacity to interact with PRC2 is expected to be conserved, and they seem to have a similar pattern of expression14. Altogether, this questions on the function of this protein in non-rodent eutherians. Besides, while this study focused on early development, further investigations on the consequences that EZHIP deletion could have later on development for the embryos that survive to term is required. It is indeed well documented that alteration of DNA-methylation-dependent imprinting could have major phenotype after embryologic development2. Finally, publicly available data sets suggest that Ezhip is expressed, to some extent, in placenta and some specific area of the brain. It will be interesting to determine the function it could have in these organs.
Methods
Histone Methyl transferase assays
StrepTag-EZHIP, recombinant PRC2 (F-EZH2 + SUZ12 + EED + RABP48) and cofactors (StrepTag-AEBP2 a.a. 294–503 and F-JARID2 a.a. 1-530) were all expressed in SF9 cells infected with the corresponding baculovirus described in refs. 14,41. Recombinant EZHIP or PRC2 were resuspended in BC300 + Proteases inhibitors and, purified on Strep-Tactin or M2-beads respectively. H3 and H3K27me3 peptides (Eurogentec) correspond to amino acids 20 to 32 of histone H3 and were added to a final concentration of 40 μM. Octamers were refolded starting from bacterially expressed histones (Xenopus sequences, recovered from inclusion bodies) and purified on a S200 sizing column. Nucleosomes were reconstituted with circular plasmid DNA by progressive salt dialysis. For HMT assays, PRC2 and AEBP2/JARID2 were added to the reaction at equimolar concentration. 200 ng of PRC2 and 1μg of chromatin/octamer were used by default. Incubation was performed in HMT buffer (50 mM Tris pH 8.5, 2.5 mM MgCl2 and 2.5 mM DTT,) plus 3H-SAM (Perkin Elmer) for 30mn at 30°c. Thre reaction was stopped by addition of 5X-loading buffer, runned on 4–15% SDS-PAGE (Biorad), transferred to PVDF membrane, stained with coomassie blue and exposed on Phosphor screen Tritium (Cytiva) overnight.
Sucrose gradient
Insert corresponding to an array of 3 nucleosomes (Widom positioning sequence 3×206 nt) was purified from the plasmid backbone by sizing column (Sephacryl S-500, Cytiva) and used for DNA interaction and chromatin refolding by salt dialysis. DNA, chromatin, EZHIP and PRC2, 2μg each, were incubated together prior to sucrose gradient in HEB buffer (25 mM Hepes, 40 mM KCL, 0.2 mM EDTA, 1 mM DTT). After incubation, samples were loaded on sucrose gradients previously formed with a gradient maker (Biocomp) and centrifugated on an SW60ti at 60000 g for 15hrs. Gradients were fractionated manually (250μL/fraction, 16 fractions). Elutions were analyzed either on agarose gel (DNA) or by Dot Blot (anti-strep for EZHIP and anti-Flag for recombinant PRC2).
Mouse EZHIP antibody & Western blot procedure
Antigen for antibody production (6X-His-mEZHIP a.a. 1-415) was expressed in SF9 cells infected by the corresponding baculovirus. The protein was purified on Ni-NTA Agarose (native purification). Rabbits were immunized using a standard protocol (immunization/bleed alternated every 2 weeks, Covalab). Final bleed was affinity purified on antigen covalently bound to beads (AffiGel-10 Gel, Biorad), eluted with 0.2 M glycine•HCl, pH 2.6, and stored in 40% glycerol 0,1 M Tris-HCl pH 8.0. Validation of the antibody diluted 1/4000 was performed by western analysis of nuclear extracts from Hela cells transiently expression either the murine form or the human form of Ezhip cDNA. For nuclear extract preparation, cells were incubated with buffer A (10 mM Hepes pH 7.9, 2.5 mM MgCl2, 0.25 M sucrose, 0.1% NP40, 0.5 mM DTT, 1 mM PSMF) for 10 min on ice, centrifuged at 6800 g for 10 min, resuspended in buffer B (25 mM Hepes pH 7.9, 1.5 mM MgCl2, 700 mM NaCl, 0.5 mM DTT, 0.1 mM EDTA, 20% glycerol), sonicated and centrifuged at 20800 g for 15 min. As a control for human EZHIP detection, antibody hEZHIP (HPA006128) from Sigma-Aldrich was used.
Mouse oocyte and embryo collection (natural mating)
Animal care and use for this study were performed in accordance with the recommendations of the European Community (2010/63/UE). Mice were hosted on a 12 h light–12 h dark cycle at an ambient temperature of 22 ± 2 °C and 40–70% humidity with free access to food and water in the pathogen-free Animal Care Facility. All experimental protocols were approved by the ethic committee of the Institut Curie CEEA-IC118 under the APAFIS #1470-2018040917413626-v1 given by French national authority in compliance with the international guidelines. The Ezhip-KO mouse line was established previously14 and is maintained by mating heterozygous females on C57Bl/6 J (B6J) males purchased from Charles River. CAST Ei/J strain was maintained in house. The Xist-KO lane maintained in C57Bl/6J and enabling paternal deletion by crossing Xist-/Y male with female of interest was previously described28,42.
For embryos obtained by natural mating (all figures but 2a and 5b), the detection of the vaginal plug was designed day 0.5 dpc (day post coitum). Oocytes at Germinal Vesicle stage from 8- to 15-week-old Ezhip KO/KO (matKO) or Ezhip WT/WT or KO/WT (Ctr) females. Briefly, ovaries were transferred in M2 medium (Sigma Aldrich M7167) containing 2 μm milrinone (Sigma Aldrich M4659) and were punctured with a 25 Gauge needle. The cumulus cells were removed from the oocytes using a narrow glass pipette, wash in M2 medium and briefly incubated in acid Tyrod solution (Sigma Aldrich T1788) to remove the zona pellucida and washed in 0.2% BSA/ PBS. Approximately 100 oocytes (each genotype) were used for further analysis. To maximize the use of each female, we proceed to collect oocytes from ovaries at the same time as preimplantation or E6.5 embryos. Ezhip-matKO females or Ezhip-Ctr were mated either with CAST Ei/J males (CUT&RUN; RNA seq) or C57BL6/J males (Immunofluorescence). Two cell stage (2 C stage) or blastocyst stage embryos were collected by flushing the reproductive tracts in M2 medium at 37 hr or 80 hr post mating. Zona pellucida and polar bodies were removed with acid Tyrod treatment. Approximately 30 embryos (2 C stage) or 20 embryos (blastocyst, for both sexes) were used for analysis. Post-implantation 6.5 embryos were isolated from the maternal deciduas recovered in the uterus of the pregnant mouse. E6.5 extraembryonic ectoderm was obtained by roughly dissecting out the ectoplacental cone and the epiblast part of each embryo in M2 medium. Dissected part when washed in cleaned drops of M2 medium before usage. GVs or embryos were immediately used for CUT&RUN, immunofluorescence or RNA seq experiments, precise numbers of oocytes/embryos are provided in Supplementary Table 1.
Immunofluorescence (natural mating)
Oocytes and preimplantation embryos were processed as in ref. 43. Primary antibody incubation was performed at 4 °C overnight. mEZHIP antibody was diluted 1/800, its specificity was controlled on early preimplantation Ezhip-matKO embryos. Rabbit anti-H3K27me3 (C36B11, Cell Signaling Technology 9733) was used 1/ 400. After washes, the samples were incubated with a 1/500 dilution of Alexa Fluor A488 donkey anti rabbit IgG (Life technologies) at RT for 1 h. After washes, the samples were mounted between glass coverslip and slide in Vectashield with DAPI solution (Vector Laboratories H1200-10). Imaging was performed on an inverted confocal microscope (Zeiss) LSM700 with a Plan apoDICII (numerical aperture1.4) 63x oil objective. Z-sections were taken every 1 μm and using the Zen software (Zeiss).
Mouse superovulation
For Figs. 2a and 5b/c, embryos were isolated from superovulated female mice. This was performed in the animal facility at the Institut Curie with permission by the institutional veterinarian overseeing the operation (APAFIS numbers 39490- 2022111819233999 v2). The animal facilities are operated according to international animal welfare rules.
Superovulation of female mice was induced by intraperitoneal injection of five international units of pregnant mare’s serum gonadotropin (Ceva, Syncropart), followed by intraperitoneal injection of five international units of human chorionic gonadotropin (MSD Animal Health, Chorulon) 44–48 h later. Embryos were recovered at E1.5 by flushing oviducts and at early blastocyst stage by flushing oviducts and uteri with 37 °C homemade KSOM Hepes44 (NaCl (Honeywell Research Chemicals, Fluka Cat31434), KCl (Sigma, BioReagent P540 LifeGlobal, ZEKS-050),KH2PO4 (Avantor Science Central, 26936.293), Na Lactate (Sigma, L4263), Na pyruvate (Sigma P4562), CaCl2⋅ 2H2O (FisherScientific, C79-500), MgSO4⋅ 7H2O (Sigma, 230391), Na4EDTA⋅ 4H2O (Sigma, E-5391) MEM essential AA solution (FisherScientific GIBCO, #11130-036), non-essential AA solution (FisherScientific GIBCO, #11140-035), L-Glutamine (FisherScientific GIBCO, #25030-032), D(+) glucose (Sigma, G-6152), NaHCO3 (Sigma, S-5761), Penisilin and Streptomycin (FisherScientific GIBCO, 15140122), Hepes (Sigma, H4034)) using a modified syringe (Acufirm, Berlin, Germany, 1400 LL 23).
Immunofluorescence after superovulation
2-cell stage embryos were fixed in 2% PFA (Electron Microscopy Sciences, 15710) for 10 min at 37 °C, washed in PBS, and permeabilized with 0.5% Triton X-100 (Euromedex, 2000-C) in PBS (PBT) at room temperature before being placed in blocking solution (PBT with 3% BSA) at 4 °C for 2–4 h. Primary antibody against H3K27me3 (C36B11, Cell Signaling Technology 9733) was applied at 1:400 in blocking solution at 4 °C overnight. After several washes in PBT at room temperature, embryos were incubated with the secondary antibody Alexa Fluor 546 anti-rabbit (Invitrogen, A11010) and Alexa Fluor 488 phalloidin (Invitrogen, A12379) each at 1:200 and DAPI (Sigma, D9542) at 1:100 in blocking solution at room temperature for 1 h. Embryos were washed in PBT and imaged immediately after. Imaging was performed using an inverted Zeiss Observer Z1 microscope with a CSU-X1 spinning disc unit (Yokogawa). Excitation was achieved using 488 and 561 nm laser lines through a 63x/ 1.2 C Apo Korr water immersion objective. Emission was collected through 525/50 and 595/50 band pass filters onto an ORCA-Flash 4.0 camera (C11440, Hamamatsu).
Quantification of the H3K27me3 signal
For each cell, the signal intensity was measured in a representative 83 μm2 and 44 μm2 circular area for 2 cells embryos and blastocysts, respectively, of the equatorial nuclear plane and in a directly adjacent cytoplasmic area. Both cells were measured and averaged in 2 cells embryos, and 10 cells were measured and averaged in blastocysts. Mitotic and apoptotic cells were excluded from analysis. We then calculated the nuclear to cytoplasmic ratio.
SmRNA FISH after superovulation
The embryos were recovered at early blastocyst stage and placed in homemade KSOM (as above without Hepes) supplemented with 0.1% BSA (Sigma, A3311) in 10 μL droplets covered in mineral oil (Sigma, M8410). To remove the zona pellucida, embryos were incubated in Tyrode’s solution (Sigma, T1788) for 10 s. After washes with KSOM media, embryos were transferred onto coverslips previously coated in Denhardt’s solution, dried for 30 min at room temperature, after all excess liquid was removed. Once dried, the embryos were fixed for 20 min in 4% PFA and subsequently rinsed three times in PBS.
smRNA-FISH against XIST was performed using ACD RNAscope technology (RNAscope® Multiplex Fluorescent Detection Kit v2 and Xist probe Bio-techne #418281-C3) according to the manufacturer’s instructions. We removed the H2O2 and the Protease III steps to not disrupt the embryos attachment. After permeabilization with 0.5% Triton-X100 in PBS for 10 min on ice, embryos were incubated 2 h at 40° in probe-containing solution. Then, they were washed 2 times for 2 min each with the ACD Wash Buffer. For the amplifier (Amp1, Amp2, Amp3) and fluorophore labelling steps (HRP, TSA+Cy5, HRP blocker), embryos were incubated at 40° for 15/30 min, with washes between each step. After the final wash, embryos were incubated with DAPI solution for 1 min at room temperature, and the coverslips were mounting with VECTASHIELD® PLUS (Vector Laboratory H-1900).
Imaging embryos after smRNA FISH was performed using a Zeiss Axio Imager epifluorescence microscope equipped with the ApoTome.2 module (Zeiss), with a 40x/1.3 OIL DICII PL APO (UV) VIS-IR (420762-9800) objective. Z-sections were taken at 0.2 μm intervals, and analysis was performed using ImageJ. Male and female embryos were identified by counting the Xist signal in the nucleus. In WT embryos, males did not exhibit Xist clouding, whereas females displayed one cloud per cell. Regarding mutant embryos, different profiles were observed. The first one comprised cells with one or two Xist clouds per cell, and a small proportion of cells had no clouds. This were classified as female embryos. The second profile contained cells with a single Xist cloud, as well as a significant proportion of cells without cloud. These were considered to be male embryos.
Single cell or single embryo RNA seq
GVs, 2 C, blastocysts and E6.5 embryos RNA seq were prepared as described in ref. 45. Sexes of the embryos were assessed based on Xist and Eif3s3y expression. Library construction was performed as in ref. 46 using the Accel-NGS 2S Unique Dual Indexing DNA library kit (Swift Biosciences). The following PCR program was used: 98 °C 45 °9 °C 15 s, 60 °C 10 s and 68 °C 1 min. Steps two and three were repeated respectively 6 times (oocyte, 2-cell and blastocyst) or 8 times (6.5 dpc Extraembryonic Ectoderm samples) the specified number of times. Library sizes distribution was assessed with Agilent 4200 Bioanalyzer with D5000 ScreenTapes (Agilent Technologies). Libraries were sequenced with 100 bp paired end reads on a NovaSeq 6000 S1 (Illumina).
CUT&RUN
WT and Ezhip-matKO CUT&RUN on H3K27me3 were performed as described previously47 with a few adaptations. LoBind 1,5 mL Eppendorf tubes were pre-coated with pre-coat buffer (150 mM NaCl, 10% BSA, 1X Protease Inhibitor Cocktail (PIC) and ddH2O) and kept on ice. 20 μl Concavalin A-coated beads per sample was resuspended in 1.5 mL of Binding Buffer (20 mM Hepes-KOH pH 7.6, 10 mM KCl, 1 mM CaCl2, 1 mM MgCl2 and ddH2O). Beads were resuspended in 150 μl of binding buffer per sample. Cells were gently resuspended in 300 μl Nuclear Extraction Buffer that was added to the pre-coated tubes and incubated for precisely 5 min on ice. 150 μl of the prepared beads was added per sample and incubated on a rotating rack at 5–7 rpm. Samples were quickly pulsed on a microcentrifuge and all liquid was removed. Beads were resuspended in 1 mL Blocking Buffer per sample (20 mM Hepes-KOH pH 7.6, 150 mM NaCl, 0.5 mM spermidine, 0.1% BSA, 1X PIC, 2 mM EDTA and ddH2O) and homogenized by rotating 10 times. Samples were quickly pulsed on a microcentrifuge and incubated for 5 min at room temperature. After a quick pulse on the microcentrifuge, samples were placed on a magnetic rack and liquid was withdrawn. Beads were resuspended in 200 μl of Wash Buffer (20 mM Hepes-KOH pH 7.6, 150 mM NaCl, 0.5 mM spermidine, 0.1% BSA, 1X PIC and d ddH2O). Primary antibody was diluted at 1:100 for a final concentration of 1:200, and 200 μl of primary antibody solution was added to the samples. Samples incubated rotating at 4 °C for 2,5 h at 5–10 rpm. After washing, beads were resuspended in 200 μl of Wash Buffer. pA-MNase (Recombinant Protein Purification Platform of the Institut Curie) was diluted at 1:250 in wash buffer for a final concentration of 1:400, 200 μl of pA-MNase solution was added per sample and incubated rotating for 1 hour at 4 °C at 5–10 rpm. Samples were resuspended in 150 μl wash buffer and chilled on ice water bath for 5 min. Targeted digestion was started by adding 3 μl of 100 mM CaCl2 to each sample and incubated on ice for 30 min. The reaction was stopped by adding 150 μl of 2X Stop Buffer (200 mM NaCl, 20 mM EDTA, 4 mM EGTA, 50 μg/mL RNase A, 40 μg/mL glycogen and 1 pg/mL Drosophila S2 spike-in DNA). Samples were incubated at 37 °C for 20 min to allow digested fragments to be released and centrifuged at 16.000 g for 5 min at 4 °C. Each sample was placed on a magnetic rack and supernatant was transferred to a new pre-coated tube. Chromatin was released by adding 2.5 μl of 20 mg/ml proteinase K and 3 μl of 10% SDS and incubated at 70 °C for 30 min. DNA was extracted using phenol/chloroform followed by chloroform extraction and precipitated with 0.5 μl 20 mg/ml glycogen and 900 μl absolute ethanol and incubated on ice for 20 min. Samples were centrifuged for 20 min at 16.000 g at 4 °C and supernatant was aspirated carefully. DNA pellets were washed with 85% ethanol and left to air-dry until ethanol was evaporated and resuspended in 40 μl 1 mM Tris-HCl pH 8.0 and 0.1 mM EDTA and proceeded to library preparation. Library construction was performed using the Accel-NGS 2S Unique Dual Indexing DNA library kit (Swift Biosciences) with the following amplification program: 45 seconds on 98 °C, 21 cycles of 10 seconds 98 °C, 15 s 60 °C and 1 min 68 °C. End hold at 4 °C. Library sizes distribution was assessed with Agilent 4200 Bioanalyzer with D5000 ScreenTapes (Agilent Technologies). Libraries were sequenced with 100 bp paired end reads on a NovaSeq 6000 S1 (Illumina). Embryos number for each analysis is provided in Supplementary Table 1.
RNA-Seq data processing
The fasta file of the mouse reference genome mm10 was first masked with the CAST/EiJ strain-specific SNPs, as described by the Sanger Mouse Genomes Project (v.5 SNP142), using SNPsplit (v0.3.2)48 so that all polymorphic sites would be masked by the ambiguity nucleobase ‘N’. Adapters and low-quality bases (<Q20) were removed from reads with TrimGalore (v0.4.0) and Cutadapt (v1.8.2)49. Trimmed reads were then aligned to the “N-masked” reference genome using the gene annotation from GENCODE (Release 23/GRCm38.p6) and the STAR (v2.7.0a) aligner50 with the parameters --outFilterMultimapNmax 1 -–outFilterMismatchNmax 999 --seedSearchStartLmax 20 --outFilterScoreMinOverLread 0 --outFilterMatchNminOverLread 0 --outFilterMatchNmin 30 --outSAMattributes NH HI NM MD --alignEndsType EndToEnd. To assess the quality of each RNA-Seq sample, we calculated three parameters: 1) library size, or the number of mapped reads per cell; 2) number of expressed genes; and 3) total amount of RNA, quantified as the sum of Log2(TPM + 1) values for all genes in each sample. Only samples with values for each of these three parameters above a threshold, determined as the median − 4*MAD (Median Absolute Deviation), were kept for subsequent analyses. An additional quality control step was applied to E6.5 extra-embryonic ectoderm (ExE) samples: we calculated a median Z-score for the gene expression values of epiblast, visceral endoderm, and ExE lineage markers, as defined previously51. Samples in which the median Z-score for epiblast markers was higher than that of ExE markers were discarded, as tissue contamination could significantly interfere with the allele-specific analysis of X-linked expression. Reads spanning “N-masked” polymorphic sites were assigned to the maternal or paternal genomes using SNPsplit (v0.3.2)48. Reads overlapping heterozygous SNPs were quantified using SAMtools (v1.9)52 mpileup. Only SNPs with at least 10 overlapping reads were considered informative and kept for subsequent analyses. For the analysis of gene expression, reads overlapping exons (Release 23/GRCm38.p6) were counted using FeatureCounts (Subread v1.5.1)53 using total BAM files.
CUT&RUN-Seq data processing
The N-masked mm10 fasta file was concatenated to the Drosophila reference genome dm6 and indexed with Bowtie2 (v2.2.5)54 bowtie2-build. Adapters and low-quality bases (<Q20) were removed from the CUT&RUN-Seq reads with TrimGalore (v0.4.0) and Cutadapt (v1.8.2). Trimmed fastq files of individual samples were then aligned against the concatenated mouse:Drosophila reference genomes using the Bowtie2 aligner (v2.2.5) with default parameters. PCR duplicates were discarded using Picard tools (v1.97) and reads overlapping the mouse genome were assigned to either the maternal or the paternal genomes using SNPsplit (v0.3.2). The sex of H3K27me3 CUT&RUN-Seq blastocyst samples was verified by means of three parameters: 1) ratio of chrY versus total reads, 2) fraction of chrX reads assigned to the maternal genome, and 3) fraction of autosomal reads assigned to the maternal genome. Bam files were merged according to genotype and, when available, sex using SAMtools merge (v1.9). Parental-specific bigwig files were generated using Deeptools bamCoverage (v3.0.2)55 with the following parameters: --normalizeUsingRPKM –ignoreForNormalization chrX chrY –samFlagInclude 64 –binSize 50 –extendReads. Reads overlapping problematic, blacklisted regions (wgEncodeDacMapabilityConsensusExcludable.bed)56 were excluded from the computation of coverage with the option --blackListFileName. A scale factor was calculated on Drosophila spike-in reads binned into genome-wide non-overlapping 10 kb bins using DESeq257, and the same factor was given as the parameter ‘–scaleFactor’ for both allelic signals over each analysis. Average plots of signal were generated with Deeptools computeMatrix (v3.0.2) and in-house scripts.
To evaluate the effect of the Ezhip-matKO on H3K27me3 deposition in oocytes, we first identified chromatin fragments of significant enrichment in WT samples using csaw58. Total CUT&RUN-Seq reads were binned into genome-wide 10 kb sliding windows (Csaw), and only windows overlapping autosomes with enrichment levels 2-fold or higher over background were selected. Windows laying at a maximum distance of 20 kb were merged. We also sought to identify H3K27me3-rich regions in oocytes profiled by H3K27me3 ChIP-Seq of the GSE76687 dataset4, using the same criteria. The lists of PRC2 target regions of the two datasets were intersected and explored for differential enrichment in matKO vs WT oocytes.
To investigate the impact of the Ezhip matKO on the dynamics of H3K27me3 during early embryogenesis, we first identified genomic regions of significant enrichment in WT oocytes, 2 C, and blastocysts using csaw. Parental-specific CUT&RUN-Seq reads were binned into genome-wide 10 kb sliding windows, and only windows overlapping autosomes with enrichment levels 2-fold or higher over background were selected. Windows laying at a maximum distance of 20 kb were merged. We next performed the same analysis using H3K27me3 ChIP-Seq samples of the GSE76687 dataset; more specifically, those profiling GV oocytes, the 2-cell stage, and inner cell mass. The lists of H3K27me3-rich regions of the two datasets were intersected to generate a consensus collection of 3,942 genomic fragments. These fragments were divided into 3 clusters according to their dynamic behavior and parental bias using soft clustering implemented by mfuzz59.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The Gene Expression Omnibus (GEO) accession number for CUT&RUN is GSE278811 and for the RNAseq GSE278534. The following data set were exploited for this manuscript (GSE129029) and (GSE136093). Source Data are provided with this paper. Request for materials should be addressed to raphael.margueron@curie.fr Source data are provided with this paper.
References
Xu, Q. & Xie, W. Epigenome in Early Mammalian Development: Inheritance, Reprogramming and Establishment. Trends Cell Biol. 28, 237–253 (2018).
Tucci, V., Isles, A. R., Kelsey, G., Ferguson-Smith, A. C. & Erice Imprinting Group Genomic Imprinting and Physiological Processes in Mammals. Cell 176, 952–965 (2019).
Inoue, A., Jiang, L., Lu, F., Suzuki, T. & Zhang, Y. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 547, 419–424 (2017).
Zheng, H. et al. Resetting Epigenetic Memory by Reprogramming of Histone Modifications in Mammals. Mol. Cell 63, 1066–1079 (2016).
Chen, Z., Djekidel, M. N. & Zhang, Y. Distinct dynamics and functions of H2AK119ub1 and H3K27me3 in mouse preimplantation embryos. Nat. Genet. 53, 551–563 (2021).
Mei, H. et al. H2AK119ub1 guides maternal inheritance and zygotic deposition of H3K27me3 in mouse embryos. Nat. Genet. 53, 539–550 (2021).
Inoue, A., Chen, Z., Yin, Q. & Zhang, Y. Maternal Eed knockout causes loss of H3K27me3 imprinting and random X inactivation in the extraembryonic cells. Genes Dev. 32, 1525–1536 (2018).
Inoue, A., Jiang, L., Lu, F. & Zhang, Y. Genomic imprinting of Xist by maternal H3K27me3. Genes Dev. 31, 1927–1932 (2017).
Loda, A., Collombet, S. & Heard, E. Gene regulation in time and space during X-chromosome inactivation. Nat. Rev. Mol. Cell Biol. 23, 231–249 (2022).
Chen, Z., Yin, Q., Inoue, A., Zhang, C. & Zhang, Y. Allelic H3K27me3 to allelic DNA methylation switch maintains noncanonical imprinting in extraembryonic cells. Sci. Adv. 5, eaay7246 (2019).
Matoba, S. et al. Noncanonical imprinting sustains embryonic development and restrains placental overgrowth. Genes Dev. 36, 483–494 (2022).
Pajtler, K. W. et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. (Berl.) 136, 211–226 (2018).
Piunti, A. et al. CATACOMB: An endogenous inducible gene that antagonizes H3K27 methylation activity of Polycomb repressive complex 2 via an H3K27M-like mechanism. Sci. Adv. 5, eaax2887 (2019).
Ragazzini, R. et al. EZHIP constrains Polycomb Repressive Complex 2 activity in germ cells. Nat. Commun. 10, 3858 (2019).
Hübner, J.-M. et al. EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro-Oncol. 21, 878–889 (2019).
Jain, S. U. et al. PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat. Commun. 10, 2146 (2019).
Jain, S. U. et al. H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2. Mol. Cell 80, 726–735.e7 (2020).
Skene, P. J., Henikoff, J. G. & Henikoff, S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 13, 1006–1019 (2018).
Du, Z. et al. Polycomb Group Proteins Regulate Chromatin Architecture in Mouse Oocytes and Early Embryos. Mol. Cell 77, 825–839.e7 (2020).
Collombet, S. et al. Parental-to-embryo switch of chromosome organization in early embryogenesis. Nature 580, 142–146 (2020).
Firulli, A. B., McFadden, D. G., Lin, Q., Srivastava, D. & Olson, E. N. Heart and extra-embryonic mesodermal defects in mouse embryos lacking the bHLH transcription factor Hand1. Nat. Genet. 18, 266–270 (1998).
Garrick, D. et al. Loss of Atrx affects trophoblast development and the pattern of X-inactivation in extraembryonic tissues. PLoS Genet 2, e58 (2006).
Itoh, M. et al. Role of Gab1 in heart, placenta, and skin development and growth factor- and cytokine-induced extracellular signal-regulated kinase mitogen-activated protein kinase activation. Mol. Cell. Biol. 20, 3695–3704 (2000).
Jeon, S. Y. et al. Hypoxia-induced downregulation of XIAP in trophoblasts mediates apoptosis via interaction with IMUP-2: implications for placental development during pre-eclampsia. J. Cell. Biochem. 114, 89–98 (2013).
Lee, B.-K. et al. Fosl1 overexpression directly activates trophoblast-specific gene expression programs in embryonic stem cells. Stem Cell Res 26, 95–102 (2018).
Borensztein, M. et al. Contribution of epigenetic landscapes and transcription factors to X-chromosome reactivation in the inner cell mass. Nat. Commun. 8, 1297 (2017).
Marahrens, Y., Loring, J. & Jaenisch, R. Role of the Xist gene in X chromosome choosing. Cell 92, 657–664 (1998).
Marahrens, Y., Panning, B., Dausman, J., Strauss, W. & Jaenisch, R. Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev. 11, 156–166 (1997).
Stafford, J. M. et al. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci. Adv. 4, eaau5935 (2018).
Souroullas, G. P. et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat. Med. 22, 632–640 (2016).
Romero, P. et al. EZH2 mutations in follicular lymphoma distort H3K27me3 profiles and alter transcriptional responses to PRC2 inhibition. Nat. Commun. 15, 3452 (2024).
Blackledge, N. P. & Klose, R. J. The molecular principles of gene regulation by Polycomb repressive complexes. Nat. Rev. Mol. Cell Biol. 22, 815–833 (2021).
Lu, C. et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352, 844–849 (2016).
Fan, H. et al. BAHCC1 binds H3K27me3 via a conserved BAH module to mediate gene silencing and oncogenesis. Nat. Genet. 52, 1384–1396 (2020).
Fan, H. et al. A conserved BAH module within mammalian BAHD1 connects H3K27me3 to Polycomb gene silencing. Nucleic Acids Res 49, 4441–4455 (2021).
Zhang, Y. et al. Corepressor protein CDYL functions as a molecular bridge between polycomb repressor complex 2 and repressive chromatin mark trimethylated histone lysine 27. J. Biol. Chem. 286, 42414–42425 (2011).
Pasini, D. et al. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res 38, 4958–4969 (2010).
Drosos, Y. et al. NSD1 mediates antagonism between SWI/SNF and polycomb complexes and is required for transcriptional activation upon EZH2 inhibition. Mol. Cell 82, 2472–2489.e8 (2022).
Okamoto, I. et al. Eutherian mammals use diverse strategies to initiate X-chromosome inactivation during development. Nature 472, 370–374 (2011).
Chu, C. et al. Analysis of developmental imprinting dynamics in primates using SNP-free methods to identify imprinting defects in cloned placenta. Dev. Cell 56, 2826–2840.e7 (2021).
Sanulli, S. et al. Jarid2 Methylation via the PRC2 Complex Regulates H3K27me3 Deposition during Cell Differentiation. Mol. Cell 57, 769–783 (2015).
Borensztein, M. et al. Xist-dependent imprinted X inactivation and the early developmental consequences of its failure. Nat. Struct. Mol. Biol. 24, 226–233 (2017).
Ancelin, K. et al. Maternal LSD1/KDM1A is an essential regulator of chromatin and transcription landscapes during zygotic genome activation. eLife 5, e08851 (2016).
Biggers, J. D., McGinnis, L. K. & Raffin, M. Amino Acids and Preimplantation Development of the Mouse in Protein-Free Potassium Simplex Optimized Medium1. Biol. Reprod. 63, 281–293 (2000).
Pérez-Palacios, R., Fauque, P., Teissandier, A. & Bourc’his, D. Deciphering the Early Mouse Embryo Transcriptome by Low-Input RNA-Seq. Methods Mol. Biol. Clifton NJ 2214, 189–205 (2021).
Tang, F. et al. RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nat. Protoc. 5, 516–535 (2010).
Hainer, S. J., Bošković, A., McCannell, K. N., Rando, O. J. & Fazzio, T. G. Profiling of Pluripotency Factors in Single Cells and Early Embryos. Cell 177, 1319–1329.e11 (2019).
Krueger, F. & Andrews, S. R. SNPsplit: Allele-specific splitting of alignments between genomes with known SNP genotypes. F1000Research 5, 1479 (2016).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10 (2011).
Dobin, A. & Gingeras, T. R. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinforma. 51, 11.14.1–11.14.19 (2015).
Wen, J. et al. Single-cell analysis reveals lineage segregation in early post-implantation mouse embryos. J. Biol. Chem. 292, 9840–9854 (2017).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinforma. Oxf. Engl. 25, 2078–2079 (2009).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinforma. Oxf. Engl. 30, 923–930 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Ramírez, F., Dündar, F., Diehl, S., Grüning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–W191 (2014).
Amemiya, H. M., Kundaje, A. & Boyle, A. P. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep. 9, 9354 (2019).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Lun, A. T. L. & Smyth, G. K. csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res 44, e45 (2016).
Futschik, M. E. & Carlisle, B. Noise-robust soft clustering of gene expression time-course data. J. Bioinform. Comput. Biol. 3, 965–988 (2005).
Acknowledgements
High-throughput sequencing was performed by the NGS platform of the Institut Curie supported by grants ANR-10-EQPX-03 and ANR-10-INBS-09-08 from the Agence Nationale de le Recherche (investissements d’avenir) and by the Canceropôle Ile-de-France. We thank the Institute Curie Animal Facilities for animal welfare and husbandry, the Cell and Tissue Imaging Platform – PICT-IBiSA (member of France–Bioimaging – ANR-10-INBS-04) for technical input and the Recombinant Protein platform. Work in the lab of R.M. was supported by la FRM (Fondation pour la Recherche Médicale, SPF20150934266) and the ANR (ANR- 18-CE12-016). S.D. was supported by a PhD fellowship from la Ligue contre le Cancer and a fourth PhD year from la FRM. J-L.M. is supported by the Institut Curie, the Centre National de la Recherche Scientifique, the Institut National de la Santé Et de la Recherche Médicale, and is funded by grants from the European Research Council Consolidator Grant ERC-2022-CoG 101087656 and the Agence Nationale de la Recherche ANR-23-CE13-0020. We thank Dr. M. Borensztein, R. Galupa, N. Servant, L. Syx, M. Wassef and D. Holoch for valuable comments on the manuscript and Dr. C. Rodrigues for his help with data submission to GEO.
Author information
Authors and Affiliations
Contributions
J-L.M., K.A., and R.M. designed the experiments with input from all the authors. S.D., A.P., J.B., C.M., and K.A. performed the experiments. L.R. performed the bioinformatics analysis. V.R. contributed to the sequencing. B.A. helps with mouse husbandry. A.D. helps with image processing and analysis. All authors contributed to data analysis. K.A. and R.M. prepared the manuscript with input from all the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ozren Bogdanovic, Kenjiro Shirane and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Diop, S., Richart, L., Petitalot, A. et al. H3K27me3-dependent imprinting and transcriptional regulation in early mouse embryos requires EZHIP-mediated restriction of PRC2 activity. Nat Commun 17, 1758 (2026). https://doi.org/10.1038/s41467-026-68467-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68467-4
