
Abstract
Conventional storage and retrieval of nucleic acid specimens, particularly unstable RNA, rely on costly cold-chain infrastructure and inefficient robotic handling, inhibiting large-scale nucleic acid archives needed for global genomic biobanking. We introduce a scalable room-temperature storage system with minimal physical footprint that enables database-like queries on encapsulated, barcoded, and pooled nucleic acid samples. Queries incorporate numerical ranges, categorical filters, and combinations thereof, advancing beyond previous demonstrations of single-sample retrieval or Boolean classifiers. We evaluate this system on ninety-six mock SARS-CoV-2 genomic samples barcoded with theoretical patient data including age, location, and diagnostic state, demonstrating rapid, scalable retrieval. We further demonstrate storage and sequencing of human patient-derived nucleic acid samples, illustrating applicability to clinical genomic analysis. By avoiding freezer-based storage and retrieval, this approach scales to millions of samples without loss of fidelity or throughput, enabling large-scale pathogen and genomic repositories in under-resourced or isolated regions of the US and worldwide.
Similar content being viewed by others
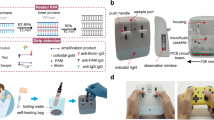

Introduction
Large-scale collection, transport, storage, and retrieval of nucleic acids is essential to enable numerous advanced biotechnological application areas, including population-scale disease tracking1, precision genomic medicine2,3,4, forensics5, and global ecological record-keeping6,7. In particular, genomic DNA and RNA collected from large and diverse patient cohorts empower both pathogen tracing and genomic medicine by enabling the prediction of disease-onset likelihood, as well as informing personalized treatment plans4,8,9,10,11,12. In general, intact native nucleic acid samples provide the most complete representation of genomic information, including epigenetic marks, such as methylation patterns in DNA and genomic viral RNA, as well as modifications to mRNA and lncRNA13,14.
Comprehensive nucleic acid analyses rely on both short-read and long-read sequencing platforms to generate the most detailed and accurate genomic data. These platforms, along with gold standard assays like mass spectrometry for analyzing epigenomic and RNA modifications15, can be both time- and cost-prohibitive. As a result, most samples are preserved in cold storage for future analysis rather than being fully processed immediately. For example, the UK Biobank has collected approximately 500,000 patient samples, but genetic characterization has been limited to a large pre-selected set of genetic markers. Portions of the original blood samples were preserved at either − 80 °C or − 180 °C to enable future analyses16,17.
This long-term storage strategy offers several advantages. It reduces upfront costs, allows researchers to take advantage of future improvements in sequencing technology, and enables sequencing efforts to focus on samples that prove relevant to research questions of interest, which may only become apparent long after sample collection. Yet despite its advantages, storage of intact nucleic acid samples is also a costly option. Samples require energy-intensive cold-chain infrastructure for sample preservation and, once sequencing is desired, transport to centralized analysis facilities18,19,20. This infrastructure burden is particularly challenging for RNA samples, which are highly susceptible to degradation without stringent preservation protocols21,22. Even after reaching analysis facilities, samples may require prolonged low-temperature storage while awaiting comprehensive analyses. Further, the throughput and efficiency of sample recall from large-scale, automated freezer systems are limited by mechanical factors such as robotic automation speed.
As epidemiology, pathogen surveillance, personalized medicine, and ecological conservation efforts attempt to scale to worldwide sample collection, the aforementioned challenges create significant technological barriers that limit access to nucleic acid samples from under-resourced regions both in low- and high-income nations23. This contributes to severely limited participation in rare diseases research, where understanding complex genetic traits and disease associations requires the analysis of tens of millions or more intact DNA and RNA samples from diverse global populations24,25,26,27,28. Similarly, pathogen monitoring and ecological preservation efforts at a global scale become prohibitively cumbersome and costly. Thus, there is an urgent need for low-cost, low-energy, and scalable storage infrastructures that preserve DNA and RNA at the point of collection while simultaneously enabling ambient transport and efficient sample retrieval for downstream genomic analyses.
Traditional biosample storage methods, which rely on barcoded tubes stored in freezers or liquid nitrogen tanks, face significant challenges in cost and practicality as collections scale into the millions. For these vast biosample databases, sophisticated systems are required for efficient sample search and retrieval, in addition to continuous energy consumption and high-cost storage infrastructure. While automation partially alleviates these barriers, these biosample databases remain limited by their low storage density and use of sequential rather than parallel or multiplexed sample access.
In contrast, molecular-based approaches enable in principle the pooling of millions to billions of unique nucleic acid biosamples per tube, effectively creating a highly dense biosample database. Retrieval of specific samples or sets of related samples can be achieved using biochemical approaches such as PCR29, magnetic pulldown30, or fluorescence-activated sorting (FAS)31,32 that use molecular labels including primers, affinity tags, and fluorescent dyes. Primers and affinity tags are often made from DNA, capitalizing on the innate specificity of DNA hybridization to ensure precision and scalability of retrieval. These methods simultaneously process the entire pool of biosamples, which improves retrieval efficiency significantly by executing millions to billions of concurrent molecular search and retrieval operations in solution to far surpass the capabilities of conventional manual or robotic search-and-retrieval of individual tubes. For example, we previously demonstrated a system of silica-encapsulated biosamples labeled with DNA barcodes. The DNA barcodes encoded Boolean metadata about each sample, such as the species of origin, with each barcode sequence associated with a single metadata value (e.g., E. coli or Homo sapiens). This framework facilitated precise identification of samples using fluorescent probes to detect each barcode, while silica encapsulation effectively preserved biosamples at room temperature and prevented unintended interactions with the biosamples that could interfere with biochemical retrieval. With this framework, we demonstrated Boolean retrieval in two studies: one using a prototypical image database stored in plasmid DNA32, and another with a pool of genomic DNA and RNA samples31. Retrieval was demonstrated to have high specificity, with successful retrieval of target samples comprising just 1 in 106 of the total sample pool32. However, this and prior molecular labeling systems29,30,33 remain limited to simple Boolean queries and do not effectively perform numerical range searches (e.g., for time or age ranges) or categorical searches that are required for efficient, large-scale biospecimen retrieval and are routinely performed on modern digital databases using Structured Query Language (SQL).
As an example, consider labeling samples with barcodes to indicate patient ages between 0 and 99. Previous barcoding schemes would associate each possible age value with one of 100 distinct barcode sequences31,32. A simple numerical range query for samples from patients aged 50–74 would require checking for the presence of 25 different barcodes, requiring 9 stages of fluorescent sorting, assuming up to three barcodes may be checked at each stage. Moreover, the queries essential for genomics and epidemiological applications routinely involve selection based on multiple patient criteria, diagnostic conditions, and/or time periods, further compounding the number of sorting stages required. Considering the time taken and selection error compounded at each sorting stage, this type of barcoding scheme quickly becomes impractical for complex queries. This renders these prior molecular labeling systems unsuitable for large-scale genomic databases, which contain tens to hundreds of millions or even billions of samples, and where such queries become essential.
To enable biosample database implementations that approach the capabilities of modern digital databases, in the current work, we develop a scalable biosample database that permits efficient metadata queries for numerical ranges, such as ranges of dates or ages; categories, such as cities or countries; and previously demonstrated Boolean classifications, such as symptomatic/asymptomatic. Central to our approach is the introduction of a type-aware schema that maps each metadata field to a compact, composable set of barcodes (rather than one barcode per value), enabling these queries via barcode presence readout. In this framework, sets of barcodes efficiently encode values across distinct data types, providing a high-level abstraction over the raw binary information encoded by the presence or absence of each barcode sequence. Using a synthesized database of model SARS-CoV-2 genomes, we demonstrate the effectiveness of our query system in a simulated pathogen outbreak scenario, where passengers entering a major international airport are comprehensively swabbed for SARS-CoV-2 to track the pandemic (Fig. 1a). Each sample simultaneously encodes age, vaccination status, presence of symptoms, and flight information (flight number, month and year, and place of origin). We performed retrospective epidemiological and immunological analyses with queries of increasing complexity, including queries for a particular health status, for three distinct age ranges, and for simultaneous matches to criteria for date range, location, and health status. These examples illustrate a general-purpose SQL-like query language, permitting arbitrary logical expressions composed of numerical range, categorical, and Boolean metadata criteria. This work thereby demonstrates a scalable storage system that supports the expressive query capabilities of modern digital databases (Fig. 1b), while eliminating cold-chain logistics through silica encapsulation for long-term RNA sample preservation at room temperature31,32. This framework addresses bottlenecks in nucleic acid storage and retrieval, offering broad applications in molecular diagnostics, pathogen surveillance, and ecological preservation.

a Simulated scenario of sample collection at Boston Logan airport with subsequent pooling and nucleic acid extraction, encapsulation, and barcoding of samples using our proposed molecular filesystem. b Workflow for querying and analyzing samples within a molecular database, shown side-by-side with generic database operations. Some image elements taken from NIAID NIH BioArt Source66,67,68.
Results
Database construction
Ninety-six mock patient samples were separately encapsulated in 5 µm silica microcapsules (Fig. 1a). These microcapsules simulated samples drawn from passengers flying into Logan International Airport in Boston, Massachusetts, and subsequently stored archivally for future diagnostic or epidemiological testing, if needed. Each sample consisted of at most one variant of the SARS-CoV-2 genome using either synthetic P.1 and B.1.1.7 variants and a unique internal 85-nt barcode whose purpose was to aid validation of microcapsule retrieval by making possible downstream identification of de-encapsulated microcapsules in a pool. Variants were quasi-randomly assigned to each sample, with approximately a 15% chance of some variant being present. In contrast to our previous work31,32, where each encapsulation reaction contained a single nucleic acid species such as plasmid DNA32, or various other individual nucleic acids like mammalian and bacterial genomic DNA, total RNA from human cells, or SARS-CoV-2 RNA31, the current work employed co-encapsulation of multiple nucleic acid types within each reaction. Specifically, the nucleic acids introduced into each encapsulation reaction consisted of ~1015 copies of 85-nt ssDNA internal barcodes that uniquely identified each patient sample for purposes of retrieval validation and, for designated samples, ~107 copies of 5 kb fragments of synthetic SARS-CoV-2 RNA. Because the encapsulation process relied solely on charge interactions between the negatively charged phosphate backbone of nucleic acids, we expected successful incorporation of all nucleic acids irrespective of length or type (Supplementary Fig. 1), as previously demonstrated31. Subsequent validation using qPCR and RT-qPCR confirmed efficient nucleic acid encapsulation and de-encapsulation, yielding an estimated average of approximately 1 copy of the SARS-CoV-2 genome and ~108 copies of the internal barcode per microcapsule.
Database labeling and querying
The ideal SARS-CoV-2 genomic database would label each sample with metadata features, such as its unique identifier, patient health status, sample acquisition date, and flight origin. For our example use case, several features were chosen to describe a variety of relevant metadata, including patient age and month and year of the arrival flight, which are examples of numerical metadata; vaccination status, which is an example of Boolean metadata; and flight number and city of origin, which are examples of categorical metadata. For each microcapsule, each feature value was encoded into a set of barcodes to be displayed on its exterior (Fig. 1a), with a total of either 13 or 14 unique 25-nt ssDNA barcodes per microcapsule. These barcodes were derived from a library of 240,000 orthogonal 25-mers34, which were computationally designed and experimentally validated for orthogonality to ensure minimal cross-hybridization even when many distinct barcode sequences were used within a pooled system. Differing encoding strategies were employed based on the type of metadata (numerical, Boolean, or categorical), which enabled type-specific queries of each feature, such as queries for numerical ranges. More detail about each encoding and retrieval strategy is given in Methods.
For numerical metadata, we encoded each numerical value as a sequence of digits in a mixed-radix representation, corresponding to a sequence of barcodes with one barcode per digit. Queries for an exact numerical match are performed by probing for every barcode in its representation, while ranges of various sizes are possible by omitting one or more of the less significant digits (Fig. 2). Categorical metadata were encoded using distinct combinations of k barcodes for each possible value, so that the value of k determined the number of barcodes required to identify a sample’s specific feature value. This approach scales to accommodate an extensive number of feature values. Boolean metadata, on the other hand, were straightforward, with the presence or absence of a single barcode indicating the feature’s state.

a Each query is composed of criteria on numerical, Boolean, and categorical metadata. b Each metadata feature corresponds to a set of barcodes, with different encoding strategies depending on data type. In this example, the numerical metadata date is represented by one barcode per digit for a total of seven barcodes (two for the month, three for the day, and two for the last two digits of the year). The year is in base 10 while the month and day are represented using a mixed-radix representation combining bases 3 and 4, with the place value of each digit indicated by a subscript. Ranges of contiguous dates are specified by omitting one or more barcodes (see Supplementary Fig. 6 for additional details). Note that only barcodes for the month and year were attached and queried experimentally in this paper. For the categorical metadata city, each possible value is represented by a distinct subset of three barcodes. The Boolean metadata symptomatic is indicated by the presence (True) or absence (False) of a single barcode. c Any query corresponds to a corresponding logical expression comprising AND, OR, NOT operations on several barcodes. This logical expression then guides the dye-labeling strategy, search grouping, and dispensing of dye-labeled DNA barcodes for sample selection. Selected samples are then retrieved using optical sorting. Some image elements taken from NIAID BioArt Source66,67.
When performing a query, the search string is first translated into a DNA barcode list (Fig. 2). This list not only facilitates the query but also informs the selection of unique dyes needed for FAS. For NOT logic, the barcode that denotes the unwanted condition is tagged with a fluorescent dye. During sorting, microcapsules that do not show this fluorescence are selected, thus excluding the condition indicated by the dye. OR logic employs a single dye for all relevant barcodes, selecting samples with the matching dye. For AND logic, each query barcode is tagged with a distinct dye, and only samples displaying all unique dyes are selected. This intricate logic necessitates a careful selection of dyes and a strategic grouping of search parameters, ensuring precise sample retrieval.
Immunological case study
To demonstrate the application of a large-scale molecular database with advanced search query capabilities, we performed several database queries on the synthetic database of 96 SARS-CoV-2 samples hypothetically collected from airplane passengers entering Boston, MA. We designed search queries to demonstrate the breadth of the queries enabled by the sample labeling approach, as well as to show how an actual database of this type could be used to answer valuable retrospective epidemiological and immunological questions.
We began with the immunological question of whether specific SARS-CoV-2 variants were present in asymptomatic passengers. To answer this question, we used a query for when the Boolean feature symptomatic was false. For each sample, the barcode bc_symptomatic was present when this feature was true, and absent when this feature was false. Thus, our query NOT symptomatic should retrieve exactly those samples not displaying this barcode.
The presence of the barcode bc_symptomatic on each microcapsule was determined using a fluorescent probe combined with fluorescence-associated sorting (FAS). However, in comparison to previous work using FAS for barcode detection on microcapsules31,32, the number of distinct barcodes attached per microcapsule was increased from 3 to up to 14, leading to a proportional decrease in the copy number of each barcode per microcapsule. To compensate for this copy number reduction, we used fluorescence amplifiers routinely used to detect low copy numbers of RNA in cells in flow cytometry35 (Fig. 3a). The pool of microcapsules was mixed with this amplifier probe and subsequently passed through a FAS instrument, which generates a stream of droplets. Fluorescence from each droplet was measured, and selected populations were defined using fluorescence intensity to distinguish microcapsules with and without the fluorescent probe attached. The number of particles measured for each sample showed a bimodal distribution between the retrieved and non-retrieved microcapsules for the NOT symptomatic query (Fig. 3b). The low-intensity population was separated from the rest of the pool for subsequent de-encapsulation and sequencing.

a For this query, the probe included a region complementary to barcode bc_symptomatic followed by a repeating sequence that allowed 3-fold fluorescence amplification. b Histogram of FAS results showing 93% of microcapsules with low fluorescence, indicating the absence of the bc_symptomatic barcode. c AUC across read depths for the NOT symptomatic selection. Bars represent mean AUC across three independently sorted replicate samples, with individual replicate data points shown. Source data are provided as a Source Data file. d Internal barcode read counts distributions from retrieved samples across varying sequencing depths, where each point represents the number of counts per barcode per replicate (total of three independent sorting replicates). These distributions, depicted with box-and-whisker plots, compare true positive reads from retrieved barcodes that correspond to ground truth positive samples, i.e., for this selection, samples that are truly NOT symptomatic, and false positive reads, i.e., reads from retrieved barcodes that correspond to ground truth negative samples. For each sequencing depth, the total read counts from all sorting replicates are 279 true positive read counts and 9 false positive read counts. In each box-and-whisker plot, boxes span the first and third quartiles, with the center line indicating the median. Whiskers indicate maxima and minima of points within 1.5 times the interquartile range. Source data are provided as a Source Data file. Data plots were made using Matplotlib54. Some image elements taken from NIAID NIH BioArt Source66.
To validate correct retrieval, sequencing reads of the 85-nt internal barcodes were matched to the known sequences to identify the samples in the sequencing data. We quantified retrieval performance using several metrics. Receiver operator characteristic (ROC) analysis on the internal barcode read counts was performed to compute the area under the ROC curve (AUC). The AUC value provides a threshold-independent measure reflecting the system’s overall ability to rank target samples above non-target samples, with 1.0 indicating every target sample is enriched relative to every non-target sample, and 0.5 indicating performance no better than random chance (i.e., a randomly selected target sample is equally likely to be enriched or not relative to a randomly selected non-target sample). For the NOT symptomatic query, where the target (non-symptomatic samples) constituted a high proportion (93 of 96 samples, or ~96.9%) of the initial pool (Fig. 3b), the mean AUC over 3 replicates was consistently high, approaching 1.0 with increasing read depth (Fig. 3c). Figure 3d further details the retrieval performance by presenting the distributions of internal barcode read counts for the NOT symptomatic query at different sequencing depths. These distributions indicate that read counts from true positive barcodes were generally higher than those from false positive barcodes, with improved separation between the two distributions as sequencing depth increased.
Next, we demonstrated how range queries on patient age could be used to explore if certain age groups were more susceptible to different SARS-CoV-2 variants36. Age metadata was encoded numerically using a base-5 representation of three digits with place values 25, 5, and 1. Our conversion of numerical representations to barcodes is described in more detail in Supplementary Fig. 6. We considered three age range queries of different size: age = 0 (Fig. 4a), 15 ≤ age < 20 (Fig. 4e), and 50 ≤ age < 75 (Fig. 4i). Range queries of other sizes or positions are also possible by combining ranges using AND, OR, and NOT logic. For the narrow query age = 0, we selected samples labeled with barcodes bc_age_x25_seq0, bc_age_x5_seq0, and bc_age_x1_seq0 using the same type of multi-stranded branched probes as previously described, labeled with fluorophores Atto 565, Alexa Fluor 647, and Alexa Fluor 750, respectively (Fig. 4b). For the moderate-range query 15 ≤ age < 20, we selected samples labeled with barcodes bc_age_x25_seq0 and bc_age_x5_seq3, using probes labeled with fluorophores Atto 565 and Alexa 647 (Fig. 4f). For the query of broad range 50 ≤ age < 75, we selected samples labeled with the barcode bc_age_x25_seq2 using a probe labeled with fluorophore Atto 565 (Fig. 4j).

a–d Query age = 0. a Schematic representation of the query and retrieval process. b Representative FAS histograms for the query age = 0, showing sequential selection based on barcodes age_x25_seq0 (Atto 565), age_x5_seq0 (Alexa Fluor 647), and age_x1_seq0 (Alexa Fluor 750). c AUC values across read depths for the query age = 0. d Internal barcode read counts distributions from retrieved samples across varying sequencing depths for the query age = 0, separated by true positive read counts (n = 3 at each sequencing depth) and false positive read counts (n = 285). e–h Query 15 ≤ age < 20. e Schematic representation of the query and results. f Representative FAS histograms for the query 15 ≤ age < 20, showing sequential selection based on barcodes age_x25_seq0 (Atto 565) and age_x5_seq3 (Alexa Fluor 647). g AUC values across read depths for the query 15 ≤ age <2 0. h Internal barcode read counts distributions from retrieved samples across varying sequencing depths for the query 15 ≤ age < 20, separated by true positive read counts (n = 12) and false positive read counts (n = 276). i–l Query 50 ≤ age < 75. i Schematic representation of the query and retrieval process for the query 50 ≤ age < 75. j Representative FAS histograms for the query 50 ≤ age < 75, showing selection based on barcode age_x25_seq2 (Atto 565). k AUC values across read depths for the query 50 ≤ age < 75. l Internal barcode read counts distributions from retrieved samples across varying sequencing depths for the query 50 ≤ age < 75, separated by true positive read counts (n = 63) and false positive read counts (n = 225). AUC plots show mean values across three independently sorted replicate samples, with individual data points shown. Whisker plots display individual barcode read counts per replicate (3 independent sorting replicates). In each box-and-whisker plot, boxes span the first and third quartiles, with the center line indicating the median. Whiskers indicate maxima and minima of points within 1.5 times the interquartile range. Source data for bar plots and box-and-whisker plots are provided as a Source Data file. Data plots were made using Matplotlib54.
In each case, sequencing the internal 85-nt barcodes confirmed accurate sample retrieval. ROC analysis yielded perfect AUC values (1.0) across all read depths for the first two queries, age = 0 (Fig. 4c) and 15 ≤ age < 20 (Fig. 4g). The broader query, 50 ≤ age < 75, also showed good performance, with AUC values improving from approximately 0.8 to over 0.9 with increasing read depth (Fig. 4k). Examination of the internal barcode read count distributions (Fig. 4d, h, l) provided further insight. For the narrower queries (age = 0, Fig. 4d; and 15 ≤ age < 20, Fig. 4h), a clear and consistent separation was observed between the higher read counts of true positive barcodes and the very low read counts of false positive barcodes across all sequencing depths, indicating excellent discrimination. The broader query (50 ≤ age < 75, Fig. 4l) exhibited more overlap between true positive and false positive read count distributions compared to the narrower queries, particularly at lower sequencing depths.
Epidemiological case study
We then sought to demonstrate how a composite query involving multiple metadata types can be used to understand the transmission of SARS-CoV-2 infection from specific areas, date ranges, and flight cities37. For the epidemiological case study, we illustrated two aspects of our database: first, efficient representation and querying of categorical features; and second, the composition of several smaller queries into arbitrarily complex logical expressions. This was performed via the retrieval of all samples for passengers flying from Chicago between July and September 2020, who were either symptomatic or unvaccinated. This was equivalent to the query (symptomatic OR NOT vaccinated) AND flight_city = Chicago AND 6 ≤ arrival_month ≤ 8 AND arrival_year = 2020, which combines queries on two numerical features, one categorical feature, and two Boolean features. Such a query necessitated examining eight barcodes: bc_vaccinated, bc_symptomatic, bc_city_seq0, bc_city_seq3, bc_city_seq4, bc_flight_month_x3_seq2, bc_flight_year_x10_seq2, and bc_flight_year_x1_seq0, making it one of the most complex queries tested on any molecular database to date, both semantically and in terms of the number of barcodes tested. To further exhibit the flexibility of our approach to a variety of fluorescent channels and probe design methodologies, we selected a new set of dyes for this query, which modified the bandwidth of our dye markers (Fig. 5a). Specifically, we transitioned from Atto 565, Alexa Fluor 647, and Alexa Fluor 750 to Atto 488, Atto 565, and Alexa Fluor 647. The reduced brightness of Atto 488 relative to the other dyes necessitated the use of branched probe designs that amplify fluorescence signals, similar to branched probed designs used to improve the relative brightness of low-copy targets in cell imaging38,39. This strategy allowed the amplification of the net fluorescent signal by increasing the number of dye markers per barcode.

a Three-stage selection schematic showing sequential application of query criteria with corresponding FAS histograms. b Representative scatterplots from the first round and histograms from the second and third rounds of FAS. Top (round 1): Scatterplots assessing vax_seq0 (Atto 565) and symptomatic_seq0 (Atto 488) against flight_month_x3_seq2 (Alexa Fluor 647). Middle (round 2): Histograms for city selection using city_seq0 (Atto 488), city_seq3 (Atto 565), and city_seq4 (Alexa Fluor 647). Bottom (round 3): Histograms for year selection using flight_year_x10_seq2 (Atto 488) and flight_year_x1_seq0 (Atto 565). c AUC across read depths for the combined query. AUC data for each sequencing depth show mean values (n = 3 independent sorting experiments). Source data are provided as a Source Data file. d Internal barcode read counts distributions from retrieved samples across varying sequencing depths for the combined query, separated by true positive read counts (n = 6) and false positive read counts (n = 282). In each box-and-whisker plot, boxes span the first and third quartiles, with the center line indicating the median. Whiskers indicate maxima and minima of points within 1.5 times the interquartile range. Source data are provided as a Source Data file. Data plots were made using Matplotlib54. Some image elements taken from NIAID NIH BioArt Source66.
This query was performed over three FAS passes, using probes with fluorophores Atto 488, Atto 565, and Alexa Fluor 647 for bc_vaccinated, bc_symptomatic, and bc_flight_month_x3_seq2, respectively, for the first pass (Fig. 5b, top). The sorted populations were stripped of their fluorescent probes using a denaturation buffer. For the second round of selection, fluorophores Atto 488, Atto 565, and Alexa Fluor 647 were used for selecting bc_city_seq0, bc_city_seq3, bc_city_seq4, respectively (Fig. 5b, middle). Again, the previous probes were removed prior to subjecting the sorted population to the next round of selection. For the final FAS pass, Atto 488 and Atto 565 were used for bc_flight_year_x10_seq2 and bc_flight_year_x1_seq0, respectively (Fig. 5b, bottom). Sequencing of the 85-nt internal barcodes after all three passes indicated correct enrichment of all on-target samples (AUC = 1.0) (Fig. 5c). Consistent with this performance, the internal barcode read count distributions (Fig. 5d) showed a clear and consistent separation between the high read counts from the on-target samples and the low read counts from the off-target samples across all sequencing depths. The success of this demonstration illustrates two important features of our approach: first, the ability to implement a single molecular database query that describes criteria spanning many features, such as numerical ranges, categorical values, and multiple Boolean conditions; and second, the ability to split complex queries over several FAS passes without loss of retrieval fidelity.
SARS-CoV-2 sequencing
Identifying the dominant SARS-CoV-2 variant is essential for assessing the virulence of emerging strains, forecasting outbreaks, and expediting vaccine development40. We aimed to identify the predominant SARS-CoV-2 variant in our queries, given that our samples were encapsulated with either the Alpha or Gamma variants. After sorting 100,000 to 700,000 microcapsules for each selection, we sequenced samples that showed a positive result from a specifically designed end-point tiling amplicon PCR for SARS-CoV-2 (ARTIC protocol)41 yielding the expected distribution of fragment lengths (Supplementary Fig. 3). Computational demultiplexing using Freyja42 revealed that all samples showed mostly Alpha variants followed by Gamma variants and other variants that were not assigned by Freyja (Fig. 6a). In all cases, the ratio of expected Alpha to Gamma abundance: 8:3 for selections from Fig. 3, 2:1 for selections from Fig. 4c, and 1:0 for selections from Fig. 5, closely matched the measured abundances in Fig. 6a, further providing support to the high retrieval precision observed using an orthogonal sequencing approach.

a For the three database queries NOT symptomatic, 50 ≤ age < 75, and (symptomatic OR NOT vaccinated) AND flight_city = Chicago AND 6 ≤ arrival_month ≤ 8 AND arrival_year = 2020, the variants in each sample were quantified. These variants should correspond to the synthetic Alpha or Gamma that had been encapsulated in each of the 96 mock patient samples. Each bar height indicates mean abundance values calculated across three independently sorted replicates. Source data are provided as a Source Data file. b Results for sequencing of clinical SARS-CoV-2 samples. Dark and light-colored circles represent each duplicate sequencing run for each sample. Source data are provided as a Source Data file. Data plots were made using Matplotlib54.
To demonstrate the viability of encapsulation for storage and recovery of clinical SARS-CoV-2 samples, we applied our approach to five distinct patient-derived samples. Each sample contained different sub-lineages of the Omicron variant, allowing us to gauge our ability to detect small variations in the viral genomes present during encapsulation through the comparison of called variants of each sample with and without encapsulation. For each sample, we successfully recalled the sub-lineages of Omicron variants (Supplementary Table 2). Further examination of the data indicated that the precision and recall of variant calling for the encapsulated samples were affected by the sequencing coverage (Fig. 6b), which we attribute to the low copy numbers of SARS-CoV-2 retrieved from encapsulation resulting in amplicon dropouts (Supplementary Fig. 4 and Supplementary Fig. 5). The dropouts observed in the low-copy clinical samples (Supplementary Fig. 4 and Supplementary Fig. 5) appeared stochastic and were not consistently biased against longer amplicons, a pattern characteristic of low template input effects rather than systematic RNA degradation during processing43,44,45. Crucially, despite these low-template effects in some clinical samples, overall RNA integrity was sufficient for successful variant identification using the short-read ARTIC protocol for both our synthetic retrieved samples (Fig. 6a) and the recalled Omicron sub-lineages from the clinical samples (Supplementary Table 2).
Discussion
The large-scale, global collection of genomic DNA and RNA promises a plethora of new data relevant to health and security sectors, ranging from pathogen surveillance to personalized medicine. Although centralized laboratories for genomic analysis have enabled sequencing of nucleic acid samples even in low-resource settings, the continuous energy requirements and physical constraints of cold-chain storage and transportation remain major accessibility barriers to the creation of large-scale global nucleic acid repositories23, indicating the need for scalable, low-cost long-term storage facilities with efficient sample retrieval capabilities. In this work, we demonstrated an intuitive yet powerful sample labeling strategy that significantly expands querying capabilities within a pooled molecular database, rendering it analogous to searching common digital file databases such as public datasets hosted by Google BigQuery, Microsoft Azure, and Amazon Web Services46,47,48. The nucleic acid database query language can accommodate arbitrary logical combinations of ranged queries, categorical queries, and truth queries on features that have been encoded into barcodes displayed on each nucleic acid specimen, encapsulated within a microcapsule for long-term stability. In our demonstration of the search query language, we showed how this nucleic acid database could be applied to answer several examples of retrospective epidemiological and immunological questions by analyzing sequencing results from cohorts retrieved from a database of simulated SARS-CoV-2 samples.
The effectiveness and reliability of such a query language depend on the high specificity of the underlying molecular recognition and retrieval process. Our FAS system of retrieval differs from other DNA-based information systems, such as those using PCR amplification. In PCR-based systems, even rare initial non-specific binding or mispriming events can be exponentially amplified over subsequent PCR cycles, introducing false positives and imposing practical limits on usable primer sequence space. Previous studies have shown the number of usable barcodes for PCR-based retrieval may be as low as 14,000 to avoid off-target binding29. In contrast, our retrieval based on direct hybridization of fluorescent probes allows the use of a large library of 240,000 barcode sequences originally designed for use with microarrays34. This is possible because our FAS approach involves no enzymatic amplification of the initial probe-barcode binding event; consequently, weak or transient off-target interactions typically result in fluorescence signals below our detection threshold and are thus rejected without propagation or amplification. Furthermore, whereas additional PCR cycles can exacerbate false positives, additional cycles of FAS can progressively enrich on-target samples, improving the purity of the final retrieved samples.
In this paper, we illustrate the utility of our query language and the specificity of our FAS-based retrieval with a demonstration on a database of 96 samples with up to 14 barcodes per microcapsule. Our system of barcoded microcapsules allows all 96 samples to be consolidated into a single tube, resulting in a hundredfold reduction in storage space while maintaining a broad range of sophisticated search capabilities, including combinations of numerical range, categorical, and Boolean queries. This scalability and efficiency in sample management have direct implications for enhancing biosurveillance strategies as proposed by the Nucleic Acid Observatory49. By enabling the pooling of thousands or millions of samples into fewer tubes, our approach offers a significant footprint reduction over methods that require a separate vessel for each sample, thereby streamlining the process of monitoring and responding to pathogenic threats. Consider the traditional storage of 1 million nucleic acid samples. Storing these samples in – 80 °C freezers, each accommodating 40,000 samples, would require 25 freezers. In contrast, the microcapsule architecture, capable of retrieval from a pool of 10,000 distinct nucleic acids in just one tube at room temperature, as we have previously shown31,32, would only require 100 tubes for storage. An entire tube can be queried in around 15 min, assuming a FAS rate of 1000 microcapsules per second and a redundancy of 100 microcapsules per sample. This streamlined approach not only offers a potential solution to the challenges outlined by the Nucleic Acid Observatory in deploying biosurveillance approaches but also underscores the potential for our technology to facilitate rapid and efficient global health responses.
Beyond the immediate benefits of consolidation and advanced search, our approach also incorporates features crucial for robust, long-term archive management and utility. First, as we demonstrated in previous work31,32, the FAS-based retrieval system allows for the recovery of non-target populations of the pooled archive after each query. This feature ensures that the overall sample collection remains intact and available for subsequent, unrelated queries. Second, our system permits quality control checks beyond the standard pre-encapsulation assessment of nucleic acid integrity and quantity via the co-encapsulated 85-nt internal DNA barcodes within each sample. These internal barcodes can be periodically interrogated via simple PCR—akin to fixity checks in digital data storage—to confirm sample presence and accessibility within the pooled archive without needing to amplify or sequence the primary encapsulated material. This enables efficient, low-cost monitoring of archive integrity over time. Post-retrieval, these same internal barcodes can validate the success and specificity of each query. When coupled with the room-temperature nucleic acid stability endowed by silica encapsulation31, this comprehensive approach to sample preservation and quality control significantly alleviates the long-term operational burdens and costs associated with traditional cold-chain storage.
The successful demonstration herein sets the stage for broader application and further refinement. Future work will seek to characterize per-step efficiencies during multi-round FAS, particularly for very complex queries, and perform comprehensive techno-economic analyses. Such studies will more precisely define throughput capabilities, understanding the optimum balance between the number of samples aggregated per tube against search time, and cost considerations for extremely large archives under high query loads, enabling quantitative comparisons with freezer farms as implemented by, for example, Azenta Life Sciences50. Continued refinement of these operational aspects, potentially incorporating hierarchical storage strategies as well as custom selection and sorting instrumentation, as discussed, may be crucial for facilitating the widespread dissemination and adoption of this genomic banking and querying system. In addition, we hope to investigate further improvements to this barcoding scheme in future studies. For example, more advanced barcoding schemes could incorporate other data types and search criteria, such as text metadata with substring matching, or the use of non-orthogonal barcode sequence design as demonstrated previously for similarity-based searching51.
This approach represents a significant advance in expanding our ability to store, organize, and access nucleic acids, implementing the ability to perform the search functions that are essential for modern digital databases. Moreover, with the ongoing transformation of immunoassays52 and spatial tissue data53 into DNA molecules, we anticipate that the encapsulation and barcoding approach that we have demonstrated here can be used to store and query a comprehensive range of genomic, transcriptomic, and proteomic data. Leveraging these capabilities in future work could generalize the application of our approach from pathogen surveillance to related fields such as personalized medicine and ecological conservation. Finally, the prospect of encoding digital data, such as health records, into DNA, envisages a future where vast biological information could be efficiently stored, marking a significant leap forward in the compact and versatile storage of biological information in the palm of the hand.
Methods
Statistics and reproducibility
No statistical method was used to predetermine sample size. All experiments were performed in at least three independent biological replicates, unless otherwise stated. For Fig. 6b, two technical replicates are shown for each clinical sample; no statistical analyses or error estimates were derived from these data. No data were excluded from the analyses. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Quantitative values such as area under the receiver operating characteristic curve (AUC) and read count distributions were computed directly from sequencing data without inferential statistical testing. Replicates yielded consistent results across independent experiments, confirming reproducibility of the molecular retrieval and sequencing workflows.
Data analysis was performed using Python 3.13.3 with pandas (v2.2.3) and NumPy (v2.2.5). ROC curves and AUC values were calculated using scikit-learn (v1.6.1). Figures were generated using Matplotlib (v3.10.3)54 and Seaborn (v0.13.2). Custom analysis scripts are available in https://doi.org/10.5281/zenodo.17402438.
General materials
All DNA oligonucleotides (oligos)—including internal barcodes (Supplementary Data 2), splint adapters, 5’-amino-modified DNA barcodes (Supplementary Data 3), dye-labeled probes, preamplifier probes, amplifier sequences (Supplementary Data 1), master forward and reverse primers (Supplementary Table 1), random hexamers, and 20-mer oligodeoxythymidine—were synthesized and processed by Integrated DNA Technologies (IDT). Specifically, 5’-amino-modified barcodes in Echo 384 polypropylene microplates and internal barcodes in 96-deep well plates were purchased as desalted and delivered in nuclease-free water at 1000 μM and in 1 × TE at 500 μM, respectively. Dye-labeled probes were selected for their brightness (quantum yield × molar absorption coefficient at the excitation wavelength for each detection channel) while minimizing fluorescence spillover, with the final selections being Atto 488, Atto 565, Alexa Fluor 647, and Alexa Fluor 750. These dye-labeled probes, along with adapter sequences and branch sequences, were received in 1 × TE at concentrations of 100 μM, 33 μM, and 10 μM, respectively. The master primers were desalted and resuspended at 100 μM with nuclease-free water, while random hexamers and 20-mer oligodeoxythymidine were purified using ion-exchange high-performance liquid chromatography and resuspended at 50 μM with nuclease-free water. SARS-CoV-2 RNA controls were sourced from Twist Bioscience with catalog numbers 103909 and 104044. All oligos were stored at –20 °C. Silica particles of 5 μm diameter with hydroxy-terminated surfaces (catalog number: DNG-B017) were obtained from Creative Diagnostics, and N-[3-(Trimethoxysilyl)propyl]-N,N,N-trimethylammonium chloride (TMAPS; 50% methanol; catalog number: H66414) was acquired from Alfa Aesar. Chemicals such as tetraethyl orthosilicate (TEOS), N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (AEAPTS), N-methyl-2-pyrrolidone (NMP), isopropanol, and ethanol were sourced from Millipore Sigma, bearing catalog numbers: 131903, 8.19172, 270458, 278475, and 459836, respectively. DBCO-PEG5-tetra ester (1260-10) and azidoacetic acid N-hydroxysuccinimide ester (1070-100) were from Click Chemistry Tools. Additionally, carbonate buffer (500 mM, pH 9.0; catalog number: J63899.AK) and saline sodium citrate (SSC, 20 ×; catalog number: 15557044) were purchased from Thermo Fisher, while dextran sulfate (50% in water; catalog number: S4030) came from Millipore Sigma, 10% Tween 20 from VWR (catalog number: 97063-980), formamide from VWR (catalog number: 97062-006), and 5 M sodium chloride from VWR (catalog number: 97062-858).
Internal barcode generation
A subset of primers, 700 in total, from the validated 240,000 primer library34 was checked for alignment against SARS-CoV-2 using BLAST55. The alignment XML output file from BLAST was then parsed to create a list of primers orthogonal to the SARS-CoV-2 genome. The resulting primer list was further filtered by selecting primers with 60–65 °C melting temperature using the melting temperature module in Biopython (version 1.85).
Internal barcodes were generated by first picking a master primer pair from the filtered primer list, forward: 5’–GGCTATGAGACTGTTCGCTAATCAC–3’ and reverse: 5’–CCCTTTGTGGGCACAGTTTAGTCTC–3’, which flanked a unique barcode taken from the primer list. Five-nucleotide randomer spacers (N) were also added between the master primers and the unique barcode to increase the sequence diversity of the internal barcodes for downstream sequencing. Together, the 85-nucleotide internal barcode sequence structure is GGCTATGAGACTGTTCGCTAATCACNNNNNUUUUUUUUUUUUUUUUUUUUUUUUUNNNNNCCCTTTGTGGGCACAGTTTAGTCTC, where U is the unique barcode sequence. The full list of internal barcode sequences is given in Supplementary Data 2.
96-well encapsulation
Ammonium-functionalized silica particles were prepared by hydrolyzing 100 μl TMAPS in a suspension containing 100 mg of silica particles in ethanol at room temperature for 24 h under constant agitation (1500 rpm) using a BioShake iQ thermal mixer, followed by three sequential washes in ethanol. The ammonium-functionalized particles are stored in isopropanol.
For each well in a Nunc 96 deep-well polypropylene plates (Thermo Fisher; catalog number: 278752), a total of 2 mg of ammonium-functionalized silica particles and 800 μl of 0.1% (v/v) Tween 20 in nuclease-free water were added. A total of 5 nanomoles of each internal barcode were added to their respective wells. Ten million copies of SARS-CoV-2 standards were added to each randomly selected well; the list of SARS-CoV-2 variants of concern (VOC) associated with each sample are given in Supplementary Data 5. A volume of 10 μl of TMAPS and 5 μl of TEOS were then added to each well. The plate was then covered with a chemically resistant silicone mat (Axygen; catalog number: AM-2ML-RD-S) and shaken for four days at 1500 rpm using a BioShake iQ thermal mixer (Bulldog Bio; catalog number: 1808-0506).
Encoding of features with DNA barcodes
A full list of sequences for each DNA barcode is given in Supplementary Data 3. The mapping between feature values and DNA barcodes is given in Supplementary Data 6. For each feature, a set of barcodes was allocated from which a subset was drawn to encode each feature value. The sets of barcodes for each feature were disjoint from each other. The encoding strategy for feature values differed based on the type of the metadata (numerical, Boolean, or categorical), which enabled type-specific queries of each feature, such as queries for feature values matching particular numerical ranges.
Numerical features
Numerical features were encoded using a mixed-radix number system (i.e., a sequence of digits with the base allowed to vary between positions) (Supplementary Fig. 6a). A position with base n was allocated n distinct barcode sequences, one for each of the n possible digit values at that position. Thus, a feature value represented by k digits was encoded on each microcapsule with a collection of k distinct barcodes. Ranges of varying size could be specified by allowing some number of the least significant digits to vary (i.e., using wildcards at these digits) (Supplementary Fig. 6b). Experimentally, any numerical range specified in this manner can be retrieved by omitting the corresponding complementary probes during sorting. The base at each position was chosen to provide a good compromise between compression (number of barcodes required on each microcapsule) and the variety of the range sizes that could be represented.
Boolean features
Each Boolean feature was encoded using a single barcode assigned to that feature, similar to barcoding previously used to indicate image content in a database of images32. When the value of a Boolean feature was TRUE for a sample, the barcode was displayed on the microcapsule; a value of FALSE was indicated by the absence of that barcode.
Categorical features
For each categorical feature, we used a combinatorial number system56 to associate each possible feature value with a distinct k-combination drawn from a chosen set of n barcodes, where k is the number of distinct barcodes used to represent this feature on each microcapsule. The number of possible feature values that may be represented is \(({{n}\atop{k}})\), which grows rapidly with both n and k. The combinatorial number system provides a method to associate each possible k-combination with a unique integer value between 0 and \(({{n}\atop{k}})-1\). Each feature value was assigned a unique numerical index in that range, from which the corresponding k-combination of barcodes was determined. The values of k and n were chosen to provide a reasonable compromise between feature width (the number of barcodes required on a microcapsule to represent its feature value) and the number of barcodes that needed to be allocated for this feature.
Barcoding of individual samples and pooling
After encapsulation, the plate was centrifuged for 1 minute at 1000 × g. The supernatant was removed, then backfilled with 1000 μl of 0.1% (v/v) Tween 20. A volume of 10 μl of AEAPTS was then added. The plate was then covered with a chemically resistant silicone mat and shaken for 1 day at 1500 rpm using a BioShake iQ thermal mixer.
To wash the encapsulated microparticles after amino modification, the plate was centrifuged for 1 min at 1000 × g. The supernatant was removed and then backfilled with 1000 μl of NMP. The washing step was repeated thrice and finally resuspended with 500 μl of NMP. A mass of 1 mg of azidoacetic acid N-hydroxysuccinimide ester was added to each well. The plates were then re-sealed with a chemically resistant silicone mat and shaken for 3 h at 1500 rpm using a BioShake iQ thermal mixer. After azide modification, the previous wash steps were repeated, and the microparticles were finally resuspended in 500 μl of NMP. A mass of 0.5 mg of DBCO-PEG5-tetrafluorophenyl ester was added to each well, re-sealed with a chemically resistant silicone mat, and shaken for 5 h at 1500 rpm using a BioShake iQ thermal mixer. The plate was washed with NMP thrice and then resuspended with 200 μl of NMP.
Barcode combinations were dispensed in a 96-well plate using an Echo 550 liquid handler and then transferred to the 96-deep-well plate containing the encapsulated microparticles. To each well of the 96 deep-well plates was added 800 μl of 50 mM carbonate buffer. The plate was re-sealed with a chemically resistant silicone mat and then shaken for 12 h at 1500 rpm using a BioShake iQ thermal mixer. The barcoded microparticles were centrifuged at 1000 × g then the supernatant was removed. The resulting pellet was washed with 20 mM Tris, 1 mM EDTA, and 0.1% (v/v) Tween 20 through repeated centrifugation, removal of the supernatant, and redispersing of the supernatant with 20 mM Tris, 1 mM EDTA, and 0.1% Tween 20 for three times. The microparticles were finally redispersed in 1000 μl of 20 mM Tris, 1 mM EDTA, and 0.1% (v/v) Tween 20. A volume of 500 μl from each well was taken and pooled together to create the sample library.
A full list of the metadata associated with each sample is given in Supplementary Data 4.
Selection of microparticles
An aliquot of 500 μl of the sample library was placed in a 1.5 ml tube and then centrifuged at 1000 × g to sediment the microparticles. The supernatant was removed, then the microparticles were re-dispersed with 200 μl of hybridization buffer (10 × SSC, 10% (v/v) dextran sulfate, 10% (v/v) formamide, and 0.05% (v/v) Tween 20). The hybridization buffer was optimized to create stringent conditions that minimized non-specific binding by destabilizing weak or mismatched duplexes. Separately, equivolume of barcode probes, adapters, and fluorescent probes, and 1 × SSC were pre-hybridized using the following method: 98 °C for 10 s, 40 °C for 5 min, 20 °C for 2 min. A volume of 20 μl of prehybridized probe solutions were then added to the microparticle suspension. The resulting mixture was shaken at 1500 rpm for 15 min at 35 °C using a BioShake iQ thermal mixer, then centrifuged at 1000 × g to sediment the microparticles. The supernatant was removed, and the microparticles were re-dispersed in 1000 μl of the sorting buffer, composed of 1 × SSC and 0.05% (v/v) Tween 20, to further remove any non-specifically bound probes. The microparticle sedimentation and washing steps were repeated thrice. The microparticles were finally resuspended in 1000 μl of sorting buffer. The fluorescently labeled microparticles were sorted using a Sony SH800 sorter equipped with a 100 μm sorting chip.
FAS were performed using a Sony SH800 cell sorter with a 100-µm sorting chip and three fluorescence detection channels. Forward and side scatter were first used to gate singlet microcapsules, excluding debris and aggregates. Fluorescence intensities for Atto 488, Atto 565, Alexa Fluor 647, and Alexa Fluor 750 were analyzed in two-dimensional scatterplots for each dye combination. Gates were established manually using unstained and single-stained controls to define positive and negative populations for each barcode-specific probe (Supplementary Fig. 15). All gating thresholds and instrument settings were held constant for every query and replicate. Because fluorescence profiles were indistinguishable between sorts performed under identical conditions, a representative gating dataset was recorded once and used as the reference for all subsequent experiments.
In the multi-pass selection process, existing fluorescent probes from previously sorted populations were meticulously removed before initiating subsequent rounds. Populations from an earlier selection were initially centrifuged in a 1.5 ml tube at 1000 × g for 20 s, after which the sheath buffer was gently discarded. Next, 1000 µl of a denaturation buffer, comprised of 0.2 M NaOH in 90% formamide and pre-heated to 70 °C, was added. This mixture was swiftly vortexed for 5 s and then incubated at 70 °C in a BioShake iQ thermal mixer for 15 min. Following this incubation period, another 20-second centrifugation at 1000 × g was performed then denaturation buffer was discarded. Then, 1000 µl of a denaturation wash buffer, composed of 0.05% Tween 20 and pre-heated to 70 °C, was added. After a brief 5-second vortex and a 20 s centrifugation at 1000 × g, the denaturation wash buffer supernatant was carefully siphoned off. Finally, 200 µl of hybridization buffer was added, preparing the sample for the next selection phase.
Sorted microparticles were de-encapsulated using 10 μl of electronics-grade 5:1 buffered oxide etch (VWR, catalog number: JT5192-3) and then diluted to 50 μl with nuclease-free water. The released samples were immediately used for Illumina sequencing library preparation.
Internal barcode validation using short-read sequencing
A volume of 1 μl of 50 μM of combined master forward (5’–TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGNNNNNGGCTATGAGACTGTTCGCTAAT*C*A*C–3’) and reverse (5’–GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGAGACTAAACTGTGCCCACAAA*G*G*G–3’) primers with three consecutive phosphorothioates from the 3’-end, 44 μl of nuclease-free water, and 50 μl of repliQa HiFi ToughMix (Quantabio, catalog number: 95200-500) were added to 5 μl of the released sample. The samples were amplified for 20 cycles using the manufacturer’s protocol then purified using 1 × of AMPure XP beads (Beckman Coulter, catalog number: A63881). Samples were eluted from the magnetic beads using 22 μl of 20 mM Tris with 0.05% (v/v) Tween 20. Concentration of the amplified sample was measured using Qubit fluorescence assay (Thermo Fisher, catalog number: Q33231). Ten nanograms from the first PCR amplification were then taken to cycle-limited indexing PCR. Indexing PCR includes 10 μl of an indexing primer set from IDT® for Illumina® DNA/RNA UD Indexes (Illumina, catalog number: 20027213) or Nextera™ DNA CD Indexes (Illumina, catalog number: 20018708) and repliQa HiFi ToughMix as PCR master mix. Indexed samples were cleaned using 1 × of AMPure XP beads and then quantified using quantitative PCR (qPCR). Final pooled libraries were then sequenced using an Illumina 2000 (800 pM loading concentration) using a P1 flow cell with 150 × 2 reads, 50–80% human genome spike-in for nucleotide diversity, and 2% PhiX internal standard.
Internal barcode analysis and performance metric calculation
Retrieval performance was assessed by analyzing internal barcode sequencing data at varying read depths (103, 104, 105, and 106 reads) across three independent biological replicates. Sequencing reads were aligned to reference barcode sequences using the Biopython PairwiseAligner package (version 1.85) with stringent parameters (match: + 1, mismatch: − 20, gap open: − 2, gap extend: −2). Alignments exceeding a score threshold of 35 were classified as matches and tabulated for downstream analysis.
For each query, we calculated the area under the receiver operating characteristic curve (AUC) to quantify discrimination performance independent of threshold selection. Complementing this analysis, distributions of read counts for true positive and false positive barcodes were generated and analyzed to visualize the separation between correctly retrieved target barcodes and any incorrectly retrieved non-target barcodes. True positive barcode counts were defined as reads matching the internal barcodes of known target samples present in the retrieved population. Conversely, false positive barcode counts were defined as reads matching the internal barcodes of known non-target samples that were nonetheless detected in the retrieved population.
Synthetic SARS-CoV-2 sequencing
A portion of the released samples were then processed for sequencing SARS-CoV-2 samples using NEBNext® ARTIC SARS-CoV-2 FS Library Prep Kit for Illumina (New England Biolabs, catalog number: E7658) using the VarSkip primers with several modifications. Complementary DNA synthesis was performed using SuperScriptTM IV (catalog number: 18091200), 12 μl of released sample, and 1 μl of 50 μM 20-mer oligodeoxythymidine and 50 μM random hexamers for primers. Reverse transcription reactions were incubated at 50 °C for 1 h. Finally, amplicons were amplified for 40 cycles.
Resulting libraries following the NEBNext® ARTIC SARS-CoV-2 FS Library Prep protocol were quantified using qPCR and then sequenced on a Nextseq 2000 (750 pM loading concentration) using a P1 flow cell with 150 × 2 reads and 1–10% PhiX internal standard.
Sequencing reads were first aligned with the SARS-CoV-2 Wuhan sequence (NC_045512.2) using minimap257 (2.24-r1122). Human-readable sequence alignment maps were converted to binary alignment maps using samtools58 (v1.13). Variant calling was performed using LoFreq59 (v2.1.5). Resulting variant calls were then used to demix the SARS-CoV-2 variants for each sample using Freyja42 (v1.5.3).
Encapsulation of clinical SARS-CoV-2 samples
Copy numbers of the SARS-CoV-2 virus were measured upon receipt using probe-based quantitative reverse transcription polymer chain reaction (New England Biolabs; catalog number: M3019; Thermo Fisher; catalog number: A45583), detecting the N1 gene (Integrated DNA Technologies; catalog number: 10006713), and using synthetic SARS-CoV-2 Alpha variant as positive controls for the calibration curve (Twist Bioscience; catalog number: 103907). A sample volume of 1 µL was used for each qRT-PCR reaction.
To encapsulate, 500 µL of each sample was added to individual 1.5 mL tubes, then diluted to 1000 µL using nuclease-free water. A mass of 1 mg of trimethylammonium-functionalized silica microparticles was then added to the solution. After mixing for 5 seconds using a vortex mixer, 10 µL of 50% N-[3-(trimethoxysilyl)propyl]-N,N,N-trimethylammonium chloride in methanol (TMAPS; Alfa Aesar; catalog number: H66414) and 10 µL of tetraethoxysilane (Millipore Sigma; catalog number: 333859) were then added. The resulting mixture was shaken using a BioShake iQ thermal mixer at 1600 rpm for 4 days, then centrifuged at 1000 × g for 30 s to pellet the encapsulated samples. The supernatant was carefully removed, then 1000 µL of 2% (v/v) 2-azido-N-[3-(triethoxysilyl)propyl]acetamide in ethanol was added. The resulting mixture was further mixed for 16 h at room temperature.
The azido-modified encapsulated samples were pelleted and washed twice with N,N-dimethylacetamide (DMAc; Millipore Sigma; catalog number: 185884), then the particles were re-dispersed with 1000 µL DMAc. A mass of 0.5 mg of DBCO-dPEG®₁₂-tetrafluorophenyl ester (Quanta Biodesign; catalog number: 11366) were added to each azide-modified encapsulated sample then the resulting mixture was shaken at 1600 rpm using a BioShake iQ thermal mixer for 1 h at 40 °C.
The tetrafluorophenyl-modified encapsulated samples were pelleted using a centrifuge at 1000 × g for 30 s and washed twice with DMAc, then the particles were re-dispersed with 100 µL DMAc. A volume of 900 µL of 500 mM phosphate buffer (Thermo Fisher; catalog number: J60825.AP) was added. Then, a volume of 10 µL of each DNA barcode as amino-modified DNA oligonucleotides 500 µM in nuclease-free water were added to each sample. Three barcodes for each sample were used to add complexity to the library. The table below shows the DNA barcode assigned to each sample Table 1.
After 16 hours of mixing on a thermomixer at room temperature, the barcoded encapsulated samples were pelleted using a centrifuge at 1000 × g for 30 s, washed twice with 1000 µL of hybridization buffer, then finally resuspended with 1000 µl of hybridization buffer. Barcoded encapsulated samples were kept at room temperature.
Clinical SARS-CoV-2 sequencing
Ten µl of unencapsulated and 500,000 microparticles of encapsulated clinical SARS-CoV-2 samples were prepared for sequencing using NEBNext® ARTIC SARS-CoV-2 FS Library Prep Kit for Illumina (New England Biolabs, catalog number: E7658) using the VarSkip primers with several modifications. Encapsulated RNA was released from microparticles with 25 µl of 5:1 buffered oxide etch, and 12 µl was desalted using a 7k MWCO Zeba column (Thermo Fisher; catalog number: 89878). To remove any residual DNA fragments, all samples were first subjected to DNAse treatment (Thermo Fisher; catalog number: 11766051). First-strand complementary DNA synthesis was performed using SuperScript IV, 12 µL of the released sample or unencapsulated sample, and 20-mer oligodeoxythymidine and random for primers. Reverse transcription reactions were incubated at 50 °C for 1 h. Finally, amplicons were amplified for 40 cycles.
Following the rest of the NEBNext® ARTIC SARS-CoV-2 FS Library Prep protocol, the resulting libraries were quantified using qPCR and then sequenced on a Nextseq 2000 (800 pM loading concentration) using a P3 flow cell with 150 × 2 reads and 2–20% PhiX internal standard.
Sequencing reads were aligned using bwa60 (v.0.7.17-r1188). Sequence alignment maps were then converted to binary alignment maps using samtools58 (v1.13). Variant calling, variant filtering, and consensus generation were performed using GATK61 (v4.6.1.0). Duplicates from binary alignment maps were filtered using GATK MarkDuplicates, then variant calling was performed using GATK HaplotypeCaller. Variants were filtered using GATK VariantFiltration. First, variants with a QualByDepth (QD) value less than 2.0 were excluded, using a filter tag QD2. QD provides a normalized variant confidence score by the depth of sample reads supporting a variant. Variants with a raw quality score (QUAL) below 30.0 were discarded, labeled under the QUAL30 filter. Strand Odds Ratio (SOR), a metric that denotes the symmetry of the variant’s presence in both forward and reverse reads, was also considered. Variants with an SOR greater than 3.0 were filtered out and marked with the SOR3 tag. This ensures that the variant is supported by both forward and reverse reads and isn’t an artifact from a potential strand bias. Further, Fisher Strand (FS) values, which indicate strand bias, exceeding 60.0 led to excluding the respective variants, tagged under the FS60 filter. Variants with a Mapping Quality (MQ) less than 40.0, indicative of the overall alignment quality of reads supporting a given variant, were filtered out and designated with the MQ40 tag. Normalization of filtered variant calling files and variant overlap analyses between encapsulated and unencapsulated samples were performed using BCFtools27 (v1.13). True positive (TP) variant counts were directly inferred from the overlapping VCF, while the false positive (FP) and false negative (FN) counts were derived by subtracting TP from the encapsulated and unencapsulated normalized variant calling files, respectively. Precision was computed as the proportion of TP relative to the sum of TP and FP, and recall was derived as the proportion of TP relative to the sum of TP and FN. Finally, SARS-CoV-2 lineages were analyzed using NextClade62 (v2.12.0).
Ethics statement
This study was reviewed by the Massachusetts Institute of Technology Committee on the Use of Humans as Experimental Subjects (COUHES), which determined that it does not involve human subjects as defined in the U.S. Federal Regulations 45 CFR 46 and therefore does not require IRB review or approval. This study did not involve human participants as defined under U.S. Federal Regulations 45 CFR 46. The work used de-identified residual clinical SARS-CoV-2 samples provided by an external diagnostic laboratory solely for methodological validation. No identifiable information, sex, gender, age, or other participant metadata were collected or accessible to the investigators. Accordingly, no informed consent, participant recruitment, or compensation was required. Sex and gender were not considered in the study design because no participant-level data were available. All research was performed in accordance with relevant institutional guidelines and regulations for the handling of de-identified or synthetic nucleic acid samples.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw sequencing data from human-derived samples have been deposited in the NCBI BioProject database under accession number PRJNA1344794: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1344794. Processed match counts to each internal barcode for each experiment are available on Zenodo at https://doi.org/10.5281/zenodo.1050134763. Raw datasets are available on Zenodo at https://doi.org/10.5281/zenodo.1751619164. Figure source data are provided in this paper.
Code availability
Data analysis scripts with processed outputs are archived on Zenodo and are available at https://doi.org/10.5281/zenodo.1050134763 and on the GitHub repository https://github.com/lcbb/BiosampleSQL under the MIT license. The version of this repository associated with this publication is archived on Zenodo and is accessible at https://doi.org/10.5281/zenodo.1740243865.
References
Kreier, F. The myriad ways sewage surveillance is helping fight COVID around the world. Nature https://doi.org/10.1038/d41586-021-01234-1 (2021).
Collins, F. S. & Varmus, H. A New Initiative on Precision Medicine. N. Engl. J. Med. 372, 793–795 (2015).
Vargas, A. J. & Harris, C. C. Biomarker development in the precision medicine era: lung cancer as a case study. Nat. Rev. Cancer 16, 525–537 (2016).
Tarazona, S., Arzalluz-Luque, A. & Conesa, A. Undisclosed, unmet and neglected challenges in multi-omics studies. Nat. Comput. Sci. 1, 395–402 (2021).
Lee, S. B. et al. Assessing a novel room temperature DNA storage medium for forensic biological samples. Forensic Sci. Int. Genet. 6, 31–40 (2012).
Ryder, O. A., McLaren, A., Brenner, S., Zhang, Y.-P. & Benirschke, K. DNA Banks for endangered animal species. Science 288, 275–277 (2000).
Brandies, P., Peel, E., Hogg, C. J. & Belov, K. The value of reference genomes in the conservation of threatened species. Genes 10, 846 (2019).
Kieffer, C., Genot, A. J., Rondelez, Y. & Gines, G. Molecular computation for molecular classification. Adv. Biol. 7, 2200203 (2023).
Zhang, D. Y. & Seelig, G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 3, 103–113 (2011).
Lopez, R., Wang, R. & Seelig, G. A molecular multi-gene classifier for disease diagnostics. Nat. Chem. 10, 746–754 (2018).
Zhang, C. et al. Cancer diagnosis with DNA molecular computation. Nat. Nanotechnol. 15, 709–715 (2020).
Yin, F. et al. DNA-framework-based multidimensional molecular classifiers for cancer diagnosis. Nat. Nanotechnol. 18, 677–686 (2023).
Roundtree, I. A. & He, C. RNA epigenetics—chemical messages for posttranscriptional gene regulation. Curr. Opin. Chem. Biol. 30, 46–51 (2016).
Kan, R. L., Chen, J. & Sallam, T. Crosstalk between epitranscriptomic and epigenetic mechanisms in gene regulation. Trends Genet. 38, 182–193 (2022).
Helm, M. & Motorin, Y. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat. Rev. Genet. 18, 275–291 (2017).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Elliott, P., Peakman, T. C. & Biobank, U. K. The UK Biobank sample handling and storage protocol for the collection, processing and archiving of human blood and urine. Int. J. Epidemiol. 37, 234–244 (2008).
Bull, R. A. et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nat. Commun. 11, 6272 (2020).
Minogue, T. D., Koehler, J. W., Stefan, C. P. & Conrad, T. A. Next-generation sequencing for biodefense: biothreat detection, forensics, and the clinic. Clin. Chem. 65, 383–392 (2019).
Whitmore, L. et al. Inadvertent human genomic bycatch and intentional capture raise beneficial applications and ethical concerns with environmental DNA. Nat. Ecol. Evol. 7, 873–888 (2023).
Opitz, L. et al. Impact of RNA degradation on gene expression profiling. BMC Med. Genomics 3, 36 (2010).
Gallego Romero, I., Pai, A. A., Tung, J. & Gilad, Y. RNA-seq: impact of RNA degradation on transcript quantification. BMC Biol. 12, 42 (2014).
Mendy, M. et al. Biospecimens and Biobanking in Global Health. Glob. Health Pathol. 38, 183–207 (2018).
Ziyatdinov, A. et al. Genotyping, sequencing and analysis of 140,000 adults from Mexico City. Nature 622, 784–793 (2023).
Wall, J. D. et al. The GenomeAsia 100K project enables genetic discoveries across Asia. Nature 576, 106–111 (2019).
Naslavsky, M. S. et al. Whole-genome sequencing of 1,171 elderly admixed individuals from Brazil. Nat. Commun. 13, 1004 (2022).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Bick, A. G. et al. Genomic data in the All of Us Research Program. Nature 627, 340–346 (2024).
Organick, L. et al. Random access in large-scale DNA data storage. Nat. Biotechnol. 36, 242–248 (2018).
Tomek, K. J. et al. Driving the scalability of DNA-based information storage systems. ACS Synth. Biol. 8, 1241–1248 (2019).
Banal, J. L. & Bathe, M. Scalable nucleic acid storage and retrieval using barcoded microcapsules. ACS Appl. Mater. Interfaces 13, 49729–49736 (2021).
Banal, J. L. et al. Random access DNA memory using Boolean search in an archival file storage system. Nat. Mater. 20, 1272–1280 (2021).
Organick, L. et al. Probing the physical limits of reliable DNA data retrieval. Nat. Commun. 11, 616 (2020).
Xu, Q., Schlabach, M. R., Hannon, G. J. & Elledge, S. J. Design of 240,000 orthogonal 25mer DNA barcode probes. Proc. Natl. Acad. Sci. USA 106, 2289–2294 (2009).
Porichis, F. et al. High-throughput detection of miRNAs and gene-specific mRNA at the single-cell level by flow cytometry. Nat. Commun. 5, 5641 (2014).
Goldstein, E., Lipsitch, M. & Cevik, M. On the effect of age on the transmission of SARS-CoV-2 in households, schools, and the community. J. Infect. Dis. 223, 362–369 (2021).
Fauver, J. R. et al. Coast-to-coast spread of SARS-CoV-2 during the Early Epidemic in the United States. Cell 181, 990–996 (2020).
Kishi, J. Y. et al. SABER amplifies FISH: enhanced multiplexed imaging of RNA and DNA in cells and tissues. Nat. Methods 16, 533–544 (2019).
Player, A. N., Shen, L.-P., Kenny, D., Antao, V. P. & Kolberg, J. A. Single-copy gene detection using branched DNA (bDNA) in situ hybridization. J. Histochem. Cytochem. 49, 603–611 (2001).
Tao, K. et al. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 22, 757–773 (2021).
Bei, Y. et al. Overcoming variant mutation-related impacts on viral sequencing and detection methodologies. Front. Med. 9, 989913 (2022).
Karthikeyan, S. et al. Wastewater sequencing reveals early cryptic SARS-CoV-2 variant transmission. Nature 609, 101–108 (2022).
Lagerborg, K. A. et al. Synthetic DNA spike-ins (SDSIs) enable sample tracking and detection of inter-sample contamination in SARS-CoV-2 sequencing workflows. Nat. Microbiol. 7, 108–119 (2022).
Kubik, S. et al. Recommendations for accurate genotyping of SARS-CoV-2 using amplicon-based sequencing of clinical samples. Clin. Microbiol. Infect. 27, 1036.e1–1036.e8 (2021).
Rosenthal, S. H. et al. Development and validation of a high throughput SARS-CoV-2 whole genome sequencing workflow in a clinical laboratory. Sci. Rep. 12, 2054 (2022).
BigQuery public datasets. Google Cloud https://cloud.google.com/bigquery/public-data.
Open Datasets Documentation - Tutorials, API reference - Azure - Azure Open Datasets. https://learn.microsoft.com/en-us/azure/open-datasets/.
Open Data on AWS. https://aws.amazon.com/opendata/.
The Nucleic Acid Observatory Consortium. A global nucleic acid observatory for biodefense and planetary health. Preprint at arXiv:2108.02678 (2021).
Azenta Life Sciences. Cryogenic Storage Solutions in Life Sciences. https://www.azenta.com/learning-center/resources/cryogenic-storage-solutions-life-sciences-comprehensive-guide-decision-making (2024).
Bee, C. et al. Molecular-level similarity search brings computing to DNA data storage. Nat. Commun. 12, 4764 (2021).
Eldjarn, G. H. et al. Large-scale plasma proteomics comparisons through genetics and disease associations. Nature 622, 348–358 (2023).
Zhao, T. et al. Spatial genomics enables multi-modal study of clonal heterogeneity in tissues. Nature 601, 85–91 (2022).
Hunter, J. D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 9, 90–95 (2007).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Knuth, D. E. The Art of Computer Programming, Volume 4, Fascicle 2: Generating All Tuples and Permutations. (Addison-Wesley, 2005).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Wilm, A. et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 40, 11189–11201 (2012).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Aksamentov, I., Roemer, C., Hodcroft, E. & Neher, R. Nextclade: clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 6, 3773 (2021).
Berleant, J. D., Banal, J. L., Rao, D. K. & Bathe, M. Enabling global-scale nucleic acid repositories through versatile, scalable biochemical selection from room-temperature archives. Zenodo https://doi.org/10.5281/ZENODO.10501347 (2025).
Berleant, J. D., Banal, J. L., Rao, D. K. & Bathe, M. Full datasets from: Enabling global-scale nucleic acid repositories through versatile, scalable biochemical selection from room-temperature archives. Zenodo https://doi.org/10.5281/ZENODO.17516191 (2025).
Berleant, J. D., Banal, J. L., Rao, D. K. & Bathe, M. lcbb/BiosampleSQL: Publication release. Zenodo https://doi.org/10.5281/ZENODO.17402438 (2025).
NIAID Visual & Medical Arts. Eppendorf Tube. NIAID NIH BIOART Source. bioart.niaid.nih.gov/bioart/143 (2024).
NIAID Visual & Medical Arts. 96 Well Plate. NIAID NIH BIOART source. bioart.niaid.nih.gov/bioart/7 (2024).
NIAID Visual & Medical Arts. Next gen sequencer. NIAID NIH BIOART source. bioart.niaid.nih.gov/bioart/386 (2024).
Acknowledgements
M.B. and J.D.B. were supported by the Office of Naval Research (N00014-21-1-4013), the Army Research Office (ICB Subaward KK1954), and the National Science Foundation (CBET-1729397, OAC-1940231, and CCF-1956054). Additional funding to M.B. was provided through the National Science Foundation (CCF-2403100) and to J.D.B. through a National Science Foundation Graduate Research Fellowship (Grant No. 1122374). J.L.B. acknowledges support in part by the National Science Foundation SBIR Phase I 2136447, UCSF Parnassus Flow CoLab RRID:SCR_018206, DRC Center Grant NIH P30 DK063720, UCSF Center for Advanced Technology at Mission Bay, and Illumina. This research was also supported by a core center grant from the National Institute of Environmental Health Sciences, National Institutes of Health (P30-ES002109). We are grateful to T.B. Schardl and C.E. Leiserson (MIT CSAIL) for useful discussions on DNA barcoding. We thank G. Paradis, M. Jennings, and M. Griffin of the Flow Cytometry Core at the Koch Institute at the Massachusetts Institute of Technology (MIT) for flow sorting assistance. We are grateful to Delaware Diagnostics Labs for providing us de-identified clinical SARS-CoV-2 samples. We thank G. Tikhorimov for providing access to a Beckman Coulter Labcyte Echo 550. We are grateful to Ella Maru Studio, Inc. for assistance in creating the airport schematic in Fig. 1a.
Author information
Authors and Affiliations
Contributions
M.B., J.D.B., and J.L.B. conceived the sample storage system. J.D.B. designed the sample barcoding scheme and query language architecture. J.L.B. designed and implemented sample synthesis, FAS selection, and post-processing after selection of mock patient samples, and encapsulation and barcoding of clinical SARS-CoV-2 samples. D.K.R. prepared samples for sequencing and analyzed the data. J.L.B. and J.D.B. performed data analysis after querying and calculation of summary statistics. M.B. supervised the entire project. All authors contributed equally to the writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The Massachusetts Institute of Technology has filed a patent related to this work on behalf of J.L.B., M.B., J.D.B., and additional inventors (US Patent App. 17/836,726). J.L.B. and M.B. are co-founders and equity shareholders of Cache DNA, Inc. (Cache). J.L.B. is an employee of Cache and an independent contractor of OpenAI. D.K.R. was an intern at Cache for the period of this work.
Peer review
Peer review information
Nature Communications thanks Fajia Sun and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Berleant, J.D., Banal, J.L., Rao, D.K. et al. Enabling global-scale nucleic acid repositories through versatile, scalable biochemical selection from room-temperature archives. Nat Commun 17, 2807 (2026). https://doi.org/10.1038/s41467-026-69402-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69402-3
