
Abstract
Stem cell differentiation dynamically remodels and stiffens the extracellular matrix (ECM), generating stage-specific biomechanical cues that guide tissue development. However, conventional biomaterials, designed to mimic mature ECM stiffness, neglect its spatiotemporal heterogeneity due to their static, non-evolvable nature. Herein, we develop a cell-programmed adaptative contraction (CPAC) hydrogel that enables mesenchymal stem cells (MSCs) to actively remodel their microenvironment through alkaline phosphatase (an early osteogenic marker)-mediated hydrophilic-to-hydrophobic transition and contraction of microgels. This cell-programmed remodeling establishes local mechanical heterogeneity and promotes osteogenesis through a positive feedback loop. Mechanistically, the evolving matrix enhances mechanotransduction-related microRNA expression, suppresses EZH2, and reduces H3K27 trimethylation to active osteogenic transcription. In vivo, MSC-laden CPAC hydrogels significantly enhance the repair of rat cranial defects. These findings introduce a paradigm of cell-instructed, dynamically evolvable biomaterials that recapitulate the adaptive nature of native ECM to orchestrate stem cell fate and tissue morphogenesis.
Similar content being viewed by others

Introduction
During early bone development, mesenchymal stem cells (MSCs) play a critical role in the continuous remodeling of the extracellular matrix (ECM), a process essential for guiding their differentiation into osteoblasts and driving bone formation1,2,3,4. The non-mineralized early developing bone ECM is loosely crosslinked and mechanically homogeneous5,6,7,8,9. As development progresses (Fig. 1a), the dynamic interactions between cells and the ECM lead to progressive restructuring and maturation of ECM due to increasing level of molecular aggregation and cross-linking, thereby transforming the ECM into a complex and heterogeneous structure comprising both the stiff components (e.g., collagen fibrils) and relatively soft elements (e.g., polysaccharide ground substances)10,11,12,13,14. The biochemical modifications of ECM by cell-secreted markers such as enzymes drive this dynamic remodeling and generate the spatially heterogeneous mechanical signals of ECM that are critical for regulating stem cell fate in a timeline that matches the cellular development timescale15,16,17,18,19,20,21.

a Schematic showing the natural ECM network changed from homogeneous to heterogeneous during skull development. b Schematic showing the network heterogenization of MSCs-laden NC hydrogel (left), NCLS hydrogel (middle), and CPAC hydrogel (right) after ALP exposure. c Mechanism of heterogeneous network formation induced by ALP-mediated phase transition in CPAC hydrogels. d Proposed signaling pathways involved MSC-driven active remodeling of the extracellular matrix and enhanced osteogenesis.
Previous studies have revealed that the biomaterials with prefabricated stiffness promote the commitment of stem cells towards osteogenesis through the activation of mechanotransduction signaling, such as the YAP/TAZ-dependent pathways22,23. However, few studies have demonstrated the ECM-emulating biomaterials that can dynamically remodel into mechanically heterogeneous structures in response to stem-cell-secreted biomarkers to timely provide the complex mechanical cues necessary for guiding the development of complex tissues such as bone based on stem cell differentiations. Previous approaches, such as sequential cross-linking of hydrogels using enzymatic or photoinitiated crosslinkings, have sought to replicate the dynamic stiffening process inherent to natural ECM24,25. However, the temporal and spatial resolution of these artificial stiffening mechanisms remains limited in the absence of cellular participation, leading to potential misalignment with the spatiotemporal requirements of stem cell differentiations. Therefore, the development of a biomimetic hydrogel that can dynamically respond to key differentiation markers of stem cells to undergo the cell-programmed heterogeneous remodeling in both space and time can potentially enhance the differentiation of stem cells and regeneration of tissue defects.
In this study, we develop a microgel-integrated hydrogel system capable of evolving into the ECM-mimetic heterogeneous structure in response to alkaline phosphatase (ALP, a key early osteogenesis marker of stem cells), termed cell-programmed adaptive contraction (CPAC) hydrogel. Unlike conventional static microgels, the phosphatase-responsive microgel is designed to be enzymatically modified by ALP secreted by the encapsulated MSCs to undergo a hydrophilic-to-hydrophobic transition and volume contraction, leading to biomimetic structural heterogenization of the CPAC hydrogel with local network stiffening (Fig. 1b). This cell-driven remodeling closely recapitulates native bone ECM development and overcomes the spatiotemporal limitations of previous artificial stiffening strategies. This strategy promotes the mechanosensing and osteogenesis of encapsulated MSCs and, therefore, further elevates the ALP expression, thereby establishing a positive feedback loop that reinforces the osteogenic differentiation through mechanotransduction (Fig. 1c). Notably, the cell-triggered hydrogel local stiffening alters the expression of mechanotransduction-related microRNAs (miRNAs) by the encapsulated MSCs. These mechanosensitive miRNAs target and suppress the expression of EZH2, a key histone methyltransferase, thereby reducing H3K27 trimethylation (H3K27me3) and enhancing the transcriptional activation of osteogenic genes (Fig. 1d). This mechanotransduction–epigenetic coupling enabled by the CPAC hydrogel significantly enhances the osteogenic commitment of MSCs and in situ healing of critical-sized cranial defects in rats. Our findings highlight the critical importance of cell-driven biomaterial adaptation in establishing a spatiotemporally coordinated inductive biomechanical microenvironment for guiding choreographed stem cell differentiation and tissue regeneration. The CPAC hydrogel thus demonstrates a cell-instructed biomaterial system capable of undergoing dynamic, ALP-mediated remodeling to form mechanically heterogeneous structures that promote MSC osteogenesis through mechanotransduction–epigenetic coupling. This work advances the design of biomaterials that respond adaptively to cellular cues, offering a step toward more physiologically relevant ECM mimetics.
Results and discussion
Synthesis of phosphatase-responsive contractive microgels as the building blocks of CPAC hydrogels
We first designed and synthesized the phosphorylated hydroxypropyl methacrylate (Phos-HPMA) microgels capable of phosphatase-responsive hydrophilic-to-hydrophobic transition mediated by enzymatic activity (Fig. 2a). Specifically, the Phos-HPMA monomer was synthesized via phosphorylation of HPMA using phosphorus oxychloride26, preserving the integrity of the carbon-carbon double bond, as evidenced by the characteristic vinyl proton resonances at δ 5.7 and 6.1 ppm in the 1H NMR spectrum (Supplementary Fig. 1). Successful phosphorylation was further confirmed by the presence of a distinct peak in the ³¹P NMR spectroscopy (Supplementary Fig. 2). This phosphorylated monomer was subsequently copolymerized with N, N’-methylenebisacrylamide (MBA) and acrylic acid via reverse emulsion polymerization, forming a crosslinked microgel network with controlled composition and uniform morphology. As a control, phosphorylated hydroxyethyl acrylate (Phos-HEA) microgels were synthesized following a similar protocol, with a comparable phosphorylation degree validated by NMR (Supplementary Figs. 3 and 4). HPMA possesses two additional methyl groups and one extra methylene group compared to HEA, which confers enhanced hydrophobicity to HPMA, particularly in its polymerized form. Upon phosphorylation and subsequent microgel fabrication, both Phos-HPMA and Phos-HEA microgels initially demonstrate comparable hydrophilic characteristics. However, under sustained ALP activity, enzymatic cleavage of phosphate groups occurs, leading to a progressive increase in hydrophobicity of Phos-HPMA microgels accompanied by volume contraction. To evaluate the ALP-triggered responsiveness, an equal mass of Phos-HPMA and Phos-HEA microgels was incubated in phosphate-buffered solution (PBS) or 1 U/mL ALP solution (37 °C, 72 h). Inverted microscopy revealed a striking size reduction in Phos-HPMA microgels upon ALP treatment, with the average diameter shrinking from 170 μm (as observed in PBS) to 100 μm (in ALP) (Fig. 2b, c). In contrast, the Phos-HEA microgels retained their size (168–165 μm) under both conditions, confirming their enzymatic inertness. Post-lyophilization scanning electron microscopy (SEM) observation further corroborated these findings, revealing substantial deswelling of Phos-HPMA microgels post-ALP exposure, consistent with phosphate ester hydrolysis-induced contraction (Fig. 2d). To ensure seamless integration of the microgels with the network of the proposed cell-laden CPAC hydrogels, all microgels were functionalized with polymerizable vinyl groups, facilitating covalent linkage during network formation (Supplementary Figs. 5 and 6).

a Synthetic route of Phos-HPMA microgels. b Microscopic images and c diameter changes of Phos-HEA and Phos-HPMA microgels after 72 h in PBS and ALP solution (scale bar = 100 μm). Data are presented as mean values ± SD, n = 15 independent microgels, ns not significant, ****p < 0.0001 (two-tailed Student’s t-test). d SEM images of Phos-HEA and Phos-HPMA microgels before and after ALP treatment (scale bar = 20 μm). Each experiment was repeated three times independently with similar results. e Schematic illustration of the cell-secreted ALP-induced internal network heterogenization in CPAC hydrogels. f SEM and g confocal images showing the changes in hydrogel network structure after ALP treatment (scale bar = 100 μm). Each experiment was repeated three times independently with similar results. h Time-dependent variation of storage modulus in NC and CPAC hydrogels after ALP treatment. Data are presented as mean values ± SD, n = 3 independent hydrogels, ns not significant, *p < 0.05, ****p < 0.0001 (two-tailed Student’s t-test). i Heatmaps displaying the local effective Young’s modulus distribution in CPAC hydrogels after 72 h of treatment with (i) PBS, (ii) ALP, and (iii) MSC encapsulation. Data are presented as mean ± SD; ns not significant; ****P < 0.0001. Source data are provided as a Source data file.
ALP-mediated microgel contraction and local stiffening generate the mechanically heterogeneous structures of CPAC hydrogel
We next fabricated the CPAC hydrogel by photopolymerizing a precursor solution containing p-tert-butylphenyl-modified hyaluronic acid (HA-TP), gelatin, mono-acryloyl cyclodextrin (Ac-β-CD), and Phos-HPMA microgels. The successful synthesis of HA-TP and AC-β-CD was demonstrated by 1NMR spectroscopy (Supplementary Figs. 7 and 8). The obtained CPAC hydrogel comprised covalently bonded microgels embedded within the supramolecular inter-microgel network, stabilized by the dynamic host–guest complexation between TP and β-CD (Fig. 2e). This design enables enzyme-responsive structural reorganization, where the contraction of the Phos-HPMA microgels triggered by cell-secreted ALP can induce inter-microgel network reorganization and structural heterogenization in the CPAC hydrogel. The non-contractile control hydrogel (NC hydrogel) was fabricated by replacing the Phos-HPMA microgels with the Phos-HEA microgels, which lack enzymatic responsiveness. Both hydrogels were formulated with identical gelatin concentrations to ensure consistent cell-adhesive ligand density. Rheological characterization demonstrated that CPAC and NC hydrogels exhibited comparable initial viscoelastic profiles, with a storage modulus (G’) of ~1 kPa, a loss modulus (G”) of ~0.2 kPa (Supplementary Fig. 9a), and Young’s modulus (E) of ~2.5 kPa (Supplementary Fig. 9b). Stress relaxation kinetics showed near-identical characteristic for half of the peak stress relaxation times (τ1/2 = 135 ± 6.85 s) for CPAC and NC hydrogels, confirming that microgel incorporation did not perturb the network dynamics (Supplementary Fig. 9c). Upon ALP treatment, SEM images demonstrated the substantial thinning of the inter-microgel network in the CPAC hydrogel compared to the NC hydrogel (Fig. 2f). Confocal microscopy images of the FITC-labeled hydrogel matrices (green fluorescence) further supported this observation, showing increased fluorescence intensity surrounding the non-fluorescent microgels (black voids) post-ALP treatment, revealing the enhanced local matrix density and structural heterogeneity (Fig. 2g). Rheological characterization documents progressive increase in bulk storage modulus for CPAC hydrogels, achieving 3 kPa after 72 h of ALP exposure, while NC specimens maintained their original bulk stiffness (~1 kPa) (Fig. 2h). Notably, nanoindentation mapping of the CPAC hydrogels revealed a striking increase in local stiffening at the microgel sites (2-fold increase, a 10 kPa stiffness surge), while the surrounding inter-microgel network (red zones) softened due to the microgel contraction-induced thinning of the local network. In contrast, no such changes in local stiffness were observed in the NC hydrogels (Fig. 2(i, ii); Supplementary Fig. 10). Moreover, the theoretical analysis of CPAC hydrogel deformation also confirmed the formation of the heterogeneous structure within the hydrogel, with stiffness increasing radially near the Phos-HPMA microgel surface (Supplementary Fig. 11). To validate the specific response of CPAC hydrogel to ALP, identical sets of CPAC hydrogel were treated with ALP/ALP inhibitor and acid phosphatase, respectively. It was observed that compared to the ALP-treated group, the internal structures of these two groups did not exhibit significant changes (Supplementary Fig. 12a–c), indicating that ALP is the primary factor responsible for the formation of the heterogeneous structure in CPAC hydrogel. We further examined the responsiveness of the hydrogels to cell-secreted ALP by encapsulating osteogenically-induced MSCs in the CPAC and NC hydrogels. As shown in Supplementary Fig. 12d, ALP levels in both groups gradually increased during culture, but CPAC hydrogels exhibited a markedly higher and sustained ALP release. This elevated ALP secretion strongly correlates with the heterogeneous stiffening pattern observed in CPAC hydrogels. After 3 days of culture, nanoindentation mapping (Fig. 2(iii)) showed a similar pattern of heterogeneous stiffening and softening within the CPAC hydrogel treated with exogenous ALP (local effective moduli ranged from ~0.5 kPa in softer regions to ~14 kPa in stiffer regions). This stiffening is driven by cell-mediated microgel contraction, confirming the autonomous remodeling capacity of the material. In contrast, the MSC-laden NC hydrogels showed no significant changes in local stiffness (Supplementary Fig. 10(iii)). Notably, CPAC hydrogels exhibited comparable behavior in vivo, as ALP secreted by MSCs after implantation reached sufficient levels to drive microgel contraction and the formation of heterogeneous structures within the hydrogel (Supplementary Fig. 13). Moreover, the CPAC hydrogel demonstrated rapid structural recovery within 60 s following 60% strain-induced fracture (Supplementary Fig. 9d), confirming robust self-healing behavior driven by reversible TP/β-CD interactions. The hydrogel also retained structural integrity after extrusion through a 21-gauge needle (Supplementary Fig. 14), supporting potential for minimally invasive delivery. This reversible nature of the host–guest interactions in the bulk network enables the adaptive remodeling by facilitating stress redistribution and network reorganization in response to microgel contraction. Additionally, we evaluated the mechanical stiffness and heterogeneous structure of the CPAC hydrogel in both parallel and perpendicular directions. As shown in the Supplementary Fig. 15, the CPAC hydrogel exhibits the same Young’s modulus (~11 kPa), consistent compressive stress–strain curves, and the local effective moduli ranged (from ~0.5 kPa in softer regions to ~14 kPa in stiffer regions), indicating that CPAC hydrogels are mechanically isotropic at the bulk level both in vitro and in vivo. Furthermore, to validate the stability of CPAC hydrogel under complex in vivo conditions, we incorporated variations in pH and temperature into the evaluation. As shown in Supplementary Fig. 16, CPAC hydrogels were able to maintain stable formation of heterogeneous structures within physiological ranges of pH (7.2–7.8) and temperature (33–39 °C) for 3 days, with no significant impact on the hydrogel structure. These findings collectively confirm that both the exogenous and cell-secreted ALP can trigger the contraction of Phos-HPMA microgels, thereby inducing the heterogeneous restructuring and associated local stiffness changes in the CPAC hydrogel.
The cell-programmed local stiffening in CPAC hydrogel promotes osteogenic differentiation of MSCs
Before examining osteogenic differentiation, we first evaluated whether CPAC hydrogel supports the proliferation and survival of encapsulated MSCs. Compared to the NC group, CPAC hydrogels exhibited a higher number of EdU-positive cells, indicating an increase in newly synthesized DNA (Supplementary Fig. 17a). Consistently, live/dead staining confirmed robust cell viability (Supplementary Fig. 17b), and CCK-8 assays further verified significantly higher proliferation rates in the CPAC group relative to NC (Supplementary Fig. 17c). These results suggest that the heterogeneous structure formed during microgel contraction not only provides space for cell expansion but also creates a favorable microenvironment that sustains MSC proliferation. Furthermore, the results of Actin staining and the statistical analysis of cell spreading area for MSCs (Supplementary Fig. 18) indicated that MSCs spread over a larger area in the CPAC hydrogel. Subsequently, we evaluated the impact of cellular ALP-induced local stiffening in CPAC hydrogel on the osteogenesis of encapsulated MSCs by analyzing the expression of key osteogenic markers in encapsulated MSCs. After 3 days of culture, PCR results revealed significantly upregulated expression of crucial osteogenic genes, including runt-related transcription factor 2 (Runx2), ALP, Col 1, and osteopontin (OPN) in MSCs cultured in CPAC hydrogels compared to those in NC hydrogels (Fig. 3a). The 7-day PCR data also demonstrated that MSCs exhibited a higher degree of osteogenic differentiation within the CPAC hydrogel over longer culture periods (Supplementary Fig. 19). Immunofluorescence staining, Masson’s trichrome staining and ALP color development kit confirmed enhanced protein expression of Col 1 and ALP in the CPAC hydrogel group, with greater fluorescence intensity than in the NC hydrogel group (Fig. 3b–d, Supplementary Figs. 20 and 21). Consistently, WB analysis further verified the increased synthesis of Col 1 and ALP proteins in the CPAC hydrogel group (Fig. 3e). The above results indicate that the ALP-triggered matrix remodeling and heterogeneous stiffness distribution in CPAC hydrogel can better facilitate stellate spreading and osteogenic differentiation of encapsulated MSCs.

a Relative gene expression of selected osteogenic markers (Runx2, ALP, Col 1, OPN) after 3 days of osteogenic induction. Data are presented as mean values ± SD, n = 3 independent hydrogels, ns not significant, **p < 0.01, ****p < 0.0001 (two-tailed Student’s t-test). Immunofluorescent staining of b Col 1 and c ALP in NC and CPAC hydrogels, respectively (scale bar = 75 μm). d Quantification of Col 1 and ALP immunofluorescence intensity in the two hydrogels. Data are presented as mean values ± SD, n = 10 independent hydrogels, ns not significant, ****p < 0.0001 (two-tailed Student’s t-test). e Western blot analysis of Col 1 and ALP in the two hydrogels. Data are presented as mean values ± SD, n = 3 independent hydrogels, ns not significant, **p < 0.01 (two-tailed Student’s t-test). f Schematic illustration of MSC-driven internal network heterogenization during cell culture within CPAC hydrogels. Source data are provided as a Source data file.
Mechanistically, ALP secreted by differentiating MSCs catalyzes the degradation of phosphorylated Phos-HPMA microgels, leading to structural contraction and localized stiffening within the CPAC hydrogel matrix (Fig. 3f). The resulting mechanical heterogeneity creates spatially confined high-stiffness microdomains that promote cytoskeletal reorganization and focal adhesion formation—key drivers of mechanotransduction. This enhanced mechanical signaling upregulates osteogenic transcription factors (e.g., Runx2 and Col 1), reinforcing ALP secretion and accelerating matrix heterogenization through a positive feedback loop. This self-amplifying mechanochemical cascade distinguishes the CPAC hydrogel as a responsive biomimetic matrix capable of undergoing cell-programmed restructuring to promote cellular functions.
The cell-triggered local stiffening of CPAC hydrogel promotes osteogenesis by activating mechanotransduction
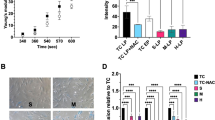
To further isolate the pro-osteogenic effects of cell-programmed active matrix remodeling from passive modulus variations, we engineered another control hydrogel system incorporating non-degradable, high-modulus N,N-dimethylacrylamide (DMAA) microgels into the dynamic hydrogel network (Fig. 4a). This non-contractile hydrogel with localized stiffening (NCLS) platform enabled differentiation of the contribution of MSC-driven matrix remodeling from intrinsic stiffness gradients. As shown in Supplementary Fig. 10b(i), the initial effective Young’s modulus of NCLS hydrogels matched the peak modulus of CPAC hydrogels, and this modulus remained unchanged upon exposure to ALP or after 3 days of MSC culture (Supplementary Fig. 10b(ii, iii)). We subsequently evaluated the expression of Col 1 and ALP across hydrogel types (Fig. 4b, c). CPAC hydrogels exhibited the highest Col 1 and ALP expression compared to all groups, while NCLS hydrogels showed no significant difference in osteogenic marker expression relative to NC hydrogels.

a Schematic illustration of the NC, NCLS, and CPAC hydrogel networks. Immunofluorescent staining showing the b Col 1 and c ALP expression in different hydrogel groups (scale bar = 20 μm). Immunofluorescent staining of mechanotransduction signaling proteins, d pFAK, e Paxillin, and f pMyosin IIa in MSCs cultured on different hydrogel substrates, respectively (scale bar = 20 μm). g Quantification of immunofluorescence intensity for the osteogenic marker proteins (Col 1 and ALP) and mechanotransduction signaling proteins (pFAK, Paxillin, and pMyosin IIa) in different hydrogel groups. Data are presented as mean values ± SD, n = 15 independent cells, ns not significant, ****p < 0.0001 (two-way ANOVA). h Effects of different hydrogel substrates on the spread area of MSCs, n = 15 independent cells. Data are presented as mean ± SD; ns not significant; ***p < 0.001 (two-way ANOVA). Source data are provided as a Source data file.
To explore the underlying mechanotransduction pathways, we assessed the phosphorylation of focal adhesion kinase (FAK) at tyrosine 397 (pFAK) and myosin IIa at serine 1943 (pMyosin IIa). Immunofluorescence analysis demonstrated markedly higher pFAK, paxillin, and pMyosin IIa levels in CPAC hydrogels compared to control groups (Fig. 4d–f), with quantitative intensity profiling confirming these observations (Fig. 4g). Additionally, morphometric analysis revealed a significant increase in MSC spreading within CPAC hydrogels (Fig. 4h), suggesting increased cell—matrix interactions. Collectively, these findings confirm that the dynamic, heterogeneous matrix remodeling within CPAC hydrogels promotes osteogenic differentiation through enhanced mechanotransduction. The increased expression of pFAK, paxillin, and pMyosin IIa underscores the activation of mechanosensitive signaling cascades. This supports the established mechanobiology paradigm, where matrix stiffening and dynamic remodeling trigger cytoskeletal tension, reinforcing focal adhesion formation and osteogenic commitment27. These findings highlight that the enhanced mechanotransduction observed in CPAC hydrogels stems from cell-programmed active matrix remodeling, demonstrating the critical role of dynamic matrix heterogeneity in guiding osteogenic outcomes.
Mechano-epigenetic regulation governs MSC osteogenesis in CPAC hydrogels
MiRNAs such as miR-124, miR-214, and miR-26a are established regulators of cellular mechanotransduction, influencing chromatin remodeling through the EZH2/H3K27me3 axis28. To investigate their role in osteogenic differentiation within CPAC hydrogels, we compared miRNA expression between hydrogel groups (Fig. 5a). Cells encapsulated in CPAC hydrogels exhibited significantly elevated levels of miR-124-3p, miR-214, and miR-26a compared to NC controls. This upregulation correlated with a marked reduction in the expression of EZH2 and its downstream target, H3K27me3, in CPAC hydrogels (Fig. 5b). Immunofluorescence analysis further confirmed these findings, showing diminished EZH2 and H3K27me3 fluorescence intensity in CPAC hydrogels compared to NC hydrogels (Fig. 5c–f). ChIP-qPCR then demonstrated that H3K27me3 was enriched at the RUNX2 promoter (Fig. 5i), and this reduction was accompanied by increased RUNX2 transcription (Fig. 3a) and enhanced protein expression of Col 1 and ALP (Fig. 5g). In addition, to further validate the mechanistic link, we incorporated siRNA-based inhibitors of miR-124, miR-214, and miR-26a into the CPAC hydrogel cultures. Western blot analysis demonstrated that the inhibition of these miRNAs markedly increased the protein levels of EZH2 and H3K27me3, while simultaneously reducing the expression of osteogenic markers ALP and Col 1 (Supplementary Fig. 22). These results demonstrate that the cell-programmed matrix heterogenization in CPAC hydrogel establishes an epigenetic landscape conducive to osteogenic differentiation through miRNA-mediated suppression of the EZH2/H3K27me3 axis.

a Comparison of the expression levels of miR-124-3-1, miR-214, and miR-26a in MSCs cultured in different hydrogels. Data are presented as mean values ± SD, n = 3 independent hydrogels, ns not significant, **p < 0.01, ****p < 0.0001 (two-tailed Student’s t-test). b Western blot and quantitative analysis of EZH2 and H3K27me3 expression in different hydrogels. Data are presented as mean values ± SD, n = 3 independent hydrogels, ns not significant, **p < 0.01, ****p < 0.0001 (two-tailed Student’s t-test). c–f Immunofluorescent staining and statistical analysis of EZH2 and H3K27me3 in different hydrogel groups (scale bar = 75 μm). Data are presented as mean values ± SD, n = 10 independent hydrogels, ns not significant, ****p < 0.0001 (two-tailed Student’s t-test). g Immunofluorescent staining of EZH2, H3K27me3, Col 1, and ALP in NC hydrogels before and after EZH2 inhibitor treatment (scale bar = 75 μm). h Quantification of immunofluorescence for Col 1 and ALP in NC hydrogels before and after EZH2 inhibitor treatment. Data are presented as mean values ± SD, n = 10 independent hydrogels, ns not significant, ****p < 0.0001 (two-tailed Student’s t-test). i ChIP-qPCR analysis showing the occupancy of H3K27me3 at the promoter site of RUNX2. Data are presented as mean values ± SD, n = 3 independent hydrogels, ns not significant, ****p < 0.0001 (two-tailed Student’s t-test). j Schematic illustration of the cellular pathway through which mechano-stimulation generated by the CPAC hydrogel regulates osteogenic gene expression. Source data are provided as a Source data file.
To further validate the role of EZH2 in MSC osteogenic differentiation, we pharmacologically inhibited EZH2 activity in NC hydrogels using a selective EZH2 methyltransferase inhibitor. After 3 days of cell culture, immunofluorescence staining revealed a substantial decrease in EZH2 and H3K27me3 expression, accompanied by a significant increase in Col 1 and ALP (Fig. 5g). The fluorescence intensity of Col 1 and ALP was notably higher in the inhibitor-treated group compared to the untreated controls (Fig. 5h), reinforcing the role of EZH2 suppression in promoting osteogenic differentiation. Collectively, our findings support a mechano-epigenetic regulatory model in which MSC-driven formation of locally stiffened microdomains within CPAC hydrogels activates cellular mechanotransduction pathways. This mechanical stimulation upregulates the expression of miR-124-3p, miR-214, and miR-26a, which in turn suppress EZH2 synthesis28,29,30,31, reducing H3K27 trimethylation in the nucleus and enhancing the expression of osteogenic genes32 (Fig. 5j). The resultant increase in ALP secretion further promotes localized matrix heterogenization through enzymatic microgel contraction, establishing a positive feedback loop that reinforces the osteogenic differentiation of MSCs encapsulated in the heterogeneous hydrogel.
MSC-laden CPAC hydrogels promote bone regeneration in an animal critical defect model
Before evaluating the therapeutic efficacy of dynamic heterogeneous CPAC hydrogel in facilitating bone regeneration, we first assessed the long-term stability of the hydrogel both in vitro and in vivo. As shown in Supplementary Fig. 23, the hydrogel underwent gradual degradation over 4 weeks, consistent with the expected degradation trend of implantable materials. However, the heterogeneous structure in the remaining non-degraded regions remained stable (Supplementary Fig. 24). This indicates that the CPAC hydrogel can stably provide the heterogeneous structural extracellular microenvironment required by MSCs during bone defect repair. Moreover, to evaluate the therapeutic efficacy of dynamic heterogeneous CPAC hydrogel in facilitating bone regeneration, we employed a rat calvarial defect model. Both CPAC and NC hydrogels encapsulated with rat mesenchymal stem cells (rMSCs) were cultured in osteogenic medium for 3 days before being transplanted into the rat cranial defects (Fig. 6a). As shown by micro-CT imaging and quantitative analysis, the CPAC group promoted the regeneration process in the early (4 weeks) and late (8 weeks) stages of bone repair (Fig. 6b, Supplementary Fig. 25), revealing denser new bone formation than that in the NC group and untreated (“Blank”) group. Specifically, at 8 weeks postoperatively, the BV (Bone volume) of the CPAC hydrogel group measured at 11.42 ± 0.87 mm3 (Fig. 6c, Supplementary Fig. 26a, b), which were 4.7-, 3.1-, and 2.4-fold greater than those of the Blank, Control, and NC hydrogel groups, respectively. The BV/TV (bone volume/total volume) and BS/BV (bone surface/bone volume) values exhibited similar trends, further demonstrating markedly higher new bone volume in the CPAC hydrogel group compared to the other groups (Fig. 6d, e, Supplementary Fig. 26c, d). Histological staining provided additional evidence of enhanced bone matrix formation. Hematoxylin and eosin (H&E) staining and Masson’s trichrome staining revealed that the CPAC hydrogel group presented significantly greater new bone and fibrous tissue matrix formation than other groups (Fig. 6f, g, Supplementary Fig. 26e, f). Moreover, immunohistochemical (IHC) staining of OCN, a key osteogenic marker, showed markedly higher expression in the CPAC hydrogel group than in other groups, confirming enhanced osteoblastic activity (Fig. 6h, Supplementary Fig. 26g). Collectively, these results establish that CPAC hydrogels promote robust bone regeneration, highlighting the potential of CPAC hydrogels for improving critical bone defect repair.

a Schematic illustration of MSCs-loaded NC and CPAC hydrogel implantation in rat cranial bone defects. b Micro-CT images showing bone defects at 8 weeks post-implantation for the Blank, Hydrogel, NC, and CPAC hydrogel groups. Quantification of c bone volume (BV), d bone volume/total volume (BV/TV), and e bone surface area/total volume (BS/TV) in the bone defect sites after 8 weeks, respectively. Data are presented as mean values ± SD, n = 3 independent SD rats, ns not significant, *p < 0.05, **p < 0.01, ****p < 0.0001 (one-way ANOVA). f H&E staining, g Masson’s trichrome staining of the bone defects in the Blank, NC, and CPAC hydrogel groups, respectively (scale bar = 200 μm). h OCN immunohistochemical staining in the Blank, NC, and CPAC hydrogel groups (scale bar = 100 μm). Each experiment was repeated three times independently with similar results. Data are presented as mean ± SD; **p < 0.01, ***p < 0.001, ****p < 0.0001. Source data are provided as a Source data file.
While the CPAC hydrogel demonstrates robust osteoinductive performance, some limitations remain. The mechanical heterogeneity of the system arises from cell-secreted ALP activity rather than predefined gradients, and the precise spatial control of stiffness may vary with cell density or metabolic activity. In addition, long-term mechanical stability and remodeling behavior under complex in vivo conditions warrant further investigation.
In this work, we developed a cell-programmed heterogeneous dynamic hydrogel system, termed CPAC hydrogel, which replicates the mechano-microenvironment essential for MSC osteogenic commitment. This construct leverages host–guest interaction-mediated dynamic networks to mimic native ECM remodeling capabilities faithfully. Moreover, MSC-directed dephosphorylation of microgel components enables spatiotemporal modulation of network heterogeneity through precisely controlled hydrophilic-hydrophobic phase transitions. The resultant CPAC hydrogel establishes a locally stiffened matrix that promotes the osteogenic differentiation of encapsulated MSCs by activating the mechanotransduction signaling pathways. Notably, the structural dynamics and heterogeneity within the CPAC hydrogel upregulated mechanosensitive miRNA expression in MSCs, thereby suppressing EZH2-mediated histone H3K27 trimethylation for enhanced osteogenesis. Functional validation through a critical-sized rat calvarial defect model confirmed the therapeutic efficacy of MSC-laden CPAC hydrogels in facilitating cranial bone regeneration. These findings establish the critical role of biomimetic physical cues in next-generation regenerative matrix design, establishing CPAC hydrogel as a promising platform for bone tissue engineering.
Methods
Protocol approval
The experimental procedures in this study were conducted in strict compliance with relevant ethical guidelines. The use of animals was reviewed and approved by the Animal Care and Use Committee of South China University of Technology (Permit Number 2023088).
Materials
Sodium hyaluronate (HA-Na, Mw = 4 kDa) was purchased from Bloomage Freda Biopharm (China). Dowex was purchased from sigma, tetrabutylammonium hydroxide di-tertbutyl dicarbonate (Boc Anhydride), sodium chloride (NaCl), β-cyclodextrin (β-CD), 4-(t-Butyl)-phenylacetic acid, 4-dimethylaminopyridine (DMAP), 2-hydroxyethyl methacrylate, 2-hydroxypropyl methacrylate, N,N-dimethylacrylamide, phosphorus oxychloride, span 80, cyclohexane, N,N’-methylene diacrylamide, acrylic acid, and 3-Buten-1-amine hydrochloride were purchased from J&K Chemicals (China). (USA). PBS, Dulbecco’s modified Eagle medium, fetal bovine serum (FBS), penicillin/streptomycin, alpha-minimum essential medium (α-MEM), and L-glutamine were purchased from Gibco (USA). The Calcein-AM, propidium iodide, Cell Counting Kit-8 (CCK-8), DAPI, TRIzol, Alkaline Phosphatase (ALP) Assay Kit, H&E staining kit, were purchased from Beyotime.
Synthesis of phosphorylated hydroxyethyl acrylate (Phos-HEA) and phosphorylated hydroxypropyl methacrylate (Phos-HPMA)
First, phosphorus oxychloride (7.46 g, 1.1 eq) was mixed with tetrahydrofuran (33 mL) and transferred into a 250 mL round-bottom flask under a nitrogen atmosphere. The flask was then placed in an ice bath for cooling. 2-hydroxyethyl acrylate (5.14 g, 1 eq) and triethylamine (4.93 g, 1.1 eq) were mixed with tetrahydrofuran (7 mL) and added into a dropping funnel under nitrogen. The solution was slowly dripped into the round-bottom flask. After 3 h of thorough reaction, 7.5 mL of water was added to the dropping funnel and slowly dripped into the flask to complete the hydrolysis. The reaction mixture was heated to room temperature and washed three times with saturated sodium chloride solution. The tetrahydrofuran (THF) was removed by rotary evaporation, yielding a light brown oily product (Phos-HEA). HPMA (6.38 g, 1 eq) was used to synthesize the Phos-HPMA following the same procedure.
Preparation of Phos-HEA microgel, DMAA microgel, and Phos-HPMA microgel
Phos-HEA (280 mg, 1 eq) was weighed and dissolved in 10 mL of H2O. Acrylic acid (10 μL, 0.1 eq) was added to the solution. N,N-dimethylbisacrylamide (MBA, 22 mg, 0.1 eq) was then dissolved in the solution, followed by the addition of a KPS solution (KPS (7 mg, 0.018 eq) dissolved in 100 μL of DI water). The reaction mixture was maintained under a nitrogen atmosphere at a stirring speed of 1200 rpm, and then mixed with 40 mL of cyclohexane and 900 μL of Span 80 to form a homogeneous emulsion. The emulsion was heated to 70 °C and stirred for 2 h. Afterward, the mixture was centrifuged, washed five times with anhydrous ethanol/DI water, and freeze-dried to obtain white Phos-HEA microgel. DMAA (142 mg, 1 eq) and Phos-HPMA (320 mg, 1 eq) were used to prepare the DMAA microgel and Phos-HPMA microgel, respectively, following the same procedure.
Preparation of Ac-Phos-HEA microgel, Ac-DMAA microgel, and Ac-Phos-HPMA microgel
Phos-HEA (1 g) was weighed and dispersed in 50 mL of H2O. Nitrogen gas was bubbled through the solution for 30 min to remove oxygen. EDC (3.68 g, 19.2 mmol) and NHS (552 mg, 4.8 mmol) were weighed and dissolved in the above solution to activate the carboxyl groups on the microgel surface. The solution was stirred for 2 h, after which 3-buteneamine hydrochloride (258 mg, 2.4 mmol) was added. The pH of the solution was adjusted to 8 using triethylamine. The reaction was carried out under a nitrogen atmosphere at room temperature for 12 h. The reaction mixture was placed in a 3500 Da dialysis bag and dialyzed for 5 days. Finally, the product was freeze-dried, yielding 75%. The DMAA microgel (1 g) and Phos-HPMA microgel (1 g) were used to prepare the Ac-DMAA microgel and Ac-Phos-HPMA microgel following the same procedure.
Synthesis of 4-(t-Butyl)-phenylacetic acid modified hyaluronic acid (HA-TP)
A solution of sodium hyaluronate (HA‑Na, 2.0 g) in deionized water was treated with Dowex resin (6.0 g) for ion exchange, then neutralized with tetra‑n‑butylammonium hydroxide (TBAOH) to obtain an aqueous HA‑TBA solution at pH 7.0. After freeze‑drying, the product was redissolved in dimethyl sulfoxide (DMSO). HA‑TBA (1.0 g, 1.4 mmol in terms of disaccharide repeating units, 1 eq.), 4‑(tert‑butyl)phenylacetic acid (0.8 g, 4.2 mmol, 3 eq.), and 4‑dimethylaminopyridine (DMAP, 0.13 g, 1.05 mmol, 0.75 eq.) were dissolved in anhydrous DMSO (100 mL) at 25 °C. Under nitrogen atmosphere and at 25 °C, di‑tert‑butyl dicarbonate (BOC₂O) was added dropwise. The mixture was warmed to 45 °C and stirred for 24 h. The resulting reaction mixture was dialyzed successively against DMSO, NaCl solution, and deionized water to remove low‑molecular‑weight impurities. Finally, the purified solution was freeze‑dried to afford the product as a white solid.
Synthesis of Ac-β-CD
Ac-β-CD was synthesized following established procedures from the literature33. In a typical preparation, β-cyclodextrin (β-CD, 10 g) was dissolved in dimethylformamide (DMF, 150 mL) and triethylamine (TEA, 7 mL). The solution was cooled to 0 °C with stirring. Acryloyl chloride (5 mL) was then introduced, and the reaction was allowed to proceed for 6 h. Following the reaction, the mixture was filtered to eliminate the precipitated trimethylamine hydrochloride salt. The filtrate was concentrated to roughly 20 mL via rotary evaporation. This concentrated solution was subsequently added dropwise into acetone (600 mL), which induced the formation of a white solid. The precipitate was collected, washed thoroughly with acetone, and finally subjected to freeze-drying to obtain the product.
Preparation of NC, NCLS, and CPAC hydrogel
For the NC hydrogel, HA-TP (1%), gelatin (3%), Ac-β-CD (5%), and Ac-Phos-HEA microgel (5%) were dissolved in PBS (900 μl). After complete dissolution, the solution was allowed to stand overnight. Subsequently, 100 μl of 0.5 wt% lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) was added, and polymerization was initiated under blue light (λ = 405 nm, 350 mW/cm²) for 5 min to obtain the NC hydrogel. For the NCLS and CPAC hydrogels, the Ac-Phos-HEA microgels were replaced with an equal mass of Ac-DMAA microgels and Ac-Phos-HPMA microgels, respectively, while keeping all other conditions identical.
3D cell culture within hydrogels
The human mesenchymal stem cells (hMSCs) were purchased from REGEN-αGEEK Biotechnology Co. Ltd (a 65-year-old male donor). Primary hMSCs were cultured in αMEM supplemented with 16.7% FBS (Excel), 1% penicillin-streptomycin (Gibco), and 1% L-glutamine (Gibco), and passaged to passage 4 (P4) under conditions of 37 °C and 5% CO2. After 5 min of digestion with 0.05% trypsin (Gibco), the cells were collected. Following cell counting, the cells were placed into individual 1.5 mL Eppendorf tubes and centrifuged at 290 × g for 3 min, with the supernatant discarded. For each type of hydrogel, the encapsulation density of hMSCs was 1 × 10⁷ cells per mL of hydrogel precursor solution (NC, NCLS, or CPAC). After mixing, 50 µL of precursor solution containing hMSCs was crosslinked under blue light (λ = 405 nm) for 5 min and transferred into a 24-well plate. Constructs were continuously cultured in either growth or osteogenic medium (10 mM β-glycerophosphate, 50 µg/mL ascorbic acid 2-phosphate, and 100 nM dexamethasone). For the EZH2 inhibitor group, the inhibitor (Cat: HY-148458, MCE) was added to osteogenic medium at a final concentration of 10 μM. Constructs were cultured in the inhibitor-containing osteogenic medium for 12 h, after which the medium was replaced with fresh osteogenic medium for continued culture.
Microgel shrinkage quantification
Microgel shrinkage was quantified by capturing images of Phos-HEA and Phos-HPMA microgels before and after ALP treatment using an inverted microscope. Fifteen microgels were randomly selected from each group, and their diameters were measured using Nano Measure software (Version 1.2). The shrinkage of the microgels was quantified using Nano Measure (Version 1.2)
Fabrication and rheological characterization of the NC and CPAC hydrogels
Rheological characterizations were performed using a TA Discovery HR-30 hybrid rheometer. Samples were formed in situ by pipetting a 50 µL monomer solution between the bottom HR-30 hybrid rheometer plate and an 8 mm flat plate, and the experiment was initiated after photopolymerization (λ = 365 nm, 396 mW/cm2). Evolution experiments were performed at 37 °C. Time sweep measurements were conducted at 37 °C with a fixed strain of 1% and a frequency of 10 rad/s for 120 s. For the alternating strain step shear test, the temperature was maintained at 37 °C, with a frequency of 10 rad/s. The strain was alternated between 60% (45 s) and 1% (45 s) for three full cycles to obtain the step shear profile. The stress relaxation properties of the samples were measured from compression tests of the gel discs (8 mm in diameter, 2 mm thick). The gel discs were compressed to 15% strain with a deformation rate of 0.3 mm s−1 and held for 300 s to obtain the stress relaxation curve. The Young’s modulus was then calculated as the slope of the linear region of the resulting stress‑strain curve. Each sample group was tested independently three times to ensure reproducibility.
Phos-HPMA microgel mediated the heterogeneous distribution of matrix stiffness in the CPAC Hydrogel
Nanoindentation technology was applied to characterize the Young’s modulus distribution of both hydrogels under underwater conditions via a Piuma nanoindenter (Optics 11, Netherlands). Specifically, the hydrogels were maintained in PBS at 37 °C for 1 h before being loaded onto the plate. We randomly selected a 350 × 350 μm square matrix area on the surface of the samples and performed nanoindentation measurements at intervals of 50 μm. A total of 8 × 8 valid Young’s modulus data points were obtained, forming a heatmap. The selected areas were random, and the measurements were repeated three times.
Real-time quantitative PCR
Total RNA was isolated from cultured samples using TRIZOL reagent (Vazyme, R401-01) following the manufacturer’s protocol. RNA concentration and purity were assessed by spectrophotometric measurement (NanoDrop 300, Thermo Fisher Scientific). First-strand cDNA synthesis was performed using PrimeScript RT Master Mix (Takara, RR014A) according to the manufacturer’s specifications. Quantitative PCR was carried out using TB Green Premix Ex Taq (Takara, RR820A) on a LightCycler 480 instrument (Roche Diagnostics). The thermal cycling protocol comprised: initial denaturation at 95 °C for 30 s, followed by 40 cycles of amplification (95 °C for 5 s, 60 °C for 30 s), and a final dissociation stage (95 °C for 15 s, 60 °C for 1 min, and 95 °C for 15 s). GAPDH served as the endogenous control for normalization. Relative gene expression levels were determined using the comparative 2−ΔΔCT method. Primer sequences are provided in Supplementary Table 1.
Immunofluorescence staining
Following a brief PBS rinse, specimens were fixed in 4% paraformaldehyde (Aname, #153799) for 30 min at ambient temperature. After two additional PBS washes, samples were permeabilized and blocked for 2 h at room temperature using PBS containing 3% bovine serum albumin (Millipore, #S30) and 0.25% Triton X-100 (Sigma, #T8787). Primary antibody incubation was carried out overnight at 4 °C in blocking buffer. Unbound antibodies were removed through three washes with dilution buffer, followed by 1-h incubation with Alexa Fluor 488-conjugated secondary antibody (1:200 dilution). For cytoskeletal and nuclear visualization, samples were subsequently treated with phalloidin (1:400, 1 h) and DAPI (1:100, 30 min). Fluorescence imaging was performed using a Zeiss LSM780 confocal system, with quantitative analysis of fluorescence intensity conducted using ImageJ software (NIH).
The primary antibodies used were as follows: anti-Col 1 (Cat.#14695-1-AP, ProteinTech, 1:500), anti-ALP (Cat.#DF6225, Affinity, 1:500), anti-FAK (phospho Y397) (Cat.# 66258-1-lg, ProteinTech, 1:500), anti-paxillin (Cat.# 10029-1-lg, ProteinTech, 1:500), anti-phospho-MYH9 (Ser1943) (Cat.# AF4425, Affinity, 1:500), anti- EZH2 (Cat.# 21800-1-AP, ProteinTech, 1:500), anti- H3K27me3 (Cat.# A2363, Abclonal, 1:500).
The secondary antibodies used were as follows: CoraLite488-conjugated goat anti-mouse IgG (H + L) (Cat.#SA0013-1, ProteinTech, 1:500), CoraLite488-conjugated goat anti-rabbit IgG (H + L) (Cat.#SA0013-2, ProteinTech, 1:500), and goat anti-rabbit Alexa Fluor 594 (Cat.#S0006, Affinity, 1:500).
Western blot
Cellular proteins were lysed using ice-cold RIPA buffer containing 1 mM phenylmethylsulfonyl fluoride. Nuclear and cytoplasmic fractions were isolated following the supplier’s protocol (Beyotime, Cat.#P0013B). Protein concentrations were determined using a bicinchoninic acid assay, followed by heat denaturation at 98 °C for 15 min. Protein samples (20 µg) were resolved on 10% SDS‒PAGE gels, initially at 80 V for 30 min and subsequently at 120 V for 80 min. Separated proteins were transferred onto PVDF membranes at 300 mA for 120 min. Membranes were blocked with 5% nonfat milk for 1 h at room temperature, followed by incubation with primary antibodies at 4 °C overnight. HRP-conjugated secondary antibodies were applied for 1 h at room temperature. Protein bands were detected using an enhanced chemiluminescence substrate (Epizyme, Cat. #SQ201) and imaged with a Bio-Rad system (Tanon, Shanghai, China). Band intensities were quantified using ImageLab software (Bio-Rad). A fixed rectangular selection tool was applied to each target band, and local background signals were subtracted to obtain net intensities. To normalize for loading differences, target protein signals were adjusted relative to internal controls (e.g., GAPDH) within the same lane. Triplicate experimental data were processed using GraphPad Prism 9.0 and expressed as mean ± SD. Statistical comparisons were performed via unpaired two-tailed Student’s t-test, with significance set at p < 0.05.
Chromatin immunoprecipitation (ChIP) and ChIP-qPCR
Cells were fixed with 1% formaldehyde and subsequently quenched with glycine. After washing with PBS, the fixed samples were suspended in cell lysis buffer (5 mM PIPES-KCl, pH 8.0, 85 mM KCl, 0.5% NP40), and the collected nuclei were stored at −80 °C as nuclear pellets or nuclear lysates dissolved in nuclei lysis buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% SDS). For H3K27me3 ChIP, lysates were thawed and sonicated with Sonifier (BRANSON) to obtain chromatin fragments of 300–1000 bp, and 10-fold-diluted with ChIP dilution buffer (16.7 mM Tris-HCl pH 8.0, 1.2 mM EDTA, 0.01% SDS, 1.1% Triron100×, 167 mM NaCl). After double-dilution with a sonication buffer (90 mM Hepes pH 7.9, 220 mM NaCl, 10 mM EDTA, 1% NP40, 0.2% sodium deoxycholate, 0.2% SDS), nuclei were homogenized with a Bioruptor (Tosho denki). After washing, a complex of nuclear proteins/DNAs and antibodies against H3K27me3 (Proteintech; 39157; 5ug used) was retrieved with Protein A- and Protein G-Dynabeads (Life Technologies). After the cross-linking was reversed, chromatin fragments were treated with RNase A and proteinase K. DNA was purified with phenol-chloroform extraction or Ampure XP (Beckman Coulter). In the ChIP-qPCR analyses, the values from the immunoprecipitated samples were normalized to input DNA. Primer sequences are listed in Supplementary Table 2.
Rat skull defect repair model
All experimental procedures involving animals were conducted in accordance with protocols approved by the Animal Experimentation Ethics Committee of the South China University of Technology (AEC No. 2023088). Nine 8-week-old male Sprague-Dawley (SD) rats were acclimatized for 7 days under controlled conditions (24–26 °C, 70% humidity). The rats were randomly allocated into four groups (n = 3 per group): (1) NC hydrogel, (2) CPAC hydrogel, (3) CPAC hydrogel without MSCs (Control), and (4) non-treated control (Blank). Following institutional guidelines, anesthesia was induced via intramuscular injection of a ketamine-xylazine mixture (0.2% ketamine, 0.1% xylazine in saline). Aseptic surgical exposure of the calvarium was performed, and two 5 mm diameter critical-sized defects were created in each rat skull. In each animal, one defect was filled with the hydrogel sample, while the contralateral defect served as an internal control. Primary rMSCs were expanded to passage 3 (P3) prior to encapsulation in hydrogels. Both NC and CPAC hydrogels, each containing 5 × 106 cells, were preconditioned in osteogenic medium for 3 days before implantation into the calvarial defects. The surgical site was subsequently closed via suturing. Following an 8-week recovery period, the rats were euthanized for analysis. Skull specimens were initially subjected to micro-computed tomography (micro-CT) scanning, followed by decalcification in 10% ethylenediaminetetraacetic acid (EDTA, pH 7.4) for 2 months in preparation for immunohistochemical evaluation.
Quantification of micro-CT
Micro-CT Scanning and Analysis: Upon euthanasia, rat calvarial bones were isolated and fixed in 4% paraformaldehyde. Fixed samples were scanned using a Micro-CT system (Quantum GX2, Perkinelmer AG) under the following conditions: 70 kV for 140 ms with a resolution setting of 25 μm and 1024 × 1024 pixels. Three-dimensional reconstruction and subsequent data analysis of the defect region were performed to quantify bone regeneration. Key outcomes included bone volume (BV), bone volume/tissue volume (BV/TV), and bone surface/tissue volume (BS/TV).
Immunohistochemical staining
The skull specimens were first fixed in 4% paraformaldehyde for 48 h to ensure proper preservation of tissue morphology and antigens. Following fixation, the samples underwent mild decalcification with 10% EDTA solution (pH 7.4, 25 °C) for 6 weeks. Tissue dehydration was performed through a graded ethanol series (30–100%), with subsequent xylene clearing and paraffin embedding. Serial sections (4 μm thickness) were prepared for histological evaluation.
Tissue sections were subjected to H&E and Masson’s trichrome staining according to standard protocols, followed by coverslipping with mounting medium. For immunohistochemical detection, sections were incubated overnight at 4 °C with primary antibodies targeting osteocalcin (OCN; 1:100 dilution, DF12303, Affinity). Following primary antibody incubation, sections were treated with HRP-conjugated secondary antibody (room temperature, 30 min) and developed with diaminobenzidine substrate (10 min, room temperature). All stained sections were permanently mounted for microscopic evaluation.
Theoretical analysis of CPAC hydrogel deformation
Using elasticity theory, we analyzed how the hydrogel deforms in response to changes in the radius of the medium ball. Let the ball initially have radius \({R}_{0}\), which then changes to \(R\). Due to strong interactions at the interface, the surrounding hydrogel is perturbed accordingly. As a continuous elastic material, the hydrogel’s deformation can be described in spherical coordinates by the displacement components \(\left({u}_{r},{u}_{\theta },{u}_{\varphi }\right)\). Because of the spherical symmetry, only the radial displacement is nonzero, giving \({u}_{\theta }=0\) and \({u}_{\varphi }=0\).
According to elasticity theory, the equilibrium is expressed as:
where \(E\) is the Young’s modulus. This system is subject to two boundary conditions: (1) At infinity (\(r\to \infty\)), the deformation vanishes, i.e., \({u}_{\infty }=0\); (2) At the ball’s surface (\(r={R}_{0}\)), the radial displacement equals the radius change, i.e., \({u}_{0}=R-{R}_{0}\). Solving Eq. (1) under these conditions, we can obtain the expression for the radial displacement.
Generally, the Young’s modulus is assumed to be a fixed value (\(E={E}_{0}\)), then the solution of Eq. (1) yields:
The above equation shows that the hydrogel deforms radially around the ball, with greater deformation arising from larger radius changes.
For cases like the experiments conducted here, \(E\) is not fixed. When the hydrogel deforms, its local properties, such as polymer structures and interactions, change, which in turn alters \(E\). The relationship between \(E\) and hydrogel deformation may be complex, but in the small deformation limit, we assume \(E\) is proportional to the deformation. Therefore, \(E\) can be approximated as a linear function,\(E={E}_{0}+k{u}_{r}\), where \({E}_{0}\) is the Young’s modulus of the undeformed hydrogel, and \(k\) is a constant that characterizes the dependence of \(E\) on \({u}_{r}\). Solving Eq. (1) with this assumption yields:
where \(b={R}_{0}^{2}\left[k{\left(R-{R}_{0}\right)}^{2}+{E}_{0}\left(R-{R}_{0}\right)\right]\). Note that \(r\ge {R}_{0}\) in the above equation. This expression shows that the Young’s modulus varies radially around the medium ball. As \(r\) decreases, \(E\) increases, meaning the Young’s modulus is greater closer to the ball’s surface. Equation (3) matches well with our experimental data, Supplementary Fig. 11a. By fitting the experimental data, we determine \({E}_{0}=270.26{{\mathrm{Pa}}}\), and \(k=-21.26{{\mathrm{Pa}}}/{{\mathrm{\mu m}}}\) or \(-2.13\times {10}^{7}{{{\rm{N}}}}/{{{{\rm{m}}}}}^{3}\).
Solving Eq. (1) also provides the analytical solution for the radial displacement:
Equation (4) shows that the hydrogel deforms radially around the ball, with greater deformation arising from larger radius changes (Supplementary Fig. 11b).
Methods for ALP inhibitor use
ALP inhibitor (Beyotime, Cat# ST019) was prepared as a 10 mM stock solution using sterile 1× PBS. For experiments, the inhibitor was added to the culture medium at a final concentration of 0.1 mM and applied for 72 h. For the control group, an equal volume of PBS was added.
Methods for ALP activity quantification and the CCK-8 assay
The ALP activity in the culture medium was quantified using a commercial colorimetric assay kit (Beyotime, Cat# P0321S) based on the hydrolysis of p-nitrophenyl phosphate (pNPP). Specifically, after the designated treatments, the culture medium was collected and centrifuged to remove debris. Then, 50 µL of the supernatant was mixed with 50 µL of the pNPP substrate working solution in a 96-well plate and incubated in the dark at 37 °C for 30 min. The enzymatic reaction was terminated by adding the stop solution provided in the kit. The absorbance of the yellow reaction product, p-nitrophenol, was immediately measured at 405 nm using a microplate reader. A standard curve was established using p-nitrophenol standards, and the ALP activity was calculated for each sample based on the absorbance readings. All assays were performed in triplicate.
Cell viability was assessed using the Cell Counting Kit-8 (CCK-8; Beyotime, Cat# C0038). Following the designated treatments, 100 µL of the CCK-8 reagent was directly added to each well (900 µL culture medium with one hydrogel). The plate was then incubated at 37 °C in a humidified incubator with 5% CO2 for 2 h. After incubation, the absorbance of each well was measured at a wavelength of 450 nm using a microplate reader. Wells containing culture medium and CCK-8 reagent without cells were used as blanks for background subtraction. Cell viability was calculated as a percentage relative to the untreated cells (set as 100%), using the following formula:
where As is the absorbance of the sample (treated cells), Ac is the absorbance of the control (untreated cells), and Ab is the absorbance of the blank. All experiments were performed with three independent replicates. Day 0 indicates the viability of MSCs before encapsulation, while day 3 represents the viability after MSCs were loaded in NC and CPAC hydrogels for 3 days.
Methods for ALP activity quantification
The alkaline phosphatase (ALP) activity in the culture medium was quantified using a commercial colorimetric assay kit (Beyotime, cat# P0321S) based on the hydrolysis of p-nitrophenyl phosphate (pNPP). The specific procedure was as follows: After the designated treatments, the culture medium was collected and centrifuged. Then, 50 µL of the supernatant was mixed with 50 µL of the pNPP substrate working solution in a 96-well plate and incubated in the dark at 37 °C for 30 min. The enzymatic reaction was terminated by adding the stop solution provided in the kit. The absorbance of the yellow reaction product, p-nitrophenol, was immediately measured at 405 nm using a microplate reader. A standard curve was generated using p-nitrophenol standards, and the ALP activity was calculated for each sample based on the absorbance readings. All assays were performed in triplicate.
Methods for EdU incorporation assays and live/dead staining
The alkaline phosphatase (ALP) activity in the culture medium was quantified using a commercial colorimetric assay kit (Beyotime, cat# P0321S) based on the hydrolysis of p-nitrophenyl phosphate (pNPP). The specific procedure was as follows: After the designated treatments, the culture medium was collected and centrifuged. Then, 50 µL of the supernatant was mixed with 50 µL of the pNPP substrate working solution in a 96-well plate and incubated in the dark at 37 °C for 30 min. The enzymatic reaction was terminated by adding the stop solution provided in the kit. The absorbance of the yellow reaction product, p-nitrophenol, was immediately measured at 405 nm using a microplate reader. A standard curve was generated using p-nitrophenol standards, and the ALP activity was calculated for each sample based on the absorbance readings. All assays were performed in triplicate.
For live/dead staining, a staining solution was prepared in 1× PBS containing Calcein-AM and propidium iodide (PI), each at a final concentration of 1 µM. The hydrogels cultured for 3 days in a 24‑well plate were carefully removed and transferred to a new plate, and 1 mL of the prepared staining solution was added to each well. The plate was then incubated at 37 °C in the dark for 30 min. Following incubation, the hydrogels were washed three times with 1× PBS. Finally, imaging was performed using a confocal laser scanning microscope with detection channels set to 488 nm and 594 nm.
Methods for in vivo implantation model and sample processing for ALP, nanoindentation and mass quantification
According to the previously described method for creating critical-size bone defects in rat calvaria, hydrogels loaded with rMSCs were implanted into the calvarial defect areas. The implanted hydrogels were retrieved at different time points: before implantation (0 h), and at 24 h and 72 h after implantation. The retrieved hydrogels were added to 1 mL of 1× PBS, thoroughly homogenized using a tissue grinder, and then centrifuged. The alkaline phosphatase (ALP) activity in the hydrogel was quantified using a commercial colorimetric assay kit (Beyotime, cat# P0321S) based on the hydrolysis of p-nitrophenyl phosphate (pNPP). The specific procedure was as follows: 50 µL of the supernatant was mixed with 50 µL of the pNPP substrate working solution in a 96-well plate and incubated in the dark at 37 °C for 30 min. The enzymatic reaction was terminated by adding the stop solution provided in the kit. The absorbance of the yellow reaction product, p-nitrophenol, was immediately measured at 405 nm using a microplate reader. A standard curve was generated using p-nitrophenol standards, and the ALP activity was calculated for each sample based on the absorbance readings. All assays were performed in triplicate.
For nanoindentation testing, the extracted hydrogels were fixed onto glass slides. An 8×8 matrix of points, with each point spaced 50μm apart, was randomly selected on the hydrogel surface. The effective Young’s modulus at each point in the matrix was measured using a nanoindenter.
For the degradation experiment, the implanted hydrogels were retrieved at different time points, freeze-dried, and weighed (recorded as WX). Taking the mass of the freeze-dried hydrogel sample before implantation as W0, the degradation rate of the hydrogel was calculated as follows:
All samples in each group were tested in three independent experiments to ensure reproducibility.
Method for alkaline phosphatase color development kit
After fixation with paraformaldehyde, the hydrogel was cryosectioned into 8 μm thin slices. The resulting slices were washed three times with PBS. A working solution was prepared according to the manufacturer’s instructions of the BCIP/NBT Alkaline Phosphatase Color Development Kit (Beyotime, cat# C3206). The sections were fully covered with the working solution and incubated at room temperature in the dark for 30 min. The color development reaction was terminated by washing twice, and images were captured using an inverted microscope.
Statistics and reproducibility
Experiments included triplicate samples (n = 3), and results are shown as mean ± standard deviation. Statistical significance was determined using GraphPad Prism (version 9.0.0) and Origin (version 2018). Comparisons between two groups were analyzed by an independent t-test, while comparisons across more than two groups were analyzed by one-way ANOVA with Tukey’s multiple comparison test. In all analyses, statistical significance was defined as p < 0.05.
Data availability
The data supporting the findings from this study are available within the article, Supplementary Information, or Source data file. Source data are provided with this paper. Source data is available for Figs. 2c, h, i, 3a, d, e, 4g, h, 5a–c, f, h and 6c–e, and Supplementary Figs. 9a–d, 10a(i, ii, iii), b(i, ii, iii), 12a–d, 13a–c, 15a–l, 16a(i, ii, iii, iv), b, c(i, ii, iii, iv), d(i, ii, iii, iv), e, f, 17c, 18b, 19, 23, 24a(i, ii, iii), b(i, ii, iii) and 25b–d in the associated source data file. Source data are provided with this paper.
References
Feng, Q. et al. Engineering the cellular mechanical microenvironment to regulate stem cell chondrogenesis: Insights from a microgel model. Acta Biomater. 113, 393–406 (2020).
Even-Ram, S., Artym, V. & Yamada, K. M. Matrix control of stem cell fate. Cell 126, 645–647 (2006).
Li, Z. et al. Metabolite-dependent m6A methylation driven by mechanotransduction-metabolism-epitranscriptomics axis promotes bone development and regeneration. Cell Rep. 44, 115611 (2025).
Yang, C., Tibbitt, M. W., Basta, L. & Anseth, K. S. Mechanical memory and dosing influence stem cell fate. Nat. Mater. 13, 645–652 (2014).
Prince, E. & Kumacheva, E. Design and applications of man-made biomimetic fibrillar hydrogels. Nat. Rev. Mater. 4, 99–115 (2019).
Zhao, J. et al. Liquid–liquid phase separation-mediated cellular-scale compartmentalization of hydrogel covalent cross-linking promotes microtubule-based mechanosensing. J. Am. Chem. Soc. 147, 14336–14347 (2025).
Theocharis, A. D., Skandalis, S. S., Gialeli, C. & Karamanos, N. K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 97, 4–27 (2016).
Zauchner, D. et al. Synthetic biodegradable microporous hydrogels for in vitro 3D culture of functional human bone cell networks. Nat. Commun. 15, 5027 (2024).
Moharrer, Y. & Boerckel, J. D. Tunnels in the rock: dynamics of osteocyte morphogenesis. Bone 153, 116104 (2021).
Fang, F. et al. The role and applications of extracellular vesicles in osteoporosis. Bone Res. 12, 4 (2024).
Khatib, N. S. et al. Mechanoregulatory role of TRPV4 in prenatal skeletal development. Sci. Adv. 9, eade2155 (2023).
Goossens, P. et al. Integrating multiplex immunofluorescent and mass spectrometry imaging to map myeloid heterogeneity in its metabolic and cellular context. Cell Metab. 34, 1214–1225.e6 (2022).
Florencio-Silva, R., Sasso, G. R. D. S., Sasso-Cerri, E., Simões, M. J. & Cerri, P. S. Biology of bone tissue: structure, function, and factors that influence bone cells. Biomed. Res. Int. 2015, 1–17 (2015).
Franz-Odendaal, T. A., Hall, B. K. & Witten, P. E. Buried alive: How osteoblasts become osteocytes. Dev. Dyn. 235, 176–190 (2006).
Discher, D. E., Janmey, P. & Wang, Y. Tissue cells feel and respond to the stiffness of their substrate. Science 310, 1139–1143 (2005).
Lutolf, M. P. & Hubbell, J. A. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat. Biotechnol. 23, 47–55 (2005).
Khetan, S. et al. Degradation-mediated cellular traction directs stem cell fate in covalently crosslinked three-dimensional hydrogels. Nat. Mater. 12, 458–465 (2013).
Yang, B. et al. Enhanced mechanosensing of cells in synthetic 3D matrix with controlled biophysical dynamics. Nat. Commun. 12, 3514 (2021).
Kloxin, A. M., Kasko, A. M., Salinas, C. N. & Anseth, K. S. Photodegradable hydrogels for dynamic tuning of physical and chemical properties. Science 324, 59–63 (2009).
Liu, L. et al. Cyclic stiffness modulation of cell-laden protein–polymer hydrogels in response to user-specified stimuli including light. Adv. Biosyst. 2, 1800240 (2018).
Hörner, M. et al. Phytochrome-based extracellular matrix with reversibly tunable mechanical properties. Adv. Mater. 31, 1806727 (2019).
Yang, J. et al. Photo-tunable hydrogels reveal cellular sensing of rapid rigidity changes through the accumulation of mechanical signaling molecules. Cell Stem Cell 32, 121–136.e6 (2025).
Killaars, A. R., Walker, C. J. & Anseth, K. S. Nuclear mechanosensing controls MSC osteogenic potential through HDAC epigenetic remodeling. Proc. Natl. Acad. Sci. USA 117, 21258–21266 (2020).
Li, X. et al. Dynamic stiffening hydrogel with instructive stiffening timing modulates stem cell fate in vitro and enhances bone remodeling in vivo. Adv. Healthc. Mater. 12, 2300326 (2023).
Guvendiren, M. & Burdick, J. A. Stiffening hydrogels to probe short- and long-term cellular responses to dynamic mechanics. Nat. Commun. 3, 792 (2012).
Phillips, D. J., Wilde, M., Greco, F. & Gibson, M. I. Enzymatically triggered, isothermally responsive polymers: reprogramming poly(oligoethylene glycols) to respond to phosphatase. Biomacromolecules 16, 3256–3264 (2015).
Doyle, A. D., Carvajal, N., Jin, A., Matsumoto, K. & Yamada, K. M. Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility-dependent adhesions. Nat. Commun. 6, 8720 (2015).
Xu, S. et al. Flow-dependent epigenetic regulation of IGFBP5 expression by H3K27me3 contributes to endothelial anti-inflammatory effects. Theranostics 8, 3007–3021 (2018).
Kumar, S., Kim, C. W., Simmons, R. D. & Jo, H. Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: mechanosensitive athero-miRs. Arterioscler. Thromb. Vasc. Biol. 34, 2206–2216 (2014).
Italiano, A. Role of the EZH2 histone methyltransferase as a therapeutic target in cancer. Pharmacol. Ther. 165, 26–31 (2016).
Vella, S. et al. MicroRNA-101 is repressed by EZH2 and its restoration inhibits tumorigenic features in embryonal rhabdomyosarcoma. Clin. Epigenet. 7, 82 (2015).
Zhang, Y. X. et al. Dynamic and distinct histone modifications of osteogenic genes during osteogenic differentiation. J. Biochem. 158, 445–457 (2015).
Feng, Q. et al. Mechanically resilient, injectable, and bioadhesive supramolecular gelatin hydrogels crosslinked by weak host-guest interactions assist cell infiltration and in situ tissue regeneration. Biomaterials 101, 217–228 (2016).
Acknowledgements
This work was financially supported by the National Key Research and Development Program (2024YFB3814900, K.Z.). This work was supported by the National Science Foundation of China (52433010, L.B.; 32271385, K.Z.; 52303184, J.Z.). This work was funded by Guangdong Basic and Applied Basic Research Foundation (2025B1515020018, K.Z.; 2025A1515012981, J.Z.). This work was financially supported by the GJYC program of Guangzhou (2024D03J0004, L.B.; 2024D01J0084, K.Z.). This work was supported by the Fundamental Research Funds for the Central Universities (2024ZYGXZR086, K.Z.; 2024ZYGXZR068, J.Z.; 2025ZYGXZR015, L.B.). This work was supported by the Guangdong Provincial Key Area R&D Program (2024B0101080001, Y.Z.).
Author information
Authors and Affiliations
Contributions
Conceptualization: Q.L., H.L., J.Z., K.Z., L.B.; methodology: Q.L., H.L., W.G., H.Y., Z.L., Y.H., Y.F., S.W.; investigation: Q.L., H.L., H.Y., Z.L., Y.H., S.W.; visualization: Q.L., H.L., H.Y.; supervision: J.Z., K.Z., L.B.; writing—original draft: Q.L.; writing—review and editing: J.Z., Y.Z., K.Z., L.B.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hongbing Jiang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ling, Q., Li, H., Zhao, J. et al. Mechanically heterogeneous hydrogel with cell-programmed network restructuring promotes tissue regeneration by mechano-epigenetic modulation. Nat Commun 17, 2184 (2026). https://doi.org/10.1038/s41467-026-69004-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69004-z
