
Abstract
Photothermal dry reforming of methane, a promising approach to producing syngas, typically uses oxide materials and is mediated by lattice oxygen. However, it is limited by the balance of the oxygen-releasing/replenishing loop. Here, we present a methane activation pathway free of lattice oxygen on a N-Ni/NiCo@C catalyst. Direct involvement of reactive oxygen species (O*/OH*) released from CO2 promotes effective CH4 activation and eliminates coke deposition on oxophilic NiCo sites. Control experiments and density functional theory calculations show that forming a C-N-Ni structure is essential for improving catalytic performance. This is achieved by strengtheni ng the electronic interaction between the NiCo nanoparticles and the carbon layer, thereby boosting the elemental steps of CH4 dehydrogenation and CH* oxidation and preventing metal oxidation. The optimal catalyst exhibits a high light-to-chemical energy efficiency of 52%, maintaining stability during 200 hours of continuous operation at a mild temperature of 540 °C. This work demonstrates that highly efficient and stable photothermal dry reforming of methane can be achieved by bypassing lattice oxygen.
Similar content being viewed by others
Introduction
Methane dry reforming (DRM), which converts the two main greenhouse gases (CH4 and CO2) into syngas (CO and H2), is a promising way of reducing carbon emissions and producing a valuable feedstock for industrial processes1. However, traditional thermally driven DRM requires high operating temperatures (700−1000 °C) to overcome the endothermic nature of the reaction (ΔH298K = 247 kJ mol-1), which leads to severe catalyst deactivation via metal sintering and coking, particularly for Ni- and Co-based catalysts2,3. Recent advancements in light-driven DRM offer a promising way to address these issues by using photothermal effects to reduce operating temperatures and improve energy efficiency4,5. Nevertheless, this approach faces challenges such as low light utilization efficiency (typically <10% under standard solar irradiation) and significant coking at moderate temperatures (400−600 °C)6.
The abundance and mobility of active oxygen species are crucial for achieving anti-coking photothermal DRM (PDRM). This is because all C-H bonds in CH4 are typically cleaved at oxygen sites by photogenerated holes7,8,9. Traditionally, metal oxide-based catalysts (e.g., CeO2, β-Ga2O3, WO3) have been employed for the DRM process, enabling carbon species to interact directly with lattice oxygen (OL) (C* + OL → CO)10,11,12,13. Therefore, the oxygen release capability of these catalysts plays a key role in mitigating coke-induced deactivation. However, the strong metal–oxygen interactions in metal oxides severely restrict the mobility of lattice oxygen, thereby impeding coke consumption and reducing DRM efficiency14. Furthermore, the excessive release of OL during prolonged DRM operation results in metal segregation and accelerates catalyst deactivation15. Ideally, an approach should be developed that bypasses the involvement of lattice oxygen to overcome this limitation. It is well established that substantial active oxygen species (O*/OH*) are formed during CO2 conversion in the DRM process (CO2 → CO + O*, CO2 + H* → CO + OH*). Additionally, Ni/Co exhibits a strong binding affinity towards oxygen-containing radicals due to its high oxophilicity16,17. Thus, the adsorbed O*/OH* species over the Ni/Co sites can be effectively utilized for CH4 activation, thereby substituting the role of OL. However, the limited electron transfer efficiency around the Ni/Co sites makes these metals susceptible to oxidation deactivation by O*/OH* species6,18. The main challenge in addressing this issue lies in constructing an efficient, directional electron transfer channel to enhance electron density around the metal sites.
Recently, metal-organic framework (MOF)-derived materials have emerged as potential candidates for efficient, stable, light- or thermally driven catalysis19,20. MOF-templated synthesis can produce metal centers that are highly dispersed and coated with a carbon layer. This provides broad light absorption, high photothermal efficiency, and especially fast electron conduction20,21,22,23,24. Notably, electrocatalytic studies have revealed that tuning the N-ligands during MOF synthesis can facilitate the formation of metal-N-C structures that exhibit unique capabilities in electron modulation, intermediate regulation, and metal anchoring in room-temperature catalysis25,26. This special structural feature means that MOF-derived catalysts could be useful for directed electron transfer in PDRM. However, their applications in photothermal catalysis remain unexplored, as high-temperature pyrolysis converts metal-N-C structures into metal@NC structures19,27,28, which fail to preserve the intrinsic electronic and catalytic properties of the metal-N coordination sites.
Here, we present the design of an efficient, stable NiCo-MOF-derived N-Ni/NiCo@C catalyst featuring C-N-Ni coordination. This optimized catalyst achieves a light-to-chemical energy efficiency (LTCEE) of 52% at 2.8 W cm−2 of light irradiation, producing H2 and CO at rates of 423.3 and 487.4 mmol h−1 g−1, respectively. Crucially, it maintains stable H2/CO ratios for 200 h at the coke-sensitive temperature of 540 °C, outperforming N-free and single-metal samples, as well as NiCo/Al2O3, through synergistic N-Ni coordination, alloying, and a carbon-coated structure. The C-N-Ni coordination acts as a directional electron transfer pathway that regulates electron distribution within the NiCo alloy. This promotes the activation of C-H bonds at the Ni sites and drives the activation of CO2 at the Co sites. Additionally, photoexcited electrons accumulate at the N-Ni sites via the carbon coating, which effectively prevents the NiCo metal from being deactivated by reactive oxygen species (OH*/O*) during the DRM process. Notably, abundant oxygen radicals oxidize CH* to CHO* and CO under the influence of photogenerated holes, thereby eliminating carbon deposition and inhibiting the water-gas shift reaction (RWGS). The precise construction of the Ni-N-C sites provides a feasible strategy for achieving efficient, stable photothermal DRM under mild conditions.
Results and discussion
Synthesis and structural characterization of catalysts
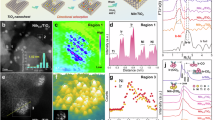
A series of MOF-derived NiCo nanoalloys (denoted N0, N1, and N2) with varying nitrogen-doping levels was synthesized using a one-step microwave hydrothermal method. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images reveal a well-defined spherical morphology, with an average diameter of 344 nm. These nanospheres are further decorated with uniformly dispersed 7.2 nm nanoparticles (Fig. 1a, b and Supplementary Fig. 1a, b). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) analysis (Fig. 1c, d) shows that these nanoparticles are coated in graphitic carbon and have a lattice fringe spacing of 0.206 nm, which is consistent with the (111) crystal plane of a NiCo alloy22. Elemental mapping and line-scanning profiles confirm the homogeneous spatial distribution of Ni and Co atoms of the MOF precursor and MOF-derived catalyst, as well as the high dispersion of the nanoparticles (Fig. 1e and Supplementary Fig. 2–4). By contrast, the reference samples N0 and N2 exhibit inferior nanoparticle dispersion and larger average NiCo particle sizes of 11.8 and 35.8 nm, respectively (Supplementary Fig. 5 and 1c, d).

a SEM image. b HRTEM image, c HAADF-STEM image. d The FITT image of the highlighted part in c of the catalyst. e HRTEM-EDS elemental mapping. f The compositions of the sample measured by ICP-OES, the arrows point to the Y-axis. g XRD patterns of the catalysts with different doping amounts and their locally enlarged patterns, the dotted lines denote the peak range and shift trend.
Results from inductively coupled plasma-optical emission spectroscopy (ICP-OES) show that as the dosage of the N-ligand increases in N0, N1, and N2, so does the N doping content, rising from 1.5 wt.% to 2.4 wt.% and 3.0 wt.%. The N detected in N0 is likely to come from the DMF solvent (Fig. 1f and Supplementary Table 1). An increased N doping content was found to lead to a significantly decreased Ni content, ranging from 28.3 wt.% in N0 to 12.0 wt.% in N1 and 3.1 wt.% in N2, while the Co content showed minimal change. Consequently, the Co/Ni molar ratio increased from 1.2 in N0 to 2.1 in N1 and 8.4 in N2. Notably, the Co/Ni ratio of 1.2 in N0 is similar to the equal amounts of Ni and Co precursors. Therefore, the sharp decrease in Ni content suggests that a higher dosage of N-ligand forms a coordination bond with Ni, inhibiting the formation of metallic Ni during catalyst synthesis.
Figure 1g shows the X-ray diffraction (XRD) patterns of the N0, N1 and N2 samples. The peak at 26° corresponds to graphitic carbon (PDF #41-1487), which is consistent with the observations made using HAADF-STEM (Fig. 1c). Peaks at 44.3°, 51.6° and 76.0° align with cubic metallic nickel (PDF #04-0805) and/or cobalt (PDF #15-0806). The enlarged peak at approximately 44.3° is positioned between the standard peaks of pure Co (111) and Ni (111) (Fig. 1g, inset), which confirms the formation of a NiCo alloy22,29. Notably, this peak gradually shifts towards higher angles with increasing N-doping, approaching the Ni (111) position. Meanwhile, the graphitic carbon peak remains unshifted (Supplementary Fig. 6). Typically, the peak shift in NiCo alloys tends to approach the increased metal22, while our ICP-OES data show significant Ni depletion. This suggests that N-doping introduces lattice strain, causing the lattice spacing to decrease. This could significantly alter the electronic distribution of Ni-Co nanoalloys and potentially enhance their photothermal catalytic performance.
Catalytic performance for DRM
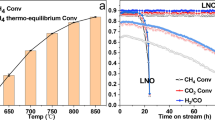
The catalytic performance of catalysts with varying levels of N-doping was evaluated at 600 °C under 2.8 W cm−2 light irradiation (Fig. 2a). The H2 and CO formation rates exhibited a volcano-type trend as the N content increased, peaking at 561.8 and 597.4 mmol gcat−1 h−1, respectively, with an H2/CO ratio of 0.94. Dark conditions at the same temperature yielded significantly lower rates and H2/CO ratios (Supplementary Fig. 7a). Under full-spectrum illumination, the optimal N1 catalyst exhibited higher CH4 and CO2 conversions across 330–600 °C compared to thermal-only conditions, exceeding thermal equilibrium limits (Fig. 2b). The corresponding H2/CO production rates and ratios in photo-enhanced DRM were also higher than that in thermal DRM (Supplementary Fig. 7b, c), confirming the photothermal synergy.

a Light-enhanced H2/ CO production and ratios on catalysts prepared with different N-ligand addition, the arrow points to the right Y-axis. b CH4 and CO2 conversions of N1 under photothermal (P) and thermal conditions (T) at different temperatures, error bars represent the standard deviation (SD) from three independent experiments. c Arrhenius plots of H2 in photothermal catalysis and thermo-catalysis by N1. d Production selectivity of N1 under photothermal and thermal conditions at different temperatures, error bars represent the SD from three independent experiments. e LTCEEs of N1 under different temperatures. f LTCEEs of samples prepared with different N-ligand addition at 510 °C. g Stability test of N1, N0 and reference sample (ref., Ni1Co2/Al2O3) in light and dark conditions, the arrows point to the right Y-axis. Reaction condition: 20 mg catalyst, P = 1 atm, 300 W xenon light source, light intensity: 2.8 W cm−2, feed gas: CO2/CH4/Ar=5/5/40 mL, GHSV = 150,000 cm3 gcat−1 h−1.
To clarify the effect of changes to the Co/Ni ratio on N doping (Fig. 1f), a control sample (Ni1Co2@C) with the same metal composition but no external N ligand was prepared. Its H2 production rate was approximately 50% lower than that of N1 and slightly below that of N0, emphasizing the importance of N doping rather than the metal ratio as the primary activity enhancer in this study (Supplementary Fig. 7d). The apparent activation energies (Ea) calculated via Arrhenius plots revealed that the photothermal Ea values for H2 formation were 93.7, 52.6 and 188.7 kJ mol−1 for N0, N1 and N2, respectively, which are significantly lower than their corresponding thermal Ea values of 152.3, 160.4 and 222.8 kJ mol−1 (Fig. 2c and Supplementary Fig. 7e, f). The lowest Ea of N1 suggests that it possesses the optimized active sites for photoelectric activation. Selective analysis shows that the selectivity of CO production in photothermal catalysis and thermal catalysis remains almost unchanged (Fig. 2d). However, light irradiation significantly enhances the selectivity of H2 production, indicating that photothermal catalysis optimizes the reaction pathway related to CH4 activation more effectively. This observation has been further explored in the mechanism study.
The LTCEE was calculated using the data in Fig. 2b to serve as a metric for the catalyst’s capacity to convert light energy into chemical energy30. As shown in Fig. 2e, the LTCEE exhibited strong temperature dependence for N1, peaking at 510 °C. This suggests that enhanced performance does not solely result from photoelectric activation; rather, suitable temperatures enable optimal light utilization, which is essential for minimizing energy input in practical applications. Figure 2f shows the LTCEE of all samples at 510 °C with different amounts of N doping. The LTCEE values correlate with the catalytic performance of these samples, and N1 exhibits the highest efficiency of 52%, competitive to that of previously reported solar-driven systems7,11,12,13,18,30,31,32,33,34 (Supplementary Table 2). These results suggest that the appropriate amount of N doping plays a decisive role in enhancing the LTCEE of the catalyst and the performance of PDRM.
For practical applications, especially in PDRM, the stability of the catalyst is of vital importance, as carbon deposition often leads to rapid deactivation. Notably, N1 exhibited high activity and stability for 200 h at 540 °C during continuous testing (Fig. 2g). Specifically, the H2/CO ratio increased slightly from 0.8 to 0.84 under light and from 0.7 to 0.73 in the dark. CH4 conversion declined by only 0.04% per hour under light versus 0.06% in the dark, while CO2 conversion decreased by 0.08% in both conditions. This indicates that light mainly stabilizes active sites for CH4 activation. In contrast, N0 exhibited significant deactivation, with CO2 and CH4 conversion losses of 1.8% and 1.2% per hour, respectively. Furthermore, the conversion rate and H2/CO ratio of the reference sample, Ni1Co2/Al2O3, both showed a sharp decline within 50 h. A detailed study of the spent N1 catalyst after 200 h was conducted and compared with the fresh sample. As shown in Supplementary Fig. 8, no phase change, loss of active sites, or increased amorphous carbon was observed. These results confirm that the N-Ni/NiCo@C structure significantly enhances the stability of high-loading NiCo catalysts in DRM by resisting deactivation via structural protection and active site stabilization.
Identification of active sites
To elucidate the influence of N-doping on atomic coordination, we conducted X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) studies on the N1 catalyst, alongside the reference samples N0 and N2. The Ni K-edge XANES spectra of these samples shift towards Ni2+ character with increasing N-doping (Fig. 3a), though this is distinct from the spectrum of NiO. EXAFS analysis revealed an increase in Ni-N scattering at 1.2 Å from N0 to N2, together with a metallic Ni-Ni/Co peak at ~2.15 Å. This confirms the presence of dual coordination environments (Fig. 3b). Consistently, the 2.2 Å maximum in wavelet transform (WT) analysis confirms Ni–Ni/Co coordination, while the gradually emerging peak at approximately 1.2 Å directly indicates increased Ni–N bonding (Fig. 3d and Supplementary Fig. 9a−c). Regarding the Co K-edge XANES spectra (Supplementary Fig. 9d), all spectra matched the Co foil, indicating metallic Co states. Co K-edge EXAFS showed prominent ca. 2.15 Å Co-Co coordination peaks with a right shift in N2/N1/N0 (Fig. 3c), suggesting NiCo nanoalloy formation35, consistent with the results of XRD and energy-dispersive X-ray spectroscopy (EDS) mapping. The corresponding WT analysis revealed a ca. 2.2 Å Co–Co coordination feature, with a faint ca. 1.25 Å signal in N-doped samples (Supplementary Fig. 9e–h), implying N-induced Co ion formation.

a Ni K-edge XANES spectra. Ni (b) and Co (c) K-edge EXAFS spectra, the highlighted parts represent the different metal coordination peaks. d Wavelet transform of Ni K-edge EXAFS. N 1 s (e) and Ni 2p (f) XPS spectra, the dotted line denotes the peak shift trend. g Schematic representation of the atomic structures of the NiCo alloy and N-Ni/NiCo surfaces and their corresponding charge density difference analysis, the circles focus on the active sites. h Adsorption energy diagram of CH4 and CO2 at different adsorption sites on the NiCo alloy and N-Ni/NiCo surfaces.
X-ray photoelectron spectroscopy (XPS) analysis reveals significant electronic modulation in NiCo nanoalloys (Fig. 3e and Supplementary Fig. 10). The Ni 2p spectra show that the Ni2+ peak (856.2 eV) shifts by −0.77 eV from N0 to N1, with an additional shift of −0.50 eV to N2. This positive shift increases the electron density at the Ni sites, creating electron-rich Niδ⁺ (0<δ < 2) species that accelerate the kinetics of H⁺ reduction for enhanced H2 evolution. This suggests that Ni-N coordination could increase the surface energy and improve the reactivity of the Ni sites36. The Co 2p spectra show a decrease in Co0 intensity (778.5 eV) with N-doping (Supplementary Fig. 10); this aligns with increases in ionic coordination as determined by EXAFS. The distribution of oxygen species further supports catalyst activation, with oxygen vacancies (OA, 531.9–532.3 eV) dominating surface oxygen (69.9–75.7%), thereby promoting CO2 adsorption37. N 1 s XPS analysis reveals that the predominance of pyridinic-N (398.8 eV) in N1 (62%) exceeds that in N2 (57%) and N0 (54%) (Fig. 3f and Supplementary Fig. 10). Pyridinic-N has been reported to favor CH4/CO2 activation19,27; thus, the optimal performance of N1 is likely related to the prevalence of Ni-N-C sites in this material.
The EXAFS and XPS analyses above have confirmed that the Ni-N coordination site increases the electron density around the Ni sites. Density functional theory (DFT) calculations were performed to further explore the effects of N doping on charge transfer in NiCo nanoparticles. For the pure NiCo surface (Fig. 3g), the average metal-metal bond length is ~2.49 Å. N-doping (N-Ni/NiCo) reduces the distances between neighboring sites to 2.42 Å (Ni-Ni), and 2.40 Å (Co-Co), indicating lattice compression and consistent with the XRD peak shifts shown in Fig. 1g. Differential charge analysis revealed electronic transfer dynamics between Co and Ni. On the undoped surface, the cobalt marked with a circle acts as an electron acceptor (1.23e), while nickel releases 1.11e. Conversely, on the N-Ni/NiCo surface, the electron transfer pathway has changed. The Co atom marked with a circle loses 0.31 e, Ni releases only 0.21 e, and the adjacent N atoms gain approximately 1.10e. This indicates that N significantly promotes the accumulation of electrons around Ni, which is consistent with the enhanced Ni electron density observed in the XPS results. This result assists in proving the role of Ni-N sites in regulating the electronic environment of NiCo.
To investigate the role of Ni-N sites in the DRM reaction, we first examined reactant adsorption and activation via temperature-programmed desorption (TPD)38. In the CH4-TPD experiment (Supplementary Fig. 11a, b), the amount of CO produced increased sequentially from N0 to N2, while CH4 desorption decreased. N2 exhibited the maximum CO yield among them, indicating that CH4 activation is dominated by the abundance of Ni-N sites. Conversely, the amount of CO2 desorbed and the amount of CO derived from it progressively diminished across the series during CO2-TPD (Supplementary Fig. 11c, d), which correlates with reduced metallic NiCo content. This confirms that CO2 activation primarily relies on NiCo alloy sites.
Due to the variety of active sites (Ni, Ni-N, and Co) present in the investigated catalyst, it is difficult to precisely determine their individual contributions to the adsorption and activation of CH4 and CO2. Therefore, idealized surface models of the NiCo alloy and the N-Ni/NiCo catalyst were created, and the adsorption energies of CH4 and CO2 on these active sites were calculated using DFT. As shown in Fig. 3h, four types of active sites were considered: Ni and Co on NiCo, and Ni-N and Co(N) on N-Ni/NiCo. The results show that the CO2 adsorption energies are in the following order: Ni-N (−2.66 eV) > Co(N) (−2.64 eV) > Ni (−0.78 eV) > Co (−0.60 eV). This suggests that N-doping increases the CO₂ adsorption energy on the NiCo surface by a factor of 3.4–4.4. Furthermore, N-doping reduces the adsorption energy difference between the Ni and Co sites from 0.18 eV to 0.02 eV, favoring CO2 activation at the Co sites. Similarly, for CH4 adsorption, the energies follow the sequence Ni-N (−1.87 eV) > Ni (−1.18 eV) > Co (N) (-0.89 eV) > Co (−0.72 eV), indicating that N-doping also promotes CH4 activation predominantly mediated by Ni sites rather than Co sites. Therefore, the N-Ni/NiCo surface promotes a more effective functional differentiation, providing more Ni sites for CH4 activation and more Co sites for CO2 activation. These findings clearly demonstrate the pivotal role of N doping in regulating the electronic structure and catalytic properties of the active Ni and Co sites. The N-Ni site remarkably enhances reactant adsorption capacity and optimizes the distribution of CH4 and CO2 adsorption across different sites, which is consistent with the TPSR results. This synergistic effect is advantageous for achieving high catalytic efficiency and stability.
In situ investigation for reaction mechanism
In order to investigate the mechanistic effects of light on N1, the surface intermediates were monitored using in situ diffuse reflectance Fourier transform spectroscopy (DRIFTS). Under dark conditions with a CH4/CO2/Ar flow during temperature ramping from 300 to 540 °C (Fig. 4a), the following characteristic peaks were observed over the N1 sample: gas-phase CH4 (3014 and 1302 cm−1) and CO2 (2360 and 2340 cm−1) vibrations8,18, adsorbed bidentate carbonate (b-CO32−, 1701 cm−1) and monodentate carbonate (m-CO32−, 1350 cm−1). The intensified O-H stretches (3700 and 1600 cm−1) indicated the RWGS reaction. Upon illumination, the adsorption of CH4/CO2 intensified markedly, and the carboxylate peak (1707 cm−1) strengthened, while the -CHx species (1460 cm−1) emerged8, confirming the enhanced chemisorption of CO2 and CH4. Illumination produced a carboxyl intermediate (-COOH, 1538 cm-1)39, along with the disappearance of the water signal. This suggests that H* (from CH4 dissociation) reacts with CO2 to form -COOH, which has been reported to decompose readily into CO and HO* radicals40. This reaction pathway is distinct from conventional CO2 splitting into CO and O*39, thus favoring the conversion of CO2. Furthermore, the oxidative HO* could promote the oxidation of hydrocarbon intermediates (-CHx)40, thereby increasing the H2/CO product ratio and improving coking resistance18.

In situ DRIFTS over N1 catalyst in the dark (a) and light irradiation (b) in the atmosphere of CO2, CH4, and Ar (10/10/80 vol%) mixture from 300 to 540 °C, dotted lines mark the positions of the peaks. c In situ DRIFTS over N1 catalyst in the dark and light irradiation in the atmosphere of CH4 and Ar (10/90 vol%) mixture at 540 °C. NAPXPS spectra of Ni 2p (d), Co 2p (e), O 1 s (f) and N 1 s (g) with and without illumination.
To further investigate the influence of light on the activation of CH4 and CO2, the N1 catalyst was tested individually under a 10% CH4/Ar and a 10% CO2/Ar atmosphere, as shown in Fig. 4c and Supplementary Fig. 12. In the dark, a strong gaseous CH4 peak and weak CO2 signals were observed, suggesting that CH4 was partly oxidized by surface oxygen species. Upon illumination, the CO2 signal intensified, indicating that more surface C* species resulting from CH4 dehydrogenation were oxidized. New bands emerged in the 1500–1600 cm−1 region, suggesting enhanced CH4 activation. Notably, the conversion of CH4 to CO2 is reported to mitigate coking during the DRM process8. However, for CO2 activation under a 10% CO2/Ar atmosphere (Supplementary Fig. 12), the adsorption peak showed negligible differences between dark and light conditions. This suggests that light primarily promotes CH4 activation of. This is consistent with previous DFT results, which highlight the main enhancement in CH4 activation over the N1 catalyst.
In situ near-ambient pressure XPS (NAPXPS) studies of PDRM over the N1 catalyst reveal light-mediated stabilization mechanisms. After 2 h of DRM in the dark at 540 °C (Fig. 4d), the Ni0/Ni2+ ratio decreased from 0.8 to 0.4, while the Co0/Co2+ ratio dropped from 1.0 to 0.5. Under illumination, these ratios increased to 1.0 and 1.1, respectively, confirming that light preserves the metallic states that are critical for reactant activation. This is because oxidized metal sites promote agglomeration and exhibit poor activation capacity41,42. Together with in situ DRIFTS results, metal oxidation appears to be related to the CO2 conversion process, whereby CO2 adsorption on metallic sites oxidizes the metal and generates CO. In contrast, light enables an alternative pathway by promoting the formation of COOH* intermediates, which decompose into CO and OH* without oxidizing the metal sites (Fig. 4b). The O 1 s NAPXPS spectra further support this argument (Fig. 4e). The O 1 s spectrum of the fresh catalyst exhibits four peaks at 529.0, 531.7, 533.0 and 535.0 eV, which are assigned to oxygen vacancies (Ov), surface O-C, adsorbed O=C and H2O8, respectively. After 2 h of dark DRM reaction, the intensity of the Ov peak dropped sharply while the intensity of the O=C peak increased. This suggests that CO2-adsorbed species occupied metal-Oᵥ sites, leading to elevated metal valence states. The increased H2O peak compared to the initial spectrum indicates the occurrence of the RWGS reaction. In contrast, PDRM induced negligible changes to the Ov and O=C peaks, and the H2O peak disappeared, confirming that light suppresses both metal oxidation and RWGS. Furthermore, the N 1 s spectra (Fig. 4f) also changed after a 2-h DRM reaction in the dark. There was a positive shift in the N-metal peak, a significant decrease in the N-pyridine peak, and the emergence of a graphitic N peak. Importantly, illumination prevents these degradations. Overall, the light-induced new pathway preserves active sites and suppresses RWGS, thereby endowing N1 with exceptional PDRM activity, H2 selectivity, and catalyst stability.
Quasi-in situ isotope labeling using 12CH4 and 13CO2 was further employed to trace the CH4/CO2 degradation pathways in DRM/PDRM. Gas chromatography (GC) analysis identified three peaks at 2.5, 2.8, and 3.6 min, with temperature-dependent shifts confirming 13C incorporation (Supplementary Fig. 13a, b). The mass spectra (MS) of the three peaks (P1, P2, P3) at 540 °C under illumination were analyzed. For P1 (Fig. 5a), the peaks at m/z = 12, 16, and 28 correspond to 12CO originating from 12CH4, while the peaks at m/z = 13, 1,6 and 29 correspond to 13CO from 13CO2. Additionally, peaks corresponding to the intermediate product formaldehyde (H12CHO, m/z = 30, 31) were detected. All the peaks in P2 correspond to the ion fragments of 12CH4 (Fig. 5b). In P3 (Fig. 5c), the main reactant 13CO2 (m/z = 13, 16, 29, 45) was observed alongside peaks of 12CO2 (m/z = 12, 16, 28, 44). In this system (12CH4 + 13CO2), 12C-containing substances can only originate from 12CH4 or the graphitic carbon in the MOF material. Thus, a control experiment was carried out in a 13CH4 + 13CO2 atmosphere, which yielded no 12CO2 (Fig. 5d). This rules out the participation of catalyst carbon and confirms the conversion process from CH4 to CO2. Quantitative analysis of this transformation (Fig. 5d) demonstrated light-enhanced and temperature-dependent formation of 12CO2 (m/z = 44) from 12CH4. Notably, unlike the previously reported carbon exchange mechanism between reactants (i.e., CO2 and CH4) in the PDRM reaction8, this unidirectional CH4-to-CO2 conversion suppresses coking induced by CH4 cracking and reduces H2 consumption by CO2.

Mass spectra of CO (a), CH4 (b), and CO2 (c) over N1 at 540 °C under light irradiation using an equivalent amount of 12CH4 and 13CO2 as the feeding gas. 12C-labeled species as a function of reaction temperature over N1 using equivalent amounts of 12CH4 and 13CO2 in dark (d) and under light (e). f The production of 12CO2 in different reaction systems. Reaction conditions: 10 mg of catalyst and 300 W Xe lamp with a light intensity of 2.8 W cm−2.
The reaction pathway can be further elucidated by examining the temperature-dependent evolution of 12CH4 and 13CO2. From 300 to 570 °C under illumination (Fig. 5e), the intensity of 13CH4 decreased rapidly while the concentrations of 12CO and 12CO2 increased. The H12CHO intermediate first increased and was then completely converted to CO/H2/CO243. In the dark reaction (Fig. 5f), a slower tendency for CH4 degradation and product formation tendency was observed, with H12CHO remaining stable at high temperatures. This identifies HCHO as the key intermediate species in the conversion of CH4 to CO2. Conventional pathways involve stepwise CH4 dehydrogenation followed by oxidation via catalyst lattice oxygen44, which risks coking if oxidation is lacking. Crucially, our catalysts contain no lattice oxygen; thus, the detection of HCHO implies an alternative route whereby CHx* intermediates from CH4 cracking are oxidized by oxygen radicals to form HCHO before complete mineralization. Among these reactive oxygen species, there should be O* from CO2 deoxygenation and OH* from *COOH decomposition. Supplementary Fig. 13c, d show how the temperature affects the way 13C-labeled substances evolve in PDRM and DRM. Compared with DRM, light significantly promoted the conversion of 13CO2 to 13CO, which is consistent with the activity test results. Furthermore, no formic acid species were detected in the gas phase; however, COOH* was observed in the in situ DRIFTS. This may be due to the strong nucleophilicity of COOH*, causing them to adsorb strongly at the metal sites.
DFT calculation for catalytic mechanism
DFT calculations were performed to elucidate the energetics of CH4 (Fig. 6a) and CO2 (Fig. 6b, c) dissociation at the NiCo alloy and N-Ni/NiCo interfaces. Figure 6a shows the CH4 dehydrogenation pathway on both surfaces (CH4 → CH4* → CH3* → CH2* → CH*). At the NiCo interface, the activation barriers for the formation of CH3*, CH2*, and CH* are 0.08, 2.13, and 1.08 eV, respectively. CH3* → CH2* constitutes the rate-determining step (RDS). In contrast, the N-Ni/NiCo exhibits lower barriers of −0.27, 0.90, and −1.11 eV respectively, indicating that N doping markedly enhances CH4 activation. The subsequent CH* → C* step, which is responsible for carbon accumulation, has a negligible barrier of 0.01 eV on NiCo, but rises to 2.09 eV on N-Ni/NiCo. This proves that N incorporation significantly suppresses coking, which is in agreement with catalyst lifetime tests. Furthermore, the literature reports that both CH* and CH2* can react with oxygen radicals (OH* and O*) to yield CO. CH* + OH* → CHOH* less favorable from a kinetic perspective than CH2* + OH* → CH2OH*40,45. In our calculations, CH2* readily converts to CH* on N-Ni/NiCo with a ΔE of −1.11 eV. GC-MS detection of formaldehyde also supports the formation of CO via the reaction CH* + O*. Therefore, Fig. 6a shows the calculated transition state results for CHO* formation and subsequent dehydrogenation: CH* → CHO* → CO* → CO (g). The barriers for these three steps on NiCo are −0.08, 2.13, and 1.08 eV respectively; all of these values exceed the corresponding values on N-Ni/NiCo (−0.10, −2.13 and −1.08 eV). This demonstrates that N-Ni/NiCo promotes not only CH* oxidation, but also its subsequent dehydrogenation to form CO. These calculation results prove that the CH4 → CHO* → CO pathway is energetically favored for DRM syngas production at these interfaces.

Gibbs free energy diagrams of CH4 conversion (a) and CO2 conversion (b, c) on the pristine NiCo and N-Ni/NiCo surface, the dashed box marks the carbon accumulation step in the reaction process. d Schematic illustration of catalyst structures and reaction mechanisms for the light-driven DRM reaction, the dashed circle marks the electron transfer between atoms.
In situ DRIFTS under illumination revealed the formation of *COOH on the surface, implying that CO2 reacts with H* to form *COOH. Reportedly, *COOH may either hydrogenate to form the undesired intermediate HCOOH in the RWGS reaction, or cleave to produce CO and OH*, with the latter facilitating CH4 activation40. Supplementary Fig. 14 shows that *COOH hydrogenation to HCOOH requires a ΔE of −1.25 eV on NiCo, but only 0.34 eV on N-Ni/NiCo, which highlights the latter’s superior ability to suppress the RWGS reaction. Conversely, *COOH cleavage into CO and OH* is highly exergonic (ΔE = −2.61 e V) on N-Ni/NiCo versus −1.43 eV on NiCo (Fig. 5b), identifying CO2 → *COOH → CO + OH* as the preferred CO2 activation pathway. Another common CO2 activation pathway is direct deoxygenation to CO39, which occurs with an energy barrier of −0.49 eV on N-Ni/NiCo; this is significantly lower than the −0.13 eV required on NiCo (Fig. 6c). This indicates that N doping could enhance CO2 deoxygenation too, which is consistent with the superior DRM performance of N-Ni/NiCo.
Based on the above research results, Fig. 6d shows a schematic diagram of the PDRM mechanism. In the NiCo alloy, Ni is the main site for activating CH4 and CO2. The higher electronegativity of Co leads to electrons transferring from Ni to Co. This forms Ni sites with electron deficiency, which reduces Ni’s ability to activate C-H bonds. This makes the dehydrogenation of CH4 the rate-limiting step of the DRM reaction. In contrast, the N-Ni sites adjust the electron transfer path under the influence of lattice strain and the electron-withdrawing effect. This inhibits charge transfer from Ni to Co and makes Co sites more effective at activating CO2, while Ni sites mainly activate CH4. When illuminated, photogenerated electrons in the carbon layer transfer to the N-Ni and adjacent Ni-Co sites. This enhances the activation of CH4 on N-Ni and inhibits the oxidation of Ni-Co by CO2. This process generates a large number of surface oxygen species (O*/OH*), which react with CH* with the assistance of photogenerated holes to form the key intermediate *CHO and ultimately convert to CO/CO2. This new reaction pathway, combined with efficient directional charge transfer, gives the N-Ni/NiCo@C catalyst an extremely high LTCEE, excellent photothermal catalytic performance, an improved H2/CO ratio, and superior stability. This highlights the advantage of using active oxygen species directly to activate CH4 and the key role of engineering the electron structure in optimizing the photothermal catalytic pathway.
In summary, we have demonstrated the effectiveness and durability of a photothermal DRM process using the N-Ni/NiCo@C catalyst. Such impressive performance is made possible by the directional transfer of photogenerated electrons under the influence of the electron-withdrawing C-N-Ni site. Following the doping of the NiCo alloy with N, the electronegativity difference between Ni and Co narrowed significantly, enabling them to act as activation sites for CH4 and CO2, respectively, in the DRM reaction. Experiments and theoretical simulation results indicate that the released reactive oxygen species can directly oxidize CH* to CHO, which then converts to CO or CO2. This crucial step not only reinforces DRM but also blocks the dehydrogenation of CH* to carbon deposition. Furthermore, the N-Ni sites trap the photogenerated electrons around the Ni/Co interface, thereby preventing the metal from being oxidized by oxygen species. This is necessary to ensure long-term catalytic durability. Specifically, N-Ni/NiCo@C exhibits an excellent H2/CO production rate of 452.3/527.6 mmol h−1·g-1 and a selectivity of 0.95, while remaining stable for 200 h. Furthermore, a remarkable LTCEE of 52% is achieved with the optimized catalyst. This work presents a novel approach to designing efficient catalysts for PDRM and emphasizes the importance of a directional electron transfer path in synergizing photothermal effects, optimizing the electronic structure, and anti-coking mechanisms.
Methods
Material synthesis
The NiCo-MOF was synthesized using the microwave hydrothermal method. Specifically, 0.362 g Ni (NO3)2·6H2O (AR, Sinopharm Chemical Reagent Co., LTD), 0.3662 g Co (NO3)2·6H2O (99.99%, Aladdin), 0.262 g 1,3,5-benzene tricarboxylic acid (Beijing Bailingwei Technology Co., LTD), 3.275 g polyvinylpyrrolidone (Aladdin) and variable 1,10-phenanthroline (98.0%, Macklin) were dissolved in 75 mL N, N-dimethylformamide (DMF), and stirred with a magnetic stirrer for 30 min, after which they were transferred to a 250 mL autoclave. After sealing, the autoclave was installed in a microwave hydrothermal chamber. The temperature program was set to raise the temperature to 150 °C, followed by a 1.5 h reaction at this temperature. Upon completion, the autoclave was naturally cooled to room temperature before being taken out. The resulting product was centrifuged and filtered, washed three times with DMF, and dried at 60 °C in an oven for 8 h to obtain NiCo-MOF powder. The as-prepared NiCo-MOF powder was calcined at 700 °C for 2 h feed with 200 mL/min Ar to yield N-doped carbon-coated catalyst (N-Ni/NiCo@C). The addition amount of 1,10-phenanthroline was controlled at 0, 0.203, 0.406, 0.609, and 0.816 g. The obtained samples were sequentially labeled as N0, N0.5, N1, N1.5, and N2.
DRM performance measurements
DRM performance measurements were carried out under ambient pressure in a fixed-bed continuous flow quartz reactor with a quartz window at the middle of the furnace for light irradiation. For each test, 20 mg of catalyst was loaded and held in place by quartz wool. The reaction gas (10% CH4/10% CO2/80% Ar) was continuously introduced into the reactor with a flow rate of 50 mL min−1, giving a weight hour space velocity (WHSV) of 150,000 mL/(g h). A 300 W xenon lamp (PLS-SXE300E, Perfect Light) was employed as the light source to drive the DRM reaction. The products were evaluated by an online gas chromatograph (GC-2014ATFSPL, Ar carrier, Shimadzu) equipped with two flame ionization detectors (FIDs) and a thermal conductivity detector (TCD). The bulk temperature of the catalyst powder was detected using a thermocouple.
Quantification of reaction rates:
where ni represent molar flow rate of gas (mol h−1), Xi represents the gas volume fraction detected by TCD, rCO and rH2 are the production rates of CO and H2 (mol g h−1), F is the original flow rate of the gas (L/min), and m is the weight of catalyst (g).
According to Arrhenius formula, the logarithm of the H2 reaction rate (In rH2) was plotted against the reciprocal of temperature (1/T), and the activation energy (Ea) was obtained from the following slope:
R is the molar gas constant (8.314 J mo1−1 K−1), A is the pre-factor, and T is the thermodynamic temperature (K) of each temperature gradient.
LTCEE calculation: Δr(H2) = r(H2)PDRM − r(H2)DRM, Δr(CO) = r(CO)PDRM − r(CO)DRM, Δr(H2) = 2α-β and Δr(CO) = 2α + β were written, where α and β represent the reaction rates of DRM and RWGS, respectively.
LTCEE is defined as:
where ΔHDRM (247 KJ mol−1) and ΔHRWGS (41 KJ mol−1) are the standard reaction enthalpy of DRM and RWGS reaction, respectively, and P refers to the power of irradiation.
Catalyst characterizations
XRD patterns were recorded on a Rigaku-Smartlab 9KW from Japan with Cu-Kα radiation (λ = 1.54178 Å). Morphology and EDS element mapping were recorded by SEM, HRTEM and HAADF-STEM. XPS was performed on a Thermo Scientific ESCALAB 250Xi spectrometer with Al Kα as the X-ray source. XAFS spectroscopy with the Co K-edge and Ni K-edge was tested on the TableXAFS-500A using a 1.6 kW X-ray light source. The elemental content data are collected using ICP-OES by UTLIMA-2 (JY, France) and ONH836TOC (LECO). Raman spectra (PicRaman M3, timegate) are used to analyze the coking of the samples. TGA analysis was carried on the NETZSCH TG 209 F1 Libra TGA to determine the amount of carbon deposited.
CH4-TPD measurements
CH4-TPD measurements were carried out on AutoChem2920 (Micromeritics). Typically, 100 mg of catalyst was placed in a quartz tube reactor and treated under He flow at 300 °C for 60 min to remove impurities. After cooling to 40 °C, the catalyst was exposed to 10% CH4/90% Ar. Then, the catalyst bed temperature was increased from 40 to 700 °C (ramping rate, 10 °C min−1) under He flow. The desorption products were detected by the mass spectrometry. CO2-TPD experiments were performed in a similar process except using CO2 flow.
In situ DRIFTS measurements
The in situ DRIFTS measurements were performed using a Thermo Scientific IS50 Fourier-transform spectrometer equipped with a Harrick diffuse reflectance accessory. In the test of CO2 and CH4 adsorption and activation, the fresh catalyst was first purged with Ar flow for 30 min at room temperature to remove impurities. Then the fresh catalyst was exposed to 10% CH4/10% CO2/80% Ar flow to collect the DRIFTS spectra for CO2 and CH4 adsorption. After adsorption saturation, the system was exposed to light irradiation (PLS-SXE300E, Perfect light), and the temperature data were recorded continuously. The DRIFTS for respective CO2 and CH4 adsorption and activation were performed in a similar process except using 10% CO2/90% Ar flow and 10% CH4/90% Ar, respectively.
In situ NAP-XPS measurements
In situ NAP-XPS measurements were performed on Thermo Scientific ESCALAB 250Xi spectrometer with Al Kα as the X-ray source. The fresh catalyst was first treated at ultrahigh vacuum (up to 10−9 mbar). After that, the system was purged with 0.5 mbar CO2 and 0.5 mbar CH4 for 2 h at 540 °C to collect the data. The in-situ NAP-XPS for PDRM was performed in a similar process, except adding 300 W Xe lamp irradiation.
Quasi in-situ 13C-labeled isotope labelling experiments
Quasi in-situ 13C-labeled isotope labelling experiments were performed in the same reactor with activity test by using two different reactant systems as feeding gas, including equivalent amounts of 12CH4 + 13CO2 (I), 13CH4 + 13CO2 (II). In a typical test, 10 mg of fresh catalyst was used. After sealing the reactor, the reactor was first purged with Ar flow for 30 min at 300 °C and then equivalent amounts of 13C-labeled substances (i.e., Systems I or Systems II) were introduced with the temperature increasing from 300 to 600 °C at a rate of 10 °C/min in dark or under 300 W Xe lamp irradiation, the products were analyzed using a GC-MS (8860-5977 C, He carrier, Agilent) and the data collected continuously.
DFT calculations
Based on density functional theory (DFT), a series of theoretical calculation methods were employed in this study46, including the Vienna ab initio simulation package (VASP), the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) framework, the projector-augmented wave (PAW) method, the Monkhorst–Pack k-point grid, and the DFT + U correction method. The energy convergence threshold for self-consistent iteration was 10−6 eV, and the residual force for ionic relaxation needed to be below 0.02 eV Å−1. It should be noted that the PBE-GGA-based DFT approach possesses inherent limitations, including the neglecting nuclear quantum effects, inadequate characterization of van der Waals interactions, and restriction to ground-state properties in the absence of supplementary extensions.
The catalyst model was constructed using a 3 × 3 supercell for the (111) facet of CoNi alloy (N0)2, with a 15 Å vacuum layer inserted to prevent interactions between adjacent periodic images. To simulate N atom incorporation, four surface metal atoms on the CoNi (111) surface were substituted with N atoms, forming the N-Ni/CoNi (111) structure (N1).
The adsorption energy (ΔEads) of surface species on N1 or N0 surfaces was calculated using the following equation:
where ΔEtotal, Eadsorbate, and Esurface denote the total energy of the adsorbed surface species, the energy of the adsorbate in the gas phase, and the energy of the clean N1/N0 surfaces, respectively.
The Gibbs free energy (ΔG) for gas molecules (CH₄, CO₂) and adsorbates was calculated using the following equation:
where E is the total structural energy from DFT calculations, ZPE is the zero-point energy, and S is the vibrational entropy.
Charge density differences were used to analyze electron density and electron cloud distribution changes for the substituted metal atoms on N1 and N0 surfaces. The charge density difference was calculated using the following equation:
where ρtotal, ρsurface, and ρM represent the charge densities of the substituted surface, clean surface, and substituted metal atom, respectively.
Data availability
The processed data generated in this study are provided in the main text, supplementary information, and Source Data files. Source data are provided in this paper. DFT models are provided in Supplementary data 1 file. Source data are provided with this paper.
References
Song, Y. et al. Dry reforming of methane by stable Ni-Mo nanocatalysts on single-crystalline MgO. Science 367, 777–781 (2020).
Zhou, T. et al. Ultrafine metal nanoparticles isolated on oxide nano-islands as exceptional sintering-resistant catalysts. Nat. Mater. 24, 891–899 (2025).
Zhu, Q. et al. Enhanced CO2 utilization in dry reforming of methane achieved through nickel-mediated hydrogen spillover in zeolite crystals. Nat. Catal. 5, 1030–1037 (2022).
Shoji, S. et al. Photocatalytic uphill conversion of natural gas beyond the limitation of thermal reaction systems. Nat. Catal. 3, 148–153 (2020).
Song, C., Wang, Z., Yin, Z., Xiao, D. & Ma, D. Principles and applications of photothermal catalysis. Chem Catal. 2, 52–83 (2022).
Wang, Z. et al. Reveal and correlate working geometry and surface chemistry of Ni nanocatalysts in CO2 reforming of methane. Mater. Today 79, 16–27 (2024).
Li, J. et al. Activating lattice oxygen in perovskite ferrite for efficient and stable photothermal dry reforming of methane. J. Am. Chem. Soc. 147, 14705–14714 (2025).
Xiong, H. et al. Highly Efficient and selective light-driven dry reforming of methane by a carbon exchange mechanism. J. Am. Chem. Soc. 146, 9465–9475 (2024).
Wang, H. et al. Facilitating the dry reforming of methane with interfacial synergistic catalysis in an Ir@CeO2-x catalyst. Nat. Commun. 15, 3765 (2024).
Yan, G. et al. Reaction product-driven restructuring and assisted stabilization of a highly dispersed Rh-on-ceria catalyst. Nat. Catal. 5, 119–127 (2022).
Rao, Z. et al. Insights into the nonthermal effects of light in dry reforming of methane to enhance the H2/CO ratio near unity over Ni/Ga2O3. ACS Catal. 11, 4730–4738 (2021).
Xie, T. et al. Enhanced photothermal catalytic performance of dry reforming of methane over Ni/mesoporous TiO2 composite catalyst. Chem. Eng. J. 429, 132507 (2022).
Yang, Y. et al. Light-induced redox looping of a rhodium/CexWO3 photocatalyst for highly active and robust dry reforming of methane. Angew. Chem. Int. Ed. 61, e202200567 (2022).
He, J. et al. Subsurface A-site vacancy activates lattice oxygen in perovskite ferrites for methane anaerobic oxidation to syngas. Nat. Commun. 15, 5422 (2024).
Guo, C. et al. Light-driven metal exsolution-redissolution of high-entropy oxide enabling high-performance dry reforming of methane. Adv. Mater. 37, 2500928 (2025).
Tu, W., Ghoussoub, M., Singh, C. V. & Chin, Y.-H. C. Consequences of surface oxophilicity of Ni, Ni-Co, and Co clusters on methane activation. J. Am. Chem. Soc. 139, 6928–6945 (2017).
Li, X. et al. Cobalt single-atom catalysts with high stability for selective dehydrogenation of formic acid. Angew. Chem. Int. Ed. 59, 15849–15854 (2020).
Rao, Z. et al. Light-reinforced key intermediate for anticoking to boost highly durable methane dry reforming over single atom Ni active sites on CeO2. J. Am. Chem. Soc. 145, 24625–24635 (2023).
Ma, J. et al. Rational design of N-doped carbon-coated cobalt nanoparticles for highly efficient and durable photothermal CO2 conversion. Adv. Mater. 35, e2302537 (2023).
Alli, R. D., Zhou, R., Mohamedali, M. & Mahinpey, N. Effect of thermal treatment conditions on the stability of MOF-derived Ni/CeO2 catalyst for dry reforming of methane. Chem. Eng. J. 466, 143242 (2023).
Liang, T.-Y. et al. Bimetallic metal-organic framework-derived hybrid nanostructures as high-performance catalysts for methane dry reforming. ACS Appl. Mater. Interfaces 12, 15183–15193 (2020).
Liu, X. et al. Highly efficient solar-driven CO2-to-fuel conversion assisted by CH4 over NiCo-ZIF derived catalysts. Fuel 310, 122441 (2022).
Alli, R. D. & Mahinpey, N. Influence of organic ligand and nickel loading on the performance of MOF-derived catalysts for dry reforming of methane. Fuel 361, 130756 (2024).
Komarala, E. P. et al. Tune and turn the pyrolysis of metal organic frameworks towards stable supported nickel catalysts for the dry reforming of methane. Appl. Surf. Sci. 666, 160388 (2024).
Wang, P. et al. Atomically dispersed cobalt catalyst anchored on nitrogen-doped carbon nanosheets for lithium-oxygen batteries. Nat. Commun. 11, 1576 (2020).
Lu, Q. et al. Encapsulating cobalt nanoparticles in interconnected N-doped hollow carbon nanofibers with enriched Co-N-C moiety for enhanced oxygen electrocatalysis in Zn-Air batteries. Adv. Sci. 8, 2101438 (2021).
Jiang, Z. et al. Pyridinic N and carbon defects synergistically promote methane dry reforming to syngas catalyzed by Co/N-CNTs. Fuel 337, 127136 (2023).
Zhang, Y. et al. Combined steam and CO2 reforming of methane over Co-Ce/AC-N catalyst: effect of preparation methods on catalyst activity and stability. Int. J. Hydrogen Energy 47, 2914–2925 (2022).
Wu, S. et al. Formation of NiCo alloy nanoparticles on Co-doped Al2O3 leads to high fuel production rate, large light-to-fuel efficiency, and excellent durability for photothermocatalytic CO2 reduction. Adv. Energy Mater. 10, 2002602 (2020).
Yao, Y. et al. Highly efficient solar-driven dry reforming of methane on a Rh/LaNiO3 catalyst through a light-induced metal-to-metal charge transfer process. Adv. Mater. 35, e2303654 (2023).
Lorber, K. et al. On the mechanism of visible-light accelerated methane dry reforming reaction over Ni/CeO2-x catalysts. Appl. Catal., B 301, 120745 (2022).
Zhang, Z.-Y. et al. Photo-enhanced dry reforming of methane over Pt-Au/P25 composite catalyst by coupling plasmonic effect. J. Catal. 413, 829–842 (2022).
Zhang, Z.-Y. et al. Promoted solar-driven dry reforming of methane with Pt/mesoporous-TiO2 photo-thermal synergistic catalyst: performance and mechanism study. Energy Convers. Manage. 258, 115496 (2022).
Liu, H. et al. Light assisted CO2 reduction with methane over SiO2 encapsulated Ni nanocatalysts for boosted activity and stability. J. Mater. Chem. A 5, 10567–10573 (2017).
Ma, W. et al. Encapsulated Co-Ni alloy boosts high-temperature CO2 electroreduction. Nature 641, 1156–1161 (2025).
Sandoval-Diaz, L. et al. Metastable nickel-oxygen species modulate rate oscillations during dry reforming of methane. Nat. Catal. 7, 161–171 (2024).
Li, J. et al. Tuning adsorbate-mediated strong metal-support interaction by oxygen vacancy: A case study in Ru/TiO2. Angew. Chem. Int. Ed. 63, e202407025 (2024).
Shao, B. et al. Synergistic promotions between CO2 capture and in-situ conversion on Ni-CaO composite catalyst. Nat. Commun. 14, 996 (2023).
Li, Q. et al. Suppressive strong metal-support interactions on ruthenium/TiO2 promote light-driven photothermal CO2 reduction with methane. Angew. Chem. Int. Ed. 62, e202300129 (2023).
Cheng, F., Duan, X. & Xie, K. Dry reforming of CH4 /CO2 by stable Ni nanocrystals on porous single-crystalline MgO monoliths at reduced temperature. Angew. Chem. Int. Ed. 60, 18792–18799 (2021).
Li, Y. K., Müller, F., Schöllkopf, W., Asmis, K. R. & Sauer, J. Gas-phase mechanism of O·-/Ni2+-mediated methane conversion to formaldehyde. Angew. Chem. Int. Ed 134, e202202297 (2022).
Ye, D. et al. Anti-sintering Ni-W catalytic layer on reductive tungsten carbides for superior high-temperature CO2 reduction. Adv. Mater. 37, 2504431 (2025).
Yu, X., De Waele, V., Löfberg, A., Ordomsky, V. & Khodakov, A. Y. Selective photocatalytic conversion of methane into carbon monoxide over zinc-heteropolyacid-titania nanocomposites. Nat. Commun. 10, 700 (2019).
Hu, Q. et al. Extraordinary catalytic performance of nickel half-metal clusters for light-driven dry reforming of methane. Adv. Energy Mater. 13, 2300071 (2023).
Guo, Y.-P., Li, W.-Y. & Feng, J. Reaction pathway of CH4/CO2 reforming over Ni8/MgO(100). Surf. Sci. 660, 22–30 (2017).
Meng, Y. et al. Curvature-influenced electrocatalytic NRR reactivity by Heme-like FeN4-site on carbon materials. Molecules 30, 1670 (2025).
Acknowledgements
This work is financially supported by Eastern Institute of Technology, Ningbo. We also acknowledge support by the Young Innovative Talent of Yongjiang Talent Project (2023A-387-G) to H.Z., the China Postdoctoral Science Foundation (2024M751550) to T.P., and the Ningbo City Natural Science Foundation (2024J155) to T.P. Y.X. acknowledges financial support from the National Natural Science Foundation of China (NSFC) (22232003).
Author information
Authors and Affiliations
Contributions
T.P. performed all the experimental work. W.X. completed all the DFT calculations. T.P. and H.Z. designed the research and co-wrote the manuscript in discussion. H.D., Y.X., Z.C., and H.Z. discussed the data and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Sibo Wang, Jinlong Zhang, who co-reviewed with Shiqun Wu and the other anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pan, T., Xu, W., Deng, H. et al. A lattice oxygen-free design for efficient and stable photothermal methane dry reforming. Nat Commun 17, 2151 (2026). https://doi.org/10.1038/s41467-026-68898-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68898-z
