
Abstract
Prostate cancer lineage plasticity is associated with changes in DNA methylation and enhancer of zeste homolog 2 (EZH2) activity. How these epigenetic programs functionally interact to modulate transcriptional reprogramming in neuroendocrine prostate cancer (NEPC) is not well understood. In this study, we demonstrate that hypomethylated regions of DNA preferentially accumulate the repressive mark, H3K27me3. We established an NEPC mouse model with deletion of Ezh2 in the background of Pten and Rb1 loss plus human MYCN overexpression. Deletion or pharmacological inhibition of EZH2 in NEPC murine or patient-derived models leads to a genome-wide rewiring of DNA methylation, characterized by hypomethylation and upregulation of neuroendocrine-lineage genes along with hypermethylation and repression of polycomb repressive complex 2 (PRC2) targets. On the other hand, deletion of DNA methyltransferase 1 (DNMT1) results in significant changes in H3K27me3 distribution, particularly affecting bivalent promoters bearing both H3K27me3 and active H3K4me3 marks. In NEPC models, neuroendocrine-lineage genes are repressed upon DNMT1 deletion associated with increased H3K27me3. Conversely, in prostate adenocarcinoma models, DNMT1 deletion leads to de-repression of neuroendocrine lineage genes with a loss of H3K27me3 marks. Our findings reveal a functional interplay between two repressive epigenetic machineries that mediates lineage plasticity in prostate cancer.
Similar content being viewed by others
Introduction
Acquired change in DNA methylation has been proposed as a mechanism underlying epigenetic plasticity in several cancer types1. In prostate cancer, the DNA methylome is altered during the transition from benign prostate to localized prostate cancer2, and later during disease progression and castration resistance3,4; this is especially apparent in the subset of prostate adenocarcinomas that transform histologically to neuroendocrine prostate cancer (NEPC)2,5. Methylation changes are dynamic, with preclinical studies pointing to a tumor suppressor role of the DNA methyltransferase DNMT1 in early-stage prostate cancer, in contrast to an oncogenic function of DNMT1 in later stage prostate cancer, corresponding with distinct changes in the global methylome and transcriptome6. The importance of DNA methylation dynamics during normal development and cell lineage fate decisions points to DNA methylation having a critical function in lineage plasticity7,8,9, yet the mechanisms by which DNA methylation changes drive lineage transitions in prostate cancer are not well understood.
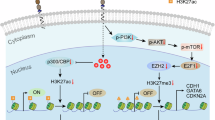
DNA methylation can directly repress gene expression but can also cooperate with other epigenetic pathways to modulate downstream transcriptional activity10,11,12. The enhancer of zeste homolog 2 (EZH2) protein is a catalytic subunit of the polycomb repressive complex 2 (PRC2) and catalyzer of the repressive histone 3 lysine 27 tri-methylation (H3K27me3) mark that also acts to suppress downstream transcriptional activity and has been implicated in prostate cancer lineage plasticity13,14,15,16. EZH2 is overexpressed in castration-resistant prostate cancer (CRPC) including NEPC and is an emerging therapeutic target13,16,17,18,19,20. We have previously shown how differential activity of PRC2 can result in modulation of neuroendocrine (NE)-associated lineage transcription factors facilitating NEPC reprogramming13,16. Evidence from embryonic stem cell (ESC) studies suggests that DNA hypomethylation at CpG islands during the transition to a ground-state pluripotency state redirects PRC2 occupancy leading to increased H3K27me3 levels similar to what is observed in DNMT knockout cell lines21,22,23. Conversely, H3K27me3 depletion can lead to an increase in DNA methylation in ESCs24. The compensatory relationship between DNA methylation and EZH2 has been shown in some cancer types during acquired treatment resistance25,26 or immune evasion27,28,29, where continued transcriptional repression upon DNA methylation inhibition is alleviated by an EZH2 inhibitor. How DNA methylation and EZH2/PRC2 activity interact to modulate prostate cancer lineage plasticity has not been well characterized.
Here, we sought to address the potential crosstalk between DNA methylation and EZH2 in advanced prostate cancer. We demonstrate an overlap in DNA methylation and H3K27me3 profiles that is dynamic following genetic and pharmacological perturbations of either. Using mouse and patient-derived models, we highlight the compensatory roles of these two repressive mechanisms during progression of CRPC towards a NE-lineage state.
Results
Overlap of hypo-methylated DMRs and H3K27me3 marked genes in NEPC
Consistent with the distinct methylation patterns observed in NEPC patient tumors compared with castration-resistant prostate adenocarcinoma (PRAD)17, the PRAD-to-NEPC transitioning murine model that lacks Pten and Rb1 and harbors human MYCN overexpression (PRN mice) also acquires DNA methylation changes during progression to NEPC30. NEPC tumors from patients and PRN mice, as well patient-derived NEPC organoids and androgen-deprived PRAD cells with similar genomic alterations (MYCN (gene encoding N-Myc) overexpression and RB1 loss), all exhibit overexpression of DNA methylation regulators including DNMT1 compared to their PRAD counterparts (Fig. S1A–F). Analysis of differentially methylated regions (DMRs) that distinguish NEPC and PRAD tumors in PRN mice revealed 548 hypo-methylated (hypo-DMR) and 589 hyper-methylated (hyper-DMR) regions in NEPC as compared to PRAD foci; 67% of these DMR-associated genes had similar methylation changes in NEPC versus castration-resistant PRAD patient-derived organoids (PDOs, Fig. 1A). Of these, 41% of hyper- and 54% of hypo-DMRs were also common with patient tumor derived DMRs from Zhao et al. (Fig. 1B)4. While differences are expected due to inter-tumoral heterogeneity, overall, these data support substantial consistency of DNA methylation patterns in NEPC versus castration-resistant PRAD across patients and models. The distribution of hypo- and hyper-DMRs was similar across different genomic locations and chromosomes (Fig. S2A, B). The murine-defined gene-sets associated with DMRs when applied to patient datasets31,32 also distinguished samples according to histological subtype, further highlighting the clinical relevance of the defined DMRs (Fig. S2C). Notably, gene set enrichment analysis (GSEA) of PRN-defined DMRs revealed an enrichment for hypo-DMRs associated with bivalent promoters, neurogenesis, and PRC2-related pathway genes as compared to hyper-DMRs (Fig. 1C). We observed similar enriched pathways at the common patient-defined and PDO-defined 274 hypo- and 116 hyper-DMRs that were at promoters (Fig. 1D). Gene body and intergenic DMRs from PRN and PDOs had a similar pathway enrichment (Fig. S2D–G). Bivalent promoters contain both repressive H3K27me3 and activating histone 3 lysine 4 tri-methylation (H3K4me3) marks rendering genes in a poised state which is critical for lineage specification during normal tissue development33. Significant enrichment of PRC2 target genes across NEPC patients and models pointed to a potential interplay between DNA methylation and PRC2 activity.

A Volcano plot showing differentially methylated regions (DMRs) in PRN NEPC tumors (n = 2) compared to PRN PRAD tumors (n = 2) highlighting common DMR-associated genes from patient CRPC tumors (NEPC and PRAD). B Volcano plot showing differentially methylated regions (DMRs) in NEPC PDOs (n = 3; MSKPCa10, MSKPCa4 and WCM154) compared to PRAD PDOs (n = 2; WCM1358 and MSKPCa2) highlighting common DMRs from patient CRPC tumors (NEPC and PRAD). C Pathway analysis of enriched gene sets for genes associated with hypo- and hyper-methylated regions in PRN NEPC tumors (n = 2) using GSEA. The False Discovery Rate (FDR) q value was calculated using the default permutation-based method in GSEA. D Pathway analysis of enriched gene sets for genes associated with hypo- and hyper-methylated regions in NEPC PDOs (n = 3; MSKPCa10, MSKPCa4 and WCM154) compared to PRAD PDOs (n = 2; WCM1358 and MSKPCa2) that are common with DMRs from patient CRPC tumors (NEPC and PRAD) using GSEA. The False Discovery Rate (FDR) q value was calculated using the default permutation-based method in GSEA.
To further characterize this potential interplay, we profiled genome-wide H3K27me3 marks in PRN mice and observed a distinct H3K27me3 profile in NEPC tumor foci compared to PRAD (Fig. 2A). Overall, 38% of the H3K27me3 marks in PRAD were lost following the transition to NEPC, and NEPC foci acquired 51% of new H3K27me3 marks. There was increased deposition of H3K27me3 at hypo-DMRs compared with hyper-DMRs, which was not specific for any genomic location (e.g., promoter, gene body or intergenic regions, Fig. 2B and S3A, B). Similarly, we identified increased occupancy of H3K27me3 at the patient tumor hypo-DMRs described in Zhao et al.4 in patient-derived models of NEPC compared with PRAD (Fig. 2C, D, and S3C). This relationship was further validated using EPIC-array-based DMR data of NEPC patient-derived xenografts (Fig. S3D)32,34. Extending this analysis to an additional 9 NEPC and 9 CRPC PRAD patient tumors that we characterized by H3K27me3 CUT&RUN16,17, we observed negative correlation between H3K27me3 levels and DNA methylation at promoter regions (Fig. 2E)4. Of note, NE-lineage associated genes (e.g., PROX1, INSM1, ASCL1) were enriched for both repressive marks in PRAD and luminal-lineage genes (e.g., AR, REST) were enriched for both marks in NEPC associated with their lower expression levels in PRAD and NEPC, respectively (Fig. 2E, Fig. S3E).

A Venn diagram depicting overlap of H3K27me3 peaks in NEPC (n = 2) and PRAD (n = 2) in PRN murine model. B H3K27me3 deposition in PRN NEPC (n = 2) and PRAD (n = 2) at differentially hypo- and hyper-methylated regions of PRN NEPC compared to PRAD tumors. C Cumulative H3K27me3 deposition (top) and methylation signal (bottom and heatmap) from NEPC patient-derived organoids (n = 3; WCM154, WCM1078, WCM1262) at hypo- and hyper-methylated regions from patient CRPC tumors (NEPC and PRAD). D Cumulative H3K27me3 deposition (top) and methylation signal (bottom and heatmap) from AR-negative NEPC-positive (n = 5) and AR-positive NEPC-negative (n = 5) LuCaP patient-derived xenografts at hypo- and hyper-methylated regions from patient CRPC tumors (NEPC and PRAD). E Scatter plot with differentially enriched 10 kb genome-wide bins of H3K27me3 log2FPM values in NEPC (n = 9) and castration resistant PRAD (n = 9) samples on the x-axis16 and fold change of DNA methylation of castration resistant PRAD (n = 95) and NEPC (n = 5) on the y-axis4; pale pink, luminal-lineage transcription factors highly repressed in NEPC are labelled; burgundy, NE-lineage transcription factors highly repressed in PRAD are labelled; r value (r = −0.08394051) corresponding p value (p value < 2.2e-16) calculated from Pearson’s product-moment correlation analysis; gray line, linear regression; gray band, standard error of regression.
Overall, our findings in different models and patient tumors demonstrate an increased deposition of H3K27me3 at DNA hypo-DMRs regions in NEPC as compared to PRAD supporting an interaction of DNA methylation and PRC2-mediated epigenetic programs.
DNA methylation changes upon EZH2 deletion
To functionally characterize this interaction, we deleted Ezh2 in PRN mice. For this, we crossed mice harboring Ezh2 floxed alleles (Ezh2f/f) with PRN mice and generated PRNE mice (Pb-Cre4+/-;Ptenf/f;Rb1f/f;LSL-MYCN+/+;Ezh2f/f) with prostate epithelial cells having triple homozygous knockout of Pten, Rb1 and Ezh2 and an induction of human MYCN upon Cre recombinase activation (Fig. 3A, B). Upon Ezh2 deletion, PRNE mice still developed NEPC and had a similar survival as PRN mice, but had a shorter survival compared with heterozygous mice (PRNEhet (Pb-Cre4+/-;Ptenf/f;Rb1f/f;LSL-MYCN+/+;Ezh2f/+)) with one wild-type EZH2 allele (Fig. S4A, B). Histological evaluation of PRNEhet mice (n = 2) demonstrated only 30% of the tumor foci to be NEPC at 12 weeks compared to the 95% of NEPC foci in PRNE mice at a similar age (Fig. S4B, C). These data suggest that deleting both EZH2 alleles does not prevent aggressive NEPC progression.

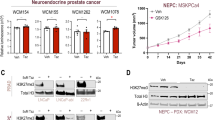
A Schematic of crossing PRN mice (Pb-Cre4+/-;Ptenf/f;Rb1f/f;LSL-MYCN+/+) with Ezh2 floxed mice to generate PRNE (Pb-Cre4+/-;Ptenf/f;Rb1f/f;LSL-MYCN+/+;Ezh2f/f) murine model. Figure created in BioRender. Singh, R. (2025) https://BioRender.com/tg01n0q. B H&E images of AR-negative NEPC primary prostate tumors from PRN and PRNE murine models and immunohistochemical staining for AR, INSM1, EZH2 and H3K27me3. Scale bar: 20 μm. Images are representative of two independent tumors for each genotype. C Bar plots showing the percentage of PRN-defined hypo- and hyper-methylated DMRs with changes in DNA methylation in NEPC tumors of PRNE (n = 2) compared to NEPC tumors of PRN mice (n = 2). Two-tailed ANOVA was performed to compare the between groups showing differential or no methylation change across different genomic context. D Volcano plot showing differentially methylated regions (DMRs) in PRNE NEPC tumors (n = 2) compared to PRN NEPC tumors (n = 2). E Volcano plot showing differentially methylated regions (DMRs) in tazemetostat (EZH2i)-treated MSKPCa10 NEPC organoids compared to vehicle-treated controls. Inset: Western blot showing reduced H3K27me3 in tazemetostat (EZH2i)-treated MSKPCa10 NEPC organoids. Representative western blot from three independent experiments is shown.
Evaluation of DNA methylation changes at the clinically relevant PRN-defined DMRs (Fig. 1A) in the PRNE model revealed that 52% hypo-DMRs from the PRN model were hyper-methylated in the absence of Ezh2 while 25% of PRN hyper-DMRs were hypo-methylated in PRNE (Fig. 3C). Deletion of Ezh2 resulted in a substantial change in the global DNA methylation profile (3635 hypo- and 1754 hyper-DMRs; FDR < 0.05) in NEPC tumors from PRNE mice compared to PRN mice (Fig. 3D). Of the DMRs, 20% of hypo-DMRs and 60% of hyper-DMRs were marked by H3K27me3 in NEPC tumors in PRN. This data suggests that the majority of EZH2 deletion-induced DNA methylation changes occur at regions that were differentially methylated during disease progression and were marked by H3K27me3 in the EZH2-intact NEPC model.
To examine whether pharmacological inhibition of EZH2 influences the DNA methylome, we treated the NEPC PDO MSKPCa10, which was used to define DMRs (Fig. 1B), with the EZH2 inhibitor (EZH2i) tazemetostat or vehicle control for 14 days (Fig. 3E) and evaluated for genome-wide DNA methylation changes after treatment. We observed 185 hyper- and 681 hypo-DMRs after tazemetostat as compared to vehicle-treated organoids (Fig. 3E). When evaluating the genomic distribution of DMRs upon EZH2 silencing or inhibition, we found that, for both models, hyper-methylated regions were enriched at promoters as compared to hypo-methylated regions (Fig. S4D). We further observed similar hypo-methylation at neuron-related genes (e.g., Onecut2, Ntng2, and Slitrk2) and hyper-methylation at H3K27me3 targets (e.g., Gata5, Kcnt1, Hoxb3, and Uncx) in tazemetostat-treated NEPC PDO as observed in PRNE NEPC tumors compared to PRN NEPC (Figure S4E). Interestingly, 76% of the hyper-methylated promoters in PRNE mice were marked by H3K27me3 in NEPC tumors of PRN mice, supporting the compensatory role of the DNA methylation machinery upon EZH2 loss.
Hypo-methylation after EZH2 inhibition is associated with neuroendocrine differentiation
Next, we sought to understand which genes and pathways were specifically impacted by differential DNA methylation upon EZH2 silencing or inhibition. We performed integrative analyses using differentially expressed genes and DMRs in PRNE NEPC tumors compared to PRN NEPC tumors and identified 648 downregulated genes that were hyper-methylated and 676 upregulated genes that were hypo-methylated (Fig. 4A). Gene-set enrichment analyses with DMRs at promoters and intergenic regions overlapping with known enhancers, revealed that upregulated genes with hypo-DMRs in PRNE NEPC tumors were enriched at genes associated with neural differentiation and nervous system development, while downregulated genes with hyper-DMRs were enriched at genes associated with canonical PRC2 targets and bivalent promoters (Fig. 4B). Additionally, gene-sets related to NEPC derived from Beltran et al.17 and Tsai et al.35 were strongly enriched in PRNE as compared to PRN suggesting that deletion of Ezh2 may induce NE-lineage genes further owing to changes in DNA methylation patterns (Fig. 4C). Genes such as Foxa1, Onecut2 and Gata5 showed a compensatory phenomenon where deletion of Ezh2 led to hyper-methylation at the regions covered by H3K27me3 in PRN mice with wildtype Ezh2, leading to further downregulated expression in PRNE NEPC (Fig. 4D, E). In contrast, upon Ezh2 deletion, Foxa2 was upregulated and hypo-methylated at its distal enhancer-like region following Ezh2 deletion. This region is covered by H3K27me3 in PRN NEPC tumors (Fig. 4D, E). We performed similar analyses of the acquired DMRs in MSKPCA10 NEPC PDO cells following tazemetostat treatment. Consistent with genetic perturbation, gene set enrichment analysis with hypoDMRs at the promoters and intergenic regions overlapping with enhancers showed enrichment at neurogenesis related pathways following EZH2 inhibition. However, PRC2 target genes as well as bivalently marked promoters and intergenic regions were enriched with both hypo- and hyper-DMRs upon EZH2 inhibition. (Fig. 4F).

A Volcano plot showing PRNE DMRs with expression changes compared to PRN NEPC tumors. Adjusted p value calculated and log2 fold change was calculated using default method in DeSeq2. B GSEA pathway analysis for promoter and intergenic PRNE DMRs with concordant expression changes. The False Discovery Rate (FDR) q value was calculated using the default permutation-based method in GSEA. C Z-score normalized FPKM mRNA expression of published NEPC signature genes (Beltran et al. and Tsai et al.) in PRN PRAD tumors (n = 5 biological replicates), PRN NEPC tumors (n = 8 biological replicates) and PRNE NEPC tumors (n = 2 biological replicates). Box plot represents 25% − 75% percentile with median denoted as a line. Whiskers extend +/−1.5× interquartile range. Data points that lie outside the whiskers are plotted individually. Two-sided Wilcoxon test was performed for statistical analyses. D IGV tracks showing overlap of H3K27me3 and PRNE- and PRN-defined DMRs at the promoters of Foxa1, Gata5, Onecut2 and Foxa2. E Log2 Fold change in expression for FOXA1, GATA5, ONECUT2 and FOXA2 in PRNE NEPC tumors (n = 2) compared to PRN NEPC (n = 2). F GSEA pathway analysis for promoter and intergenic DMRs in tazemetostat (EZH2i)-treated MSKPCa10 NEPC organoids. The False Discovery Rate (FDR) q value was calculated using the default permutation-based method in GSEA.
Taken together, these data suggest that changes in DNA methylation upon EZH2 silencing or inhibition leads to a further induction of NE-related genes, pointing to a potential compensatory mechanism for DNA methylation to occupy regions that were PRC2 targets.
DNMT silencing or inhibition leads to redistribution of H3K27me3
Given that EZH2 impacts DNA methylation, we posited that DNA methylation changes could also impact EZH2/PRC2 activity. Silencing of DNMT has been associated with redistribution of H3K27me3 repressive marks during normal development, and an intact methylome is required for PRC2-mediated gene repression10. Using CRISPR-Cas9, we knocked out DNMT1 in NEPC PDO (WCM154) and the CRPC cell line 22Rv136 (Fig. S5). Analysis of H3K27me3 signal in 10-kb genome-wide regions revealed 27,390 regions to be enriched and 38,741 regions to be depleted for H3K27me3 upon DNMT knockout (KO) in WCM154 as compared to control (Fig. S6A); this differential distribution of H3K27me3 was not specific for any genomic location (e.g., promoter, intron, or exon) (Fig. S6B, C). In 22Rv1, silencing of DNMT1 led to enrichment in 19,724 and depletion in 23,386 10-kb regions of H3K27me3 levels (Fig. S6D) with a significant level of depletion of H3K27me3 at gene promoters (sgGFP, 15.1%; sgDNMT1, 4.7%) (Fig. S6E).
Next, we sought to understand how re-distribution of H3K27me3 upon DNMT1 KO impacts gene expression. Focusing only on promoter regions, we identified 303 upregulated genes and 242 downregulated genes that were associated with depletion or enrichment of H3K27me3, respectively, after DNMT1 silencing in NEPC organoids (Fig. 5A). Gene ontology analyses revealed that downregulated genes were related to neurogenesis processes as compared to upregulated genes impacted by changes in H3K27me3 (Fig. 5B, and S7A). Curated gene-set (C2) enrichment analyses identified H3K27me3-enriched downregulated genes with high-CpG-density promoters that are bivalent in the brain37 (Fig. 5C). Investigating the epigenetic profile of H3K27me3 and H3K4me3 at the downregulated genes confirmed the marked impact on bivalent promoters upon DNMT1 silencing, particularly with a notable loss of H3K4me3 accompanying the enrichment of H3K27me3 (Fig. 5D). NEPC-related transcription factors such as POU3F2 (encoding BRN2) and LHX2 were among the genes with prominent bivalent promoters that were dysregulated upon DNMT1 silencing and associated with transcriptional repression (Fig. 5E). Homer motif analyses of differentially regulated bivalent-genes did not reveal motifs of classical transcription factors implicated in lineage plasticity (Figure S7B, C).

A Volcano plot of differentially expressed genes in WCM154 DNMT1 knockout (sgDNMT1) vs control (sgGFP) (n = 3/condition); light blue, significantly downregulated genes upon DNMT1 knockout (log2FoldChange < 0; p-value < 0.05); orange, significantly upregulated genes upon DNMT1 knockout (log2FoldChange > 0; p-value < 0.05); violet, downregulated genes with enrichment of H3K27me3 in promoter; green, upregulated genes with depletion of H3K27me3 in promoter. All statistical analyses used Wilcoxon two-sided tests. B Gene ontology analysis of genes downregulated with H3K27me3 enrichment p value < 0.05 and Log2FoldChange < 0; size, -log10FDR; violet-black gradient, gene ratio. The False Discovery Rate (FDR) q value was calculated using the default permutation-based method in GSEA. C C2 curated oncogenic gene set analysis of genes downregulated with H3K27me3 enrichment p value < 0.05 and Log2FoldChange < 0; size, -log10FDR; violet-black gradient, gene ratio. The False Discovery Rate (FDR) q value was calculated using the default permutation-based method in GSEA. D H3K27me3 (orange) or H3K4me3 (blue) CUT&RUN from WCM154 sgGFP and sgDNMT1 was analyzed and plots show profiles and heatmaps are shown of normalized peaks at ±3 kb from the transcriptional start site (TSS) or transcriptional end site (TES) of upregulated (green) and downregulated (violet) genes upon DNMT1 knockout with changes in H3K27me3. E Screenshots from Integrative Genomics Viewer of CUT&RUN of IgG or H3K27me3 or H3K4me3 levels in WCM154 sgGFP and sgDNMT1 at the genomic loci of indicated genes confirming changes in bivalency; gray, IgG; orange, H3K27me3; blue, H3K4me3. F Venn diagram showing overlap of H3K27me3 peaks in decitabine treated tumors (n = 3) and vehicle treated tumors (n = 3) in NEPC xenografts. G Cumulative H3K27me3 and H3K4me3 deposition in decitabine treated (n = 3) and vehicle treated NEPC tumors (n = 3) at bivalently marked peaks from DNMT1 deleted NEPC cells.
We explored whether DNMT inhibition has a similar effect on H3K27me3 re-distribution by performing a short-term in vivo treatment study with the pan-DNMT inhibitor decitabine in NEPC patient-derived xenografts. A significant shift in H3K27me3 deposition was observed with 57,333 H3K27me3-enriched and 23,980 H3K27me3-depleted peaks in decitabine-treated tumors compared to vehicle (Fig. 5F; and Fig. S8A). Consistent with DNMT1 deletion, decitabine-treated tumors showed a decrease in promoter bivalency (increase in H3K27me3 deposition, decrease in H3K4me3), (Fig. 5G). This included promoters associated with neuronal development/signaling (e.g. CDX2, NTRK2, PTPN7 and STON2). The regions that had a decrease in bivalency following DNMT1 deletion or decitabine treatment overlap with DMRs observed in patient tumors (Fig. S8B).
In the CRPC cell line 22Rv1, DNMT1 silencing led to 307 upregulated and 49 downregulated genes impacted by the re-distribution of H3K27me3 (Fig. 6A). There was a negative correlation between baseline DNA methylation and H3K27me3 levels genome-wide, though no correlation between baseline DNA methylation status and H3K27me3 levels at promoters of differentially expressed genes (Fig. S9A, B). In contrast to the downregulated genes in NEPC, upregulated genes in 22Rv1 were related to neurogenesis and neuron development pathways (Fig. 6B, and S10A). Among the upregulated genes, Curated GSEA (C2) also revealed an enrichment of genes with bivalent promoters in the brain (Fig. 6C). H3K4me3 deposition was increased upon loss of DNMT1 at promoters with a marked loss of H3K27me3 associated with their induction (Fig. 6D). In this case, NEPC-related genes POU3F2 and LHX2 had inverse effects on their epigenetic profile; in other words, their bivalent promoters were depleted of H3K27me3 which may have contributed to their de-repression upon DNMT1 silencing (Fig. 6E). Homer analyses of the promoters of differentially regulated genes impacted by changes in H3K27me3 identified potential transcriptional regulators influencing gene expression including ASCL2 binding sites in genes upregulated upon DNMT1 silencing in 22Rv1 (Figure S10B, and C).

A Volcano plot of differentially expressed genes in 22Rv1 DNMT1 knockout (sgDNMT1) vs control (sgGFP) (n = 3/condition); light blue, significantly downregulated genes upon DNMT1 knockout (log2FoldChange < 0; p value < 0.05); orange, significantly upregulated genes upon DNMT1 knockout (log2FoldChange > 0; p value < 0.05); violet, downregulated genes with enrichment of H3K27me3 in promoter; green, upregulated genes with depletion of H3K27me3 in promoter. All statistical analyses used Wilcoxon two-sided tests. B Gene ontology analysis of genes upregulated with H3K27me3 enrichment p value < 0.05 and Log2FoldChange > 0; size, -log10FDR; green-black gradient, gene ratio. The False Discovery Rate (FDR) q value was calculated using the default permutation-based method in GSEA. C C2 curated oncogenic gene set analysis of genes upregulated with H3K27me3 enrichment p value < 0.05 and Log2FoldChange < 0; size, -log10FDR; green-black gradient, gene ratio. The False Discovery Rate (FDR) q value was calculated using the default permutation-based method in GSEA. D H3K27me3 (orange) or H3K4me3 (blue) CUT&RUN from 22Rv1 sgGFP and sgDNMT1 was analyzed and plots show profiles and heatmaps are shown of normalized peaks at ±3 kb from the transcriptional start site (TSS) or transcriptional end site (TES) of upregulated (green) and downregulated (violet) genes upon DNMT1 knockout with changes in H3K27me3. E Screenshots from Integrative Genomics Viewer of CUT&RUN of IgG or H3K27me3 or H3K4me3 levels in 22Rv1 sgGFP and sgDNMT1 at the genomic loci of indicated genes confirming changes in bivalency; gray, IgG; orange, H3K27me3; blue, H3K4me3.
To investigate the potential therapeutic relevance of the PRC2-DNMT interaction, we conducted pilot studies co-targeting DNMT and EZH2, both in combination and sequentially. Unfortunately, all in vivo studies had to be discontinued due to toxicity associated with dosing and administration (Figure S11). Further research is needed to optimize dosing strategies, evaluate safety, and assess the efficacy of combination therapy in prostate cancer preclinical models.
Overall, these data support a crosstalk between DNMT1 and PRC2-mediated H3K27me3 distribution patterns. DNMT1 silencing leads to downregulation of NE-related genes and pathways in NEPC and an upregulation of similar pathways in PRAD.
Discussion
A crosstalk between DNA methylation and EZH2 has been reported across various biologic contexts, during development as well as in cancer38,39,40. A physical interaction between the PRC2 complex and DNMT can facilitate the deposition and co-occurrence of both repressive marks (H3K27me3 and DNA methylation) in the same genomic regions38,39,40. This physical interaction has been argued to be important for de novo DNA methylation in regulation of gene expression as well as for stable inheritance of repressive epigenetic regions for maintenance of cellular identity38,40. While active histone marks may play active or passive roles in transcription41,42,43, high DNA methylation in the nucleosomes has been shown to generally repress PRC2 recruitment44,45. Indirect mechanisms linking PRC2 and DNMTs have also been reported; for instance, PRC2 recruitment is antagonized by the presence of H3K36me2/3 which is a critical mark for DNA methylation44,46,47,48. An interplay between EZH2 and the histone methyltransferase NSD2 has been reported in prostate cancer which may also influence DNA methylation patterns49,50. However, the degree of overlap and potentially compensatory interaction between DNA methylation and PRC2 deposition of H3K27me3 during prostate cancer lineage plasticity has not been fully characterized. While our results do not show a substantial number of regions with both H3K27me3 and DNA hypermethylation, suggestive of a physical interaction between PRC2 and DNMT, we demonstrate a robust functional interplay between the two epigenetic machineries regulating transcriptional networks governing NEPC lineage plasticity.
The PRC2 complex plays a critical role in maintaining hypomethylation at DNA methylation valleys near key developmental genes conserved across vertebrates12. Specifically at promoter regions, H3K27me3 enrichment is negatively correlated with DNA methylation at CpG-dense regions22. Our data shows that H3K27me3 is also markedly enriched at DNA hypomethylated regions in NEPC as compared to PRAD. The genes harboring enrichment of H3K27me3 at hypomethylated regions are related to NEPC (e.g., FOXA2) further supporting a role for the PRC2-DNMT crosstalk in NEPC progression.
Upon deletion of Eed rendering PRC2 inactive in mouse embryonic cells, a prior study observed an induction of DNA methylation at previously PRC2 occupied regions resulting in maintenance of transcriptional repression12. Here, we show similar data in PRAD lineage plasticity models engineered with drivers of NEPC (MYCN induction and RB1 loss) and NEPC models, where genetic silencing or pharmacological inhibition of EZH2 leads to a significant redistribution of genome-wide DNA methylation. More specifically, this was associated with hyper-methylation of PRC2 target genes suggesting a potential compensatory effect that may be critical for the maintenance of a neuroendocrine-lineage state.
In contrast, an intact DNA methylome is critical for proper polycomb-mediated gene repression, as DNA hypo-methylation following DNMT1 deletion leads to widespread redistribution of H3K27me3 and ectopic gene expression in mouse somatic cells10. Consistent with these findings, our data across prostate cancer models shows that silencing of DNMT leads to dramatic changes in genome-wide H3K27me3. Promoters harboring strong bivalency in embryonic stem cells can lose H3K27me3 in cancer and become susceptible to DNA hyper-methylation11. Here, we show that DNMT loss in PRAD leads to induction of genes related to neuroendocrine lineage harboring bivalent promoters. On the other hand, loss of DNMT in NEPC leads to repression of neuroendocrine lineage genes harboring bivalent promoters with enrichment of H3K27me3 and depletion of H3K4me3. While our downstream HOMER analyses of differentially regulated bivalent genes suggest possible mechanisms of recruitment through certain transcription factors (e.g., ASCL2), further work is needed to confirm physical interactions between DNMT1 and/or PRC2 to thoroughly elucidate the molecular mechanisms of regulation of bivalent promoters.
The extent of DNA methylation changes we observed when EZH2 is perturbed, and the redistribution of H3K27me3 upon DNMT inhibition, was quite dramatic. To our knowledge, previous studies exploring the interplay between other histone modifiers and DNA methylation have not reported such substantial alterations in DNA methylation following the silencing of the respective histone modifiers of interest51. For instance, LSD1, which demethylates H3K4me1/2, is known to play a critical role in maintaining DNA methylation52. However, knockout of LSD1 in mouse embryonic stem cells resulted in hypo-methylation of only about 1000 regions and did not significantly impact global gene expression programs related to stemness53. Similarly, other histone modifiers, such as G9A, which mediates H3K9me1/2, have been found to be dispensable for the maintenance of imprinted DNA methylation54. More importantly, the changes in epigenetic and transcriptional profiles we observe after genetic silencing of EZH2 or DNMT1 are particularly related to prostate cancer lineage plasticity underscoring the clinical relevance of this interplay.
While further research is needed to establish the role of co-targeting EZH2 and DNMT in NEPC, this epigenetic crosstalk has potential clinical implications. In colon cancer, treatment with DNMT inhibitors resulted in the accumulation of H3K27me3, limiting the efficacy of the inhibitors27. Interestingly, DNMT inhibition increases H3K27me3 in bivalent enhancers, necessitating combinatorial treatment with EZH2 inhibitors for effective target gene activation27. A recent study on the EZH1-EZH2 dual inhibitor, valemetostat in clinical trials for adult T cell leukemia/lymphoma demonstrated its ability to reduce tumor size and provide durable clinical responses by disrupting the chromatin structure associated with H3K27me355. However, long-term treatment led to resistance, with tumor cells developing either on-target EZH2 mutations that restore chromatin structure and H3K27me3 expression or compensatory epigenetic repressive mechanisms, including elevated DNMT expression resulting in DNA hypermethylation at regions with loss of H3K27me355. Given that EZH2 is a highly sought-after target in prostate cancer with several inhibitors in clinical trials, it becomes critical to investigate compensatory mechanisms that might limit efficacy to EZH2 inhibitors and/or lead to acquired resistance.
Methods
Our research adhered to all relevant ethical regulations, with the study protocol approved by the institutional IACUC. All mouse experiments were conducted in accordance with protocols approved by the WCM-IACUC (protocol no. 2008-0019).
Human cell lines
The prostate cancer cell line, LNCaP and 22Rv1 was obtained from ATCC and authenticated using short tandem repeat (STR) analysis. The cells were grown in RPMI 1640 medium (Gibco, 11875-093) supplemented with 10% fetal bovine serum (Gemini, 900–108) and 1% penicillin/streptomycin (Gibco, 15140-122). For acute and chronic androgen deprived experiments, cells were cultured in phenol red–free RPMI medium (Gibco, 11835-030) supplemented with 5% charcoal-stripped serum (Gibco, A33821-01) and 1% penicillin/streptomycin, for 4 and 14 days respectively.
Patient-derived organoids
Established patient-derived organoids, WCM154, WCM1078, WCM1262 and WCM1358 were maintained as previously described18. The NEPC patient-derived organoid line (MSKPCa10) was a generous gift from Dr. Yu Chen’s lab (Memorial Sloan Kettering Cancer Center, New York) and grown in human organoid media as previously described56. CRPC lines were supplemented with 10 nM DHT (5α-dihydrotestosterone, Sigma-Aldrich, D-073).
Genetically engineered mouse models
Mouse experiments were performed as per protocols approved by the WCM-IACUC (protocol no. 2008-0019). All mice were bred on the same mixed genetic background (C57BL6/129 × 1/SvJ). Previously published PRN (Pb-Cre4+/-;Ptenf/f;Rb1f/f;LSL-MYCN+/+)30 mice were crossed with transgenic mice harboring Ezh2 floxed alleles (a kind gift from Dr. David Goodrich at Roswell Park Comprehensive Cancer Center) to generate a new transgenic PRNE (Pb-Cre4+/-;Ptenf/f;Rb1f/f;LSL-MYCN+/+;Ezh2f/f) model. The mice carried a CAG-LSL-MYCN human transgene at the Rosa26 locus and a Cre recombinase under prostate-specific probasin promoter. Expression of Cre results in removal of lox-stop-lox (LSL) cassette and expresses MYCN under chicken actin promoter, simultaneously knocking out Pten, Rb1 and Ezh2 floxed alleles. For analysis of differentially methylated regions, NEPC vs. PRAD tumors (n = 2 pairs) of PRN mice and PRNE vs. PRN NEPC tumors (n = 2 pairs) were used. All mice were housed in specific pathogen-free facility maintained at 21.5±1 °C temperature and 30-70% humidity in 12 h dark-light cycle with access to food and water.
Knockout cell lines
Previously published LNCaP cells engineered with N-Myc induction57 were transduced with lentiviral particles to knockout RB1. The cells were infected with 500 MOI of viral particles and antibiotic selection was carried out with 1μg/mL of puromycin for two weeks. Two guide RNAs for RB1 and two non-targeting controls were used (Table S2). Efficiency of RB1 knockout was assessed by analyzing DNA sequence around the CRISPR edit sites and comparing it with Western blots for RB1. The guide with most efficient knockout was used for further downstream experiments. WCM154 and 22Rv1 cells with DNMT1 knockout have been described previously36. For stable DNMT1 knockout in PRN organoids, 500 MOI of viral particles were used followed by spinoculation and antibiotic selection with 1μg/mL of puromycin for two weeks. Three guide RNAs for DNMT1 and three non-targeting controls were used (Table S2). Adenovirus based Cre transduction was performed for both PRN and PRNE 3D-orgnoids in matrigel as per our previously published protocol30. Efficiency of Cre-induction was validated by western blots.
In vivo inhibitor studies in patient-derived xenografts
NEPC patient-derived organoid cells (1 × 106 cells diluted in 100 μl of 1:1 PBS and Matrigel) were injected subcutaneously in the flanks of nude (NU/J) mice (Jackson Laboratories, #002019, RRID: IMSR_JAX:002019). Tumor growth was measured by calipers every day for 1 week. Mice were randomly placed in treated or vehicle groups (n = 5 each). Once the tumors measured 150mm3, 0.8 mg/kg of decitabine was administered to mice in treated group by intraperitoneal injection once daily for 7 days. Equal volume of PBS was injected to mice in vehicle groups. Tumors were harvested at the end of treatment. Immunohistochemistry for DNMT1 and H3K27me3 ChIP-Seq was performed on the harvested tumors (n = 3 pairs).
Sub-cutaneously implanted NEPC xenografts (PDXs) were castrated when tumor size reached 75mm3 and treated with decitabine (0.8 mg/kg) and EPZ011989 (250 mg/kg), an EZH2 inhibitor when tumor size reached 150mm3. Decitabine was administered once daily for a month by intraperitoneal injection while EPZ011989 was administered twice daily starting at day 8 for 21 days by oral gavage. Control mice were treated with equal volume of vehicle. Tumor volume was assessed every other day using caliper measurements for both vehicle (n = 4) and decitabine+EPZ011989 (n = 2) treated mice. All other mice in the treatment group were sick and had to be euthanized following our animal protocol before the end of treatment timepoint.
In vitro inhibitor studies in cell lines and patient-derived organoids
MSKPCa10 (NEPC) were treated with 5 μM tazemetostat, an EZH2 inhibitor for 14 days. Treatment efficacy was confirmed by H3K27me3 Western blotting at the end of treatment before performing RRBS. For cell viability assay using Cell titer Glo, MSKPCa10 and MSKPCa2 (CRPC) organoids were treated with tazemetostat (5 μM) and/or decitabine (5 μM) for 14 days while LNCaP cells with RB1 deletion and MYCN overexpression was treated similarly for 6 days. For growth response assay using incucyte, 3D organoids of PRN, PRNE and PRN with DNMT1 KO were treated with varying doses of decitabine or tazemetostat for 10 days. Media was replenished every 48 h for all experiments.
Western blotting
Protein lysates were prepared in RIPA buffer (Thermo Scientific, 89901) supplemented with a cocktail of protease and phosphatase inhibitor (Thermo Scientific, 78428/78430). DC assay was used for protein quantification and 30 μg of protein was resolved in 4-15% of midi-Protean TGX gel (BioRad, 4561084) and transferred onto a PVDF membrane (Thermo Scientific, IB24002). Membranes were blocked in 5% non-fat milk (Bio-Rad, 1706404) or bovine serum albumin (BSA) (Sigma-Aldrich, A7906) in Tris-Buffered Saline Tween-20 (TBST) (Thermo Scientific, AAJ77500K8) for 1 h at room temperature, followed by incubation with primary antibodies overnight at 4 °C (Table S1). Membranes were then incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature and visualized by chemiluminescence detection method (Millipore, WBLUF0500). For blotting H3K27me3, proteins lysates were prepared in a lysis buffer with 110 mM SDS, 100 mM DTT, 80 mM Tris-HCl pH6.9 and 10% glycerol. RCDC assay was used for quantification of the H3K27me3-specific protein lysate.
ChIP-Seq
PRAD and NEPC foci were identified from hematoxylin-eosin stained slides of OCT blocks of primary tumors, reviewed by pathologist. The corresponding foci in OCT blocks were cored using 2 mm biopsy punch and tissues were dissociated using pestle and motor homogenizer, followed by syringes in cold PBS. Dissociated cells were crosslinked in 1% methanol-free formaldehyde (Thermo Scientific, 28908) for 15 min at 37 °C. Crosslinking was quenched with 2.5 M glycine for 5 min at room temperature. Washed cell pellets were resuspended in 1 mL of lysis buffer (50 mM Tris HCl pH 8, 0.5% SDS, 10 mM EDTA with protease and phosphatase inhibitors; Thermo Fisher Scientific, 78428/78430) and lysed for 20 min at 4 °C. Collected nuclei were resuspended in a second lysis buffer (10 mM Tris HCl pH 7.5, 150 mM NaCl, 0.1% SDS, 1 mM EDTA, 1% NP-40, 1% sodium deoxycholate, with protease and phosphatase inhibitors; Thermo Fisher Scientific, 78428/78430) and sonicated in Bioruptor Pico, Diagenode for 12 mins (30 sec on / 30 sec off). To the sheared chromatin, equal volumes of specific antibody and respective IgG control antibody was added and incubated overnight at 4 °C. The immunoprecipitated chromatin was washed with several buffers and eluted in 300 µL of fresh elution buffer (100 mM NaHCO3 and 1% SDS). Reverse crosslinking was performed with 45 mM Tris pH 7, 170 mM NaCl at 65 °C overnight, followed by RNase and proteinase K treatment and DNA was purified using the NucleoSpin Gel and PCR Clean-up Kit (Macherey-Nagel, 740609) as per manufacturer’s recommendation. Libraries with purified immunoprecipitated DNA fragments were generated using the Hyper Prep Kit (Kapa Biosystems, KK8502). Library quality and fragment size distribution was checked in DNA High Sensitivity Bioanalyzer (Agilent, 5067-4626). Equimolar concentration of immunoprecipitated and input DNA fragments were pooled and sequenced for paired-end 50 bp in Illumina NovaSeq 6000 instrument at Weill Cornell Medicine Genomics core facility.
ChIP-Seq data analysis
Quality of raw sequencing reads was assessed by FastQC (Babraham Bioinformatics). Adapters and low-quality reads (Phred score <20) were removed using TrimGalore. High quality reads were aligned to mm10 or hg38 genome using Bowtie2. Reads with mapping quality score <20 were removed from downstream analysis. PCR duplicates were removed using Picard MarkDuplicates. Peak calling was performed using the sonicated input DNA as control by MACS2 algorithm with a q value of <0.05. For H3K27me3, broad peak module of MACS2 was used. Enriched ChIP-seq peak regions were annotated to mm10/hg38 genomic features using HOMER annotatePeak.pl. Known or de novo transcription factor motifs were identified using HOMER findMotifs.pl +/−100 bp from each peak. Unique and overlapping peaks between conditions were defined using BEDTools, keeping at least 1 bp overlap for overlapping peaks. For each condition, two replicates were used and normalized to their input control before merging. The combined signal from both replicates were used to generate enrichment profile plots and heatmap using deepTools.
CUT&RUN
CUT&RUN studies were performed with 100,000 cells in duplicates using a kit from Epicypher adhering to manufacturer’s protocol using antibodies against IgG or H3K4me3 (Epicypher, cat. no. 13-0041, 0.5 μg per reaction) or H3K27me3 (Invitrogen, cat. no. MA511198, 0.5 μg per reaction). Automated library-preparation and sequencing was performed by Molecular Biology Core Facilities in Dana-Farber Cancer Institute. Sequencing reads were mapped to hg38 human genome reference using bwa and visualized using deepTools as profile plots and heatmaps. Calculation of H3K27me3 signals in genome-wide 10 kb bins has been described previously16.
Reduced representation bisulfite sequencing (RRBS)
DNA methylation profiling was performed on biopsy punched tumor tissues of histopathologically confirmed foci of PRAD and NEPC from mouse primary tumor. DNA was extracted using Nucleospin Tissue kit (Macherey Nagel, 740952). For DNA methylation profiling in acute and chronic androgen deprived LNCaP NMYC induced and RB1 knockdown cells or scrambled shRNA control cells (n = 2 replicates), DNA was extracted after 4 and 42 days of androgen deprivation. For baseline DNA methylation profile from patient-derived organoids, DNA was extracted from cell pellets of each. For EZH2 knockout lines, DNA was extracted from cell pellets after 14 days of nucleofection and validating the knockout. Library preparation for RRBS libraries, sequencing, and post-processing of the raw data was performed at Epigenomics Core at Weill Cornell Medicine. The resulting libraries were normalized, pooled, multiplexed, and clustered on flow cells and sequenced for 100 cycles of single-end reads on an Illumina NovaSeq 6000 instrument.
Differential methylation analysis
Single-end bisulfite-converted reads were aligned using Bismark following trimming and adapter removal by TrimGalore. Methylkit was used for identifying differentially methylated sites (DMSs). Sites covered by at least 10 reads, at least 25% difference in methylation between conditions and 5% False Discovery Rate (FDR) were considered as significantly differentially methylated.
DMRcate was used to analyze differentially methylated regions (DMRs). CpG files generated by methylkit were normalized for coverage and sites covered by at least 10 reads were used for differential analysis. The comparison was made between two pairs of PRN NEPC Vs. PRN PRAD tumors and between NEPC PDOs (n = 3) Vs. CRPC PDOs (n = 2). A minimum of 5 consecutive CpGs were used to define a DMR and 5% FDR was used to identify significant DMRs. Homer was used to annotating the significant DMRs to promoters/TSS ( ± 2000 bp from TSS and TSS), gene body (exons, introns, 5’-UTR and 3’-UTR), intergenic regions and CpG islands. Only the intergenic regions that overlapped with known enhancers were considered in downstream gene set enrichment analysis. Known enhancers or enhancer-like signatures were adopted from Vista Enhancers, ENCODE 3 ENC + EPD enhancer gene map and ENCODE cis-regulatory elements in UCSC genome browser. Pathway analysis of the annotated genes was performed using MSigDB (HALLMARK, C2, C6 and GO gene sets) in Gene Set Enrichment Analysis.
Immunohistochemistry
All immunohistochemistry and hematoxylin and eosin (H&E)-stained slides were prepared at Center for Translational Pathology at Weill Cornell Medicine. Formalin fixed tissue was embedded in paraffin blocks and sectioned. Sections were de-paraffinized and endogenous peroxidase was inactivated. Antigen retrieval was accomplished by the Bond Epitope Retrieval Solution 1 (Leica Biosystems, AR9961) at 99–100 °C for 30 min. The sections were incubated with primary antibody for 25 min, post-primary for 15 min, and polymer for 25 min ending with colorimetric development with diaminobenzidine (DAB) for 10 min using the Bond Polymer Refine Detection Kit (Leica Biosystems, DS9800). Hematoxylin and eosin (H&E)-stained slides were also prepared from frozen OCT-embedded tumor tissue blocks, used for identifying NEPC and PRAD foci in tumor tissues for ChIP and methylome experiments. All histological evaluations and quantifications (including hematoxylin and eosin (H&E)-stained and IHC images) were performed by a board-certified, genitourinary pathologist (B. Robinson) who was blinded to animal genotypes and follow criteria that have previously been described30.
Statistical analysis
All statistical analysis was done in GraphPad Prism. Replicates were included and a 5% false discovery rate was considered for identifying significant results. All graphs and plots were prepared in Prism, R studio or deeptools. For heatmap, Euclidean method was used for distance calculation while Ward2 was used for clustering.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data generated in this study have been deposited in the Gene Expression Omnibus (GEO) under accession numbers: GSE289247 (H3K27me3 and H3K4me3 ChIP-Seq of PRN murine model and DNMT1-inhibited NEPC PDXs), GSE289248 (RNASeq of PRNE model), GSE289249 (RRBS or PRN and PRNE models, EZH2-deleted LNR cells and EZH2-inhibited MSK-PCa10), GSE286958 (RNASeq of DNMT1-deleted WCMC154 and 22Rv1) and GSE286954 (H3K27me3 and H3K4me3 CUT&RUN of DNMT1-deleted WCMC154 and 22Rv1). Methylome data from one pair of PRN NEPC and PRAD tumors were downloaded from our previous publication (GSE151581). EPIC-array-based methylation data from patient samples and LuCaP PDX samples were obtained from GSE227853. H3K27me3 ChIP-Seq data from LuCaP PDXs were obtained from GSE161948. All plasmid constructs used in this study are available upon request. Source data are provided with this paper.
Code availability
No custom code was generated or used in this study. All analyses were performed using standard software and tools, which are referenced in the Methods section.
References
Flavahan, W.A., Gaskell, E., and Bernstein, B,E. Epigenetic plasticity and the hallmarks of cancer. Science https://doi.org/10.1126/science.aal2380.ncbi.nlm.nih.gov/pubmed/28729483 (2017).
Lin, P. C. et al. Epigenomic alterations in localized and advanced prostate cancer. Neoplasia 15, 373–383 (2013).
Chen, S. et al. The cell-free DNA methylome captures distinctions between localized and metastatic prostate tumors. Nat. Commun. 13, 6467 (2022).
Zhao, S. G. et al. The DNA methylation landscape of advanced prostate cancer. Nat. Genet 52, 778–789 (2020).
Franceschini, G. M. et al. Noninvasive detection of neuroendocrine prostate cancer through targeted cell-free DNA methylation. Cancer Discov. 14, 424–445 (2024).
Kinney, S. R. et al. Opposing roles of Dnmt1 in early- and late-stage murine prostate cancer. Mol. Cell Biol. 30, 4159–4174 (2010).
Luo, C. et al. Global DNA methylation remodeling during direct reprogramming of fibroblasts to neurons. Elife. 8, e40197 (2019).
Sharma, A., Klein, S. S., Barboza, L., Lohdi, N. & Toth, M. Principles governing DNA methylation during neuronal lineage and subtype specification. J. Neurosci. 36, 1711–1722 (2016).
Stricker, S. H. & Götz, M. DNA-methylation: master or slave of neural fate decisions? Front Neurosci. 12, 5 (2018).
Reddington, J. P. et al. Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of Polycomb target genes. Genome Biol. 14, R25 (2013).
Dunican, D. S. et al. Bivalent promoter hypermethylation in cancer is linked to the H327me3/H3K4me3 ratio in embryonic stem cells. BMC Biol. 18, 25 (2020).
Li, Y. et al. Genome-wide analyses reveal a role of Polycomb in promoting hypomethylation of DNA methylation valleys. Genome Biol. 19, 18 (2018).
Dardenne, E. et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 30, 563–577 (2016).
Davies, A. et al. An androgen receptor switch underlies lineage infidelity in treatment-resistant prostate cancer. Nat. Cell Biol. 23, 1023–1034 (2021).
Ku, S. Y. et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83 (2017).
Venkadakrishnan, V. B. et al. Lineage-specific canonical and non-canonical activity of EZH2 in advanced prostate cancer subtypes. Nat. Commun. 15, 6779 (2024).
Beltran, H. et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med 22, 298–305 (2016).
Puca, L. et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat. Commun. 9, 2404 (2018).
Mosquera, M. J. et al. Extracellular matrix in synthetic hydrogel-based prostate cancer organoids regulate therapeutic response to EZH2 and DRD2 inhibitors. Adv. Mater. 34, e2100096 (2022).
Fong, K. W. et al. PALI1 promotes tumor growth through competitive recruitment of PRC2 to G9A-target chromatin for dual epigenetic silencing. Mol. Cell 82, 4611–4626 (2022).
McLaughlin, K. et al. DNA methylation directs polycomb-dependent 3d genome re-organization in naive pluripotency. Cell Rep. 29, 1974–1985.e6 (2019).
Brinkman, A. B. et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res 22, 1128–1138 (2012).
Kumar, B. & Elsässer, S. J. Quantitative multiplexed ChIP reveals global alterations that shape promoter bivalency in ground state embryonic stem cells. Cell Rep. 28, 3274–3284.e5 (2019).
van Mierlo, G. et al. Integrative proteomic profiling reveals PRC2-dependent epigenetic crosstalk maintains ground-state pluripotency. Cell Stem Cell 24, 123–137.e8 (2019).
Lee, HM. et al. Epigenome reprogramming through H3K27 and H3K4 trimethylation as a resistance mechanism to DNA methylation inhibition in BRAFV600E-mutated colorectal cancer. Clin Cancer Res. 30, 5166–5179 (2024).
Dimopoulos, K. et al. Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol. Oncol. 12, 180–195 (2018).
Chomiak, A. A. et al. Select EZH2 inhibitors enhance viral mimicry effects of DNMT inhibition through a mechanism involving NFAT:AP-1 signaling. Sci. Adv. 10, eadk4423 (2024).
Deblois, G. et al. Epigenetic switch-induced viral mimicry evasion in chemotherapy-resistant breast cancer. Cancer Discov. 10, 1312–1329 (2020).
Li, Y. et al. Histone methylation antagonism drives tumor immune evasion in squamous cell carcinomas. Mol. Cell 82, 3901–3918.e7 (2022).
Brady, N. J. et al. Temporal evolution of cellular heterogeneity during the progression to advanced AR-negative prostate cancer. Nat. Commun. 12, 3372 (2021).
Qiu, X. et al. Response to supraphysiological testosterone is predicted by a distinct androgen receptor cistrome. JCI Insight. 7, e157164 (2022).
Nguyen, H. M. et al. LuCaP prostate cancer patient-derived xenografts reflect the molecular heterogeneity of advanced disease an–d serve as models for evaluating cancer therapeutics. Prostate 77, 654–671 (2017).
Bernstein, B. E. et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326 (2006).
Patel, R. A. et al. Characterization of HOXB13 expression patterns in localized and metastatic castration-resistant prostate cancer. J. Pathol. 262, 105–120 (2024).
Tsai, H. K. et al. Gene expression signatures of neuroendocrine prostate cancer and primary small cell prostatic carcinoma. BMC Cancer 17, 759 (2017).
Yamada, Y. et al. Targeting DNA methylation and B7-H3 in RB1-deficient and neuroendocrine prostate cancer. Sci. Transl. Med. 15, eadf6732 (2023).
Meissner, A. et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770 (2008).
Vire, E. et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439, 871–874 (2006).
McGarvey, K. M. et al. Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res 66, 3541–3549 (2006).
Schlesinger, Y. et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet 39, 232–236 (2007).
Adelman, K. Nascent transcription as a predictor and driver of histone modifications. Nat. Genet 54, 223–224 (2022).
Wang, Z. et al. Prediction of histone post-translational modification patterns based on nascent transcription data. Nat. Genet 54, 295–305 (2022).
Wang, H. et al. H3K4me3 regulates RNA polymerase II promoter-proximal pause-release. Nature 615, 339–348 (2023).
Li, H. et al. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 549, 287–291 (2017).
Jermann, P., Hoerner, L., Burger, L. & Schubeler, D. Short sequences can efficiently recruit histone H3 lysine 27 trimethylation in the absence of enhancer activity and DNA methylation. Proc. Natl. Acad. Sci. USA 111, E3415–E3421 (2014).
Yuan, W. et al. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 286, 7983–7989 (2011).
Laugesen, A., Hojfeldt, J. W. & Helin, K. Molecular mechanisms directing PRC2 recruitment and H3K27 methylation. Mol. Cell 74, 8–18 (2019).
Schmitges, F. W. et al. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 42, 330–341 (2011).
Asangani, I. A. et al. Characterization of the EZH2-MMSET histone methyltransferase regulatory axis in cancer. Mol. Cell 49, 80–93 (2013).
Parolia, A. et al. NSD2 is a requisite subunit of the AR/FOXA1 neo-enhanceosome in promoting prostate tumorigenesis. Nat. Genet 56, 2132–2143 (2024).
Atlasi, Y. & Stunnenberg, H. G. The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet 18, 643–658 (2017).
Wang, J. et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet 41, 125–129 (2009).
Malla, S. et al. The scaffolding function of LSD1 controls DNA methylation in mouse ESCs. Nat. Commun. 15, 7758 (2024).
Zhang, T. et al. G9a/GLP complex maintains imprinted DNA methylation in embryonic stem cells. Cell Rep. 15, 77–85 (2016).
Yamagishi, M. et al. Mechanisms of action and resistance in histone methylation-targeted therapy. Nature 627, 221–228 (2024).
Gao, D. et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 159, 176–187 (2014).
Berger, A. et al. N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J. Clin. Invest 129, 3924–3940 (2019).
Acknowledgements
Some figures were created with BioRender.com. R.S. is supported by DoD PCRP Early Career Investigator Award (W81XWH2110090) and Urology Care Foundation Research Scholar Award (AUA 228082-01). V.B.V. is supported by DoD PCRP Early Career Investigator Award (W81XWH2210197), National Cancer Center Postdoctoral Fellowship Award, and Prostate Cancer Foundation Young Investigator Award (23YOUN15). N.J.B. is supported by NIH/NCI (K22CA269707), American Cancer Society (PF-20-119-01-TBE), and Weill Cornell Medicine JumpStart Research Career Development Award. D.S.R. is supported by NIH-NCI (R01CA274963, R01CA230913, WCM SPORE P50CA211024) and DoD PCRP (W81XWH-17-1-0652). H.B. is supported by the Prostate Cancer Foundation, DoD PCRP (W81XWH-17-1-0653) and NIH/NCI (R37CA241486-01A1, DF/HCC SPORE P50 CA272390-01, WCM SPORE P50 CA211024-01A1).
Author information
Authors and Affiliations
Contributions
Conceptualization: R.S., V.B.V., D.S.R., H.B. Methodology: R.S., V.B.V., E.I., Y.Y., M.A.B., B.D.R. Software: R.S., V.B.V., E.I., M.A.B., Validation: R.S., V.B.V., E.I., Y.Y., N.J.B., K.D., R.G., M.A.B., A.G.P., B.D.R. Formal Analysis: R.S., V.B.V., E.I., M.A.B. Investigation: R.S., V.B.V., E.I., Y.Y., N.J.B., K.D., R.G., M.A.B., A.G.P., B.D.R. Resources: R.S., V.B.V., M.C.H., L.M., M.Y.T., B.D.R., D.S.R., H.B. Data Curation: R.S., V.B.V., E.I., B. H., M.C.H., M.A.B. Writing – Original Draft: R.S., V.B.V., D.S.R., H.B. Writing – Review & Editing: R.S., V.B.V., E.I., Y.Y., N.J.B., K.D., R.G., M.A.B., A.G.P., M.Y.T., B. H., M.C.H., L.M., B.D.R., D.S.R., H.B. Visualization: R.S., V.B.V., E.I., M.A.B. Supervision: R.S., V.B.V., M.Y.T., M.C.H., L.M., D.S.R., H.B. Project administration: R.S., V.B.V., D.S.R., H.B. Funding acquisition: R.S., V.B.V., D.S.R., H.B.
Corresponding authors
Ethics declarations
Competing interests
D.S.R. has received research funding (to institution) from Janssen, Eli Lilly and Chimerix. M.C.H. served as a paid consultant/received honoraria from Pfizer and Astra Zeneca and has received research funding from Merck, Novartis, Genentech, Promicell and Bristol Myers Squibb. H.B. has served as consultant/advisory board member for Janssen, Astra Zeneca, Merck, Pfizer, Amgen, Bayer, Novartis, Daiichi Sankyo, Abbvie, and has received research funding (to institution) from Janssen, Bristol Myers Squibb, Circle Pharma, Daiichi Sankyo, Novartis, Abbvie. All other authors declare no potential conflicts of interest.
Peer review
Peer review information
Nature Communications thanks Peter Stålberg, who co-reviewed with Elham Barazeghi, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Singh, R., Venkadakrishnan, V.B., Imada, E. et al. Crosstalk between EZH2 and DNA methylation mediates neuroendocrine prostate cancer lineage plasticity. Nat Commun 17, 2992 (2026). https://doi.org/10.1038/s41467-026-69308-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69308-0
