
Abstract
The evolution of SARS-CoV-2, coupled with its immune evasion mechanisms, underscores the urgent need for antiviral strategies beyond vaccination. The papain-like protease (PLpro) is a dual-functional enzyme essential for viral replication and suppression of host innate immunity. Here, we present the rational design and characterization of YL1004, a tricyclic oral PLpro inhibitor demonstrating robust, cross-variant SARS-CoV-2 antiviral activity and favorable pharmacokinetic properties. YL1004 potently inhibits the enzymatic function of PLpro, disrupts deubiquitination and deISGylation processes, and restores antiviral immune signaling cascades. Notably, YL1004 suppresses the replication of SARS-CoV-2 wildtype, Delta and Omicron variants. Additionally, it is also effective against the Mpro E166V recombinant SARS-CoV-2 strain, which confers resistance to nirmatrelvir. In the lethal SARS-CoV-2 infection model using K18-hACE2 mice, YL1004 confers complete protection to animal survival, significantly reduces viral load in nasal turbinate and lung tissues, and alleviates virus-induced pathological tissue damages. Co-crystal structural analysis revealed a distinctive binding mode, enhancing target engagement through expanded hydrophobic interactive interface and additional hydrogen bonding interactions. Collectively, these findings establish YL1004 as a promising therapeutic candidate, harnessing dual antiviral and immunomodulatory mechanisms to combat SARS-CoV-2 and emerging variants.
Similar content being viewed by others

Introduction
The COVID-19 pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has resulted in over 777 million infections and more than 7.08 million deaths as of March 20251. While COVID-19 vaccines have in part mitigated the virus’s capacity in causing severe infections2,3, the emergence of Omicron and its sublineages with significant immune escape has showed substantially reduced sensitivity to therapeutic antibodies and undermined vaccine efficacies4,5,6,7. Given SARS-CoV-2’s anticipated long-term circulation and pandemic potential8, developing alternative antiviral strategies beyond vaccination remains crucial for addressing both current and future threats.
Papain-like protease (PLpro) of SARS-CoV-2 mediates the cleavage of coronavirus polyprotein pp1a and pp1ab at the N-terminal to release Non-structural protein 1-3 (Nsp1-3), which is required for virus transcription and replication. For example, Nsp1 of SARS-CoV-2 has been implicated in shutdown of the host translation machinery, thus suppressing the host innate immune responses while allowing efficient expression of the viral proteins to facilitate virus replication9,10. Interestingly, apart from its proteolytic activity PLpro is a multi-functioned viral protein, which also plays critical roles in deubiquitination and deISGylation11,12,13,14,15. Inhibition of PLpro offers dual therapeutic benefits by simultaneously suppressing viral replication and restoring host innate immunity. Therefore, PLpro represents a promising therapeutic target for the treatment of SARS-CoV-2 infection. Despite successful development of inhibitors targeting other viral components - including RNA-dependent RNA polymerase (RdRp) inhibitors (Remdesivir16, Molnupiravir17, Renmindevir18, Azvudine19) and main protease (Mpro) inhibitors (Nirmatrelvir20, Ensitrelvir21, Simnotrelvir22, and Leritrelvir23), PLpro inhibitor development has proven challenging. Earlier PLpro inhibitors demonstrated either suboptimal antiviral activity or unfavorable pharmacokinetic profiles, leading to clinical trial failures24,25,26,27,28,29,30,31,32,33. Recent advances with orally active PLpro inhibitors (Jun1268234, PF-0795747235, GZNL-P3636, WEHI-P837 and Jun1329638) in murine SARS-CoV-2 models are promising.
In this investigation, we report the design and characterization of YL1004, a potent and highly selective SARS-CoV-2 PLpro inhibitor featuring a tricyclic scaffold and favorable pharmacokinetic properties. YL1004 demonstrated potent antiviral efficacy against all evaluated SARS-CoV-2 variants (Delta, Omicron JN.1, and Omicron KP.3) and nirmatrelvir-resistant SARS-CoV-2 mutant. Mechanistically, YL1004 directly targets PLpro and effectively neutralizes PLpro-dependent immune evasion, underscoring its strong antiviral potential and selectivity. In JN.1-infected K18-hACE2 transgenic mice, oral treatment with YL1004 significantly reduced viral burdens and also ameliorated virus-induced pathologies in the nasal turbinate and lung tissues of infected mice. Strikingly, therapeutic treatment with YL1004 conferred full protection to the infected mice against lethal SARS-CoV-2 Delta challenge. Overall, these findings position YL1004 as a promising clinical candidate for developing next-generation antiviral therapies targeting SARS-CoV-2.
Results
Discovery of PLpro inhibitors with a tricyclic scaffold
To identify novel hit compounds for PLpro-targeted drug design, we first screened an in-house chemical library of ~35,000 compounds using a fluorescence resonance energy transfer (FRET) assay. The library consists of structurally diverse small molecules derived from our previous antiviral discovery efforts, as well as compounds with known activity against other targets such as proteases and kinases that were developed in house. These molecules include both covalent and noncovalent scaffolds with functionalities suitable for engaging cysteine proteases. Compounds that inhibited PLpro activity by more than 70% were considered primary hits. Five compounds demonstrated PLpro inhibitory activity with 50% inhibition concentration (IC50) values below 50 μM (Supplementary Fig. 4). Among these, Hit-1 (1a), featuring a unique tricyclic scaffold, emerged as the most promising lead compound with an IC50 of ~ 26.82 μM.
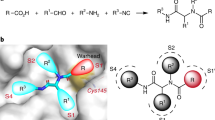
To guide structural optimization, we compared 1a with reported PLpro inhibitors24,25,26,27,28,29,30,31,32,33. Interestingly, molecular superposition revealed partial overlap between the tricyclic core of 1a (blue) and the benzamide group of classical PLpro inhibitor GRL061724 (pink) (Fig. 1a, b)39. Leveraging this observation, we matched up 1a with GRL0617 via a fragment merging strategy to design and synthesize enantiomeric compounds 2a and 2b (Fig. 1a). The (S)-enantiomer 2b exhibited significantly higher potency (IC50 = 98.6 nM) than its (R)-counterpart 2a (IC50 = 5.77 μM), demonstrating a 50-fold activity difference (Fig. 1c). Differential scanning fluorimetry (DSF) assays further confirmed direct binding to PLpro, with ΔTm values of 3.45 °C for 2a and 10.04 °C for 2b (Fig. 1d).

a Chemical structures of Hit-1 (1a), 2a, 2b, 2c 2 d and design strategy of tricyclic PLpro inhibitors by rational fragment merging. IC50 values were determined using a FRET-based enzymatic assay measuring viral polyprotein cleavage inhibition, and ΔTm values were obtained from differential scanning fluorimetry (DSF). b 1a (blue) partially overlaps with the benzamide group of classical PLpro inhibitor GRL0617 (pink) through molecular superposition. c Dose-activity curves of 2a, 2b, 2c, 2 d and GRL0617 against SARS-CoV-2 PLpro in the FRET assay. d Differential scanning fluorimetry analysis of the effect of 2a, 2b, 2c, 2 d and GRL0617 on SARS-CoV-2 PLpro stability. Exposure of hydrophobic residues monitored by an increase in relative fluorescence units (RFUs). Curves represent the average of three experiments. e Structural optimization regions of compound 2b and the molecular structure and drug properties of YL1004. IC50 values were determined by FRET enzymatic assay. Cytotoxicity CC50 was determined in Vero E6 cells with 72-hour incubation. Data in a, c, d, and e are presented as mean ± SD (n = 3 biological replicates), with the exception of the GRL0617 treated group in d (n = 2). Images in (b) were processed by using PyMOL (https:// pymol.org).
Notably, the tricyclic phenylamino group in 2b differed structurally from the phenylmethyl group in GRL0617. To this end, we synthesized compound 2 d, which differs from GRL0617 only by the replacement of the methyl group with an amino group on the benzene ring. Enzymatic evaluation revealed a substantial decrease in activity for 2 d (IC50 = 10.97 ± 0.33 μM) relative to GRL0617. Correspondingly, differential scanning fluorimetry (DSF) indicated reduced thermal stabilization (ΔTm = 2.67 ± 0.33 °C), suggesting weaker binding affinity. This loss of activity may be attributed to the absence of both the N-methylpiperazinyl group and the (S)-tricyclic core, which appear essential for properly orienting the anilino moiety to form hydrogen bonds with PLpro. To further probe the role of the N-methylpiperazine substitution, we prepared analog 2c by introducing this group into the 2 d scaffold. Compound 2c showed improved potency (IC50 = 337.7 nM) and a greater thermal shift (ΔTm = 9.13 °C) compared to 2 d, yet its activity remained 3.4-fold lower than that of the (S)-enantiomer 2b. The significant activity difference between 2a and 2b further highlights the importance of the tricyclic scaffold’s stereochemistry for PLpro inhibition. Together, these results underscore the synergistic contribution of the (S)-tricyclic core and the anilino modification to inhibitor binding (Fig. 1a, c, d).
Subsequent optimization focused on modifying the R1, R2, and R3 groups of the tricyclic core (Fig. 1e), yielding 47 derivatives. A complete list of the designed tricyclic PLpro inhibitors is shown in Supplementary Fig. 1–3, with representative examples in Table 1. Initial R1 modifications while fixing R2/R3 produced five analogs (2e-i; Supplementary Fig. 1). Notably, the geminal-cyclopropyl-substituted compound 2i showed a 5-fold potency improvement (IC50 = 18.4 nM) over 2b. However, compound 2i exhibited moderate cytotoxicity in Vero E6 cells (CC50 = 74.06 μM), suboptimal oral PK (AUC(0-t) = 9291 μg/L·h at 100 mg/kg), and CYP3A4 inhibition (IC50 = 1.87 μM), prompting further optimization. Next, R2 optimization with fixed R1 (cyclopropyl) and R3 generated nine analogs (3a–i; Supplementary Fig. 1). Unfortunately, none of these improved PLpro inhibition. Finally, R3 modifications while retaining R1/R2 yielded 34 compounds (4a–l, 5a–h, Supplementary Fig. 2; 6a–n; Supplementary Fig. 3), with 24 achieving IC50 ≤ 30 nM. Among them, compound 4b emerged as the most potent derivative, showing a 121-fold potency increase (IC50 = 12.9 nM) over GRL0617. Nevertheless, 4b retained cytotoxicity (CC50 = 38.99 μM) and CYP inhibition (CYP3A4 IC50 = 1.67 μM; CYP2C9 IC50 = 0.685 μM), likely due to metabolic toxicity from the naphthalene 4-methyl group.
Based on the molecular superposition results (Fig. 1b), which indicated that the naphthalene 4-position points toward the solvent-exposed region, we hypothesized that chemical modification at this site would minimally affect the compound’s inhibitory activity against PLpro. This hypothesis was corroborated by our structure-activity relationship (SAR) data, which demonstrated that derivatives modified at this position generally maintained IC50 values against PLpro below 50 nM. Consequently, we introduced diverse aromatic and heterocyclic substituents at this position to further optimize pharmacokinetic properties. Several derivatives (2i, 4e, 4k, 4 l, 6k, 6 f, 6 g, 6 h, 6i) achieved balanced activity, reduced cytotoxicity, and improved CYP profiles (Table 1). These compounds would undergo further evaluation to identify a clinical development candidate.
Selection of YL1004 as the candidate compound for further studies
Building on structure-activity relationship (SAR) insights and preliminary safety profiles, we systematically evaluated nine prioritized derivatives (2i, 4e, 4k, 4 l, 6k, 6 f, 6 g, 6 h, 6i) alongside the positive control Jun12682. MTT assays across four cell lines (VeroE6, BEAS-2B, HUVEC, and 293 T) revealed that compound 6 f (designated as YL1004) exhibited the most favorable safety profile, demonstrating significantly reduced cytotoxicity in VeroE6 cells (CC50 = 226.90 μM) compared to Jun12682 (CC50 = 59.34 μM) (Supplementary Table. 2). Pharmacokinetic (PK) studies in Balb/c mice following oral administration (100 mg/kg) further highlighted YL1004’s advantages, with an oral AUC of 79,818 h·ng·mL⁻¹ with 61.2% bioavailability (F), outperforming all other derivatives (Supplementary Table. 3). Additionally, cross-species pharmacokinetic evaluation in rats showed that YL1004 maintained favorable PK properties, attaining an oral AUC of 14,723 h·ng·mL⁻¹ at 10 mg/kg with 62.9% bioavailability (Supplementary Table. 4). Enhanced target engagement was further confirmed by differential scanning fluorimetry (DSF), where YL1004 induced a dose-dependent increase in PLpro thermal denaturation temperature (ΔTm = 18.07 °C at equivalent concentrations), surpassing Jun12682 (Supplementary Fig. 5). Consistent with the DSF findings, tight binding kinetic analysis using the Morrison equation yielded a Kᵢ of 2.3 ± 0.3 nM for YL1004, further substantiating its high inhibitory potency (Supplementary Table. 5).
Comprehensive profiling of YL1004 highlighted its favorable drug-like properties: promising metabolic stability in human liver microsomes (in vitro clearance = 20.07 mL/min/kg, T1/2 = 57.75 min; Supplementary Table. 6); moderate plasma protein binding (89.57%; Supplementary Table. 7); optimal solubility (134.22 μg/mL); and superior permeability (Papp = 8.17 × 10-6 cm/s in MDCK monolayers) (Supplementary Table. 8). Safety assessments further demonstrated YL1004’s high selectivity, showing very weak or no activity against 377 human kinases (<50% inhibition at 10 μM; Supplementary Table. 9), negligible effects on major cytochrome P450 isoforms (CYP1A2, CYP2C8, CYP2C9, CYP2D6, and CYP3A4; IC50 > 30 μM; Supplementary Table. 10), and no significant inhibition of the hERG channel (IC50 > 50 μM). Collectively, these findings underscore YL1004’s low risk of drug-drug interactions and cardiotoxicity.
In summary, YL1004 integrates target selectivity, robust safety, favorable pharmacokinetics, and balanced drug-like properties, positioning it as a promising therapeutic clinical candidate for further in-depth studies.
Co-crystal structure of SARS-CoV-2 PLpro in complex with YL1004
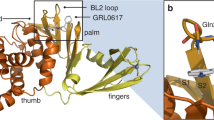
To obtain the co-crystal structure of PLpro in complex with YL1004, we generated a catalytically inactive mutant (PLpro-Cys111Ser) and determined the complex structure at 2.7 Å resolution (Fig. 2a-d). The SARS-CoV-2 PLpro consists of an N-terminal ubiquitin-like (Ubl) domain and a catalytic domain comprising thumb, palm, and finger subdomains (Fig. 2b). The catalytic triad (Cys111, His272, and Asp286) is positioned at the palm-thumb interface. YL1004 binds within the pocket formed by the flexible blocking loop 2 (BL2) and S3/S4 protein subsites (Fig. 2c). The N-methylpiperazinyl moiety of YL1004 forms a 2.9 Å hydrogen bond with the side chain of Glu167, while the aniline group establishes a 3.1 Å hydrogen bond with Tyr264 (Fig. 2d). Additionally, the piperazinyl-benzotetrahydropyran moiety participates in extensive hydrophobic interactions with Leu162, Gly163, Asp164, and Tyr268. The central amide group makes hydrogen bonds with Gln269 (2.9 Å) and Asp164 (2.6 Å), respectively. The cyclopropyl group forms hydrophobic interactions with the side chains of Pro248, Tyr264 and Thr301. Furthermore, the naphthyl ring, which is sandwiched between Met208, Pro247, Pro248 and Tyr268, forms a T-shaped π-π stacking interaction with Tyr268 and hydrophobic contacts with residues Met208, Pro247 and Pro248 (Fig. 2d). The tetrahydropyran moiety also contributes to hydrophobic interactions with residue Pro248.

a Chemical structure of YL1004. b, Cartoon view of SARS-CoV-2 PLpro, color-coded by domain: Ubl domain (green), thumb domain (yellow), palm domain (pink), and finger domain (purple). The zinc ion is shown as a sphere (wheat). The N and C termini of PLpro are labeled. c, YL1004 interacts with the BL2 loop. YL1004 is represented as a violet stick model, and the 2Fo - Fc density map (gray mesh, σ = 1.5) is shown. d, Interactions between YL1004 and PLpro. Hydrogen bonds are displayed as black dashed lines. e-g, Comparative analysis of YL1004 and GRL0617 (PDB code: 7JRN) binding modes. Superimposed structures of PLpro (white)-YL1004 (violet) and PLpro-GRL0617 (blue). The hydrogen bonds of GRL0617 are displayed as yellow dashed lines. The structural displacement of the naphthyl ring is shown (arrow). Figure b-g were generated using PyMOL (http://www.pymol.org).
The binding mode of YL1004 differs from that of several reported representative PLpro inhibitors, including GRL061724 (Fig. 2e, f), Jun1268234, PF-0795747235, GZNL-P3536, and WEHI-P437 (Supplementary Fig. 6a–d). Taking GRL0617 as an example, the strategic amino group substitution on the benzamide moiety of YL1004 forms additional hydrogen bonds with Tyr264, thereby significantly enhancing its binding affinity for PLpro (Fig. 2e). Although both compounds utilize the central amide for hydrogen bonding with Asp164 and Gln269, YL1004 exhibits shorter bond distances, suggesting enhanced stabilization (Fig. 2f). Furthermore, the methyl-to-cyclopropyl substitution in YL1004 enhances hydrophobic interactions. The extended tetrahydropyran moiety enhances hydrophobic interactions with Pro248 and promotes an upward shift (1.2 Å) of the naphthyl ring, facilitating additional hydrophobic interactions with Met208 (Fig. 2g).
YL1004 antagonizes PLpro-mediated innate immune suppression
Coronavirus PLpro is a multifunctional viral protease that hydrolyses viral polyprotein precursors. Recent studies have revealed that PLpro acts as an immunoregulatory enzyme that exerts deubiquitinating (DUB) and deISGylating activities, removing ubiquitin and ISG15 conjugates from key signaling mediators such as IRF3 to suppress type I interferon and NF-κB signaling and facilitate viral immune evasion40,41. We investigated whether YL1004 could offset these immunosuppressive effects by specifically inhibiting PLpro.
To measure the effects of YL1004 on the DUB and deISGylating activities of PLpro, we first used Ub-AMC and ISG15-AMC as fluorogenic substrates in FRET-based enzyme kinetic assays. YL1004 significantly reduced PLpro -catalyzed hydrolysis with IC50 values of 22.9 nM and 43.6 nM against Ub-AMC and ISG15-AMC, respectively, surpassing the potency of the reference compound Jun12682 (IC50 = 87.0 nM and 124.0 nM) (Fig. 3a). Consistent with the IC50 values of YL1004, tight binding kinetic analysis using the Morrison equation yielded Kᵢ values of 4.2 nM for deubiquitination and 39.3 nM for deISGylation, respectively (Supplementary Table. 11). To verify these findings in cells, HEK293T cells were co-transfected with plasmids expressing PLpro and ubiquitin. As shown in Fig. 3b, PLpro reduced the level of ubiquitinated proteins, whereas YL1004 restored ubiquitination in a dose-dependent manner, demonstrating that YL1004 inhibits PLpro-mediated deubiquitination. Remarkably, 1 µM YL1004 was more potent than 10 µM Jun12682 in suppressing the DUB activity of PLpro (Fig. 3b). In parallel, A549 cells stimulated with interferon-α (IFN-α) to induce ISGylation showed reduced ISG15-conjugated proteins upon PLpro expression. YL1004 co-treatment reversed this effect in a dose-dependent manner, indicating potent inhibition of PLpro-driven deISGylation (Fig. 3c).

a IC50 curves of YL1004 in inhibiting PLpro hydrolysis of Ub-AMC (left) and ISG-AMC (right), n = 3. b In-cell deubiquitinating activities of PLpro, HEK293T cells were transfected with plasmids encoding PLpro-Flag, Ub-HA, and indicated compounds alone or in combination. Then cell lysates were subjected to immunoblotting with anti-HA, anti-Flag, and anti-GAPDH antibodies. c HEK293T cells were treated with or without IFN-α for 48 h. Then the cell extracts were incubated with PLpro and indicated compounds, followed by immunoblotting analysis with anti-ISG15 and anti-PLpro antibody. d Counter screening of YL1004 in inhibiting common human DUBS hydrolysis of Ub-AMC (n = 2). e In cell specificity of YL1004 for PLpro over human DUBs. Lines 1 ~ 6: An anti-HA Western blot of lysed HEK293T cells treated with HA-Ub-VS in the presence of N-ethyl-maleimide (NEM, positive control inhibitor) or test compounds. Lines 7 ~ 12: PLpro was added to cell lysate before covalent modification by HA Ub-VS, showing that YL1004 eliminated PLpro-based modification. f–i YL1004 antagonized PLpro suppression on NF-κB (f), ISRE (g), IFN-β (h) or IRF3 (i) activation. Dual-luciferase reporter gene assays were performed in HEK293T cells (n = 3). Data in (a), (d), and (f-i) were shown as mean ± sd. Experiments (b), (c), and (e) were repeated three times independently with similar results. The samples were derived from the same biological source, and the corresponding gels and blots were processed in parallel to ensure consistency.
Given PLpro’s structural homology with human deubiquitinating enzymes (DUBs), we also assessed the selectivity of YL1004. FRET assays demonstrated that, at concentrations up to 50 μM, YL1004 suppressed the activities of common host DUBs by less than 50%, indicating a high degree of specificity (Fig. 3d). In a parallel investigation using the active-site probe HA-Ub–vinyl sulfone (VS), the positive control N-ethylmaleimide (NEM) broadly blocked DUB labeling in HEK293T cells, whereas YL1004 selectively interfered only with PLpro labeling, showing negligible impact on other DUBs (Fig. 3e). Moreover, in HEK293T cells lacking PLpro expression, YL1004 treatment did not alter IFN-α–induced ISGylation, reinforcing the notion that YL1004 does not interfere with host ubiquitination or ISGylation processes (Fig. 3c).
To elucidate the structural basis for YL1004’s selectivity, we performed structural alignments between PLpro and ten human DUBs. The calculated Cα RMSD values ranged from 9.95 to 31.52 Å (based on all Cα atoms, Supplementary Fig. 7), indicating substantial overall structural divergence. With respect to the substrate-binding pockets, UCHL1 adopts a configuration completely distinct from that PLpro (Supplementary Fig. 7b, c). Of note, among the tested DUBs, USP2 possesses a substrate-binding pocket most closely resembling that of PLpro (Supplementary Fig. 7a), however, a loop spanning residues 503–516 would cause steric clashes with the naphthyl ring and the tetrahydropyran moiety of YL1004 (Supplementary Fig. 7d, e). This structural incompatibility provides a mechanistic explanation for the lack of USP2 inhibition by YL1004. Overall, these findings could provide structural insight into the selectivity of YL1004 toward human USPs.
SARS-CoV-2 PLpro was implicated in antagonizing the host type I interferon and NF-κB pathways14. To this end, we further performed luciferase-based reporter assays to characterize the efficacies of YL1004 on the activation of IFN and NF-κB signalling with the expression of PLpro. Our luciferase reporter assays demonstrated that PLpro suppressed TNF-α–induced NF-κB activation, IFN-α–induced ISRE activation, and poly(I:C)–induced IFNB1/IRF3 promoter activity, whereas YL1004 counteracted these suppressions and restored the activation of the evaluated signaling pathways by targeting PLpro (Fig. 3f–i). To more comprehensively elucidate the underlying mechanism of YL1004 treatment, we performed transcriptomic profiling in PLpro-transfected cells with or without YL1004 treatment. Poly(I:C) was used as a surrogate for viral RNAs to stimulate innate immune responses. Our data indicated that PLpro expression broadly repressed genes involved in innate immunity (e.g., IFI6, ISG20, TNFRSF10D), autophagy (ULK4, SESN1), and mitochondrial stress response (FDXR, RASSF1), suggesting a concerted suppression of antiviral defenses (Fig. 4a). GO enrichment confirmed inhibition of immune-related pathways, including NF-κB and interleukin-7 signaling (Fig. 4b), consistent with functional immune suppression. YL1004 reversed these effects, restoring genes linked to immune activation (IFITM10, NFKBIB, CD86), oxidative stress control (GCLM, FDXR), and autophagy (DRAM1, MAP2K5). Enriched terms included “interleukin-1β production”, “NF-κB activation”, and “immunoglobulin response” (Fig. 4c). These findings indicate that YL1004 reactivates suppressed antiviral programs at transcriptional level.

a Heatmap of representative genes significantly regulated by YL1004 with/without PLpro expression in the context of poly(I:C). b GO enrichment analysis of genes downregulated by PLpro. c GO enrichment analysis of genes upregulated by YL1004. Data were obtained from two biological replicates.
Together, our findings indicate that YL1004 effectively antagonizes PLpro-dependent innate immune suppression, therefore further potentiating its antiviral efficacies. Further evaluation of its drug-like properties and efficacy is warranted to confirm its promise as an antiviral agent.
YL1004 protects against multiple SARS-CoV-2 variants and the nirmatrelvir-resistant SARS-CoV-2 mutant
To examine the in vitro antiviral potency of YL1004, we infected Calu3 with wildtype SARS-CoV-2 and variants (Delta, Omicron JN.1 and Omicron KP.3). Given the potency of YL1004 shown in enzymatic assays, we decided to perform 4-fold serial dilution starting from 10 µM. Our data suggested YL1004 dose-dependently decreased the subgenomic RNA of the viral envelope gene (sgE RNA) in Calu3 against wildtype and all tested SARS-CoV-2 variants. For wildtype, Delta and JN.1, YL1004 decreased viral gene copies by over 1 log at 2.5 µM (Fig. 5a). To better describe the in vitro antiviral efficacy of YL1004, we performed side-by-side comparison with Jun12682 we found YL1004 suppressed the replication of SARS-CoV-2 more efficiently at 2.5 and 10 µM when compared to Jun12682 (Supplementary Fig. 8). Additionally, we also examined its efficacy against the recombinant SARS-CoV-2 carrying the E166V substitution in Nsp5, which predominantly contributes to nirmatrelvir-resistance42,43,44. Our data showed that YL1004 inhibited the nirmatrelvir-resistant NSP5-E166V-rSARS-CoV-2 as effectively as that against Omicron KP.3. To examine the efficacy of YL1004 in reducing infectious viral progenies, we performed plaque reduction assays against the matching SARS-CoV-2 strains. In keeping with our findings with viral gene copy quantification, YL1004 efficiently decreased the infectious viral titers in SARS-CoV-2-infected Calu3, with the EC50 ranging from 0.08 µM to 0.50 µM (Fig. 5b). Based on the corresponding cytotoxicity values obtained in the same Calu3 cellular background (CC50 = 296.33 µM), the calculated selectivity indices (CC50/EC50) of YL1004 ranged from approximately 102 to 103 across tested viral variants, confirming a wide therapeutic window and favorable cellular safety profile (Supplementary Fig. 9).

a Quantification of the subgenomic envelope (sgE) gene in Calu3 cells (n = 6) infected with wildtype SARS-CoV-2 and Delta, Omicron (JN.1 and KP.3) variants and NSP5-E166V-carrying recombinant SARS-CoV-2 in the presence or absence of YL1004. Lysates were harvested at 24 hpi. (for wildtype, Delta and JN.1) and 48 hpi. (for KP.3 and NSP-E166V recombinant SARS-CoV-2) one-step reverse transcription and quantitative polymerase chain reaction (RT-qPCR) analysis. b VeroE6-TMPRSS2 cells (n = 4) were infected with 100 PFU wildtype SARS-CoV-2 and wildtype SARS-CoV-2 and Delta, Omicron (JN.1 and KP.3) variants and NSP5-E166V-carrying recombinant SARS-CoV-2 in the presence or absence of serially diluted YL1004. Cells were fixed at 48 (for wildtype and Delta) or 72 hpi. (for JN.1, KP.3 and NSP-E166V recombinant SARS-CoV-2) for visualization of plaque formation. Number of plaques were normalized to those recovered from supernatants with vehicle treatment only. IC50 was determined using nonlinear regression model (normalized response without variable slope). Each data point represents one biological repeat. Data represent mean ± SD from the indicated number of biological replicates, obtained from two or three independent experiments. Statistical significance was determined using one-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test for p value adjustment in multiple comparisons (a). * represented P < 0.05 and ** represented P < 0.01. NS, not statistically significant. WT, wildtype SARS-CoV-2.
Importantly, the EC50 of YL1004 against NSP5-E166V-rSARS-CoV-2 was 1.37 µM, suggesting its antiviral efficacy against the nirmatrelvir-resistant virus was largely not affected. In sharp contrast, E166V mutation caused up to 288-fold increase in the EC50 in vitro when treated with nirmatrelvir45. In conclusion, our in vitro data demonstrates that YL1004 potently protects against multiple SARS-CoV-2 variants and that YL1004 maintains its strong antiviral efficacy against the nirmatrelvir-resistant mutant virus.
YL1004 rescued infected mice from lethal SARS-CoV-2 challenge
To explore the in vivo antiviral potency of YL1004 against severe SARS-CoV-2 infection, we used the K18-hACE2 transgenic mouse model, which supports lethal SARS-CoV-2 challenge46,47. We first examined whether the in vivo pharmacokinetic exposure of YL1004 was sufficient to support antiviral activity. As shown in Supplementary Fig. 10, following oral administration of YL1004 at 100 mg/kg, plasma concentrations remained above the in vitro EC50 and EC90 values for more than 24 h and approximately 16 h, respectively. These data indicate that the in vivo antiviral efficacy of YL1004 is supported by sufficient systemic exposure. We next intranasally challenged the transgenic mice with 10000 PFU SARS-CoV-2 Omicron JN.1 and harvested the nasal turbinate and lung tissues at 3 days post infection (dpi.) for quantification of viral sgE RNA copies. Mice in the treatment groups were orally treated twice per day with 100 mg/kg of drug. We found that YL1004 treatment decreased viral loads in the nasal turbinates by 3.6-fold (P = 0.0457) when compared to vehicle group (Fig. 6a). Notably, YL1004 decreased viral gene copies by 26.1-fold (P = 0.0049) in the infected lung tissues, which is more potent than Jun12682 (4.6-fold decrease, P = 0.0132) (Fig. 6a). Consistently, YL1004 also lowered the infectious viral titers in the nasal turbinate lung tissues by 21- (P = 0.0107) and 13.4-fold (P = 0.0119), respectively (Fig. 6b). In addition to the virological assessments, we further evaluated whether YL1004 treatment might rescue mice from lethal SARS-CoV-2 challenge. Therefore, we infected the transgenic mice with the sublethal dose of SARS-CoV-2 Delta variant (1250 PFU/mouse). Oral treatment with YL1004 or vehicle was initiated at 1 h post virus challenge and was given twice per day. The infected mice treated with YL1004 experienced less body weight loss when compared to both vehicle (P = 0.0298) and Jun12682 (P = 0.0032) (Fig. 6c). In the survival analysis, only one out of ten mice (1/10, 10% survival) in the control group survived, which is in stark contrast with mice in the experiment group that all challenged subjects (10/10, 100% survival, P < 0.0001) were rescued by YL1004 therapeutic treatment at 14 dpi. (Fig. 6d). We continued to examine the expression of viral antigen in lung tissues of the infected transgenic mice. While multi-focal patches of viral nucleocapsid proteins were detected by immunohistochemistry staining (indicated with black arrows) in the control mice, viral antigen was rarely detected in the YL1004-treated mouse lungs (Fig. 6e). Besides, pathological findings including inflammatory infiltrations, alveolar septa thickening hemorrhage, alveolar hyperinflation and alveoli collapse were extensively identified in the control mouse lungs (Fig. 6f). In comparison, virus-imposed pathological changes were significantly alleviated in the YL1004-treated mice and only mild alveolar deformation was found (Fig. 6f). Together, we conclude that YL1004 demonstrates potent in vivo antiviral efficacies as evidenced by the marked reduction of viral burdens, viral antigen expression and virus-induced pathological lesions in target organ tissues. Importantly, YL1004 is capable of rescuing infected animals from lethal SARS-CoV-2 challenge.

a,b 8- to 12-week-old female and male K18-hACE2 transgenic mice (n = 10) were intranasally challenged with 10000 PFU SARS-CoV-2 Omicron JN.1 strain. Mice in the treatment groups were orally treated with 100 mg/kg/dose YL1004 or Jun12682 twice per day. Control mice were treated with matching solvent only. Nasal turbinate and lung tissues were harvested at 3 dpi. for virological assessment by a RT-qPCR and b plaque assays. c,d 8- to 12-week-old female K18-hACE2 transgenic mice (n = 10) were intranasally challenged with 1250 PFU SARS-CoV-2 Delta strain. c Body weight and d survival of the infected mice were monitored for 14 days or until death of the animal. (e), Representative images of immunohistochemistry (IHC) staining which shows viral antigen expression (red) in the nasal turbinate and lung tissues of Omicron JN.1-infected mice (n = 3) at 3 dpi. f Representative images of haematoxylin-eosin (H&E) staining with the nasal turbinate and lung tissues of Omicron JN.1-infected mice (n = 3) at 3 dpi. Pathological findings were indicated as follows: inflammatory infiltrations (yellow dashed circle), alveolar collapse (cross) haemorrhage (asterisk), alveolar thickening (open arrowhead). Scale bar represents 200 µm and 100 µm under 10x and 20x magnification, respectively. Enlarged area was identified with dashed rectangle. Statistical significances were determined using Brown-Forsythe and Welch one-way ANOVA tests with Dunnett’s multiple comparisons test for P value adjustment in multiple comparisons (a,b), one-way ANOVA test (paired) with the Geisser-Greenhouse correction for sphericity (c) and log-rank (Mantel-Cox) tests (d). Each data point represents one biological repeat. Data represents mean ± SD from the indicated number of biological repeats. Data were obtained from three independent experiments. * represented P < 0.05 and ** represented P < 0.01. **** represented P < 0.0001. WT, wildtype SARS-CoV-2; Veh, vehicle.
Discussion
The ongoing emergence of SARS-CoV-2 variants continues to challenge conventional therapeutic strategies, highlighting the critical need for cross-variant SARS-CoV-2 activity antivirals targeting conserved viral elements. Among these, the papain-like protease (PLpro) stands out as a dual-functional viral enzyme essential for both polyprotein processing and evasion of host innate immunity. While the clinical translation of RdRp16,17,18,19 and Mpro20–23 inhibitors has shown considerable success, efforts to target PLpro has faced persistent hurdles due to limited potency, suboptimal pharmacokinetics, and inadequate immune reactivation.
In this study, we report YL1004, an orally bioavailable PLpro inhibitor, discovered through fragment-based drug discovery (FBDD). YL1004 features a tricyclic scaffold that confers nanomolar enzymatic potency, strong metabolic stability, and excellent oral pharmacokinetics, while exhibiting high selectivity over host deubiquitinases and a favorable safety profile. Its potent antiviral spectrum and well-balanced drug-like properties render it a compelling candidate for further clinical development.
Structural elucidation of the PLpro–YL1004 complex reveals a unique, multidimensional binding mode. Compared to the classical inhibitor GRL0617, YL1004 achieves enhanced engagement through a tripartite interaction strategy: (1) the tricyclic aniline moiety forms two key hydrogen bonds with Glu167, Tyr264; (2) the central amide group stabilizes the scaffold via additional hydrogen bonds with Asp164 and Gln269; and (3) hydrophobic substituents such as the naphthyl and tetrahydropyran rings anchor the inhibitor through π–π stacking and nonpolar interactions with Met208, Pro247, and Pro248. Notably, the conformational shift induced by the tetrahydropyran group expands the hydrophobic interface and reorients the naphthyl ring to establish novel contacts. This intricate network of polar and apolar interactions enables high-affinity, selective for PLpro inhibition.
Functionally, YL1004 exerts a dual antiviral mechanism by simultaneously suppressing PLpro’s protease activity and reversing its immune-suppressive effects. Cellular and transcriptomic analyses demonstrate that YL1004 restores antiviral immune pathways, including interferon signaling, autophagy, and mitochondrial stress response, which are broadly repressed by PLpro. Reporter assays further confirmed YL1004’s ability to recover NF-κB, IRF3, and IFNB1-driven transcriptional activity. This dual action, including the direct inhibition of viral replication and reactivation of host immunity—positions YL1004 as a distinct and effective antiviral modality.
Importantly, YL1004 shows potent activity against wild-type and SARS-CoV-2 variants, including Delta, JN.1, KP.3, and the nirmatrelvir-resistant NSP5-E166V mutant. In contrast to Mpro inhibitors whose efficacy is compromised by resistance mutations, YL1004 retains potency against the E166V variant, highlighting its orthogonal and complementary antiviral potential. In a lethal challenge model, YL1004 not only reduced viral loads and inflammation in the lung but also provided complete survival protection, outperforming the current benchmark PLpro inhibitor Jun12682.
To further demonstrate the comparative advantages of YL1004 over previously reported representative PLpro inhibitors (Jun12682 and PF-07957472), we synthesized both Jun12682 and PF-07957472 and evaluated representative drug-like property parameters under identical experimental conditions (Supplementary Table. 12). Our standardized testing reveals that YL1004 (IC50 = 17.5 nM) exhibits a PLpro IC50 approximately two-fold higher than Jun12682 (IC50 = 32.1 nM), while matching PF-07957472 (IC50 = 16.9 nM) in potency. In Vero CC50 (μM) cytotoxicity assays, YL1004 (CC50 = 226.90 μM) displays the lowest toxicity among tested compounds(Jun12682 CC50 = 59.34 μM, PF-07957472 CC50 = 70.07 μM), indicating a favorable safety profile. For CYP3A4 inhibition (IC50, μM), YL1004 shows the weakest inhibitory activity against this critical metabolic enzyme. Importantly, while literature cites CYP3A4 IC50 > 30 μM for both Jun12682 and PF-07957472, our experimental results contradict these values. In hERG cardiac safety assessments, YL1004 demonstrates exceptional safety margins (IC50 > 50 μM), surpassing PF-07957472 (28 μM), no hERG inhibition data is reported for Jun12682. Finally, pharmacokinetic (PK) studies in Balb/c mice following oral administration (100 mg/kg) confirm YL1004 achieves the highest oral AUC of 79,818 h·ng·mL⁻¹, outperforming Jun12682 (AUC(0-t) = 73121 h·ng·mL⁻¹) and demonstrating significant superiority over PF-07957472 (AUC(0-t) = 25218 h·ng·mL⁻¹). Additionally, cross-species pharmacokinetic evaluation in rats showed that YL1004 maintained favorable PK properties, attaining an oral AUC(0-t) of 14,723 h·ng·mL⁻¹ at 10 mg/kg, demonstrating favorable cross-species pharmacokinetic properties. In contrast, Jun12682 shows markedly reduced exposure (AUC(0-t) = 2236 h·ng·mL⁻¹) in rats, indicating significant species-specific differences in its pharmacokinetic behavior.
In summary, YL1004 represents a new generation of PLpro-targeted antivirals, offering structural innovation, cross-variant SARS-CoV-2 efficacy, immunomodulatory capacity, and resistance resilience. Its tricyclic architecture, multidimensional engagement with PLpro, and dual-action mechanism address critical unmet needs in coronavirus drug development. Future investigations may explore YL1004’s application in combination therapies, pan-coronaviral targeting, and prophylactic use in immunocompromised populations. These findings mark a significant advancement in host-directed antiviral strategies and pandemic preparedness.
Methods
Ethical Statement
All procedures related to animal in pharmacokinetic (PK) study and in vivo toxicity study were performed according to the guidelines approved by the Institutional Animal Care and Use Committee of West China Hospital, Sichuan University (20211063 A). The use of animals in antiviral study complied with all relevant ethical regulations and was approved by the Committee on the Use of Live Animals in Teaching and Research of The University of Hong Kong (24–128). The animals were kept in cages with individual ventilation under 65% humidity and an ambient temperature of 21 °C–23 °C and a 12 h–12 h day–night cycle for housing and husbandry. All donors of the virus strains involved in this paper have provided written consent and the work was approved by the Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster. All infectious experiments involving MERS-CoV, SARS-CoV-2 and SARS-CoV-1 followed the approved standard operating procedures of the Biosafety Level 3 facility at the Department of Microbiology, Hong Kong University
Compounds, cell lines, and viruses
The compounds (1a, 2a-h, 3a-i, 4a-l, 5a-h, 6a-n) shown in Fig. 1 and Fig. 2 were synthesized in our internal facilities. And the detailed synthesis methods and characterization data of these are listed in the supplementary materials (Supplementary Table. 1). MDCK (Cell Bank, Chinese Academy of Sciences, Shanghai, SCSP-5258), HUVEC (CCTCC, Wuhan, GDC0635), BEAS-2B (Cell Bank, Chinese Academy of Sciences, Shanghai, GNHu27), VeroE6 (ATCC, CRL-1586), and HEK293T (Cell Bank, Chinese Academy of Sciences, Shanghai, GNHu17) cells were maintained in Dulbecco’s Modified Eagle medium (DMEM, Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 unit/mL penicillin and 100 μg/mL streptomycin. Calu3 cells obtained from ATCC were maintained in DMEM/F12 supplemented with 10% FBS, 1% P/S. VeroE6-TMPRSS2 cells were obtained from the Japanese Collection of Research Bioresources (JCRB) Cell Bank and cultured also in 10% FBS, 1% P/S DMEM and 1 mM (1%) sodium pyruvate.
WT SARS-CoV-2 HKU-001a (GenBank: MT230904), B.1.617.2/Delta (GenBank: OM212471), B.1.1.529/Omicron JN.1 (GISAID: EPL_ISL_18841631) and KP.3 (GISAID: EPI_ISL_19535068) were isolated from patients with laboratory confirmed COVID-19 in Hong Kong48. Recombinant SARS-CoV-2 with nirmatrelvir resistance was constructed as we previously reported49. Briefly, the cDNA from ancestral SARS-CoV-2 HKU001a was assembled into the pSMART-BAC vector (E2621S, NEB, USA), followed by site-directed mutation of E166V in NSP5 which was confirmed by Sanger sequencing. The recombinant virus was rescued by transfection of the infectious clone into VeroE6-TMPRSS2 cells using Lipofectamine 3000 (L3000015, Thermo Fisher Scientific). All viruses were cultured in VeroE6-TMPRSS2 cells and titrated with plaque assays.
Protein Expression and Purification
The gene encoding SARS-CoV-2 (MN908947) papain-like protease (PLpro; ORF1ab residues 1564 - 1877) with an N-terminal His tag and TEV protease cleavage site was synthesized by General Biosystems following E. coli codon optimization. Using the recombinant plasmid as a template, site-directed mutagenesis PCR was performed with primers 5’-agataataatagttatctggcaaccgcactgctgaccctg-3’ and 5’-tgccagataactattattatctgcccacttaatactggtc-3’ (mutated codons shown in italics) to mutate the catalytic cysteine to serine (PLpro-Cys111Ser). The PCR product was digested with DpnI (TakaRa) and transformed into E. coli DH5α competent cells (Novagen). Single colonies were selected, cultured, and sequenced for validation.
Subsequently, the PLpro or PLpro-Cys111Ser recombinant plasmid was transformed into E. coli BL21(DE3) competent cells (Novagen). Cells were cultured until OD600 reached ~ 0.8, followed by induction with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 18 °C for 18 h. Cells were harvested by centrifugation at 7656 x g, and the pellets were resuspended in Buffer A (20 mM HEPES pH 7.5, 10 mM imidazole, 500 mM NaCl, 5 mM DTT and 10% glycerol). After sonication on ice, lysates were centrifuged at 38,759 xg (4 °C) to collect the supernatant. Target proteins were enriched via a single Ni-NTA resin column and eluted with Buffer B (20 mM HEPES pH 7.5, 500 mM imidazole, 500 mM NaCl, 5 mM DTT and 10% glycerol). The His tag was removed by TEV protease digestion, and the solution was exchanged into Buffer C (20 mM HEPES pH 7.5, 150 mM NaCl and 5 mM DTT). A second single Ni-NTA resin column purification step yielded tag-free PLpro or PLpro-Cys111Ser. Final purification was achieved by gel filtration (Superdex 75 Increase 10/300 GL, GE Healthcare) in Buffer C. The protein purity of PLpro or PLpro-Cys111Ser was confirmed by SDS-PAGE.
Crystallization and Structure Determination
The PLpro-Cys111Ser (~ 10 mg/mL) was incubated with compound YL1004 at a 1:10 molar ratio overnight at 4 °C. Crystals were grown at 4 °C using the sitting-drop vapor diffusion method with 1 μL protein-ligand mixture + 1 μL reservoir solution. Initial crystals were observed under condition No. 7 from the Wizard Classic 1: 10% (w/v) PEG8000, 100 mM MES/Sodium hydroxide pH 6.0, 200 mM Zinc acetate. High-quality crystals were obtained through optimized condition: 6% (w/v) PEG8000, 100 mM MES/Sodium hydroxide pH 6.0, 200 mM zinc acetate and 20% PEG400. These crystals were subsequently harvested and flash-cooled in liquid nitrogen.
Diffraction data for the PLpro-Cys111Ser-YL1004 complex was collected at beamline BL19U1 of the Shanghai Synchrotron Radiation Facility (SSRF) using a wavelength of 0.97861 Å. The data were processed with XDS1 and scaled using CCP4 Aimless50. The crystal structure was solved by molecular replacement with CCP4 Molrep51, using the PLpro structure (PDB: 7YBG) as the search model. Iterative refinement was performed with Phenix.refine52 and CCP4 Refmac53, followed by manual model adjustments in Coot54. Supplementary Table. 13 presents the diffraction dataset and statistics of final refinement.
Enzyme Activity Assays
FRET-based assay for PLpro and other DUBs enzymatic activity characterization. The reaction buffer contained 20 mM HEPES (pH 7.5).
For viral polyprotein cleavage, 22.5 μL PLpro (200 nM final) was preincubated with 2.5 μL compounds in black 96-well plates (WHB) for 10 min at RT. Reaction initiated with 25 μL Z-RLRGG-AMC substrate (20 μM final). Fluorescence (Ex360/Em460) monitored using CLARIOstar microplate reader (BMG Labtech). Initial linear-phase readings (7–8 cycles) analyzed by simple linear regression in GraphPad 10.0 to calculate V0. Inhibition (%) normalized to DMSO control and IC50 determined via dose-response modeling.
In the primary screen, each compound was tested at a final concentration of 30 µM. Inhibition was normalized to DMSO controls, and compounds showing more than 70% inhibition were defined as primary hits. To ensure hit specificity and chemical stability, known PAINS were excluded. Compounds containing reactive or unstable moieties, such as free aldehydes, quinones, or redox active groups were manually reviewed and deprioritized. Autofluorescent compounds were identified by measuring compound only emission signals and excluded when interference was observed. Selected compounds were further analyzed by LC–MS to confirm structural stability under assay conditions (Supplementary Table. 14).
For deISGylation and deubiquitination, 8.82 μL PLpro or other DUBs (indicated concentration) preincubated with 0.18 μL compounds in black 384-well plates (Corning) for 10 min at RT. Reaction initiated with 9 μL ISG-AMC or Ub-AMC (0.25 μM final). Fluorescence monitored with 20 initial linear-phase cycles. V0 and IC50 calculated as above
Differential scanning fluorimetry assay
PLpro (2 μM final) was mixed with compounds in white 8-strip PCR tubes using 20 mM HEPES (pH 7.5) buffer (10 μL total volume). Reactions contained 1 μL 10000× Orange dye (Sigma). Fluorescence was monitored during 1.5 °C/min heating (20 °C–95 °C) using a BioRad CFX96 RT-PCR system. Melting temperature (Tm) and thermal shift (ΔTm) were calculated via Boltzmann model in GraphPad 10.0.
Cytotoxicity assay
VeroE6/BEAS-2B/HUVEC cells were seeded in clear 96-well plates (100 μL/well) and cultured overnight. Serial compound dilutions (500 μM start, 2-fold steps) in 100 μL medium were added, with DMSO controls and blank medium. After 72 h treatment, 20 μL MTT (5 mg/mL in saline; Sigma) was added per well. Plates were incubated 1–2 h at 37 °C until DMSO controls showed purple formazan precipitation. After dissolving formazan with 50 μL DMSO/well, absorbance at 560 nm was measured using CLARIOstar Plus. CC50 values were calculated via dose-response modeling in GraphPad 8.0.
PK properties in vivo
Male Balb/c mice (6–8 weeks, 18–25 g) and SD (Sprague Dawley) rats (age: 7-11 weeks) were received test compounds prepared in 5% DMSO/5% HS-15/20% PEG300/70% saline (Supplementary Table. 3 and 4). Blood samples collected in K2-EDTA tubes at designated times were centrifuged to obtain plasma. Compound concentrations determined by LC-MS/MS were used for PK parameter calculation (Phoenix WinNonlin 7.0).
In vivo toxicity study
YL1004 was evaluated in Balb/c mice (6-8 weeks, 16-25 g, 50% male/female) formulated in 5% DMSO/3% HS-15/20% PEG300/75% sterile water. Acute toxicity: single 250 mg/kg oral dose; repeat-dose: 100 mg/kg twice daily for 14 days. Animals were monitored for 28 days (dosing + post-treatment) for weight loss, appetite, activity, and behavior.
Western Blot Analysis
For deubiquitination activity assessment, HEK293T cells were transfected with plasmids encoding Ub-HA and Flag-PLpro (or empty pcDNA3.1 vector) and treated with compounds for 36 h. Immunoblotting utilized primary antibodies: HA-tag (Cell Signaling Technology, #3724, 1:1000, rabbit), DYKDDDDK Tag (Cell Signaling Technology, #14793, 1:1000, rabbit), and GAPDH (Proteintech, 60004-1-Ig, 1:1000, mouse).
DeISGylation activity was evaluated in IFN-α (Sigma, SRP4595)-stimulated A549 cells treated with PLpro and/or compounds for 48 h. Proteins were resolved on 10% SDS-PAGE gels and probed with ISG15 (Thermo Fisher, #703131, 1:1000, rabbit), PLpro (LifeSensors, #AB602, 1:250, chicken), and GAPDH (Proteintech, 60004-1-Ig, 1:1000, mouse).
For HA-Ub-VS DUB labeling, HEK293T cell lysates (150 mM NaCl, 0.1% NP40, 50 mM Tris-HCl, 1 mM DTT, pH 8.0) were incubated with HA-Ub-VS probe in reaction buffer (150 mM NaCl, 50 mM Tris-HCl, 1 mM DTT, pH 7.5) alongside PLpro and/or compounds. Samples were analyzed by 10% SDS-PAGE, followed by immunodetection with HA-tag (Cell Signaling Technology, #3724, 1:1000, rabbit), PLpro (LifeSensors, #AB602, 1:250, chicken), and GAPDH antibodies.
Luciferase Reporter Assay
HEK293T cells were co-transfected with plasmids encoding NF-κB-Luc/Renilla/Flag-PLpro, ISRE-Luc/Renilla/Flag-PLpro, or IRF3-Luc/IFNB1-Luc/Renilla/Flag-PLpro. Transfected cells were compound-treated and stimulated with TNF-α (MCE HY-P7058), IFN-α (Sigma SRP4595), or poly(I:C) (MCE HY-107202), respectively. Dual-luciferase activity was quantified using the Dual Luciferase Reporter Assay Kit on a CLARIOstar plate reader (BMG Labtech).
Transcriptomics
HEK293T cells were transfected with plasmids encoding Flag-PLpro (or vector), treated with the indicated compounds, and stimulated with poly(I:C) (MCE, HY-107202) according to the experimental design. Cells were harvested 18–24 h after treatment for RNA extraction. Total RNA was extracted using TRIzol Reagent (Thermo Fisher, cat. no. 15596026) and treated with DNase I (NEB, cat. no. M0303L). RNA purity and concentration were assessed using NanoDrop OneC and Qubit 3.0 (Qubit RNA BR Assay kit, Thermo Fisher, cat. no. Q10210), respectively, and RNA integrity was evaluated using LabChip GX Touch (Revvity). RNA-seq library preparation, high-throughput sequencing (DNBSEQ-T7, PE150), and data analysis were performed by Seqhealth Technology Co., Ltd. (Wuhan, China) using the KC Digital mRNA Library Prep Kit with a 12-nt unique molecular identifier to reduce PCR duplication bias.
Evaluation of in vitro antiviral activity
SARS-CoV-2 or rSARS-CoV-2 stock viruses were diluted and premixed with the tested compounds at designated concentrations to infect Calu3 cells at 0.01 MOI. At 2 hpi., the inoculum was removed and replaced with supernatants supplemented with inhibitors. Cells were incubated at 37 °C until sample harvest. At 24 hpi. supernatants were harvested for infectious viral titration with plaque assays. Cell lysates were harvested for quantification of viral gene copies by RT-qPCR with specific probe and primers (Forward: CGATCTCTTGTAGATCTGTTCTC; Reverse: ATATTGCAGCAGTACGCACACA; Probe: FAM- ACACTAGCCATCCTTACTGCGCTTCG -BBQ).
SARS-CoV-2 infection in K18-hACE2 mice
8- to 12-week-old male and female K18-hACE2 transgenic mice anaesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine were intranasally inoculated with 1250 PFU SARS-CoV-2 Delta (for survival study) or 1000 PFU SARS-CoV-2 Omicron JN.1 (for sample harvest). For oral delivery, all drugs were formulated with 10% DMSO, 10% Kolliphor HS 15, 30% polyethylene glycol 400 (PEG 400) in normal saline and given to the animals at 100 mg/kg twice per day. Nasal turbinate and lung tissues were harvested at 3 dpi. for virological assessment by RT-qPCR and plaque assays. For survival study, body weight and survival of the infected mice will be monitored for 14 days or until death of the animal as we previously reported55.
RNA extraction and RT-qPCR
Viral RNA was extracted from infected cells using QIAsymphony RNA Kit (931636, Qiagen, Germany). Viral RNA from mice lung and nasal turbinate samples were extracted with the RNeasy Mini kit (74106, Qiagen). After RNA extraction, RT-qPCR was performed using QuantiNova Probe RT-PCR Kit (208354, Qiagen) or QuantiNova SYBR Green RT-PCR Kit (208154, Qiagen) with the LightCycler 480 Real-Time PCR System (Roche).
Plaque assays
To determine the infectious virus titres, supernatants from infected cells or organ tissues were ten-fold serially diluted and inoculated into monolayered VeroE6-TMPRSS2 cells with 2 h incubation at 37 °C, followed by 1% low-melting point agarose overlay (16520050, Thermofisher, USA). The cells were further incubated for 48 h (for SARS-CoV-2 wildtype and Delta) or 72 h (for Omicron JN.1 and KP.3) before fixation with 4% formaldehyde for visualization with 0.5% crystal violet diluted in 25% ethanol/distilled water.
Histology and immunohistochemistry staining
Harvested lung tissues from infected mice were fixed with 10% neutral buffered formalin with overnight incubation. Fixed tissues were processed, embedded with paraffin and sectioned into tissue slides at 5 µm thickness. SARS-CoV-2 nucleocapsid protein in the tissue slides was detected using in-house rabbit polyclonal anti-SARS-CoV NP antibody, followed by incubation with biotinylated rabbit anti-guineapig IgG (H + L) (ab6770, Abcam, UK) or biotinylated goat anti-rabbit IgG (H + L) secondary antibody (BA-1000, Vector laboratories, USA). Specificity of the inhouse primary antibody was validated as previously described56. Color development was carried out with VECTASTAIN® ABC-AP Kit and VectorRed substrate kit (AK-5000 and SK-5100, Vector Laboratories) according to the manufacturer’s instructions. The nuclei counterstaining was performed by Gill’s haematoxylin followed by mounting with Vectamount permanent mounting medium. For H&E staining, the tissue sections are stained with Gill’s haematoxylin (H-3401-500, Vector Laboratories) and eosin-Y. All images are acquired by Olympus BX53 light microscope.
Statistical analysis
All statistical analyses were performed using GraphPad Prism versions 7.0 and 8.0. Data in the figures are presented as mean ± SD (standard deviation). The number of biological replicates (n) is indicated in the corresponding figure legends and table footnotes, and each biological replicate was measured in at least duplicate as technical replicates. Statistical comparison between different groups was performed by one-way analysis of variance (ANOVA) or log-rank (Mantel-Cox) test. All t-test performed in this study is two-tailed. Differences were considered statistically significant when P < 0.05 (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Structure factors and atomic coordinates of SARS-CoV-2 PLpro in complex with YL1004 are available in the Protein Data Bank under accession code 9U7D (https://www.rcsb.org/structure/9U7D). Transcriptomics sequencing data presented in Fig. 4 have been deposited in GSA-Human (National Genomics Data Center) under accession code HRA015503 (https://ngdc.cncb.ac.cn/gsa-human/). All other data supporting the findings of this study are available from the corresponding author. Source data are provided with this paper.
References
W. H. O. World Health Organization (WHO). https://covid19.who.int (WHO, 2025).
Harvey, W. T. et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 19, 409–424 (2021).
Chemaitelly, H. & Abu-Raddad, L. J. Waning effectiveness of COVID-19 vaccines. Lancet 399, 771–773 (2022).
Willett, B. J. et al. SARS-CoV-2 Omicron is an immune escape variant with an altered cell entry pathway. Nat. Microbiol. 7, 1161–1179 (2022).
Cao, Y. et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 602, 657–663 (2022).
Iketani, S. et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature 604, 553–556 (2022).
Liu, L. et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature 602, 676–681 (2022).
Saad-Roy, C. M. et al. Immune life history, vaccination, and the dynamics of SARS-CoV-2 over the next 5 years. Science 370, 811–818 (2020).
Banerjee, A. K. et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 183, 1325–1339.e21 (2020).
Schubert, K. et al. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 27, 959–966 (2020).
Frieman, M., Ratia, K., Johnston, R. E., Mesecar, A. D. & Baric, R. S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-κB signaling. J. Virol. 83, 6689–6705 (2009).
Devaraj, S. G. et al. Regulation of IRF-3-dependent Innate Immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 282, 32208–32221 (2007).
Bailey-Elkin, B. A. et al. Crystal structure of the Middle East respiratory syndrome coronavirus (MERS-CoV) papain-like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J. Biol. Chem. 289, 34667–34682 (2014).
Shin, D. et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587, 657–662 (2020).
Harcourt, B. H. et al. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 78, 13600–13612 (2004).
Williamson, B. N. et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature 585, 273–276 (2020).
Wahl, A. et al. SARS-CoV-2 infection is effectively treated and prevented by EIDD-2801. Nature 591, 451–457 (2021).
Xie, Y. et al. Design and development of an oral remdesivir derivative VV116 against SARS-CoV-2. Cell Res. 31, 1212–1214 (2021).
Zhang, J.-L. et al. Azvudine is a thymus-homing anti-SARS-CoV-2 drug effective in treating COVID-19 patients. Sig Transduct. Target Ther. 6, 414 (2021).
Owen, D. R. et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 374, 1586–1593 (2021).
Unoh, Y. et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J. Med. Chem. 65, 6499–6512 (2022).
Jiang, X. et al. Structure-based development and preclinical evaluation of the SARS-CoV-2 3C-like protease inhibitor simnotrelvir. Nat. Commun. 14, 6463 (2023).
Chen, X. et al. Preclinical evaluation of the SARS-CoV-2 Mpro inhibitor RAY1216 shows improved pharmacokinetics compared with nirmatrelvir. Nat. Microbiol 9, 1075–1088 (2024).
Ratia, K. et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA 105, 16119–16124 (2008).
Báez-Santos, Y. M. et al. X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J. Med. Chem. 57, 2393–2412 (2014).
Rut, W. et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: a framework for anti–COVID-19 drug design. Sci. Adv. 6, eabd4596 (2020).
Welker, A. et al. Structure-activity relationships of benzamides and isoindolines designed as SARS-CoV protease inhibitors effective against SARS-CoV-2. ChemMedChem 16, 340–354 (2021).
Shan, H. et al. Development of potent and selective inhibitors targeting the papain-like protease of SARS-CoV-2. Cell Chem. Biol. 28, 855–865.e9 (2021).
Ma, C. et al. Discovery of SARS-CoV-2 papain-like protease inhibitors through a combination of high-throughput screening and a flipGFP-based reporter assay. ACS Cent. Sci. 7, 1245–1260 (2021).
Zhao, Y. et al. High-throughput screening identifies established drugs as SARS-CoV-2 PLpro inhibitors. Protein Cell 12, 877–888 (2021).
Shen, Z. et al. Design of SARS-CoV-2 PLpro inhibitors for COVID-19 antiviral therapy leveraging binding cooperativity. J. Med. Chem. 65, 2940–2955 (2022).
Tan, H., Hu, Y., Jadhav, P., Tan, B. & Wang, J. Progress and challenges in targeting the SARS-CoV-2 papain-like protease. J. Med. Chem. 65, 7561–7580 (2022).
Sanders, B. C. et al. Potent and selective covalent inhibition of the papain-like protease from SARS-CoV-2. Nat. Commun. 14, 1733 (2023).
Tan, B. et al. Design of a SARS-CoV-2 papain-like protease inhibitor with antiviral efficacy in a mouse model. Science 383, 1434–1440 (2024).
Garnsey, M. R. et al. Discovery of SARS-CoV-2 papain-like protease (PLpro) inhibitors with efficacy in a murine infection model. Science AdvAnceS 10, 4288 (2024).
Lu, Y. et al. Discovery of orally bioavailable SARS-CoV-2 papain-like protease inhibitor as a potential treatment for COVID-19. Nat. Commun. 15, 10169 (2024).
M. Bader, S. et al. A novel PLpro inhibitor improves outcomes in a pre-clinical model of long COVID. Nat. Commun. 16, 2900 (2025).
Jadhav, P. et al. Design of quinoline SARS-CoV-2 papain-like protease inhibitors as oral antiviral drug candidates. Nat. Commun. 16, 1604 (2025).
Fu, Z. et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 12, 488 (2021).
Xiong, Y. et al. The substrate selectivity of papain-like proteases from human-infecting coronaviruses correlates with innate immune suppression. Sci. Signal 16, eade1985 (2023).
Cao, D. et al. The SARS-CoV-2 papain-like protease suppresses type I interferon responses by deubiquitinating STING. Sci. Signal 16, eadd0082 (2023).
Iketani, S. et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 613, 558–564 (2023).
Duan, Y. et al. Molecular mechanisms of SARS-CoV-2 resistance to nirmatrelvir. Nature 622, 376–382 (2023).
Shuai, H. et al. An orally available Mpro/TMPRSS2 bispecific inhibitor with potent anti-coronavirus efficacy in vivo. Nat. Commun. 16, 6541 (2025).
Zhu, Y. et al. In vitro selection and analysis of SARS-CoV-2 nirmatrelvir resistance mutations contributing to clinical virus resistance surveillance. Sci. Adv. 10, eadl4013 (2024).
Zheng, J. et al. COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature 589, 603–607 (2021).
Shuai, H. et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 603, 693–699 (2022).
Liu, Y. et al. Lineage-specific pathogenicity, immune evasion, and virological features of SARS-CoV-2 BA.2.86/JN.1 and EG.5.1/HK.3. Nat. Commun. 15, 8728 (2024).
Chu, H. et al. An orally available Mpro/TMPRSS2 bispecific inhibitor with potent anti-coronavirus efficacy in vivo. Res. Sq rs.3.rs-5454588 (2024) https://doi.org/10.21203/rs.3.rs-5454588/v1.
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. D. Biol. Crystallogr. 69, 1204–1214 (2013).
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D. Biol. Crystallogr. 66, 22–25 (2010).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D. Biol. Crystallogr. 68, 352–367 (2012).
Kovalevskiy, O., Nicholls, R. A., Long, F., Carlon, A. & Murshudov, G. N. Overview of refinement procedures within REFMAC 5: utilizing data from different sources. Acta Crystallogr D. Struct. Biol. 74, 215–227 (2018).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D. Biol. Crystallogr 66, 486–501 (2010).
Chu, H. et al. Coronaviruses exploit a host cysteine-aspartic protease for replication. Nature 609, 785–792 (2022).
Shuai, H. et al. Emerging SARS-CoV-2 variants expand species tropism to murines. EBioMedicine 73, 103643 (2021).
Acknowledgements
This research was supported by National Key R&D Program of China [2023YFF1204905 (S.Y.)], National Natural Science Foundation of China [82130104, T2221004 (S.Y.), 82473844 (J. L.)], 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University [ZYGD23006 (S.Y.) and ZYYC24004 (J.L.)], New Cornerstone Science Foundation [NCI202338 (S.Y.)], the Major Project of Guangzhou National Laboratory [GZNL2024A01005 (S.Y.)], the Health and Medical Research Fund [COVID1903010-14, 23220522, CID-HKU1-5 (H.C.)], the Food and Health Bureau, The Government of the HKSAR; the General Research Fund [17118621, 17119122 (H.C.)], Collaborative Research Fund [C7103-22G (H.C.)] and Theme-Based Research Scheme [T11-709/21-N (H.C.)], the Research Grants Council of the HKSAR; InnoHK initiative of the Innovation and Technology Commission of the HKSAR Government; National Key Research and Development Program of China [projects 2021YFC0866100 and 2023YFC3041600 (H.C.)]; General Programme, Guangdong Provincial National Science Foundation, China [2023A1515012325, 2023A1515011891 (H.C.)], Sichuan Science and Technology Program [(2024NSFSC1287 (C.H.) and 2026NSFSC0570(J.L.)]. Postdoctoral Fellowship Program of CPSF [GZB20250829 (J.Q.)]. China Postdoctoral Science Foundation [2025M783575 (J.Q.)]. Natural Science Foundation of Sichuan Province [2026NSFSC1745 (J.Q.)]. We thank the staff members of BL18U1 and BL19U1 beamlines at the National Facility for Protein Science in Shanghai, and Qianlun Pu from the Core Facilities of West China Hospital (Sichuan, China) for great supports. We thank Prof. Minghai Tang for LC-MS analysis assistance. We also acknowledge the support from Dr. Gallant Ho on the Gallant Ho Outstanding Young Professorship.
Author information
Authors and Affiliations
Contributions
S.Y., H.C., and J.L. conceived the research; S.Y. and J.N. performed the drug design; J.N., C.H., and Y.L. with the assistance of S.Z., R.M., and Q.H. performed the chemical synthesis; R.Z., J.Q., X. D., and J. S. performed gene expression, protein purification and crystallization experiments; R.Z. and J.L. collected the data, determined the crystal structure, and analyzed the inhibition mechanism; J.Q. and Y.C. with the assistance of L.Z. and L.D. performed high-throughput screening, enzymatic activity and inhibition assays, IC50 measurements, DSF assays, cytotoxicity assays and in vivo toxicity assays; H.S., L.D., C.Y., J.H., W.G., and L.W. performed in vitro and in vivo antiviral assays; S.Y., J.L., and H.C. with the assistance of J.N., H.S., J.Q., and R.Z. wrote and revised the manuscript. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
All authors declares no competing interests.
Peer review
Peer review information
Nature Communications thanks Hao Huang, who co-reviewed with Yuxian Xiong, Brian Sanders, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nan, J., Shuai, H., Qiao, J. et al. YL1004 is a SARS-CoV-2 papain-like protease inhibitor with immunomodulatory and antiviral activity in mice. Nat Commun 17, 2035 (2026). https://doi.org/10.1038/s41467-026-68795-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68795-5
