
Abstract
Polygenic scores (PGS) quantify genetic contributions to complex traits, yet existing single- and multi-ancestry methods lack multi-dimensional evaluation within a unified framework. Here, we benchmarked 13 state-of-the-art PGS methods across 36 traits in UK Biobank European and African samples. The prediction performance, computational efficiency, the number of variants, and the impact of different linkage disequilibrium (LD) reference sizes were thoroughly assessed for each method. Results of single-ancestry methods demonstrate that LDpred2 has superior performance across a broad spectrum of complex traits in terms of accuracy and computational efficiency; however, other methods remain valuable for specific traits. For multi-ancestry methods, PRS-CSx and X-Wing have comparable performance, whereas LDpred2-multi outperforms both. Notably, we find that increasing the panel size of the LD reference significantly elevates PGS performance for sample sizes below 1,000, and it reaches a plateau when it exceeds 5,000 samples. Furthermore, implementing PGS calculation methods requires considerable technical effort and resource allocation. To support easy use of these PGS methods, we developed a user-friendly online computing platform, PGS-hub, that integrates all evaluated methods and is pre-configured with ancestry-stratified LD panels. This resource enables a scalable and harmonized PGS computation platform for the PGS community.
Similar content being viewed by others

Introduction
Genome-wide association studies (GWAS) have revolutionized our understanding of the genetic underpinnings of complex traits and diseases. Over the past decades, GWAS have identified thousands of genetic variants associated with a wide array of traits, ranging from common diseases such as coronary artery disease and type 2 diabetes to quantitative traits such as height and body mass index1,2,3,4,5,6,7. These discoveries have significantly advanced our ability to dissect the genetic architecture of traits that do not follow Mendelian inheritance8, shedding light on the polygenic nature of most complex diseases.
Despite these advances, the challenge remains that each genetic variant typically contributes only modestly to the overall genetic risk1,4,5,7. This has led to the widespread adoption of polygenic scores (PGS), which aggregate the effects of numerous genetic variants across the genome into a single risk metric9,10,11,12. By summing the estimated effect sizes of genetic variants, weighted by the number of risk alleles, PGS provides a powerful tool for quantifying an individual’s genetic predisposition to complex diseases and traits13,14.
The development of PGS methods has rapidly accelerated, resulting in a proliferation of sophisticated computational approaches designed to enhance predictive accuracy15,16. These methods leverage a variety of statistical frameworks, including, but not limited to, clumping and thresholding algorithms, Bayesian hierarchical models, and continuous shrinkage priors, to account for the complex genetic architecture underlying polygenic traits. Approaches such as P + T17,18,19,20,21,22,23, LDpred24,25, and PRS-CS26 have become prominent due to their distinct strategies for handling linkage disequilibrium (LD) and optimizing SNP effect size estimation, but a broader spectrum of methodologies—including Lassosum27,28, SDPR29, SBayesR30, and other emerging methods31,32—has also gained recognition for their flexibility and performance across diverse genetic architectures and study designs. In addition, recent efforts in multi-ancestry PGS methods (e.g., PRS-CSx and X-wing) couple genetic effects across populations via shared shrinkage priors to improve the utility of PGS in diverse populations33,34. The heterogeneity in computational strategies and predictive power across populations underscores the need for systematic benchmarking to assess these methods critically. By comparing their relative strengths and weaknesses across diverse contexts, we can better understand how to optimize polygenic prediction in both ancestry-specific and trans-ethnic cohorts, which has become a high priority for the research community.
Indeed, several independent comparisons of PGS methods already exist35,36,37,38,39,40,41,42. For instance, Ni et al. evaluated 10 PGS methods for psychiatric disorders in European individuals39. Several studies expanded comparisons across diverse ancestries35,37,38,40, traits35,36,37,38,40, biobanks37, and inclusion of machine-learning prediction methods40. However, many studies mainly focus on comparisons of traditional predictive accuracy metrics and fail to explore the practical performance impacts of different LD panel sizes, SNP numbers, and trait architectures. Furthermore, although limited studies have begun to establish PGS computing services41,42, a high-performance publicly available web-based platform that integrates various methods into a unified framework could reduce the burden on users of setting up and optimizing individual computational pipelines, thereby enhancing accessibility and reproducibility in PGS research.
In this study, we introduce PGS-hub, a web-based platform that provides an automated pipeline supporting a diverse range of PRS methods under uniform data-processing protocols, thereby minimizing methodological variability and facilitating direct and rigorous method comparisons. Moreover, we utilize PGS-hub to systematically evaluate 13 PGS methods (including Ensemble strategies for both single- and multi-ancestry methods) across 36 complex traits, leveraging data from both European and African populations in the UK Biobank. We thoroughly investigate the performance of different methods across predictive metrics, LD reference panel sizes, SNP numbers, trait architectures, and computational costs. We further consider ensemble strategies (both for single- and multiple-ancestry methods) and the multi-source independently optimized PGS (LDpred2-multi) for cross-ancestry evaluation. Overall, our work offers insights that address an important gap in the field and enhance the utility of PGS in diverse populations.
Results
Overview of the PGS-hub platform
To enable the convenient and efficient computation of multiple PGS methods simultaneously, we developed a PGS calculation platform called PGS-hub (Fig. 1). This platform includes a front-end web interface and a back-end parallelized computing infrastructure. PGS-hub currently integrates ten popular PGS methods, including eight single-ancestry methods (P + T21, Lassosum228, LDpred225, MegaPRS (non-annotation version)31, PRS-CS26, SBayesR30, SBLUP32, SDPR29), and two multi-ancestry methods (PRS-CSx33 and X-Wing34) (Supplementary Table 1).

The PGS-hub platform is designed with a web front-end and a back-end. Users use the browser function to upload GWAS summary statistics and select the specific ancestry of 1000 Genome LD references, or upload their self-defined LD references in a pgen file. Users next select the particular methods and parameters for submission. The submissions are processed in a WDL workflow and executed by the Cromwell execution engine. A Slurm server is used to design and allocate tasks. Results of SNP effect sizes for the given traits, using selected methods and parameters, are packed into compressed folders (.zip) for users to download. EUR European, AFR African, EAS East Asian, SAS South Asian.
To enhance user experience, PGS-hub offers several user-friendly features: (1) automatic conversion of user inputs into the appropriate file formats for different PGS methods; (2) support for custom parameter settings tailored to each PGS method; (3) pre-computed and integrated LD reference panels for four ancestries (European, African, East Asian, and South Asian) based on data from the 1000 Genomes Project; and (4) support for uploading and utilizing user-defined LD reference panels. Thus, users need to upload GWAS summary statistics once, select multiple PGS methods, and run computations on the PGS-hub platform, ultimately obtaining SNP weights adjusted by the selected PGS methods.
To ensure efficient parallel computing and easy migration across diverse computing environments, we implemented these pipelines using the workflow description language (WDL), virtualized various PGS methods with Docker, and used Slurm and Cromwell as scheduling and execution engines to manage computing tasks. This implementation also supports local deployment on various HPC clusters and portability across different computing environments. To facilitate downstream PGS computation and comparison, we established equivalent processing pipelines to standardize outputs of different PGS methods, creating a unified downstream pipeline for seamless integration and comparison of results. This design framework also allows for the flexible incorporation of new PGS methods in the future.
Overview of the benchmark analysis
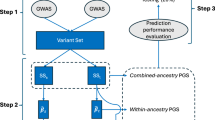
For our benchmark analysis workflow (Fig. 2), we compared 13 PGS methods using GWAS summary statistics from 36 complex traits—excluding samples from UK Biobank participants—including eight methods for single-ancestry modeling (P + T, Lassosum2, LDpred2, MegaPRS (non-annotation version), PRS-CS, SBayesR, SBLUP, SDPR), three methods for cross-ancestry modeling (PRS-CSx, X-Wing, LDpred2-multi (Combined LDpred2 results from different ancestries, Methods)), and one Ensemble strategy for combination of all the single- or multiple- ancestry methods respectively (Methods).

Prediction evaluation statistics (36 complex traits) based on single- and multi-ancestry PGS methods were derived from European and African-sourced GWAS summary statistics and 1000 Genomes Project LD references (European and African samples). Among them, 6 complex traits were selected to build polygenic scores derived from different sample sizes of LD reference panels, ranging from 50 to 20,000, from the 1000 Genomes Project whole-genome sequencing dataset and UK Biobank genomic imputation dataset (INFO >0.8). Nine single-ancestry PGS methods (P + T, LDpred2, Lassosum2, PRS-CS, SDPR, SBLUP, MegaPRS (non-annotation version), SBayesR, and Ensemble method) and four multi-ancestry PGS methods (PRS-CSx, X-Wing, LDpred2-multi, and Ensemble method) were benchmarked in UK Biobank European and African individuals. Individuals were equally divided into the training dataset (188,745 Europeans and 4107 Africans) and the testing dataset (188,745 Europeans and 4107 Africans), where the training dataset was used for parameter optimization and the testing dataset was used for evaluating the performance of PGS methods. The benchmark was evaluated based on predictive accuracy, risk stratification value, performance across different LD sample sizes and SNP numbers, and computational cost (CPU hours and maximum memory used).
We employed European GWAS summary statistics from 19 complex diseases and 17 quantitative traits, which covered 12 disease categories defined by the International Classification of Diseases 10 (ICD-10) (Methods), and six African GWAS summary statistics from three complex diseases and three quantitative traits. To obtain an unbiased estimate of evaluation performance, we manually curated the GWAS summaries so that the GWAS derivation samples didn’t overlap with UK Biobank participants. Moreover, the same number of high-quality SNPs were chosen from the overlap between the 1000 Genomes Project (European or African samples) and Hapmap3 SNPs to create LD references, which were then transformed into formats specific to each PGS method to ensure consistent evaluation (Methods). Three complex diseases and three quantitative traits were selected to build polygenic scores derived from different sample sizes of LD reference panels ranging from 50 to 20,000 from the 1000 Genome WGS dataset and UK Biobank genomic imputation dataset (INFO >0.8).
PGS based on eight single-ancestry methods, two multi-ancestry methods, and one Ensemble strategy (each ensembled from single or multi-methods) were validated and evaluated in UK Biobank cohorts. UK Biobank 377,490 unrelated European individuals were randomly partitioned into two equal parts (1 : 1 ratio): training dataset (188,745 individuals) and testing dataset (188,745 individuals), and similarly UK Biobank 8214 African individuals were randomly divided into training dataset (4107 individuals) and testing dataset (4107 individuals), which training dataset was used for parameters optimization and testing dataset was used for evaluation of the performance of PGS methods. The benchmark analysis was conducted to assess predictive accuracy (AUC, odds ratio), risk stratification value, computational cost, and the performance effect of different LD panel sizes and SNPs quantity (Fig. 2).
Comparison and evaluation of single-ancestry PGS methods integrated on the PGS-hub platform in the UK Biobank
We compared and evaluated the performance properties of single-ancestry PGS methods for 36 complex traits in multiple ways. For 19 complex diseases, predictive accuracy was evaluated using the area under the receiver operating characteristic curve (AUC) and odds ratio per standard deviation (OR/SD) of PGS, while predictive accuracy in 17 quantitative traits was measured by Pearson R2 (squared Pearson correlation between two sets of residuals: one from a linear regression model of the specific PGS on covariates (age, sex, genotyping array, and top ten ancestry principal components), and the other from a linear regression model of the quantitative trait value on covariates (age and sex)) and incremental R2 (change in the R2 of a regression model when adding the PGS as a predictor to the model) (Methods).
Results revealed that different PGS methods performed differently across complex traits (Figs. 3 and 4, Supplementary Fig. 2, and Supplementary Tables 5–7, 9–11). We first compared and selected the best PGS method for each trait. The methods that produced the highest OR/SD in each disease and the highest Pearson R2 in each quantitative trait were selected as the best methods. As shown in Fig. 3a, the performance of the best method in each disease was impressive, ranging from 1.236 of LDpred2 for stroke (OR/SD, 95% CI 1.19–1.29) to 2.315 of LDpred2 for schizophrenia (OR/SD, 95% CI 2.10–2.56), with a median of 1.692 (Fig. 3a and Supplementary Table 7). LDpred2 outperformed other methods in 14 out of 19 diseases. SDPR ranked after LDpred2, and performed best in 3 out of 19 diseases (Fig. 3b). In general, Lassosum2, PRS-CS, and SDPR had robust and stable performance of accuracy second only to LDpred2, while SBayesR, SBLUP, and MegaPRS (non-annotation version) had poor accuracy compared to average performance (P + T) in complex diseases (Fig. 4a, c, Supplementary Tables 5–7, and Supplementary Fig. 1). Predictive accuracy of best PGS methods in quantitative traits was more variable, ranging from 0.010 of LDpred2 for estimated glomerular filtration rate (Pearson R2, 95% CI 0.008–0.012) to 0.285 of PRS-CS for height (Pearson R2, 95% CI 0.281–0.287), with a median of 0.060 (Fig. 3c and Supplementary Table 10). LDpred2 outperformed other methods in 9 out of 17 quantitative traits. Lassosum2 and PRS-CS outperformed other methods in 3 out of 17 quantitative traits, respectively. SDPR and SBayesR performed best for the remaining two traits (Fig. 3c, d). Similarly, LDpred2, Lassosum2, PRS-CS, and SDPR had overall better performance than other methods in nearly all the quantitative traits, while SBayesR and MegaPRS (non-annotation version) showed relatively worse performance, although SBayesR displayed the best performance in Docosahexaenoic acid trait (Fig. 4b, d and Supplementary Tables 9–11).

a The method that produced the highest odds ratio per standard deviation (OR/SD) in each disease was chosen as the best method. The median OR/SD of PGS with 95% confidence interval for 19 complex diseases was assessed in logistic regression models adjusted for age, sex, genotyping array, and the first ten principal components of ancestry in the testing group of N = 188,745 independent UK Biobank unrelated European individuals. b PGS methods’ performance of frequency as the best method across diseases. Bars represent the times chosen as the best method across 19 diseases. c The method that produced the highest Pearson R2 in each quantitative trait was chosen as the best method. The median Pearson R2 with 95% confidence interval was calculated using Pearson’s correlation coefficient on the residuals from a linear regression model of the specific PGS with age, sex, array, and the first ten principal components, with a linear regression model of the particular quantitative trait outcome with age and sex. Evaluations were conducted in N = 188,745 independent UK Biobank unrelated European testing individuals. d PGS methods’ performance of frequency as the best method across quantitative traits. Bars represent the times chosen as the best method across quantitative traits.

a Box plot of each PGS method’s AUC performance across diseases evaluated in the UK Biobank European testing group (N = 188,745). In each box plot, the central horizontal line represents the mean of each PGS method’s AUC performance across diseases. The box spans the interquartile range (IQR, 25th and 75th percentiles), and whiskers extend to the minimum and maximum values within 1.5 × IQR from the lower and upper quartiles. Data points outside this range are considered outliers. b Box plot of each PGS method’s Pearson R2 performance across quantitative traits evaluated in the UK Biobank European testing group (N = 188,745). The horizontal line inside the box represents the mean of each PGS method’s Pearson R2 performance across quantitative traits, with box plot elements defined as in (a). c Barplot of each PGS method’s AUC performance across diseases. Each colored bar represents a specific method. X-axis: PCa prostate cancer, BCa breast cancer, CAD coronary artery disease, PD Parkinson’s disease, SCZ schizophrenia, POAG primary open-angle glaucoma, HF heart failure, LCa lung cancer, COPD chronic obstructive pulmonary disease, T2D type 2 diabetes, PsO psoriasis, CD Crohn’s disease, SLE systemic lupus erythematosus, RA rheumatoid arthritis, VTE venous thromboembolism, BP bipolar disorder. d Barplot of each PGS method’s Pearson R2 performance across quantitative traits. Each colored bar represents a specific method. X-axis: HT height, HDL high-density lipoprotein cholesterol, LDL low-density lipoprotein cholesterol, TC total cholesterol, ApoB apolipoprotein B, RC remnant cholesterol, BMI body mass index, n-3 omega-3 fatty acids, ApoA1 apolipoprotein A1, HbA1c glycated hemoglobin HbA1c, PG phosphoglycerides, DHA docosahexaenoic acid, n-6 omega-6 fatty acids, SM sphingomyelins, Ca calcium, eGFR estimated glomerular filtration rate (creatinine based).
We further used formal statistical comparisons, including bootstrapping with 1000 resamples and pairwise generalized least squares (GLS) tests (Methods), to assess whether differences in performance of various methods were significant across non-independent traits. For each trait, we demonstrate that the best method we identified was statistically significantly superior to the second-best method, with a P value <0.01 for all traits (Supplementary Table 13). We further used pairwise GLS tests to compare the overall performance of different methods, accounting for the non-independence among the 36 traits. Results showed LDpred2 exhibited significantly higher performance than all other methods, except for Lassosum2 and PRS-CS (Supplementary Table 14 and Supplementary Fig. 2). We also examined the performance of the auto algorithm with the grid algorithm we used for LDpred2 and PRS-CS, demonstrating that the grid outperforms the auto algorithm for most traits (Supplementary Table 15).
Moreover, we characterized the performance of the methods across different trait architectures, such as heritability and polygenicity. We found a general trend where higher heritability is associated with higher prediction performance, with LDpred2, SDPR, and PRS-CS consistently emerging as top-performing methods for these high-heritability traits (For example, LDSC estimated Crohn’s disease heritability = 0.421, best OR/SD = 2.052 by LDpred2; LDSC estimated height heritability = 0.122, best Pearson R2 = 0.285 by PRS-CS; Supplementary Table 16). Notably, when examining traits with high heritability but concurrently high polygenicity, such as Rheumatoid arthritis (LDSC estimated heritability 0.513 and polygenicity 0.35, respectively), LDpred2 maintained the highest performance OR/SD = 1.362, although high polygenicity attenuated predictive performance for all methods (Supplementary Table 16).
For African individuals, we further conducted benchmark analyses of eight single-ancestry methods using GWAS summary statistics from African-ancestry studies (independent of the UK Biobank), focusing on three diseases and three quantitative traits (Methods) due to the limited availability of African-specific GWAS data. As shown in Supplementary Fig. 3, the efficacy of all single-ancestry methods was significantly lower in African than in European populations, with relatively comparable performance across most methods. For disease traits, LDpred2 and SDPR performed better, as indicated by higher AUC and OR/SD compared to other methods. For example, in Coronary artery disease, LDpred2 achieved the highest OR/SD of 1.115; for Type 2 diabetes, SDPR achieved the superior OR/SD of 1.308. For quantitative traits, the basic method P + T showed unexpectedly favorable performance. For example, P + T achieved the highest Pearson R2 of 0.032 in HDL among all evaluated methods (Supplementary Tables 17 and 18).
Ensemble method boosts predictive power
Next, we generated an Ensemble method by linearly regressing the best scores derived from each method for each trait (Methods), and compared it with the eight single-ancestry methods to evaluate their predictive accuracy across traits. Results showed that the Ensemble method outperformed the existing eight single-ancestry methods in all 36 complex diseases and quantitative traits (Fig. 4a–d, Supplementary Tables 8, 12, and Supplementary Fig. 1). For 19 complex diseases, the total nine methods achieved relatively comparable AUC, with significantly higher in prostate cancer and lower in bipolar disease, ranging from 0.547 (SBayesR for bipolar disease) to 0.898 (LDpred2 for prostate cancer; 0.900 of Ensemble method for prostate cancer), with median 0.713 (Figs. 3a, 4a, c and Supplementary Tables 6–8). Further, performance of OR/SD of nine methods across diseases was more impressive and variable, with highest in schizophrenia (LDpred2: 2.31, 95% CI 2.10–2.56; Ensemble method: 2.36, 95% CI 2.14–2.61.) and lowest in stroke (SBayesR: 1.01, 95% CI.), with median 1.695 (Supplementary Fig. 1 and Supplementary Tables 6–8). For 17 quantitative traits, Pearson R2 displayed the highest in height with 0.285 (PRS-CS, 95% CI 0.281–0.287; Ensemble method: 0.291, 95% CI 0.288–0.295), and with a median 0.050 across quantitative traits (Figs. 3c, 4b, d and Supplementary Tables 10–12). Incremental R2 performed proportionally to Pearson R2 for each trait (Supplementary Tables 8 and 10).
Evaluation of risk stratification values
Another crucial component in evaluating the PGS method for disease prediction is the risk stratification values in the general population. We calculated the OR of the top 10% versus the bottom 10% of PGS, the OR of the top 10% versus the middle 20% of PGS, and the OR of the bottom 10% versus the middle 20% of PGS for single-ancestry and Ensemble methods in 19 diseases (Fig. 5 and Supplementary Table 19). The scope of OR for top vs bottom 10% was ranging from 1.046 (SBayesR for venous thromboembolism) to 16.897 (SDPR for schizophrenia; Ensemble method 17.903 for schizophrenia). Among them, the Ensemble method, LDpred2, PRS-CS, and Lassosum2 showed larger differences between the top and bottom deciles to identify individuals at high risk in most diseases. The OR of the top 10% vs the middle 20% was ranging from 1.052 (SBayesR for primary open angle glaucoma) to 4.725 (LDpred2 for schizophrenia; Ensemble method 5.099 for schizophrenia). OR for the top 10% vs the bottom 10% and OR of the top 10% vs the middle 20% were consistently high for some diseases (e.g. schizophrenia, prostate cancer, coronary artery disease, psoriasis), which could mean the high-risk individuals of these kinds of diseases include a large number of cases or low-risk groups include very few cases. Furthermore, the OR of the bottom 10% vs the middle 20% was also evaluated to assess the protective property of each method across diseases. As expected, Ensemble method, LDpred2, PRS-CS, and Lassosum2 showed better protective performance, while SBayseR and MegaPRS (non-annotation version) displayed the highest ratios, indicating a more distinct distribution of predicted genetic risk values across PGS methods (Supplementary Table 19).

Heatmap of the odds of being a case in the top 10% of PGS versus the odds of being a case in the bottom 10% or middle 20% of the PGS distribution for each method across 19 diseases. The values correspond to risk-stratification levels. PsO psoriasis, SCZ schizophrenia, PCa prostate cancer, CD Crohn’s disease, SLE systemic lupus erythematosus, BCa breast cancer, CAD coronary artery disease, PD Parkinson’s disease, HF heart failure, LCa lung cancer, COPD chronic obstructive pulmonary disease, T2D type 2 diabetes, RA rheumatoid arthritis, VTE venous thromboembolism, BP bipolar disorder, POAG primary open-angle glaucoma. Analyses were conducted in the UK Biobank European testing cohort (N = 188,745). Rows correspond to diseases, and columns correspond to single-ancestry PGS methods.
Evaluation of LD reference panel sizes and SNP numbers on PGS performance
Recent studies have suggested that the larger sample size of the LD reference panel provides more precise estimates of the correlation structure between genetic variants24, indicating that the sample size of the LD reference panel can influence the performance of risk prediction. To demonstrate that, we first built LD references of about 1.1 million SNPs (overlap with Hapmap3 loci) which inferred with high-coverage (~30X) WGS data from 50, 100, 250, and 500 individuals of 1000 Genomes Project and 1000, 5000, 10,000, and 20,000 individuals in the UK Biobank genomic imputation dataset (INFO >0.8) (Methods), and compared their performances from eight single-ancestry methods for predicting three diseases and three quantitative traits in European ancestry (Method).
As shown in Fig. 6 and Supplementary Fig. 4, upgrades were provided from 50 to 1000 or 5000 sample sizes of LD reference panels for precise risk prediction in most traits (for Asthma, CAD, T2D, and BMI, the significant upgrade sustained to 1000; for HDL, the significant upgrade sustained to 5000 sample sizes of LD references), and the accuracy performance became stable across LD sample sizes above 1000 or 5000 individuals for prediction of most traits from most PGS methods, although the range of performance improvement varied among different methods across different traits (Supplementary Tables 21 and 22). For example, as the LD panel size increased, AUC improved most in Asthma, ranging from 0.600 in the LD reference panel of 50 individuals to 0.631 in the LD reference panel of 20,000 individuals (Lassosum2 method) (Supplementary Fig. 4a and Supplementary Table 21). OR/SD improved the most in coronary artery disease, ranging from 1.570 resulting from 50 of LD panel size to 1.978 for 20000 of LD panel size (PRS-CS method) (Supplementary Table 21).

a Average AUC of single-ancestry methods across different sample sizes of LD references in asthma, coronary artery disease, and type 2 diabetes. X-axis: different sample sizes of LD references, ranging from 50 to 20,000. Y-axis: value of AUC. b Average Pearson R2 of single-ancestry methods across different sample sizes of LD references in body mass index, glycated hemoglobin HbA1c, and high-density lipoprotein cholesterol. X-axis: different sample sizes of LD references, ranging from 50 to 20,000. Y-axis: value of Pearson R2. All the results are based on the evaluation in the UK Biobank European testing cohort (N = 188,745).
For quantitative traits, Pearson R2 improved most in the HDL trait, ranging from 0.007 derived from the 50 of the LD panel size to 0.210 of the LD sample size of 20,000 (SDPR method) (Supplementary Fig. 4f and Supplementary Table 22). SBayesR showed unstable, inaccurate performance compared with other methods, likely due to a computational limitation in the inherent model for prior inference when using extracted information from different sample sizes of LD references.
We further built African LD reference panels of varying sizes (50–5000) in the UK Biobank to examine the effect of panel size on PGS performance in African populations. Results showed that even LDpred2 exhibited reduced unstable performance in African populations, and varying LD panel sizes significantly influenced all methods. This instability may stem from the relatively low statistical power of existing African GWAS (Supplementary Fig. 5 and Supplementary Table 24).
As the above results were derived from the LD reference of 1.1 million Hapmap3 SNPs, we continue to construct LD references of 50–20,000 panel size, utilizing more SNPs for each trait in European ancestry (~6.6 million SNPs, Methods) to study the performance across different SNP numbers. Since most PGS methods can’t bear such a large number of SNPs, we only tested the effect of different SNP numbers using the PRS-CS and MegaPRS (non-annotation version) methods. Interestingly, we found that generating PGS from 6.6 million SNPs performed worse in accuracy than generating PGS from 1.1 million SNPs for both methods, probably owing to noise or offsets introduced by PGS construction using too many SNP loci (Supplementary Fig. 6 and Supplementary Table 23). Moreover, consistent with the observations from PRS-CS and the non-annotation version of MegaPRS, MegaPRS (incorporating BLD-LDAK annotations) further confirmed that increasing the number of SNPs from 1.1 million to 6.6 million did not improve predictive performance (Supplementary Table 23).
Evaluation of the computational cost of single-ancestry methods
Computational cost is another criterion for evaluating the performance of PGS methods. To demonstrate this, we evaluated the computational efficiency of eight single-ancestry PGS methods by recording the number of CPUs used, the running time, and the maximum memory usage during SNP effect size estimation for each method computing each trait (Methods, Supplementary Tables 27 and 28). We found that P + T ran fastest, taking an average of 0.84 CPU hours to complete 24 parameter settings for each trait, while SBayesR and PRS-CS ran slower by taking an average of 271.5 and 213.2 CPU hours (about 10–14 h runtime for 20 CPUs) to complete only one parameter setting for each trait, respectively (Fig. 7a and Supplementary Tables 27 and 28). In general, P + T, MegaPRS (non-annotation version), SBLUP, Lassosum2, and LDpred2 are computationally efficient (Fig. 7a and Supplementary Tables 27, 28). To measure memory usage, we used Slurm to record the memory usage of the computation workflows for different PGS methods. The results showed that P + T was the most memory-efficient, with an average maximum memory usage of 873.9 MB during computation per trait. MegaPRS (non-annotation version), SBLUP, LDpred2, and Lassosum2 were also memory-efficient, while SDPR, SBayesR, and PRS-CS were memory intensive but still less than 20 GB (Fig. 7b and Supplementary Tables 27 and 28).

a Total CPU hours of single-ancestry methods for each trait evaluated in UK Biobank European testing datasets (N = 188,745). X-axis: single-ancestry methods. Y-axis: Total CPU hours (in log scale). b Maximum memory usage of single-ancestry methods of each trait evaluated in UK Biobank European testing datasets (N = 188,745). X-axis: single-ancestry methods. Y-axis: Maximum memory usage (MB). Statistics of computational time and memory for each method are recorded by completing all corresponding parameter settings, except for PRS-CS and SBayesR, which complete one parameter calculation before proceeding to another; thus, the computational cost of PRS-CS and SBayesR was counted by completing only one parameter setting. In each box plot, the central horizontal line represents the median value across traits. The box indicates the interquartile range (IQR, 25th–75th percentiles), and whiskers extend to the most extreme values within 1.5×IQR from the box. Points beyond the whiskers represent individual observations (a, b).
The computational costs of eight single-ancestry methods using different sample sizes of LD reference data were also evaluated. For the generation of 50–20,000 sample sizes of LD references, the total CPU hours and memory usage were higher consumed with increasing sample size of LD references for most methods, while SDPR’s memory usage was highest in 50 and 100 of LD panel size (more than 160 GB), probably indicating SDPR’s Bayesian nonparametric framework, which struggles to stabilize LD matrix estimation with tiny sample sizes (Supplementary Table 20).
For the computational time of eight single-ancestry PGS methods across different LD sample sizes for six complex traits, P + T and SBLUP exhibited the largest runtime increases as LD panel sizes increased. This is expected since both approaches rely on on-the-fly LD estimation, which scales approximately linearly with the number of reference individuals and SNPs. The dynamic computation of pairwise LD statistics significantly increased their computational burden at larger panel sizes. CPU hours stabilized for methods that use pre-computed LD reference panels (e.g., PRS-CS, etc.). Some methods (e.g., SDPR) exhibited difficulty converging for LD panel sizes of 50 and 100 and required longer computation time than for larger LD panel sizes (Supplementary Fig. 7 and Supplementary Tables 20–22).
Comparison and evaluation of multi-ancestry PGS methods integrated on the PGS-hub platform in the UK Biobank
PGS evaluation statistics were derived from PRS-CSx, X-Wing, and LDpred2-multi for three complex diseases and three quantitative traits in UK Biobank European and African individuals, using external European- and African-sourced GWAS summary statistics (Fig. 2 and Supplementary Tables 2 and 3). Scores of LDpred2-multi were generated by a linear combination of the best LDpred2 scores for each ancestry in each trait (we call this method LDpred2-multi; Methods). The ensemble multi-ancestry method generated PGS by combining the best scores from each multi-ancestry method (PRS-CSx and X-Wing). As evaluation in single-ancestry methods, we equally divided the UK Biobank European unrelated individuals and African individuals into training and testing datasets, and the training dataset was used for parameter optimization for each ancestry, and the testing dataset was used for comparing and evaluating the performance of multi-ancestry PGS methods (Fig. 2, Methods).
We compared and evaluated the performance of PRS-CS, X-Wing, LDpred2-multi, and the multi-ancestry Ensemble method across six complex traits in European- and African-ancestry populations, then selected the best multi-ancestry method for each trait in each ancestry. As shown in Fig. 8, for three complex diseases, LDpred2-multi outperformed other multi-ancestry methods (including the Ensemble multi-ancestry method) with the highest AUC and OR/SD in all diseases in both European and African ancestries. PRS-CSx and X-Wing methods achieved comparable AUC and OR/SD performance, and PRS-CSx displayed slightly higher accuracy in Asthma and coronary artery disease, while X-Wing showed superior performance in type 2 diabetes (Fig. 8a and Supplementary Table 25). Similarly, for three quantitative traits, LDpred2-multi displayed exceptional performance in Pearson R2 across both European and African ancestry. PRS-CSx showed higher Pearson R2 than X-Wing in body mass index and high-density cholesterol in European ancestry, while the opposite performance trend was displayed in African ancestry, as X-Wing outperformed PRS-CSx in these two quantitative traits (Fig. 8b and Supplementary Table 26). As expected, all three multi-ancestry methods performed worse on African ancestry than on European ancestry.

a AUC performance of PRS-CSx, X-Wing, LDpred2-multi, and Ensemble method for asthma, coronary artery disease, and type 2 diabetes in European and African ancestry. b Pearson R2 of PRS-CSx, X-Wing, LDpred2-multi, and Ensemble method for body mass index, glycated hemoglobin HbA1c, and high-density lipoprotein cholesterol in European and African ancestry. All the results were evaluated in the UK Biobank European and African testing individuals (N = 188,745 and 8214) for (a, b).
In addition, for computational cost, LDpred2-multi was undoubtedly the most efficient, as it leverages and combines LDpred2 results across two ancestries. Besides, PRS-CSx displayed significantly less CPU hours and memory usage than X-Wing, with average CPU hours of 1635.06 (PRS-CSx) versus 3035.76 (X-Wing) and maximum usage of 1835.26 MB (PRS-CSx) vs 4536.36 MB (X-Wing) upon completing seven parameter settings in each trait (Supplementary Tables 27 and 28).
Annotations significantly increase PGS performance
Notably, PGS generated from annotated MegaPRS (BLD-LDAK annotations) displayed significantly higher performance compared to MegaPRS non-annotation counterparts, achieving superior performance relative to the best-performing non-annotation methods (LDpred2, PRS-CS, Lassosum2, and SDPR) for 12 out of 19 disease traits and 8 out of 17 quantitative traits (Supplementary Tables 29 and 30). Similarly, another PGS method—leveraging functional genomic annotations—SBayesRC, according to our supplementary analysis, showed superior performance to many existing non-annotation methods (five diseases and seven quantitative traits) (Supplementary Tables 31 and 32). However, since this version of PGS-hub focuses on foundational PRS models without functional annotations, functional-enabled PGS methods will be systematically integrated in subsequent versions of PGS-hub.
Discussion
The advent of advanced sequencing technologies and the establishment of population-scale biobanks have significantly enhanced our ability to understand the genetic factors contributing to disease risk and quantitative traits. PGS has emerged as a powerful tool for aggregating risk alleles into a single measure to predict complex traits. Despite the widespread development of diverse PGS methodologies, their inconsistent computation and practical evaluation across ancestries have presented challenges for standardization and clinical translation.
In this study, we introduce the PGS-hub platform, which integrates a comprehensive suite of ten cutting-edge PGS methods tailored for single- and multi-ancestry contexts. The web-based platform provides an automated pipeline that supports a wide range of PRS methods under standardized data-processing protocols, minimizing methodological variability and facilitating direct method comparisons. We further utilize PGS-hub to conduct a thorough benchmarking analysis of different methods across predictive accuracy, risk stratification, computational efficiency, and the influence of varying LD reference panel sizes and the number of SNPs included in PGS generation for 36 complex traits in the UK Biobank. Moreover, we have extended the scope of our investigation by incorporating multi-ancestry analyses (encompassing European and African populations), ensemble strategies (applied to both single- and multi-ancestry methods), and multi-source independently optimized PGS (LDpred2-multi) for cross-ancestry evaluation and improvement from a practical perspective. These efforts address critical gaps in current PGS research, particularly in its application across diverse populations, and provide actionable insights to enhance PGS’s utility in broader contexts.
PGS-hub utility
PGS methods are computationally intensive, require complex parameter optimization, and involve diverse input formats, which complicates efforts to compute and compare their performance35,36,37,38,39,40,43. Limited studies have tried to provide pipeline resources to facilitate PGS calculation41,42. Based on these, we are committed to delivering an exceptional web-based platform, PGS-hub, with an automated and standardized framework. This platform streamlines the process by allowing users to submit their GWAS summary statistics and LD references, including user-defined LD references, and perform both single-ancestry and multi-ancestry PGS calculations online. PGS-hub significantly reduces the burden on users of setting up different computational pipelines and minimizes methodological variability, thereby enhancing accessibility and reproducibility in PGS research.
Evaluation of predictive accuracy for single- and multi-ancestry methods across ancestries
Single-ancestry PGS methods have matured significantly, yet their effectiveness still varies across genetic architectures and population contexts. In European ancestry, our evaluation confirms that the single-ancestry method LDpred2 generally outperforms others across many traits, consistent with prior studies within a reference-standardized framework in one or broader biobank36,37,40, making it the preferred choice for diverse applications. Notably, methods such as Lassosum2, PRS-CS, and SDPR also excel in specific traits, while simpler models like P + T deliver moderate yet stable performance. However, methods such as MegaPRS (non-annotation version) and SBayesR, despite showing strengths for certain traits39, often lag in broader applications, emphasizing the need for careful method selection based on the target phenotype. We further demonstrate and recommend that users prioritize the grid-tuning algorithm of LDpred2 and PRS-CS when individual-level data is available, as they provide better predictive performance to the auto algorithm.
In the African population, due to the smaller GWAS sample size, there are expected declines in the predictive performance of all methods. However, LDpred2, Lassosum2, PRS-CS, and SDPR still outperform other methods, indicating their superior ability to adapt to diverse complex genetic structures and population backgrounds. Notably, P + T outperforms other methods on specific quantitative traits, such as HbA1c and HDL, indicating the advantages of P + T in the methodological strategy for this scenario.
Multi-ancestry methods, though historically less explored, are gaining prominence as trans-ancestry GWAS studies expand12,44. Our analysis highlights that high-performing single-ancestry methods can be adapted for cross-ancestry prediction. For example, LDpred2-multi, which synthesizes ancestry-specific LDpred2 scores, consistently outperforms tailored multi-ancestry methods like PRS-CSx and X-Wing across most traits. The results are consistent with Pain, O38, which indicates that linear combinations of independently optimized scores consistently outperform current jointly optimized multi-ancestry methods by comparison of PRS-CS multi and PRS-CSx, it is suggested that leveraging the strengths of ancestry-specific methods within a combined framework holds promise for enhancing multi-ancestry predictions.
Method performance across trait architectures
Performance across different trait architectures (e.g., heritability and polygenicity) reveals nuanced patterns that further inform method selection. We observe a general positive association between trait heritability and predictive performance, as in Systemic lupus erythematosus and Crohn’s disease, yielding robust OR/SD values via SDPR and LDpred2. This highlights the capacity of top-performing methods to capitalize on stronger genetic signals. Notably, traits with high heritability but elevated polygenicity, such as Rheumatoid arthritis, exhibited attenuated predictive performance, likely due to the cumulative effects of numerous small-effect variants, which are inherently challenging to model. However, LDpred2 also maintained superiority in highly polygenic traits, which may be due to the strength of its sophisticated modeling of linkage disequilibrium and effect-size shrinkage, enabling robust performance across diverse genetic architectures.
Ensemble strategies: enhancing predictive power
One of the most notable contributions of this study is the evaluation of ensemble approaches, which combine multiple single-ancestry or multiple-ancestry PGS methods to optimize performance. By aggregating predictions from diverse algorithms, ensemble methods achieved superior accuracy and broader applicability across all tested traits. Specifically, the single-ancestry ensemble approach demonstrated remarkable enhancements in predictive performance. Notably, this approach excelled in highly heritable traits like schizophrenia and coronary artery disease, with the ensemble PGS achieving a median OR/SD increase of 5–10% over the best standalone method.
The performance of LDpred2-multi further illustrates the benefits of synthesizing single-ancestry predictions. By combining the best-performing LDpred2 scores across ancestries, LDpred2-multi consistently outperformed standalone methods in accuracy and computational efficiency, indicating that linear combinations of independently optimized scores outperform the current jointly optimized cross-ancestry PGS method while being substantially more computationally efficient. This hybrid approach also offers a partial answer to a critical question in the field: How should researchers reconcile discrepancies in risk predictions across algorithms? By synthesizing diverse methodologies, ensemble approaches provide a more stable and reliable prediction framework. Similarly, Patel, et al.12 have demonstrated the power of ensemble strategies in a multi-ancestry setting by integrating polygenic risk scores across multiple ancestries and traits to develop a state-of-the-art coronary artery disease PGS. Their study highlights the potential of combining multi-trait and multi-ancestry data to elevate predictive accuracy further, aligning closely with our findings.
However, the benefits of ensemble methods come with trade-offs. Most Ensemble strategies require significant computational resources, increased processing time, and careful weight optimization for combining predictions. While they deliver the best overall performance, their adoption in resource-limited settings may be constrained. Researchers must balance these considerations against the marginal gains in accuracy, tailoring their approach to the specific needs of their study.
Stratified risk identification
A key strength of PGS lies in its capacity for population risk stratification. Such stratification approaches are particularly valuable in research aimed at isolating high-risk subpopulations, even when methods vary in their performance. For example, while the Ensemble method consistently achieves high predictive accuracy across traits (e.g., AUC and OR/SD metrics), its relative performance in identifying differences in risk stratification is not consistently superior. For example, LDpred2 displays high ORs in top versus middle and bottom deciles for schizophrenia and psoriasis, while SDPR outperforms LDpred2 in specific stratification comparisons for schizophrenia. Similarly, MegaPRS (non-annotation version) achieves the best predictive performance for psoriasis overall but is surpassed by LDpred2 in OR/SD for specific decile comparisons. These findings emphasize the importance of evaluating PGS methods not only for their overall predictive capacity but also for their stratification utility, which can vary based on trait-specific characteristics and method design.
LD panel size and SNP quantity associate with predictive power
Our findings reveal key insights into the influence of LD reference panel size and SNP quantity on PGS predictive performance. We observe that increasing the LD reference panel size from European ancestry significantly enhances predictive accuracy, with notable improvements up to ~1000 or 5000 individuals. Beyond this threshold, the rate of improvement diminishes, and performance stabilizes. When the panel size exceeds 5,000 individuals, most PGS methods exhibit a plateau in performance. This stabilization may occur owing to a too-large panel size in the LD reference, primarily increasing the coverage of rare-variant genetic variation, whereas the SNPs used for PGS calculations in our benchmark are predominantly common variants derived from GWAS. However, for African ancestry, we found that the performance of most stable methods, such as LDpred2, Lassosum2, and PRS-CS, was unstable and significantly influenced by LD panel sizes. This instability may stem from multiple factors: the relatively low statistical power of existing African GWAS, which limits the reliability of genetic association signals, and the limited adaptability of current methods to low-power datasets from underrepresented populations, such as African ancestry groups.
Interestingly, an excessive number of SNPs reduces predictive efficacy. This phenomenon likely results from the inclusion of weak-effect loci, introducing noise that offsets potential gains. These results underscore the critical need to balance the inclusion of comprehensive genetic information with methodological efficiency to optimize prediction accuracy. These findings are consistent with those of Zheng, et al.45 in certain scenarios.
Computational costs
Computational cost remains a pivotal factor influencing the applicability of PGS methods, particularly in large-scale studies or resource-constrained environments. For example, linear mixed-model-based methods with global shrinkage are more efficient, as their analytic solutions expedite computatio16. Approximate inference algorithms consistently reduce memory demands compared to exact inference techniques within similar modeling frameworks16. Additionally, multi-threading or parallel processing can enhance computational efficiency across various methods. Within our study, methods such as P + T, Lassosum2, and LDpred2 emerge as computationally efficient, requiring significantly less CPU time and memory for parameter optimization. Conversely, Bayesian methods like PRS-CS and SBayesR incur substantial computational demands, highlighting the trade-off between accuracy and resource consumption. Our work underscores the need to account for computational requirements when selecting methods, particularly when evaluating PGS for diverse traits and cohorts.
Limitations and future directions
Despite advancements in PGS methodologies, substantial challenges persist. The heterogeneity in disease genetic architecture, encompassing variations in heritability46, polygenicity47, and environmental interplay48, complicates the generalizability of PGS. Similarly, algorithmic differences across PGS methods—ranging from Bayesian priors to linear regression frameworks—necessitate careful consideration of trade-offs among accuracy, computational cost, and resource availability. Furthermore, a recent study evaluating polygenic scoring methods across five biobanks found greater variation between biobanks37, suggesting the need for broader comparison beyond methodologies and population backgrounds.
While this version of the PGS-hub platform establishes a standardized, reproducible framework for benchmarking widely used PGS methods under a unified LD reference structure, several limitations remain. First, the present benchmarking focuses on models without functional annotations. Future releases will integrate annotation-informed methods and more broadly include summary-statistic-based tuning strategies, such as the X-Wing LEOPARD algorithm, which achieves performance comparable to individual-level tuning without requiring access to individual-level data. Second, more advanced cross-ancestry modeling strategies will be incorporated. Third, although African ancestry data were integrated, the representation of non-European populations remains limited. Expanding cross-ancestry coverage will be essential for improving the equity and portability of polygenic prediction.
Overall, PGS-hub serves as a foundational tool to address these challenges, facilitating systematic benchmarking and minimizing methodological variability. Continued refinement and adoption of platforms like PGS-hub will be instrumental in driving the clinical translation of PGS research.
Methods
Construction and availability of the PGS-hub platform
PGS-hub platform integrates ten PGS methods, in which eight single-ancestry PGS methods: (P + T21, Lassosum228, LDpred225, MegaPRS (non-annotation version)31, PRS-CS26, SBayesR30, SBLUP32, SDPR29) and two multi-ancestry methods (PRS-CSx33 and X-Wing34) to compute multiple PGS methods with a range of parameters at one time. The PGS-hub platform used Java for its back-end, while the front-end was built with Vue, HTML, CSS, and JavaScript. Users can upload their GWAS summary statistics in the prescribed format, which must include all necessary data for calculating various PGS methods. This data should encompass the specific chromosome, SNP ID, SNP position, reference allele, alternative allele, reference allele frequency, effect of the reference allele, standard error, P value of the effect, the sample size, effect sample size (Neff = 4/(1/Ncase + 1/Ncontrol)49, among them the SNP ID should be formatted as “chr:position:sorted(ref:alt)” with hg38 genome build. For details on usage and file format, please refer to the tutorial on the website. For linkage disequilibrium (LD) references, users can choose European, African, South Asian, and East Asian LD references derived from 1000 Genome Project50 integrated on the PGS-hub platform or upload their self-defined LD references as well. PLINK51,52 format (.pgen) files are required to minimize file size while uploading LD references.
All GWAS summary statistics and LD references are first verified for data format compliance and reformatted according to the selected PGS method before any computational analysis begins. Users can choose PGS methods and set the corresponding parameters according to the website guidelines. After submission, their computational jobs are executed on the backend of the PGS-hub platform. To ensure uniform processing across all PGS methods, all uploaded data undergo standardized processing pipelines, written in the Workflow Description Language (WDL) and executed using the Cromwell execution engine53. This approach enhances the modularization and adaptability of various PGS methods on the platform, ensuring efficient operation and computation. Each method has also been dockerized for convenient implementation within a docker container. Moreover, a Slurm server54,55 was implemented to effectively manage the allocation and scheduling of computing resources for multiple tasks. The output files, which include each SNP effect size for chosen methods, are packed into compressed folders (.zip) for users to download.
To ensure data privacy and security, PGS-hub incorporates robust protocols for handling user-submitted data, including GWAS summary statistics and LD reference panels. All uploaded data, raw inputs, intermediate files, and computation results are automatically deleted 30 days after the last access, with users able to trigger immediate deletion via a dedicated dashboard function. Data transmission and storage are secured using industry-standard encryption (TLS 1.3 for transmission, AES-256 for storage) on restricted-access servers, in accordance with the institutional security policies of the National Genomics Data Center, China National Center for Bioinformation. In compliance with data transfer agreements and global regulations (e.g., GDPR, China’s Data Security Law), PGS-hub requires users to confirm they hold necessary permissions for data upload and processing, while the platform architecture minimizes handling of individual-level raw genotypes for custom LD panels, the data conversion is processed in isolated environments, and never stores user input files. Identifiable information is strictly prohibited. Transparency is ensured through a detailed data privacy policy and transfer agreement on the platform, prompting users to agree and confirm data ownership and compliance during registration, and strengthening responsible data use.
The PGS-hub platform is available via application to the National Genomics Data Center (NGDC) at the Beijing Institute of Genomics, Chinese Academy of Sciences/China National Center for Bioinformation (https://ngdc.cncb.ac.cn/pgs-hub/#/home).
GWAS summary data
Thirty-six GWAS summary statistics, including 19 diseases and 17 quantitative traits, were gathered from participants of European ancestry, detailed in Supplementary Tables 2 and 3, which covered 12 categories of diseases according to the International Classification of Diseases, 10th Revision (ICD-10)56. These categories include the circulatory system, dermatologic, digestive, endocrine/metabolic, genitourinary, hematopoietic, mental disorders, musculoskeletal, neoplasms, neurological, respiratory, eye, and accessory organ diseases. For cross-population PGS methods, we evaluated three diseases and three quantitative traits in European- and African-ancestry populations, and the GWAS summary statistics are detailed in Supplementary Tables 2 and 3.
All the GWAS summary statistics were manually examined to ensure that the GWAS derivation samples did not overlap with UK Biobank individuals. The multi-allelic, strand-ambiguous, low minor allele frequency (<0.01), and low UK Biobank imputation quality (INFO <0.3) variants are removed. All the GWAS summary statistics were liftover to the hg38 genome build for PGS computation. We uniformly retained the MHC/HLA region across all methods, recognizing that uniform removal across all analyses could potentially underestimate predictive performance for non-immune traits.
Phenotype definition and partition of training and testing data in the UK Biobank
We used 377,490 unrelated Europeans and 8214 Africans from the UK Biobank to evaluate PGS methods. The samples with quality controlled by four steps: (1) Self-report gender is the same as inferred gender; (2) No heterozygous missingness outlier; (3) No putative sex chromosome aneuploidy; (4) The missing rate is less 5% for high-quality markers. After quality control, we used hold-out validation to randomly partition the entire UKB dataset into two equal parts (1:1 ratio) in a single step. Specifically, 50% of the samples were assigned to the training dataset for parameter optimization of polygenic risk score (PGS) methods, and the remaining 50% were allocated to the independent testing dataset for evaluating model performance.
The diagnosis of each disease was based on a composite of data from self-reports in an interview with a trained nurse, electronic health record (EHR) information, including inpatient ICD-10 diagnosis codes, and Office of Population and Censuses Surveys (OPCS-4) procedure codes. Phenotype definitions for each trait were detailed in Supplementary Table 4.
All UK Biobank participants provided written informed consent at enrollment, and the study was conducted under UK Biobank’s ethical approval (Application Approval Number: 89885).
Build linkage disequilibrium reference panels
High-coverage (~30X) whole genome sequencing data from the 1000 Genomes Project50, which contains 503 European and 661 African individuals, was used to build the linkage disequilibrium (LD) reference for each PGS method. The overlapping SNPs from the 1000 Genomes Project and HapMap3 were used, resulting in a total of 1,112,773 and 1,077,912 common SNPs retained in the European and African populations, respectively. These SNPs were then mapped to the hg38 genome build using the CrossMap 0.6.457 liftover tool and formatted to meet the specific requirements of each PGS method. To investigate the impact of LD reference panel sample size on PGS method performance, we constructed LD reference panels for European and African ancestry at varying sample sizes, including 50, 100, 250, and 500 individuals from the 1000 Genomes Project (high-coverage ~30X WGS data) and 1000, 5000, 10,000, and 20,000 individuals from the UK Biobank genomic imputation dataset (INFO >0.8). For African ancestry, the LD panel size is limited to 5000 individuals due to the small sample size of African ancestry individuals in the UK Biobank. These LD references were built using ~1.1 million SNPs overlapping with Hapmap3 loci to ensure consistency in SNP selection across panels.
Furthermore, to assess the effect of larger SNP loci included in PGS generation, we extended our analysis by constructing LD references with sample sizes ranging from 50 to 20,000 of European ancestry using whole-genome associated SNPs (~6.6 million SNPs) for six traits (three diseases and three quantitative traits), then formatted to meet the specific requirements of PRS-CS and MegaPRS (non-annotation version).
PGS methods and parameter settings
Single-ancestry PGS methods
P value pruning and thresholding (P + T)21 scores were built using a P value and LD-driven clumping procedure implemented in PLINK252. P + T PGS scores were created over a range of P value (1, 0.5, 0.05, 5 × 10−4, 5 × 10−6, 5 × 10−8) and R2 (0.2, 0.4, 0.6, 0.8) thresholds, for a total of 24 P + T-based candidate scores for each trait.
Lassosum228 is a smoothed version of the upgraded Lassosum penalty for fitting integrated risk models using summary statistics. Scores were built over a range of lambda (a sequence of 30 values, logarithmically spaced between 0.1 and 0.001) and delta (0.001,0.01, 0.1, 1), with a total of 120 Lassosum2-based candidate scores for each trait.
LDpred225 calculates a posterior mean effect size for each variant based on a prior and subsequent shrinkage based on the variants’ LD patterns in a reference population. The LDpred2 scores were derived from various proportion (p) of presumed causal variants, p = 1.0 × 10−4, 1.8 × 10−4, 3.2 × 10−4, 5.6 × 10−4, 1.0 × 10−3, 1.8 × 10−3, 3.2 × 10−3, 5.6 × 10−3, 1.0 × 10−2, 1.8 × 10−2, 3.2 × 10−2, 5.6 × 10−2, 1.0 × 10−1, 1.8 × 10−1, 3.2 × 10−1, and 5.6 × 10−1; the scale of heritability, s = 0.7, 1, and 1.4; and whether to apply sparse LD matrix or not, for a total of 96 LDpred2-based candidate scores for each trait. We also generate PGS from the auto algorithm of LDpred2.
MegaPRS (non-annotation version)31 employs the BayesR variant of the model along with the genome-wide complex trait analysis (GCTA) heritability model to infer the prior variance for each genetic marker’s effect size. As per the MegaPRS guideline, we set the model option to “mega” and set skip-cv “YES” to output all 45 models after calculating h2 (heritability) by the LDAK software. In total, MegaPRS (non-annotation version) has 45 candidate scores for each trait.
PRS-CS26 utilizes a high-dimensional Bayesian regression framework by placing a continuous shrinkage (CS) prior to SNP effect sizes. Scores were calculated with the adjustable gamma-gamma prior a, b, and global shrinkage parameter phi, with parameter settings of a = 1, b = 0.5, phi = 1.0 × 10−6, 1.0 × 10−5, 1.0 × 10−4, 1.0 × 10−3, 1.0 × 10−2, 1.0 × 10−1, 1. For each trait, there are seven PRS-CS-based candidate scores. We also calculate PGS using the auto-PRS-CS algorithm.
SBayesR30 builds polygenic risk prediction using Bayesian multiple regression on summary statistics. SBayesR scores were created with MCMC iterations = 10,000 and burn-in = 2000, as recommended by the SBayesR developers. There is one SBayesR-based candidate score for each trait.
SBLUP32 scores were generated using the default summary-statistic-based best linear unbiased prediction model, with heritability calculated via LDSC56 and COJO-SBLUP analysis was performed.
SDPR29 is a Bayesian nonparametric method for predicting complex traits using summary statistics. It does not rely on specific parametric assumptions on the effect size distribution. The SDPR-based scores were derived from the default settings.
MegaPRS (annotated version) was applied using the BLD-LDAK annotation model58, which estimates posterior mean effect sizes by integrating annotation-informed priors with LD-adjusted shrinkage. We constructed a grid of 45 annotation-based parameter combinations by varying the LDAK weight-scaling factor, the annotation-specific heritability coefficients, and the shrinkage penalties governing effect-size sparsity. For each parameter set, MegaPRS computed SNP effects using summary statistics and an ancestry-matched LD reference panel, yielding 45 MegaPRS candidate scores per trait.
SBayesRC45 was applied using the default parameter settings recommended by the developers. The method models SNP effect sizes under a Bayesian regression framework that incorporates functional annotations into the prior variance while performing block-wise LD-adjusted shrinkage. Using genome-wide summary statistics and an ancestry-matched LD reference panel, SBayesRC jointly estimates variant effects via iterative posterior updating within LD blocks. Under this default configuration, we generated a single annotation-informed SBayesRC polygenic score for each trait.
Cross-population PGS methods
PRS-CSx33 extends the PRS-CS framework by coupling genetic effects across populations via a shared continuous shrinkage prior, enabling more accurate effect size estimation by sharing information between summary statistics and leveraging LD diversity across populations. Similar to PRS-CS, PRS-CSx scores were calculated with the adjustable gamma-gamma prior a, b, and global shrinkage parameter phi, with parameter settings of a = 1, b = 0.5, phi = 1.0 × 10−6, 1.0 × 10−5, 1.0 × 10−4, 1.0 × 10−3, 1.0 × 10−2, 1.0 × 10−1, 1. For each trait and each population, there are seven PRS-CSx-based candidate scores.
X-Wing34 quantifies local genetic correlations for complex traits across populations, employs an annotation-dependent estimation procedure to amplify correlated genetic effects, and combines multiple population-specific PGS into a unified score. Similar to PRS-CSx, X-Wing scores were calculated by the flexible parameter’s gamma-gamma prior a, b, and global shrinkage parameter phi, with parameter settings of a = 1, b = 0.5, phi = 1.0 × 10−6, 1.0 × 10−5, 1.0 × 10−4, 1.0 × 10−3, 1.0 × 10−2, 1.0 × 10−1, 1. For each trait and each population, there are seven X-Wing-based candidate scores.
LDpred2-multi is defined as a linear combination of the best PGS derived from LDpred2 in each ancestry, forming the final PGS of LDpred2-multi for evaluation in each ancestry. LDpred2-multi leveraged the optimized LDpred2 parameters for each trait.
Ensemble of PGS scores
The ensemble score for single- or multi-ancestry is derived from linearly combining the best polygenic scores derived from different methods for a given trait. The combined score for the ith individual is:
where βj are the weights obtained from the regression analysis, xij represents the jth PGS for the i-th individual, and n represents the number of linearly combined algorithms. The Ensemble strategy is the same for both single- and multi-ancestry methods.
PGS training and testing
Polygenic risk scores for a range of parameters for each PGS method were computed by multiplying the genotype dosage of each risk allele by its respective weight, then summing across all variants for each training sample using the PLINK2 software52. For the testing data, the PGS with the best parameter selected from the training dataset for each method was computed in the same way. For each PGS method (single- and multi-ancestry), parameter optimization was performed, and the selection of the best-performing PGS for each trait was exclusively based on the training dataset. Ensemble models combined the best score from each method trained on the training dataset. The testing dataset was used solely for the final evaluation and comparison of method performance, ensuring independence and validity.
For each complex disease, we identified the optimal PGS parameter in the training data by selecting the one that maximized the area under the receiver operating characteristic curve (AUC) in a logistic regression model. This model included the disease-specific candidate PGS, age, sex, genotyping array, and the first ten principal components of ancestry as predictors. For each quantitative trait, the best parameter score with the best capacity of prediction accuracy was determined based on the Pearson correlation square (R2), which calculates the square of Pearson correlation between the residuals from two linear regression models: one modeling each PGS against age, sex, genotyping array, and the first ten principal components of ancestry, and another modeling with each quantitative trait value as the outcome, adjusting for enrollment age and sex as covariates. Additionally, for the selected best PGS parameter, we computed incremental R², which measures the increase in the proportion of phenotypic variance explained (R²) when PGS is added to a baseline model that includes only age, sex, genotyping array, and the first ten principal components of ancestry. The performance in the testing dataset was evaluated using the same regression model.
To assess the risk-stratification capability of PGS methods for disease prediction, we calculated odds ratios (ORs) across PGS percentile groups in the general population. Specifically, for each of the 19 diseases, we computed the OR of the top 10% versus the bottom 10% of PGS, the OR of the top 10% versus the middle 20% of PGS, and the OR of the bottom 10% versus the middle 20% of PGS for both single-ancestry PGS methods and the Ensemble method. For each disease, PGS distributions were standardized within the population, and individuals were stratified into deciles based on their PGS values. Logistic regression models were used to estimate the odds ratio (OR) of disease risk for each percentile comparison, adjusting for age, sex, genotyping array, and the first ten principal components of ancestry as covariates. OR estimates were obtained separately for each PGS method and reported with 95% confidence intervals (CI). The OR values were used to evaluate the ability of PGS methods to distinguish high-risk individuals (top 10%) from low-risk individuals (bottom 10%) and to assess predictive strength within the population (top 10% vs. middle 20%). Additionally, we examined the OR of the bottom 10% vs. the middle 20% to evaluate the protective effect of low genetic risk across diseases. Statistical analyses were performed using R (version 4.1.2) with the logistic regression function.
For statistical comparisons to assess whether differences in performance of various methods are significant. For each trait, we conducted a comprehensive post-hoc analysis using bootstrapping (1000 resamples) on the testing dataset to generate confidence intervals and P values for comparisons between the top-performing methods on key metrics (OR/SD for diseases, Pearson R2 for quantitative traits). The null hypothesis for the P value computation was that the two methods had equal chances of being superior. For comparisons of method performance across non-independent traits, pairwise GLS tests were used to assess metric (OR/SD for diseases, Pearson R2 for quantitative traits) differences between methods, with GLS tests of the mean difference in standardized Z-scores across traits. (Z-scores were standardized from key metrics OR/SD for diseases and Pearson R2 for quantitative traits). We used the GLS-derived test statistic and its corresponding normal-approximation P value to evaluate whether two PRS methods differ significantly in their mean performance across non-independent traits. False discovery rate (FDR) correction was applied across all pairwise comparisons. Notably, for approximately half of the traits (e.g., coronary artery disease, apolipoprotein A1, calcium), the best method outperformed the second-best method in all 1000 bootstrap resamples, yielding P value <1 × 10−300.
Heritability and polygenicity
We calculated the heritability and polygenicity for all 36 complex traits (19 diseases and 17 quantitative traits) included in our study. Heritability was estimated using the “snp_ldsc” function from the “bigsnpr” R package, while polygenicity was computed based on the SBayesR model implemented in the GCTB software using trait-specific GWAS summary statistics. We then systematically evaluated the relationship between heritability/polygenicity and the performance of various PRS methods, as measured by OR/SD for diseases and Pearson R² for quantitative traits.
Evaluation of LD reference panel sizes and SNP number on PGS performance
To evaluate the impact of LD reference panel sample size on PGS performance, we assessed PGS prediction accuracy using different LD reference panels constructed for European and African ancestry. PGS performance was evaluated across all eight single-ancestry PGS methods for predicting three complex diseases (asthma, coronary artery disease, and type 2 diabetes) and three quantitative traits (BMI, HDL, and HbA1c) in European ancestry. For African ancestry, three stable single-ancestry methods (LDpred2, Lassosum2, and PRS-CS) were evaluated for the same six complex traits.
For each LD reference panel, PGS models were trained using GWAS summary statistics and subsequently evaluated in an independent UK Biobank testing dataset following an identical training–testing framework across panels. PGS performance was assessed using the AUC and OR/SD for complex diseases and Pearson’s R² for quantitative traits. To investigate the effect of LD reference panel sample size, PGS performance was compared across LD panels constructed using increasing numbers of individuals (50, 100, 250, 500, 1000, 5000, 10,000, and 20,000), each based on 1.1 million HapMap3-overlapping SNPs.
Additionally, to examine the impact of SNP number, we repeated the analysis using LD reference panels ranging from 50 to 20,000 individuals, constructed from ~6.6 million whole-genome associated SNPs in European ancestry. Since most PGS methods cannot efficiently process such a large SNP set, the effect of larger SNP panels on PGS performance was evaluated exclusively using PRS-CS and MegaPRS (with/without annotation).
Measurement of computational cost
The number of CPUs, computation time, and memory usage for each PGS method were monitored using the Cromwell (Version 86) execution engine53 configured in the Slurm job scheduler system (Version slurm-wlm 22.05.8)54,55. The WDL pipelines for each method set the number of CPUs used. Slurm measured the computation time and memory usage. Memory usage was measured as the maximum memory footprint during the WDL workflow computation for each PGS method. CPU hours were measured as the total of running time (CPU numbers multiplied by running hours). The runtime reported for each method is the average total computation time across all parameter configurations.
Software and tools
Data analyses were performed using PLINK2 (v2.0), R (v4.1.2), and Python (v3.7). Polygenic score computation and related analyses used the following software/tools/packages (versions in parentheses): bigsnpr (v1.7.1), GCTB (v2.04.3), GCTA (v1.94.1), SDPR (v0.9.1), PRS-CS (v1.0), PRS-CSx (v1.0), and LDAK (v5.2). Statistical analyses and model evaluation were conducted using pROC (v1.18.5), dplyr (v1.1.2), and tidyr (v1.3.0). Figures were generated using ggplot2 (v3.3.5), seaborn (v0.12.2), matplotlib (v3.6.2), sci_palettes (v1.0.0), and pandas (v1.5.2). Genome build conversion was performed using CrossMap (v0.6.4). Workflow execution used workflow description language (WDL) pipelines run with Cromwell (v86) and scheduled using Slurm (slurm-wlm v22.05.8), with each method containerized using Docker.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The individual-level genotype and phenotype data used in this study were obtained from the UK Biobank. These data were available under restricted access due to participant consent and data governance requirements. Access can be obtained by qualified researchers through formal application to the UK Biobank (https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access), subject to approval by the UK Biobank and relevant institutional review boards. The raw individual-level UK Biobank data were protected and are not publicly available due to data privacy and ethical restrictions. This research was conducted using the UK Biobank resource under application approval number 89885 and approved by the Beijing Institute of Genomics, Chinese Academy of Sciences (China National Center for Bioinformation) institutional review board. The genome-wide association study (GWAS) summary statistics used in this study are publicly available from established community repositories, including the GWAS Catalog (https://www.ebi.ac.uk/gwas/; accession numbers: GCST003724, GCST004988, GCST004132, GCST006085, GCST90019017, GCST000679, GCST005831, GCST004773, GCST90132700, GCST90132701, GCST90012622, GCST90132694, GCST90132719, GCST90002244, GCST000817, GCST90132736, GCST90132737, GCST90132742, GCST90132747, GCST90131435, GCST009057, GCST90002242, and GCST90101746), the Global Biobank Meta-analysis Initiative (https://www.globalbiobankmeta.org/), FinnGen (https://www.finngen.fi/en/access_results), the Global Lipids Genetics Consortium (http://csg.sph.umich.edu/willer/public/glgc-lipids2021), dbGaP (https://www.ncbi.nlm.nih.gov/gap/; accession numbers: phs000876.v1.p1, phs001672, and phs001672.v11.p1), the International Parkinson Disease Genomics Consortium (https://www.pdgenetics.org/), the Psychiatric Genomics Consortium (https://pgc.unc.edu/), the CARDloGRAMplusC4D (http://www.cardiogramplusc4d.org/data-downloads/), and the Genes & Health (https://www.genesandhealth.org/research/scientific-data-downloads). The processed summary-level data and derived resources generated in this study are provided in the Supplementary Information. In addition, the PGS-hub platform developed in the present study is publicly available at https://ngdc.cncb.ac.cn/pgs-hub/#/home. Access to the PGS-hub platform requires user registration and agreement to the Terms and Conditions and the Data Privacy Policy and Transfer Agreement.
Code availability
The code supporting the findings of this study is publicly available at https://github.com/xinyu-c9/PGS-hub_offline. To ensure reproducibility and long-term accessibility, a citable, archived version of the code has been deposited in Zenodo (https://doi.org/10.5281/zenodo.17994509)59. In addition, a Code Ocean capsule is provided to facilitate computational reproducibility (https://doi.org/10.24433/CO.5394565.v1).
References
Boyle, E. A., Li, Y. I. & Pritchard, J. K. An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186 (2017).
Visscher, P. M. et al. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22 (2017).
Loos, R. J. F. 15 years of genome-wide association studies and no signs of slowing down. Nat. Commun. 11, 5900 (2020).
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569 (2010).
Makowsky, R. et al. Beyond missing heritability: prediction of complex traits. PLoS Genet. 7, e1002051 (2011).
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
Génin, E. Missing heritability of complex diseases: case solved? Hum. Genet. 139, 103–113 (2020).
Plomin, R., Haworth, C. M. & Davis, O. S. Common disorders are quantitative traits. Nat. Rev. Genet. 10, 872–878 (2009).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018).
Khera, A. V. et al. Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell 177, 587–596.e589 (2019).
Mujwara, D. et al. Integrating a polygenic risk score for coronary artery disease as a risk-enhancing factor in the pooled cohort equation: a cost-effectiveness analysis study. J. Am. Heart Assoc. 11, e025236 (2022).
Patel, A. P. et al. A multi-ancestry polygenic risk score improves risk prediction for coronary artery disease. Nat. Med. 29, 1793–1803 (2023).
Wray, N. R., Kemper, K. E., Hayes, B. J., Goddard, M. E. & Visscher, P. M. Complex trait prediction from genome data: contrasting EBV in livestock to PRS in humans: genomic prediction. Genetics 211, 1131–1141 (2019).
Meuwissen, T. H., Hayes, B. J. & Goddard, M. E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157, 1819–1829 (2001).
Choi, S. W., Mak, T. S. & O’Reilly, P. F. Tutorial: a guide to performing polygenic risk score analyses. Nat. Protoc. 15, 2759–2772 (2020).
Ma, Y. & Zhou, X. Genetic prediction of complex traits with polygenic scores: a statistical review. Trends Genet. 37, 995–1011 (2021).
Wray, N. R., Goddard, M. E. & Visscher, P. M. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 17, 1520–1528 (2007).
Purcell, S. M. et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752 (2009).
Dudbridge, F. Power and predictive accuracy of polygenic risk scores. PLoS Genet. 9, e1003348 (2013).
Wray, N. R. et al. Research review: polygenic methods and their application to psychiatric traits. J. Child Psychol. Psychiatry 55, 1068–1087 (2014).
Euesden, J., Lewis, C. M. & O’Reilly, P. F. PRSice: polygenic risk score software. Bioinformatics 31, 1466–1468 (2015).
Chatterjee, N., Shi, J. & García-Closas, M. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat. Rev. Genet. 17, 392–406 (2016).
Privé, F., Vilhjálmsson, B. J., Aschard, H. & Blum, M. G. B. Making the most of clumping and thresholding for polygenic scores. Am. J. Hum. Genet. 105, 1213–1221 (2019).
Vilhjálmsson, B. J. et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet. 97, 576–592 (2015).
Privé, F., Arbel, J. & Vilhjálmsson, B. J. LDpred2: better, faster, stronger. Bioinformatics 36, 5424–5431 (2021).
Ge, T., Chen, C. Y., Ni, Y., Feng, Y. A. & Smoller, J. W. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 10, 1776 (2019).
Hahn, G. et al. A smoothed version of the lassosum penalty for fitting integrated risk models using summary statistics or individual-level data. Genes https://doi.org/10.3390/genes13010112 (2022).
Privé, F., Arbel, J., Aschard, H. & Vilhjálmsson, B. J. Identifying and correcting for misspecifications in GWAS summary statistics and polygenic scores. HGG Adv. 3, 100136 (2022).
Zhou, G. & Zhao, H. A fast and robust Bayesian nonparametric method for prediction of complex traits using summary statistics. PLoS Genet. 17, e1009697 (2021).
Lloyd-Jones, L. R. et al. Improved polygenic prediction by Bayesian multiple regression on summary statistics. Nat. Commun. 10, 5086 (2019).
Zhang, Q., Privé, F., Vilhjálmsson, B. & Speed, D. Improved genetic prediction of complex traits from individual-level data or summary statistics. Nat. Commun. 12, 4192 (2021).
Robinson, M. R. et al. Genetic evidence of assortative mating in humans. Nat. Hum. Behav. 1, 0016 (2017).
Ruan, Y. et al. Improving polygenic prediction in ancestrally diverse populations. Nat. Genet. 54, 573–580 (2022).
Miao, J. et al. Quantifying portable genetic effects and improving cross-ancestry genetic prediction with GWAS summary statistics. Nat. Commun. 14, 832 (2023).
Gunn, S. et al. Comparison of methods for building polygenic scores for diverse populations. HGG Adv. 6, 100355 (2025).
Pain, O. et al. Evaluation of polygenic prediction methodology within a reference-standardized framework. PLoS Genet. 17, e1009021 (2021).
Monti, R. et al. Evaluation of polygenic scoring methods in five biobanks shows larger variation between biobanks than methods and finds benefits of ensemble learning. Am. J. Hum. Genet. 111, 1431–1447 (2024).
Pain, O. Leveraging global genetics resources to enhance polygenic prediction across ancestrally diverse populations. HGG Adv. 6, 100482 (2025).
Ni, G. et al. A comparison of ten polygenic score methods for psychiatric disorders applied across multiple cohorts. Biol. Psychiatry 90, 611–620 (2021).
Wang, C., Zhang, J., Veldsman, W. P., Zhou, X. & Zhang, L. A comprehensive investigation of statistical and machine learning approaches for predicting complex human diseases on genomic variants. Brief. Bioinform. https://doi.org/10.1093/bib/bbac552 (2023).
Yang, S. et al. PGSFusion streamlines polygenic score construction and epidemiological applications in biobank-scale cohorts. Genome Med. 17, 77 (2025).
Yang, S. & Zhou, X. PGS-server: accuracy, robustness and transferability of polygenic score methods for biobank scale studies. Brief. Bioinform. https://doi.org/10.1093/bib/bbac039 (2022).
Kulm, S., Mezey, J. & Elemento, O. Benchmarking the accuracy of polygenic risk scores and generative methods. Preprint at medRxiv https://doi.org/10.1101/2020.04.06.20055574 (2020).
Ge, T. et al. Development and validation of a trans-ancestry polygenic risk score for type 2 diabetes in diverse populations. Genome Med. 14, 70 (2022).
Zheng, Z. et al. Leveraging functional genomic annotations and genome coverage to improve polygenic prediction of complex traits within and between ancestries. Nat. Genet. 56, 767–777 (2024).
Guo, W. et al. Polygenic risk score and heritability estimates reveals a genetic relationship between ASD and OCD. Eur. Neuropsychopharmacol. 27, 657–666 (2017).
Wang, Y., Tsuo, K., Kanai, M., Neale, B. M. & Martin, A. R. J. A. r. o. b. d. s. Chall. opportunities developing more generalizable polygenic risk scores 5, 293–320 (2022).
Visscher, P. M., Yengo, L., Cox, N. J. & Wray, N. R. Discovery and implications of polygenicity of common diseases. Science 373, 1468–1473 (2021).
Grotzinger, A. D., Fuente, J., Privé, F., Nivard, M. G. & Tucker-Drob, E. M. Pervasive downward bias in estimates of liability-scale heritability in genome-wide association study meta-analysis: a simple solution. Biol. Psychiatry 93, 29–36 (2023).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Voss, K., Auwera, G. A. V. d. & Gentry, J. Full-stack genomics pipelining with GATK4 + WDL + Cromwell. F1000Res. 6, 2–4 (2017).
Jette, M. A. & Wickberg, T. Architecture of the Slurm Workload Manager. In Job Scheduling Strategies for Parallel Processing (eds Klusáček, D., Corbalán, J. & Rodrigo, G. P.) Lecture Notes in Computer Science, Vol. 14283, 3–23 (Springer, Cham, 2023).
Yoo, A. B., Jette, M. A. & Grondona, M. SLURM: Simple Linux Utility for Resource Management. In Job Scheduling Strategies for Parallel Processing (eds Feitelson, D., Rudolph, L. & Schwiegelshohn, U.) Lecture Notes in Computer Science, Vol. 2862, 44–60 (Springer, Berlin Heidelberg, 2003).
DiSantostefano, J. International classification of diseases 10th revision (ICD-10). J. Nurse Pract. 5, 56–57 (2009).
Zhao, H. et al. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007 (2014).
Speed, D. & Balding, D. J. SumHer better estimates the SNP heritability of complex traits from summary statistics. Nat. Genet. 51, 277–284 (2019).
Xinyu, C. PGS-hub_offline: code for analyses reported in this paper. Zenodo https://doi.org/10.5281/zenodo.17994509 (2025).
Acknowledgements
Dr. Wang is supported by the Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDA0460400, and the Pioneering Action Grants of the Chinese Academy of Sciences, National Natural Science Foundation of China, Grant No. 82470352. This research has been conducted using the UK Biobank Resource (Application Approval Number: 89885), and we thank the volunteers participating.
Author information
Authors and Affiliations
Contributions
M.W., G.Z., F.W., and X.C. conceived and designed the study. X.C., F.W., Y.A., X.Y., T.X., Q.C., R.Z., K.Y., and K.Z. performed the comparative analyses. H.Z., J.H., and Y.Z. developed and implemented the website. F.W. and X.C. drafted the manuscript. F.W., X.C., and M.W. critically revised the manuscript. A.C.F. and G.Z. contributed important intellectual content. F.W. and M.W. supervised the study. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
Dr. Fahed reports being a co-founder of Goodpath and Avigena, and serving as a scientific advisor to MyOme, Arboretum Health, HeartFlow, and Aditum Bio. These entities had no role in the design of the study, data collection, analysis, interpretation of the results, or the writing of the manuscript, and are not directly related to the subject matter of this research. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, X., Wang, F., Zhao, H. et al. Comprehensive benchmarking single and multi ancestry polygenic score methods with the PGS-hub platform. Nat Commun 17, 2014 (2026). https://doi.org/10.1038/s41467-026-68599-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68599-7
