
Abstract
Bacteriophage–antibiotic coadministration is increasingly used for refractory infections, yet the in vivo interactions among phages, bacteria, antibiotics, and host immunity remain poorly defined. We report a longitudinal, multiomic case analysis of a male in his seventies with cystic fibrosis (CF) experiencing an acute-on-chronic pulmonary exacerbation caused by multidrug-resistant (MDR) Pseudomonas aeruginosa. After colistin discontinuation due to nephrotoxicity, ciprofloxacin was initiated, with an intravenous two-phage cocktail introduced days later. Distinct mucoid and nonmucoid bacterial subpopulations associated differentially with antibiotic versus phage exposure, consistent with nonoverlapping selective pressures. Phage activity was temporally constrained, with one phage dominating early bacterial and genomic signals before attenuating after approximately seven days, despite continued genomic detectability. In contrast, the second phage showed no evidence of productive activity. This asymmetry coincided with phage-reactive humoral immunity: pre-existing IgM was associated with lack of recoverability of one phage, while treatment-associated IgM emergence temporally tracked attenuation of the dominant phage. Although phage-resistant variants arose during therapy, they showed limited expansion relative to susceptible populations. These findings define a mechanistic framework—chemobiotherapy—in which chemical and biological antimicrobials coordinate through ecological and immunologic complementarity rather than direct pharmacologic synergy.
Similar content being viewed by others


Introduction
Pseudomonas aeruginosa pulmonary infection in people with cystic fibrosis (CF) exemplifies a debilitating, therapy-refractory chronic disease sustained by continuous antibiotic-selective pressure1. In the remodeled CF airway, Pseudomonas forms biofilms, diversifies into mucoid and resistant variants, and establishes a dynamic equilibrium with the host that resists eradication2,3. As these populations evolve toward multidrug-resistant (MDR) and extensively drug-resistant (XDR) phenotypes, pulmonary exacerbations become more frequent, driving irreversible lung function decline and reduced quality of life1,4. While CF transmembrane conductance regulator (CFTR) modulator therapies have transformed care by restoring ion transport and improving outcomes, they fail to clear chronic lung infections that remain the main driver of inflammation and decline. Likewise, gene-editing and mRNA-based therapies aim to correct or bypass the CFTR defect, but they do not resolve the entrenched Pseudomonas colonization that characterizes advanced disease5,6,7.
Bacteriophage (phage) therapy offers a precise, self-amplifying biological strategy to control bacterial infections that persist despite intensive antibiotic treatment. By exploiting bacterial machinery for replication, phages maintain activity in proportion to pathogen load and extend their lytic reach into biofilms and other protected niches where chemical agents face substantial limitations8. Phages remain active in MDR and antibiotic-rich environments and can drive adaptive trade-offs that diminish bacterial virulence or restore antibiotic sensitivity9. Their capacity to coevolve with bacterial hosts confers therapeutic durability, providing adaptive plasticity without the need for continual reformulation10. Collectively, these advantages make phage–antibiotic combination therapy a pragmatic approach for clinical application, with phages acting as adjuvants that extend or diversify antibiotic activity. Such regimens achieve efficacy through two complementary mechanisms: immediate pharmacologic synergy that accelerates pathogen clearance, and evolutionary steering that applies coordinated selective pressures to limit bacterial escape while promoting less-fit or antibiotic-sensitive variants. However, this framework overlooks the biological and spatial heterogeneity that underlies most refractory infections.
Chronic P. aeruginosa infection, whether in CF, wounds, or other long-standing disease states, exemplifies how spatial and phenotypic heterogeneity sustains persistence. Within the host, compartmentalized clonal lineages diversify through mutation, horizontal gene exchange, and phase variation, generating subvariants that differ in metabolic activity, virulence, stress tolerance, and antibiotic susceptibility3,11. The predominance of dormant and slow-growing cells insulates the population from uniform pharmacologic pressure, fragmenting antibiotic efficacy and reinforcing tolerance rather than outright resistance2,12. Persistent host inflammation and immune remodeling further partition the airway into microenvironments that resist both antibiotics and immune clearance13,14. In practice, such heterogeneity is managed primarily through pharmacologic intensification (higher doses, prolonged courses, sequential regimens) and occasionally supplemented with biofilm-disrupting adjuncts or host-directed interventions15. While these strategies can transiently reduce bacterial burden, they rarely impose unified selective pressures across the infection’s heterogeneous landscape, limiting durable resolution.
Phage–antibiotic coadministration provides a tractable system for examining how chemical and biological antimicrobials act within the immune-modulated environment of advanced lung disease. We conducted a longitudinal, integrative analysis of intravenous phage–antibiotic co-therapy in an adult in their seventies with CF during an acute-on-chronic pulmonary exacerbation caused by P. aeruginosa. This approach resolved bacterial population structure, phage dynamics, and host immune responses over time. Rather than exerting uniform effects throughout the infected lung, antimicrobial pressures were distributed across dynamically structured microbial subpopulations and constrained by host immunity and temporal dynamics. These observations define a mechanistic model, termed chemobiotherapy, in which phages and antibiotics act as coordinated but nonsynergistic selective forces whose effects are constrained and partitioned by microbial ecology and host immune modulation within a heterogeneous infection landscape.
Results
Rapid functional recovery under phage–antibiotic cotherapy
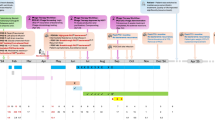
A male with CF (c.3849 + 10kbC > T and c.2012delT genotypes) in their 70 s was admitted with a severe pulmonary exacerbation caused by P. aeruginosa infection consolidated in the left upper lobe (Fig. 1b). Prognosis at presentation was poor given his advanced disease, age, and comorbidities (Table S1). Sputum cultures identified two coexisting P. aeruginosa subpopulations with distinct antibiotic patterns. Mucoid isolates were uniformly susceptible to β-lactams, aminoglycosides, and fluoroquinolones, whereas nonmucoid isolates were broadly MDR to carbapenems, piperacillin–tazobactam, aminoglycosides, and fluoroquinolones (Fig. 1c; Table S2). Given these susceptibility profiles, colistin plus ceftazidime was initiated as first-line therapy, alongside inhaled hypertonic saline, DNase, bronchodilators, and supplemental oxygen (Fig. 1a). After six days of treatment, serum creatinine rose to 3.8 mg dL−1, promoting discontinuation of colistin due to acute kidney injury (AKI; Fig. S1). Despite early cessation, lung function improved, with FEV₁ (forced expiratory volume in one second) increasing from 0.92 L (27% predicted) to 1.39 L (36% predicted) (Fig. 1d).

a Timeline of clinical events (gray) and intravenous (IV) antimicrobial treatments (colored) for pulmonary exacerbation in an elderly patient with CF. Time is shown relative to the first IV phage dose (vertical dashed line, day 0); the second dashed line marks the end of all IV therapies (day 15). First-line colistin + ceftazidime was discontinued early due to acute kidney injury (AKI). b Posterior-to-anterior chest x-rays at hospitalization (left) and immediately before co-therapy (right) show persistent left-upper-lobe consolidation (yellow arrow). The reduced but unresolved opacity underscores the need for continued treatment. c Heatmap of minimum inhibitory concentrations (MICs) for patient sputum isolates (values in Table S2). Mucoid colonies (top) were largely susceptible, whereas nonmucoid colonies (bottom) were extensively drug-resistant. ND indicates MIC not determined d Forced expiratory volume in 1 s (FEV₁, green) and percent-predicted FEV₁ (ppFEV₁, blue) over time. The dashed line denotes the mean baseline FEV₁ (1.49 ± 0.06 L).
During the subsequent antibiotic-free interval, pulmonary infiltrates persisted (Fig. 1b), and FEV₁ remained below the pre-exacerbation baseline (~1.5 L; Fig. 1d), consistent with ongoing infection (Fig. 1b). Following partial renal recovery (Fig. S1), ciprofloxacin was initiated on day –4 followed by intravenous phage co-therapy with P1A and PYO2 on day 0 (Fig. 1a; further details see Supplementary Results and Figs. S2, S3). The staggered regimen was designed to minimize potential drug-phage antagonism and enable close safety monitoring16,17,18. Ciprofloxacin alone improved FEV₁ to 1.66 L (43% predicted) by day 0 (Fig. 1d), after which phage dosing commenced at 2 × 10⁷ PFU as a test infusion, escalating to 2 × 10⁸ PFU mL−1 every 8 h for 14 days, consistent with standard durations for pulmonary antibiotic therapy19. Coadministration was well tolerated with no adverse events, and renal function improved throughout second-line treatment. By day 3, FEV₁ had increased to 1.74 L (50% predicted), a ~ 5% gain over antibiotic monotherapy and 7% above pre-exacerbation baseline (Fig. 1d). At this point, the patient was discharged to complete outpatient therapy via a peripherally inserted central catheter (PICC). Lung function plateaued for 10 days, declined modestly near the end of cotherapy, and stabilized thereafter at ~1.4 L (38% predicted), representing ~90% recovery of pre-exacerbation lung function. The patient’s sustained gains despite advanced CF, comorbidities, prior AKI, and acute MDR infection underscore the potential of adjunctive phage therapy to restore clinical stability where conventional options are exhausted.
Phage-antibiotic exposure accelerated removal of severe airway obstruction
Addition of phage therapy to ongoing antibiotic treatment was associated with rapid relief of airway obstruction, the principal driver of functional decline in CF1,20. This additive effect was evident from changes in forced vital capacity (FVC; Fig. 2a). At admission, FVC was 0.92 L (37% predicted), consistent with severe obstruction. Treatment with colistin-ceftazidime improved FVC to 2.63 L (54% predicted) before discontinuation due to renal toxicity. During the subsequent ciprofloxacin phase, lung function rose more gradually (0.07 L day−1) to 3.30 L (66% predicted). After initiation of phage therapy, the rate of improvement more than doubled, with FVC rising by 0.18 L within 24 h and peaking nearly 0.5 L (~10% predicted) above the pre-exacerbation baseline. The transient increase in FVC above the chronic baseline suggested that co-therapy temporarily improved airway caliber beyond the patient’s established level of obstruction. FVC then declined modestly to 3.00 L, –5% from peak predicted, after treatment cessation (Fig. 2a). This reduction was within expected post-exacerbation variability and likely reflected partial re-accumulation of mucus and inflammation rather than loss of therapeutic benefit14,21. In contrast, the inflated FEV₁/FVC ratio (57% during ciprofloxacin monotherapy) fell to 52% by the end of co-therapy and 48% post-treatment (Fig. 2b). This suggests that while co-therapy increased lung volume and temporarily relieved airway obstruction, chronic damage of the small-airways continued to restrict expiratory flow, preventing full recovery of lung function22.

a, b Spirometric evidence of improved airway mechanics. a Forced vital capacity (FVC, green) and percent-predicted FVC (ppFVC, blue) over time. Dashed line indicates mean baseline FVC = 3.00 ± 0.12 L. b FEV₁/FVC ratio, representing the relationship between airflow and volume, expressed as percent change over time. The declining ratio during treatment reflects persistent small-airway damage limiting full recovery. c Quantitative computed tomography (qCT) chest images in the axial lung window after 2 days of ciprofloxacin alone (left) and one day post-coadministration (right). Reduced opacity in the left upper lobe indicates decreased inflammation. d Bar graphs showing reduced lobar air-trapping percentages following co-therapy. Values are reported for total lung (TL), right lung (RL), left lung (LL), right upper lobe (RUL), right lower lobe (RLL), left upper lobe (LUL), and left lower lobe (LLL). e Mid-maximal expiratory flow (MMEF, L s−1) derived from spirometry confirms partial small-airway clearance. f Principal-component analysis (PCA) biplot of 16 dimensionally reduced patient data sets integrating pulmonary, hematologic, and renal indices. Each circle represents a single day’s standardized data, colored by treatment phase; hierarchically defined clusters are circled. Arrows indicate variable loadings (see Table S5). Vector directionality and sample convergence toward baseline indicate an additive contribution of phage therapy relative to antibiotic monotherapy.
Chest computed tomography (CT) imaging corroborated the spirometric findings. Between days –2 and 16, parenchymal opacities in the left upper lobe—the primary site of infection—diminished, and airway margins became more distinct following co-therapy (Fig. 2c). Across the lungs, air trapping declined by 4.5%, with corresponding increases in inspiratory (+0.37 L) and expiratory (+0.16 L) volumes (Fig. 2d; Table S3). Improvements were widespread but regionally variable. The right lower lobe showed the greatest decline in air trapping (–5.1%), followed by the left upper lobe (–3.9%), and left lower lobe (–2.2%). The right upper lobe changed minimally (–1.9%) but showed an apparent doubling of emphysema score (11.1% to 19.9%). This likely reflects chronic structural changes that became more apparent as inflammation resolved rather than evidence of new parenchymal loss23,24. Coadministration therefore reduced obstruction across multiple lobes, with the greatest improvement in infected regions, while irreversible airway remodeling limited full functional recovery. This structural improvement was mirrored functionally by an increase in maximum mid-expiratory flow (MMEF) from 0.60 to 0.65 L s−1 (Table S3; Fig. 2e), reflecting transient enhancement of small-airway conductance before partial rebound.
Despite advanced disease and severe airflow obstruction at presentation, spirometry improved rapidly with co-therapy, and the patient was discharged after three days with stable oxygenation and preserved pulmonary function. Multivariate mapping of pulmonary (FEV₁, FVC, MMEF), renal (creatinine), and hematologic (WBC, platelets) indices showed that lung function recovery dominated overall clinical variance (Fig. 2f; Tables S4, S5). PC1 (43.5%) aligned with FEV₁ and airflow, and PC2 (27.9%) with FVC and ventilatory volume, while renal and hematologic measures contributed minimally. The multivariate profile reflected the treatment response, showing airflow improvement with colistin–ceftazidime, regression during the antibiotic holiday, recovery with ciprofloxacin, and a peak under phage–ciprofloxacin treatment. The latter produced the largest displacement along both PCs and reflected the strongest combined gains in airflow and volume (Fig. 2f). Because ciprofloxacin and co-therapy samples clustered along the same recovery vector, directional continuity suggests an additive contribution from phages. Post-treatment samples partially converged toward baseline, showing that the benefits of coadministration were substantial but not fully sustained.
Co-therapy reduces Pseudomonas burden with transient airway diversification
As lung function recovered, co-therapy also reshaped the airway microbiome. At baseline, metagenomic and culture profiling showed near complete dominance by Pseudomonas (~94% of bacterial reads), with minor abundances of Klebsiella (2.1–3.8%) and other genera (≤3.4%) (Figs. 3a, S4; Table S6). By day −69, these secondary taxa had nearly disappeared, and the community collapsed into uniform Pseudomonas dominance (>99%). Estimating bacterial load per host cell using human genome equivalents25,26,27,28 showed an eightfold increase in Pseudomonas abundance from approximately 15 to 167 cells per human cell. This transition to ecological simplification and pathogen overgrowth is characteristic of an impending exacerbation detectable nearly 70 days before the clinical flare29.

a–c Quantification of bacterial dynamics in sputum. a Relative abundance of Pseudomonas in metagenomes. b Absolute abundance normalized to human reads, revealing bacterial load reductions otherwise obscured by dominance. c Layered area chart of mucoid (blue) and nonmucoid (yellow) subpopulations, expressed as colony-forming units (CFU) per mL of sputum. Persistent layering of mucoid behind nonmucoid counts indicates phenotype dominance. Vertical dashed lines mark key events: ciprofloxacin initiation (day –4), phage initiation (day 0), and end of co-therapy (day 14). d Heatmap of isolate susceptibilities to phages P1A, PYO2, and ciprofloxacin (CIP), separated by phenotype and collection day. Plaque formation indicates sensitivity (yellow); absence indicates resistance (maroon). Asterisks denote isolates analyzed by automated antibiotic susceptibility testing (AST; values in Table S2).
During co-therapy, absolute Pseudomonas abundance peaked at approximately 211 cells per human cell and then declined more than tenfold by day 7 (Fig. 3b). This inflection coincided with a shift from bacterial proliferation toward clearance and with the onset of physiological recovery (Fig. 2a), indicating that treatment achieved net reversal of infection dynamics. The decline in Pseudomonas burden was followed by a transient rebound in community diversity, with enrichment of Klebsiella (8.4%), Staphylococcus (2.3%), and other genera (~5%), before Pseudomonas re-established dominance (~95%) at a lower, chronic level after treatment ceased (Fig. 3a; Table S6). These dynamics suggest that CF airway infection occupies a rigid, pathogen-dominated state that can be transiently displaced under co-therapy pressure but rapidly rebounds once treatment ends.
Selective collapse of MDR Pseudomonas reshapes infection ecology
Phage-antibiotic treatment rapidly rebalanced the Pseudomonas population, reversing antibiotic-driven competitive release and restoring dominance of the less virulent mucoid lineage (Fig. 3c). Five days before phage initiation, mucoids dominated at ~10⁸ CFU mL−1, while nonmucoids were nearly 1000-fold fewer. Under ciprofloxacin monotherapy, mucoids declined modestly (~0.6 log, 15%), whereas nonmucoids expanded nearly 100-fold, consistent with competitive release in which partial suppression of mucoids permitted MDR variants to proliferate and drive the infection toward a more aggressive, acute-like state30. Phage addition abruptly reversed this trajectory. Within three days, nonmucoids collapsed by ~3 logs, and mucoids declined more steeply than under ciprofloxacin alone. Neither population was eradicated, however, and sequential rebounds of nonmucoids (day 7) and mucoids (day 12) marked waning therapeutic pressure (Fig. 3c). By the end of therapy, total bacterial burden had stabilized near 10⁷ CFU mL−1 and consisted almost entirely of mucoid variants ( ~ 99%). Phage treatment selectively reduced the emergent MDR subpopulation, forcing an ecological reversion of the infection to a chronic, antibiotic-susceptible state dominated by less virulent mucoid cells.
Partial recovery of both lineages despite ongoing therapy, coinciding with rebound phases observed in vivo, suggested adaptive restructuring within surviving populations. Twelve isolates collected between days −538 and 0 were uniformly sensitive to both therapeutic phages (Fig. 3d), whereas contemporaneous mucoids were inherently phage-insensitive, consistent with capsule-mediated tolerance2,31. By day 2, 1 of 3 mucoid isolates exhibited sensitivity to PYO2, indicating selective amplification of a minor pre-existing subpopulation rather than de novo adaptation. This was consistent with a modest improvement of mucoid suppression after phage addition (Fig. 3c). In contrast, nonmucoid isolates adapted under phage pressure, acquiring P1A resistance on day 13 (1 of 3 isolates) and by day 15 in about half the isolates (Fig. 3d), coinciding with the rebound in sputum nonmucoids (Fig. 3c). All nonmucoids remained PYO2-sensitive, suggesting that although both phages target LPS, their distinct binding sites or infection dynamics constrained cross-resistance. These divergent trajectories highlight the complementary activities of the cocktail, with P1A exerting potent but resistance-prone pressure and PYO2 providing sustained coverage with limited escape.
Antibiotic susceptibilities remained stable throughout therapy, indicating that population restructuring reflected phage activity rather than antibiotic adaptation. Mucoid isolates retained mixed fluoroquinolone sensitivity (56% resistant), and nonmucoids remained uniformly resistant. For all other antibiotic classes, profiles were unchanged, with mucoids pan-susceptible and nonmucoids MDR (Table S2; Fig. S5). No evidence of phage-associated conversion or transfer of antibiotic resistance was detected. Consistent MIC values across timepoints further support that lineage turnover resulted from ecological selection rather than genetic acquisition of new resistance determinants. Together with stable drug susceptibilities, the rapid loss of the MDR subpopulation indicates that phage pressure, not antibiotic activity, was the dominant selective force during co-therapy.
Phage pressure elicits variant-dependent extinction and adaptive diversification
Phage pressure emerged as the dominant evolutionary force within a long-standing CF airway infection, eliminating highly susceptible variants while promoting diversification among survivors. The exacerbation arose from within-host evolution of an entrenched P. aeruginosa infection rather than reinfection by a new strain (Table S7). All sputum isolates belonged to the CF-adapted ST244 clonal complex32, which had diversified into coexisting mucoid and nonmucoid populations. Two derivative sequence types, ST4739 and ST4740, were unique to this patient and corresponded to MDR nonmucoid variants that evolved from the ancestral ST244 background. Each carried nonsynonymous mutations in mutL (DNA repair) and trpE (tryptophan biosynthesis), consistent with hypermutation and metabolic shifts12,33,34. Core-genome comparisons showed that this diversification occurred within a single lineage capable of generating both mucoid and nonmucoid phenotypes (Fig. S4). The coexistence of ancestral and patient-specific lineages shows that the chronic infection remained genetically and phenotypically heterogeneous under sustained antibiotic and host selection, creating a complex exacerbation with diverse targets for co-therapy.
Within the nonmucoid population, four temporally ordered clades captured distinct responses to alternating phases of antibiotic and phage exposure (Fig. 4a). The earliest lineage (Clade 1) persisted from pre-therapy isolates (day −538 and −440) through day 162, indicating a long-term airway reservoir. An intermediate lineage (Clade 2), present immediately before phage administration (day −5), was not recovered after only 3 days of co-therapy, consistent with phage-mediated clearance of highly susceptible variants. This lineage was less genetically tractable, lacking adaptations found in persistent clades, for instance, in acetate metabolism (acoA)12 (Fig. 4b, Supplementary Data 1). A new short-lived lineage (Clade 3) appeared on day 14 as a phage-sensitive subpopulation (Fig. 3d) but was not recovered in later sampling (Fig. 4a). All three isolates within this clade were found to have excised 1 of 3 longitudinally intact prophages35 from their genomes (Fig. 4b, Table S8), likely the result of overwhelming co-treatment stress which drove reduced isolate pathogenicity. In contrast, the long-persistent Clade 4 lineage, present in baseline isolates (days −370 and 0), diversified quickly during therapy to generate P1A-resistant descendants that dominated for more than 100 days (Fig. 4a). Although all nonmucoid variants carried mutL mutations characteristic of hypermutators33,36, only Clade 4 acquired resistance, indicating that elevated mutation rate alone did not confer escape. The turnover of lineages within the stable ST244 background demonstrates that phage pressure, rather than antibiotics, was the dominant evolutionary driver reshaping the nonmucoid infection.

a Phylogenetic tree of 22 nonmucoid accessory genomes. Isolates were outgrouped by ancestral isolate –538a and grouped into four clades. b Circular plots of variants mapped to the –538a reference genome, ordered chronologically and colored by clade in (a). Red bars indicate nonsynonymous mutations (single-nucleotide polymorphisms [SNP]. insertions [INS], and deletions [DEL] in lipopolysaccharide (LPS) biosynthesis, envelope, and metabolic genes. The five outermost rings denote P1A-resistant isolates. c Boxplots comparing planktonic growth of Clade 4 nonmucoids in phage-free media. Boxes represent interquartile ranges of area under the growth curve (AUC), lines mark medians, whiskers denote minima and maxima, and individual replicates (n = 4) are shown as points. One-way ANOVA with Tukey multiple comparisons of AUC were performed. ****denotes adjusted p < 0.0001. d Phylogenetic tree of nine mucoid accessory genomes, outgrouped by ancestral strain –440b and separated into three clades. e Circular plots of variants mapped to the –440b genome, arranged chronologically and color-coded by clade from (d). Mutations predominantly occurred in efflux transporters, outer membrane, and alginate synthesis genes. f Schematic summarizing lineage trajectories across relative resistance and fitness space. Each bubble represents a clade, scaled by isolate density and colored as in panels (a, d). Phage-driven evolution of nonmucoids (Clades 2–4) increased resistance at a cost to fitness, whereas mucoid lineages remained stable within the low-risk, antibiotic-susceptible range. Together, these pressures shifted the population toward a lower-risk, chronic configuration. For both trees, scale bar = 0.1 nucleotide substitutions per site; bootstrap values > 0.2 are indicated at nodes.
Resistance in the phage-susceptible Clade 4 lineage arose through remodeling of the lipopolysaccharide (LPS) structure, consistent with altered P1A binding rather than receptor loss. Mutations clustered within core LPS biosynthetic and modification genes, including rfe, wbpA, wbpG, ugd, galU, lpxP, and mlaA/mlaE (Fig. 4b; Supplementary Data 1), consistent with stepwise modification of surface molecules involved in P1A attachment37. These resistant variants exhibited a hyperpigmented phenotype characteristic of pyomelanin overproduction (Fig. S6) but lacked large chromosomal deletions typically associated with this trait37,38. In addition, genome-wide variant mapping showed that P1A-resistant isolates accumulated ~2% fewer mutations than phage-sensitive counterparts (799 vs. 832 per genome; Supplementary Data 1), suggesting that phage pressure contributed specific, LPS-related modification while host factors conferred numerous other deleterious mutations characteristic of chronic CF colonization39. Additional substitutions in pchE, pchF, and pchR indicate compensatory shifts in iron metabolism and redox balance, likely stabilizing envelope function under phage stress (Fig. 4b; Supplementary Data 1). Recurrent mutations in rfe, ugd, and pchR across independent isolates support convergent selection for coordinated remodeling of surface and metabolic networks.
These molecular changes carried a measurable fitness cost. P1A-resistant isolates grew 25–40% slower than their ancestral, phage-susceptible counterparts (Fig. 4c), reflecting the potential energetic burden of cell surface remodeling and redox compensation12. Yet these variants persisted through therapy and recovery, suggesting that phage pressure favored a slow-growing, stress-tolerant phenotype over complete clearance. Together, pyomelanin overproduction, envelope stabilization, and reduced antibiotic susceptibility define a persistence-adapted state. This aligns with prior reports that phage resistance in P. aeruginosa often emerges via diffuse remodeling of cell-surface architecture rather than receptor loss, as large deletions impose prohibitive fitness costs10,40,41. P1A resistance in Clade 4, therefore, represents an energetically expensive but stabilizing adaptation, which shifted P. aeruginosa toward a chronic, therapy-refractory phenotype.
Whereas nonmucoid Clade 4 variants adapted through LPS remodeling, the coexisting mucoid population evolved along a distinct ecological path. The earliest lineage persisted from day −440 through day 29, accumulating accessory gene mutations in mexF (efflux pump), nodT (outer membrane), and proP (major facilitator superfamily (MFS) transporters) consistent with long-term adaptation to antibiotic stress (Fig. 4d, Table S9). A second lineage, represented by isolates from days −370, −321, 11, and 15, captured transitional variants arising under sustained therapy (proP, nodT) and intermittent phage exposure (wzz, LPS synthesis). In contrast, a third lineage appeared only after the onset of coadministration and expanded during recovery (day 29 isolate). The post-treatment dominance of this lineage suggests that co-therapy reshaped the airway environment to favor pre-existing mucoid variants tolerant to both antibiotics (proP, nodT, emrA, araJ, adeB relating to efflux transporters) and phages (algE, alginate production). Despite turnover among mucoid lineages, antibiotic susceptibility remained unchanged (Table S2), indicating that reduced treatment effectiveness reflected population restructuring rather than acquisition of new resistance determinants. Together, these divergent evolutionary paths show that phage pressure did not simply select for resistance but redirected bacterial fitness toward low growth and stress-adapted states. Figure 4f presents a temporal schematic of infection reversion, showing reduction of the aggressive MDR nonmucoid lineage and reoccupation of the airway by residual mucoids at lower density.
Divergent fates of therapeutic phages in the CF airway
Rising Pseudomonas density before treatment indicated microbiome instability in the airway, suggesting that fluctuations in viral abundance could reveal how endogenous phages respond to pathogen overgrowth and influence therapeutic outcomes. Between days –321 and –69, Litunavirus members declined from 84 to 57% of viral reads, while Pakpunavirus and Pbunavirus increased from 5 to 23% and 8 to 15%, respectively (Fig. 5a; Table S6). This redistribution among Pseudomonas-infecting phages paralleled the rise in bacterial host density preceding exacerbation (Fig. 3b). Because the therapeutic phages PYO2 and P1A belonged to these same genera (99% nucleotide identity to Litunavirus and ~30% to Pakpunavirus; Table S10), treatment effectively introduced close relatives of pre-existing airway phages. Within three days of intravenous dosing, total viral reads surged and the airway virome collapsed to near-complete Pakpunavirus dominance (>98%), indicating rapid airway replication of viable P1A virions (Fig. 5a; Table S6 and S11). In contrast, Litunavirus became scarce, consistent with PYO2 failing to reach the airways and adequately amplify despite repeated dosing (Tables S6, S11).

a Stacked bar plots showing the relative abundance of Pseudomonas-infecting and other viral taxa in sputum before, during, and after co-therapy. Therapeutic phages P1A (Pakpunavirus; blue) and PYO2 (Litunavirus; red) are highlighted alongside endogenous airway phage genera (Pbunavirus, Phikzvirus, Phikmvvirus, and others). Viral taxa comprising <1% of total reads are grouped as “other” (black). b, c Temporal dynamics of therapeutic phages in sputum. b Host-normalized absolute abundance of P1A and PYO2. P1A abundance increased sharply by day 3 to ~141 virions per human cell, consistent with in situ replication. c Phage-to-bacterium ratio (multiplicity of infection, MOI) in sputum. Despite a ~10-fold reduction in P1A reads after day 3, concurrent bacterial decline maintained a mean MOI near 0.8 throughout co-therapy. d, e Serum evidence of phage-specific immune responses. d In vitro phage neutralization, measured as efficiency of plating (EOP). Neutralization increased over the course of treatment, resulting in near-complete loss of both phages by two weeks. Data are presented as mean ± SD (n = 4). One-way ANOVA with Dunnett’s multiple comparisons to day 0. Asterisks denote adjusted p values (*<0.05; ** <0.01). e Anti-phage IgM responses measured by ELISA. IgM against PYO2 rose rapidly and remained elevated, consistent with pre-existing cross-reactive immunity, whereas anti-P1A IgM appeared later and increased gradually. Data are presented as mean ± SD (n = 3). One-way ANOVA with Dunnett multiple comparisons to day 0. Asterisks denote adjusted p values (*< 0.05; ** <0.01; *** <0.001). f, g Hematologic indicators of systemic inflammation during treatment. f White blood cell (WBC) counts and g platelet counts measured from whole blood. Dashed lines denote baseline means (WBC = 5,375 ± 1,250 cells µL−1; platelets = 2.18 × 105 ± 1.02 × 104 cells µL−1). Mild transient increases correspond to the onset of anti-phage IgM induction.
The Pakpunavirus bloom coincided with the sharp decline in nonmucoid Pseudomonas, consistent with P1A-driven clearance dynamics (Fig. 3c). This dominance reflected strong lytic pressure exerted by P1A during the early high-burden phase of treatment, climbing to ~9.2 × 10⁶ PFU mL−1 of sputum and roughly 141 virions per human cell, levels exceeding those achievable by intravenous delivery alone (Fig. 5b). As bacterial density was cleared, P1A abundance also decreased because fewer susceptible hosts were available to sustain self-replication. After achieving a multiplicity of infection (MOI) of approximately 0.8 within the first few days and persisting through day 29, self-dosing phage activity could constrain the residual nonmucoid population even after treatment ceased (Fig. 5c).
After nine days of dosing, Pakpunavirus declined to 14% of viral reads, while Litunavirus reappeared (~4%), and endogenous Pbunavirus expanded from 1 to 78% (Fig. 5a; Table S6). This shift marked a turnover in phage dominance as the airways stabilized under treatment. The rise of Pbunavirus, whose representative phage E215 was more active against mucoids than either therapeutic phage (Fig. S5), likely tracked the growing mucoid population (Fig. 3c). Late in therapy, additional Pseudomonas-associated phages from the Phikzvirus, Bruynoghevirus, and Phikmvvirus genera appeared, indicating further diversification of the airway virome. By the end of treatment and in the following weeks, Pakpunavirus partially rebounded (~40% of viral reads), Pbunavirus declined (~68% to 37%), and Litunavirus modestly increased (~2% to 7%), while the minor phages together accounted for ~9% (Fig. 5a; Table S6). These transitions show that the post-therapy airway virome became more heterogeneous as previously undetected Pseudomonas phages re-emerged from the endogenous background, while the bacterial community remained overwhelmingly Pseudomonas (Fig. 3a). This divergence reflects the narrow host ranges and variable sensitivities of coexisting Pseudomonas variants, allowing different phages to expand on residual subpopulations and yielding a more heterogeneous virome and a new ecological steady state after treatment.
Virome-primed innate humoral immunity restricts phage activity before bacterial adaptation
Phages initially reduced nonmucoid Pseudomonas populations, but titers either failed to amplify or declined after an early peak even as bacterial abundance and susceptibility remained stable (Figs. 5a–c, 3c–d). These dynamics indicate that host clearance mechanisms, rather than bacterial resistance, underpinned the mid-treatment attenuation of phage activity. By day 29, circulating infectivity, measured by testing patient serum against the therapeutic phages, had declined to near the detection threshold (efficiency of plating ≈ 0.02 for P1A; 0.006 for PYO2), indicating near-loss of systemic activity (Fig. 5d). Among the two therapeutic phages, PYO2 exhibited the earliest and steepest decline in activity. Infectivity decreased more than threefold within the first week and remained almost undetectable thereafter (Fig. 5d). This rapid loss coincided with a tenfold rise in anti-PYO2 IgM by day 7 that persisted through day 29, while IgG remained at baseline (Figs. 5e, S7). The timing and magnitude of this response indicate that PYO2 was neutralized almost immediately by pre-existing cross-reactive antibodies, likely acquired through prior environmental exposure to related Litunavirus members. Having a pre-existing humoral immunity, which intensified over time, likely dampened in situ replication of PYO2 (Fig. 5c) despite the continued presence of susceptible Pseudomonas, thereby shortening its therapeutic window.
By contrast, P1A showed a slower rate of decline consistent with a delayed, de novo immune response. Circulating infectivity remained high throughout the first treatment week, corresponding to strong airway amplification (Fig. 5d) and a three-log reduction of nonmucoid Pseudomonas (Fig. 3c). A modest reduction in circulating activity appeared by day 7, about 15% below baseline, followed by a pronounced decline by day 13 and a 93% loss by day 15 (Fig. 5d). This coincided with the first measurable rise in anti-P1A IgM, which increased fourfold by day 15 and reached about sevenfold above baseline by day 29, while IgG remained low and nonspecific (Figs. 5e, S7). The delayed onset and progressive rise in IgM indicate that P1A was initially tolerated but gradually neutralized as a primary antibody response developed. During the first phase of therapy, P1A expanded quickly but then declined as humoral responses emerged, consistent with antibody-mediated neutralization of circulating and airway P1A infectivity (Fig. 5a–c).
Consistent with systemic immune activation42, white blood cell and platelet counts rose modestly during the first week of coadministration (WBC 6.7 × 103 to 7.9 × 103 cells mL−1; platelets 2.16 × 105 to 2.34 × 105 cells mL−1; Fig. 5f–g; Table S4). These hematologic changes coincided with the emergence of anti-phage IgM (Fig. 5e) and occurred while lung function continued to improve (Fig. 1d), indicating transient immune activation during phage exposure without evidence of inflammatory adverse events. However, host immunity also constrained phage activity throughout the co-therapy phase, with the timing and magnitude of these responses defining the therapeutic window and endpoint of treatment.
Discussion
Our findings provide mechanistic insight into coordinated phage–antibiotic treatment within the heterogeneous CF airway. Chronic P. aeruginosa infection in CF epitomizes the paradox of disease control without cure, whereby antibiotics and immunity suppress pathogen outgrowth yet entrench its persistence. Phage therapy breaks this stalemate by introducing self-renewing agents that parasitize bacterial subpopulations beyond antibiotic reach. Whether phages can truly function as biological therapeutics within complex host environments has remained an open question. Achieving sufficient delivery, amplification, and persistence despite bacterial resistance and immune constraints has been a key challenge43. In this study, intravenous phage–antibiotic coadministration fulfilled these criteria, producing rapid functional recovery accompanied by obstruction clearance and bacterial reduction following last-resort antibiotic failure in a patient with advanced CF. Co-therapy reset the infection from acute-on-chronic towards its pre-exacerbation equilibrium, consistent with other compassionate-use cases in CF44,45,46,47. The improvement was mechanistically linked to in situ phage replication that preyed upon the proximal driver of the exacerbation, a nonmucoid MDR lineage, highlighting phages as active therapeutic agents rather than passive antibiotic adjuncts in the diseased lung. This shows the capacity of phages to function as self-replicating biologics within the human airway, achieving targeted ecological reorganization even under conditions of advanced disease and immune constraint. Yet the durability of this response was dampened by emerging immune containment and bacterial host resistance, underscoring that efficacy during chronic infection depends as much on immunological and ecological context as on phage–bacteria compatibility. The following sections outline how ecological, immunologic, and evolutionary interactions shape system-level control under phage–antibiotic coadministration.
Clinical recovery arose from reorganization of the infection ecosystem rather than additive antimicrobial effects. Ciprofloxacin suppressed the dominant mucoid population, relieving obstruction and inflammation, but permitting competitive release of an aggressive MDR nonmucoid lineage that expanded beneath apparent improvement. In situ phage replication imposed localized selective pressure on this emergent population, driving its collapse by roughly an order of magnitude and initiating ecological steering that remodeled the infection community41,48,49,50. The infection then stabilized at a lower, host-contained burden, characterized by reduced growth potential and attenuated virulence. Though compositionally altered, this renewed equilibrium was functionally analogous to the stability preceding the exacerbation. Temporal alignment of phage amplification, MDR collapse, and physiological recovery indicates that phage activity, not cumulative exposure to drug and phages, was the principal driver of improvement44,51,52. The parallel emergence of anti-phage antibodies, bacterial resistance, and expansion of endogenous airway phages demonstrates that host, viral, and bacterial adaptations together reinforced the ecological reset, yielding a transient but self-limiting equilibrium compatible with clinical stability. This stabilization likely reflected not only microbial reorganization but also partial recalibration of the host inflammatory milieu, consistent with the functional recovery observed clinically.
As Pseudomonas density declined and obstruction eased, the CF lung transitioned from therapeutic dependence to physiological self-governance. Sustained pulmonary gains, together with stable hematologic indices apart from a transient rise in leukocytes and platelets, reflected coordinated resolution of infection and inflammation rather than simple suppression. Co-therapy suppressed hyperinflammatory activity and allowed the system to re-equilibrate around the reduced mucoid Pseudomonas burden that could be maintained without further intervention. Mechanistically, this restorative phase represents the terminal stage of immunophage synergy53. The sharp contraction of the acute MDR nonmucoid lineage and consolidation of the chronic mucoid population marked the ecological transition toward stability. Transient airway diversification during recovery further indicates that phage activity drove the infection below the threshold needed to sustain inflammatory cycling, reopening a critical window for host defense recalibration14,20,54. The immune system then reinforced this reorganization by containing both residual mucoid and nonmucoid subpopulations, consistent with the plateau in P1A titers and stable pulmonary indices during follow-up. This sequence of phage-driven reduction, ecological rebalancing, and immune consolidation completed an externally initiated yet internally stabilized infection control within the CF airway. Recovery, therefore, depended less on bacterial eradication and more on coordinated activity among drugs, phages, and host immunity to restore ecological and physiological balance.
This biphasic, spatially segregated response defines a therapeutic framework we term chemobiotherapy, in which chemical and biological agents act through coordinated mechanisms to address independent bacterial subpopulations and navigate host-mediated efficacy limits. Conventional combination therapies built on pharmacologic synergy or adjuvancy are constrained by additive logic, boosting potency but frequently failing to address the residual ecological, spatial, and immunologic features of chronic, therapy-refractory infection55,56. These strategies continue to treat infection as a fixed target rather than an evolving system shaped by microbial dynamics, host responses, and ecological feedback. Chemobiotherapy instead assigns drugs and phages distinct, nonredundant roles across compartments of the infection ecosystem. Chemical agents provide rapid, broad, and predictable clearance for immediate stabilization, while phages deliver self-amplifying, niche-penetration, host-responsive control that maintains selective pressure as bacterial populations fluctuate within the ecosystem3,10,39,41. The goal is mechanistic complementarity rather than simple synergy, integrating distinct therapeutic actions to overcome modality-specific constraints and reestablish physiological homeostasis. Comparable frameworks have emerged in oncology, where chemotherapeutic and immune biologics act coordinately to reprogram tumor microenvironments, converting cytoreduction into durable immune control and functional recovery57,58,59. Taken together, chemobiotherapy represents a conceptual framework derived from this case that redirects therapy from isolated targets to the infection ecosystem and explains how durable control can emerge through coordinated reprogramming of the environment in favor of the host.
The durability of chemobiotherapy in this case hinged on its negotiation with host immunity. Though neutralizing antibody responses in phage therapy are typically delayed (for two to six weeks) and dominated by IgG45,60,61,62, this patient exhibited atypical kinetics. Neutralization occurred within days, was mediated exclusively by IgM, and coincided with loss of phage activity. Although phages were administered intravenously, they likely reached the airway through inflammatory transudation and increased vascular permeability characteristic of advanced CF63. The near-immediate loss of PYO2 therefore suggests recognition by a pre-existing, T cell–independent IgM pool localized to the chronically infected lung. This cross-reactive antibody population was likely imprinted by the patient’s own virome through continuous exposure to related Litunavirus members. It may function as a natural buffer that regulates phage antigen load and preserves mucosal immune homeostasis64,65. Conversely, the collapse of P1A after its peak amplification coincided with the emergence of de novo IgM, indicating that robust phage replication provided sufficient antigenic stimulus to elicit a primary humoral response. This self-limiting dynamic, in which intense replication drives IgM induction and subsequent neutralization, likely defines the temporal boundary of effective therapy and underscores that efficacy depends on balancing replication with immune containment. Crucially, the absence of IgG class switching likely reflects the combined effects of short antigen persistence, due to rapid phage clearance, and the CF-associated impairments in CD4⁺ T-cell and B-cell maturation required for IgG production13,65,66.
While acknowledging the limited causal inference inherent to a single-patient compassionate-use case and the constraints on assessing long-term outcomes and cellular immunity46,61, this study provides a multiomic view of phage–bacterium–host dynamics in an acute-on-chronic airway infection. Coherence across clinical, ecological, and immune trajectories supports phage-driven reorganization of the infection ecosystem and provides mechanistic insight into system-level therapeutic processes. Although these findings define key principles in which the virome functions as an immunologic gatekeeper and therapy acts through coordinated ecological and immune reorganization, further validation in multi-patient studies will be required to confirm and generalize this framework. Nevertheless, the emerging concepts of chemobiotherapy and immunophage synergy offer a systems logic that may extend beyond P. aeruginosa and CF. Many chronic or relapsing infections, such as Staphylococcus aureus osteomyelitis, Mycobacterium abscessus lung disease, biofilm-associated wounds, and enteric infections including Salmonella and Clostridioides difficile, exhibit ecological partitioning that separates bacterial subpopulations by niche or phenotype67,68,69. In these settings, chemical and biological agents could act across complementary compartments to achieve more durable resolutions. Defining how timing, dose, and immune modulation shape these cooperative dynamics will be central to translating chemobiotherapy and immunophage synergy from mechanistic principles to therapeutic practice.
In conclusion, this case illustrates how phage–antibiotic treatment succeeded by reorganizing infection ecology under stringent clinical and biological constraints. Despite advanced disease, MDR, systemic delivery, and rapid immune containment, coordinated phage replication and bacterial adaptation reorganized the infection into a stable, host-contained state. More broadly, this study supports the principle that durable efficacy in chronic infection depends not on maximal antimicrobial potency, but on synchronizing delivery, replication, and immune tolerance within the patient’s ecological and immunologic landscape. Together, these findings establish a systems-level principle that can inform the rational design and translation of phage-based therapeutics for chronic infection.
Methods
Ethics statement
Written informed consent was obtained from the patient. The intervention was provided under a compassionate use framework and approved by the U.S. Food and Drug Administration and the Institutional Review Board of the University of California, San Diego (IRB #802083). The study was conducted in accordance with the principles of the Declaration of Helsinki and complied with the CARE guidelines for reporting clinical cases.
Patient health metrics and pulmonary function tests
Pulmonary function and hematology tests were performed at the University of California, San Diego Medical Hospital. Clinical microbiology, hematology, and radiographic imaging were conducted as part of routine care and follow-up. At each visit, height, weight, body mass index, urine, and spirometry were recorded. Spirometry was performed according to American Thoracic Society guidelines and normalized using the Hankinson equations70. Forced expiratory volume in one second (FEV₁), forced vital capacity (FVC), and maximum mid-expiratory flow (MMEF) were measured, and percentage predicted values (ppFEV₁ and ppFVC) were calculated as described previously71. Quantitative computed tomography (CT) was performed at UC San Diego Medical Hospital, and % emphysema, % air trapping, inspiratory volume, and expiratory volume were quantified using an automated CT algorithm23. Urine samples were analyzed for creatinine concentration.
Bacterial strains and isolate collection
Pseudomonas aeruginosa laboratory strains PAK (CP020659.1) and PAO1 (NC_002516.2) were used to propagate phages P1A and PYO2, respectively. Laboratory strains were cultured overnight (18–24 h) in Luria broth (LB) at 37 °C with shaking. Clinical P. aeruginosa isolates were obtained from patient sputum samples using selective cetrimide agar and were subsequently streak-purified to isolate single colony morphologies. Patient bacterial isolates were then grown in LB at 37 °C with shaking for in vitro sensitivity testing and DNA isolation. Isolates are named for their isolation date, relative to the first day of phage treatment (day 0). For long-term storage, isolates were grown to OD600 > 0.5, mixed with 50% glycerol (final, 25% v/v) and frozen at −80 °C.
Phage strains and clinical production
Phages P1A and PYO2 were sequenced and confirmed not to contain known toxins, harmful and antibiotic resistance genes, as well as lysogeny-related mechanisms. Phage production was performed as previously described by Luong et al.16. In brief, each phage’s enrichment was performed by inoculating bacterial cultures with an MOI of 0.01 to yield an overnight (18–24 h) 3 L lysate. The amplified phages were sterilized via double centrifugation and filtration at cutoffs of 0.8, 0.45, and 0.22 µm. The 3 L lysate was concentrated using a 100 kDa Vivaflow crossflow filtration cassette (Sartorius), further removing endotoxins, exotoxins, and other potential contaminants, and then diafiltered with sterile cold phosphate buffer saline (PBS) before final concentration to >1011 PFU mL−1. Intact phage particles were further separated with cesium chloride (CsCl) density-gradient ultracentrifugation at 100,000 × g for 24 h, followed by 3 rounds of dialysis in fresh cold PBS to remove CsCl. Final LPS scrubbing was performed with Pierce™ High-Capacity Endotoxin Removal Spin Columns.
Phage quantification & susceptibility testing
Phage concentrations were monitored throughout the purification process and for in vitro experiments using the spot titering method16. For each sample, tenfold serial dilutions of phage stock were prepared in a microplate and spotted in triplicate onto bacterial-seeded agar plates. Spots were allowed to fully dry before plates were incubated for 18–24 h at 37 °C. Quantification was performed immediately after incubation. For susceptibility testing, 4 µL of phage concentrates (diluted to 108 and 106 PFU mL−1) were spotted onto bacterial-seeded agar plates. After allowing spots to dry for approximately 15 min, the plates were incubated at 37 °C for 24 h. The presence or absence of plaques was recorded for each bacterial isolate.
Phage master stocks and preparation of therapeutic doses
Phage master stocks were quantified by spot titration, yielding 5.88 × 1011 PFU mL−1 and 453 endotoxin units (EU) mL−1 for P1A, and 5.81 × 1011 PFU mL−1 and 8920 EU mL−1 for PYO2. These values reflect concentrated production stocks. Phage identity was confirmed by PCR using strain-specific primers (5′–3′): P1A forward, TCTCTACCCTCTGGCCTTCC; P1A reverse, CCGTAATCCCGTAGCCACTC; PYO2 forward, GCACCAAGAACAAGGTGCAG; and PYO2 reverse, GTTCCGGTTGCTTGGTTTCC. The two phages were combined to create therapeutic doses containing 2 × 108 PFU total (1 × 108 PFU per strain) in 1 mL of pyrogen-free Dulbecco’s phosphate-buffered saline (dPBS). Final therapeutic doses were diluted, processed, and release-tested to meet USP 85 endotoxin limits ( < 1 EU mL−1 kg−1 body weight). The solution was 0.22 µm filtered, sterile fill-finished into single-dose sealed glass vials, stored protected from UV light, and transported at 4 °C.
Sterility and endotoxin testing
FOCUS Laboratories (Allentown, PA, USA) confirmed that each phage production batch met sterility standards (USP 71) containing no microbes and determined (USP 85) that each phage dose contained <1 EU mL−1 kg−1 of patient body weight.
Phage cytotoxicity testing
Phage cytotoxicity was evaluated using the CellTiter-Glo® 2.0 Assay (Promega) according to the manufacturer’s protocol and as previously described16. A549 lung epithelial cells (ATCC CCL-185) were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco) supplemented with 10% fetal bovine serum at 37 °C in 5% CO₂. Cells were seeded at approximately 2 × 104 per well in 96-well plates and incubated for 24 h. Purified phage stocks were diluted to 10⁹ PFU mL−1 and added at a phage-to-cell ratio of approximately 1000. Sodium dodecyl sulfate (1%) and phosphate-buffered saline (PBS) served as positive and negative controls, respectively. After 24 h incubation, CellTiter-Glo reagent was added, and plates were mixed for 2 min, followed by a 10 min incubation at 22 °C. Luminescence was measured using a BMG Labtech microplate reader.
Automated antibiotic susceptibility testing
Sputa sample colonies were grown and tested for susceptibility by the UCSD Clinical Microbiology using VITEK® 2 (BioMérieux) for most antibiotics and disk diffusion for ceftolozane-tazobactam. MIC cutoffs were based on CLSI breakpoints (2022). Additional ciprofloxacin sensitivity testing of isolated nonmucoid and mucoid isolates from patient sputa was tested in liquid culture at a range of 16 µg mL−1 to 0.03 µg mL−1. MIC was determined by the lowest concentration with no bacterial growth, indicated by an OD600 < 0.3.
Sputum collection and DNA extraction
Spontaneously expectorated sputum was collected in a sterile container and briefly stored at 4 °C until DNA extraction. The freeze-boil method described by Silveira et al.72 was used for metagenomic DNA extraction. In short, homogenized sputum was stored in a temperature-resistant cryovial and subjected to five freeze-boil cycles (5 min at –80 °C and 100 °C, respectively). After liquefaction, the sample was centrifuged to remove bulk cell debris, and the supernatant was processed using the NucleoSpin Microbial DNA Isolation Kit (Machery-Nagel) according to the manufacturer’s protocol.
Sequencing, metagenomic taxonomic classification, and metagenomic abundances
Metagenomic DNA libraries were prepared by SeqCenter (Pittsburgh, PA, USA) using the Nextera DNA Flex kit (Illumina) and sequenced on an Illumina NextSeq 550 platform to generate paired-end reads. Reads were quality filtered using fastp (v.0.19.4)73 and taxonomically classified with Kraken2 (v.2.1.3)74 under default parameters. Classification results were visualized in Pavian (v.1.0)75 to quantify classified and unclassified reads per taxon. Relative abundance was calculated as the proportion of reads assigned to each taxon among all classified reads. Absolute bacterial abundance was estimated following Pust et al.25, by scaling the diploid human genome length to millions, multiplying by normalized bacterial read counts (per million reference base pairs), and dividing by the human read count. For phage quantification, reads were mapped to the P1A and PYO2 genomes using bbmap (v.35.85) (minid = 0.95)76, and species-specific read counts were normalized to phage genome length and divided by the human read count. In situ multiplicity of infection (MOI) was calculated as the ratio of P1A absolute abundance to that of Pseudomonas reads at each time point.
Data standardization and principal component analysis
Variables including FEV1, FVC, FEV1/FVC, ppFVC, ppFEV1, MMEF, creatinine, WBC, and platelets were standardized to have a zero mean and unit variance in preparation for principal component analysis (PCA). PCA was conducted using the prcomp function with centering and scaling to reduce the dataset to principal components that capture the most important patterns and variance. The first two principal components were visualized using the ggplot2 package (v.3.4.3). Hierarchical clustering analysis (HCA) was performed using the hclust function, based on a distance matrix derived from PCA scores. Ward’s method (ward.D2) was applied to minimize within-cluster variance by squaring dissimilarities before updating clusters77. All PCA and dendrogram functions were sourced from the R stats package (v.3.6.2).
Quantification of total microbial load
Sputum samples were archived with 50% glycerol and stored at −80°C until colony quantification. Archival sputum glycerol stocks were scraped to obtain 50 μL samples, which were then serially diluted in sterile dPBS. After dilution, 650 μL of each dilution series was spread onto selective cetrimide agar plates and incubated at 37 °C for 24 h. Total CFU’s were counted and categorized into nonmucoid and mucoid phenotypes. Plating was repeated in triplicate for each sputum sample (n = 3).
Pangenome phylogenies
After quality control, high-quality reads were de novo assembled into contigs using SPAdes (v.4.0.0)78 with default parameters. Assemblies were annotated with Prokka (v.1.14.5)79 using P. aeruginosa PAO1 (NC_002516.2) as the reference for gene prediction and functional annotation. Sequences were separated by mucoid and nonmucoid phenotype for subpopulation core genome alignments using Roary (v.3.11.2)80. Two phylogenies were generated: (i) a core genome tree based on multiple sequence alignment of 49 conserved genes defined by clusters of orthologous groups (COG) families, and (ii) an accessory genome tree derived from binary gene presence–absence comparisons. Maximum-likelihood phylogenies were inferred with FastTree2 (v.2.2.0)81 using a generalized time-reversible (GTR) model and 1000 iterations. The oldest available ST244 clinical isolate (PABL047; RefSeq GCF_003411805.2) served as the outgroup for core genome analysis, while the earliest patient isolates (−538a, nonmucoid; −440b, mucoid) were used as outgroups for accessory genome alignments. Trees were visualized with iTOL (Interactive Tree of Life, v.6.0.0)82.
Multilocus sequence typing (MLST) analysis
FASTA files of each assembled isolate were single sequence queried against the PubMLST Pseudomonas aeruginosa typing database against all loci (acsA, aroE, guaA, mutL, nuoD, ppsA, trpE)83. Isolated data were recorded, and the database was accessed to find additional information about the ST244, ST4739, and ST4740 profiles.
Single-nucleotide polymorphism (SNP) analysis using breseq
For the first isolated nonmucoid (–538a) and mucoid (–440b) strains, cleaned reads were further processed and de novo assembled using SpAdes (v. 4.0.0), and the genome for PAO1 (NC_002516.2) was used for reference-guided scaffolding using RagTag (v.2.1.0). The final polished assembly was annotated using bakta (v.1.11.4; database V5.1_2024-01-19)84. Variant analysis was performed using breseq (v.0.39.0). Cleaned forward reads for the remaining 21 nonmucoid and 8 mucoid isolates (as processed by fastp) were run in the breseq pipeline against their respective root isolate using default settings for all analyses. All detected SNPs were retained for downstream analysis. The resulting genome difference (.gd) files were then filtered to remove synonymous mutations using the gdtools utility program. Biopython (v.1.78) and Matplotlib (v.3.10.0) were used to visualize selected variant distributions along the circular reference genome, SNPs were plotted by genomic position, with annotated coding regions overlaid to highlight variant localization within gene boundaries.
Prophage prediction and excision analysis in clinical isolates
For the first root isolates (–538a and –440b), prophage prediction was performed using Phaster85 with default settings. Concatenated FASTA files were used from their polished RagTag assemblies. Breseq outputs were scanned for deletions in their chromosomal positions relative to either mucoid or nonmucoid roots to determine excision from isolates. No prophage excision was detected in mucoid isolates (Fig. 4e), and excision from nonmucoid isolates is as indicated in Fig. 4b.
Growth kinetics
Nonmucoid isolates from clade 4 lineage were grown overnight in LB at 37 °C. Replicates (n = 4) were initiated from independent CFUs, pelleted, and resuspended in sterile Milli-Q water. In a 96-well microplate, 2 × LB and 2 × 106 CFU were added. The plate was incubated at 37 °C with orbital shaking in a microplate reader (BMG Labtech) for 24 h, with absorbance taken every 6 min. Bacterial densities were verified using the spot method. Area under the curve was calculated by fitting OD600 data using the Growthcurver R package (v.0.3.1)86.
Phage strain mapping and read summarization to metagenomes
Specific mapping of therapeutic phages P1A and PYO2 to fastp trimmed metagenome reads was performed using bbmap (v.35.85)76. A minimum alignment identity cut-off of 0.95 was used (minid = 0.95). Reads aligned from bbmap were further parsed through featureCounts (v.2.1.1)87 to assign aligned reads to P1A and PYO2 genomic features. Both P1A and PYO2 were re-annotated using bakta (v.1.11.4; database V5.1_2024-01-19) for updated gene assignments. Classified reads were further manually assigned based on structural or functional groups, with non-canonical phage genes designated as “other”.
Serum preparation and neutralization testing
Phages P1A and PYO2 were diluted to 1 × 106 PFU mL−1 in sterile dPBS. Collected serum was diluted in sterile dPBS to achieve a final concentration of 10%. Phages were then added to the serum to reach a final concentration of 1 × 105 PFU mL−1. The phage-serum mixture was incubated at room temperature for 24 h. After incubation, phage concentrations were determined by serial dilution and plating onto their respective isolation host strains using the spot plaque titer assay. Plates were incubated overnight at 37 °C, and plaques were counted to quantify phage recovery. Efficiency of plating (EOP) is calculated relative to the titer after 24 h incubation with pre-treatment sera (day 0, 100%) for each phage.
Enzyme-linked immunosorbent assay (ELISA)
Sera were screened for anti-phage IgG and IgM using an indirect ELISA adapted from Dedrick et al.61. Archival sera were aliquoted upon receipt, stored at −80 °C, and thawed once for assay. Enzyme immunoassay plates (Corning) were coated with either sterile carbonate–bicarbonate buffer (Sigma) or purified phages (P1A or PYO2, 1 × 108 PFU per well) and incubated for 24 h at 4 °C. Plates were washed five times with PBST (Corning dPBS + 0.05% Tween-20), blocked with PBST + 3% milk, and incubated for 20 h at 4 °C. Serially diluted patient sera (10−2 – 10−7) were added and incubated for 20 h at 4 °C. After washing, secondary antibodies (goat anti-human IgG Fc-HRP from ThermoFisher Cat# A18823 Lot# 98-106-082423; or goat anti-human IgM heavy chain-HRP from ThermoFisher Cat# A18841 Lot# 77-145-012422) were diluted 1:10,000 and incubated for 1 h in the dark at 21 °C. Plates were washed twice with PBST and three times with PBS before the addition of 3,3′,5,5′-tetramethylbenzidine (TMB; Sigma). After 8 min of incubation in the dark at 21 °C, the reaction was stopped with 0.16 M sulfuric acid. Absorbance was read at 450 nm (signal) and 570 nm (background) using a microplate reader. Results were plotted against serum dilution and fit to a logistic curve. Phage-coated wells were tested in triplicate, and non-phage controls in duplicate. For normalization of IgM intensity, OD₄₅₀ values from ‘no-phage’ controls were subtracted from corresponding phage-coated readings.
Statistics
Statistical analyses were performed, and graphs were generated using R Studio (3.6.2) and GraphPad Prism software (v.10.1.1). For principal component analysis, statistical analyses followed SAS standards.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Sequencing data generated in this study have been deposited in the NCBI Sequence Read Archive under accession code PRJNA1266762. Source data underlying all figures are provided with this paper as a Source Data file. Certain clinical metadata generated in this study are subject to access restrictions due to patient privacy protections and the terms of institutional ethical approval and informed consent. These data are therefore not publicly available. Access for academic research purposes may be granted upon reasonable request to the corresponding author, subject to institutional review board approval and applicable privacy regulations. Source data are provided with this paper.
References
Bell, S. C. et al. The future of cystic fibrosis care: a global perspective. Lancet Respiratory Med. 8, 65–124 (2020).
Malhotra, S., Limoli, D. H., English, A. E., Parsek, M. R. & Wozniak, D. J. Mixed communities of mucoid and nonmucoid Pseudomonas aeruginosa exhibit enhanced resistance to host antimicrobials. mBio 9, e00275–e00318 (2018).
Clark, S. T. et al. Phenotypic diversity within a Pseudomonas aeruginosa population infecting an adult with cystic fibrosis. Sci. Rep. 5, 10932 (2015).
Cramer, N., Klockgether, J. & Tümmler, B. Microevolution of Pseudomonas aeruginosa in the airways of people with cystic fibrosis. Curr. Opin. Immunol. 83, 102328 (2023).
Sousa, A. A. et al. Systematic optimization of prime editing for the efficient functional correction of CFTR F508del in human airway epithelial cells. Nat. Biomed. Eng. 9, 7–21 (2025).
Rowe, S. M. et al. Inhaled mRNA therapy for treatment of cystic fibrosis: Interim results of a randomized, double-blind, placebo-controlled phase 1/2 clinical study. J. Cyst. Fibros.: Off. J. Eur. Cyst. Fibros. Soc. 22, 656–664 (2023).
Nichols, D. P. et al. Pharmacologic improvement of CFTR function rapidly decreases sputum pathogen density, but lung infections generally persist. J. Clin. Invest. 133, e167957 (2023).
Usui, M., Yoshii, Y., Thiriet-Rupert, S., Ghigo, J.-M. & Beloin, C. Intermittent antibiotic treatment of bacterial biofilms favors the rapid evolution of resistance. Commun. Biol. 6, 275 (2023).
Kunisch, F. et al. Targeting Pseudomonas aeruginosa biofilm with an evolutionary trained bacteriophage cocktail exploiting phage resistance trade-offs. Nat. Commun. 15, 8572 (2024).
Betts, A., Gifford, D. R., MacLean, R. C. & King, K. C. Parasite diversity drives rapid host dynamics and evolution of resistance in a bacteria-phage system. Evolution 70, 969–978 (2016).
Weimann, A. et al. Evolution and host-specific adaptation of Pseudomonas aeruginosa. Science 385, eadi0908 (2024).
Pedersen, B. H. et al. Metabolic specialization drives reduced pathogenicity in Pseudomonas aeruginosa isolates from cystic fibrosis patients. PLoS Biol. 22, e3002781 (2024).
Hector, A. et al. Regulatory T-Cell impairment in cystic fibrosis patients with chronic Pseudomonas infection. Am. J. Respir. Crit. Care Med. 191, 914–923 (2015).
Fischer, B. M. et al. Increased expression of senescence markers in cystic fibrosis airways. Am. J. Physiol.-Lung Cell. Mol. Physiol. 304, L394–L400 (2013).
Qin, S. et al. Pseudomonas aeruginosa: pathogenesis, virulence factors, antibiotic resistance, interaction with host, technology advances and emerging therapeutics. Signal Transduct. Target. Ther. 7, 199 (2022).
Luong, T., Salabarria, A.-C., Edwards, R. A. & Roach, D. R. Standardized bacteriophage purification for personalized phage therapy. Nat. Protoc. 15, 2867–2890 (2020).
Champagne-Jorgensen, K., Luong, T., Darby, T. & Roach, D. R. Immunogenicity of bacteriophages. Trends Microbiol. 31, 1058–1071 (2023).
Lopes, A., Pereira, C. & Almeida, A. Sequential combined effect of phages and antibiotics on the inactivation of Escherichia coli. Microorganisms 6, 125 (2018).
Goss, C. H. et al. A randomized clinical trial of antimicrobial duration for cystic fibrosis pulmonary exacerbation treatment. Am. J. Respir. Crit. Care Med. 204, 1295–1305 (2021).
Waters, V. J. et al. Factors associated with response to treatment of pulmonary exacerbations in cystic fibrosis patients. J. Cyst. Fibros. 14, 755–762 (2015).
Liou, T. G. et al. Airway inflammation accelerates pulmonary exacerbations in cystic fibrosis. iScience 27, 108835 (2024).
Willmering, M. M. et al. Sensitive structural and functional measurements and 1-year pulmonary outcomes in pediatric cystic fibrosis. J. Cyst. Fibros. 20, 533–539 (2021).
Hasenstab, K. A. et al. Automated CT staging of chronic obstructive pulmonary disease severity for predicting disease progression and mortality with a deep learning convolutional neural network. Radiol. Cardiothorac. imaging 3, e200477 (2021).
Koo, M. C. et al. Quantitative computed tomography and visual emphysema scores: association with lung function decline. ERJ Open Res. 9, 00523–2022 (2023).
Pust, M.-M. et al. The human respiratory tract microbial community structures in healthy and cystic fibrosis infants. npj Biofilms Microbiomes 6, 61 (2020).
Pienkowska, K. et al. The cystic fibrosis upper and lower airway metagenome. Microbiol Spectr. 11, e0363322 (2023).
Moran Losada, P. et al. The cystic fibrosis lower airways microbial metagenome. ERJ Open Res. 2, 00096–2015 (2016).
Tang, G. et al. Metagenomic estimation of absolute bacterial biomass in the mammalian gut through host-derived read normalization. mSystems 10, e00984–00925 (2025).
Widder, S. et al. Microbial community organization designates distinct pulmonary exacerbation types and predicts treatment outcome in cystic fibrosis. Nat. Commun. 15, 4889 (2024).
Cramer, N. et al. Competitive fitness of Pseudomonas aeruginosa isolates in human and murine precision-cut lung slices. Front. Cell Infect. Microbiol. 12, 992214 (2022).
Roach, D. R., Sjaarda, D. R., Castle, A. J. & Svircev, A. M. Host exopolysaccharide quantity and composition impact Erwinia amylovora bacteriophage pathogenesis. Appl. Environ. Microbiol. 79, 3249–3256 (2013).
Flores-Vega, V. R., Partida-Sanchez, S., Ares, M. A., Ortiz-Navarrete, V. & Rosales-Reyes, R. High-risk Pseudomonas aeruginosa clones harboring β-lactamases: 2024 update. Heliyon 11, e41540 (2025).
Oliver, A. Mutators in cystic fibrosis chronic lung infection: prevalence, mechanisms, and consequences for antimicrobial therapy. Int. J. Med. Microbiol. 300, 563–572 (2010).
Palmer, G. C., Jorth, P. A. & Whiteley, M. The role of two Pseudomonas aeruginosa anthranilate synthases in tryptophan and quorum signal production. Microbiol. (Read.) 159, 959–969 (2013).
Kyrkou, I. et al. Pseudomonas aeruginosa maintains an inducible array of novel and diverse prophages over lengthy persistence in cystic fibrosis lungs. FEMS Microbiol. Lett. 372, fnaf017 (2025).
Oliver, A. & Mena, A. Bacterial hypermutation in cystic fibrosis, not only for antibiotic resistance. Clin. Microbiol. Infect. 16, 798–808 (2010).
Schumann, A. R., Sue, A. D. & Roach, D. R. Hypoxia increases the tempo of phage resistance and mutational bottlenecking of Pseudomonas aeruginosa. Front. Microbiol. 13, 905343 (2022).
Hocquet, D. et al. Pyomelanin-producing Pseudomonas aeruginosa selected during chronic infections have a large chromosomal deletion, which confers resistance to pyocins. Environ. Microbiol. 18, 3482–3493 (2016).
Cramer, N., Fischer, S., Hedtfeld, S., Dorda, M. & Tümmler, B. Intraclonal competitive fitness of longitudinal cystic fibrosis Pseudomonas aeruginosa airway isolates in liquid cultures. Environ. Microbiol. 22, 2536–2549 (2020).
Scanlan, P. D. et al. Coevolution with bacteriophages drives genome-wide host evolution and constrains the acquisition of abiotic-beneficial mutations. Mol. Biol. Evol. 32, 1425–1435 (2015).
Wright, R. C. T., Friman, V. P., Smith, M. C. M. & Brockhurst, M. A. Resistance evolution against phage combinations depends on the timing and order of exposure. mBio 10, e01652–01619 (2019).
Kinsey, G. R., Li, L. & Okusa, M. D. Inflammation in acute kidney injury. Nephron Exp. Nephrol. 109, e102–e107 (2008).
Yang, Q., Le, S., Zhu, T. & Wu, N. Regulations of phage therapy across the world. Front Microbiol 14, 1250848 (2023).
Chan, B. K. et al. Personalized inhaled bacteriophage therapy for treatment of multidrug-resistant Pseudomonas aeruginosa in cystic fibrosis. Nat. Med. 31, 1494–1501 (2025).
Bernabéu-Gimeno, M. et al. Neutralizing antibodies after nebulized phage therapy in cystic fibrosis patients. Med 5, 1096–1111.e1096 (2024).
Tamma, P. D. et al. Safety and microbiological activity of phage therapy in persons with cystic fibrosis colonized with Pseudomonas aeruginosa: study protocol for a phase 1b/2, multicenter, randomized, double-blind, placebo-controlled trial. Trials 23, 1057 (2022).
Hahn, A. et al. Bacteriophage therapy for pan-drug-resistant Pseudomonas aeruginosa in two persons with cystic fibrosis. J. Invest. Med. High Impact Case Rep. 11, 23247096231188243 (2023).
Lourenço, M. et al. The spatial heterogeneity of the gut limits predation and fosters the coexistence of bacteria and bacteriophages. Cell Host Microbe 28, 390–401.e395 (2020).
Hsu, B. B. et al. Dynamic modulation of the gut microbiota and metabolome by bacteriophages in a mouse model. Cell Host Microbe 25, 803–814.e805 (2019).
Seed, K. D. et al. Evolutionary consequences of intra-patient phage predation on microbial populations. eLife 3, e03497 (2014).
Dedrick, R. M. et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 25, 730–733 (2019).
Weiner, I. et al. Phage therapy with nebulized cocktail BX004-A for chronic Pseudomonas aeruginosa infections in cystic fibrosis: a randomized first-in-human trial. Nat. Commun. 16, 5579 (2025).
Roach, D. R. et al. Synergy between the host immune system and bacteriophage is essential for successful phage therapy against an acute respiratory pathogen. Cell Host Microbe 22, 38–47 e34 (2017).
Tarique, A. A. et al. Differential expression of genes and receptors in monocytes from patients with cystic fibrosis. J. Cyst. Fibros. 18, 342–348 (2019).
Minato, Y. et al. Mutual potentiation drives synergy between trimethoprim and sulfamethoxazole. Nat. Commun. 9, 1003 (2018).
Kumar, V. et al. Antibiotic adjuvants: a synergistic tool to combat multi-drug resistant pathogens. Front Cell Infect. Microbiol. 13, 1293633 (2023).
West, H. et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 20, 924–937 (2019).
Haddad, R. et al. Induction chemotherapy followed by concurrent chemoradiotherapy (sequential chemoradiotherapy) versus concurrent chemoradiotherapy alone in locally advanced head and neck cancer (PARADIGM): a randomised phase 3 trial. Lancet Oncol. 14, 257–264 (2013).
Chen, L. et al. Camrelizumab vs placebo in combination with chemotherapy as neoadjuvant treatment in patients with early or locally advanced triple-negative breast cancer: The CamRelief randomized clinical trial. JAMA 333, 673–681 (2025).
Żaczek, M. et al. Antibody production in response to Staphylococcal MS-1 phage cocktail in patients undergoing phage therapy. Front. Microbiol. 7, 1681 (2016).
Dedrick, R. M. et al. Potent antibody-mediated neutralization limits bacteriophage treatment of a pulmonary Mycobacterium abscessus infection. Nat. Med. 27, 1357–1361 (2021).
Stellfox, M. E. et al. Bacteriophage and antibiotic combination therapy for recurrent Enterococcus faecium bacteremia. mBio 15, e03396–03323 (2024).
Rodriguez-Miguelez, P. et al. Evidence of microvascular dysfunction in patients with cystic fibrosis. Am. J. Physiol.-Heart Circ. Physiol. 310, H1479–H1485 (2016).
Ouchida, R. et al. Critical role of the IgM Fc receptor in IgM homeostasis, B-cell survival, and humoral immune responses. Proc. Natl. Acad. Sci. 109, E2699–E2706 (2012).
Bojanowski, C. M., Lu, S. & Kolls, J. K. Mucosal immunity in cystic fibrosis. J. Immunol. 207, 2901–2912 (2021).
Berg, M. et al. Evidence for altered immune-structural cell crosstalk in cystic fibrosis revealed by single-cell transcriptomics. J. Cyst. Fibros. 24, 849–860 (2025).
García-Betancur, J.-C. et al. Cell differentiation defines acute and chronic infection cell types in Staphylococcus aureus. eLife 6, e28023 (2017).
Lam, L. H. & Monack, D. M. Intraspecies competition for niches in the distal gut dictates transmission during persistent Salmonella infection. PLOS Pathog. 10, e1004527 (2014).
Shaw, L. P. et al. Children with cystic fibrosis are infected with multiple subpopulations of Mycobacterium abscessus with different antimicrobial resistance profiles. Clin. Infect. Dis. 69, 1678–1686 (2019).
Hankinson, J. L., Odencrantz, J. R. & Fedan, K. B. Spirometric reference values from a sample of the general U.S. population. Am. J. Respir. Crit. Care Med. 159, 179–187 (1999).
Conrad, D. J. et al. Median regression spline modeling of longitudinal FEV1 measurements in cystic fibrosis (CF) and chronic obstructive pulmonary disease (COPD) patients. PLOS ONE 12, e0190061 (2017).
Silveira, C. B. et al. Multi-omics study of keystone species in a cystic fibrosis microbiome. Int. J. Mol. Sci. 22, 12050 (2021).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Breitwieser, F. P. & Salzberg, S. L. Pavian: interactive analysis of metagenomics data for microbiome studies and pathogen identification. Bioinformatics 36, 1303–1304 (2019).
Bushnell, B. BBMap: a fast, accurate, splice-aware aligner. (Lawrence Berkeley National Lab.(LBNL), Berkeley, CA (United States), 2014).
Murtagh, F. & Legendre, P. Ward’s hierarchical agglomerative clustering method: which algorithms implement Ward’s criterion?. J. Classification 31, 274–295 (2014).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput Biol. 19, 455–477 (2012).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Page, A. J. et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693 (2015).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLOS ONE 5, e9490 (2010).
Letunic, I. & Bork, P. Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82 (2024).
Jolley, K. A., Bray, J. E. & Maiden, M. C. J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res 3, 124 (2018).
Schwengers, O. et al. Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb. Genom. 7, 000685 (2021).
Arndt, D. et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44, W16–W21 (2016).
Sprouffske, K. & Wagner, A. Growthcurver: an R package for obtaining interpretable metrics from microbial growth curves. BMC Bioinforma. 17, 172 (2016).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general-purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2013).
Acknowledgments
We thank members of the BIT Lab for helpful discussions and technical support. We are grateful to G. Burkeen for assistance with sample handling, J. Mielke for administrative support, R. Cook for bioinformatics guidance, and F. Rohwer for study feedback. This work was supported by the ARCS Foundation Scholarship (TL), the Cystic Fibrosis Foundation (KCJ), and the Conrad Prebys Foundation (DRR).
Author information
Authors and Affiliations
Contributions
Conceptualization, TL, DJC, and DRR; Methodology, TL, LK, KCJ, JAM, and DJC; Formal analysis and data curation, TL and LK; Investigation, TL, LK, KCJ, JAM, and DJC; Visualization, TL and DRR; Writing – original draft, TL and DRR; Writing – review and editing, all authors; Supervision, DJC and DRR; Resources and funding acquisition, DP, DJC, and DRR.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Sebastien Boutin, Paul Turner and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luong, T., Kharrat, L., Champagne-Jorgensen, K. et al. Ecological partitioning enables phage–antibiotic cooperation in a human Pseudomonas infection. Nat Commun 17, 2615 (2026). https://doi.org/10.1038/s41467-026-69247-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69247-w
