
Abstract
The discovery of KRAS G12C inactive state inhibitors represented a significant advancement in the field of precision oncology. While inactive state inhibition shows promise in patients, Switch II (SWII)-binding inhibitors targeting both inactive and active states remain an underdeveloped therapeutic modality. Here, we describe the discovery of such KRAS G12C dual inhibitors that bind the SWII allosteric site using a chemically differentiated warhead to covalently modify both the KRAS G12C inactive and active states. Co-crystal structures reveal that these inhibitors perturb a key water-mediated hydrogen bonding network and trigger allosteric remodeling of the GTP-bound protein surface and SWI that prevents binding to downstream effectors. Consistent with simultaneous targeting of the active and inactive states, dual inhibitors provide rapid covalent target engagement and suppression of MAPK signaling. However, they demonstrate similar efficacy in cellular and in vivo models when compared to inactive state-selective ones despite faster target inactivation. Furthermore, both inhibitor classes show similar cellular efficacy in the presence of growth factors that drive KRAS, wt NRAS, and wt HRAS to the active state. These data provide the first detailed account of targeting both the active and inactive states of KRAS G12C and highlight the absence of a mechanistic advantage in contexts dependent on prolonged target inhibition.
Similar content being viewed by others

Introduction
KRAS is the most frequently mutated oncogene, and KRAS mutations are well-validated drivers of oncogenesis1. As such, significant effort has been focused on developing direct inhibitors of KRAS and KRAS mutants2. KRAS G12C, in particular, has been the subject of intense research and numerous drug development programs owing to the presence of its nucleophilic mutant cysteine3,4,5. Shokat and coworkers initially demonstrated that this cysteine residue could be productively engaged by an electrophilic warhead, inducing a small molecule binding pocket proximal to the Switch II (SWII) loop of the protein6. These tool molecules preferentially modify the GDP-bound inactive state of the mutant protein and disrupt effector interaction with KRAS, with little binding or activity against the oncogenic, GTP-bound active state. However, because KRAS G12C cycles between these two states, over time these irreversible inactive state inhibitors can fully modify the total protein pool by sequestering it in an inhibitor-bound inactive state, preventing downstream signaling and, ultimately, tumor proliferation7. Currently, two KRAS G12C mutant selective inhibitors that target the inactive state have been approved by the FDA8,9,10, with many more undergoing clinical development11,12,13,14,15,16,17,18,19. While these approved inhibitors have shown clinically meaningful improvement in progression-free survival, it has been widely proposed that targeting both the active and inactive states of KRAS G12C would circumvent therapeutic resistance to inactive state-selective inhibitors driven by genomic and non-genomic mechanisms that increase the cellular pool of GTP-loaded KRAS G12C20,21,22,23. Indeed, next-generation inhibitors have begun to emerge that operate under this principle and can target the oncogenic active state directly24,25.
In this study, we report our efforts to target both KRAS G12C states and disclose the discovery and characterization of SWII-binding covalent dual inhibitors, predicated on the identification of a differentiated class of covalent warhead.
Results
Design and discovery of compound 2
As allosteric agents, current SWII inhibitors bind KRAS in the presence of a nucleotide, which is located proximal to the site of covalent attachment. Targeting the active state with its bound GTP represents a significantly increased challenge over inhibiting the GDP-bound inactive state, as the γ-phosphate of GTP protrudes into the induced binding site. However, computational modeling based on published KRAS G12C GDP-inhibitor bound complexes suggested that a subtle change in warhead trajectory could be enough to sidestep the γ-phosphate and transform an inactive state-selective inhibitor into a dual active and inactive state inhibitor. Thus, in contrast to reported optimization efforts for inactive state-selective inhibitors that have focused on improving SWII binding elements, we turned our attention to modification of the covalent warhead itself.
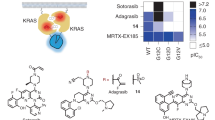
We selected adagrasib (1) as the starting point for this exploration and synthesized a series of warhead-modified analogs intended to maintain productive contacts in the induced binding pocket while accommodating either GDP or GTP bound to KRAS G12C (Fig. 1a). KRAS G12C active state activity was assessed in a protein-protein disruption assay using KRAS G12C bound to GMPPNP, a nonhydrolyzable mimetic of GTP, and the RAS-binding domain (RBD) of its native binding partner BRAF. Compounds that functionally inhibit the active state would be expected to prevent the binding of effectors, like RAF RBD. From these studies, two chemically-related compounds with substituted acrylamide warheads, 2 and 3, emerged that demonstrated dose-dependent disruption of the RAS/RAF interaction (IC50: 138 nM and 120 nM, respectively) (Fig. 1b). To confirm these compounds’ continued ability to inhibit the inactive state of KRAS G12C as well, a SOS1-mediated GDP nucleotide exchange assay was deployed, in which compounds that successfully bind and sequester inactive KRAS G12C would be expected to inhibit GDP exchange. Both compounds 2 and 3 maintained comparable inactive state activity to adagrasib (1) in the nucleotide exchange assay (1 IC50: 12 nM, 2 IC50: <15 nM, 3 IC50: <15 nM) (Fig. 1c), even with their more highly substituted acrylamide warheads9,26,27. Intrigued by this unexpected finding and having successfully executed on our design of dual inhibitors, we next endeavored to understand the scope of this differentiated warhead class and the structural factors leading to active state inhibition.

a Chemical structures of analogs prepared. b RAS/RAF disruption potency of analogs. c GDP exchange inhibition potency of analogs. For Figs. 1b and 1c, IC50s for 2, 3, 4, and 5 represent the average of two independent experiments; IC50s for 1, 6, 7, 8, and divarasib represent the average of three independent experiments. Where applicable, data are given as mean ± SD. Source data are provided as a Source Data file.
Nucleotide accommodation and allosteric remodeling govern active state inhibition
To assess the generality of our approach, we applied our differentiated warhead to additional known KRAS-targeting architectures. First, we synthesized warhead analogs of sotorasib8, an FDA-approved KRAS G12C inactive state-selective inhibitor, and divarasib16,28, a KRAS G12C inactive state-selective inhibitor currently undergoing Phase III clinical trials. These analogs (4 and 5, respectively) maintained inactive state potency and showed a dose-dependent decrease of GDP nucleotide exchange activity comparable to their parent compound (sotorasib IC50: 25 nM, 4 IC50: 290 nM; divarasib IC50: 10 nM, 5 IC50: 6 nM). However, unlike adagrasib analogs 2 and 3, analogs 4 and 5 showed minimal disruption of the RAS/RAF interaction (4 IC50: 5.2 μM, 5 IC50: 4.3 μM). A third analog in this series, 6, was synthesized based on the disclosure of MRTX-1133, a noncovalent KRAS G12D-selective dual inhibitor with binding elements informed by 129. Analog 6 showed potent dose-dependent disruption of the RAS/RAF interaction (IC50: 4 nM) and GDP nucleotide exchange inhibition (IC50: 7 nM). Taken together, these results show that accommodating the bound GTP nucleotide is necessary, but not sufficient, for potent active state inhibition.
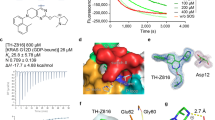
Reasoning that the other factor(s) determining active state potency could be associated with structural aspects of inhibitor binding, we obtained a co-crystal structure of compound 2 and GMPPNP-bound KRAS G12C (Fig. 2a). This structure revealed that 2 bound in the induced SWII pocket of KRAS G12C. The tetrahydropyridopyrimidine core made a key hydrogen bond to His95, a residue not shared by HRAS or NRAS; a cation-π interaction between the pyrrolidine group and His95 reinforced that contact. The nitrile group displaced a bound water molecule (W1) proximal to Gly10 and Thr58. These interactions aligned with those observed in the published co-crystal structure of adagrasib (1) and GDP-bound KRAS G12C, indicating that the SWII allosteric pocket forms similarly in GMPPNP-bound KRAS G12C (Supplementary Fig. 1)9.

a Co-crystal structure of compound 2 and GMPPNP-bound KRAS G12C. Key contacts between 2 and the protein are highlighted (PBD: 9NZM). b Omit Fo-Fc map contoured at 3 r.m.s.d. of warhead region indicating cysteine attachment at the α-position of the warhead. Inset: mechanistic comparison of this warhead’s reactivity to that of a standard acrylamide. c Co-crystal structure of compound 2 and KRAS G12C overlaid with a crystal structure of KRAS interacting with the RBD of BRAF (yellow, PDB:6XI7). The red and blue arrows indicate the difference in Glu37 and Arg68 positioning, respectively, between the two structures. d Co-crystal structure of compound 6 and GDP-bound KRAS G12C (PDB: 9NZN). e SWII (cyan)-SWI (purple) region of the GDP-bound KRAS G12C and compound 6 co-crystal structure, revealing a bidentate salt bridge interaction between Glu37 and Arg68.
Examination of the warhead region of the structure (Fig. 2b) suggested an origin for the special properties of these covalent inhibitors. Compound 2 reacted with cysteine at the α-position of the warhead with concomitant loss of fluoride, a previously undescribed mechanism of covalent inhibition. Mass spectrometry corroborated this mode of covalent modification, as incubating 2 with either GDP- or GMPPNP-bound KRAS G12C led to adducts lacking the fluoride (Supplementary Fig. 2). This reactivity stands in stark contrast to that seen with acrylamide warheads of known KRAS G12C inhibitors, which react with cysteine exclusively at the β-position. We attribute this divergent reactivity to the unique electronic characteristics of these warheads. The presence of an electron-poor heterocycle in conjugation with the acrylamide fluoro-alkene enables vinylogous nucleophilic aromatic substitution reactivity, which occurs preferentially over standard conjugate addition reactivity.
Finally, we scanned the protein surface for conformational changes, focusing particularly on the Switch I (SWI) loop, which is known to be the point of interaction with effectors, like RAF (Fig. 2c). We noticed that an allosteric remodeling of the SWI loop had occurred upon compound binding, as evidenced by an interaction between Glu37 and Arg68. A similar interaction was present in the published co-crystal structure of adagrasib (1) and GDP-bound KRAS G12C, but not in the structure of sotorasib and GDP-bound KRAS G12C. We obtained a co-crystal structure of 6 and GDP-bound KRAS G12C to ascertain whether binding of that inhibitor also led to formation of the Glu37-Arg68 interaction (Fig. 2d). Once again, the covalent warhead had reacted with cysteine at the α-position. Excitingly, in this structure, the Glu37-Arg68 interaction was more pronounced, with a bidentate salt bridge existing between the two residues (Fig. 2e).
Drawing this allosteric modification back to inhibitor structure, we hypothesized that perturbation of key water molecule W1 by the inhibitor led to the observed remodeling of the SWI loop and, accordingly, binding to the active state. Both 2 and 6 have chemical groups (a nitrile group and an alkyne group, respectively) known to interact with W1: the nitrile group of 2 displaces W1, while the alkyne group of 6 stabilizes W1 through a nonclassical hydrogen bond (Supplementary Fig. 3)9,29. To test this hypothesis, we prepared a nitrile-bearing analog of 5, which itself does not contain a group to interact with W1 and does not disrupt the RAS/RAF interaction. Gratifyingly, compound 7 demonstrated potent dose-dependent RAS/RAF disruption, with over 250-fold increased potency compared to compound 5 (7 IC50: 16 nM; 5 IC50: 4.3 μM). Likewise, compound 8, an analog of 6, also with an alkyne group, showed significant active-state potency (IC50: 133 nM). Together, these data suggested that two factors are required for potent active state inhibition:
(1) a trajectory that minimizes negative interactions with the γ-phosphate of GTP and
(2) functional group(s) that interact with W1, stabilizing the Gly10/Thr58 pocket and favoring formation of the Glu37-Arg68 salt bridge.
Compounds 6 and 8 are potent, selective KRAS G12C dual inhibitors
Armed with this robust structural understanding of active state inhibition, we next interrogated the potency and selectivity profiles of compounds 6 and 8, the two lead dual inhibitors. A full kinetic evaluation was undertaken for 8 (Fig. 3a), which showed it to be a rapid inhibitor of both the GDP-bound inactive state KRAS G12C (kinact/KI > 79,000 M−1 s−1) and GMPPNP-bound active state KRAS G12C (kinact/KI > 8,000 M−1 s−1), in line with biochemical data. This is in contrast to divarasib, which is reported to inhibit only GDP-bound inactive state KRAS G12C (kinact/KI: 710,000 M−1 s−1) (Supplementary Fig. 4)30,31. Furthermore, a chemical reactivity study of compound 8 with glutathione (72% remaining after 30 min) predicted a lack of promiscuity with cysteine-based nucleophiles. This result was mirrored by other compounds in the series (e.g., 6 and 7), indicative of a general trend with this warhead motif (Supplementary Table 1).

a Observed rate of covalent adduct formation (kobs) plotted against compound concentration for 8. Blue circles represent rates of adduct formation with GMPPNP-bound KRAS G12C; gold squares represent the same for GDP-bound KRAS G12C. Covalent occupancy was measured in two independent experiments; averaged values were used to calculate the plotted kobs values. b Cellular potencies of dual inhibitors 6 and 8, and inactive state-selective inhibitor divarasib in MIA PaCa-2, a KRAS G12C mutant PDAC cell line. pERK inhibition was measured after 2 h of compound treatment, and proliferation inhibition after 5 d. Data are from three independent experiments and are presented as mean ± SD. c Cellular potencies of 6 and 8 compared to divarasib and RMC-6291 across a panel of G12C- (left) and non-G12C- (right) mutant cell lines. IC50s in the G12C-mutant cell lines are the average of two independent experiments for 6, 8, divarasib, and, where applicable, RMC-6291 and are presented as mean ± SD. IC50s for 6 and 8 in the non-G12C-mutant cell lines are the average of three independent experiments and are presented as mean ± SD. d Reactive cysteine profiling of compound 8 (left) and divarasib (right). Dots in the blue quadrant represent cysteine-containing peptide fragments found to be significantly reduced in samples treated with inhibitor (fold change < 2 and adj. p-value < 0.01); dots in the red quadrant represent cysteine-containing peptide fragments found to be significantly exposed in samples treated with inhibitor (fold change > 2 and adj. p-value < 0.01). For each protein, moderated t-statistics were computed using empirical Bayes shrinkage of variance estimates. All p-values correspond to two-sided tests. Where indicated, p-values were adjusted for multiple comparisons using the Benjamini–Hochberg false discovery rate (FDR) procedure. Adjusted p-values (FDR) are reported unless stated otherwise. The fragment for KRAS G12C is highlighted in purple. Source data are provided as a Source Data file.
We next assessed compounds 6 and 8, along with positive control divarasib, for on-target activity in KRAS G12C cell lines. KRAS G12C dual inhibitors potently decreased phosphorylation of ERK1/2 (pERK), a well-known signaling node downstream of KRAS, in MIA PaCa-2, a KRAS G12C mutant cell line (Fig. 3b). Compound 8 showed comparable pERK potency (IC50: 0.4 nM) to divarasib (IC50: 0.3 nM). A similar trend was observed in these compounds’ ability to inhibit cellular proliferation in this model, with compounds 6 and 8 exhibiting potency comparable to divarasib (6 IC50: 0.1 nM; 8 IC50: 1.6 nM; divarasib IC50: 0.6 nM).
To build further support for on-target KRAS G12C inhibition, we profiled 6 and 8, along with inactive state-selective KRAS G12C inhibitor divarasib and RMC-6291, a clinical-stage KRAS G12C active state tricomplex inhibitor that does not bind SWII, in a panel of cell lines containing both KRAS G12C mutants and non-G12C mutants (Fig. 3c). Compounds 6 and 8 showed robust and potent activity in a panel of KRAS G12C mutant cell lines with a cell line sensitivity profile similar to KRAS G12C inactive state-selective inhibitor divarasib. The tricomplex inhibitor RMC-6291 also showed potent growth inhibition across the subset of KRAS G12C mutant cell lines evaluated. In non-G12C mutant cell lines (BxPC3, SW1990, HPAC), the ability of 6 and 8 to inhibit cellular proliferation was attenuated ~10,000-fold (6 average IC50: 5.1 μM; 8 average IC50: 6.3 μM), highlighting these compounds’ selectivity. Additionally, we assessed compounds of interest by mass spectrometry-based global cysteinome profiling in H358 cells, a KRAS G12C mutant line (Fig. 3d), which evaluated over 22,000 cysteines in the human proteome within 6900 protein families, and found that compound 8 showed selectivity for the Cys12 of KRAS G12C over the majority of other peptides identified, including Cys 80 of KRAS and NRAS (Supplementary Fig. 5). Divarasib likewise selectively modified KRAS G12C, though it decreased identification of more cysteine-containing peptides than did compound 8. Overall, these data demonstrate that compounds 6 and 8 are potent, selective KRAS G12C inhibitors and validate this warhead as a viable targeting motif with suitable kinetics of covalent inhibition.
Cellular kinetics of dual state inhibition
Next, we evaluated the cellular kinetic profile of dual active- and inactive-state targeting. In contrast to inactive state-selective inhibitors, dual state inhibitors are anticipated to bypass the need for active-to-inactive state conversion and potentially show faster KRAS G12C inhibition in cells. We measured the dissociation between KRAS G12C and its effector, RAF, in cells using a KRAS G12C/RAF RBD (RAS/RAF) nanoBRET assay (Fig. 4a). In this assay, RMC-6291 showed very rapid (t1/2 < 5 min) disruption in line with its reported KRAS G12C active state activity. Compound 6 behaved similarly to RMC-6291 and demonstrated very rapid (t1/2 = 9 min) cellular RAS/RAF disruption. Compound 8 disrupted the cellular RAS/RAF interaction more slowly (t1/2 = 56 min) than compound 6. Both compounds demonstrated more rapid RAS/RAF disruption than the inactive state-selective inhibitor divarasib, a finding that is consistent with biochemical measurements.

a Kinetics of cellular RAS/RAF disruption for dual inhibitors 6 and 8, inactive state-selective inhibitor divarasib, and active state-selective inhibitor RMC-6291. Data are an average of two independent experiments and are presented as mean ± SD. b Time course of pERK inhibition of 8 in MIA PaCa-2 cells from 30 min to 2 h. Data are an average of two independent experiments are presented as mean ± SD. c Time course of target engagement of 6, 8, divarasib, and RMC-6291 with KRAS G12C in MIA PaCa-2 cells from 5 min to 2 h at concentrations between 0.1 nM and 1000 nM. Data are an average of two independent experiments and are presented as mean ± SD. d Suppression of MAPK transcriptional signaling in H1792 cells by 6 and divarasib at 24 and 48 h. The boxplots show the medians with the interquartile range (box) and range (whiskers). Pairwise comparisons were performed using two-sided t-tests (DMSO vs compound 6; DMSO vs divarasib; and compound 6 vs divarasib). At 24 h: p = 0.000034, 0.00055, and 0.0013, respectively; at 48 h: p = 0.0016, 0.0093, and 0.026, respectively. Significance levels are indicated as follows: * = p < 0.05, ** = p < 0.01, and *** = p < 0.001. Source data are provided as a Source Data file.
We investigated the rate of KRAS G12C covalent adduct formation and inhibition of downstream pERK signaling in MIA PaCa-2. Compound 8 inhibited pERK signaling rapidly, and the extent of inhibition increased over time (Fig. 4b). A cellular target engagement assay was developed based on a SMaSh assay, where an unoccupied target protein was labeled by a biotinylated probe followed by detection with streptavidin32. Compounds 6 and 8 demonstrated rapid endogenous KRAS G12C covalent adduct formation in cells (Fig. 4c). Complete KRAS G12C engagement (>95%) occurred earlier for dual inhibitor 6 than for divarasib (5 min vs. 1 h) at 1 µM. Finally, we assessed the transcriptional readout of pERK inhibition via global RNAseq in H1792 cells (Fig. 4d). Compound 6 showed on-target MAPK suppression after 24 and 48 h. Furthermore, compound 6 demonstrated deeper suppression of MAPK pathway genes at 24 and 48 h compared to divarasib. Additional gene expression analysis of qPCR examining 10 genes associated with MAPK signaling established that compound 8 also demonstrated deeper suppression of the MAPK pathway at 24 h (Supplementary Fig. 6, Supplementary Table 2). These results confirm that dual inhibitors are able to inhibit KRAS G12C and its signaling more rapidly than inactive state-selective inhibitors, consistent with simultaneous dual targeting that does not require GTP-to-GDP cycling.
Compound 8 is efficacious in vivo
We assessed the impact of KRAS G12C dual inhibition in vivo in both pharmacodynamic (PD) and efficacy experiments using KRAS G12C xenograft models. Compound 8 was selected for these studies, given its superior oral bioavailability relative to compound 6 (Supplementary Table 3). Compound 8 and divarasib were studied in KRAS G12C mutant mouse xenograft models, measuring KRAS G12C covalent adduct formation as a direct indicator of target engagement and inhibition of phosphorylation of ERK as a downstream PD marker. In mice bearing MIA PaCa-2 tumors, compound 8 demonstrated dose- and time-dependent KRAS G12C target engagement that correlated with robust pERK suppression relative to total ERK (Fig. 5a). Significantly, at the 50 and 150 mg/kg doses, compound 8 exhibited over 85% KRAS G12C covalent engagement and approximately 50% pERK suppression at 1 h post-treatment. In contrast, at 1 h post-treatment, the KRAS G12C inactive state-selective inhibitor divarasib showed no more than 70% target engagement. By 7 h post-treatment, maximum target engagement had been reached for all inhibitors, and >90% target engagement was observed at the highest tested doses. These results parallel the cellular studies that showed faster kinetics of covalent target engagement for dual inhibitors but equivalent KRAS G12C target engagement over time for both classes of inhibitors (Fig. 5b)33,34. Compound 8 was also tested in an H2122 KRAS G12C mutant mouse xenograft model, which has been reported to be less sensitive to KRAS G12C inhibition35. Similar results were seen here as with the MIA PaCa-2 model, with compound 8 demonstrating rapid KRAS G12C target engagement and deep pERK suppression at early time points (Supplementary Fig. 7). Compound 8 was advanced into in vivo KRAS G12C tumor efficacy studies to assess the impact of KRAS G12C dual inhibition on tumor growth. In the MIA PaCa-2 xenograft model, deep tumor regressions were observed with compound 8 at once daily oral doses of 15 mg/kg or greater (Fig. 5c). At the highest dose tested, 150 mg/kg daily (QD), compound 8 drove complete responses in 6 of 8 animals (Fig. 5d). Additionally, compound 8 and divarasib were taken into an efficacy study in a KRAS G12C-mutant NSCLC patient derived xenograft (PDX) model, LUN055, reported to be resistant to KRAS G12C inactive state inhibitors but sensitive to active state inhibition (Fig. 5e)24. After 21 days of treatment with compound 8 at 150 mg/kg twice daily (BID), tumor growth inhibition of 88% was observed, along with high levels of target engagement and pERK suppression. Comparable efficacy results were seen in the LUN055 PDX model with the inactive state-selective inhibitor divarasib, indicating a lack of mechanistic differentiation. The observation that both compounds achieved similar maximum covalent target engagement provides indirect evidence that resistance in this PDX model is not driven by increased pools of GTP-loaded G12C that are inaccessible to inactive state-selective inhibitors. Overall, these data demonstrate that dual inhibitors are efficacious in vivo, but with no apparent advantage over inactive state inhibition.

a Pharmacokinetic/pharmacodynamic (PK/PD) assessment of compound 8 and divarasib in a MIA PaCa-2 mouse xenograft model Top: plasma concentrations and pERK inhibition at 1, 3, 7, and 24 h for 8 and divarasib. Bottom: Target engagement for 8 and divarasib with KRAS G12C at 1, 3, 7, and 24 h (n = 3 biological replicates per dose level per time point). Data are given as mean ± SEM. b Comparison of free drug vs target engagement of 8 and divarasib at 1 h (top) and 7 h (bottom). c Efficacy study of compound 8 in a MIA PaCa-2 mouse xenograft model showing change in tumor volume over the 21 days of treatment (n = 8 biological replicates per group). Data are presented as mean ± SEM. d Waterfall plot of tumor volumes at the conclusion of the study. e Efficacy study of compound 8 and divarasib in an LUN-055 mouse xenograft model showing change in tumor volume of 21 days of treatment (left), end of study target engagement (center), and end of study pERK suppression (right) (n = 5 biological replicates for compound 8; n = 6 biological replicates for divarasib). Source data are provided as a Source Data file.
H- and NRAS contribute to growth factor-mediated resistance
It has been proposed that a further mechanistic difference between active state- and inactive state-selective inhibitors can be seen by assessing the impact of cellular stimuli on pathway inhibition24. Specifically, growth factors like HGF and EGF are ubiquitous in the tumor microenvironment, promote tumor growth, and confer clinical resistance to MAPK inhibition by shifting RAS nucleotide loading to the GTP state36,37,38. Inhibitors with potent active state activity may be expected to be impacted to a lesser extent by this pathway activation than inactive state-selective inhibitors.
We investigated the effect of growth factors on KRAS G12C active state inhibitors, including SWII dual inhibitor 8, tricomplex active state-selective inhibitor RMC-6291, and panRAS tricomplex active state-selective inhibitor RMC-6236. As reported in the literature, EGF did not lessen the ability of RMC-6291 to inhibit the proliferation of LU6524. However, EGF significantly reduced the potency of compound 8 and divarasib and did so to a similar extent for both (greater than 40-fold) (Fig. 6a). An analogous result was seen in H1792 cells in the presence of HGF, with both 8 and divarasib showing a greater than 10-fold decrease in potency (Fig. 6b). However, in this cell line, RMC-6291 showed a 6-fold decrease in potency as well, and RMC-6236 showed a 3-fold decrease in potency. Moving to H1373 cells (Fig. 6c, top), treatment with HGF led to a 100-fold reduction in potency for RMC-6291 and a > 2000-fold reduction in IC50 for both 8 and divarasib. However, a < 10-fold reduction in IC50 for RMC-6236 was observed. These data suggest that targeting the active state of KRAS G12C does not prevent EGF- and HGF-induced resistance. Our observations above indicate that receptor tyrosine kinase (RTK) ligands can drive resistance to multiple classes of KRAS G12C selective inhibitors, which suggests to us that—perhaps unexpectedly—RTK-mediated activation of wild-type HRAS and NRAS may be the operative mechanism of resistance in this context39. The smaller fold-increase seen with RMC-6236 reinforces this hypothesis and suggests potential benefits of panRAS inhibition in overcoming this resistance mechanism. We also observed an increase of GTP-bound K- and NRAS, but not HRAS, in LU65 treated with EGF (Supplementary Fig. 8). To further interrogate this possibility, we established an HRAS/NRAS double knockout (DKO) H1373 isogenic cell. Deletion of HRAS and NRAS greatly diminished the effect of HGF-mediated resistance to all KRAS G12C inhibitors (Fig. 6c, bottom). In contrast to wild-type HRAS/NRAS-intact cells (Fig. 6c, top), KRAS G12C SWII dual, inactive state-selective, tricomplex active state-selective inhibition, and tricomplex pan-RAS inhibition showed minimal change in potency in the presence of HGF in this context (10-fold, 10-fold, less than two-fold, and less than five-fold, respectively). We also observed less fold-increase of pERK IC50 in H1373 H/NRAS DKO cells in the presence of HGF when treated with compound 8 (Supplementary Fig. 9). These data reveal the importance of RTK-mediated HRAS and NRAS activation as a mechanism of resistance for KRAS G12C inhibitors and suggest further avenues of exploration for combating growth factor-mediated resistance, including SOS1 inhibition40. Indeed, trametinib, a MEK inhibitor, showed only modest reduction in potency in the presence of EGF and HGF in all cell lines tested, a finding that may be attributed to trametinib’s ability to block signaling downstream of K-, H-, and NRAS (Supplementary Table 4).

a Effect on LU65 proliferation IC50s with EGF treatment for dual inhibitor 8, inactive state-selective inhibitor divarasib, active state-selective inhibitor RMC-6291, and panRAS inhibitor RMC-6236. Data are from two independent experiments and are presented as mean ± SD. b Effect on H1792 proliferation IC50s with HGF treatment for the same collection of compounds. Data are from two independent experiments and are presented as mean ± SD. c Top: Effect on H1373 proliferation IC50s with HGF treatment for 8, divarasib, RMC-6291 and RMC-6236. Bottom: Attenuation of growth factor-mediated resistance using an H- and NRAS double knockout isogenic cell line. Data are from two independent experiments and are presented as mean ± SD. Source data are provided as a Source Data file.
Discussion
Prior to the FDA approval of KRAS G12C inactive state-selective inhibitors, KRAS mutants were considered undruggable drivers of oncogenesis. Although that perspective has largely changed, the development of SWII-binding dual active- and inactive-state covalent inhibitors has lagged behind that for inactive state-selective inhibitors, possibly due to the initial assertion that the SWII binding pocket is not accessible in active state KRAS G12C6,41. In this work, we report the strategic design of irreversible inhibitors capable of potently binding both the active and inactive states of KRAS G12C based on a chemically differentiated trisubstituted acrylamide warhead.
Structure-activity relationship studies told a nuanced story and indicated that merely accommodating the γ-phosphate of GTP with this novel warhead was not sufficient for active state potency. Co-crystal structures of 2 and 6 revealed that these active inhibitors induced significant conformational changes in the protein’s SWI loop, a region critical for effector binding. This allosteric remodeling could be traced back to those compounds’ interaction with a conserved water network near Gly10/Thr58, specifically with their perturbation of a single water molecule (W1). Other contacts of dual inhibitors with the protein, however, were largely similar to those of inactive state-selective inhibitors and other chemical matter that binds to KRAS G12C. Based on these similarities, it is likely that previously observed mutations, especially at H95 and Y9621, would also significantly attenuate the binding of dual inhibitors. Such structural insight provided a blueprint for the optimization of SWII dual inhibitors, leading to the identification of 8, which was characterized along with 6 in further studies.
In cellular assays, dual inhibitors demonstrated rapid and robust inhibition of KRAS G12C signaling in line with dual targeting, as evidenced by decreased phosphorylation of ERK1/2 (pERK) and potent growth inhibition in KRAS G12C mutant cell lines. The kinetic advantage of dual targeting relative to inactive state-selective targeting was further validated in multiple orthogonal assays, where faster KRAS G12C target engagement and more rapid disruption of the KRAS G12C-RAF interaction were observed. While these findings highlight an area where dual inhibitors may offer improvements over inhibitors that target the inactive state, over time, both dual and inactive state-selective inhibitors yielded similar levels of maximum target engagement, pERK suppression, and antiproliferative activity. Collectively, our results therefore indicate no advantage to having dual state inhibition over inactive state inhibition alone.
The impact of growth factor-induced resistance on KRAS G12C inhibition was also investigated. Unexpectedly, growth factor receptor activation was found to drive resistance to all mechanisms of action tested, irrespective of G12C active or inactive state-selectivity. The extent of this effect was cell line and growth factor-specific. Importantly, RTK-mediated activation of wild-type H- and NRAS appeared to be driving much of this resistance in the H1373 cell line tested, as opposed to increased KRAS G12C GTP loading, a finding consistent with other reports39. The observed resistance in the presence of EGF and HGF suggested that additional strategies, such as combination therapies targeting nodes upstream of RAS (e.g., SOS1), other RAS isoforms, or downstream MAPK pathway components, may be necessary to fully overcome this challenge40.
In vivo, compound 8 exhibited sufficient oral bioavailability to demonstrate rapid target engagement and deep pERK1/2 suppression in multiple xenograft models. Significant tumor regressions in a KRAS G12C mutant mouse xenograft model were also observed, as was significant growth inhibition in a KRAS G12C PDX model. While compound 8’s activity against both active and inactive state KRAS G12C yielded rapid covalent target engagement and pERK suppression in vivo, this failed to translate to improved maximum pathway suppression, target engagement, or antitumor activity relative to inactive-state inhibitor divarasib. These observations in a PDX model with de novo resistance to KRAS G12C inhibition provided additional evidence to support that resistance to inactive-state inhibitors is not driven by accumulation of GTP-loaded active-state KRAS G12C.
In conclusion, our studies establish the feasibility of KRAS G12C dual inhibitors based on the discovery of a differentiated class of covalent warheads. A detailed understanding of compound-mediated allosteric modulation of SWI provided a robust framework for improving the potency of KRAS G12C inhibitors, which were used to interrogate outstanding hypotheses in the field regarding active state inhibition. While dual inhibition conclusively led to more rapid target engagement than an inactive state-selective inhibitor, a more complex narrative emerged in other contexts. Resistance data generated in cells treated with RTK-activating growth factors were consistent with the hypothesis that wild-type H- and NRAS activation, as opposed to an increased pool of GTP-bound KRAS G12C, is the dominant driver of RTK-mediated resistance to SWII-binding KRAS G12C inhibitors. Likewise, although the in vitro and in vivo rate of target engagement was improved with dual state inhibitors, this did not translate into improved overall pathway suppression or antitumor efficacy. Taken together, the preclinical data described herein indicate that potency against GTP-loaded KRAS G12C does not provide meaningful differentiation from inactive state-selective inhibitors across multiple assay systems, including xenograft efficacy models, cellular proliferation, and growth factor-mediated resistance models. Whether these findings will translate to patients with KRAS G12C tumors is currently unknown.
Methods
Chemistry
All reactions were performed in Chemglass reaction vials affixed with pressure release caps under ambient atmosphere, unless otherwise indicated (Supplementary Note 1). Reactions that required heating were carried out in temperature-controlled heating blocks, unless otherwise indicated. Air- and moisture-sensitive liquids were transferred via syringe. Volatile solvents were removed under reduced pressure rotary evaporation below 35 °C. Reaction progress was monitored by LCMS (Shimadzu Nexera X2) or by analytical thin-layer chromatography (TLC). TLC was performed using glass plates pre-coated with silica gel (250 μm thickness, 10 μm particle size, Millipore Sigma) and impregnated with a fluorescent indicator (254 nm), and plates were visualized by exposure to ultraviolet light (UV). Automated column chromatography was performed on CombiFlash® NextGen 300+ machines using pre-packed columns of silica gel (RediSep® Normal-phase Silica Flash Columns).
All solvents and reagents, including reported KRAS G12C inhibitors divarasib, RMC-6291, and panRAS inhibitor RMC-6236, were purchased at the highest commercial grade available and used as received, without additional purification.
Proton nuclear magnetic resonance (1H NMR) spectra were recorded on Bruker Avance 500 (500 MHz), or Bruker Avance NEO 600 (600 MHz) spectrometers at 23 °C, or a Bruker Avance 500 (500 MHz) spectrometer equipped with a BBO cryoprobe at 27 °C. Proton-decoupled carbon nuclear magnetic resonance (13C NMR) spectra were recorded on a Bruker Avance 500 (126 MHz) spectrometer equipped with a BBO cryoprobe at 27 °C. Fluorine nuclear magnetic resonance (19F NMR) spectra were recorded on a Bruker Avance 500 (471 MHz) spectrometer at 23 °C. Proton chemical shifts are expressed as parts per million (ppm, δ scale) and are referenced to residual protium in the NMR solvent. Carbon chemical shifts are expressed as parts per million (ppm, δ scale) and are referenced to the NMR solvent. Fluorine chemical shifts are expressed as parts per million (ppm, δ scale) and are not additionally referenced. Data are represented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), coupling constant (J) in Hertz (Hz), and integration.
Analytical UPLC/MS was conducted with two orthogonal methods on an Xbridge C18 column with dimensions 2.1 mm × 50 mm and 1.7 μm particle size at 50 °C. Method 1: Gradient over 3 min of 0 → 100 %B; mobile phase A: MeCN/H2O (5:95) with 0.05% TFA, and mobile phase B: MeCN/H2O (95:5) with 0.05% TFA. Method 2: Gradient over 3 min of 0 → 100 %B; mobile phase A: MeCN/H2O (5:95) with 10 mM ammonium acetate; mobile phase B: MeCN/H2O (5:95) with 10 mM ammonium acetate. Purities were assessed by UV absorbance (220 nm) and 1H NMR and were ≥ 95%, unless otherwise indicated.
KRAS G12C nucleotide exchange assay
Homogeneous time-resolved fluorescence (HTRF) nucleotide exchange assays were performed to measure the activity of compounds against the inactive form of KRAS G12C. Compounds were prepared as 11-point serial dilutions in DMSO and dispensed into 384-well plates. Recombinant biotinylated, GDP-loaded KRAS G12C and Tb cryptate-labeled streptavidin (Revvity) were incubated with compounds at room temperature for 20 min in assay buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM MgCl2, 0.0025% Igepal-CA630, 1 mM DTT, 100 µg/mL BSA). BODIPY-labeled GDP (ThermoFisher) and recombinant SOS1 were added for final assay conditions of 20 nM KRAS G12C, 0.5 nM streptavidin-Tb cryptate, 400 nM BODIPY-labeled GDP, 10 nM SOS1, and 1% DMSO. The 8 µL reactions were incubated at room temperature for 30 min. The HTRF signal of each well was measured using a PerkinElmer Envision plate reader, and the signal ratios (λem 520/ λem 495) were calculated. IC50 values were calculated from the dose-response curves. For compounds 2 and 3, concentrations up to 50 nM were used to determine IC50 due to fluorescence interference between the compound and the BODIPY fluorophore on KRAS G12C-bound GDP.
KRAS G12C-RAF disruption assay
KRAS G12C-RAF disruption HTRF assays were performed to measure the activity of compounds against the active form of KRAS G12C. Compounds were prepared as 11-point serial dilutions in DMSO and dispensed into 384-well plates. Recombinant biotinylated, GMP-PNP-loaded KRAS G12C was treated with compounds at room temperature for 20 min in assay buffer (50 mM Tris, pH 7.5, 100 mM NaCl, 1 mM MgCl2, 1 mM DTT, 100 ug/mL BSA). Recombinant, 5-FAM-labeled GST-BRAF1 RBD was added, followed by a 20-min incubation at room temperature. Tb cryptate-labeled streptavidin (Revvity) was added, and the 8 μL reaction mixtures were incubated for 3 h. Final assay conditions were 5 nM KRAS G12C, 9 nM 5-FAM-labeled GST-BRAF1 RBD, 0.25 nM streptavidin-Tb cryptate, and 1% DMSO. The HTRF signal of each well was measured using a PerkinElmer Envision plate reader, and the signal ratios (λem 520/ λem 495) were calculated. IC50 values were calculated from the dose-response curves.
Protein crystallography
See Supplementary Note 2 for a full description of crystallographic methods employed.
Determination of kinact/KI
Compounds of interest were pre-dispensed in dose titration into an empty 384 W microtiter plate by an Echo-655 (Beckman) liquid handler. In-house recombinant KRAS G12C (1-169) TVMV-BioP-His (Biotinylated) GDP or GMP-PNP was diluted to 1 μM in buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 5 mM MgCl2. Covalent reaction progression was initiated with a dragonfly liquid handler (SPT Labtech) as 45 uL protein reagent was dispensed into the pre-printed microtiter plate, followed by time-dependent quench with formic acid (0.05% final concentration). At the conclusion of the reaction, the microtiter plate was sealed and centrifuged at 300 x g for 1 min. Samples were analyzed by a Sciex 6500 + QQQ Mass Spectrometer equipped with a Lead Sampler (LS-1 – Sound Analytics) sampling robotic system and a Protein BEH C4 column. Full scan MS data was collected, and then the intact protein charge envelope (and compound adduct) was deconvoluted/integrated with PM-Intact Mass (Protein Metrics v.5.0). Covalent occupancy curves were fitted with an exponential curve fitting algorithm followed by kinact /KI fits of the kobs vs. compound concentration with a Michaelis-Menten curve fit in GraphPad Prism.
Chemical reactivity with glutathione assay
Glutathione (5 mM) was added to 100 mM potassium phosphate buffer (pH 7.4) containing 20% acetonitrile at 37 °C (n = 2). To initiate the reaction, 1 μM test article was added, and 100 μL of the sample was removed. The reaction was quenched by the addition of IS 0.05 μM Tolbutamide 95/5% ACN/water with 0.1% formic acid, and samples were frozen immediately at designated timepoints (0 and 0.5 h). Samples were thawed prior to analysis by LCMS/MS.
Cell proliferation assay
For the 2D proliferation assay, 500-1000 cells/well were seeded into 384-well plates (Corning#: 3765), and for the 3D ULA proliferation assay, 500–1000 cells/well were seeded into 384-well plates (Sbio #: MS-9384WZ). If growth factor is used in the assay, 100 ng/mL of human epidermal growth factor (ThermoFisher: PHG0315) or human hepatocyte growth factor (ThermoFisher: PHG0324) was added to the culture medium. Cells were treated for 5 days, and the viability signal was obtained using CellTiter-Glo® 2.0 Cell Viability Assay (Promega#: G9243) for 2D proliferation assay and CellTiter-Glo® 3D Cell Viability Assay (Promega#: G968B) for 3D proliferation assays per manufacturer’s instructions. IC50 values were determined using GraphPad Prism.
pERK immunofluorescence assay
Cells (2000-5000 cells per well) were plated in 384-well plates (Revvity 6057602) and incubated overnight. Cells were treated with compound (2 h) and fixed with paraformaldehyde (4%) for 15 min. Cells were washed three times (PBS), permeabilized (0.1% Triton X-100) for 15 min, and washed three times (PBS). Cells were blocked for 1 h at room temperature (5% normal goat serum, 0.2 M glycine, PBS) followed by incubation with pERK antibody (CST-4370S, 1:250) overnight at 4 °C. Cells were washed three times (PBS) and stained with anti-rabbit secondary antibody (Invitrogen A-11034, 1:1000), Hoechst 33342 (Invitrogen H3570, 1:1000), and CellMask (Invitrogen H32721, 1:15000) for 1 h at room temperature. Cells were washed three times (PBS) prior to imaging (Opera Phenix). Average pERK intensity per cell was measured (n = 2), and IC50 values were calculated from the dose-response curve using GraphPad Prism.
Cellular KRAS:RAF disruption assay
Stable cell lines (HEK293T) were generated to co-express RAF1 and KRAS G12C, as indicated, using the BiBRET vector (Promega). Engineered cell lines were suspended in media (OptiMEM I Reduced Serum Medium, no phenol red, 4% FBS). HaloTag NanoBRET 618 ligand (Promega) was added per the manufacturer’s instructions, plated in 96-well plates (10,000 cells/well), and allowed to rest at 37 °C, 5% CO2 overnight. NanoBRET NanoGlo Vivazine substrate (Promega) was added per the manufacturer’s instructions and incubated for 1 h at 37 °C, 5% CO2 to equilibrate substrate luminescence. Cells were treated with compounds, and NanoBRET signal was measured every 5 min up to 2 h (PerkinElmer Envision). The signal ratio (λem 618/ λem 460) was calculated, and the half-life was determined using GraphPad Prism.
Cellular KRAS G12C covalent target engagement assay
MIA PaCa-2 (ATCC) cells (25,000 cells/well) were plated in 96-well plates. Cells were treated with compounds and incubated for the indicated duration. Cells were washed with PBS once, and MSD Tris lysis buffer (MSD) with 1x protease/phosphatase inhibitor (ThermoFisher) was added to each well, followed by incubation at 4 °C with gentle rocking for 30 min. Lysate was collected, and a biotinylated KRAS G12C probe (see Supplementary Note 1) was added to a final concentration of 10 µM, followed by incubation at 23 °C for 2 h. Labeled lysate was transferred to 96-well MSD plates pre-coated with 2 µL/mL RAS antibody (Abcam ab271848), followed by incubation at 23 °C for 1 h with shaking at 500 rpm. Plates were washed with MSD wash buffer three times. 1 µg/mL Streptavidin SULFO-TAG Labeled (MSD) in Diluent 100 (MSD) was added to each well, followed by incubation for 30 min. Plates were washed with MSD wash buffer three times. MSD Read buffer (MSD) was added, and the MSD signal was measured. 10 µL of cell lysate was added with CellTiter-Glo® 2.0 Cell Viability Assay (Promega#: G9243) to measure cell viability. MSD signal was normalized with the cell viability signal. The percent of target engagement was calculated based on values from treated wells and vehicle control wells.
Cysteinome proteomics
H358 cells (ATCC) (0.5 million per well) were seeded in 6-well plates and treated with compounds at 0.1 µM for 4 h with n = 5. Cells were lysed in PBS pH 7.4 with sonication. Lysate was treated with 500 µM desthiobiotin iodoacetamide for 1 h at 23 °C in the dark42. Tryptic lysates were prepared using the Preomics iST-BCT 96 sample kit in conjunction with their Phoenix cartridge for peptide cleanup. After clean-up, the dried samples were resuspended in 100 mM TEAB, pH 7.4 and desthiobiotinylated peptides were enriched with Dynabeads Streptavidin MyOne T1 for 1 h at 23 °C and eluted with 0.1% formic acid in 50% acetonitrile/water. Solvent was evaporated, and samples were resuspended in 0.1% formic acid in water. Samples were analyzed with timsTOF pro2 coupled with Evosep One, and the data was acquired with Data Independent Acquisition Parallel Accumulation Serial Elution Fragmentation mode (DIA-PASEF). Database search and relative quantification was conducted with DIA-NN (v.1.8.1) with an experiment-specific spectral library. Peptide data obtained from DIA-NN was subjected for differential expression analysis with the limma R package.
RNAseq
H1792 cells (ATCC CRL-5895) were seeded in 6-well plates and treated with compounds at 0.7 nM for 24, 48 and 72 h with n = 3. Cell pellets were collected. Bulk RNA sequencing was conducted at Azenta Life Sciences (South Plainfield, NJ, USA). Briefly, RNA was extracted from frozen cell pellets of 1 M cells using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). Strand-specific RNA sequencing library was prepared by using NEBNext Ultra II Directional RNA Library Prep Kit for Illumina following the manufacturer’s instructions (NEB, Ipswich, MA, USA). Sequencing was performed on an Illumina NovaSeq instrument according to the manufacturer’s instructions, using 2 x 150 bp Paired-End (PE) reads configuration and targeting 50 M reads per sample. Paired-end reads were analyzed using the Seven Bridges platform (Seven Bridges Genomics). FASTQ reads were aligned to the reference genome GRCh38 using STAR43 with default parameters and transcriptome abundance estimated through RSEM v1.1.1344 with Ensembl GRCh38 v91 gene annotation. EdgeR45 was then used for library normalization and log CPM conversion with a prior pseudocount of 1. GSVA46 and custom R scripts were used to quantify the 10-gene MAPK pathway activity score signature47. Statistical comparisons of GSVA enrichment scores were made using Welch’s T-tests.
Active RAS pull-down
LU65 cells (JCRB0079; 6 million per 100 mm dish) were seeded and cultured under standard conditions. Cells were stimulated with 100 ng/mL human epidermal growth factor (EGF; ThermoFisher, PHG0315) for 30 min. After treatment, cells were collected and washed with ice-cold PBS. Active RAS was isolated using the Active Ras Detection Kit (Cell Signaling Technology, CST-8821) according to the manufacturer’s instructions. Briefly, cell lysates were prepared and incubated with GST-Raf1-RBD conjugated glutathione resin for 1 h at 4 °C with gentle rocking. The resin was washed three times with the provided wash buffer, and bound proteins were eluted with the reducing sample buffer. GTP-bound KRAS, HRAS, and NRAS were detected by Western blotting using isoform-specific antibodies (Santa Cruz Biotechnology: KRAS—sc-30, HRAS—sc-31, NRAS—sc-35)48.
Quantitative PCR
H1792 cells were seeded and cultured under standard conditions. Cells were treated with compounds at 0.7 nM for 24 h. After treatment, cell pellets were collected, and total RNA was extracted using the miRNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Reverse transcription was performed using the SuperScript IV VILO Master Mix (ThermoFisher Scientific, 11756050). Quantitative PCR (qPCR) was carried out using TaqMan gene expression probes and TaqMan Fast Advanced Master Mix (ThermoFisher Scientific, 4444557). The expression of 10 genes—identical to those profiled in the RNA-seq experiment for MAPK pathway activity quantification—was analyzed47.
PK
The PK of compound 8 was evaluated in female CD-1 mice (body weight 25 to 30 g). Animal studies strictly adhered to Institutional Animal Care and Use Committee (IACUC) protocols. Two groups of animals (n = 3 per group) received 8 either as an intravenous (IV) bolus injection (1 mg/kg, 5 mL/kg) via the jugular vein or by oral gavage (30 mg/kg, 10 mL/kg). The compound was formulated in solution containing 80% 20 mM citrate buffer (pH 4) and 20% CAPTISOL for both administrations. Mice had free access to food and water. For the IV study, blood samples were obtained at 0.083, 0.25, 0.5, 1, 3, 5, 7, and 24 h post dose. For oral dosing, blood samples were collected at 0.25, 1, 3, 5, 7, and 24 h post dose. Blood samples (~0.3 mL) for all studies were collected into K3EDTA-containing tubes and then centrifuged at 4 °C (1500 to 2000 x g) to obtain plasma, which was stored at –20 °C until quantitative analysis of the parent by LCMS/MS.
In vivo
In vivo CDX studies were conducted at the Bristol Myers Squibb (BMS) facility in Cambridge, Massachusetts, managed by Charles River Accelerator and Development Lab (CRADL) staff. Animal studies strictly adhered to Institutional Animal Care and Use Committee (IACUC) protocols. Mice were provided sterilized food and water ad libitum and housed in ventilated cages with a twelve-hour light cycle.
MIA PaCa-2 cells were cultured in vitro with 90% DMEM + 10% FBS (Gibco #10564011, Gibco #10082147) and split at a sub-cultivation ratio of 1:10. NCI-H2122 cells were cultured in vitro with 90% RPMI1640 + 10% FBS (Gibco #11875135, Gibco #10082147) and split at a sub-cultivation ratio of 1:10. Cells were implanted targeting a low passage number. Larger studies used cell factories (Corning #CLS3311) to achieve the desired quantity of cells prior to in vivo implantation. Xenograft tumors were established in vivo by injecting MIA PaCa-2 or NCI-H2122 cells (5 × 10^6) suspended in PBS (Gibco #14190144) containing 50% Matrigel (Corning #CLS354234) into the right flank of female athymic Crl:NU(NCr)-Foxn1nu -homozygous nude mice (Charles River #409).
Efficacy
Treatment was initiated when average tumor volumes were ~200–250 mm3. Tumor volume and body weight were measured at least twice a week. At no point did tumor burden exceed the IACUC maximum of 2000 mm3. Tumor volume was measured with calipers and calculated using 0.5 x Length x Width2 equation.
Percent tumor growth inhibition (%TGI) was calculated using the following equation: \(\left(\frac{\frac{{{TV}}_{T}}{{{TV}}_{T0}}-\frac{{{TV}}_{C}}{{{TV}}_{c0}}}{1-\frac{{{TV}}_{C}}{{{TV}}_{c0}}}\right)x100\%\) where \({{TV}}_{T}\)= tumor volume of treatment group and \({{TV}}_{C}\)= tumor volume of vehicle group.
Sampling of peripheral whole blood was performed via tail snip after first dose, at steady state, and end of study, that confirmed consistent concentrations of drug in mouse plasma. When possible, tumor samples were collected at the end of the study and snap frozen. All samples were stored at −80 °C until submitted for downstream PK and PD analysis.
In vivo PK/PD
Treatment was initiated when average tumor volumes were ~400–600 mm3. These tumor volumes did not exceed the IACUC maximum burden of 2000 mm3. Animals were randomized into groups of n = 3 per timepoint. Tumor samples were collected at the indicated time point after oral dosing. Tumor fragments were collected and frozen immediately in liquid nitrogen. Terminal peripheral blood sampling was performed by cardiac puncture. All samples were stored in a −80 °C freezer until the respective analysis was conducted.
Dosing
All oral dosing was performed at a dosing volume of 10 mL/kg. Compound 8 was formulated in 0.5% (v/w) PS80; 20% (w/w) Captisol; 79.5% (w/w) Citrate Buffer, 20 mM pH = 4.0 (solution). Divarasib was formulated with 99.5% H2O; 0.5% METHOCELA 4 M (suspension). For efficacy studies, the formulation was made every 7 days and stored at 4 °C.
In vivo ex vivo analysis
Tumor fragment lysate
Tumor fragment lysates were created using homogenization tubes (Bertin Corp P000922LYSK0A.0). 500 μL of lysis buffer from the Phospho/Total ERK1/2 Whole Cell Lysate Kit (Meso Scale Discovery; Cat# K15107D) was added to each tube. Protease and phosphatase inhibitors were used at a 2x concentration. Fragments were immediately homogenized using the Cryolys Evolution (P000671-CLYS2-A.0) until uniformly homogeneous (Frequency of 30/sec; 30 s*3 cycles). After homogenization, tubes were placed on ice for 15 min, then centrifuged for 15 min at 250 x g at 4 °C. Supernatant was aliquoted, and lysate protein concentrations were quantified using the BCA assay kit (Pierce; Cat# 23225). Excess lysate was stored at −80 °C.
Quantifying pERK and total ERK in tumor lysates
Protein concentration for each sample was normalized to 1 μg/μL for measurement of Phospho/Total ERK1/2 ratios. The Phospho/Total ERK1/2 Whole Cell Lysate Kit (Meso Scale Discovery; Cat# K15107D) manufacturer-suggested protocol was followed as described.
Quantifying target engagement in tumor lysates
Protein concentration for each sample was normalized to 1.2 μg/μL, and the protocol was followed as mentioned previously in the “Cellular KRAS G12C covalent target engagement assay” Materials and Methods section.
LUN055 PDX efficacy study
The in vivo study with the LUN055 KRASG12C-mutant NSCLC PDX tumor model was conducted at GenenDesign in Shanghai, China. Animal studies strictly adhered to Institutional Animal Care and Use Committee (IACUC) protocol #23051001. 6–8-week-old female BALB/c nude mice from Vital River were enrolled in the study and were implanted with passage 6 LUN055 fragments and housed in ventilated cages in an SPF facility.
Group randomization and treatment were initiated when the average tumor volume was ~200 mm3. Animals received either vehicle or treatment on a BID dosing regimen (8 + 16 h dosing). Sampling of peripheral whole blood was performed via tail vein collection. At the end of the study, tumor samples were collected 7 h after the final dose. At no point did tumor burden exceed the IACUC maximum of 2000 mm3. All samples were stored in a −80 °C freezer until the respective analysis was conducted. Subsequent analysis was performed at the BMS facility in Cambridge, Massachusetts.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The coordinate data for cocrystal structures have been deposited in the Protein Data Bank (PDB) under accession codes 9NZM [https://doi.org/10.2210/pdb9NZM/pdb] and 9NZN [https://doi.org/10.2210/pdb9NZN/pdb]. Mass spectrometry data for reactive cysteine profiling are available at the PRoteomics IDEntifications Database (PRIDE) under accession code: PXD062227. RNAseq data have been uploaded to the Sequence Read Archive (SRA) under project ID: PRJNA1244353. Source data are provided with this paper.
References
Jančík, S., Drábek, J., Radzioch, D. & Hajdúch, M. Clinical relevance of KRAS in human cancers. J. Biomed. Biotechnol. 2010, 150960 (2010).
Uprety, D. & Adjei, A. A. KRAS: from undruggable to a druggable cancer target. Cancer Treat. Rev. 89, 102070 (2020).
Janes, M. R. et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172, 578–589.e517 (2018).
Ostrem, J. M. L. & Shokat, K. M. Targeting KRAS G12C with covalent inhibitors. Annu. Rev. Canc. Biol. 6, 49–64 (2022).
Kwan, A. K., Piazza, G. A., Keeton, A. B. & Leite, C. A. The path to the clinic: a comprehensive review on direct KRAS(G12C) inhibitors. J. Exp. Clin. Cancer Res. 41, 27 (2022).
Ostrem, J. M., Peters, U., Sos, M. L., Wells, J. A. & Shokat, K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–555 (2013).
Patricelli, M. P. et al. Selective inhibition of oncogenic kras output with small molecules targeting the inactive state. Cancer Discov. 6, 316–329 (2016).
Canon, J. et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019).
Fell, J. B. et al. Identification of the clinical development candidate MRTX849, a covalent KRAS(G12C) inhibitor for the treatment of cancer. J. Med. Chem. 63, 6679–6693 (2020).
Lanman, B. A. et al. Discovery of a covalent inhibitor of KRAS(G12C) (AMG 510) for the treatment of solid tumors. J. Med. Chem. 63, 52–65 (2020).
Kettle, J. G. et al. Structure-based design and pharmacokinetic optimization of covalent allosteric inhibitors of the mutant GTPase KRAS(G12C). J. Med. Chem. 63, 4468–4483 (2020).
Broker, J. et al. Fragment optimization of reversible binding to the switch II pocket on KRAS leads to a potent, in vivo active KRAS(G12C) inhibitor. J. Med. Chem. 65, 14614–14629 (2022).
Kettle, J. G. et al. Discovery of AZD4625, a covalent allosteric inhibitor of the mutant GTPase KRAS(G12C). J. Med. Chem. 65, 6940–6952 (2022).
Li, J. et al. A phase I/II study of the first-in-human trial of JAB-21822 (KRAS G12C inhibitor) in advanced solid tumors. J. Clin. Oncol. 40, 3089–3089 (2022).
Lorthiois, E. et al. JDQ443, a structurally novel, pyrazole-based, covalent inhibitor of KRAS(G12C) for the treatment of solid tumors. J. Med. Chem. 65, 16173–16203 (2022).
Xu, J. et al. Atroposelective negishi coupling optimization guided by multivariate linear regression analysis: asymmetric synthesis of KRAS G12C covalent inhibitor GDC-6036. J. Am. Chem. Soc. 144, 20955–20963 (2022).
Kettle, J. G. et al. Discovery of AZD4747, a potent and selective inhibitor of mutant GTPase KRAS(G12C) with demonstrable CNS penetration. J. Med. Chem. 66, 9147–9160 (2023).
Ma, X. et al. Discovery of MK-1084: an orally bioavailable and low-dose KRASG12C inhibitor. J. Med. Chem. 67, 11024–11052 (2024).
Zhang, J. et al. D3S-001, a KRAS G12C inhibitor with rapid target engagement kinetics, overcomes nucleotide cycling, and demonstrates robust preclinical and clinical activities. Cancer Discov. 14, 1675–1698 (2024).
Xue, J. Y. et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 577, 421–425 (2020).
Awad, M. M. et al. Acquired resistance to KRAS(G12C) inhibition in cancer. N. Engl. J. Med. 384, 2382–2393 (2021).
Zhao, Y. et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature 599, 679–683 (2021).
Tanaka, N. et al. Clinical acquired resistance to KRAS(G12C) inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov. 11, 1913–1922 (2021).
Schulze, C. J. et al. Chemical remodeling of a cellular chaperone to target the active state of mutant KRAS. Science 381, 794–799 (2023).
Sharma Alok, K. et al. Revealing the mechanism of action of a first-in-class covalent inhibitor of KRASG12C (ON) and other functional properties of oncogenic KRAS by 31P NMR. J. Biol. Chem. 300, 105650 (2024).
Xia, G. et al. A chemical tuned strategy to develop novel irreversible EGFR-TK inhibitors with improved safety and pharmacokinetic profiles. J. Med. Chem. 57, 9889–9900 (2014).
Zhao, B. et al. Design, synthesis and biological evaluation of AZD9291 derivatives as selective and potent EGFRL858R/T790M inhibitors. Eur. J. Med. Chem. 163, 367–380 (2019).
Sacher, A. et al. Single-agent divarasib (GDC-6036) in solid tumors with a KRAS G12C mutation. N. Engl. J. Med. 389, 710–721 (2023).
Wang, X. et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J. Med. Chem. 65, 3123–3133 (2022).
Purkey, H. Discovery of GDC-6036, a clinical stage treatment for KRAS G12C-positive cancers. In AACR Annual Meeting. (New Orleans, 2022).
Srinivasan, B. Diffusion limit and the reactivity/affinity conundrum: implications for optimization and hit finding for irreversible modulators. J. Med. Chem. 68, 13137–13147 (2025).
Labenski, M. T. et al. SMaSh: a streptavidin mass shift assay for rapidly quantifying target occupancy by irreversible inhibitors. Biochemistry 60, 2915–2924 (2021).
Strelow, J. M. A perspective on the kinetics of covalent and irreversible inhibition. SLAS Discov. 22, 3–20 (2017).
Weiss, A. et al. Discovery, preclinical characterization, and early clinical activity of JDQ443, a structurally novel, potent, and selective covalent oral inhibitor of KRASG12C. Cancer Discov. 12, 1500–1517 (2022).
Chakraborty, A. et al. AZD4625 is a potent and selective inhibitor of KRASG12C. Mol. Cancer Ther. 21, 1535–1546 (2022).
Wang, W. et al. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin. Cancer Res 15, 6630–6638 (2009).
Straussman, R. et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 487, 500–504 (2012).
Wilson, T. R. et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 487, 505–509 (2012).
Ryan, M. B. et al. KRAS(G12C)-independent feedback activation of wild-type RAS constrains KRAS(G12C) inhibitor efficacy. Cell Rep. 39, 110993 (2022).
Sudhakar, N. et al. The SOS1 inhibitor MRTX0902 blocks KRAS activation and demonstrates antitumor activity in cancers dependent on KRAS nucleotide loading. Mol. Cancer Ther. 23, 1418–1430 (2024).
Zheng, Q. et al. Denitrogenative alkylation of K-Ras(G12D) Inhibits Oncogenic Signaling In Cancer Cells. J. Am. Chem. Soc. 147, 24785–24792 (2025).
Kuljanin, M. et al. Reimagining high-throughput profiling of reactive cysteines for cell-based screening of large electrophile libraries. Nat. Biotechnol. 39, 630–641 (2021).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 12, 323 (2011).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinforma. 14, 7 (2013).
Wagle, M.-C. et al. A transcriptional MAPK Pathway Activity Score (MPAS) is a clinically relevant biomarker in multiple cancer types. NPJ Precis. Oncol. 2, 7 (2018).
Foote, J. et al. A Pan-RAS inhibitor with a unique mechanism of action blocks tumor growth and induces antitumor immunity in gastrointestinal cancer. Cancer Res. 85, 956–972 (2025).
Acknowledgments
We thank the Department of Discovery Synthesis and Syngene for the preparation of synthetic intermediates. Dr. Jack L. Sloane is kindly acknowledged for his preparation of desthiobiotin iodoacetamide. Dr. Ryan Schroder is thanked for his assistance with NMR experiments. Cole Hediger and Alba Font Tello are thanked for their technical assistance. Dr. Lata Jayaraman and Dr. Andrew Bloecher are kindly acknowledged for their feedback and discussion on KRAS biology.
Author information
Authors and Affiliations
Contributions
M.L.C., Z.Z., D.B.D., R.R.M., S.K.L., X.Z., Y.A., R.M.B., S.B.B., A.R.D., D.P.D., D.M.D., L.D, J.K., C.G.L., R.P., J.Q., S. Sheriff, J.J.B., and M.L.S. designed the experiments. M.L.C., Z.Z., D.B.D., R.R.M., S.K.L., X.Z., Y.A., L.C., R.L.C., D.P.D., D.M.D., B.M.D., L.D., M.E.-S., B.E.F., K.F., C.H., J.K., C.G.L., C.M., G.A.M., K.M., M.F.P., R.P., J.Q., M.R., S. Sharma, S. Sheriff, A.K.S., J.S., N.S., R.L.T., W.V., T.W., T.Y., and D.Y. carried out the experiments and analyzed the results. R.M.B., S.B.B., A.R.D., E.L., and M.J.M. secured resources and advised the project. M.L.C. drafted the manuscript. All authors revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare the following competing financial interests: All authors are current or former employees of Bristol Myers Squibb and may also be stockholders. Bristol Myers Squibb funded all studies.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Condakes, M.L., Zhang, Z., Danahy, D.B. et al. Covalent inhibitor design confers activity against both GDP- and GTP-bound forms of KRAS G12C. Nat Commun 17, 2233 (2026). https://doi.org/10.1038/s41467-026-69003-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69003-0
