
Abstract
Irreversible electroporation (IRE) is an ablative treatment for pancreatic cancer. It utilizes high-intensity pulsed electric field (PEF) to eliminate cancer cells by irreversibly disrupting cell membranes. However, PEF intensity is distributed unevenly; and cancer cells may survive in regions where it falls below the threshold of complete ablation. We find that iron-base metal organic framework nanoparticles (MOF-Fe) sensitize pancreatic cancer cells to PEF by inducing iron overload and ferroptosis. But their efficacy is diminished by the upregulation of ferritin heavy chain 1 (FTH1), a cellular response to restore iron homeostasis. C20U4V, a proteolysis targeting chimera (PROTAC) derived from arachidonic acid, degrades FTH1 and potentiates MOF-Fe-induced ferroptosis. It is then encapsulated in reactive oxygen species (ROS)-responsive micelles. The resulting M-C20U4V, when combined with MOF-Fe, efficiently induces ferroptosis and boosts PEF ablation efficacy. Therefore, disruption of iron homeostasis represents a potential strategy to lower the risk of tumor recurrence after IRE.
Similar content being viewed by others


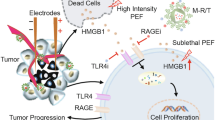
Introduction
Irreversible electroporation (IRE) is a local ablative technique for treating patients with solid tumors, including pancreatic cancer1. During ablation, electrodes are placed in or around the tumor to deliver high-intensity electric pulses. The resulting pulsed electric field (PEF) then eliminates cancer cells by inducing irreversible disruption of their cell membranes2, and promotes systemic anti-tumor immunity through the release of damage-associated molecular patterns3. However, the PEF intensity is unevenly distributed in the tumor4,5. In regions where PEF intensity falls below the threshold for completion of cell eradication, residual cancer cells may survive and ultimately drive tumor recurrence.
Current approaches to eliminating these residual cancer cells often rely on inducing apoptosis6,7,8,9,10. Therefore, their efficacy may be compromised when cancer cells harbor mutations in the apoptotic cascades. Ferroptosis is an iron-dependent, non-apoptotic form of regulated cell death that can overcome resistance to conventional chemotherapies11,12. During ferroptosis, intracellular iron catalyzes the Fenton reaction to generate reactive oxygen species (ROS); these ROS then oxidize phospholipids in cellular or organelle membranes. The excessive accumulation of oxidized phospholipids ultimately triggers cell membrane rupture and subsequent cell death. Cells have evolved sophisticated mechanisms to counteract ferroptosis via glutathione peroxidase 4 (GPX4)-dependent and -independent pathways13,14. Correspondingly, inhibitors that target these defensive mechanisms, such as erastin and 1S,3R-RLS-3 (RSL3), can induce ferroptosis in cancer cells. The clinical translation of these inhibitors, however, has yet to be achieved15.
Given that iron overload is the primary driver of ferroptosis16, numerous iron-containing nanoparticles have been developed to induce this form of cell death17. These nanoparticles are endocytosed by cancer cells and trafficked to acidic lysosomes, where they degrade and release ferrous or ferric ions. Notably, the activation of lysosomal iron has recently been shown to catalyze ROS production and initiate ferroptosis18. MIL-101 (Fe) metal-organic framework (MOF-Fe) nanoparticles could release iron in a flash fashion19, and therefore are promising candidates to induce iron overload. On the other hand, intracellular iron levels are tightly regulated by iron homeostasis20, and excess iron can be exported out of cells by ferroportin or sequestered in ferritin. Ferritin is a 24-mer protein composed of two subunits: a heavy chain (FTH1) and a light chain (FTL). FTH1 catalyzes the oxidation of ferrous into ferric ions, while FTL facilitates ferric ion biomineralization and long-term storage within ferritin21. Conversely, autophagic degradation of ferritin disrupted iron homeostasis and induced ferroptosis22, while FTH1-targeted siRNA enhanced iron nanoparticle-induced lipid oxidation23.
RNA interference reduces protein expression by disrupting mRNA translation. Changes in protein expression typically occur 24 h after siRNA transfection24. In contrast, pre-existing proteins can be degraded using proteolysis-targeting chimeras (PROTACs)25. PROTACs are bifunctional molecules composed of a target-binding moiety and an E3 ligase-binding moiety. They can recruit E3 ligases to target proteins, leading to their ubiquitylation and subsequent degradation in proteosomes. Mono- or poly-unsaturated fatty acids (MUFA and PUFA, respectively) have been reported to bind to a U-shaped pocket in ferritin and promote iron mineralization26,27. A ferritin degrader was generated as a pyroptosis inducer from oleic acid, an MUFA28. However, MUFAs confer resistance to ferroptosis29. In contrast, PUFAs can promote ferroptosis once incorporated into phospholipids and peroxidized by lipoxygenases30.
We hypothesized that disrupting iron homeostasis using MOF-Fe and a PUFA-derived ferritin-degrading PROTAC could sensitize pancreatic cancer cells to PEF. To test this hypothesis, we assessed the capacity of MOF-Fe to induce ferroptosis and enhance the efficacy of PEF ablation. FTH1 expression was genetically modulated to elucidate its role in MOF-Fe-induced ferroptosis. A PROTAC library was synthesized by conjugating PUFAs, directly or indirectly through different linkers, to a ligand for von-Hippo-Lindau (VHL) E3 ligase, (2S,4R)−1-((S)−2-Amino-3,3-dimethylbutanoyl)−4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, abbreviated as (S,R,S)-AHPC. The chimeras were screened for their ability to degrade FTH1 in the presence of MOF-Fe. C20U4V, the lead compound derived from arachidonic acid, was assessed for its ability to potentiate MOF-Fe-induced ferroptosis. Lastly, a ROS-responsive polymeric micelles formulation, M-C20U4V, was prepared. The anti-tumor efficacy of PEF, MOF-Fe, and M-C20U4V was evaluated in murine pancreatic tumor models. Our findings suggest that disruption of iron homeostasis is a potential strategy to lower the risk of tumor recurrence after IRE.
Results
PEF upregulated ferroptosis suppressor genes
Although PEF has been reported to generate ROS and cause lipid oxidation31, it is unclear whether such oxidative stress causes ferroptotic cell death. Panc-1 cells were treated with PEF (600 V/cm, 99 pulses), a setting that induced biological alterations without complete elimination of cancer cells32. C11-BODIPY is an established probe for lipid oxidation; malondialdehyde (MDA) is a product of lipid peroxidation33. PEF did not enhance C11-BODIPY oxidation34 or increase MDA concentration (Fig. 1a, b). PEF-treated cells were incubated with ferroptosis inhibitors (liproxstatin-1, abbreviated as Lip-1; ferrostatin-1, abbreviated as Ferr-1) or a reducing agent (tocopherol) at concentrations below their respective half-inhibitory concentrations (IC50, Fig. 1c and Supplementary Fig. 1a). None of these agents rescued the viability of Panc-1 or MIAPaCa-2 cells following PEF (Fig. 1d and Supplementary Fig. 1b). In terms of other cell death modes (Supplementary Fig. 1c–e), PEF-treated cells showed a higher frequency of Annexin V+ populations, an indicator of apoptosis. They also released lactate dehydrogenase (LDH) and expressed a slightly higher level of cleaved-gasdermin D (N-terminal), suggesting the occurrence of pyroptosis. There was no significant change in the expression of phosphor-MLKL and phosphor-RIPK1, two necroptosis markers.

a Flow cytometry histograms and quantification of C11-BODIPY oxidation, n = 7 independent experiments. b MDA concentration, n = 3 independent experiments. c Experiment scheme for panel (d). d Relative Panc-1 viability after PEF and incubation with Lip-1, Ferr-1, or tocopherol, n = 6 independent experiments. e GO analysis of biological process (GO_BP) for genes upregulated in the PEF + cell debris group relative to the vehicle control group. f Volcano plots of transcriptome. Differentially expressed genes (DEGs) in the ferroptosis pathway are labeled with red (upregulated) or blue (downregulated). g GSEA on the WikiPathway gene set. h Volcano plots of transcriptome. DEGs in the NRF2 pathway are labeled with red (upregulated) or blue (downregulated). Cells in panels e–h were treated with PEF and incubated with cell debris for 24 h, n = 3 independent experiments. i Immunoblots of Panc-1 cells. The samples derived from the same experiment but on different gels for NRF2, another for HMOX1, FTH1, and β-actin, were processed in parallel, n = 3 (NRF2), 4 (HMOX1), and 5 (FTH1) independent experiments. Quantification is included in Supplementary Fig. 1f. j Hematoxylin-eosin (H&E) staining of KPC-A719 tumors at 24 h after PEF ablation. The border between the viable tumors and the ablation zone is marked with a black dashed line. Residual cancer cells are indicated with black arrowheads. The experiments were independently repeated three times with similar results. Scale bar = 100 μm. k 4-HNE staining of KPC-A719 tumors and quantification, n = 8 randomly selected field-of-views (FOVs) from three independent mice per group. Scale bar = 100 μm. l Immunoblots of KPC-A719 tumors. The samples derived from the same experiment but on different gels for NRF2, another for HMOX1, FTH1, and β-actin, were processed in parallel, n = 9 (NRF2, HMOX1) or 12 (FTH1) biological replicates from three independent mice per time point. Quantification is included in Supplementary Fig. 1g. PEF intensity = 600 V/cm in cell culture or as specified for in vivo studies. Data were presented as mean ± standard error of mean (SEM). Statistical significance was determined using one-tailed ratio-paired t-test (a), one-tailed unpaired t-test (b, k), one-way ANOVA with Dunnett’s multiple comparison (d), GO analysis adjusted with Benjamini–Hochberg algorithm (e), EdgeR analysis using negative binomial generalized linear model (GLM) (f, h) or GSEA analysis (g) adjusted with Benjamini–Hochberg algorithm, and p values are indicated. Source data are provided as a Source Data file. The elements in Fig. 1c were generated using PowerPoint.
Residual cancer cells following IRE therapy have been reported to be affected by both PEF and the debris of IRE-killed cells35. Panc-1 cells were accordingly treated and their transcriptome analyzed. Upregulated genes in the treated cells were significantly enriched in gene ontology (GO) terms including “regulation of ferroptosis” and “ferroptosis (Fig. 1e). Seven genes were upregulated, and three were downregulated, in the ferroptosis pathway (WikiPathways, WP4313, Fig. 1f). Six of the upregulated genes (HMOX1, SLC7A11, GCLM, GCLC, FTL, and FTH1) are ferroptosis suppressors, and two of the downregulated ones (LPCAT3 and ACSL5) are ferroptosis drivers36. Gene set enrichment analysis (GSEA) unveiled the nuclear factor erythroid 2-related factor 2 (NRF2)-related pathways among the top-upregulated ones (Fig. 1g). NRF2 pathway (WikiPathways, WP2884) regulates cellular response to oxidative stress, and 16 genes were upregulated (Fig. 1h). The protein expression levels of NRF2, heme oxygenase 1 (HMOX1), and FTH1 gradually increased following PEF treatment in Panc-1 cells (Fig.1i and Supplementary Fig. 1f).
These findings were further verified in KPC-A719 pancreatic tumor models. Tumors treated with PEF ablation presented a necrotic core surrounded by viable tumor regions (Fig. 1j). 4-Hydroxynonenal (4-HNE) is a marker of lipid peroxidation. The frequency of 4-HNE+ pixels was similar between the viable regions of treated tumors and those of sham controls (Fig. 1k). In a separate experiment, KPC-A719 tumors were treated with PEF at a lower voltage (500 V). On day 2 post-PEF, the expression levels of HMOX1 and FTH1 increased by 13.6 and 2.6-fold, respectively (Fig. 1l and Supplementary Fig. 1g). Collectively, these results indicate that low-intensity PEF upregulated ferroptosis suppressor genes in pancreatic cancer cells instead of inducing ferroptotic cell death.
MOF-Fe transiently suppressed tumor recurrence after PEF ablation
We next asked whether MOF-Fe could induce ferroptosis and potentiate PEF. MOF-Fe nanoparticles were octahedral-shaped with a diameter of 98.4 ± 2.0 nm. Their surface ζ-potential was 31.0 ± 2.5 mV after coating with polyethylene imine (PEI) (Fig. 2a–c). Superparamagnetic iron oxide (SPIO) nanoparticles represent another class of iron-containing nanoparticles for theranostic applications and are well-documented to induce ferroptosis37,38. MOF-Fe released iron more rapidly than SPIO at pH 5.2 (Fig. 2d). MOF-Fe degradation was accompanied by size reduction, surface ruffling, and particle agglomeration (Supplementary Fig. 2a). Accordingly, Panc-1 cells formed fewer colonies with MOF-Fe than with SPIO (Fig. 2e and Supplementary Fig. 2b). FerroOrange is a ferrous ion-specific fluorescence probe39. Panc-1 incubated with MOF-Fe showed stronger FerroOrange and oxidized C11-BODIPY staining, along with higher intracellular iron concentrations (Fig. 2f–h). Lipidomics revealed that MOF-Fe elevated the levels of two oxidized and three peroxidized phosphatidylethanolamine (Fig. 2i and Supplementary Data). Conversely, Lip-1 diminished MOF-Fe-induced ROS elevation (DCFA-DA staining) and C11-BODIPY oxidation (Supplementary Fig. 2c, d). These results indicate the MOF-Fe-induced changes were indeed ferroptotic events.

a–c SEM photograph (a), size distribution (b), and ζ-potential histogram (c) of MOF-Fe, scale bar = 500 nm. The experiments were independently repeated three times with similar results. d Accumulative release of iron at 37 °C, n = 4 independent experiments. e Quantification of Panc-1 colonies after treatment with MOF-Fe or SPIO, n = 3 independent experiments. f Immunofluorescence images of FerroOrange staining and quantification of fluorescence intensity of individual cells, n = 14 (control) or 100 (SPIO and MOF-Fe) randomly selected cells (technical replicates). The experiments were independently repeated three times with similar results. Scale bar = 100 μm. g Intracellular concentrations of iron, n = 8 independent experiments. Panc-1 cells in panels (f, g) were incubated with MOF-Fe or SPIO for 24 h at 10 μg Fe/mL. h Flow cytometry histograms and quantification of C11-BODIPY oxidation, n = 3 independent experiments. i Volcano plots of lipidomics after treatment with MOF-Fe relative to vehicle control, n = 5 independent experiments. j Quantification of Panc-1 colonies after treatment with PEF and MOF-Fe, n = 3 (No PFE) or 6 (PEF) independent experiments. PEF intensity = 500 V/cm. k Iron uptake in KPC-A719 tumors and major organs at 24 h after PEF ablation, n = 4 (MOF-Fe), 5 (sham control), or 6 (PEF ablation + MOF-Fe) independent mice. l Growth curves of KPC-A719 tumors, n = 5 (sham control and PEF ablation), 7 (MOF-Fe), or 10 (PEF ablation + MOF-Fe) independent mice. m GSEA on the WikiPathway gene set, n = 3 independent experiments. n Volcano plots of proteomics. Differentially expressed proteins in the ferroptosis pathway are labeled, n = 2 independent experiments. o, Immunoblots of Panc-1 cells after treatment with MOF-Fe for various durations. The samples derived from the same experiment but on different gels for NRF2, another for HMOX1, FTH1, and β-actin, were processed in parallel, n = 3 independent experiments. Quantification is included in Supplementary Fig. 4d. Panc-1 cells in panels (i, m, n) were incubated with MOF-Fe at 3 μg Fe/mL. Data were presented as mean ± SEM. Statistical significance was determined using one-way ANOVA with Dunnett’s multiple comparison (d, f, g, l), two-tailed unpaired t-test (e, j), one-tailed ratio-paired t-test (h), multiple two-tailed unpaired t-test without correction (i), or one-tailed Welch’s test (k), GESA (m) or EdgeR analysis using negative binomial generalized linear model (GLM) (n) adjusted with Benjamini–Hochberg algorithm, and p values are indicated. Source data are provided as a Source Data file.
Panc-1 cells treated with PEF plus MOF-Fe formed fewer colonies than those with PEF or MOF-Fe alone (Fig. 2j and Supplementary Fig. 2e). The anti-tumor efficacy of PEF ablation and MOF-Fe was then investigated in vivo. PEF ablation significantly enhanced MOF-Fe retention in KPC-A719 tumors: at 24 h post-injection, tumors in the PEF ablation + MOF-Fe group contained 3.9-fold more iron than those in the MOF-Fe alone group (Fig. 2k). Tumor-bearing mice were treated as outlined in Supplementary Fig. 2f. No significant change of body weight was observed (Supplementary Fig. 2g). The combination of PEF ablation + MOF-Fe was more efficacious in tumor suppression than either monotherapy for up to 20 days from enrollment (Fig. 2l and Supplementary Fig. 2h). Tumor sections were collected on day 4 after enrollment and analyzed (Supplementary Fig. 3). H&E staining displayed extensive necrosis in the tumor of the combination group. Tumors injected with MOF-Fe showed positive staining of iron (Prussian blue). The combination group also exhibited stronger staining of 4-HNE, FTH1, and HMOX1, along with fewer Ki67+ proliferating cells than those from the other groups. These data suggest that MOF-Fe induced ferroptosis and potentiated PEF ablation in pancreatic cancer.
KPC-A719 tumors in the PEF ablation + MOF-Fe group rebounded quickly after day 20 after enrollment (Fig. 2l), indicating the presence of acquired resistance. The underlying mechanism was then investigated. NRF2-related pathways were significantly upregulated in MOF-Fe-treated Panc-1 cells (Fig. 2m), which was corroborated by the enrichment of GO term “response to oxidative stress” (Supplementary Fig. 4a) and NRF2 translocation from the cytosol into the nucleus (Supplementary Fig. 4b). Quantitative proteomics showed that MOF-Fe elevated the expression levels of six proteins in the ferroptosis pathway, all of which belong to ferroptosis suppressors (Fig. 2n). Four proteins in the ferroptosis pathway were downregulated, three of which are ferroptosis drivers. Increase in the mRNA expression levels of HMOX1 and FTH1 were verified with RT-PCR (Supplementary Fig. 4c). Immunoblots confirmed a time-dependent increase in NRF2, FTH1, and HMOX1 protein expression in MOF-Fe-treated Panc-1 and KPC-A719 cells, as well as a dose-dependent increase in FTH1 expression in Panc-1 cells (Fig. 2o and Supplementary Fig. 4d–f). Given that many NRF2-mediated genes are ferroptosis suppressors, we surmised that NRF2 signaling might underlie acquired resistance to the PEF + MOF-Fe combination treatment.
FTH1 promoted resistance to MOF-Fe-induced ferroptosis
FTH1, which sequesters excess iron in cells, belongs to both the ferroptosis and NRF2 pathways. Analysis of the GEPIA2 database40 revealed higher FTH1 expression in human pancreatic tumors than in the adjacent normal tissues. Higher FTH1 expression also correlated with worse prognosis in patients with pancreatic cancer (Supplementary Fig. 4g, h). We went on to explore the role of FTH1 in ferroptosis resistance. Deferoxamine (DFO, an iron chelator), Lip-1, and brusatol (an NRF2 inhibitor) suppressed MOF-Fe-induced FTH1 upregulation, suggesting that FTH1 was involved in NRF2-mediated anti-ferroptosis response (Supplementary Fig. 4i–l).
FTH1-overexpressing (OE) and -knockdown (KD) Panc-1 cells were established, respectively (Supplementary Fig. 5a and Fig. 3a). FTH1-overexpression diminished MOF-Fe-induced elevation of ROS (DCFH-DA+ cells%) and C11-BODIPY oxidation, and facilitated colony formation in the presence of MOF-Fe (Supplementary Fig. 5b–d). A reverse trend was observed in FTH1-KD cells (Supplementary Fig. 5e–g). Interestingly, FTH1-OE Panc-1 cells were less sensitive to erastin and RSL3 than the vector control (Supplementary Fig. 6a). FTH1 expression was then restored in FTH1-KD cells (Fig. 3b) using a modified FTH1-overexpressing plasmid, abbreviated as FTH1-OE (m). The coding sequence of this plasmid had synonymous mutations, preventing recognition by the FTH1-targeting shRNAs (Supplementary Tables 6, 8). FTH1 restoration alleviated MOF-Fe-induced ROS and C11-BODIPY oxidation, and increased the number of colonies in the presence of MOF-Fe (Fig. 3c–e and Supplementary Fig. 6b–d). Taken together, these data suggest that FTH1 promoted resistance to MOF-Fe-induced ferroptosis.

a, b Immunoblots of FTH1-KD (a) and -rescue (b) Panc-1 cells. The experiments were independently repeated three times with similar results. c Immunofluorescence images and quantification of the percentage of DCFH-DA+ cells per FOV, n = 6 randomly selected FOVs (technical replicates). The experiments were independently repeated three times with similar results. Scale bar = 100 μm. d Flow cytometry histograms and quantification of C11-BODIPY oxidation, n = 4 independent experiments. Panc-1 cells in (c, d) were treated with MOF-Fe (10 μg Fe/mL) for 48 h. e Representative images of Panc-1 colonies and quantification, n = 3 independent experiments. Data were presented as mean ± SEM. Statistical significance was determined using one-way ANOVA with Dunnett’s multiple comparison (c, e) or one-tailed ratio-paired t-test (d), and p values are indicated. Source data are provided as a Source Data file.
Arachidonic acid-derived PROTAC degraded MOF-Fe-induced FTH1
An exploratory PROTAC library was synthesized by conjugating (S,R,S)-AHPC to six fatty acids (oleic acid C18:1, linoleic acid C18:2, linolenic acid C18:3, arachidonic acid C20:4, eicosapentaenoic acid C20:5, and erucic acid C22:1) directly or through five linkers (Fig. 4a). Figure 4b illustrates the structure of C20U4V, a representative chimera prepared by directly conjugating arachidonic acid (AA) to (S,R,S)-AHPC. Chemical characterization of the chimeras is included in Supplementary Figs. 16–73, 81–109. Chimeras with IC50 values lower or close to 10 μM in both Panc-1 and MIAPaCa-2 cells were first identified using CCK-8 assay (Fig. 4c and Supplementary Table 1. C20U4V emerged as the candidate compound after another round of screening for FTH1 degradation in the presence of MOF-Fe (Fig. 4d and Supplementary Fig. 7a). C20U4V was efficiently taken up by Panc-1 cells, elevating HMOX1 and FTH1 mRNA transcription and increasing NRF2 and HMOX1 protein expression (Supplementary Fig. 7b–d). This observation was in consistence with the AA moiety in C20U4V, which has been reported to induce oxidative stress30. In the presence of MOF-Fe, C20U4V degraded FTH1 in a time- and dose-dependent manner in Panc-1, MIAPaCa-2, and KPC-A719 cells (Fig. 4e and Supplementary Fig. 7e–h).

a Schemes of PROTAC library. b Chemical structure of C20U4V. c Scattered plots of the IC50 values of the PROTAC library against Panc-1 and MIAPaCa-2 cells after 72 h treatment, n = 6 independent experiments. d Scatter plots of the change of FTH1 expression in Panc-1 cells after treatment with MOF-Fe (3 μg Fe/mL) and the PROTAC library for 12 h, n = 3 independent experiments. e Immunoblots of FTH1 degradation by C20U4V in the presence of MOF-Fe, n = 3 independent experiments. Quantification is included in Supplementary Fig. 7e. f Volcano plots of protein expression in Panc-1 cells treated for 12 h with MOF-Fe + C20U4V vs. MOF-Fe. Differentially expressed proteins in the ferroptosis pathway are labeled. MOF-Fe (3 μg Fe/mL), C20U4V (20 μM), n = 3 independent experiments. g Chemical structure of BPAA and immunoblots of FTH1 pulled down by BPAA. The samples derived from the same experiments but different gels were processed in parallel: FTH1 pulldown was detected on one gel, and the input controls of FTH1 and β-actin were run on another gel, n = 3 independent experiments. Quantification is included in Supplementary Fig. 8a. h Immunoblots of HA-tagged ubiquitin (HA-Ub) immunoprecipitated by anti-Flag antibody in the lysate of HEK-293T. The HEK-293T cells were transfected with HA-Ub and Flag-FTH1 plasmids, and treated with C20U4V (10 μM) in the presence of MG132 (10 μM) for 6 h. The samples derived from the same experiment but different gels were processed in parallel: IP detection of HA-Ub and Flag-FTH1 were run on one gel, and the input controls of HA-Ub, Flag-FTH1, and β-actin were run on another gel. The experiments were independently repeated twice with similar results. A biologically independent replicate is shown in Supplementary Fig. 9a. i FTH1 expression after incubation with MOF-Fe and AA for 48 h, n = 3 independent experiments. Quantification is included in Supplementary Fig. 9b. j FTH1 expression after incubation with MOF-Fe and (S,S,S)-C20U4V for 12 h. Quantification is included in Supplementary Fig. 9c, n = 3 independent experiments. k VHL expression in VHL-knockdown Panc-1 cells. The experiments were independently repeated twice with similar results. l FTH1 expression in NC or shVHL-knockdown cells after incubation with MOF-Fe and C20U4V for 48 h. Quantification is included in Supplementary Fig. 9d, n = 3 independent experiments. Data were presented as mean ± SEM. Statistical significance was determined using multiple one-tailed ratio-paired t-test without correction for multiple comparison (d) or EdgeR analysis using negative binomial generalized linear model (GLM) adjusted with Benjamini–Hochberg algorithm (f), and p values are indicated. Source data are provided as a Source Data file.
Quantitative proteomics revealed significant expression changes in eight ferroptosis-related proteins in MOF-Fe + C20U4V-treated Panc-1 cells versus MOF-Fe-treated cells (Fig. 4f), including FTH1 and FTL (Supplementary Fig. 7i). Given the presence of AA moiety in C20U4V, we analyzed the AA interactome in MOF-Fe-treated Panc-1 cells using BPAA, a biotin-conjugated AA derivative (Fig. 4g and Supplementary Figs. 8a, 74, 75, 110). Pull-down assay confirmed the interaction between FTH1 and BPAA. Proteomics revealed 648 proteins, including FTH1, that might have interacted with BPAA (Supplementary Fig. 8b). GO analyses of the interactome across biological process (BP), molecular function (MF), and cellular components (CC) yielded enrichment of a broad span of GO terms such as “DNA replication initiation”, “DNA helicase activity”, and “podosome” (Supplementary Fig. 8c–e). This observation was consistent with the ubiquitous roles of AA in cellular physiology. Seven ferroptosis-related and eight NRF2-related proteins were identified in the interactome, among which NQO1 and GCLM was also degraded by C20U4V (Supplementary Fig. 8f–h).
We then analyzed whether C20U4V degraded FTH1 as a PROTAC. C20U4V enhanced FTH1 ubiquitylation in HEK-293T cells transfected with HA-tagged ubiquitin (HA-Ub) and Flag-labeled FTH1 (Flag-FTH1, Fig. 4h and Supplementary Fig. 9a). No FTH1 degradation was achieved by AA or (S,S,S)-C20U4V prepared from (S,S,S)-AHPC, the inactive stereoisomer of (S,R,S)-AHPC (Fig. 4i, j and Supplementary Fig. 9b, c). Neither did C20U4V degrade MOF-Fe-induced FTH1 in VHL-KD cells (Fig. 4k, l and Supplementary Fig. 9d). In FTH1-OE Panc-1 cells (Supplementary Fig. 9e, f), C20U4V degraded FTH1 when cycloheximide (CHX) was added to inhibit de novo protein synthesis. Conversely, (S,S,S)-C20U4V abolished this FTH1 degradation. Collectively, we have demonstrated that C20U4V functioned as a PROTAC and degraded MOF-Fe-induced FTH1.
C20U4V potentiated MOF-Fe and PEF in vitro
The MOF-Fe + C20U4V combination inhibited colony formation in Panc-1, MIAPaCa-2, and KPC-A719 cells with HSA synergy scores above 10 (Fig. 5a and Supplementary Fig. 10a–f). The dual-treatment induced stronger C11-BODIPY oxidation and a higher frequency of DCFH-DA+ cells than MOF-Fe alone (Fig. 5b, c). MDA concentration was similar between the MOF-Fe and dual-treatment groups, both of which were higher than that in the vehicle control (Fig. 5d). Compared to the vehicle control, the dual-treatment upregulated 14 oxidized and 20 peroxidized phospholipids, including PC 18:0-22:6-OOH, PE 18:0-22:6-OOH, and PE 16:0-22:6-OOH (Fig. 5e). Compared to MOF-Fe, the dual treatment also upregulated nine oxidized and 14 peroxidized phospholipids (Supplementary Fig. 10g). Ferroptosis is often accompanied by mitochondria changes41. As shown in Fig. 5f–h, the aggregate (red)-to-monomer (green) ratio of JC-1 staining characterizes mitochondrial membrane potential, which was significantly lowered by all the treatments. Adenosine triphosphate (ATP) concentration was reduced by MOF-Fe or the dual-treatment. The dual-treatment also caused smaller mitochondria with disorganized cristae (yellow arrows).

a Quantification of Panc-1 colonies, n = 3 independent experiments. b Histograms and quantification of C11-BODIPY oxidation, n = 6 independent experiments. c Percentage of DCFH-DA+ cells per FOV, n = 8 (vehicle), 6 (MOF-Fe), or 7 (C20U4V, MOF-Fe + C20U4V) randomly selected FOVs (technical replicates). The experiments were independently repeated three times with similar results. d MDA content, n = 5 independent experiments. e Volcano plots of lipidomics after treatment with MOF-Fe (3 μg Fe/mL) and C20U4V (20 μM) relative to vehicle control, n = 5 independent experiments. f Immunofluorescence of JC-1 staining and quantification of aggregate-to-monomer ratio, n = 6 (vehicle), 7 (MOF-Fe, MOF-Fe + C20U4V), or 9 (C20U4V) randomly selected FOVs (technical replicates). The experiments were independently repeated three times with similar results. g Quantification of intracellular ATP, n = 5 independent experiments. h Representative TEM images of mitochondria (yellow arrows). The experiments were independently repeated three times with similar results. Scale bar = 1 μm. Panc-1 cells in panels (b–h) were treated with MOF-Fe (3 μg Fe/mL for (e, h), and 5 μg Fe/mL for (b–d, f, g)) and/or C20U4V (10 μM for (b–d, f–h), and 20 μM for (e)). i Quantification of Panc-1 colonies, n = 3 (vehicle, MOF-Fe) or 6 (PEF, PEF + MOF-Fe) independent experiments. j Immunoblots of Panc-1 cells. The samples derived from the same experiment but on different gels for NRF2, another for HMOX1, FTH1, and β-actin, were processed in parallel. Quantification is provided in Supplementary 10j. k, l GO_BP analysis for genes upregulated (k) and downregulated (l) in the PEF + MOF-Fe + C20U4V group relative to the control group. m, GSEA on the Hallmark gene set. Panc-1 cells in panels (j-m) were treated with PEF (600 V/cm) and incubated with MOF-Fe (3 μg Fe/mL) and C20U4V (10 μM) for 48 h, n = 4 (j) or 3 (k–m) independent experiments. Data are presented as mean ± SEM. Statistical significance was determined using a one-tailed ratio-paired t-test (b), one-way ANOVA with Dunnett’s multiple comparison (c, d, f, g, i), multiple two-tailed unpaired t-test without correction (e), GO (k, l), or GSEA (m) analysis adjusted with the Benjamini–Hochberg algorithm, and p values are indicated. Source data are provided as a Source Data file.
The dual-treatment sensitized pancreatic cancer cells to PEF. Panc-1 and KPC-A719 cells treated with the “PEF + MOF-Fe + C20U4V” triple-treatment formed fewer colonies than all the other groups (Fig. 5i and Supplementary Fig. 10h, i). Panc-1 cells in the triple-treatment group exhibited significantly higher NRF2 and HMOX1 protein expression than those in the dual-treatment group (Fig. 5j and Supplementary Fig. 10j). Differentially expressed genes (DEGs) between the triple-treatment group and the vehicle control were analyzed via GO analysis. Upregulated genes in the triple-treatment group were enriched in stress response-related GO terms; whereas downregulated genes were enriched in those linked to cell division and DNA replication (Fig. 5k, l). GSEA revealed ten significantly changed Hallmark gene sets (gsea-msigdb.org) when comparing the triple-treatment groups to the vehicle control (Fig. 5m and Supplementary Table 2). The “Hallmark E2F targets” gene set, which is related to the progression of the cell cycle, was downregulated with the lowest NES score (NES = −2.50, p.adjust = 1.67 × 10−9). By contrast, the “Hallmark unfolded protein response” was upregulated with the highest NES score (NES = 2.25, p.adjust = 1.21 × 10−8), indicating a high level of cellular stress.
In pancreatic cancer patient-derived organoids, the dual-treatment significantly reduced organoid number compared to the vehicle control and yielded smaller organoids than all other groups (Fig. 6a). Organoids in the vehicle control exhibited glandular structures consisting of simple cuboidal epithelium with enlarged nuclei. Those treated with MOF-Fe or C20U4V formed fewer and smaller glandular structures, which consisted of simple or stratified epithelial cells of variant sizes. In contrast, the dual-treatment abolished glandular formation, resulting in cell clusters with newly-formed lumens (Fig. 6b). Prussian Blue staining was observed in MOF-Fe and the dual-treatment groups (Fig. 6c), and organoids in the dual-treatment group exhibited significantly fewer Ki67+ cells than all other groups (Fig. 6d). The efficacy of “PEF + MOF-Fe + C20U4V” triple treatment was then examined in organoids derived from another patient. The triple-treatment produced fewer and smaller organoids than all other groups (Fig. 6e). We also evaluated C20U5V to determine whether the sensitization effect was specific to C20U4V. C20U5V was prepared by directly conjugating eicosapentaenoic acid to (S,R,S)-AHPC. It moderately degraded FTH1 in the presence of MOF-Fe (Supplementary Fig. 7a). As shown in Supplementary Fig. 11, Panc-1 cells treated with the MOF-Fe + C20U5V dual-treatment or MOF-Fe alone showed similar levels of C11-BODIPY oxidation. MOF-Fe and C20U5V synergistically inhibited colony formation in Panc-1 cells. The PEF + MOF-Fe + C20U5V triple-treatment also yielded fewer colonies than PEF or the dual-treatment.

a Bright-field images of organoids and quantification of organoid abundance, n = 8 randomly selected FOVs; and diameter, n = 39 (vehicle), 45 (MOF-Fe), or 54 (C20U4V, MOF-Fe + C20U4V) randomly selected organoids (technical replicates) after treatment with MOF-Fe (3 μg/mL) and C20U4V (3 μM). b H&E staining. c Prussian staining. d Ki67 staining and quantification, n = 15 (vehicle, MOF-Fe, C20U4V) or 13 (MOF-Fe + C20U4V) randomly selected organoids (technical replicates). e Bright-field images and quantification of organoid abundance, n = 8 randomly selected FOVs; and diameter, n = 50 (vehicle, PEF) or 49 (MOF-Fe + C20U4V, PEF + MOF-Fe + C20U4V) randomly selected organoids (technical replicates) after treatment with MOF-Fe (3 μg/mL) + C20U4V (3 μM) and/or PEF (600 V/cm). Organoids generated from patient #1 were used for panels (a–d), and those generated from patient #2 were used for panel (e). All experiments were independently repeated three times with similar results. Scale bar = 100 μm. Data were presented as mean ± SEM. Statistical significance was determined using one-way ANOVA with Dunnett’s multiple comparison (a, d, e), and p values are indicated. Source data are provided as a Source Data file.
C20U4V encapsulation in ROS-responsive polymeric micelles
C20U4V was insoluble in water and thus cannot be directly used in vivo. We previously reported that ROS levels were elevated in PEF-treated tumors42, and therefore prepared ROS-responsive polymeric micelles to encapsulate C20U4V. Methoxypolyethylene glycol was extended with ε-caprolactone via controlled ring polymerization and capped with 2-bromoisobutyryl bromide. The macroinitiator was then reacted with 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl acrylate via the atom radical polymerization to yield the resultant triblock copolymer, abbreviated as PEG-PCL-PB (Fig. 7a). Chemical characterization of the products is included in Supplementary Figs. 76–80. The average degree of polymerization of the PCL and PB blocks was about 11 and 15, respectively, which was calculated from comparing the integrals of methylene protons of the PEG block (δ = 3.38, 3.64 ppm), the methylene protons of the PCL block (δ = 1.65, 2.31, and 4.06 ppm), and the aromatic protons of the PB block (δ = 7.21, 7.73 ppm, Supplementary Fig. 80).

a Schematic illustration of PEG-PCL-PB synthesis and M-C20U4V. b, c Representative histograms of hydrodynamic diameter (b) and surface ζ-potential (c) of M-C20U4V. Experiments were independently repeated three times with similar results. d Profiles of payload release from M-C20U4V, n = 3 independent experiments. e Immunoblots and quantification of FTH1 in Panc-1 cells treated with MOF-Fe and M-C20U4, n = 3 independent experiments. f, g Quantification of Panc-1 colonies (f) and HSA synergy scores (g) after treatment with MOF-Fe or M-C20U4V (in C20U4V-equivalent concentration), n = 3 independent experiments. h, i, Biodistribution of DiR-labeled M-C20U4V in KPC-A719 tumors (h). Micelles were intravenously injected 4 h prior to PEF ablation, and the tumor and major organs were collected at 44 h after PEF ablation, n = 5 (untreated) or 7 (PEF ablation) independent mice. Representative fluorescence images (i) of major organs and tumors include: heart (He), liver (Li), spleen (Sp), lung (Lu), kidneys (Ki), stomach (St), intestines (In), bone (Bo), muscle (Mu), and tumor (Tu). j Tumor growth curves of subcutaneous KPC-A719 models. n = 6 (sham control, PEF, MOF-Fe + M-C20U4V) or 14 (PEF ablation + MOF-Fe + M-C20U4V) independent mice. k Animal survival of orthotopic KPC-A719 models, n = 6 (sham control, MOF-Fe + M-C20U4V), 7 (PEF ablation), or 13 (PEF ablation + MOF-Fe + M-C20U4V) independent mice. Data were presented as mean ± SEM. Statistical significance was determined using one-way analysis of variance (ANOVA) with Dunnett’s multiple comparison (e, j) or log-rank test (k), and p values are indicated. Source data are provided as a Source Data file. The micelle schemes in Fig.7a were generated using PowerPoint.
C20U4V was encapsulated using the nanoprecipitation method with a loading efficiency of 90% and a loading percentage of 9% by weight. M-C20U4V, the resulting formulation, exhibited an average hydrodynamic size of 78.6 ± 14.5 nm and a surface ζ-potential of −0.2 ± 4.5 mV (Fig. 7b, c). Incubation of M-C20U4V with 100 μM H2O2 accelerated the release of payload (Fig. 7d). C20U4V and M-C20U4V showed similar IC50 values in Panc-1, MIAPaCa-2, and KPC-A719 (<6 μM, Supplementary Fig. 12a). Blank micelles were non-toxic to Panc-1 or MIAPaCa-2 cells at concentrations up to 268.5 μg/mL, equivalent to 30 μM C20U4V (Supplementary Fig. 12b). M-C20U4V degraded FTH1 in MOF-Fe-treated Panc-1, MIAPaCa-2, and KPC-A719 cells over a 12–48 h time course (Fig. 7e and Supplementary Fig. 12c, d). M-C20U4V and MOF-Fe inhibited Panc-1 colony formation with an HSA synergy score above 20 (Fig. 7f, g). After labeling with DiR, a near-infrared fluorescence dye, M-C20U4V was intravenously injected into mice bearing KPC-A719 tumors (Fig. 7h, i). Its uptake value in untreated tumors was 11.2 ± 2.4%ID/g (percent of injected dose per gram of tissue). PEF ablation at 4 h after M-C20U4V injection did not significantly change its tumor uptake (14.1 ± 2.0%ID/g). Taken together, we have prepared a ROS-responsive formulation in M-C20U4V for in vivo evaluation.
MOF-Fe and M-C20U4V enhanced PEF ablation in vivo
The anti-tumor efficacy of PEF ablation, MOF-Fe, and M-C20U4V was first evaluated in subcutaneous KPC-A719 tumor models, using a treatment schedule outlined in Supplementary Fig. 12e. Growth curves of individual tumors are shown in Supplementary Fig. 12g. As shown in Fig. 7j, tumors of the MOF-Fe + M-C20U4V dual-treatment grew at a similar rate to that of the sham control. Tumors in the PEF ablation group experienced a brief growth delay for about 8 days, but then bounced back quickly. In contrast, tumors receiving the PEF ablation + MOF-Fe + M-C20U4V triple-treatment shrank to a minimal volume for about 22 days, and then grew at a slower rate than those of all other groups. Three out of 14 mice in the triple-treatment group had no visible tumors at the study’s conclusion (day 60). The average tumor size in the triple-treatment group on day 60 was significantly smaller than those in all the other groups (p < 0.0001). A similar trend of therapeutic efficacy was observed in KPC-A719 orthotopic models (Fig. 7k), which are more relevant to human malignancy43. Mice in the triple-treatment group exhibited a median survival of 27 days, significantly longer than those in the control (11 days, p < 0.0001), PEF ablation (12 days, p = 0.001), or dual-treatment (14.5 days, p = 0.005) groups. No significant change in body weight was observed in either model (Supplementary Fig. 12f, h)
Intratumoral immune cells in orthotopic KPC-A719 tumors were analyzed with flow cytometry on day 4 after enrollment (Fig. 8a–j). The gating strategy is shown in Supplementary Fig. 13a–c. Tumors in the triple-treatment group contained significantly more CD45+, CD3+T, and CD4+T cells than those in the other groups. The abundance of CD8+ and Interferon-γ+ (IFNγ+) CD8+ T cells were similar in PEF- and triple-treatment groups, both of which were higher than those in the other two groups. There was no significant difference in the CD8+ T cell-to-Treg ratio among all groups. CD8+ T cell depletion alongside the triple-treatment resulted in significantly larger tumors on day 11 post-enrollment (Supplementary Fig. 13d–h). Thus, CD8+ T cells at least partially contributed to the therapeutic efficacy of the triple-treatment.

a–j Frequency of intratumoral immune cells on day 4 after enrollment: a CD45+ cells, b CD3+ T cells: CD45+CD3+, c CD4+ T cells: CD45+CD3+CD4+, d CD8+ T cells: CD45+CD3+CD8+, e IFNγ+CD8+ T cells: CD45+CD3+CD8+IFNγ+, f CD8+ T cells/Treg ratio, Treg: CD45+CD3+CD4+CD25+Foxp3+, g dendritic cells: CD45+CD11c+MHC-II+. h–j Mean fluorescence intensity (MFI) of CD80 (g), CD86 (h), and MHC-II (i) in dendritic cells. For panels a–d, f, n = 7 (sham control), 8 (PEF ablation, MOF-Fe + M-C20U4V), and 9 (PEF ablation + MOF-Fe + M-C20U4V), independent mice. For panel e, n = 7 (sham control), 9 (PEF ablation), 10 (MOF-Fe + M-C20U4V), and 11 (PEF ablation + MOF-Fe + M-C20U4V) independent mice. For panel g–j n = 7 (sham control and PEF ablation), 8 (MOF-Fe + M-C20U4V), and 10 (PEF ablation + MOF-Fe + M-C20U4V) independent mice. k Ki67 staining, n = 16 (sham control and PEF ablation), 21 (MOF-Fe + M-C20U4V) or 22 (PEF ablation + MOF-Fe + M-C20U4V) randomly selected FOVs from 5 independent mice per group. l FTH1 staining, n = 14 (sham control), 18 (PEF ablation), 31 (MOF-Fe + M-C20U4V), or 21(PEF ablation + MOF-Fe + M-C20U4V) randomly selected FOVs from five independent mice per group. m 4-HNE staining, n = 8 (sham control), 11 (PEF ablation), or 13 (MOF-Fe + M-C20U4V, PEF ablation + MOF-Fe + M-C20U4V) randomly selected FOVs from five independent mice per group. n HMOX1 staining, n = 8 (sham control), 9 (PEF ablation, MOF-Fe + M-C20U4V), or 16 (MOF-Fe + M-C20U4V, PEF ablation + MOF-Fe + M-C20U4V) randomly selected FOVs from five independent mice per group. o Multiplex immunofluorescence staining of CD8 and IFN-γ and quantification. n = 20 randomly selected FOVs from five independent mice per group. Scale bar = 100 μm. Tumors in panels j–o were collected on day 9 after enrollment. Data were presented as mean ± SEM. Statistical significance was determined using one-way ANOVA with Dunnett’s multiple comparison (a–o), and p-values are indicated. Source data are provided as a Source Data file.
Short-term response of KPC-A719 orthotopic tumors were also examined on day 9 after enrollment. Tumors of the sham control presented a mixture of small or medium-sized irregular glands and poorly differentiated sarcoma-like tumor cells. Some tumor glands formed a cancer nest surrounded by desmoplastic stroma. Tumor cells exhibited enlarged and hyperchromatic nucleus, multiple nucleoli, and pathological mitosis (Supplementary Fig. 14). Necrotic regions were visible in tumors treated with MOF-Fe + M-C20U4V, especially around the sites of MOF-Fe accumulation. Infiltration by macrophages and neutrophils were present at the borders between the necrotic and viable regions. Both PEF and the triple treatment induced extensive areas of necrosis. Residual cancer cells were present outside the ablation zone of PEF-ablated tumors, whereas extensive infiltration by neutrophils and macrophages were observed in the triple treatment group. Tumors in the triple-treatment groups had the lowest proportion of Ki67+ proliferating cells, and the highest percentage of FTH1+, 4-HNE+ or HMOX1+ pixels among all four groups (Fig. 8k–n). The triple treatment induced IFNγ levels comparable to those from PEF ablation alone (Fig. 8o). It also resulted in a massive infiltration of CD68+ macrophages, accompanied by a reduction in CD206+ M2 macrophages (Supplementary Fig. 15a, b). Together, we have shown that the triple-treatment exhibited satisfactory anti-tumor efficacy and activated anti-tumor immunity.
Lastly, we investigated the acute toxicity of the triple treatment in mice bearing KPC-A719 tumors (Supplementary Fig. S15c, d). No pathological change was found in the heart, liver, spleen, lung, or kidneys. The indexes of liver function (alanine transaminase, aspartate transaminase), kidney function (creatine and blood urea nitrogen), and heart (creatine kinase) function were similar between treated and control mice.
Discussion
In this study, we demonstrated that low-intensity PEF upregulated ferroptosis suppressor genes in pancreatic cancer cells. Although MOF-Fe transiently suppressed tumor recurrence following PEF ablation, subsequent FTH1 upregulation quickly diminished its therapeutic efficacy. C20U4V, an arachidonic acid-derived PROTAC, enhanced MOF-Fe-induced ferroptosis via FTH1 degradation, and sensitized both pancreatic cancer cell lines and patient-derived organoids to PEF. We then developed M-C20U4V, a ROS-responsive polymeric micelles nano-formulation. Finally, the combination of MOF-Fe and M-C20U4V induced ferroptosis and potentiated PEF ablation in vivo.
The tumor recurrence rate following IRE varies across different tumor types. It remains a clinical challenge, particularly in the context of pancreatic cancer. Twenty-three out of 50 patients with pancreatic cancer developed local recurrence after IRE in the PANFIRE-2 phase II study44. In a recent analysis on a multi-institutional database, 32 (17%) out of 187 patients with pancreatic cancer had local recurrence following IRE45. Various strategies have been explored to reduce the risk of tumor recurrence, including chemotherapy, radiotherapy, immunotherapy, and targeted therapy6,7,8,9,10,35,46. However, the biological effects of these strategies primarily converge on apoptotic pathways. Cancer cells that harbor mutations in the apoptotic cascades, including those in caspases and the Bcl-2 family, may develop treatment resistance47. In this study, we explored the feasibility of lowering the risk of tumor recurrence by inducing ferroptosis in residual cancer cells. Ferroptosis has been reported to overcome resistance to conventional therapies48.
Low-intensity PEF exerts multiple biological changes without completely eliminating cancer cells. These effects include Ca2+ influx, ROS elevation, and damage to DNA, proteins, and membranes49. In this study, we found that PEF triggered NRF2-orchestrated antioxidant responses and upregulated a suite of ferroptosis-suppressor genes (Fig. 1e-l). The NRF2 pathway is also known to promote tumor aggressiveness and therapeutic resistance50. In this context, our findings help explain the observation that residual cancer cells grew more rapidly following IRE compared to their untreated counterparts51. Iron-catalyzed ROS production is essential for initiating ferroptosis. Iron-based nanoparticles can undergo endocytosis independently of transferrin receptors and are therefore promising candidates for inducing intracellular iron overload. We postulated that nanoparticles with faster iron release would be more effective at inducing iron overload and thus selected MOF-Fe for this study. Accordingly, MOF-Fe elevated intracellular ferrous ion levels to a greater extent than SPIO, and more effectively suppressed colony formation in Panc-1 cells (Fig. 2d–f). However, in IRE-treated tumor models, MOF-Fe only transiently delayed tumor recurrence (Fig. 2l). This observation aligns with other reports showing that iron-based nanoparticles exhibit limited toxicity to cancer cells, likely because intracellular iron levels are tightly regulated via iron homeostasis mechanisms52. Iron homeostasis is critical for sustaining the proper functional integrity of cells. Briefly, extracellular ferric ions bound to transferrin or hyaluronate are recognized by their respective receptors, i.e., transferrin receptor and CD44, before being endocytosed and transported into lysosomes53. Ferric ions are then released from these bound complexes and reduced to ferrous ions by members of the six-transmembrane epithelial antigen of the prostate (STEAP) family proteins. The ferrous ions are transported into the cytosol by divalent metal-ion transporter 1 (DMT1) to form the labile iron pool, which is then parcelled to various organelles to support essential physiological functions. Excess ferrous ions can be exported out of the cell by ferroportin or sequestered in ferritin for later use52.
Disruption of iron homeostasis can amplify the cytotoxicity of iron-based nanoparticles. Simultaneous downregulation of ferroportin and lipocalin-2, two proteins involved in iron export, enhanced SPIO-induced ferroptosis54. Ferumoxytol nanoparticles exhibited greater toxicity to primary acute myeloid leukemia cells that had lower ferroportin expression55. Recently, activation of lysosomal iron has emerged as an effective strategy for triggering ferroptosis and thereby eliminating drug-resistant persister cancer (DTP) cells18. Salinomycin sequestered iron in lysosomes to the point of iron overload, ultimately causing ferroptotic death in breast cancer stem cells56. These results underscore the potential of using iron-based nanoparticles to eliminate drug-resistant cancer cells, as these nanoparticles are endocytosed via the endosome-lysosome route and are often designed to release their payloads within acidic lysosomes. In addition, certain types of cancer cells exhibit iron addiction, a trait that renders them particularly vulnerable to ferroptosis. Indeed, ovarian tumor-initiating cells have been reported to accumulate excess iron and rely on iron for proliferation. Treatment with erastin effectively induced ferroptotic cell death in these cells and reduced tumor burden in vivo57.
The MOF-Fe-induced upregulation of FTH1 and FTL (Fig. 2n, o and Supplementary Fig. 7i) could be attributed to cellular responses aimed at sequestering excess iron. FTH1 expression correlated inversely with the sensitivity of Panc-1 cells to MOF-Fe-induced ferroptosis (Fig. 3 and Supplementary Figs. 5, 6). FTH1 overexpression also alleviated the cytotoxicity of erastin and RSL3. Previous reports have shown that FTH1 mitigated erastin-induced ferroptosis in neuronal cells58, and ferritin upregulation abolished ROS production in oligodendroglial cells following treatment with iron oxide nanoparticles59. In addition to iron storage, ferritin and its two subunits also regulate cancer cell plasticity via epigenetic modulation of epithelial-to-mesenchymal (EMT)-related genes21. FTH1 overexpression promoted lipid droplet formation and radiotherapy resistance60. It also stabilized hypoxia-inducible factor 1α (HIF-1α), a transcription factor that subsequently activated angiogenesis and drove tumor progression61. Consequently, FTH1 upregulation correlates with worse prognosis across many cancer types62, including pancreatic cancer (Supplementary Fig. 4g)
MOF-Fe substantially elevated FTH1 expression within 24 h (Fig. 2o). Since RNA interference typically takes 24 h to take effect24, we switched to methods that are capable of degrading pre-existing FTH1. Several natural product derivatives exhibit such activity, including artesunate, baicalin (extracted from S. baicalensis Georgi), and CT-1 (a derivative of cryptotanshinone)63,64,65. We endeavored to identify an FTH1-degrading PROTAC through rational design and screening. Arachidonic acid binds to a U-shaped pocket between two FTH1 subunits of ferritin (Kd = 60 μM) via interactions between its carboxylate group and adjacent arginine residues27. Oleic acid, an MUFA, exhibited similar interactions with FTH126. DeFer-2 was prepared using oleic acid and (S,R,S)-AHPC to degrade ferritin and trigger pyroptosis in melanoma cells28. However, exogenous MUFAs inhibit ferroptosis by suppressing ROS accumulation and reducing PUFA oxidation29. In contrast, PUFAs are susceptible to oxidation at their bis-allylic carbons. Notably, PUFA peroxidation by lipoxygenases, coupled with the presence of a labile iron pool, is actually required for ferroptosis30. Arachidonic acid-derived C20U4V degraded FTH1 in MOF-Fe-treated pancreatic cancer cells, and potentiated ferroptosis (Fig. 5). FTH1 degradation was also achieved by several other PUFA-based PROTACs (Fig. 4d and Supplementary Fig. 7a). In particular, eicosapentaenoic acid-based C20U5V exhibited efficacy similar to that of C20U4V in sensitizing pancreatic cancer cells to MOF-Fe and PEF (Supplementary Fig. 11).
The poor water solubility of C20U4V prompted us to develop a ROS-responsive nano-formulation (M-C20U4V), which accumulated in PEF-treated tumors (Fig. 7h, i). The triple combination treatment with PEF, MOF-Fe, and M-C20U4V exhibited satisfactory anti-tumor efficacy in both subcutaneous and orthotopic KPC-A719 pancreatic tumor models (Fig. 7j, k). Both PEF ablation and ferroptosis are known to stimulate anti-tumor immunity3,13. Accordingly, the triple-treatment increased tumor infiltration by CD45+ leukocytes, CD3+ T cells, CD4+ T cells, and CD8+ T cells (Fig. 8a–d). On the other hand, the impact of ferroptosis on CD8+ T cells remains ambiguous. While CD8+ T cells were susceptible to GPX4 inhibition66, their tumoricidal activity also depended on the proper function of acyl-CoA synthetase long chain family member 4 (ACSL4), a key pro-ferroptosis protein67. We observed a greater frequency of IFNγ+CD8+ T cells in tumors from the triple-treatment group compared to those from the sham control group (Fig. 8e, o), suggesting that the activity of CD8+ T cells were likely preserved.
Our study has several limitations. Firstly, the mechanism underlying FTH1 upregulation by MOF-Fe was not exhaustively investigated. Other mechanisms may also be involved, including transcriptional regulation by HIF-1α, pro-inflammatory cytokines, oncogenes, and non-coding RNAs; as well as post-translational regulation by iron regulatory proteins62. Secondly, MOF-Fe and C20U4V were not integrated on a single nanoplatform. Physical absorption of C20U4V onto MOF-Fe was limited by insufficient loading efficiency. The intrinsic oxidative capacity of MOF-Fe may also catalyze the oxidation of C20U4V during storage68. Future liposomal or lipid nanoparticles integrating excessive iron and FTH1 degraders may exhibit potential for clinical translation. It should be noted that the binding affinity of PUFA to ferritin remains suboptimal, which can be improved using more advanced techniques for targeted protein degradation, such as the degron-tagging systems.
In summary, we have shown that although MOF-Fe transiently suppressed tumor relapse following IRE, its efficacy was rapidly diminished by FTH1 upregulation. C20U4V degraded FTH1 and potentiated MOF-Fe-induced ferroptosis. The combination of MOF-Fe and M-C20U4V efficiently induced ferroptosis and enhanced the efficacy of PEF ablation in murine pancreatic tumor models. Collectively, our results suggest that disruption of iron homeostasis is a feasible strategy to reduce the risk of tumor recurrence after IRE.
Method
Research compliance
All animal studies were approved by the Institutional Animal Care and Use Committee of Huazhong University of Science and Technology, complied with the ARRIVE guidelines, and were conducted in accordance with the National Research Council’s Guide for the Care and Use of Laboratory Animals at specific pathogen-free facilities. Informed consent was obtained from all patients involved in the study. Information on sex and gender was obtained by self-reporting from human participants with consent to share at the individual level. The study on patient-derived organoids was approved by the Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology. Approval number: [2021] IEC (284), approval date: March 3, 2021. All experiments were conducted in compliance with the Helsinki Declaration.
Chemicals and reagents
All chemicals were purchased from Aladdin Biochemical Technology (Shanghai, China) or Sigma-Aldrich (MO, USA) and used as received. Ferroptosis inhibitors were obtained from TOPSCIENCE (Shanghai, China). Superparamagnetic iron oxide (SPIO) nanoparticles (average size = 7 nm, carboxylate modified) were purchased from XFNANO (Nanjing, China). All small-molecule probes used in the manuscript meet the community requirements in https://www.chemicalprobes.org/information-centre.
Synthesis and characterization of MOF-Fe
MOF-Fe was synthesized using the hydrothermal method via previous reports. Briefly, ferric chloride hexahydrate (FeCl3·6H2O, 0.675 g, 2.5 mmol) and amino terephthalic acid (0.226 g, 1.25 mmol) were dissolved in dimethyl formamide (DMF, 25 mL) along with acetic acid (1 mL). The mixture was heated in a Teflon-coated autoclave at 120 °C for 20 h. The product was collected by centrifuge, washed with DMF and ethanol, and then dried under vacuum. The particles were then dispersed in an aqueous solution of PEI (MW + 25,000, 10 mg/mL), and uncoated PEI was removed by centrifuging. The resulting nanoparticles, termed MOF-Fe, were stored at 4 °C and used within one week. Iron content was determined with an iron analysis kit (Solarbio, Beijing, China). MOF-Fe morphology and size were examined using scanning electron microscopy (SEM, Clara, TESCAN, Shanghai, China). Surface ζ-potential was measured on Malvern Zetasizer Nano-ZS (Malvern Instruments, MA, USA).
Synthesis of ferritin-degrading PROTACs
C20U4V synthesis is described as an example. (S,R,S)-AHPC monohydrochloride (70 mg, 0.16 mmol) and arachidonic acid (48 mg, 0.16 mmol) were added to a round-bottom flask and dissolved with N,N-dimethylformamide (DMF, 2 mL). After adding N, N-diisopropylethylamine (115 μL, 0.64 mmol) and HATU (124 mg, 0.32 mmol), the mixture was stirred for 2 h at room temperature and diluted with distilled water (20 mL). After extraction with ethyl acetate, the organic phase was collected, washed with brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated at 40 °C under reduced pressure. The crude product was purified using flash column chromatography on silica gel (dichloromethane/methanol = 20/1, vol/vol) to yield a colorless oil. The synthesis procedures and structure characterization (1H-NMR, 13C-NMR, and ESI mass spectra) of all the chimeras are included in the Supplementary Information.
Synthesis of ROS-sensitive MPEG-PCL-PB
Step 1: Methoxy polyethylene glycol (1.00 g, 0.2 mmol) was dissolved in anhydrous toluene (1 mL) in a round-bottom flask. ε-Caprolactone (0.23 mL, 2.2 mmol) and Sn(Oct)2 (15.2 μL, 4.7 mmol) were added, and the solution was stirred at 105 °C for 12 h under a nitrogen atmosphere. The mixture was precipitated in petroleum ether and dried in a vacuum. The product, PEG-PCL, was obtained as a white solid (1.2 g, yield = 96%).
Step 2: PEG-PCL (1.2 g, 0.2 mmol) was dissolved in DCM (5 mL) along with triethylamine (0.38 mL, 3 mmol) in an ice-water bath under a nitrogen atmosphere. 2-Bromoisobutyrl bromide (0.11 mL, 2 mmol) was dropped in. The reaction was gradually warmed to room temperature and stirred overnight. The mixture was concentrated at 40 °C under reduced pressure. The crude product was redissolved in tetrahydrofuran (THF) and reprecipitated in hexane to afford PEG-PCL-BIBB as a pale-yellow solid (1.1 g, yield = 90%).
Step 3: (4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol (8.6 g, 37 mmol) and triethylamine (6.2 mL, 44 mmol) was dissolved in dichloromethane (DCM, 30 mL) in an ice-water bath under a nitrogen atmosphere. Acryloyl chloride (3.6 mL, 44 mmol) was added dropwise. The reaction was stirred at room temperature for 2 h and quenched with water. The mixture was extracted with ethyl acetate (3 × 50 mL), and then the organic phase was washed with saturated aqueous sodium chloride solution (50 mL). The mixture was concentrated at 40 °C under reduced pressure. The crude product was purified by silica gel column chromatography (hexane/EA = 10:1, v/v) to afford 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl acrylate as a white solid (11 g, yield = 92%).
Step 4: PEG-PCL-BIBB (0.5 g, 0.077 mmol), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl acrylate (1.2 g, 3.85 mmol) and tris(2-dimethylaminoethyl)amine (Me6TREN, 40 μL, 0.15 mmol) was dissolved in anisole (2 mL). The mixture was evacuated and refilled with nitrogen for three cycles. Cuprous bromide (CuBr, 22 mg, 0.15 mmol) was added under nitrogen protection, and the mixture was stirred at 60 °C for 24 h. Anisole was removed under reduced pressure. The residue was redissolved in DCM, passed through a neutral alumina column to remove the catalyst, condensed under reduced pressure to afford PEG-PCL-PB as a pale gray solid (0.45 g). The chemical structures were confirmed by 1H-NMR (Supplementary Information).
M-C20U4V polymeric micelles
Drug-loaded micelles were prepared using the nanoprecipitation method. Briefly, MPEG-PCL-PB (100 mg) and C20U4V (10 mg) was dissolved in acetone (1 mL) and rotated in the dark for 1 h. Ultrapure water (2 mL) was added quickly to the acetone solution under vigorous vortexing, and the mixture was rotated at 100 mBar, 45 °C for 10 min to remove acetone. After dialyzing against ultrapure water at 4 °C, the final dispersion was filtered through a 0.22-μm syringe filter to yield the drug-loaded micelles M-C20U4V. Drug loading capacity was measured using high-performance liquid chromatography (E Classical 3100, EliteHPLC, Dalian, China) equipped with an ultraviolet-visible detector (wavelength = 254 nm). The micelles were dissolved in dimethyl sulfoxide (DMSO) to release the payload, filtered with a 0.22-μm filter, and eluted through a C18 reverse-phase column (Ø4.6 × 200 mm, 5-μm particles). The mobile phase was a gradient mixture of (A) ultrapure water and (B) acetonitrile, from 10%B at 0 min to 90%B at 20 min. Size and surface ζ-potential of M-C20U4V were examined on Malvern Zetasizer Nano-ZS (Malvern Instruments, MA, USA).
Cell lines and animal models
Panc-1 and MIAPaCa-2 human pancreatic cancer cell lines were purchased from the Cell Resource Center, Peking Union Medical College, the Headquarters of the National Infrastructure of Cell Line Resources. All cell lines were authenticated with short-tandem repeat (STR) profiling. KPC-A719 cells were isolated from the established tumor of a female KrasG12D/+; p53R172H/+; Pdx-1-Cre (KPC) genetically engineered mouse. Gene mutations were verified by Sanger sequencing. All cells were routinely tested for mycoplasma contamination.
Subcutaneous tumor models were established by inoculating 2 × 106 KPC-A719 cells at the lower left back of 8-week-old C57BL/6 female mice (Hubei Biont Biological Technology, Wuhan, China). Orthotopic models were established by directly injecting 5 × 105 KPC-A719 cells in 15 μL Hank’s balanced salt solution (HBSS) into the head of the pancreas10. Tumor growth was monitored using a caliper (subcutaneous model) or by palpation under anesthesia (orthotopic model). Mice were enrolled for treatment once the tumors reached 7 mm in one dimension, and those with orthotopic tumors exceeding 10 mm in one dimension were not enrolled. The maximal tumor burden for subcutaneous models permitted by the institutional review board is: maximal diameter exceeding 20 mm, the calculated tumor size above 3000 mm3 as determined by the formula: size = 0.5 × length × width2, or the tumor weight more than 15% of mouse body weight, whichever is reached first. The maximal tumor size/burden was not exceeded in the study. Mice bearing orthotopic tumors were euthanized once they reached humane endpoints defined by the National Institute of Health, including rapid weight loss, anorexia, lethargy, abnormal posture, etc.
Pancreatic cancer patient-derived organoids
Patient information, including sex and gender, is summarized in Supplementary Table 3. Sex and gender of participants were determined based on self-report. The information was collected with consent for sharing individual-level data. Disaggregated number for the individual experiments was provided in the source data. Sex or gender were not considered in the study design because pancreatic cancer occurs across the whole population. Patient-derived organoids were cultured according to a previous report69. Briefly, patient tumor tissues were minced into fragments and digested with organoid tissue digestion solution (D23013, D1 Medical Technology, Shanghai, China). Phosphate-buffered saline (PBS) was added to stop the digestion, and the cell suspension was passed through a 100-μm-mesh and centrifuged. The cell pellet was re-dispersed in PBS and mixed with diluted Matrigel (D23016, D1 Medical Technology) to a density of 100–1000 cells/μL. Fifty μL of the mixture was added to the center of one well of a 24-well plate, and incubated at 37 °C for 15 min. Each well was then added with 500 μL of organoid culture medium (K211M05, D1 Medical Technology), and the plate was returned to incubation at 37 °C.
Treatment with pulsed electric field (PEF)
Electric pulses were generated from an HW-E01 electroporation system (Huiwei Medical, Ningbo, China). For in vitro studies, electric pulses were delivered to adherent cells using a Petri Pulser electrode composed of 13 gold-plated electrodes 2 mm apart (Catalog# 45-0130, BTX Harvard Apparatus, MA, USA), or to cell suspension in an electroporation cuvette fitted with two aluminum plate electrodes 4 mm apart (Catalog# 1652081, Bio-Rad, Hercules, CA, USA). PEF parameters were: pulse intensity = 0 to 600 V/cm, pulse duration = 100 μs, pulse repetition frequency = 1 Hz, number of pulses = 99.
For in vivo studies, electric pulses were delivered using a two-needle array electrode with a 5-mm gap (Catalog# 45-0168, BTX Harvard Apparatus). PEF parameters were: pulse voltage = 500 or 1150 V, pulse duration = 100 μs, pulse repetition frequency = 1 Hz, number of pulses = 99. The electrode was inserted to completely penetrate the tumor nodule. After one round of PEF treatment, electrodes were retrieved and inserted along the orthogonal direction to deliver 99 more pulses with the same parameters. PEF in vivo at 1150 V was denoted as PEF ablation throughout the manuscript, while low-intensity PEF in other experiments was specified with the exact pulse intensity or voltage.
RNA sequencing and data analysis
RNA sequencing was performed at Novogene Co. Ltd. Total RNA was extracted from treated cells and assessed for quantity and quality using RNA Nano 6000 assay kit for the Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA). mRNA was isolated using poly-T oligo-attached magnetic beads and fragmented with divalent cations at elevated temperature in a buffer for first-strand synthesis. First-strand cDNA was prepared using random hexamer primers and M-MuLV reverse transcriptase. Second-strand cDNA was synthesized using DNA polymerase I and dNTPs. The cDNAs were then end-processed, purified on an AMPure XP system (Beckman Coulter, Beverly, CA, USA), amplified through PCR reactions, and further purified with AMPure XP beads to generate the libraries. Qualified libraries were pooled and sequenced on an Illumina NovaSeq 6000 platform. After cleaning and quality validation, paired-end clean reads of the sequencing data were aligned to the reference genome using Hisat2 (v2.05). The number of reads mapped to each gene was counted using featureCounts (v1.5.0-p3).
Differential expression analysis was performed using the edegR R package (v4.4.2). P values were adjusted via Benjamini and Hochberg’s algorithm to control the false discovery rate (FDR). Differentially expressed genes (DEGs) with a p.adjusted <0.05 and a |fold-change|>1.5 was considered statistically significant. Gene ontology (GO) enrichment analysis of DEGs were conducted using clusterProfiler R package (v4.14.4). Gene set enrichment analysis (GSEA) was performed using GSEABase R package (v1.68.0).
Quantitative proteomics analysis
Quantitative proteomics was performed at Novogene Co. Ltd. Panc-1 cells (n = 2 or 3 independent experiments specified in corresponding figure legends) were lysed under ultrasonication with DB lysis buffer composed of 8 M urea and 100 mM triethyl ammonium bicarbonate (TEAB) at pH 8.5, and centrifuged at 12,000×g for 15 min at 4 °C. The supernatant was incubated with dithiothreitol (DTT) at 56 °C for 1 h, and alkylated with iodoacetamide for 1 h. Protein quality was verified using electrophoresis. The samples were digested with trypsin in the DB lysis buffer overnight, acidified with formic acid to pH 3, and centrifuged at 12,000×g for 5 min. The supernatant was eluted through a C18 desalting column and lyophilized.
The lyophilized powder was dissolved in 0.1% formic acid, and injected into a Vanquish Neo upgraded UHPLC system equipped with a C18 pre-column (5 mm × 300 μm, 5 μm, Thermo 174500) heated at 50 °C and a C18 analytic column (PepMapTM Neo UHPLC 150 μm × 15 cm, 2 μm, Thermo ES906). The mobile phases were: A (0.1% formic acid in 100% water) and B (0.1% formic acid in 80% acetonitrile). The elution conditions were: flow rate = 2.5 mL/min, gradient = 4%B (0 min), 5%B (0.2 min), 75%B (4 min), 65% B (5.8 min), 99%B (6.2 min). The eluent was analyzed on a Thermo Orbitrap Astral mass spectrometer operated in a data-independent acquisition mode. The full scan was performed with the following parameters: scanning range = 380 to 980 m/z, primary resolution = 240,000 (200 m/z), AGC = 500%, parent ion window size = 2 m/z, number of DIA window = 300, NCE = 25%. The second scan was then acquired with the following parameters: scanning range = 150 to 2000 m/z, sub-ion resolution Astral = 80,000, maximal injection time = 3 ms.
All the raw files were analyzed using the DIA-NN library search software (version 1.9.2). The library search parameters were: a mass tolerance of 10 ppm for precursor ions and 0.02 Da for fragment ions. Cysteine was modified by alkylation, methionine by oxidation, and N-terminal modifications include acetylation, loss of methionine, and loss of both methionine and acetylation. Two missed cleavage sites were allowed at most. The search results were further filtered by retaining only credible peptide spectrum matches with a confidence level above 99%. Differential expression of proteins was analyzed using EdgeR R package (v 4.2.2), and adjusted with Benjamini–Hochberg (BH) algorithm. Differentially expressed proteins with a p value <0.1 and |fold-change| >1.3 were considered statistically significant.
Lipidomics
The lipidomics study was conducted at LipidALL Technology. Panc-1 cells were treated with MOF-Fe and/or C20U4V for 48 h (n = 5 independent experiments), homogenized in a mixture of chloroform/methanol/H2O (v/v/v = 3/6/1), and incubated at 1500 RPM for 1 h at 4 °C. Lipids were twice extracted from the mixture using chloroform, and the combined organic phase was pooled and dried in a SpeedVac under OH mode. The aqueous phase and cell pellet were also dried in a SpeedVac vacuum concentrator under H2O mode for protein quantification. Lipidomics analyses were conducted using an ExionLC-AD-Sciex QTRAP 6500 PLUS as previously reported70. Oxidized phospholipids were separated by normal phase-HPLC using a TUP-HB silica column (150 mm × 2.1 mm, 3 μm) with the following conditions: mobile phase A = chloroform/methanol/ammonium hydroxide = 89.5/10/0.5, mobile phase B = chloroform/methanol/ammonium hydroxide/water = 55/39/0.5/5.5. Individual lipid species were quantified by referencing to spiked internal standards. For each phospholipid, the statistical significance of the change between two comparison groups was determined using multiple two-tailed unpaired t-tests without correction. Differentially expressed phospholipids with a p value <0.05 and |fold-change| >1.3 were considered statistically significant.
Quantitative RT-PCR and immunoblotting
Total RNA was extracted using a FastPure cell/tissue total RNA isolation kit (Catalog# RC101-01, Vazyme, Nanjing, China), and transcribed to cDNA using HiScript II RT supermix (Vazyme). Quantitative RT-PCR was conducted using ChamQ SYBR qPCR master mix (Vazyme) with primers listed in Supplementary Table 4 on a Bio-Rad CFX96 real-time PCR system (Bio-Rad). The relative mRNA expression was calculated using the 2−ΔΔCt algorithm and normalized to that of 18S endogenous control. The original Ct values are included in the Source Data file.
Cells were lysed in radio-immunoprecipitation analysis (RIPA) buffer supplemented with protease/phosphatase inhibitors. Protein lysate was denatured, fractionated on tris-glycine polyacrylamide gels, and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% non-fat milk, and sequentially incubated with primary antibodies (Supplementary Table S5), horseradish peroxidase-conjugated secondary antibodies, and enhanced chemiluminescence substrate. The blots were scanned using a Tanon 5200 Chem-Image system (Tanon Science & Technology, Shanghai, China) and analyzed using Image J (v1.54 g).
Generation of gene knockdown and overexpression cell lines
Gene knockdown plasmids were constructed by ligating pLKO.1 vector with non-targeting shRNA (NC) or shRNAs targeting human FTH1 (shFTH1) or VHL (shVHL, Supplementary Table 6). The coding sequences of human FTH1 were amplified by RT-PCR using the corresponding primers (Supplementary Table 7) and cloned into pHAGE_puro vector. The coding sequence for FTH1 restoration (Supplementary Table 8) was cloned into the pTSBX-CMV-EF1-BSD vector. The obtained recombinant lentiviral plasmids were verified with sequencing and co-transfected with pMD2.G envelope plasmid and psPAX2 packaging plasmid into 293 T cells using polyethylene imide (PEI; Polysciences, PA, USA). After 24 h, the transfection medium was changed to complete growth medium, which was collected after another 48 h, sterile-filtered, and used as lentivirus supernatant. Panc-1 cells were transduced with the lentivirus supernatant in the presence of polybrene (4 μg/mL, Yeasen Biotechnology, Shanghai, China) and selected with puromycin (2.5 μg/mL) or blasticidin (10 μg/mL). Efficiency or gene knockdown or overexpression was confirmed by immunoblotting.
FTH1 Ubiquitylation determined with immunoprecipitation
The coding sequences of human FTH1 and ubiquitin (Ub) were amplified by RT-PCR using the corresponding primers (Supplementary Table 7) and cloned into pRK-Flag and pCMV-HA vectors, respectively. The obtained plasmids were verified by sequencing and co-transfected into HEK-293T cells for 48 h. The cells were treated with C20U4V (10 μM) for 6 h in the presence of the proteosome inhibitor MG132 (10 μM). Protein lysates were extracted using the immunoprecipitation lysis buffer. Protein A + G magnetic beads were incubated with anti-Flag antibody at 4 °C for 4 h, washed, and mixed with the protein lysate at 4 °C overnight (P2179S, Beyotime Biotech, Shanghai, China). The beads were collected on a magnetic separation rack and washed. Target proteins were dissociated from the beads and denatured by heating in 2× SDS buffer at 95 °C for 5 min, and analyzed by immunoblotting.
FTH1 pull-down assay
Biotin-PEG3-arachidonic acid (BPAA) was first synthesized. Briefly, arachidonic acid (87.3 mg, 0.29 mmol) was first activated with HATU (181.7 mg, 0.48 mmol) and DIPEA (124 mg, 0.96 mmol) in dichloromethane (DCM, 3 mL) for 10 min at room temperature. Biotin-PEG3-NH2 (100 mg, 0.24 mmol) was then added, and the mixture was stirred overnight in the dark. The crude product was condensed in vacuo and purified via flash column chromatography on silica gel (petroleum ether then DCM) to yield a brown solid (yield = 90%). The chemical structure was confirmed by 1H-NMR, 13C NMR and ESI mass spectrum (Supplementary Figs. 74, 75, 110).
For the pull-down assay, Panc-1 cells were seeded and incubated with MOF-Fe and different concentrations of BPAA for 24 h. The cells were lysed, and incubated with streptavidin-coated magnetic beads for 4 h at 4 °C. After washing, the absorbed proteins were dissociated from the beads and denatured by heating in 2× SDS buffer at 95 °C for 5 min, and analyzed by immunoblotting.
Qualitative proteomics analysis
Qualitative proteomics was conducted at Novogene Co. Ltd. Proteins were eluted off the magnetic beads from the pull-down assay, and processed in a similar procedure as in the section “Quantitative proteomics analysis”. The lyophilized samples were dissolved in 0.1% formic acid, and injected into an EASY-nLCTM 1200 nano-upgraded UHPLE system with a homemade C18 Nano-Trap column (4.5 cm × 75 μm, 3 μm). The mobile phases were: A (0.1% formic acid in 100% water) and B (0.1% formic acid in 80% acetonitrile). The elution conditions were: flow rate = 800 nL/min, gradient = 2%B (0 min), 6%B (2.5 min), 6%B (5 min), 25%B (25 min), 40%B (27 min), 80%B (29 min), 80%B (30 min). The eluent was analyzed on a timsTOF Pro2 mass spectrometer with a Captive Spray ion source (ion spray voltage = 1.4 kV, drying gas temperature = 180 °C) in a data-independent acquisition mode. The full scan was performed using the following parameters: m/z = 100–1700, ramp time = 100 ms, lock duty cycle = 100%, scan window size = 25 Da, and number of mobility window = 2. The resulting spectra were searched using Spectronaut-Pulsar with the following parameters: mass tolerance for precursor ion = 10 ppm, mass tolerance = 0.02 Da, maximum missed cleavage sites = 2. Identified proteins were analyzed using VennDiagram and GO R packages (v.4.2.2).
Immunofluorescence (IF) and Immunohistochemical (IHC), and multiplex IF staining
For IF staining, cells seeded on glass coverslips were fixed with 4% paraformaldehyde, and incubated with primary antibodies (Supplementary Table S5) and fluorescence-labeled secondary antibodies. Cell nuclei were counterstained with Hoechst 33342 or DAPI. Fluorescence images were acquired under a confocal fluorescence microscope, and analyzed with ImageJ. For IHC staining, formalin-fixed paraffin-embedded (FFPE) tumor sections were deparaffinated, hydrated, and heated in antigen-retrieval buffer (10 mM sodium citrate with 0.1% Tween-20, pH 6.0) at 95 °C for 20 min. The tumor sections were blocked with 5% bovine serum albumin (BSA), incubated with primary antibodies (Supplementary Table S5), anti-rabbit poly(horseradish peroxidase) (Catalog# SV0002, Boster Bio, Wuhan, China), and visualized via 3,3’-diaminobenzidine (DAB) reaction. Cell nuclei were counterstained with hematoxylin. Images were captured under a bright-field microscope, and analyzed using ImageJ. For multiplex IF, rehydrated FFPE sections were heated in an antigen-retrieval buffer (EDTA, pH 9.0) in a pressured heater, and blocked with 3% H2O2 and 10% goat serum. Each target was stained by sequential incubation with the respective primary antibody, goat anti-rabbit IgG H&L (HRP) (Catalog# ab205718, Abcam), and iFluor® tyramide (Supplementary Table S5). Cell nuclei were counterstained with DAPI. Images were scanned using a fluorescence scanner (Pannoramics SCAN II, 3D HISTECH, Budapest, Hungary) and analyzed.
Flow cytometry
Tumors were collected, weighed, and digested in RPMI-1640 medium supplemented with collagenase IV (1 mg/mL), hyaluronidase (0.4 mg/mL), DNase I (0.1 mg/mL), and BSA (10 mg/mL) in a 37 °C water bath for 30 min with constant shaking. The cell suspension was filtered through a 40-μm-mesh, treated with red blood cell lysis buffer, and resuspended in phosphate-buffered saline (PBS) supplemented with 2% fetal bovine serum (FBS) and 1 mM EDTA. After staining with Zombie NIR fixable viability kit (Catalog#423106, BioLegend, San Diego, CA, USA), the cells were fixed and permeabilized by adding methanol pre-chilled to −20 °C during vortexing and incubating on ice for 10 min. After washing, the cells were stained with corresponding antibodies (Supplementary Table S9), and analyzed on Beckman DxFLEX cytometer (Beckman Coulter, Brea, CA, USA). Data were processed using FlowJo Software V10 (BD Biosciences, San Jose, CA, USA).
Biodistribution of MOF-Fe and M-C20U4V
Tumors and major organs were collected 24 h after the intratumoral injection of MOF-Fe. After weighing, the samples were digested with nitric acid and dried. Iron content was measured using atomic absorption spectroscopy (Varian AA-240FS, Agilent, Wuhan, China). DiR-labeled M-C20U4V micelles were prepared in the same manner as M-C20U4V except that DiR (25 mg/mL in DMSO, 10 μL) was also added to the acetone solution. Micelles were intravenously injected into tumor-bearing mice 4 h before PEF ablation. At 48 h post-injection, major organs and tumors were collected, weighed, and measured for their fluorescence intensity on a fluorescence scanner (Pearl Trilogy, LI-COR, Shanghai, China).
Statistics and reproducibility
The details of data presentation, sample size, statistical analysis method, and significance of differences are specified in figure legends. Sample size was determined with reference to the experiment designs reported in similar previous studies. Randomization was adopted for all experiments. Researchers were blinded during data analyses since the samples were code-named. Experiments were performed in a non-blinded manner because the experiments involved multiple treatments that required clear labeling for operation and data collection. No data were excluded from the analysis. All experiments were independently repeated at least three times with consistent results. Significant difference was assessed using one-tailed or two-tailed unpaired Student’s t-test, or one-tailed ratio-paired t-test for the comparison of two data sets, and one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison for three or more data sets. Log-rank test was used for Kaplan–Meier survival analysis. Synergy score was calculated using Synergistic effects were determined using SynergyFinder Plus71. A p value less than 0.05 was considered statistically significant. Data were analyzed using Prism GraphPad 9 (GraphPad Software, San Diego, CA, USA).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the data supporting the findings of this study are presented within the article and its supplementary information. Source Data are provided with this paper. The raw RNA sequencing data are deposited in the Genome Sequence Archive in the National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences, under the accession numbers HRA009056 and HRA011295. The mass spectrometry proteomics data have been deposited at the ProteomeXchange Consortium via the iProX partner repository72 under the accession numbers PXD063472, PXD068014 [https://www.iprox.cn/page/project.html?id=IPX0013269000], and PXD068751. The complete, quantitative lipid concentration dataset for lipidomics is deposited in the Open Archive for Miscellaneous Data at the database, China National Center for Bioinformation, under the accession number OMIX013110. All deposited data are publicly accessible. Source data are provided with this paper.
References
Moir, J., White, S. A., French, J. J., Littler, P. & Manas, D. M. Systematic review of irreversible electroporation in the treatment of advanced pancreatic cancer. Eur. J. Surg. Oncol. 40, 1598–1604 (2014).
Martin, R. C. 2nd, McFarland, K., Ellis, S. & Velanovich, V. Irreversible electroporation in locally advanced pancreatic cancer: potential improved overall survival. Ann. Surg. Oncol. 20, S443–449 (2013).
Ringel-Scaia, V. M. et al. High-frequency irreversible electroporation is an effective tumor ablation strategy that induces immunologic cell death and promotes systemic anti-tumor immunity. EBioMedicine 44, 112–125 (2019).
Golberg, A., Bruinsma, B. G., Uygun, B. E. & Yarmush, M. L. Tissue heterogeneity in structure and conductivity contribute to cell survival during irreversible electroporation ablation by “electric field sinks. Sci. Rep. 5, 8485 (2015).
Edd, J. F. & Davalos, R. V. Mathematical modeling of irreversible electroporation for treatment planning. Technol. Cancer Res. Treat. 6, 275–286 (2007).
Lin, M. et al. Irreversible electroporation plus allogenic Vγ9Vδ2 T cells enhances antitumor effect for locally advanced pancreatic cancer patients. Signal Transduct. Target Ther. 5, 215 (2020).
Yu, B., Zhang, W., Kwak, K., Choi, H. & Kim, D. H. Electric pulse responsive magnetic nanoclusters loaded with indoleamine 2,3-dioxygenase inhibitor for synergistic immuno-ablation cancer therapy. ACS Appl Mater. Interfaces 12, 54415–54425 (2020).
Zhao, J. et al. Irreversible electroporation reverses resistance to immune checkpoint blockade in pancreatic cancer. Nat. Commun. 10, 899 (2019).
Han, J.-H. et al. An electro-ferroptotic nanoammunition enables image-guided, spatiotemporally controlled cancer ferroptosis induction via irreversible electroporation. Chem. Eng. J. 487, 150366 (2024).
Long, X. et al. Simultaneous delivery of dual inhibitors of DNA damage repair sensitizes pancreatic cancer response to irreversible electroporation. ACS Nano 17, 12915–12932 (2023).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Stockwell, B. R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 185, 2401–2421 (2022).
Friedmann Angeli, J. P., Krysko, D. V. & Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 19, 405–414 (2019).
Seiler, A. et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 8, 237–248 (2008).
Liu, K. et al. Ferroptosis: the entanglement between traditional drugs and nanodrugs in tumor therapy. Adv. Health Mater. 12, e2203085 (2023).
Ru, Q. et al. Iron homeostasis and ferroptosis in human diseases: mechanisms and therapeutic prospects. Signal Transduct. Target Ther. 9, 271 (2024).
Xu, W. et al. Chemical design of magnetic nanomaterials for imaging and ferroptosis-based cancer therapy. Chem. Rev. 125, 1897–1961 (2025).
Caneque, T. et al. Activation of lysosomal iron triggers ferroptosis in cancer. Nature 642, 492–500 (2025).
Zelepukin, I. V. et al. Flash drug release from nanoparticles accumulated in the targeted blood vessels facilitates the tumour treatment. Nat. Commun. 13, 6910 (2022).
Anderson, C. P., Shen, M., Eisenstein, R. S. & Leibold, E. A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 1823, 1468–1483 (2012).
Plays, M., Muller, S. & Rodriguez, R. Chemistry and biology of ferritin. Metallomics 13, mfab021 (2021).
Liu, J. et al. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem. Biol. 27, 420–435 (2020).
Wang, J. et al. Iron-siRNA nanohybrids for enhanced chemodynamic therapy via ferritin heavy chain downregulation. Angew. Chem. Int Ed. Engl. 62, e202302255 (2023).
Li, C. et al. Core role of hydrophobic core of polymeric nanomicelle in endosomal escape of siRNA. Nano Lett. 21, 3680–3689 (2021).
Bekes, M., Langley, D. R. & Crews, C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 21, 181–200 (2022).
Zanzoni, S. et al. Unsaturated long-chain fatty acids are preferred ferritin ligands that enhance iron biomineralization. Chemistry 23, 9879–9887 (2017).
Bu, W. et al. Ferritin couples iron and fatty acid metabolism. FASEB J. 26, 2394–2400 (2012).
Chen, Y. et al. Small-molecule ferritin degrader as a pyroptosis inducer. J. Am. Chem. Soc. 145, 9815–9824 (2023).
Magtanong, L. et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem. Biol. 26, 420–432 e429 (2019).
Yang, W. S. et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 113, E4966–4975 (2016).
Gabriel, B. & Teissie, J. Generation of reactive-oxygen species induced by electropermeabilization of Chinese hamster ovary cells and their consequence on cell viability. Eur. J. Biochem. 223, 25–33 (1994).
Mercadal, B. et al. Dynamics of cell death after conventional IRE and H-FIRE treatments. Ann. Biomed. Eng. 48, 1451–1462 (2020).
Chen, X. et al. International consensus guidelines for the definition, detection, and interpretation of autophagy-dependent ferroptosis. Autophagy 20, 1213–1246 (2024).
Nakamura, T. et al. Phase separation of FSP1 promotes ferroptosis. Nature 619, 371–377 (2023).
Ye, C. F. et al. Co-inhibition of RAGE and TLR4 sensitizes pancreatic cancer to irreversible electroporation in mice by disrupting autophagy. Acta Pharmacol. Sin. 46, 1757–1771 (2025).
Zhou, N. et al. FerrDb V2: update of the manually curated database of ferroptosis regulators and ferroptosis-disease associations. Nucleic Acids Res. 51, D571–D582 (2023).
Mahmoudi, M., Sant, S., Wang, B., Laurent, S. & Sen, T. Superparamagnetic iron oxide nanoparticles (SPIONs): development, surface modification and applications in chemotherapy. Adv. Drug Deliv. Rev. 63, 24–46 (2011).
Shen, Z. et al. Fenton-reaction-acceleratable magnetic nanoparticles for ferroptosis therapy of orthotopic brain tumors. ACS Nano 12, 11355–11365 (2018).
Yan, T. et al. PCK2 induces gefitinib resistance by suppresses ferroptosis in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 723, 150200 (2024).
Tang, Z., Kang, B., Li, C., Chen, T. & Zhang, Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 47, W556–W560 (2019).
Bock, F. J. & Tait, S. W. G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 21, 85–100 (2020).
Peng, H. et al. Local release of TGF-beta inhibitor modulates tumor-associated neutrophils and enhances pancreatic cancer response to combined irreversible electroporation and immunotherapy. Adv. Sci. 9, e2105240 (2022).
Qiu, W. & Su, G. H. Development of orthotopic pancreatic tumor mouse models. Methods Mol Biol. 980, 215–223 (2013).
Ruarus, A. H. et al. Percutaneous irreversible electroporation in locally advanced and recurrent pancreatic cancer (PANFIRE-2): a multicenter, prospective, single-arm, phase II study. Radiology 294, 212–220 (2020).
Woeste, M. R. et al. Optimizing patient selection for irreversible electroporation of locally advanced pancreatic cancer: analyses of survival. Front. Oncol. 11, 817220 (2021).
Yang, J. et al. Irreversible electroporation ablation overcomes tumor-associated immunosuppression to improve the efficacy of DC vaccination in a mice model of pancreatic cancer. Oncoimmunology 10, 1875638 (2021).
Kulbay, M., Paimboeuf, A., Ozdemir, D. & Bernier, J. Review of cancer cell resistance mechanisms to apoptosis and actual targeted therapies. J. Cell Biochem. 123, 1736–1761 (2022).
Zheng, H. et al. Targeted activation of ferroptosis in colorectal cancer via LGR4 targeting overcomes acquired drug resistance. Nat. Cancer 5, 572–589 (2024).
Batista Napotnik, T., Polajzer, T. & Miklavcic, D. Cell death due to electroporation - a review. Bioelectrochemistry 141, 107871 (2021).
Schmidlin, C. J., Shakya, A., Dodson, M., Chapman, E. & Zhang, D. D. The intricacies of NRF2 regulation in cancer. Semin. Cancer Biol. 76, 110–119 (2021).
Philips, P., Li, Y., Li, S., St Hill, C. R. & Martin, R. C. Efficacy of irreversible electroporation in human pancreatic adenocarcinoma: advanced murine model. Mol. Ther. Methods Clin. Dev. 2, 15001 (2015).
Roemhild, K. et al. Iron metabolism: pathophysiology and pharmacology. Trends Pharm. Sci. 42, 640–656 (2021).
Muller, S. et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. Nat. Chem. 12, 929–938 (2020).
Gao, J., Luo, T. & Wang, J. Gene interfered-ferroptosis therapy for cancers. Nat. Commun. 12, 5311 (2021).
Trujillo-Alonso, V. et al. FDA-approved ferumoxytol displays anti-leukaemia efficacy against cells with low ferroportin levels. Nat. Nanotechnol. 14, 616–622 (2019).
Mai, T. T. et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 9, 1025–1033 (2017).
Basuli, D. et al. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene 36, 4089–4099 (2017).
Wang, Y. Q. et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front. Aging Neurosci. 8, 308 (2016).
Hohnholt, M. C., Geppert, M. & Dringen, R. Treatment with iron oxide nanoparticles induces ferritin synthesis but not oxidative stress in oligodendroglial cells. Acta Biomater. 7, 3946–3954 (2011).
Tirinato, L. et al. Lipid droplets and ferritin heavy chain: a devilish liaison in human cancer cell radioresistance. Elife 10, e72943 (2021).
Wicks, E. E. & Semenza, G. L. Hypoxia-inducible factors: cancer progression and clinical translation. J. Clin. Invest. 132, e159839 (2022).
Shesh, B. P. & Connor, J. R. A novel view of ferritin in cancer. Biochim. Biophys. Acta Rev. Cancer 1878, 188917 (2023).
Yang, N. D. et al. Artesunate induces cell death in human cancer cells via enhancing lysosomal function and lysosomal degradation of ferritin. J. Biol. Chem. 289, 33425–33441 (2014).
Kong, N. et al. Baicalin induces ferroptosis in bladder cancer cells by downregulating FTH1. Acta Pharm. Sin. B 11, 4045–4054 (2021).
Liu, Y. et al. Dual ferroptosis induction in N2-TANs and TNBC cells via FTH1 targeting: a therapeutic strategy for triple-negative breast cancer. Cell Rep. Med. 6, 101915 (2025).
Drijvers, J. M. et al. Pharmacologic screening identifies metabolic vulnerabilities of CD8+ T cells. Cancer Immunol. Res. 9, 184–199 (2021).
Liao, P. et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell 40, 365–378.e366 (2022).
Guo, W., Shi, P., Feng, M. & Li, S. Performance and mechanism of the modified group regulated the MIL-101(Fe) type Fenton-like catalysts. ACS Omega 9, 32864–32872 (2024).
Grossman, J. E. et al. Organoid sensitivity correlates with therapeutic response in patients with pancreatic cancer. Clin. Cancer Res. 28, 708–718 (2022).
Lam, S. M. et al. A multi-omics investigation of the composition and function of extracellular vesicles along the temporal trajectory of COVID-19. Nat. Metab. 3, 909–922 (2021).
Zheng, S. et al. SynergyFinder Plus: toward better interpretation and annotation of drug combination screening datasets. Genom. Proteom. Bioinform. 20, 587–596 (2022).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–D1217 (2019).
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (82272838, 82073400, J.Z.), Key Research and Development Program of Hubei (2022BCA012, T.Y.), the Department of Science and Technology of Hubei Province (2024AFB572, X.L.), and Huazhong University of Science and Technology Innovation Institute (2024JYCXJJ009, L.L.). Imaging studies were performed at the Innovation and Research Center, School of Basic Medicine, Huazhong University and Technology. Animal studies were conducted in the Laboratory Animal Center at Huazhong University of Science and Technology with support from the facility staff.
Author information
Authors and Affiliations
Contributions
L.L.: conceptualization, data curation, formal analysis, writing—review and editing; S.Su: data curation and formal analysis; Z.W.: data curation; S.Sun: formal analysis, writing—review and editing; Y.W.: formal analysis and writing—review and editing; H.X.: data curation and formal analysis; X.L.: conceptualization, funding acquisition, and writing—review and editing; T.Y.: funding acquisition and writing—review and editing; J.Z.: conceptualization, data curation, formal analysis, funding acquisition, and writing—original draft.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Rafael V. Davalos, who co-reviewed with Kailee David, Sebastian Müller, Wooram Park, Rebekah White and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, L., Su, S., Wang, Z. et al. Disruption of iron homeostasis sensitizes pancreatic cancer to irreversible electroporation. Nat Commun 17, 1866 (2026). https://doi.org/10.1038/s41467-026-68585-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68585-z
