
Abstract
Hepatitis C virus (HCV) is a leading cause of chronic liver disease, cirrhosis, and hepatocellular carcinoma worldwide. Development of an E1E2-based HCV vaccine has been hindered by the difficulty of producing a soluble E1E2 (sE1E2) antigen that faithfully recapitulates the native virion-associated heterodimer. Guided by cryo-electron microscopy (cryo-EM) structures, we engineer genotype 1a H77 sE1E2 by truncating the E1 and E2 stems (Cut1), deleting a putative fusion peptide–containing region in E1 (Cut2), and stabilizing the heterodimer using diverse scaffolds. All H77 sE1E2.Cut1+2 scaffolds exhibit native-like E1–E2 association and strong binding to the broadly neutralizing antibody (bNAb) AR4A. A genotype 1a HCV-1 sE1E2.Cut1+2 variant scaffolded by a modified SpyTag/SpyCatcher (SPYΔN) is selected for in vitro and in vivo characterization, as well as further construct refinement. The structure of this HCV-1 sE1E2 construct in complex with bNAbs is determined by cryo-EM and negative-stain EM (nsEM), with an nsEM-based strategy established for antibody epitope mapping. HCV-1 sE1E2.Cut1+2.SPYΔN is displayed on self-assembling protein nanoparticles (SApNPs) to enhance immunogenicity. The HCV-1 sE1E2.Cut1+2.SPYΔN heterodimer and SApNPs bearing wildtype or modified glycans are evaluated in mice, alongside E2 core-based immunogens for comparison. Together, these results establish a framework for advancing E1E2-based HCV vaccines toward clinical development.
Similar content being viewed by others

Introduction
According to the World Health Organization (WHO), an estimated 50 million people worldwide are chronically infected with the hepatitis C virus (HCV), with approximately 1 million new infections occurring annually1. In 2022, about 240,000 deaths were reported, primarily due to cirrhosis and hepatocellular carcinoma, making HCV one of the leading causes of liver-related morbidity and mortality1. In the United States, the opioid crisis and injection drug use have contributed to more than 70,000 overdose-related deaths in 20192 and a sharp rise in acute HCV infections3. Over the past decade, direct-acting antiviral (DAA) therapies have demonstrated high efficacy in treating chronic HCV infection and achieving sustained virologic response (SVR)4,5,6. However, although DAAs improve liver function in patients with decompensated cirrhosis7,8, they do not prevent HCV reinfection or eliminate the risk of hepatocellular carcinoma9,10,11. Moreover, despite advances in diagnostic technologies12, early detection of asymptomatic HCV infection remains challenging, and treatment is often not initiated until liver damage or cirrhosis has already developed13. Thus, an effective prophylactic HCV vaccine is urgently needed to achieve the WHO’s goal of hepatitis elimination by 203014,15.
HCV belongs to the Hepacivirus genus in the Flaviviridae family, which comprises small, enveloped, positive-sense, single-stranded RNA viruses that infect rodents, canines, primates, and other species16,17,18. The HCV genome encodes a single polyprotein that is processed into seven nonstructural (NS) proteins and three structural proteins, including the capsid-forming core protein and two envelope glycoproteins (Env) anchored in the viral membrane19,20,21. HCV entry into host hepatocytes is mediated by the Env heterodimer, E1E222,23, in which E2 binds cellular receptors CD81 and scavenger receptor class B member 1 (SR-B1)24, as well as the low-density lipoprotein receptor (LDLr) as a co-receptor25, while E1 closely associates with E2 to facilitate heterodimer formation, membrane fusion, and other steps of the viral life cycle26,27. The development of effective HCV therapeutics and vaccines has been hampered by the virus’s inherent genetic diversity, driven by low polymerase fidelity and rapid replication, as evidenced by the existence of eight genotypes and 93 subtypes28. Upon HCV infection, rapid mutation of E1E2 and NS5A helps establish a population of related but distinct quasispecies29, enabling viral evasion from host neutralizing antibodies (NAbs) and contributing to resistance to DAAs30. Nonetheless, HCV infection spontaneously clears in 20-25% of cases, with both T cells and NAbs contributing to effective virus control31. However, a recent phase 1/2 trial evaluating recombinant viral vectors encoding an NS-based antigen (NSmut) failed to prevent chronic HCV infection, highlighting the limitations of T-cell-focused vaccines32. Early NAb studies33,34,35,36,37,38,39 demonstrated the critical role of broadly neutralizing antibodies (bNAbs) in protection against HCV, a finding increasingly recognized in vaccine design 40,41,42,43,44.
Structural studies of antibody-bound Env proteins have revealed how the humoral immune system recognizes sites of HCV vulnerability, forming the basis for rational vaccine design against this highly mutable virus45,46,47. Peptide/NAb complex structures48,49,50,51,52 have provided insights into key linear neutralizing epitopes on E1 and E2, enabling epitope grafting onto nonviral protein scaffolds, including nanoparticles (NPs)53,54,55. The crystal structures of two E2 ectodomains—an E2 core (E2c) from the H77 isolate (genotype 1a) stabilized by bNAb AR3C, and a truncated E2 from the J6 isolate (genotype 2a) bound to the non-NAb 2A12—marked a milestone in HCV research56,57. Despite differences in variable region 3 (VR3) and disulfide bonding patterns, both structures shared a similar fold58. The crystal structure of a more complete E2 provided further insight into this critical viral glycoprotein59. Stable E2c constructs enabled the structural characterization of human bNAbs and the definition of major E2 epitopes47,60,61, including antigenic site 412–423 (AS412), antigenic site 434–446 (AS434, part of the E2 front layer [FL]), and antigenic region 3 (AR3). A conserved E2 surface comprising most of the FL and the CD81 binding loop (CD81bl) forms a neutralizing face (NF) that represents a major target of NAb responses during infection, although epitopes outside the NF also contribute to neutralization60,62. Notably, AR3-directed bNAbs preferentially use the VH1-69 gene in humans and equivalent heavy-chain variable (VH) genes in nonhuman primates (NHPs)63,64,65,66, supporting a germline-targeting strategy for AR3-focused HCV vaccine design41. Variability in epitope conformations and antibody binding modes has also been associated with NF recognition by VH1-69–derived NAbs63,66,67. This growing body of E2 structural data has guided the optimization of soluble E2 (sE2) and E2c constructs to enhance epitope-specific NAb responses and advance E2-based NP vaccines68,69,70. While E2 remains a major vaccine target, recent high-resolution cryo-electron microscopy (cryo-EM) structures of E1E2 have, for the first time, revealed atomic details of the E1 structure and the E1–E2 interface71,72,73, signaling renewed interest in E1E2-based HCV vaccines40,74,75,76. In the full-length E1E2 structure, the E1 and E2 stems form an extended interface positioned above the viral membrane72. Scaffolding the E1 and E2 ectodomains with a heterodimeric coiled coil yielded the first soluble E1E2 (sE1E2) reactive with the interface-specific bNAb AR4A33, which requires a native-like E1−E2 interface77,78, and enabled determination of a 3.65 Å cryo-EM structure of another sE1E2 scaffold71. The recently reported dimer-of-dimer E1E2 structure73 added further complexity to HCV vaccine design. Nonetheless, these structural advances have spurred efforts to develop E1E2 NP vaccines76,79,80, although E1E2-based HCV vaccine design remains in its early stages 74.
In our previous studies, we established a rational vaccine design strategy for class I fusion viruses81,82 by combining antigen optimization, protein NP display, and glycan modification. As demonstrated for the HIV-1 Env83,84,85, filovirus glycoprotein86,87, SARS-CoV-1/2 spike88, and RSV fusion protein89, we first identified the causes of metastability through structural analysis and stabilized the glycoproteins with targeted mutations. We then displayed these antigens on multilayered, single-component self-assembling protein NPs (SApNPs), which were derived from 60-mer bacterial proteins E2p and I3-01, as multivalent vaccine candidates68,83,84,86,87,88,90,91,92. Our recent work also demonstrated the benefits of glycan trimming and oligomannose enrichment for HIV-1 and filovirus vaccines, respectively83,86. Similarly, Kulakova et al. reported a sevenfold improvement in sE2-induced NAb responses using a glycoengineering approach93. Here, we adapted this rational vaccine design strategy to HCV, an elusive virus that employs a noncanonical fusion mechanism. Using available E1E2 structures71,72 as templates, we first truncated E1 at H312 and E2 at Y701 and deleted an unstructured E1 region encompassing the putative fusion peptide (pFP) in genotype 1a H77. The resulting sE1E2.Cut1 and sE1E2.Cut1+2 constructs were scaffolded with four heterodimeric leucine zippers of different sizes and an N-terminally truncated SpyTag/SpyCatcher94 (SPYΔN). All H77 sE1E2.Cut1+2 scaffolds bound with high affinity to the bNAb AR4A33, indicating a native-like E1–E2 interface.
Building on these results, we next designed a His6-tagged sE1E2.Cut1+2.SPYΔN construct for the genotype 1a strain HCV-1 and characterized it in detail. Cryo-EM analysis was performed on this HCV-1 sE1E2 scaffold in complex with bNAbs AR3C (or HEPC7459) and AR4A, and a negative-stain EM (nsEM) approach was evaluated for epitope mapping. Five design variants, including one with the restored IGH526 epitope on E1, were assessed using the HCV-1 sE1E2 backbone. HCV-1 sE1E2.Cut1+2.SPYΔN was displayed multivalently on ferritin 24-mer and I3-01v9a 60-mer91 platforms as virus-like particle (VLP) vaccines. Finally, we immunized mice to evaluate NAb responses to these rationally designed sE1E2 vaccines with and without glycan modification. The results showed that NP display and oligomannose enrichment improved NAb elicitation. Notably, although the E2 core elicited more potent NAb responses than sE1E2, nanoparticle display reversed this pattern and conferred a clear advantage to sE1E2. Together, these findings provide a strong foundation and a promising design strategy for the development of effective E1E2-based HCV vaccines.
Results
Rational design of HCV sE1E2 scaffolds with a native-like E1–E2 interface
HCV E1E2 is a noncanonical fusion glycoprotein with an unusual heterodimeric architecture95. Cryo-EM analysis of full-length E1E2 bound to three NAbs provided the first complete view of this elusive viral glycoprotein and paved the way for rational vaccine design74. Although an adjuvanted membrane-bound E1E2 vaccine has been tested in humans96, an sE1E2 construct with a native-like E1–E2 interface would, in principle, offer a more tractable antigen for vaccine development. Recently, heterodimeric coiled coils were used to scaffold HCV E1E2 ectodomains, resulting in measurable AR4A binding77 and enabling the first cryo-EM structure of an sE1E2 heterodimer71. However, AR4A coexpression was required to stabilize the E1–E2 interface and improve folding for both full-length E1E2 and a scaffolded sE1E2 antigen in structural studies71,72, suggesting intrinsic metastability within the E1E2 ectodomains that must be addressed. Our first goal was to design a stable sE1E2 heterodimer, referred to hereafter as a “heterodimer” or “dimer,” distinct from the recently reported homodimer of E1E2 heterodimers 73.
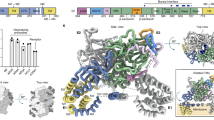
Here, we designed stable HCV sE1E2 scaffolds using a rational approach. Superposition of two cryo-EM structures71,72 revealed a similar E1E2 architecture despite different sequence backbones (Fig. 1a), with the E1 and E2 stems unresolved in the scaffolded sE1E271. The E1E2 of H77, a prototype genotype 1a isolate, was used to validate the sE1E2 construct design and heterodimeric scaffolds (Fig. 1b). For HCV Env, we hypothesized that truncation of the flexible E1 and E2 stems would enable precise structural control of sE1E2 and stabilize the E1–E2 interface, and that deletion of the unstructured pFP-containing E1 loop would reduce sE1E2 aggregation. To test this hypothesis, we truncated E1 at H312 and E2 at Y701 (termed “Cut1”) and replaced the E1 region L264–F293 with a glycine (termed “Cut2”) (Fig. 1b). For scaffolds, we further hypothesized that diverse heterodimeric coiled coils could accommodate sE1E2 and that a covalently linked heterodimer might provide the optimal protein scaffold to stabilize sE1E2 in an irreversible form. In addition to the previously reported LZ77, a 40-aa human c-Fos/c-Jun leucine zipper97, and SZ71, a 45-aa synthetic leucine zipper98, we identified a 30-aa GCP99 and a 21-aa IAAL100, both designed leucine zippers composed of acidic and basic chains. We previously used SpyTag/SpyCatcher94 to attach SARS-CoV-1/2 receptor-binding domains (RBDs) to protein NPs88. Here, we removed the unstructured N-terminus of SpyCatcher to create a covalently linked heterodimeric scaffold, termed SPYΔN. Ten constructs were designed to present H77 sE1E2.Cut1 and sE1E2.Cut1+2 on five scaffolds (LZ, SZ, GCP, IAAL, and SPYΔN), with a furin cleavage motif between sE1 and sE2 to promote native-like E1–E2 assembly (Fig. 1c and Fig. S1a). An LZ scaffold presenting H77 E1E2 ectodomains, termed sE1E2.LZ (Fig. S1a), and the original LZ-scaffolded H77 sE1E2 provided by Fuerst and co-workers77, termed sE1E2.LZorg, were included as controls. All in-house-designed sE1E2 scaffolds carried a C-terminal His6 tag to facilitate purification by immobilized metal affinity chromatography (IMAC).

a Cryo-EM structures of full-length E1E2 (PDB ID: 7T6X) and scaffolded sE1E2 (PDB ID: 8FSJ) are superimposed and shown as ribbon representations. E1 and E2 are colored magenta and blue in full-length E1E2 and orange and cyan in scaffolded sE1E2, respectively. Structurally resolved N- and C-termini, along with residues flanking the unstructured, putative fusion peptide (pFP)-containing E1 region, are labeled. b Amino acid sequences of genotype 1a H77 E1 and E2 ectodomains, highlighting stem truncation (Cut1) and replacement of the pFP-containing E1 loop with a glycine (Cut2). c Ribbon representation of a heterodimeric scaffold (top) and a schematic of the sE1E2.Cut1 (or sE1E2.Cut1+2) scaffold design (bottom). Scaffold subunits are colored pink and light blue; SpyTag002 is shown in maroon in SPYΔN. The furin cleavage motif (RRRRRR), peptide linkers (e.g., PGG and GSGS), and His6 tag are labeled. d SEC profiles of H77 sE1E2 scaffolds following transient expression in HEK293F cells and purification using a nickel column. Leftmost: sE1E2.LZ-His6; left to right: sE1E2.Cut1 (top) and sE1E2.Cut1+2 (bottom) on the LZ, SZ, GCP, IAAL, and SPYΔN scaffolds. Aggregate (A), oligomer (O), and dimer (D) peaks are labeled. SEC was performed for all constructs at least once for in vitro characterization and multiple times during protein production for animal studies. e Non-reducing SDS-PAGE analysis of H77 sE1E2 scaffolds. Leftmost: sE1E2.LZorg (Guest et al., PNAS 2021) and SEC fractions of in-house–produced sE1E2.LZ-His6 without AR4A coexpression; left to right: SEC fractions of sE1E2.Cut1 (top) and sE1E2.Cut1+2 (bottom) on the five scaffolds. Dissociated sE1 and sE2 bands are outlined with red boxes. SDS-PAGE was performed for all constructs at least once during screening, with selected constructs run on the same gel for comparison. f ELISA-derived EC50 values (µg/ml) for H77 sE1E2 scaffolds binding to bNAbs AR3C and AR4A. If OD450 < 0.5 at 10 µg/ml, binding was considered negligible and EC50 values were set to 10 µg/ml. Oligomer- and dimer-containing SEC fractions were pooled separately for ELISA analysis. Error bars represent the difference between duplicates at each concentration tested for each sample.
Eleven designed sE1E2 scaffolds were transiently expressed in 500-ml HEK293F cells, purified by IMAC using a nickel column, and analyzed by size-exclusion chromatography (SEC) on a Superdex 200 column (Fig. 1d). sE1E2.LZ showed an SEC profile with an aggregation (A) peak at ~8.2 ml and two small tailing peaks (Fig. 1d, leftmost). All sE1E2.Cut1 scaffolds, except for sE1E2.Cut1.LZ, showed improved yield with a high-molecular-weight (MW) oligomer (O) peak at ~10.6–10.8 ml and a potential dimer (D) peak at ~12.1–12.4 ml (Fig. 1d, top). Among the four leucine zippers, GCP produced the highest yield and the strongest dimer peak. SPYΔN showed a similar SEC profile to GCP, with an even more pronounced dimer peak, suggesting that dimeric sE1E2 was the predominant form with this covalently linked scaffold. Deletion of the pFP-containing E1 region (L264–F293) further improved yield and composition. As indicated by the SEC profiles, all five sE1E2.Cut1+2 scaffolds displayed a higher dimer-to-oligomer ratio, with GCP and SPYΔN showing the most evident improvement (Fig. 1d, bottom). Among these five scaffolds, sE1E2.Cut1+2.SPYΔN was the best performer, with a prominent dimer peak, only a trace amount of oligomeric species, and a relatively small aggregation peak. Next, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to analyze selected SEC fractions—two from the oligomer (O) peak and two from the dimer (D) peak for Cut1, and one from the O peak and three from the D peak for Cut1+2—across ten sE1E2 scaffolds. Two sE1E2.LZ antigens were included as controls for comparison (Fig. 1e). The LZ-scaffolded sE1E2 antigen reported by Guest et al.77, sE1E2.LZorg showed separate E1 and E2 bands but no detectable E1E2 band on the reducing gel, while the non-reducing gel displayed a large diffuse band at the top (Fig. 1e, leftmost). In contrast, sE1E2.LZ showed distinct bands on the non-reducing gel corresponding to multiple species (Fig. 1e, left). For all in-house-designed sE1E2 scaffolds, SEC fractions at ~12.1–12.4 ml produced a ~100 kDa band, consistent with the MW of a single sE1E2 dimer (D) calculated from its amino acid sequence and N-linked glycans (~3 kDa per glycan), whereas fractions at ~10.6–10.8 ml yielded a 150–250 kDa band, suggesting an sE1E2 oligomer (O). SDS-PAGE also revealed a scaffold-specific difference between the sE1E2.Cut1 and sE1E2.Cut1+2 constructs: for the four leucine zippers, dissociated E1 and E2 bands were visible on the reducing gels of sE1E2.Cut1+2 scaffolds, whereas for SPYΔN, no E1 or E2 bands were detected regardless of the E1 loop deletion. Because SPYΔN forms a covalent isopeptide bond94, only a single band remained even under reducing conditions (Fig. S1b).
We used bNAbs AR3C and AR4A in an enzyme-linked immunosorbent assay (ELISA) to validate the H77 sE1E2 scaffolds (Fig. 1f and Fig. S1c). AR3C targets a major epitope within the E2 NF formed by part of the FL (residues 426–443) and the tip of the CD81bl (residues 529-531)56, and represents a large family of AR3-directed human bNAbs of the VH1-69 origin41. AR4A is an E1E2-specific bNAb33 that recognizes an E2 epitope near the native E1–E2 interface71,72. AR4A has been used as an interface probe in binding assays to validate sE1E2 designs77,80 and as a chaperone in coexpression with E1E2 or sE1E2 to promote native-like E1–E2 association71,72,77. In the ELISA, the control sE1E2 antigen designed by Guest et al.77, sE1E2.LZorg, bound strongly to AR3C and AR4A with half-maximal effective concentration (EC50) values of 0.096 and 0.048 µg/ml, respectively, whereas the nickel-purified sE1E2.LZ-His6 bound poorly to AR3C and was barely recognized by AR4A (Fig. 1f and Fig. S1c). Among the ten designed sE1E2 scaffolds, the single sE1E2 form—expected from the cryo-EM structure and scaffold design—is the preferred species for vaccine development; other forms likely represent unintended assembly states (e.g., a dimer of heterodimers). Truncation of the E1 and E2 stems (Cut1) markedly improved bNAb binding for all five scaffolds except IAAL, yielding EC50 values of 0.072–0.148 for AR3C and 0.109–0.529 µg/ml for AR4A (Fig. 1f and Fig. S1c). Relative to sE1E2.LZorg77, the sE1E2.Cut1 scaffolds showed similar AR3C binding but weaker AR4A recognition, with EC50 values 2- to 10-fold higher. Further deletion of the pFP-containing E1 loop (Cut2) substantially increased bNAb recognition, with EC50 values for the resulting sE1E2.Cut1+2 scaffolds ranging from 0.010 to 0.027 for AR3C and from 0.007 to 0.025 µg/ml for AR4A (Fig. 1f and Fig. S1c). Based on AR4A binding alone, sE1E2.Cut1+2.GCP-His6 was the best performer, with a 7-fold higher affinity than sE1E2.LZorg77. sE1E2.Cut1+2.SPYΔN-His6 bound to AR3C and AR4A with similar affinities—3.6- and 2.0-fold higher than sE1E2.LZorg77, respectively—thus providing a balanced antigenic profile. For most sE1E2.Cut1 and sE1E2.Cut1+2 scaffolds, the oligomeric sE1E2 form showed reduced bNAb binding, likely due to steric hindrance.
Our results revealed critical elements for designing a stable, native-like sE1E2 antigen. Truncation of the sE1 and sE2 stems, together with deletion of the unstructured pFP-containing E1 loop, produced an optimal sE1E2.Cut1+2 construct for scaffolding. The beneficial effect of E1 loop deletion may be attributed to its hydrophobicity (~50%), which can promote aggregation or expose nonspecific epitopes, and to cysteines C272 and C281, which may form disulfide-linked sE1E2 oligomers with occluded bNAb epitopes. sE1E2.Cut1+2.GCP-His6 showed superior AR4A binding, suggesting that “archetypal” ~30-aa coiled coils may serve as effective scaffolds for presenting HCV sE1E2. Overall, sE1E2.Cut1+2.SPYΔN represents the most promising antigen in terms of expression yield, folding, stability, and epitope presentation.
In vitro characterization of select HCV sE1E2.Cut1+2.SPYΔN antigens
To advance an HCV vaccine construct to clinical trials, it is essential to screen diverse viral strains to identify an optimal vaccine backbone and to demonstrate that the antigen can be produced at scale using standard manufacturing systems101,102,103. Previously, two sE1E2 scaffolds derived from genotype 1a H77 and genotype 1b 1b09 were produced in Expi293F cells for in vitro characterization, cryo-EM analysis, and animal immunization71,77,78. In our recent studies, rationally designed vaccine candidates were produced in the transient Chinese hamster ovary cell (CHO) system, ExpiCHO68,83,84,86,87,88,89,90,91, providing guidance for future development in stable CHO cell lines under Good Manufacturing Practice (GMP) conditions. To evaluate compatibility with the CHO system, the five H77 sE1E2.Cut1+2 scaffolds were transiently expressed in 25-ml ExpiCHO cultures and purified by IMAC using a nickel column. Unexpectedly, the SEC profiles showed no discernible dimer peak, along with an overall low yield and the presence of lower-MW species (Fig. S1d), suggesting an incompatibility between the H77 strain and the CHO system.
To identify a suitable vaccine backbone, we extended the sE1E2.Cut1+2.SPYΔN design to genotype 1a HCV-1 and to two additional strains selected from an HCV E1E2 panel: genotype 3 UKNP3.1.2 and genotype 5 UKNP5.2.1104 (Fig. S2a). Notably, HCV-1 was used to develop the first HCV E1E2 vaccine candidate, which was GMP-produced in CHO cells and evaluated for safety and immunogenicity in a phase 1 clinical trial96. Briefly, the three sE1E2.Cut1+2.SPYΔN-His6 constructs were transiently expressed in either 500-ml HEK293F or 25-ml ExpiCHO cultures, followed by nickel purification and SEC analysis (Fig. 2a). All three strains showed higher dimer yield than H77, with HCV-1 sE1E2.Cut1+2.SPYΔN-His6 exhibiting the most favorable overall profile across cell lines. Multiple production runs of this HCV-1 sE1E2 scaffold showed overlapping dimer and high-MW peaks in the HEK293F-derived SEC profiles, but a consistent pattern of high yield and a predominant dimer peak at 13.1–13.3 ml in the ExpiCHO-derived profiles. Reducing SDS-PAGE confirmed that the fractions at 13.1–13.3 ml corresponded to the sE1E2 heterodimer, showing a band between 75 and 100 kDa on the gel (Fig. S2b). Despite its lower expression yield, the UKNP5.2.1 sE1E2 scaffold produced a distinct dimer peak in the SEC profile, suggesting that it is a suitable genotype 5 vaccine strain. Next, we applied differential scanning calorimetry (DSC) to assess the thermostability of the three sE1E2.Cut1+2.SPYΔN-His6 constructs expressed in two cell lines, for a total of six antigens (Fig. 2b). The thermostability of SpyTag/SpyCatcher has been studied in the context of a cyclized β-lactamase (BLA) fusion, with a melting temperature (Tm) of 85.4 °C reported for the reconstituted SPY domain105. Based on this study, the sharp peak at 91 °C observed in all six thermograms likely corresponds to rapid unfolding of SPYΔN, whereas the broad peaks in the 50–65 °C range may reflect melting of sE1E2, sE1, and sE2. We also observed a unique pattern for the HCV-1 sE1E2 scaffold produced in ExpiCHO cells: it exhibited two low-Tm transitions (50.1 and 58.7 °C), whereas the UKNP3.1.2 and UKNP5.2.1 scaffolds displayed a single low-Tm peak, suggesting strain-specific thermostability profiles.

a SEC profiles of HEK293F- and ExpiCHO-produced genotype 1a HCV-1, genotype 3 UKNP3.1.2, and genotype 5 UKNP5.2.1 sE1E2.Cut1+2.SPYΔN-His6 antigens. All antigens were purified using a nickel column. Results are shown for three HEK293F and four ExpiCHO production runs. b DSC thermograms of HEK293F- and ExpiCHO-produced HCV-1, UKNP3.1.2, and UKNP5.2.1 sE1E2.Cut1+2.SPYΔN-His6 antigens. All antigens were purified using a nickel column and SEC. Experimental data and Gaussian fits are shown as black dots and red lines, respectively. Thermal denaturation midpoints (Tm or Tm1–3) are labeled. c Site-specific glycan profiles for HCV-1 sE1E2.Cut1+2.SPYΔN-His6 antigens produced in HEK293F and ExpiCHO cells, with two glycan modifications (Kif and Kif/endo F) for the latter. Kifunensine (Kif) was added to ExpiCHO cell cultures to generate oligomannose-type glycans, which were trimmed by endo F1-3 to GlcNAc cores. Glycans are grouped as complex (solid pink), hybrid (pink lines), oligomannose (solid green), or unoccupied (solid gray); undetermined sites are labeled “N.D.” d ELISA-derived EC50 (µg/ml) values for unmodified, Kif-treated, and glycan-trimmed HCV-1 sE1E2.Cut1+2.SPYΔN-His6 binding to 11 NAbs and CD81-Fc. Antigens were produced in ExpiCHO cells and purified by a nickel column and SEC. When OD450 < 0.5 at 10 µg/ml, EC50 values were set to 10 µg/ml. e BLI profiles of unmodified, Kif-treated, and glycan-trimmed HCV-1 sE1E2.Cut1+2.SPYΔN-His6 binding to 10 NAbs (excluding murine NAb A4) and CD81-Fc. Sensorgrams were obtained on an Octet RED96 using a six-point antigen titration series (starting at 1000 nM, followed by 2-fold dilutions) and are shown in Fig. S2g. Peak responses at the highest concentration are summarized in a color-coded matrix (green to red).
The HCV glycan shield consists of ~15–16 glycans and masks conserved NAb epitopes on the E1E2 complex106,107. Site-specific glycan analysis has been performed for the bNAb-bound and unbound full-length AMS0232 E1E2 heterodimer using liquid chromatography-mass spectrometry (LC-MS)72. Our recent studies have demonstrated the differential effects of glycan modification on NAb responses induced by HIV-1 Env and filovirus GP vaccines83,86. Here, we examined the glycan shield on HCV-1 sE1E2.Cut1+2.SPYΔN-His6 produced in two different cell lines and subjected to two distinct glycan modifications. For the latter, kifunensine (Kif) was added to the ExpiCHO cell culture to enrich oligomannose content, and these N-glycans were further trimmed to GlcNAc stumps using endoglycosidase F1-3 (endo F1-3). Before glycan profiling, we assessed the quality of the glycan-modified antigens. In SEC, Kif had minimal impact on sE1E2 folding, yield, or purity, whereas endo F treatment shifted the dimer peak from ~13.0 to ~13.8 ml (Fig. S2c). In DSC, both Kif- and Kif/endo F-treated antigens yielded similar thermograms, with Tm values nearly identical to the unmodified material (Fig. S2d). The double-peak (Tm1/Tm2) pattern of sE1E2 unfolding appeared to be an intrinsic feature of this antigen―likely due to its strain and E1E2 sequence―when expressed in CHO cells (Fig. 2b, left and Fig. S2d). We next determined site-specific glycosylation and occupancy (Fig. 2c and Fig. S2e). Two aliquots of each antigen were digested separately with chymotrypsin and α-lytic protease to generate peptides and glycopeptides containing a single N-linked glycan site for LC-MS analysis. Both HEK293F- and ExpiCHO-produced sE1E2 displayed predominantly complex-type glycans, with slightly more oligomannose-type glycans in the ExpiCHO material. NeuGc- and NeuAc-containing glycans were absent in the ExpiCHO-produced antigen, whereas sialylated glycans were detected at every site in the HEK293F-produced sE1E2. High levels of fucosylation were observed in sE1E2 from both cell lines. As expected, Kif and Kif/endo F treatments converted most N-glycan sites into oligomannose-type glycans and GlcNAc cores, respectively (Fig. 2c, bottom). Glycan N305, which forms a salt bridge with E655 in full-length E1E272, could not be resolved by LC-MS analysis. Notably, Kif treatment yields the closest mimic of the native glycan shield on HCV virions, which is largely composed of oligomannose-type glycans 72.
HCV-1 sE1E2.Cut1+2.SPYΔN-His6 dimer antigens carrying unmodified, oligomannose-type (Kif-treated), and GlcNAc (Kif/endo F-treated) glycans were assessed against a panel of 11 NAbs, along with CD81 fused to the fragment crystallizable (Fc) region (CD81-Fc) (Table S1). These NAbs target diverse epitopes: one murine NAb recognizes an epitope on E1, whereas the others are directed against AR1–AR5 on E233,66,108. ELISA revealed a differential effect of glycan modification on sE1E2 binding (Fig. 2d and Fig. S2f). Relative to the unmodified antigen, Kif treatment modestly improved sE1E2 binding to CD81-Fc and AR3-directed NAbs, with negligible effects on other epitopes. In contrast, Kif/endo F treatment resulted in reduced binding to AR2-AR5 to varying degrees. Glycan trimming likely affected NAb binding to the E2 NF and AR4 through weakened glycan interactions and local conformational changes, respectively. The E2 NF is surrounded by N-glycans N417, N423, N430, N532, and N54060. Crystal structures showed that AR3C heavy and light chains each form a hydrogen bond with the N430 GlcNAc core56,68, and that HC1AM heavy and light chains engage the N423 and N532 GlcNAc cores through three hydrogen bonds67. These intact GlcNAc cores largely mitigated the adverse effect of glycan trimming on AR3 recognition. AR4 is an E1E2-specific epitope near the noncanonical N-glycan N695, which contains an NXV motif with ~25% occupancy72. A hydrogen bond between N695 and AR4A was observed in the cryo-EM structure of a scaffolded sE1E271, but not in the full-length E1E2 structure72. Because removal of this glycan site had a minimal impact on E1E2 antigenicity72, the 18-fold reduction in AR4A binding to endo F-treated HCV-1 sE1E2 likely reflects a conformational change at the E1–E2 interface. Biolayer interferometry (BLI) data were consistent with the ELISA results (Fig. 2e and Fig. S2g). AR5A showed the highest binding signals with plateaued dissociation. Notably, glycan trimming appeared to cause abnormalities in BLI responses at high antigen concentrations (Fig. S2g, left). Compared with the HEK293F-expressed H77 antigen (Fig. 1f and Fig. S1b), the ExpiCHO-expressed, unmodified or Kif-treated HCV-1 sE1E2.Cut1+2.SPYΔN-His6 bound to bNAbs AR3C and AR4A with up to 5.6- and 1.7-fold higher affinity, respectively, supporting its suitability as a vaccine antigen.
HCV-1 sE1E2.Cut1+2.SPYΔN emerged as a promising vaccine antigen. In addition to its high yield and purity when expressed in CHO cells, it exhibited a desirable antigenic profile, with strong binding to AR4A, multiple NAbs targeting diverse E1E2 epitopes, and CD81-Fc. Its modest thermostability could likely be improved through targeted stabilizing mutations. Most importantly, Kif treatment yielded high oligomannose content—a key feature of the E1E2 glycan shield on native HCV virions72—thereby warranting detailed in vivo evaluation.
Electron microscopy (EM) analysis of HCV-1 sE1E2.Cut1+2.SPYΔN bound to antibodies
While HCV E1E2 has resisted X-ray crystallography for decades, cryo-EM has enabled high-resolution structure determination of full-length E1E272 and scaffolded sE1E271, indicating that cryo-EM may offer critical structural insight into this elusive antigen. We first conducted cryo-EM analysis of HCV-1 sE1E2.Cut1+2.SPYΔN-His6 in complex with the fragment antigen-binding (Fab) regions of AR4A and HEPC74. A total of 5106 movies were collected on a Titan Krios 300 kV cryo-transmission electron microscope (cryo-TEM) equipped with a Gatan K3 camera. After movie processing in cryoSPARC109, particle populations of sE1E2.Cut1+2.SPYΔN-His6 bound by both antibodies were identified, confirming that the stabilized sE1E2 antigen is structurally compatible with recognition by both bNAbs. Selection of 36,896 particles for 3D classification enabled reconstruction of a density map at a moderate resolution of 6.30 Å (Fig. S3a and Table S2). Rigid-body fitting of the NAb-bound sE1E2.SZ structure (PDB ID: 8FSJ71) into the map revealed a similar overall conformation, despite differences in the E1E2 strain and construct (1b09 sE1E2 vs. HCV-1 sE1E2.Cut1+2) and scaffold (SZ vs. SPYΔN) (Fig. S3b). We next performed cryo-EM analysis of HCV-1 sE1E2.Cut1+2.SPYΔN-His6 bound to AR4A and AR3C Fabs. To mitigate orientation bias during sample vitrification, two different grid preparation platforms were used (see “Methods” section), resulting in datasets containing 19,080 and 5248 movies. After cryoSPARC processing109, two particle subsets (85,959 and 89,013 particles) were combined to generate a density map at a resolution of 4.97 Å (Fig. S3c and Table S2). This map enabled rigid-body fitting of the cryo-EM structure of sE1E2.SZ with AR4A (PDB ID: 8FSJ71) and the crystal structure of E2c with AR3C (PDB ID: 4MWF56), providing additional structural confirmation of the antigenic integrity of sE1E2.Cut1+2.SPYΔN-His6 (Fig. S3d). However, the resolutions obtained in these analyses did not permit atomic-level modeling of either complex.
Recently, we applied nsEM to obtain low-resolution (10–12 Å) models of NAb–antigen complexes to validate rationally designed viral antigens such as HIV-1 Env83, RSV F89, and ebolavirus GP86, and to identify NAb epitopes89. Here, we adapted this approach as a low-resolution alternative for antigen model building and NAb epitope mapping, using HCV NAbs with known complex structures as anchors for density fitting to determine both the position of the scaffold (e.g., SPYΔN) and the binding site of an NAb on the scaffolded sE1E2 structure. We first performed nsEM analysis of HCV-1 sE1E2.Cut1+2.SPYΔN-His6 bound to AR3C and AR4A Fabs individually and in combination. In each case, the complex was imaged on a Talos L120C TEM and processed with cryoSPARC to generate density maps suitable for rigid-body fitting (Fig. S3e). For AR3C, we generated a model complex by superimposing the crystal structure of the E2mc3-v1/AR3C complex (PDB ID: 6UYD68) onto the cryo-EM structure of a scaffolded sE1E2 (PDB ID: 8FSJ71). Upon fitting this model into the density map, we identified unoccupied density extending from the sE1E2 C-termini (E1-H312 and E2-Y701), into which the SPY domain structure (PDB ID: 4MLI94) fit well (Fig. 3a, left). This “complete” low-resolution complex confirmed that AR3C binds to the FL and CD81bl, consistent with high-resolution cryo-EM data71. Similarly, fitting the AR4A-bound sE1E2.SZ structures (PDB ID: 8FSJ71) and the SPY domain (PDB ID: 4MLI94) into the nsEM density produced a complete model for the AR4A-bound HCV-1 sE1E2.Cut1+2.SPYΔN-His6 complex (Fig. 3a, middle), confirming that AR4A binds to the E1–E2 bridging region in E2 (residues 646–701)71. Fitting both AR3C- and AR4A-bound HCV-1 sE1E2.Cut1+2.SPYΔN-His6 models into the density map of the ternary complex revealed nearly identical epitopes and angles of approach for the two Fabs, consistent with our cryo-EM analysis (Fig. 3a, right; Fig. S3c). These structural findings highlight the utility of HCV-1 sE1E2.Cut1+2.SPYΔN-His6 in nsEM-based antibody epitope mapping.

a Selected 2D class averages (top) and EM density maps from 3D reconstructions (bottom) of sE1E2 in complex with AR3C (left), AR4A (middle), or both AR3C and AR4A (right). Atomic models were fitted into the corresponding density maps to aid structural interpretation. b Selected 2D class averages (top) and 3D EM density maps (bottom) of sE1E2 bound to NAb combinations: AR1B and AR3C (left), AR5A and AR4A (middle), and AR2A, AR1B, and AR3C (right). In (a, b), maps and fitted models are shown in two orientations (rotated 180°) to provide alternative views of the sE1E2/NAb interfaces. c Epitope mapping of antigenic regions AR1–5 on the surface of HCV-1 sE1E2.Cut1+2.SPYΔN-His6. AR1, AR2, and AR5 epitope locations were inferred from negative-stain EM reconstructions of complexes with AR1B, AR2A, and AR5A, respectively. In (a–c), models were fitted into EM densities using the cryo-EM structure of engineered sE1E2.SZ bound to HEPC74 and AR4A (PDB ID: 8FSJ), the crystal structure of the E2mc3/AR3C complex (PDB ID: 6UYD), and the crystal structure of SpyTag/SpyCatcher (PDB ID: 4MLI).
We then extended this approach to AR1B, AR2A, and AR5A, whose epitopes have not been structurally defined on the native E1E2 heterodimer. Previous studies suggested that AR1B and AR2A bind E2, but their precise epitope locations remain unclear62. Using AR3C and/or AR4A Fabs as anchors, we fitted AR1B, AR2A, and AR5A Fab models into unoccupied regions within nsEM density maps generated for HCV-1 sE1E2.Cut1+2.SPYΔN-His6 bound to different NAb combinations. These models revealed putative epitope locations for each NAb on E1E2 (Fig. 3b, c, and Table S3). AR1B appeared to recognize a loop between β6 and β7, opposite the AR3C site, with 12 of 21 residues previously identified by mutagenesis34,108 falling within the nsEM-defined footprint. AR5A targeted a region near the E1–E2 interface, centered on residues R639 and L665 and extending toward the back layer (BL) of the heterodimer. Notably, both R639 and L665 were identified in earlier mutagenesis studies33,108. The model further indicated that the AR5A epitope is spatially distinct from that of AR4A, allowing both NAbs to engage E1E2 simultaneously without direct competition. However, the presence of one NAb may influence the approach angle or local interactions of the other, especially given the proximity of their footprints. This observation differs from previous mutagenesis analyses33,34,108, suggesting that some unrelated mutations may disrupt antibody binding indirectly by inducing conformational changes within the epitope. Finally, a quaternary complex containing AR1B, AR3C, and AR2A showed that AR2A binds the β11 strand, adjacent to the AR4A and AR5A epitopes, with 88% overlap with the mutagenesis-defined epitope34,108. Together, this nsEM-based epitope mapping approach provided valuable insights into how NAbs recognize antigenic regions on native E1E2.
Our nsEM analysis, supported by cryo-EM, revealed key structural and antigenic features of a rationally designed sE1E2 antigen. This SPYΔN-scaffolded sE1E2, which presents all five major ARs in a stable, native-like conformation, enables rapid epitope mapping via nsEM for antibodies isolated from infected individuals and immunized animals. The test cases of AR1B, AR2A, and AR5A serve as templates for nsEM-based NAb epitope mapping. However, this sE1E2 scaffold cannot be used to probe stem-directed NAbs, such as IGH526 38,50,72.
Impact of design variation on sE1E2 heterodimer folding, antigenicity, and structure
A central hypothesis in our structure-based sE1E2 design is that stem truncation (Cut1: E1-H312 and E2-Y701) enables precise structural control and stabilizes the E1–E2 interface through a dimeric scaffold. While the sE1E2.Cut1+2 construct performed robustly across multiple scaffolds, it was important to assess how changes in the truncation sites might affect dimer folding, structure, and antigenicity. To this end, we generated a panel of sE1E2 variants for comparative analysis. In the first set, we made subtle adjustments to the sE1 and sE2 anchoring sites by adding one or two downstream residues and modifying the linkers (Fig. 4a, left), yielding three variants (V1–V3). In the second set, we extended the sE1 and sE2 stems to include the IGH526 epitope50 and the complete E1 and E2 ectodomains (Fig. 4a, right), generating two additional variants (Ext1 and Ext2). All five constructs (Fig. S4a) were evaluated using the same experimental pipeline.

a Schematic of five sE1E2 variants derived from the sE1E2.Cut1+2.SPYΔN template. Sequence features used to adjust the anchoring sites (set 1: V1–V3) and C-terminal extensions (set 2: Ext1 and Ext2) are shown on the left and right, respectively. b SEC profiles of three anchoring-site variants (HCV-1 sE1E2.Cut1+2.SPYΔN-V1/2/3-His6; blue, left three) and two extension variants (HCV-1 sE1E2.Ext1/2.Cut2.SPYΔN-His6; red, right two). SEC traces from three production runs of HCV-1 sE1E2.Cut1+2.SPYΔN-His6 (black, dark gray, and light gray) is overlaid for comparison. All antigens were transiently expressed in 25-ml ExpiCHO cultures and purified using a nickel column. c SDS-PAGE analysis of HCV-1 sE1E2 variants. Left: non-reducing gels for anchoring-site variants V2 and V3; right: non-reducing and reducing gels for the extension variant Ext1. SEC fractions 11 and 17–19 were analyzed. Uncharacterized lower-molecular-weight bands are outlined with red boxes. d DSC thermograms of HCV-1 sE1E2 variants, including anchoring-site variants V2 (left) and V3 (middle), and extension variant Ext1 (right). Antigens were purified using a nickel column followed by SEC. Experimental data and Gaussian fits are shown as black dots and red lines, respectively; thermal denaturation midpoints (Tm or Tm1–3) are labeled. e nsEM analysis of HCV-1 sE1E2 variants. Representative 2D class averages and 3D reconstructions of HCV-1 sE1E2.Cut1+2.SPYΔN-His6 (left) and the sE1E2.Cut1+2.SPYΔN-V2-His6 variant (middle), each in complex with AR4A, and HCV-1 sE1E2.Ext1.Cut2.SPYΔN-His6 bound to AR3C and IGH526 (right). Atomic models of sE1E2 and AR4A (PDB ID: 8FSJ) and IGH526 (PDB ID: 4N0Y) were fitted into density maps to aid structural interpretation. The sE1E2.Cut1+2.SPYΔN-His6/AR4A complex is also shown in Fig. 3a. f ELISA-derived EC50 (µg/ml) values for HCV-1 sE1E2 variants binding to 12 NAbs (including IGH526) and CD81-Fc. All sE1E2 variants were produced in ExpiCHO cells and purified using a nickel column and SEC. When OD450 < 0.5 at 10 µg/ml, EC50 values were set to 10 µg/ml.
The five His6-tagged, SPYΔN-scaffolded sE1E2 variants were transiently expressed in 25-ml ExpiCHO cultures, purified using a nickel column, and analyzed by SEC (Fig. 4b). Among the three minor variants, V1 showed a substantial reduction in dimer yield, whereas V2 and V3 produced similar SEC profiles with a distinct dimer peak at ~12.8 ml and yields comparable to sE1E2.Cut1+2.SPYΔN-His6. Of the two extension variants, Ext1—which included the IGH526 epitope50 but not the full membrane-proximal external region (MPER)—retained a visible dimer peak, whereas Ext2, containing the full-length E1E2 ectodomains, did not. These results suggest that the MPER may interfere with dimer formation in the scaffolded format. Non-reducing SDS-PAGE showed clear bands between 75 and 100 kDa for V1 and V2 with minimal background (Fig. 4c), consistent with previous results for sE1E2.Cut1+2.SPYΔN-His6 (Fig. S2b). In contrast, the short extension in Ext1 produced two faint lower-molecular-weight bands (above and below 50 kDa) under both reducing and non-reducing conditions, suggesting proteolytic cleavage within the added sequence or reduced structural integrity. DSC analysis (Fig. 4d) revealed nearly identical thermograms for V2 and V3, each with two unfolding transitions at 52.5–54.5 °C (Tm1) and 89.6–90.5 °C (Tm2). Ext1, however, exhibited a broader transition between 50.0 and 57.7 °C, indicating that the C-terminal extension alters dimer stability. Because V2 and V3 behaved similarly, V2 was selected—together with Ext1—for structural characterization by nsEM (Fig. 4e). Micrographs from the earlier sE1E2.Cut1+2.SPYΔN-His6/AR4A analysis (Fig. 3a, middle) and new images of V2 in complex with AR4A Fab collected under identical conditions were processed for side-by-side comparison. The 2D class averages showed comparable complex morphology, and 3D reconstructions aligned closely with the cryo-EM model71, in which AR4A engages the E2 bridging region through its long heavy-chain complementarity-determining region 3 (HCDR3) loop, inserting as a wedge into the groove (Fig. 4e, left two panels). Ext1 in complex with IGH526 was analyzed using AR3C as an orientation reference, yielding a near-perfect fit to the cryo-EM model71. The unoccupied density extending from the C-terminus of the fitted sE1 structure corresponded to the IGH526 Fab, consistent with its defined epitope50 (Fig. 4e, right). Finally, the antigenicity of V1–V3 and Ext1 was assessed by ELISA, with IGH526 included to probe the extended epitope in Ext1 (Fig. 4f and Fig. S4b). Among the three minor variants, V2 and V3 showed NAb binding comparable to that of the parent sE1E2.Cut1+2.SPYΔN-His6, whereas V1 exhibited ~10-fold lower affinity for AR4A and AR5A, consistent with partial disruption of the E1–E2 interface. Ext1 bound most NAbs at levels similar to the parent construct and, as expected, only Ext1 was recognized by IGH526, with a moderate EC50 value of 0.21 µg/ml.
The results for these five constructs support our hypothesis that even small adjustments at the sE1E2 anchoring sites (e.g., V1) can markedly influence dimer formation, thermal stability, and the integrity of the E1–E2 interface. While Ext1 presents all known neutralizing epitopes of the virion-associated, full-length E1E2 heterodimer, its potential cleavage or reduced structural integrity observed by SDS-PAGE warrants further investigation. Therefore, sE1E2.Cut1+2.SPYΔN remains the optimal design for protein NP display and in vivo evaluation.
Rational design and in vitro characterization of HCV-1 sE1E2-presenting nanoparticles
Several strategies have been explored to develop HCV Env-based NP vaccines76. Previously, we designed a mini-E2 core, termed E2mc3, for strains from genotypes 1a and 6a, and displayed E2mc3 on a ferritin (FR) 24-mer as well as unmodified E2p and I3-01 60-mers68. The E2mc3-E2p 60-mer elicited potent NAb responses with a unique B-cell repertoire profile68. Yan et al. reported an sE2–FR NP with improved immunogenicity in mice69. Sliepen et al. designed an sE2E1 construct via permutation and displayed six different sE2E1 antigens on a two-component NP (2c-NP) platform, I53-50, for rabbit immunization80. Although this mosaic sE2E1 2c-NP induced cross-NAbs, the trimeric unit sE2E1-I53-50A failed to bind bNAbs AR4A33 and AT1618110, suggesting a non-native conformation80. Most recently, an engineered sE1E2.SZ heterodimer71, the HCV core protein, and a Toll-like receptor (TLR) 7/8 agonist were co-assembled with biodegradable polymers into nanocomplexes79. While this vaccine was highly immunogenic in mice, cryo-EM revealed an irregular shape due to random polymer assembly79. Thus, although these nanocomplexes approximate the size of a virion, they lack the ordered, symmetric assembly characteristic of native viruses. To date, no protein NP vaccine displaying a “native-like” sE1E2 heterodimer has been reported.
We aimed to develop such an sE1E2 NP vaccine by integrating sE1E2.Cut1+2.SPYΔN into FR and our multilayered 1c-SApNP platform83,87,88,91. Based on the nsEM analysis (Fig. 3), sE1E2.Cut1+2.SPYΔN adopts an elongated shape, with sE1Δ264–293 and sE2 tightly anchored to SPYΔN, which can be displayed on an SApNP by fusing the C-terminus of SpyCatcherΔN to the N-terminus of an NP subunit (Fig. 5a, left). We tested the FR 24-mer and the I3-01v9a 60-mer91 as candidate SApNP carriers. Notably, I3-01v9a has a multilayered design, with an inner layer formed by 60 locking domains (LDs) and a hydrophobic T-help core composed of 60 PADRE peptides83,87,88, and has been optimized for displaying monomeric antigens91. E2p, which has tightly clustered N-termini at each threefold axis on the NP surface, is better suited for trimer display and was therefore excluded from this study. To accommodate the non-symmetric sE1E2 shape and enhance epitope exposure, we inserted 10GS and 5GS spacers between SPYΔN and the NP subunit for FR and I3-01v9a-LD7-PADRE (or I3-01v9a-L7P91), respectively (Fig. S5a). Computational modeling estimated particle diameters of 36.9 and 47.0 nm for FR and I3-01v9a, respectively, measured at residue S432 of the E2 NF (Fig. 5a, middle and right).

a Left, schematic representation of sE1E2.Cut1+2.SPYΔN and its multivalent display on multilayered self-assembling protein nanoparticles (SApNPs); middle, surface model of HCV-1 sE1E2.Cut1+2.SPYΔN-10GS-FR, with a 36.9 nm diameter measured at residue E2-S432; right, surface model of HCV-1 sE1E2.Cut1+2.SPYΔN-5GS-I3-01v9a-LD7-PADRE (I3-01v9a-L7P), with a 47.0 nm diameter. b SEC profiles of ExpiCHO-produced, AR3C-purified HCV-1 sE1E2.Cut1+2.SPYΔN-10GS-FR with unmodified (left) and Kif-treated (right) glycans. Data are from three production runs. An EM micrograph corresponding to the ~10.6 ml SEC peak is shown. c DSC thermograms of ExpiCHO-produced, AR3C-purified HCV-1 sE1E2.Cut1+2.SPYΔN-10GS-FR with unmodified (left) and Kif-treated (right) glycans. Experimental data and Gaussian fits are shown as black dots and red lines, respectively. Thermal denaturation midpoints 1–3 (Tm1–3) and onset temperatures (Tonset) are labeled. d SEC profiles of ExpiCHO-produced, AR3C-purified HCV-1 sE1E2.Cut1+2.SPYΔN-5GS-I3-01v9a-L7P with unmodified (left) and Kif-treated (right) glycans. Data are from two production runs. An EM micrograph corresponding to the ~8–9 ml SEC peak is shown. e Particle size distributions of HCV-1 sE1E2.Cut1+2.SPYΔN-presenting FR (left) and I3-01v9a-L7P (right) with unmodified and Kif-treated glycans. Hydrodynamic diameters (Dh) were measured by DLS and are indicated. f Site-specific glycan profiles for unmodified (left) and Kif-treated (right) HCV-1 sE1E2.Cut1+2.SPYΔN-10GS-FR. Kifunensine (Kif) was added during expression to generate oligomannose-type glycans. Glycans are grouped as complex (solid pink), hybrid (pink lines), oligomannose (solid green), or unoccupied (solid gray); undetermined sites are labeled “N.D.” g ELISA-derived EC50 (µg/ml) values of unmodified (left) and Kif-treated (right) HCV-1 sE1E2.Cut1+2.SPYΔN-presenting FR and I3-01v9a binding to 11 NAbs and CD81-Fc. SApNPs were produced in ExpiCHO cells and purified using an AR3C antibody column and SEC. When OD450 < 0.5 at 10 µg/ml, EC50 values were set to 10 µg/ml. h BLI profiles of unmodified and Kif-treated HCV-1 sE1E2.Cut1+2.SPYΔN-10GS-FR SApNPs binding to 10 NAbs (excluding the murine NAb A4) and CD81-Fc. Sensorgrams were obtained on an Octet RED96 using a six-point antigen titration series (starting at 18 nM, followed by 2-fold dilutions) and are shown in Fig. S5f. Peak responses at the highest concentration are summarized in a color-coded matrix (green to red).
HCV-1 sE1E2.Cut1+2.SPYΔN-10GS-FR was transiently expressed in 100-ml ExpiCHO cultures with or without Kif. Using a previously described immunoaffinity chromatography (IAC) method86,88,91,111, we prepared an AR3C column to purify the sE1E2-presenting SApNPs. AR3C-purified protein from three production runs was analyzed by SEC on a Superose 6 10/300 column, and fractions of interest were further examined by nsEM (Fig. 5b). Regardless of Kif treatment or production run, all SEC profiles showed three distinct peaks at ~8.1, ~10.6, and ~17.0 ml, likely corresponding to aggregates, complete NPs, and unassembled subunits, respectively. nsEM analysis confirmed that the middle peak contained well-formed NPs with a recognizable FR core and surface decorations. Additional analyses were conducted to further characterize the ~8.1 ml peak relative to the NP peak at ~10.6 ml (Fig. S5b). Using either AR3C or AR4A for IAC purification, large NP aggregates or clusters were observed in the high-MW peak. DSC was used to assess the thermostability of unmodified and Kif-treated FR SApNPs (Fig. 5c). Consistent with the DSC data for individual sE1E2 antigens (Fig. 2b), the first peak represented sE1E2 unfolding (Tm1 ≈ 55 °C), whereas the second and third peaks corresponded to melting of FR (Tm2 ≈ 84 °C) and SPYΔN (Tm3 ≈ 90 °C), respectively. HCV-1 sE1E2.Cut1+2.SPYΔN-5GS-I3-01v9a-L7P was analyzed similarly by SEC and nsEM after transient expression in 150-ml ExpiCHO cultures, with and without Kif treatment, followed by AR3C purification (Fig. 5d). The yield of I3-01v9a was ~15-20-fold lower than FR across three production runs, likely reflecting the challenge of simultaneously displaying 60 native-like sE1E2 heterodimers per particle while assembling both an inner LD layer and a PADRE core. nsEM confirmed that the SEC peak at ~8–9 ml contained large particles with densely arrayed sE1E2 antigens. Due to the low yield, DSC was not performed for I3-01v9a. Reducing SDS-PAGE and blue native (BN) PAGE confirmed the MW of subunits and the purity of intact protein NPs, respectively (Fig. S5c). Both SApNPs showed bands at ~100–125 kDa on reducing gels, with the Kif-treated material migrating slightly higher. On the BN gel, FR produced a band above the 669 kDa marker, whereas I3-01v9a did not migrate from the well due to its large size. Particle size distributions for unmodified and Kif-treated FR and I3-01v9a were determined by dynamic light scattering (DLS) (Fig. 5e). Based on hydrodynamic diameter (Dh), FR averaged 33–34 nm, whereas I3-01v9a showed broader distributions centered at 59.6 and 65.2 nm for the unmodified and Kif-treated forms, respectively. The multilayered I3-01v9a 60-mer thus appeared more expanded in solution than the tightly packed FR 24-mer.
Site-specific glycosylation and occupancy were determined for unmodified and Kif-treated HCV-1 sE1E2.Cut1+2.SPYΔN-10GS-FR (Fig. 5f and Fig. S5d). Compared with individual sE1E2 (Fig. 2c), the unmodified sE1E2 FR displayed a higher proportion of oligomannose-type glycans, although complex-type glycans still predominated at some sites (Fig. 5f). The preferential binding of AR3C to Kif-treated sE1E2 (Fig. 2d) likely enriched oligomannose-type glycans in sE1E2 FR when purified using an AR3C column. Consistent with the individual sE1E2 analysis (Fig. 2c), Kif treatment effectively inhibited N-glycan processing on sE1E2 FR, producing GlcNAc2Man8 and GlcNAc2Man9 glycoforms primarily at each site with minimal impact on site occupancy. The antigenicity of HCV-1 sE1E2.Cut1+2.SPYΔN-presenting FR and I3-01v9a was assessed by ELISA (Fig. 5g and Fig. S5e). Relative to the individual sE1E2, multivalent display on the FR 24-mer improved NAb binding by up to 3.6- and 3.7-fold and CD81-Fc binding by 12.4- and 9.5-fold for the unmodified and Kif-treated forms, respectively. Notably, Kif-treated sE1E2 FR bound the E1E2-specific bNAbs AR4A and AR5A with 3.5- and 3.2-fold higher affinities, respectively, than the Kif-treated sE1E2 dimer. In most cases, I3-01v9a appeared less effective in ligand binding than FR and, in ~40% of cases, even less effective than individual sE1E2. Because of the low yield of I3-01v9a, only FR was further evaluated by BLI (Fig. 5h and Fig. S5f). FR showed the greatest improvement in binding to AR5A and RM2-01, with 2.0–2.7-fold higher signals than individual sE1E2. Kif treatment had little effect on binding signals in BLI measurements.
Our results indicate that sE1E2.Cut1+2.SPYΔN is optimal for designing HCV NP vaccines, as the covalent SPYΔN scaffold transforms sE1E2 from a metastable heterodimer into a stable monomeric unit that can be readily displayed on protein NPs. With the current construct design, the FR 24-mer with a 10GS spacer exhibited the most desirable in vitro properties, whereas the I3-01v9a 60-mer with a 5GS linker may require further optimization to improve production yield and surface display. Nonetheless, HCV-1 sE1E2.Cut1+2.SPYΔN-presenting FR and I3-01v9a SApNPs can provide valuable insights into immune responses induced by NP-based vaccines.
HCV-1 sE1E2.Cut1+2.SPYΔN heterodimer- and SApNP-induced NAb responses in mice
We assessed the immunogenicity of ExpiCHO-produced HCV-1 sE1E2.Cut1+2.SPYΔN vaccines, including a His6-tagged dimer and two SApNPs (FR and I3-01v9a-L7P), in mice (Fig. 6a). The antigens―either nickel/SEC-purified dimer or AR3C/SEC-purified SApNPs―were adjuvanted with aluminum hydroxide (AH) and a TLR9 agonist (CpG ODN 1826) to enhance vaccine-induced immune responses. Each dose contained 10 μg of antigen formulated with 100 μl of AH/CpG, for a final injection volume of 200 μl. Mice received four doses at weeks 0, 3, 6, and 9 via the intraperitoneal (i.p.) route. As reported in our previous early-stage vaccine design studies83,84,86,87,88, the i.p. route was used for its reproducibility and technical ease, while the short interval allowed rapid screening of multiple constructs. CpG was included as an add-on adjuvant based on its performance in our SARS-CoV-2 vaccine study111. Sera were collected two weeks after each immunization for HCV pseudoparticle (HCVpp) neutralization assays68. Five strains were included in the HCVpp panel: four representing each of the recently defined tiers112 and one from our previous study68. Specifically, these were UKNP5.2.1 (genotype 5, Tier 1), H77 (genotype 1a, Tier 2), UKNP4.2.2 (genotype 4, Tier 3), ED43 (genotype 4, from our previous panel68, tier unassigned), and UKNP3.1.2 (genotype 3, Tier 4). This small panel provides an initial assessment of NAb responses to the rationally designed sE1E2 constructs while offering some coverage of genotype diversity and neutralization sensitivity.

a Schematic of the mouse immunization regimen for HCV-1 sE1E2.Cut1+2.SPYΔN vaccines (n = 8 mice/group). Mice received 200 μl of antigen/AH + CpG containing 10 μg of antigen and 100 μl of adjuvant via intraperitoneal (i.p.) injection at weeks 0, 3, 6, and 9. b Longitudinal serum NAb responses induced by HCV-1 sE1E2, FR, and I3-01v9a vaccines against genotype 1a H77 HCVpp. Serum samples from weeks 0, 2, 5, 8, and 11 were tested in neutralization assays to determine 50% inhibitory dilution (ID50) titers. c Endpoint NAb responses induced by HCV-1 sE1E2, FR, and I3-01v9a vaccines against genotype 3 UKNP3.1.2, genotype 4 UKNP4.2.2 and ED43, and genotype 5 UKNP5.2.1 HCVpps. Week-11 serum was tested. NAb responses induced by unmodified, Kif-treated, and Kif/endo F-treated HCV-1 sE1E2.Cut1+2.SPYΔN dimer vaccines (produced in ExpiCHO cells) against d H77 HCVpp at weeks 2, 5, 8, and 11 and e UKNP3.1.2, UKNP4.2.2, ED43, and UKNP5.2.1 HCVpps at week 11. NAb responses induced by unmodified and Kif-treated HCV-1 sE1E2.Cut1+2.SPYΔN-10GS-FR SApNP vaccines against f H77 HCVpp at weeks 2, 5, 8, and 11, and g UKNP3.1.2, UKNP4.2.2, ED43, and UKNP5.2.1 HCVpps at week 11. NAb responses induced by unmodified and Kif-treated HCV-1 sE1E2.Cut1+2.SPYΔN-5GS-I3-01v9a-L7P SApNP vaccines against h H77 HCVpp at weeks 2, 5, 8, and 11 and i UKNP3.1.2, UKNP4.2.2, ED43, and UKNP5.2.1 HCVpps at week 11. In (b–i), ID50 titers were calculated from HCVpp neutralization assays (0–100% constraint), with geometric means labeled. Data points are shown as geometric mean ± geometric SD. Data were analyzed using one-way ANOVA followed by Tukey’s post hoc test (each time point) or two-tailed unpaired t-tests (two-group comparisons). Statistical significance is indicated as ns (not significant) and *p < 0.05. The schematic in (a) was created in BioRender. https://BioRender.com/o89v779.
We first conducted a longitudinal analysis of serum neutralization, using 50% inhibitory dilution (ID50) to assess NAb responses. Baseline (week 0) sera from all groups showed no activity against genotype-1a H77 HCVpp, and no NAb response was detected at week 2. However, ID50 titers against H77 HCVpp were observed at weeks 5, 8, and 11 (Fig. 6b and Fig. S6a, b). At week 5, FR and I3-01v9a SApNPs showed ID50 titers of 817 and 670, respectively—45.4- and 37.2-fold higher than the dimer. At week 8, NAb responses in the FR and I3-01v9a groups peaked at ID50 titers of 1253 and 1251 after three doses. In contrast, the dimer group peaked after four doses, reaching an ID50 titer of 589 at week 11, which remained lower than both SApNP groups. Week-11 sera were also tested against heterologous UKNP3.1.2, UKNP4.2.2, ED43, and UKNP5.2.1 HCVpps (Fig. 6c and Fig. S6c, d). While none of the vaccines elicited NAbs against UKNP3.1.2, the I3-01v9a 60-mer induced the highest ID50 titers of 70, 145, and 1030 against UKNP4.2.2, ED43, and UKNP5.2.1, representing 1.9-, 2.8-, and 17.4-fold increases over the dimer. These results demonstrate the benefit of NP display of a stable, native-like sE1E2 antigen in enhancing NAb responses. The heterologous ID50 titers correlated with the titer classification of Salas et al.112, with UKNP3.1.2 (Tier 4) being the most resistant and UKNP5.2.1 (Tier 1) the most sensitive to neutralization. Notably, despite the suboptimal antigenicity of the I3-01v9a 60-mer (Fig. 5g, h), its large particle size (~50 nm) may have contributed to the elicitation of cross-NAbs.
We next assessed the immunogenicity of HCV-1 sE1E2.Cut1+2.SPYΔN vaccines carrying oligomannose-type (Kif-treated) and GlcNAc core (Kif/endo F-treated) glycans in mice. We first evaluated NAb responses induced by the sE1E2 dimer vaccines against H77 HCVpp (Fig. 6d and Fig. S6e, f). None of the dimer groups showed a NAb response at week 2, but all three groups exhibited detectable ID50 titers at weeks 5, 8, and 11. Among them, the glycan-trimmed dimer elicited the lowest ID50 titers at weeks 8 and 11. The unmodified and Kif-treated dimers produced comparable ID50 values of 570-589 at week 11, which were 3.8–3.9-fold higher than those of the glycan-trimmed dimer. Week-11 sera were also assessed against UKNP3.1.2, UKNP4.2.2, ED43, and UKNP5.2.1 HCVpps (Fig. 6e and Fig. S6g, h). Consistent with H77 neutralization (Fig. 6d), the unmodified and Kif-treated dimers elicited higher ID50 titers against UKNP4.2.2, ED43, and UKNP5.2.1 than the glycan-trimmed dimer, although differences were not statistically significant. For both FR and I3-01v9a SApNPs, unmodified and Kif-treated groups yielded comparable ID50 titers across all time points and all five HCVpps tested in this mouse study (Fig. 6f–i and Fig. S6i–l). Both unmodified and Kif-treated SApNP groups peaked in NAb responses against H77 HCVpp after three doses, with ID50 values ranging from 1105 to 1275 (Fig. 6f, h). Neutralization assays also revealed measurable NAb titers in these SApNP groups against UKNP4.2.2, ED43, and UKNP5.2.1 HCVpps, consistently higher than those of the corresponding dimer groups (Fig. 6e). However, the Kif-treated I3-01v9a SApNP yielded lower ID50 titers than expected based on its unmodified counterpart and the trend observed for the FR groups (Fig. 6i). The Kif-treated sE1E2 vaccines, in both dimer and SApNP forms, produced tier-dependent ID50 patterns112, with no group showing detectable neutralization against the Tier 4 strain UKNP3.1.2.
Overall, the sE1E2 heterodimer and its SApNPs elicited NAb titers in a manner dependent on both antigen size and viral tier. Oligomannose enrichment provided a modest advantage over unmodified glycans, consistent with our filovirus vaccine study86. Kulakova et al. reported enhanced NAb titers induced by a glycoengineered sE2 with shortened glycans93, indicating that the impact of glycan modification is context-dependent (sE2 vs. sE1E2) and may reflect a trade-off between the loss of glycan-dependent epitopes and exposure of non-neutralizing sites.
Effect of experimental factors on sE1E2 heterodimer- and SApNP-induced NAb responses
We examined the potential effects of the expression system, sex-specific differences, and injection route on NAb responses induced by HCV-1 sE1E2.Cut1+2.SPYΔN vaccines. The protocol used in our earlier immunizations (Fig. 6a) was adopted to test these experimental variables (Fig. 7a). The data from additional mouse immunization experiments were compared with those from the initial evaluation of the HCV-1 sE1E2-based vaccine constructs (Fig. 6).

a Schematic of the mouse immunization regimen for HCV-1 sE1E2.Cut1+2.SPYΔN vaccines (n = 8 mice/group). Mice received 200 μl of antigen/AH + CpG containing 10 μg of antigen and 100 μl of adjuvant via intraperitoneal (i.p.) injection, or 80 μl of antigen/AH containing 10 μg of antigen and 40 μl of adjuvant via intradermal (i.d.) injection into four footpads, at weeks 0, 3, 6, and 9. NAb responses induced by unmodified, Kif-treated, and Kif/endo F-treated HCV-1 sE1E2.Cut1+2.SPYΔN dimer vaccines (produced in HEK293F cells) against b H77 HCVpp at weeks 2, 5, 8, and 11 and c UKNP3.1.2, UKNP4.2.2, ED43, and UKNP5.2.1 HCVpps at week 11. NAb responses induced by unmodified and Kif-treated HCV-1 sE1E2.Cut1+2.SPYΔN dimer vaccines (produced in ExpiCHO cells; independent mouse study) against d H77 HCVpp at weeks 2, 5, 8, and 11 and e UKNP3.1.2, UKNP4.2.2, ED43, and UKNP5.2.1 HCVpps at week 11. NAb responses induced by unmodified and Kif-treated HCV-1 sE1E2.Cut1+2.SPYΔN-10GS-FR SApNP vaccines (produced in ExpiCHO cells; independent mouse study) against f H77 HCVpp at weeks 2, 5, 8, and 11 and g UKNP3.1.2, UKNP4.2.2, ED43, and UKNP5.2.1 HCVpps at week 11. h NAb responses induced by unmodified and Kif-treated HCV-1 sE1E2.Cut1+2.SPYΔN dimer vaccines in female and male mice. Antigens were produced in ExpiCHO cells, adjuvanted with AH, and administered via footpad injections. Week-11 serum was tested against H77 HCVpp. i Sex-dependent germinal center (GC) responses induced by HCV-1 sE1E2.Cut1+2.SPYΔN adjuvanted with AH in female and male mice. Percentages and numbers of GC B cells (left) and Tfh cells (right) are shown. ID50 titers were calculated from HCVpp neutralization assays (0–100% constraint). Data points in the neutralization plots are shown as geometric mean ± geometric SD. Statistics: one-way ANOVA with Tukey’s post hoc test (each time point) or two-tailed unpaired t-test (two-group comparisons); ns (not significant), *p < 0.05, **p < 0.01. The schematic in (a) was created in BioRender. Zhang (2026) https://BioRender.com/o89v779.
We first tested the immunogenicity of HEK293F-produced HCV-1 sE1E2.Cut1+2.SPYΔN vaccines with and without glycan modification (Fig. 7b and Fig. S7a, b). Each injection dose contained 10 μg of HEK293F-produced antigen formulated with 100 μl of AH/CpG adjuvant. Sera were collected two weeks after each dose, and ID50 titers were plotted for comparison. None of the dimer groups showed any NAb response against H77 HCVpp at week 2, but ID50 titers were detected in all groups at weeks 5, 8, and 11. The glycan-trimmed dimer group elicited the lowest ID50 titers at weeks 8 and 11, whereas the Kif-treated dimer group showed the highest ID50 titers—376 and 295—at these time points. Week-11 mouse sera were also assayed against UKNP3.1.2, UKNP4.2.2, ED43, and UKNP5.2.1 HCVpps to evaluate heterologous NAb responses (Fig. 7c and Fig. S7c, d). The unmodified and Kif-treated dimer groups produced robust responses against all four HCVpps, with ID50 titers that were 2.5- to 16.2-fold higher than those of the glycan-trimmed group, although the differences were not statistically significant. NAb responses induced by sE1E2 dimers from both cell lines followed similar patterns, except in the case of UKNP3.1.2 (genotype 3, Tier 4) HCVpp neutralization. Overall, the HEK293F-produced dimers appeared to elicit more effective cross-genotype NAb responses than those from ExpiCHO production. While promising, this observation warrants further validation.
Next, we performed additional immunizations as experimental repeats to further examine the effect of glycan modification on vaccine-induced NAb responses. The ExpiCHO-produced HCV-1 sE1E2.Cut1+2.SPYΔN antigen (10 μg), either as a dimer (unmodified vs. glycan-trimmed) or as an FR SApNP (unmodified vs. oligomannose-type), was formulated with AH/CpG adjuvant and administered intraperitoneally to female mice. For the dimer, measurable ID50 titers against H77 HCVpp were detected in both unmodified and glycan-trimmed groups at weeks 8 and 11 (Fig. 7d and Fig. S7e). The unmodified dimer elicited ID50 titers of 603 and 652 at weeks 8 and 11, respectively—7.2- and 3.8-fold higher than the glycan-trimmed dimer. It also induced higher NAb titers against UKNP4.2.2, ED43, and UKNP5.2.1 HCVpps than the glycan-trimmed dimer, although the differences were not statistically significant (Fig. 7e and Fig. S7f). These results were consistent with our earlier immunization studies (Fig. 6d, e). The Kif-treated FR group showed higher ID50 titers against H77 HCVpp than the unmodified FR group at all time points, and higher titers against UKNP3.1.2, UKNP4.2.2 (statistically significant), ED43, and UKNP5.2.1 HCVpps at week 11 (Fig. 7f, g and Fig. S7g, h). The beneficial effect of oligomannose glycans was more evident in this analysis, although still observational.
Lastly, we assessed the influence of sex on vaccine-induced NAb responses113,114. A modified regimen was used to immunize female and male mice (Fig. 7a). The ExpiCHO-produced HCV-1 sE1E2.Cut1+2.SPYΔN antigen, adjuvanted with AH alone, was intradermally injected into four footpads. Each injection consisted of 80 μl of antigen-adjuvant mix containing 10 μg of dimer antigen, evenly distributed across the four footpads. This route was used hereafter to reduce experimental variability by delivering the vaccine across four injection sites with a common adjuvant, thereby facilitating baseline comparison and mechanistic analysis. Sera collected at week 11, after four injections, were tested against H77 HCVpp. Within mice of the same sex, unmodified and Kif-treated dimer groups elicited comparable NAb titers, with no statistical significance (Fig. 7h and Fig. S7i, j). Both unmodified and Kif-treated dimer vaccines induced stronger NAb responses in female mice than in male mice. Notably, the unmodified dimer group yielded an ID50 titer of 211 in female mice, 3.8-fold higher than in males. This result aligns with our previous study of stabilized RSV prefusion F trimer vaccines, where females developed stronger NAb responses than males following vaccination89. Additionally, the unmodified and Kif-treated FR groups showed higher ID50 titers than their respective dimer groups at week 11 after four intradermal doses in female mice (Fig. S7k), consistent with our earlier immunization results (Fig. 6). To gain mechanistic insight, we assessed germinal center (GC) responses by flow cytometry, focusing on T follicular helper (Tfh) and GC B cells, which are known to contribute to sex-dependent immune responses113,114,115. Five female and five male mice were immunized intradermally with 10 μg of dimer antigen mixed with AH through the four footpads. Fresh lymph node tissues collected at week 2 post-immunization were processed into single-cell suspensions and stained with an antibody cocktail. GC responses were quantified by both the frequency and absolute number of GC B and Tfh cells (Fig. 7i and Fig. S7l). Lymph node tissues from females showed similar frequencies of GC B and Tfh cells to males but exhibited a 1.6-fold higher absolute number of both GC B and Tfh cells, consistent with previous findings in the field113,114,115.
Our results provide insights into factors that may influence vaccine-induced NAb responses. The ExpiCHO-produced HCV-1 sE1E2 dimer induced a more potent NAb response against H77 HCVpp (genotype-matched), whereas the HEK293F-produced dimer elicited higher ID50 titers against HCVpps from other genotypes. Female mice generated more robust NAb responses than males, a finding supported by GC analysis, with all immunizations conducted using a standardized baseline regimen (i.e., intradermal footpad injections with AH). Using this regimen, we confirmed the beneficial effects of particulate display of sE1E2 and oligomannose enrichment.
Comparative analysis of HCV-1 sE1E2 and E2mc3 immunogens and their SApNPs
We previously reported a structurally optimized E2 core (E2mc3), which, when displayed on the unmodified E2p SApNP, elicited robust NAb responses with a unique B-cell repertoire profile68. Here, we performed mouse immunizations to compare E2mc3 with sE1E2.Cut1+2.SPYΔN and their corresponding FR SApNP constructs to inform future HCV vaccine development.
E2mc3 and E2mc3-10GS-FR, derived from the HCV-1 strain (Fig. S8a), were transiently expressed in 25- and 100-ml ExpiCHO cultures, respectively, and purified by IAC using an AR3C column. In SEC, the E2mc3 monomer eluted as a prominent peak at ~10.6 ml on a Superdex 75 column and showed a clear ~37 kDa band on the reducing SDS-PAGE gel (Fig. S8b). The E2mc3-presenting FR also expressed well, yielding a particle peak centered at ~11.8 ml on a Superose 6 column with a distinct higher-molecular-weight band (~60 kDa) on the gel (Fig. S8c). Consistent with modeling, nsEM revealed well-formed FR particles with a dense layer of surface decorations (Fig. 8a; Fig. S8c, right). Antigenicity was assessed by ELISA using four E2-specific NAbs and CD81-Fc (Fig. 8b; Fig. S8d). Both constructs displayed the expected NAb reactivity, but the FR SApNP showed markedly higher apparent affinity, with EC50 values 10.4–17.2-fold lower than those of E2mc3, reflecting a strong avidity effect. Notably, whereas E2mc3 exhibited no detectable CD81-Fc binding, E2mc3-10GS-FR showed measurable CD81 engagement with an EC50 of 0.12 μg/ml. This suggests that deletion of variable regions and stabilization of the remaining segments may alter how E2mc3 undergoes conformational changes during CD81 binding116,117, an effect that may be partially compensated by avidity on the particle surface.

a Surface model of the HCV-1 E2mc3-10GS-FR SApNP (left) and representative nsEM micrograph of ExpiCHO-expressed, AR3C/SEC-purified E2mc3-10GS-FR SApNPs (right). The FR scaffold is shown in gray, and E2mc3 in green. b ELISA-derived EC50 (µg/ml) values for HCV-1 E2mc3 and E2mc3-10GS-FR binding to four NAbs and CD81-Fc. Antigens were produced in ExpiCHO cells and purified using an AR3C antibody column and SEC. When OD450 < 0.5 at 10 µg/ml, binding was considered negligible, and EC50 values were set to 10 µg/ml. c NAb responses induced by HCV-1 E2mc3, E2mc3-10GS-FR, sE1E2.Cut1+2.SPYΔN-His6, sE1E2.Cut1+2.SPYΔN-10GS-FR, and sE1E2.Ext1.Cut2.SPYΔN-His6 vaccines against H77 HCVpp at week 11. ID50 titers were calculated from HCVpp neutralization assays (0–100% constraint). Data points are shown as geometric mean ± geometric SD. Data were analyzed using one-way ANOVA followed by Tukey’s post hoc test, or two-tailed unpaired t-tests for two-group comparisons. Statistical significance is indicated as ns (not significant).
Mice were then immunized with HCV-1 E2mc3, sE1E2.Cut1+2.SPYΔN-His6, their corresponding FR SApNPs, and sE1E2.Ext1.Cut2.SPYΔN-His6 (Fig. 4), all formulated with AH and administered intradermally through footpads (Fig. 7a). Week-11 sera from the two sE1E2 dimer groups showed the lowest ID50 titers against H77 HCVpp, whereas E2mc3 induced ~2-fold higher potency (Fig. 8c; Fig. S8e, f). SApNP display enhanced ID50 titers for both antigen types but provided the greatest boost to sE1E2, with sE1E2-10GS-FR achieving the highest mean ID50 value of 284. Week-11 sera showed negligible neutralization of UKNP3.1.2, UKNP4.2.2, ED43, and UKNP5.2.1 HCVpps (Fig. S8g), most likely due to the weak AH adjuvant, although the injection route may also have contributed. Nonetheless, this baseline study provided valuable insight: while E2mc3 was more potent in soluble form, particulate display conferred the greatest benefit to sE1E2 in eliciting autologous NAb responses. The results also suggested that a more potent adjuvant might be required to improve neutralizing breadth in future studies.
Discussion
Hepatitis C poses a significant threat to public health worldwide1,118. If acute HCV infection is not spontaneously cleared, it can progress to a persistent infection that may lead to severe liver disease119. Because early HCV infection and associated liver disease rarely present clinical signs, serious liver damage often remains undiagnosed until advanced stages. Injection drug use and opioid use have fueled the spread of HCV in North America and Europe, causing a “silent epidemic” with substantial economic, social, and healthcare burdens120. Although DAAs are effective in treating chronic HCV infection4,5,6, they cannot prevent reinfection or reverse liver damage9,10,11. Therefore, to meet the WHO goal of a 90% reduction in new hepatitis infections by 2030, the development of a safe and effective HCV vaccine remains an urgent priority.
Structure-based design has become a cornerstone of modern vaccine research121,122,123,124, accompanied by a growing trend toward using engineered protein NPs for more efficient delivery125,126,127. In HCV vaccine development, immunogens based on individual epitopes, E2, and E1E2 have been designed to induce NAb responses against diverse HCV strains and quasispecies40,45,61,62. However, the lack of structural information on the E1E2 heterodimer has long impeded the rational design of E1E2-based HCV vaccines. Recently, cryo-EM structures have been reported for full-length E1E272, scaffolded sE1E271, and a dimer of E1E273, paving the way for structure-guided vaccine design. Previously, we established a rational vaccine strategy for class I viral glycoproteins by combining antigen optimization based on metastability analysis, antigen display on multilayered SApNPs, and glycan modification86,87,88,89,90. In this study, following a conceptually similar strategy, we designed stable sE1E2 antigens and sE1E2-presenting SApNPs for in vitro and in vivo characterization to support future HCV vaccine development.
HCV E1E2 poses a unique challenge to antigen optimization74. Unlike class I viral glycoproteins, which form homotrimers of heterodimers, HCV E1E2 is an asymmetric heterodimer known for its conformational disorder. In the case of HIV-1 Env, truncation and redesign of the N-terminus of heptad repeat 1 (HR1N) eliminated a major source of metastability and stabilized the trimeric Env84,85. However, the lack of symmetry in HCV E1E2 precludes such a design strategy. Although E2 contains a compact core formed by a disulfide-locked immunoglobulin (Ig) β-sandwich fold56, E1E2 remains intrinsically flexible due to variable regions 1–3 (VR1–3) in E2 and a loosely folded E171,72, with nearly half (~48%) of the protein unstructured, as indicated by DSSP analysis128. Genetic diversity and a dense glycan shield further complicate E1E2 optimization74. Here, we undertook a two-pronged approach to address these challenges: optimizing the sE1E2 construct for improved stability and identifying a heterodimeric scaffold suited to accommodate sE1E2. For the former, while stem truncation was beneficial, deletion of the unstructured pFP-containing region in E1 proved most critical for preserving a native-like E1–E2 interface. For the latter, although a heterodimeric coiled coil of canonical length, GCP, outperformed other coiled coils, a covalently linked SPYΔN demonstrated a more balanced in vitro profile (e.g., expression yield, purity, stability, and antigenicity). Structural information on E1E271,72 guided the selection of optimal cutting sites on E1E2 and anchoring sites on each scaffold. The lead antigen, HCV-1 sE1E2.Cut1+2.SPYΔN was extensively characterized using biochemical, biophysical, antigenic, and structural methods. Design variants based on this template yielded an sE1E2 construct containing all known NAb epitopes, including the IGH526 epitope 50.
Findings from our sE1E2 design and assessment offer important insights into HCV vaccine development based on these antigens. First, the sE1E2.Cut1+2 construct design is applicable across diverse HCV strains. Genotype 1a H77 and HCV-1 were more extensively evaluated than strains from genotypes 3 and 5. Among these constructs, HCV-1 sE1E2.Cut1+2.SPYΔN-His6 showed the best performance when produced in ExpiCHO cells, making it a promising vaccine antigen. The poor CHO expression of five H77 sE1E2.Cut1+2 scaffolds suggested a strain- rather than design-specific limitation. Thus, the sE1E2.Cut1+2 construct can be used to screen other strains to identify optimal vaccine backbones. Second, sE1E2.Cut1+2 scaffolds do not require AR4A to improve expression yield or preserve a native-like E1–E2 interface. In the first sE1E2 scaffolding study that yielded positive AR4A binding, a standard expression and purification protocol was used77. In more recent studies, full-length E1E2 and another sE1E2 scaffold were coexpressed with AR4A to enhance folding and yield71,72. The intrinsic stability and high yield of sE1E2.Cut1+2 will facilitate the development of a robust downstream process for large-scale GMP production. Third, Kif treatment offers a close glycan mimic of the native E1E2 heterodimer on virions72. The transmembrane domains (TMDs) of E1 and E2 contain a signal peptide that causes membrane-bound E1E2 to be statically retained in the endoplasmic reticulum (ER), resulting in stunted glycan processing and underprocessed oligomannose-type glycans. Without TMDs, sE1E2 scaffolds expressed in HEK293F and ExpiCHO cells present mostly complex-type glycans; however, Kif treatment effectively inhibits glycan processing and restores the oligomannose-type glycan profile. The positive effect of Kif treatment on antigenicity was associated with improved immunogenicity, although the differences in NAb responses were not statistically significant. This observation is consistent with findings in our recent filovirus vaccine study86. Lastly, sE1E2.Cut1+2.SPYΔN-His6 provides an excellent E1E2 probe for nsEM-based epitope mapping. Although we were unable to determine high-resolution cryo-EM structures, we showed that antibodies with known E1E2 or E2 complex structures can serve as anchors in nsEM analysis to map the epitopes of uncharacterized antibodies onto HCV-1 sE1E2.Cut1+2.SPYΔN-His6, a native-like E1E2 probe. In low-resolution nsEM, the globular SPYΔN is more discernible than a coiled coil and provides a distinct structural feature to orient the sE1E2 heterodimer within the EM density.
HCV E1E2 also presents a unique challenge for particulate display. The difficulties stem from HCV E1E2 being a non-covalent heterodimer, which is incompatible with highly symmetric NPs presenting 2-fold, 3-fold, and 5-fold axes on the surface129. Previously, we reported that diverse class I viral glycoproteins can be displayed on 24-meric FR and 60-meric E2p and I3-01v9 by fusing the C-termini of these trimeric antigens with the N-termini of the NP subunits, which are arrayed around threefold axes on the NP surface83,84,86,87,88,92. We also redesigned the I3-01v9 SApNP (termed I3-01v9a) to optimize surface display of monomeric antigens, such as the extracellular domain of the influenza M2 protein (M2e)90,91. Recently, Sliepen et al. designed and assessed the immunogenicity of an sE2E1 2c-NP in rabbits80. The vaccine potential of this immunogen design was dampened by the inability of the sE2E1-I53-50A subunit to bind AR4A and by the assumption that HCV E1E2 may form trimers of heterodimers on the virions, which is the basis for the proposed NP display80. In addition, proteolytic cleavage between E2 and E1 may result in misfolded sE1 stumps mixed with sE2E1 antigens on the I53-50 2c-NP surface80. In this study, we overcame these challenges by displaying sE1E2.Cut1+2.SPYΔN, a covalently linked heterodimer (equivalent to a monomer) with a native-like E1–E2 interface, onto FR 24-mer and I3-01v9a 60-mer, which are optimal for presenting monomers. The in vitro characterization of HCV-1 sE1E2.Cut1+2.SPYΔN SApNPs supported our design but also indicated that a 5GS linker is suboptimal for I3-01v9a, the 60-mer, and likely contributes to difficulties in subunit folding and NP assembly. Notwithstanding, the covalently linked sE1E2.Cut1+2.SPYΔN heterodimer can be readily displayed on well-established protein NP platforms for vaccine development.
In this proof-of-concept study, we immunized mice to evaluate rationally designed sE1E2 scaffolds and SApNPs with different glycan modifications. As expected, SApNPs induced a more rapid and potent NAb response than individual dimers, irrespective of the glycan type. Despite its suboptimal linker length, the I3-01v9a 60-mer outperformed the FR 24-mer in eliciting cross-genotype bNAbs, underscoring the importance of NP size. Kif-treated immunogens also showed a moderate, although not statistically significant, advantage in NAb induction over unmodified versions, consistent with our previous findings for filovirus vaccines86. These results inform our next steps in HCV vaccine development. First, antibodies induced by sE1E2.Cut1+2.SPYΔN vaccines—both heterodimer and SApNPs with wild-type or modified glycans—will be mapped onto the heterodimer surface to define their epitopes. Single B-cell sorting combined with X-ray crystallography has previously provided critical insights into the genetic and structural features of HCV-specific NAbs in NHPs66,130. In this study, we established an nsEM-based strategy for epitope mapping using the SPYΔN-scaffolded sE1E2 antigen as a probe, validated with AR-series antibodies. Notably, our nsEM results diverged from earlier mutagenesis-based or low-resolution analyses34,108, underscoring the need for structural mapping moving forward. This sE1E2 scaffold can further serve as a probe for electron microscopy-based polyclonal epitope mapping (EMPEM)131, facilitating the direct analysis of vaccine-induced sera. Second, mechanistic studies are needed to understand how sE1E2 dimers and SApNPs interact with the immune system. We previously investigated vaccine trafficking, retention, presentation, and GC reactions in lymph nodes for four rationally designed class I viral vaccines83,86,91,111. Extending this strategy to HCV sE1E2 and SApNPs will offer key insights into how non-class I viral antigens engage the immune system. Third, the effects of glycan modification warrant further investigation. Glycan trimming enhanced NAb titers to influenza132, SARS-CoV-2133, and HIV-183 vaccines, while Kif treatment modestly improved the immunogenicity of filovirus vaccines86. Although these studies suggested a virus-dependent effect, the Kulakova study93 and our work indicate a context-specific impact on HCV vaccines, depending on the antigen. Beyond the glycoengineered CHO lines93, other expression systems with distinct glycan profiles, such as insect134 and fungal135,136 cell lines, may be evaluated for both sE2 and sE1E2 vaccine constructs. Fourth, optimized sE1E2 heterodimer and SApNP vaccines need to be assessed in other relevant animal models137,138. Previously, we identified and characterized potent NHP NAbs using VH1-69–like germline genes, suggesting that NHPs are a useful model for studying NAb responses to the NF on E266,130. Whether NHPs also generate NAbs to AR4 and AR5 remains unclear and may require additional efforts in antibody isolation and epitope mapping. Humanized mice harboring human hepatocytes, or both human immune cells and hepatocytes, could serve as more disease-relevant models to assess these rationally designed HCV vaccine candidates139,140. Lastly, the role of T-cell immunity in sE1E2 vaccine-induced responses warrants further investigation. Both CD4+ and CD8+ T-cell responses were evaluated in a phase 1/2 clinical trial of a vector-based HCV vaccine encoding mutated NS antigens32. Although the sE1E2-based vaccine is generally considered B-cell-focused45,46,60,61, cellular immunity remains equally important141,142. Detailed analyses of the interferon-γ (IFN-γ) pathway and intracellular cytokine production may help clarify the contribution of T cells in rationally designed sE1E2 vaccines or a vaccine combining sE1E2 and NS antigens.
As HCV vaccine candidates advance toward clinical development, factors such as antigen choice, carrier platform, manufacturability, and vaccination regimen must be carefully considered. Although this study establishes a promising native-like sE1E2 antigen, highly optimized E2 cores such as E2mc368 remain viable antigen choices for HCV vaccine development, as demonstrated in our comparative mouse study. While the 24-mer FR serves as a useful model, the I3-01v9a 60-mer91 provides a more suitable platform for clinical development. Notably, an HIV-1 vaccine candidate based on the I3-01v9 platform83 is currently being evaluated in two phase 1 clinical trials (ClinicalTrials.gov IDs: NCT06541093, NCT06905275). In our SApNP design, the sE1E2 antigen and I3-01v9a subunit are encoded within a single gene, enabling one-step expression and assembly—key advantages in manufacturability over multi-component NP systems. The four-dose regimen used here was intended for construct screening in mice68,83,86,87. The decline in ID50 titers from the third to fourth dose in some SApNP groups likely reflects the negative effect of a 3-week interval, as it overlaps with the up-to-8-week retention of SApNPs and, as a result, may sequester high-affinity NAb responses. In addition to the interval, our baseline study also revealed the importance of a strong adjuvant: the failure of both E2mc3 and sE1E2 immunogens to elicit cross-genotype NAb responses may be attributed to the use of AH instead of a more potent AH/CpG adjuvant in the earlier immunizations. These factors need to be adequately addressed in future preclinical studies evaluating the most promising HCV vaccine candidates.
Methods
Construct design, expression, and purification of HCV sE1E2 immunogens
The E1E2 of genotype 1a H77 (GenBank Accession: AF009606.1) was used in the initial design and evaluation of sE1E2 scaffolds. Four heterodimeric coiled coils―LZ (PDB ID: 1FOS), SZ (PDB ID: 3HE5), GCP (PDB ID: 1U2U), and IAAL (PDB ID: 1U0I)―and a covalently linked SpyTag/SpyCatcher (PDB ID: 4MLI) were used as protein scaffolds to stabilize sE1E2. The E1E2s of genotype 1a HCV-1 (GenBank Accession: M62321.1), genotype 3 UKNP3.1.2 (GenBank Accession: KU285215.1), and genotype 5 UKNP5.2.1 (GenBank Accession: KU285226.1) were used to design sE1E2.Cut1+2.SPYΔN-His6 heterodimers (Fig. S1a and Fig. S2a), but only HCV-1 was used to design sE1E2.Cut1+2.SPYΔN-presenting FR and I3-01v9a SApNPs (Fig. S5a). Five HCV-1 sE1E2 variants were generated to examine the impact of design variation on heterodimer folding, antigenicity, and structure (Fig. S4a). HCV-1 was also used to design an sE2 core and its FR SApNP, using the previously reported H77 E2mc3 construct68 as a template (Fig. S8a).
His6-tagged H77 sE1E2 scaffolds were transiently expressed in HEK293F cells. Briefly, HEK293F cells were thawed and suspended in FreeStyleTM 293 Expression Medium (Life Technologies, CA) and cultured in a shaker incubator at 37 °C, 135 rpm, with 8% CO2. When the cells reached a density of 2.0 × 106 ml−1, they were diluted to 1.0 × 106 ml−1 in 1 L of FreeStyleTM 293 Expression Medium for transfection using a polyethyleneimine (PEI)-based (Polysciences, Inc.) protocol. For each transfection, 800 μg of H77 sE1E2 scaffold plasmid and 300 μg of furin plasmid in 25 ml of Opti-MEM (Life Technologies, CA) were mixed with 5 ml of PEI-MAX (1.0 mg/ml) in 25 ml of Opti-MEM and incubated for 30 min at room temperature. Cells were then transfected with the DNA-PEI-MAX mixture and incubated at 37 °C, 135 rpm, and 8% CO2. Five days after transfection, culture supernatants containing the target protein were harvested, clarified by centrifugation at 1126 × g for 22 min, and filtered through a 0.45 μm filter (Thermo Scientific). All H77 sE1E2 scaffolds were purified using Ni Sepharose Excel resin (Cytiva). Bound proteins were eluted twice with 15 ml of 0.5 M imidazole and buffer-exchanged into Tris-buffered saline (TBS; pH 7.2). Nickel-purified H77 sE1E2 scaffolds were further purified by SEC using a Superdex 200 Increase 10/300 GL column (Cytiva).
His6-tagged H77, HCV-1, UKNP3.1.2, and UKNP5.2.1 sE1E2.Cut1+2.SPYΔN scaffolds, five His6-tagged HCV-1 sE1E2 design variants, and all HCV-1 sE1E2.Cut1+2.SPYΔN-presenting SApNPs were produced in ExpiCHO cells (Thermo Fisher). Briefly, ExpiCHO cells were thawed and incubated in ExpiCHO™ Expression Medium (Thermo Fisher) in a shaker incubator at 37 °C, 135 rpm, with 8% CO2. When the cells reached a density of 10 × 106 ml−1, ExpiCHO™ Expression Medium was added to adjust the cell density to 6 × 106 ml−1 for transfection. ExpiFectamine™ CHO-plasmid DNA complexes were prepared for 100-ml transfections according to the manufacturer’s instructions. Kifunensine (10 mg/l; Tocris Bioscience) was added at the time of transfection to inhibit α-mannosidase I and generate oligomannose-type glycans. For each transfection, 80 μg of antigen plasmid, 30 μg of furin plasmid, and 320 μl of ExpiFectamine™ CHO reagent were mixed in 7.7 ml of cold OptiPRO™ medium (Thermo Fisher). After the first feed on day 1, ExpiCHO cells were cultured according to the Max Titer protocol at 33 °C, at 115 rpm, with 8% CO2, with a second feed on day 5 (Thermo Fisher). Culture supernatants were harvested 13–14 days after transfection, clarified by centrifugation at 3724 × g for 25 min, and filtered through a 0.45 μm filter (Thermo Fisher). All sE1E2 scaffolds were purified using Ni Sepharose Excel resin (Cytiva). Bound proteins were eluted twice with 15 ml of 0.5 M imidazole and buffer-exchanged into Tris-buffered saline (TBS; pH 7.2). All HCV-1 sE1E2 SApNPs were purified using an AR3C antibody column, eluted three times each with 5 ml of 0.2 M glycine (pH 2.2), neutralized with 2 M Tris-Base (pH 9.0), and buffer-exchanged into phosphate-buffered saline (PBS; pH 7.2). Nickel-purified sE1E2 scaffolds and AR3C-purified HCV-1 sE1E2 SApNPs were further purified by SEC using a Superdex 200 Increase 10/300 GL column (Cytiva) and a Superose 6 Increase 10/300 GL column (Cytiva), respectively. Selected SEC fractions were pooled, aliquoted, and frozen in liquid nitrogen or at −80 °C until use.
HCV-1 E2mc3 and E2mc3-10GS-ferritin SApNP were produced in ExpiCHO cells using the same protocol described above for sE1E2 and its SApNPs. Both E2mc3 and the ferritin SApNP were purified using an AR3C antibody column following the same protocol used for sE1E2 SApNPs and were further purified by SEC using a Superdex 75 Increase 10/300 GL column (Cytiva) and a Superose 6 Increase 10/300 GL column (Cytiva), respectively.
Expression and purification of neutralizing antibodies (NAbs)
All 11 NAbs, except AR3C and AR4A, as well as CD81-Fc, in the panel were provided by M. Law. AR3C and AR4A IgGs were transiently expressed in ExpiCHO cells (Thermo Fisher). At 12–14 days post-transfection, the cells were centrifuged at 3724 × g for 25 min, and the supernatants were filtered using a 0.45 μm filter (Millipore). IgGs were purified using protein A affinity resin (Cytiva) and eluted in 0.3 M citric acid (pH 3.0). The pH of the elution was immediately adjusted to 7.0 by adding 2 M Tris-Base (pH 9.0). The eluate was then concentrated and buffer-exchanged into phosphate-buffered saline (PBS) using an Amicon 10 kDa filter (Millipore). IgG concentration was quantified by UV280 absorbance with theoretical extinction coefficients.
Glycan trimming by endoglycosidase F (endo F) treatment and enzyme removal
The protocol for endoglycosidase F (endo F) treatment and removal was similar to the Endo Hf protocol used in our previous study83. Endo F1, F2, and F3 were purchased from QA-Bio (catalog nos. E-EF01, E-EF02, and E-EF03, respectively). Briefly, the surface glycans of HCV-1 sE1E2.Cut1+2.SPYΔN-His6 were trimmed by mixing 1 mg of antigen with 100 μl of 5× reaction buffer (QA-Bio), 12.5 μl each of endo F1, F2, and F3, and H2O (if necessary) to a final reaction volume of 0.5 ml. The mixture was incubated at room temperature (25 °C) for 5 h to facilitate enzymatic processing. After incubation, the mixtures were passed through a Superdex 200 Increase 10/300 GL column to remove endo F1-3. Notably, to enable glycan trimming by endo F1-3, HCV-1 sE1E2.Cut1+2.SPYΔN-His6 was expressed in the presence of kifunensine (Kif). The SEC-purified fractions were pooled, aliquoted, and frozen in liquid nitrogen or at −80 °C until use.
SDS-PAGE and BN-PAGE
HCV sE1E2 scaffolds, the E2 core (E2mc3), and their SApNPs were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and blue native-polyacrylamide gel electrophoresis (BN-PAGE). Proteins were mixed with loading dye and loaded onto a 10% Tris–glycine gel (Bio-Rad) or a 4–12% Bis-Tris NativePAGE™ gel (Life Technologies). Two conditions were used in SDS-PAGE. Under non-reducing conditions, proteins were mixed with 6× Laemmli SDS sample buffer (Thermo Scientific, catalog no. J60660) and heated to 80 °C for 5 min. Under reducing conditions, proteins were mixed with Laemmli sample buffer containing reducing agent (Thermo Scientific, catalog no. J61337.AC) and boiled at 100 °C for 5 min. After loading, SDS-PAGE gels were run for 25 min at 250 V using SDS running buffer (Bio-Rad) and stained with InstantBlue (Abcam). For BN-PAGE, proteins were mixed with 4× native sample buffer and loaded onto a BN-PAGE gel. Gels were run for 2–2.5 h at 150 V using NativePAGE™ running buffer (Life Technologies), according to the manufacturer’s instructions. BN-PAGE gels were stained with Coomassie Brilliant Blue R-250 (Bio-Rad) and destained using 6% ethanol and 3% glacial acetic acid. Gel images were acquired using a ChemiDoc™ XRS+ imaging system and processed with Image Lab software (version 6.1.0, build 7, Standard Edition) (Bio-Rad).
Differential scanning calorimetry (DSC)
Melting temperature (Tm) and other thermal parameters were obtained for HCV sE1E2 scaffolds, which were expressed in either HEK293F or ExpiCHO cells and purified by a nickel column followed by SEC, and for HCV-1 sE1E2.Cut1+2.SPYΔN-10GS-FR SApNP, which was expressed in ExpiCHO cells and purified by an AR3C antibody column followed by SEC, using a MicroCal PEAQ-DSC instrument (Malvern). Briefly, purified proteins in PBS buffer were diluted to 0.5–5 μM. Thermal unfolding was monitored at a scan rate of 60 °C h⁻¹ from 20 °C to 100 °C. Data processing, including buffer correction, normalization, and baseline subtraction, was performed using MicroCal PEAQ-DSC software. Gaussian fitting was carried out using GraphPad Prism 10.3.1.
Site-specific glycan analysis
To generate site-specific glycan profiles, 60 µg aliquots of each sample were denatured for 1 h in 50 mM Tris/HCl, pH 8.0, containing 6 M urea and 5 mM dithiothreitol (DTT). Next, sE1E2 samples were reduced and alkylated by adding 20 mM iodoacetamide (IAA) and incubated for 1 h in the dark, followed by a 1 h incubation with 20 mM DTT to eliminate residual IAA. The alkylated E1E2 samples were buffer-exchanged into 50 mM Tris/HCl, pH 8.0, using Vivaspin columns (10 kDa), and two aliquots were digested separately overnight using chymotrypsin (mass spectrometry grade, Promega) or α-lytic protease (Sigma-Aldrich) at a ratio of 1:30 (w/w). The next day, peptides were dried and extracted using an Oasis HLB µElution Plate (Waters).
The peptides were dried again, resuspended in 0.1% formic acid, and analyzed by nanoLC-ESI MS with a Vanquish Neo (Thermo Fisher Scientific) system coupled to an Orbitrap Eclipse Tribrid mass spectrometer (Thermo Fisher Scientific) with stepped higher-energy collision-induced dissociation (HCD). Peptides were separated using a μPAC™ Neo HPLC Column (180 µm × 110 cm). A trapping column (PepMap 100 C18, 3 μM, 75 μM × 2 cm) was used in line with the LC prior to separation on the analytical column. LC conditions consisted of a 280-min linear gradient of 4–32% acetonitrile in 0.1% formic acid over 260 min, followed by 20 min of alternating 76% acetonitrile in 0.1% formic acid and 4% ACN in 0.1% formic acid to ensure complete elution. The flow rate was set to 300 nL/min. The spray voltage was set to 2.5 kV, and the temperature of the heated capillary was set to 55 °C. The ion transfer tube temperature was set to 275 °C. The scan range was 350–2000 m/z. The stepped HCD collision energies were set to 15, 25, and 45% and the MS2 for each energy was combined. Precursor and fragment detection were performed using an Orbitrap at a resolution of MS1 = 120,000 and MS2 = 30,000. Standard AGC target for MS1 (4e5) and MS2 (1e4), and auto injection times (MS1 = 50 ms; MS2 = 54 ms) were used.
Glycopeptide fragmentation data were extracted from raw files using Byos (version 5.5; Protein Metrics Inc.). Fragmentation data were evaluated manually for each glycopeptide; peptides were scored as true positives when correct b and y fragment ions were observed along with oxonium ions corresponding to the identified glycan. The MS data were searched using the Protein Metrics 305 N-glycan library. All charge states for a single glycopeptide were summed. Precursor mass tolerances were set to 4 ppm and fragment tolerances to 10 ppm. A 1% false discovery rate (FDR) was applied. Relative amounts of each glycan at each site, as well as the proportion of unoccupied sites, were determined by comparing the extracted ion chromatographic areas for different glycopeptides with an identical peptide sequence. Glycans were categorized according to their detected composition.
HexNAc2Hex(9−3) was classified as M9 to M3. Structures containing fucose were categorized as FM (fucosylated mannose). Complex-type glycans were classified based on the number of HexNAc subunits and the presence or absence of fucose. Core glycans refer to truncated structures smaller than M3. As this fragmentation method does not provide linkage information, compositional isomers were grouped.
Biolayer interferometry
Antigenic profiles of HCV-1 sE1E2.Cut1+2.SPYΔN-His6 heterodimer and sE1E2.Cut1+2.SPYΔN-10GS-FR SApNP were measured using Octet RED96 (FortéBio, Pall Life Sciences) against 10 NAbs in the human IgG format and CD81-Fc. All assays were performed with agitation set to 1000 rpm in FortéBio 1× kinetic buffer. The final volume for all solutions was 200 μl per well. Assays were performed at 30 °C in solid black 96-well plates (Greiner Bio-One). For all antigens, antibodies or CD81-Fc (5 μg/ml in 1× kinetic buffer) were loaded onto the surface of Anti-Human IgG Quantitation (AHQ) biosensors for 300 s. Next, a two-fold concentration gradient of antigen, starting at 1000 nM for HCV-1 sE1E2.Cut1+2.SPYΔN-His6 dimer and 18 nM for FR SApNP was prepared as a six-point dilution series. A 60-s biosensor baseline step was applied prior to measuring the association of the biosensor-bound antibody or CD81-Fc and antigen in solution for 200 s, followed by a 300-s dissociation step. Baseline drift correction was performed by subtracting the mean shift value recorded for (i) a sensor loaded with antibody or CD81-Fc but not exposed to antigen and (ii) a sensor without antibody or CD81-Fc but exposed to antigen. Octet data were processed using FortéBio’s data acquisition software v.8.1. Peak binding responses at the highest antigen concentration were summarized in a matrix and color-coded to facilitate comparison among constructs. Binding data for each antigen-antibody or antigen-CD81-Fc pair were fitted using a 1:1 binding model, and three datasets showing the best fit were grouped to determine Kon and KD values. Binding curves were plotted using GraphPad Prism 10.3.1.
Enzyme-linked immunosorbent assay (ELISA)
CostarTM 96-well, high-binding, flat-bottom, half-area plates (Corning) were first coated with 50 µl of PBS containing 0.1 µg of the appropriate HCV sE1E2 dimer, E2mc3, or SApNP antigen. The plates were incubated overnight at 4 °C and then washed five times with PBST wash buffer (PBS containing 0.05% (v/v) Tween 20). Each well was then blocked for 1 h at room temperature with 150 µl of blocking buffer consisting of 4% (w/v) blotting-grade blocker (Bio-Rad) in PBS. Next, plates were washed five times with PBST wash buffer. To evaluate antibody or CD81-Fc binding to the coating antigens, antibodies or CD81-Fc were diluted in blocking buffer to a maximum concentration of 10 µg/ml followed by a 10-fold serial dilution. For each antibody or CD81-Fc dilution, a total volume of 50 µl was added to the appropriate wells. Plates were incubated for 1 h at room temperature and then washed five times with PBST wash buffer. For secondary antibody binding, a 1:5000 dilution of goat anti-human IgG antibody (Jackson ImmunoResearch Laboratories) was used for human and rhesus macaque NAbs and CD81-Fc, whereas a 1:3000 dilution of horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG antibody (Jackson ImmunoResearch Laboratories) was used for the murine NAb A4. The secondary antibody was prepared in PBST wash buffer, and 50 µl of the diluted secondary antibody was added to each well. Plates were incubated for 1 h at room temperature and then washed six times with PBST wash buffer. Lastly, the wells were developed with 50 µl of 3,3’,5,5’-tetramethylbenzidine (TMB; Life Sciences) for 3–5 min before the reaction was stopped with 50 µl of 2 N sulfuric acid. The plates were then immediately read on a BioTek Synergy plate reader at a wavelength of 450 nm. EC50 values were calculated from full curves using GraphPad Prism 10.3.1. When OD450 absorbance values were lower than 0.5, the EC50 values were set to 10 µg/ml in Figs. 1f, 2d, 4f, 5g, and 8b to facilitate EC50 plotting and comparison.
Cryo-EM analysis of HCV-1 sE1E2.Cut1+2.SPYΔN-His6 in complex with bNAbs
Nickel/SEC-purified HCV-1 sE1E2.Cut1+2.SPYΔN-His6 heterodimer was used to form complexes with two bNAb pairs under different conditions. AR3C and AR4A Fabs were transiently expressed in ExpiCHO cells (Thermo Fisher) and purified using Protein G affinity resin (Cytiva), and eluted in 0.2 M glycine (pH 2.0). The pH of the elution was immediately adjusted to neutral by adding 2 M Tris-Base (pH 9.0). The purified HCV-1 sE1E2 antigen was incubated with a 1.5-fold molar excess of AR3C and AR4A Fabs overnight at 4 °C, followed by SEC using a Superose 6 Increase 10/300 GL column (Cytiva). HEPC74 and AR4A Fabs were transiently expressed in Expi293F GnTI- cells (Thermo Fisher) and purified by Capture Select CH1-XL Affinity Matrix (Thermo Fisher), followed by SEC with a Superdex 200 Increase 10/300 GL column (Cytiva). The purified HCV-1 sE1E2 antigen was incubated with a 3-fold molar excess of AR4A Fab for 1 h at room temperature, followed by SEC and subsequent complexation with HEPC74 Fab.
For HCV-1 sE1E2.Cut1+2.SPYΔN-His6 bound to AR3C and AR4A, an initial sample was frozen in-house using a Vitrobot Mark IV on Quantifoil R1.2/1.3 300 mesh Cu Grids (Electron Microscopy Sciences; catalog #Q350CR1.3). Grids were screened on a Talos Arctica 200 kV Cryo-TEM equipped with a Gatan K3 camera, and an initial dataset of 1430 movies was collected. Grids from the same plunge freezing session were imaged at the National Cancer Institute’s (NCI) National Cryo-EM Facility on a Titan Krios 300 kV Cryo-TEM using a Gatan K3 camera, yielding 19,080 movies. Additional grids of the complex were also frozen using a VitroJet, aiming to produce thicker ice to improve particle orientation distribution. This yielded an additional 5248 movies. The initial 19,080 movies were processed using cryoSPARC v4.5.3. Patch motion correction and Patch CTF were performed, after which exposures were filtered to 17,941 micrographs. An initial stack of 2000 micrographs was used to obtain particles for deep picking. A stack of 1,218,366 obtained by blob picking was refined by 2D and 3D classification to 113,496 particles, which were used to generate a model for Deep Picking. 8,699,347 particles were obtained with Deep Picker and were further refined to 85,959. To reduce anisotropy in the map, this dataset was combined with the VitroJet dataset, which included an additional 5248 movies. Patch motion correction and Patch CTF were performed using cryoSPARC v4.5.3109. This dataset underwent blob picking, 2D classification, and heterogeneous refinement to yield 89,013 particles. After non-uniform refinement, the datasets were combined to yield the final maps using 174,972 particles. Coordinates from PDB IDs 8FSJ71 and 4MWF56 were fit as rigid bodies into the resulting density maps using UCSF Chimera 1.16 143.
For HCV-1 sE1E2.Cut1+2.SPYΔN-His6 bound to HEPC74 and AR4A, 5106 movies were collected on a Titan Krios 300 kV Cryo-TEM using a Gatan K3 camera (NCI National Cryo-EM Facility). Patch motion correction and Patch CTF were performed using cryoSPARC v4.5.3, after which exposures were filtered to 4141 micrographs109,144. Blob Picking yielded a stack of 11,577,316 particles, which was filtered to 516,060 particles and subsequently used for template generation. 14,430,734 particles were picked by templates and were filtered to an initial stack of 280,335 particles. 3D variability analysis (3DVA) followed by 3D classification was performed to address moderate flexibility in the AR4A angle of approach and possible anisotropy, yielding a final stack of 36,896 particles. Coordinates of PDB ID 8FSJ71 were fit as a rigid body into the final maps using UCSF Chimera 1.16 143.
Negative-stain electron microscopy (nsEM)
nsEM analysis of HCV sE1E2 scaffolds and SApNPs was performed by the Core Microscopy Facility at The Scripps Research Institute. Briefly, samples were prepared at concentrations of 0.008 and 0.01 mg/ml, respectively. Carbon-coated copper grids (400 mesh) were glow-discharged, and 8 μl of each sample was adsorbed for 2 min. Next, the excess sample was then removed, and grids were stained with 2% uranyl formate for 2 min. Excess stain was wicked away, and the grids were allowed to dry. sE1E2 heterodimer samples were analyzed at 120 kV using a Talos L120C transmission electron microscope (TEM; Thermo Fisher), and images were acquired using a CETA 16 M CMOS camera under 73,000× magnification, corresponding to a pixel size of 1.93 Å per pixel with a defocus range of 0.5–2 µm. For sE1E2 and E2mc3 SApNP samples, images were collected under 52,000× magnification, yielding a pixel size of 2.05 Å at the specimen plane, with the defocus set to −1.50 µm. Computational analysis of the sE1E2/NAb images (~30 micrographs) was performed using the high-performance computing core facility at The Scripps Research Institute. Briefly, the nsEM images were converted to MRC format by EMAN2145 for further processing by cryoSPARC v4.3.0109. Micrographs were corrected for contrast transfer function (CTF) using patch CTF estimation. Particles were selected using blob and template pickers and extracted with a box size of 140 pixels for 2D classification, with minimum and maximum particle diameters of 100 and 250 Å used for blob picking. For all dimer/NAb complexes, 3D models were generated by ab initio reconstruction and refined by heterogeneous and homogeneous refinement. All nsEM images and fitted structures were rendered using UCSF Chimera143 and ChimeraX 146.
Dynamic light scattering (DLS)
Particle size distributions of HCV-1 sE1E2.Cut1+2.SPYΔN-presenting FR and I3-01v9a SApNPs were obtained using a Zetasizer Ultra instrument (Malvern). Briefly, AR3C/SEC-purified SApNPs produced in ExpiCHO cells were diluted to 0.2 mg/ml in 1× PBS. A 30 μl aliquot of each sample was loaded into a quartz batch cuvette (Malvern; catalog no. ZEN2112). Particle size measurements were performed at room temperature (25 °C) using backscattering mode. Data processing was carried out using Zetasizer Ultra software. The resulting particle size distributions were plotted using GraphPad Prism 10.3.1.
Mouse immunization and sample collection
Mouse immunization protocols followed our previous studies83,86,89,91. All animal experiments adhered to the guidelines of the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC). Protocols (#15-0002) were approved by the Institutional Animal Care and Use Committee (IACUC) of The Scripps Research Institute (TSRI). Six- to eight-week-old female BALB/c mice were purchased from The Jackson Laboratory and housed in ventilated cages in environmentally controlled rooms at 20 °C, 50% humidity, and 12-h light-dark cycles. Except for the study examining sex-specific effects and comparing with E2 core constructs, our standard “screening regimen” was used. Mice were immunized intraperitoneally (i.p.) at weeks 0, 3, 6, and 9 (3-week intervals). Each dose consisted of 200 μl of antigen/adjuvant mix containing 10 μg of vaccine antigen and 100 μl of adjuvant, including 40 µg/50 µl of CpG (oligonucleotide 1826, a TLR9 agonist from InvivoGen) adsorbed onto 50 µl of AH (InvivoGen). Blood samples were collected 2 weeks after each injection through the retro-orbital sinus using heparinized capillary tubes. Samples were centrifuged at 13,200 × g for 10 min to separate serum from the remaining blood fraction. Serum was heat-inactivated at 56 °C for 30 min and centrifuged again at 13,200 × g for 10 min to remove precipitates. For the final time point, the remaining whole-blood fraction was diluted with an equal volume of PBS and layered over 4.5 ml of Ficoll in a 15 ml SepMateTM tube (STEMCELL Technologies), followed by centrifugation at 300 × g for 10 min at 20 °C to isolate peripheral blood mononuclear cells (PBMCs). Cells were washed once with PBS, resuspended in 1 ml of ACK red blood cell lysis buffer (Lonza), washed again with PBS, and finally resuspended in 1 ml of freezing media (10% DMSO/90% FCS). Two weeks after the final bleed, spleens were harvested and passed through a 70-μm cell strainer (BD Falcon) to generate splenocytes suspensions. Splenocytes were centrifuged, washed with PBS, treated with 5 ml of ACK lysing buffer (Lonza), and frozen in 3 ml of freezing media (10% DMSO/90% FCS). For the immunogenicity studies examining sex-specific effects and comparing with E2 core constructs, a different regimen was used89,91. Six- to eight-week-old female and male BALB/c mice were purchased from The Jackson Laboratory and housed as described above. Mice were immunized at weeks 0, 3, 6, and 9 with 80 μl of antigen/adjuvant mixture containing 10 μg of vaccine antigen in 40 μl of PBS and 40 μl of AH adjuvant (InvivoGen). Intradermal (i.d.) immunization was performed via injection into four footpads (20 μl per site) using a 29-gauge insulin needle under 3% isoflurane anesthesia with oxygen. Notably, administration across four sites reduced variability in baseline comparisons and mechanistic analyses. Blood was collected from the maxillary/facial vein 2 weeks after each immunization. Serum was isolated by centrifugation at 13,200 × g for 10 min, heat-inactivated at 56 °C for 30 min, and clarified by centrifugation at 4300 × g for 10 min. Heat-inactivated serum was stored at −4 °C until use in HCVpp assays to quantify vaccine-induced NAb responses.
Pseudovirus production and neutralization assays
An HCV pseudoparticle (HCVpp) neutralization assay68 was performed to assess vaccine-induced NAb responses in mouse sera. HCVpps were generated by co-transfecting HEK293T cells with the envelope-deficient HIV-1 pNL4-3.lucR-E− plasmid (NIH AIDS Reagent Program: https://www.beiresources.org/HIV.aspx) and an expression plasmid encoding the E1E2 gene of genotype 1a H77 (GenBank ID: AF009606.1), UKNP3.1.2 (GenBank ID: KU285215.1), UKNP4.2.2 (GenBank ID: KU285222.1), ED43 (GenBank ID: GU814265.1), or UKNP5.2.1 (GenBank ID: KU285226.1) at a 4:1 ratio, using Lipofectamine 3000 (Thermo Fisher). After 72 h, pseudoviruses were harvested from the supernatant by centrifugation at 3724 × g for 10 min, aliquoted, and stored at −80 °C until use. For neutralization assays, mouse sera were serially diluted threefold starting at 1:40 and incubated with pseudoviruses at 37 °C for 1 h in white solid-bottom, half-area 96-well plates (Corning). Next, 0.8 × 104 Huh-7 cells were added to each well, and plates were incubated at 37 °C for 60–72 h. After incubation, the supernatant was removed, and the cells were lysed. Luciferase reporter gene expression was quantified by adding Bright-Glo™ Luciferase substrate (Promega), according to the manufacturer’s instructions. Luminescence data, expressed as relative light units (RLU), were measured using a BioTek microplate reader with Gen5 software. Experimental wells were compared with virus-only control wells, and background luminescence from uninfected controls was subtracted from all readings. Dose–response neutralization curves were generated by nonlinear regression with constraints (0–100%) using GraphPad Prism 10.3.1, from which ID50 values were calculated. In the baseline study comparing sE1E2 and E2 core constructs, each mouse serum at a 1:40 dilution was tested in duplicate against non-H77 HCVpps. Serial serum dilutions were not performed for these viruses because cross-genotype neutralization was too low to generate reliable titration curves.
Lymph node disaggregation, cell staining, and flow cytometry
To study sex-dependent GC responses induced by the HCV-1 sE1E2.Cut1+2.SPYΔN/AH vaccine formulation, the frequencies and absolute numbers of GC B cells (B220+GL7+CD95+) and Tfh cells (CD3+CD4+CXCR5+PD-1+) were quantified by flow cytometry. Lymph node tissue isolation, disaggregation, cell staining, and flow cytometry analysis followed protocols established in our previous studies83,91,111. Mice were intradermally injected with HCV-1 sE1E2 vaccines (four footpads, 2.5 μg per footpad) and euthanized 2 weeks after a single-dose immunization. The administered dose was 80 μl of antigen/adjuvant mixture containing 10 μg of sE1E2 antigen per mouse. Sentinel lymph nodes were collected, mechanically disaggregated, and transferred to enzyme digestion solution in 1.5 ml Eppendorf tubes containing 958 μl of Hank’s Balanced Salt Solution (HBSS; Thermo Fisher Scientific, catalog no. 14185052), 40 μl of 10 mg/ml collagenase IV (Sigma-Aldrich, catalog no. C5138), and 2 μl of 10 mg/ml DNase I (Roche, catalog no. 10104159001). Lymph node tissues were incubated at 37 °C for 30 min on a rotator and then passed through a 70μm cell strainer to obtain single-cell suspensions. Cells were centrifuged at 400 × g for 10 min and resuspended in HBSS blocking buffer containing 0.5% (w/v) bovine serum albumin and 2 mM EDTA. Cells were placed on ice for 30 min with anti-CD16/32 antibody (BioLegend, catalog no. 101302, 1:50) to block Fc receptor–mediated nonspecific binding. Cells were then transferred to 96-well V-bottom plates and stained on ice for 30 min with a pre-mixed antibody cocktail containing: Zombie NIR live/dead stain (BioLegend, catalog no. 423106, 1:100), Brilliant Violet 510 anti-mouse/human CD45R/B220 antibody (BioLegend, catalog no. 103247, 1:300), FITC anti-mouse CD3 antibody (BioLegend, catalog no. 100204, 1:300), Alexa Fluor 700 anti-mouse CD4 antibody (BioLegend, catalog no. 100536, 1:300), PE anti-mouse/human GL7 antibody (BioLegend, catalog no. 144608, 1:500), Brilliant Violet 605 anti-mouse CD95 (Fas) antibody (BioLegend, catalog no. 152612, 1:500), Brilliant Violet 421 anti-mouse CD185 (CXCR5) antibody (BioLegend, catalog no. 145511, 1:500), and PE/Cyanine7 anti-mouse CD279 (PD-1) antibody (BioLegend, catalog no. 135216, 1:500). Following antibody staining, cells were centrifuged at 400 × g for 10 min, washed with HBSS blocking buffer, and fixed with 1.6% paraformaldehyde (Thermo Fisher Scientific, catalog no. 28906) in HBSS on ice for 30 min. Cells were centrifuged again at 400 × g for 10 min and stored in HBSS blocking buffer at 4 °C until acquisition. Flow cytometry was performed on a five-laser ZE5 cell analyzer (Yeti model; Bio-Rad) equipped with Everest software at the TSRI Core Facility. Data were analyzed using FlowJo version 10, and plots were generated in GraphPad Prism version 10.3.1.
Statistical analysis
Data were collected from 8 mice per group in immunization studies and 5 mice per group in the germinal center study. HCVpp neutralization assays were performed in duplicate. Comparisons of HCV-1 sE1E2.Cut1+2.SPYΔN vaccines (e.g., sE1E2 dimer vs. SApNP) were conducted using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison post hoc test for each time point. Two-tailed unpaired t-tests were used when comparing two independent groups. Statistical significance is indicated in the figures as follows: ns (not significant), *p < 0.05, and ** p < 0.01. Graphs and statistical analyses were performed using GraphPad Prism 10.3.1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data needed to evaluate the conclusions of this research are available in the main text and Supplementary Information. The cryo-EM data have been deposited in the Electron Microscopy Data Bank (EMDB, https://www.ebi.ac.uk/emdb/) under accession codes EMD-70622 for the complex with AR3C and AR4A and EMD-70623 for the complex with HEPC74 and AR4A. The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information files. Source data are provided with this paper.
References
World Health Organization. Global Hepatitis Report 2024 (World Health Organization, 2024).
Mattson, C. L. et al. Trends and geographic patterns in drug and synthetic opioid overdose deaths - United States, 2013-2019. MMWR Morb. Mortal. Wkly Rep. 70, 202–207 (2021).
Zibbell, J. E. et al. Increases in acute hepatitis C virus infection related to a growing opioid epidemic and associated injection drug use, United States, 2004 to 2014. Am. J. Public Health 108, 175–181 (2018).
Carrat, F. et al. Clinical outcomes in patients with chronic hepatitis C after direct-acting antiviral treatment: a prospective cohort study. Lancet 393, 1453–1464 (2019).
Feeney, E. R. & Chung, R. T. Antiviral treatment of hepatitis C. BMJ 348, g3308 (2014).
Holmes, J. A., Rutledge, S. M. & Chung, R. T. Direct-acting antiviral treatment for hepatitis C. Lancet 393, 1392–1394 (2019).
Foster, G. R. et al. Impact of direct acting antiviral therapy in patients with chronic hepatitis C and decompensated cirrhosis. J. Hepatol. 64, 1224–1231 (2016).
Cheung, M. C. M. et al. Outcomes after successful direct-acting antiviral therapy for patients with chronic hepatitis C and decompensated cirrhosis. J. Hepatol. 65, 741–747 (2016).
Conti, F. et al. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J. Hepatol. 65, 727–733 (2016).
Konerman, M. A. & Lok, A. S. Hepatitis C treatment and barriers to eradication. Clin. Transl. Gastroenterol. 7, e193 (2016).
Pol, S. Lack of evidence of an effect of direct-acting antivirals on the recurrence of hepatocellular carcinoma: data from three ANRS cohorts. J. Hepatol. 65, 734–740 (2016).
Baber, A. S., Suganthan, B. & Ramasamy, R. P. Current advances in hepatitis C diagnostics. J. Biol. Eng. 18, 48 (2024).
Thursz, M. & Fontanet, A. HCV transmission in industrialized countries and resource-constrained areas. Nat. Rev. Gastroenterol. Hepatol. 11, 28–35 (2014).
Waheed, Y., Siddiq, M., Jamil, Z. & Najmi, M. H. Hepatitis elimination by 2030: Progress and challenges. World J. Gastroenterol. 24, 4959–4961 (2018).
Cox, A. L. et al. Progress towards elimination goals for viral hepatitis. Nat. Rev. Gastroenterol. Hepatol. 17, 533–542 (2020).
Pfaender, S., Brown, R. J. P., Pietschmann, T. & Steinmann, E. Natural reservoirs for homologs of hepatitis C virus. Emerg. Microbes Infect. 3, e21 (2014).
Simmonds, P. The origin of hepatitis C virus. Curr. Top. Microbiol. Immunol. 369, 1–15 (2013).
Dubuisson, J. & Cosset, F. L. Virology and cell biology of the hepatitis C virus life cycle - an update. J. Hepatol. 61, S3–S13 (2014).
Moradpour, D. & Penin, F. Hepatitis C virus proteins: from structure to function. Curr. Top. Microbiol. Immunol. 369, 113–142 (2013).
Dubuisson, J. Hepatitis C virus proteins. World J. Gastroenterol. 13, 2406–2415 (2007).
Lindenbach, B. D. & Rice, C. M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 11, 688–700 (2013).
Yost, S. A., Wang, Y. & Marcotrigiano, J. Hepatitis C virus envelope glycoproteins: a balancing act of order and disorder. Front. Immunol. 9, 1917 (2018).
Freedman, H., Logan, M. R., Law, J. L. M. & Houghton, M. Structure and function of the hepatitis C virus envelope glycoproteins E1 and E2: antiviral and vaccine targets. ACS Infect. Dis. 2, 749–762 (2016).
Feneant, L., Levy, S. & Cocquerel, L. CD81 and hepatitis C virus (HCV) infection. Viruses 6, 535–572 (2014).
Prentoe, J. & Bukh, J. Hypervariable region 1 in envelope protein 2 of hepatitis C virus: a linchpin in neutralizing antibody evasion and viral entry. Front. Immunol. 9, 2146 (2018).
Tong, Y. M., Lavillette, D., Li, Q. C. & Zhong, J. Role of hepatitis C virus envelope glycoprotein E1 in virus entry and assembly. Front. Immunol. 9, 1411 (2018).
Mazumdar, B., Banerjee, A., Meyer, K. & Ray, R. Hepatitis C virus E1 envelope glycoprotein interacts with apolipoproteins in facilitating entry into hepatocytes. Hepatology 54, 1149–1156 (2011).
Smith, D. B. et al. Flaviviridae: Hepacivirus C classification. https://ictv.global/sg_wiki/flaviviridae/hepacivirus (2022).
Tsukiyama-Kohara, K. & Kohara, M. Hepatitis C virus: Viral quasispecies and genotypes. Int. J. Mol. Sci. 19, 23 (2018).
Bukh, J. The history of hepatitis C virus (HCV): basic research reveals unique features in phylogeny, evolution and the viral life cycle with new perspectives for epidemic control. J. Hepatol. 65, S2–S21 (2016).
Grebely, J. et al. Hepatitis C virus clearance, reinfection, and persistence, with insights from studies of injecting drug users: towards a vaccine. Lancet Infect. Dis. 12, 408–414 (2012).
Page, K. et al. Randomized trial of a vaccine regimen to prevent chronic HCV infection. N. Engl. J. Med. 384, 541–549 (2021).
Giang, E. et al. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. USA 109, 6205–6210 (2012).
Law, M. et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 14, 25–27 (2008).
Zhang, P. et al. Hepatitis C virus epitope-specific neutralizing antibodies in Igs prepared from human plasma. Proc. Natl. Acad. Sci. USA 104, 8449–8454 (2007).
Stamataki, Z. et al. Hepatitis C virus envelope glycoprotein immunization of rodents elicits cross-reactive neutralizing antibodies. Vaccine 25, 7773–7784 (2007).
Broering, T. J. et al. Identification and characterization of broadly neutralizing human monoclonal antibodies directed against the E2 envelope glycoprotein of hepatitis C virus. J. Virol. 83, 12473–12482 (2009).
Meunier, J. C. et al. Isolation and characterization of broadly neutralizing human monoclonal antibodies to the e1 glycoprotein of hepatitis C virus. J. Virol. 82, 966–973 (2008).
Hadlock, K. G. et al. Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. J. Virol. 74, 10407–10416 (2000).
Gomez-Escobar, E., Roingeard, P. & Beaumont, E. Current hepatitis C vaccine candidates based on the induction of neutralizing antibodies. Viruses 15, 1151 (2023).
Chen, F., Tzarum, N., Wilson, I. A. & Law, M. VH1-69 antiviral broadly neutralizing antibodies: genetics, structures, and relevance to rational vaccine design. Curr. Opin. Virol. 34, 149–159 (2019).
Fauvelle, C. et al. Hepatitis C virus vaccine candidates inducing protective neutralizing antibodies. Expert Rev. Vaccines 15, 1535–1544 (2016).
Ball, J. K., Tarr, A. W. & McKeating, J. A. The past, present and future of neutralizing antibodies for hepatitis C virus. Antiviral Res. 105, 100–111 (2014).
Wahid, A. & Dubuisson, J. Virus-neutralizing antibodies to hepatitis C virus. J. Viral Hepat. 20, 369–376 (2013).
Guest, J. D. & Pierce, B. G. Structure-based and rational design of a hepatitis C virus vaccine. Viruses 13, 837 (2021).
Keck, M. L., Wrensch, F., Pierce, B. G., Baumert, T. F. & Foung, S. K. H. Mapping determinants of virus neutralization and viral escape for rational design of a hepatitis C virus vaccine. Front. Immunol. 9, 1194 (2018).
Kong, L., Jackson, K. N., Wilson, I. A. & Law, M. Capitalizing on knowledge of hepatitis C virus neutralizing epitopes for rational vaccine design. Curr. Opin. Virol. 11, 148–157 (2015).
Potter, J. A. et al. Toward a hepatitis C virus vaccine: the structural basis of hepatitis C virus neutralization by AP33, a broadly neutralizing antibody. J. Virol. 86, 12923–12932 (2012).
Kong, L. et al. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc. Natl. Acad. Sci. USA 109, 9499–9504 (2012).
Kong, L. et al. Structure of hepatitis C virus envelope glycoprotein E1 antigenic site 314-324 in complex with antibody IGH526. J. Mol. Biol. 427, 2617–2628 (2015).
Meola, A. et al. Structural flexibility of a conserved antigenic region in hepatitis C virus glycoprotein E2 recognized by broadly neutralizing antibodies. J. Virol. 89, 2170–2181 (2015).
Krey, T. et al. Structural basis of HCV neutralization by human monoclonal antibodies resistant to viral neutralization escape. PLoS Pathog. 9, e1003364 (2013).
He, L. et al. Approaching rational epitope vaccine design for hepatitis C virus with meta-server and multivalent scaffolding. Sci. Rep. 5, 12501 (2015).
Nagarathinam, K. et al. Epitope-focused immunogens targeting the hepatitis C virus glycoproteins induce broadly neutralizing antibodies. Sci. Adv. 10, eado2600 (2024).
Pierce, B. G. et al. Structure-based design of hepatitis C virus vaccines that elicit neutralizing antibody responses to a conserved epitope. J. Virol. 91, e01032-17 (2017).
Kong, L. et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science 342, 1090–1094 (2013).
Khan, A. G. et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 509, 381–384 (2014).
Khan, A. G., Miller, M. T. & Marcotrigiano, J. HCV glycoprotein structures: what to expect from the unexpected. Curr. Opin. Virol. 12, 53–58 (2015).
Flyak, A. I. et al. HCV broadly neutralizing antibodies use a CDRH3 disulfide motif to recognize an E2 glycoprotein site that can be targeted for vaccine design. Cell Host Microbe 24, 703–716.e3 (2018).
Tzarum, N., Wilson, I. A. & Law, M. The neutralizing face of hepatitis C virus E2 envelope glycoprotein. Front. Immunol. 9, 1315 (2018).
Fuerst, T. R., Pierce, B. G., Keck, Z. Y. & Foung, S. K. H. Designing a B cell-based vaccine against a highly variable hepatitis C virus. Front. Microbiol. 8, 2692 (2018).
Law, M. Antibody responses in hepatitis C infection. Cold Spring Harb. Perspect. Med. 11, a036962 (2021).
Tzarum, N. et al. Genetic and structural insights into broad neutralization of hepatitis C virus by human VH1-69 antibodies. Sci. Adv. 5, eaav1882 (2019).
Capella-Pujol, J. et al. Signatures of VH1-69-derived hepatitis C virus neutralizing antibody precursors defined by binding to envelope glycoproteins. Nat. Commun. 14, 4036 (2023).
Weber, T. et al. Analysis of antibodies from HCV elite neutralizers identifies genetic determinants of broad neutralization. Immunity 55, 341–354.e7 (2022).
Chen, F. et al. Functional convergence of a germline-encoded neutralizing antibody response in rhesus macaques immunized with HCV envelope glycoproteins. Immunity 54, 781–796.e4 (2021).
Tzarum, N. et al. An alternate conformation of HCV E2 neutralizing face as an additional vaccine target. Sci. Adv. 6, eabb5642 (2020).
He, L. L. et al. Proof of concept for rational design of hepatitis C virus E2 core nanoparticle vaccines. Sci. Adv. 6, eaaz6225 (2020).
Yan, Y. et al. A nanoparticle-based HCV vaccine with enhanced potency. J. Infect. Dis. 221, 1304–1314 (2020).
Pierce, B. G. et al. Structure-based design of hepatitis C virus E2 glycoprotein improves serum binding and cross-neutralization. J. Virol. 94, e00704-20 (2020).
Metcalf, M. C. et al. Structure of engineered hepatitis C virus E1E2 ectodomain in complex with neutralizing antibodies. Nat. Commun. 14, 3980 (2023).
de la Peña, A. T. et al. Structure of the hepatitis C virus E1E2 glycoprotein complex. Science 378, 263–269 (2022).
Augestad, E. H. et al. The hepatitis C virus envelope protein complex is a dimer of heterodimers. Nature 633, 704–709 (2024).
Pierce, B. G. et al. Hepatitis C virus E1E2 structure, diversity, and implications for vaccine development. Viruses 16, 803 (2024).
Garbuglia, A. R., Pauciullo, S., Zulian, V. & Del Porto, P. Update on hepatitis C vaccine: results and challenges. Viruses 16, 1337 (2024).
Toth, E. A., Andrianov, A. K. & Fuerst, T. R. Prospects for developing an hepatitis C virus E1E2-based nanoparticle vaccine. Rev. Med. Virol. 33, e2474 (2023).
Guest, J. D. et al. Design of a native-like secreted form of the hepatitis C virus E1E2 heterodimer. Proc. Natl. Acad. Sci. USA 118, e2015149118 (2021).
Wang, R. X. et al. Induction of broadly neutralizing antibodies using a secreted form of the hepatitis C virus E1E2 heterodimer as a vaccine candidate. Proc. Natl. Acad. Sci. USA 119, e2112008119 (2022).
Fuerst, T. R. et al. Virus-mimicking polymer nanocomplexes co-assembling HCV E1E2 and core proteins with TLR 7/8 agonist-synthesis, characterization, and in vivo activity. J. Funct. Biomater. 16, 34 (2025).
Sliepen, K. et al. Induction of cross-neutralizing antibodies by a permuted hepatitis C virus glycoprotein nanoparticle vaccine candidate. Nat. Commun. 13, 7271 (2022).
White, J. M., Delos, S. E., Brecher, M. & Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 43, 189–219 (2008).
Harrison, S. C. Viral membrane fusion. Nat. Struct. Mol. Biol. 15, 690–698 (2008).
Zhang, Y.-N. et al. Single-component multilayered self-assembling protein nanoparticles presenting glycan-trimmed uncleaved prefusion optimized envelope trimmers as HIV-1 vaccine candidates. Nat. Commun. 14, 1985 (2023).
He, L. et al. HIV-1 vaccine design through minimizing envelope metastability. Sci. Adv. 4, eaau6769 (2018).
Kong, L. et al. Uncleaved prefusion-optimized gp140 trimers derived from analysis of HIV-1 envelope metastability. Nat. Commun. 7, 12040 (2016).
Lee, Y.-Z. et al. Rational design of next-generation filovirus vaccines combining glycoprotein stabilization and nanoparticle display with glycan modification. Nat. Commun. 16, 11368 (2025).
He, L. L. et al. Single-component multilayered self-assembling nanoparticles presenting rationally designed glycoprotein trimers as Ebola virus vaccines. Nat. Commun. 12, 2633 (2021).
He, L. L. et al. Single-component, self-assembling, protein nanoparticles presenting the receptor binding domain and stabilized spike as SARS-CoV-2 vaccine candidates. Sci. Adv. 7, eabf1591 (2021).
Lee, Y.-Z. et al. Rational design of uncleaved prefusion-closed trimer vaccines for human respiratory syncytial virus and metapneumovirus. Nat. Commun. 15, 9939 (2024).
Zhang, Y.-N. et al. A single-component multilayered self-assembling protein nanoparticle vaccine based on extracellular domains of matrix protein 2 against both influenza A and B. Vaccines 12, 975 (2024).
Gomes, K. B. et al. Single-component multilayered self-assembling protein nanoparticles displaying extracellular domains of matrix protein 2 as a pan-influenza A vaccine. ACS Nano 17, 23545–23567 (2023).
He, L. et al. Presenting native-like trimeric HIV-1 antigens with self-assembling nanoparticles. Nat. Commun. 7, 12041 (2016).
Kulakova, L. et al. Glycoengineering of the hepatitis C virus E2 glycoprotein improves biochemical properties and enhances immunogenicity. Npj Vaccines 10, 121 (2025).
Zakeri, B. et al. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 109, E690–E697 (2012).
Penin, F., Dubuisson, J., Rey, F. A., Moradpour, D. & Pawlotsky, J. M. Structural biology of hepatitis C virus. Hepatology 39, 5–19 (2004).
Frey, S. E. et al. Safety and immunogenicity of HCV E1E2 vaccine adjuvanted with MF59 administered to healthy adults. Vaccine 28, 6367–6373 (2010).
Glover, J. N. M. & Harrison, S. C. Crystal structure of the heterodimeric bZIP transcription factor c-Fos-c-Jun bound to DNA. Nature 373, 257–261 (1995).
Thompson, K. E., Bashor, C. J., Lim, W. A. & Keating, A. E. SynZIP protein interaction toolbox: in vitro and in vivo specifications of heterospecific coiled-coil interaction domains. ACS Synth. Biol. 1, 118–129 (2012).
Marti, D. N. & Bosshard, H. R. Inverse electrostatic effect: electrostatic repulsion in the unfolded state stabilizes a leucine zipper. Biochemistry 43, 12436–12447 (2004).
Lindhout, D. A., Litowski, J. R., Mercier, P., Hodges, R. S. & Sykes, B. D. NMR solution structure of a highly stable de novo heterodimeric coiled-coil. Biopolymers 75, 367–375 (2004).
Chu, L. & Robinson, D. K. Industrial choices for protein production by large-scale cell culture. Curr. Opin. Biotechnol. 12, 180–187 (2001).
Zhao, Q. Q. et al. Production and immunogenicity of different prophylactic vaccines for hepatitis C virus (Review). Exp. Ther. Med. 24, 474 (2022).
Shukla, A. A. & Thömmes, J. Recent advances in large-scale production of monoclonal antibodies and related proteins. Trends Biotechnol. 28, 253–261 (2010).
Urbanowicz, R. A. et al. A diverse panel of hepatitis C virus glycoproteins for use in vaccine research reveals extremes of monoclonal antibody neutralization resistance. J. Virol. 90, 3288–3301 (2016).
Schoene, C., Fierer, J. O., Bennett, S. P. & Howarth, M. SpyTag/SpyCatcher cyclization confers resilience to boiling on a mesophilic enzyme. Angew. Chem. Int. Ed. 53, 6101–6104 (2014).
Lavie, M., Hanoulle, X. & Dubuisson, J. Glycan shielding and modulation of hepatitis C virus neutralizing antibodies. Front. Immunol. 9, 910 (2018).
Helle, F., Duverlie, G. & Dubuisson, J. The hepatitis C virus glycan shield and evasion of the humoral immune response. Viruses 3, 1909–1932 (2011).
Gopal, R. et al. Probing the antigenicity of hepatitis C virus envelope glycoprotein complex by high-throughput mutagenesis. PLoS Pathog. 13, e1006735 (2017).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Merat, S. J. et al. Cross-genotype AR3-specific neutralizing antibodies confer long-term protection in injecting drug users after HCV clearance. J. Hepatol. 71, 14–24 (2019).
Zhang, Y.-N. et al. Mechanism of a COVID-19 nanoparticle vaccine candidate that elicits a broadly neutralizing antibody response to SARS-CoV-2 variants. Sci. Adv. 7, eabj3107 (2021).
Salas, J. H. et al. An antigenically diverse, representative panel of envelope glycoproteins for hepatitis C virus vaccine development. Gastroenterology 162, 562–574 (2022).
Dunn, S. E., Perry, W. A. & Klein, S. L. Mechanisms and consequences of sex differences in immune responses. Nat. Rev. Nephrol. 20, 37–55 (2024).
Klein, S. L. & Flanagan, K. L. Sex differences in immune responses. Nat. Rev. Immunol. 16, 626–638 (2016).
Jacobsen, H. & Klein, S. L. Sex differences in immunity to viral infections. Front. Immunol. 12, 720952 (2021).
Stroh, L. J., Nagarathinam, K. & Krey, T. Conformational flexibility in the CD81-bindingsite of the hepatitis C virus glycoprotein E2. Front. Immunol. 9, 1396 (2018).
Kumar, A. et al. Structural insights into hepatitis C virus receptor binding and entry. Nature 598, 521–525 (2021).
World Health Organization. Accelerating Access to Hepatitis C Diagnostics and Treatment: Global Progress Report 2020 (World Health Organization, 2021).
Westbrook, R. H. & Dusheiko, G. Natural history of hepatitis C. J. Hepatol. 61, S58–S68 (2014).
Liang, T. J. & Ward, J. W. Hepatitis C in Injection-drug Users - A Hidden Danger Of The Opioid Epidemic. N. Engl. J. Med. 378, 1169–1171 (2018).
Kulp, D. W. & Schief, W. R. Advances in structure-based vaccine design. Curr. Opin. Virol. 3, 322–331 (2013).
Graham, B. S. The journey to RSV vaccines - heralding an era of structure-based design. N. Engl. J. Med. 388, 579–581 (2023).
Graham, B. S., Gilman, M. S. A. & McLellan, J. S. Structure-based vaccine antigen design. Annu. Rev. Med. 70, 91–104 (2019).
Derking, R. & Sanders, R. W. Structure-guided envelope trimer design in HIV-1 vaccine development: a narrative review. J. Int. AIDS Soc. 24, e25797 (2021).
Nguyen, B. & Tolia, N. H. Protein-based antigen presentation platforms for nanoparticle vaccines. Npj Vaccines 6, 70 (2021).
Irvine, D. J. & Read, B. J. Shaping humoral immunity to vaccines through antigen-displaying nanoparticles. Curr. Opin. Immunol. 65, 1–6 (2020).
Rappuoli, R. & Serruto, D. Self-assembling nanoparticles usher in a new era of vaccine design. Cell 176, 1245–1247 (2019).
Kabsch, W. & Sander, C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983).
Indelicato, G., Burkhard, P. & Twarock, R. Classification of self-assembling protein nanoparticle architectures for applications in vaccine design. R. Soc. Open Sci. 4, 161092 (2017).
Chen, F. et al. Antibody responses to immunization with HCV envelope glycoproteins as a baseline for B-cell-based vaccine development. Gastroenterology 158, 1058–1071.e6 (2020).
Bianchi, M. et al. Electron-microscopy-based epitope mapping defines specificities of polyclonal antibodies elicited during HIV-1 BG505 envelope trimer immunization. Immunity 49, 288–300.e8 (2018).
Wang, C. C. et al. Glycans on influenza hemagglutinin affect receptor binding and immune response. Proc. Natl. Acad. Sci. USA 106, 18137–18142 (2009).
Huang, H. Y. et al. Vaccination with SARS-CoV-2 spike protein lacking glycan shields elicits enhanced protective responses in animal models. Sci. Transl. Med. 14, eabm0899 (2022).
Korn, J. et al. Baculovirus-free insect cell expression system for high yield antibody and antigen production. Sci. Rep. 10, 21393 (2020).
Gonzalez-Hernandez, M. et al. Preclinical immunogenicity and protective efficacy of a SARS-CoV-2 RBD-based vaccine produced with the thermophilic filamentous fungal expression system Thermothelomyces heterothallica C1. Front. Immunol. 14, 1204834 (2023).
Lazo, L. et al. A recombinant SARS-CoV-2 receptor-binding domain expressed in an engineered fungal strain of Thermothelomyces heterothallica induces a functional immune response in mice. Vaccine 40, 1162–1169 (2022).
Ploss, A. & Kapoor, A. Animal models of hepatitis C virus infection. Cold Spring Harb. Perspect. Med. 10, a036970 (2020).
Burm, R., Collignon, L., Mesalam, A. A. & Meuleman, P. Animal models to study hepatitis C virus infection. Front. Immunol. 9, 1032 (2018).
Yong, K. S. M., Her, Z. & Chen, Q. F. Humanized mouse models for the study of hepatitis C and host interactions. Cells 8, 604 (2019).
Mesalam, A. A., Vercauteren, K. & Meuleman, P. Mouse systems to model hepatitis C virus treatment and associated resistance. Viruses 8, 176 (2016).
Costa, G. L. & Sautto, G. A. Exploring T-cell immunity to hepatitis C virus: insights from different vaccine and antigen presentation strategies. Vaccines 12, 890 (2024).
Thimme, R. T cell immunity to hepatitis C virus: lessons for a prophylactic vaccine. J. Hepatol. 74, 220–229 (2021).
Pettersen, E. F. et al. UCSF chimera - a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Punjani, A., Zhang, H. W. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Tang, G. et al. EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007).
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Acknowledgements
The cryo-EM analysis was supported in part by the National Cancer Institute’s National Cryo-EM Facility at the Frederick National Laboratory for Cancer Research under contract 75N91019D00024. We acknowledge K. Vanderpool, T. Fassel, and S. Henderson of the Core Microscopy Facility at TSRI for their expert assistance with negative-stain EM analysis. We thank B. Seegers and A. Saluk of the Flow Cytometry Core Facility at TSRI for their technical support with flow cytometry. This work was supported by NIH awards AI168251 (M.L., J.Z.) and AI168917 (M.L.), and in part by Ufovax/SFP-2018-1013 (J.Z.).
Author information
Authors and Affiliations
Contributions
Project design: L.H. and J.Z.; sE1E2 scaffold and SApNP design: L.H. and J.Z; sE1E2 scaffold and SApNP expression and purification: G.W., C.D., and L.H.; SDS-PAGE and BN-PAGE: G.W., C.D., Y.-Z.L., and L.H.; DSC and DSL: Y.-Z.L. and L.H.; ELISA: Y.-Z.L. and Y.-N.Z.; BLI: Y.-Z.L. and L.H.; sE1E2.LZorg expression and purification: F.G.G., L.K., E.A.T., and T.R.F.; AR3C and AR4A expression and purification: G.W., C.D., and L.H.; other NAb (and CD81-Fc) expression and purification: E.G. and M.L.; glycan profiling: M.N., J.D.A., and M.C.; cryo-EM: B.M.J. and G.O.; negative-stain EM and antibody epitope mapping: Y.-Z.L., S.-H.H., and J.Z.; mouse immunization: Y.-N.Z. (in part); HCVpp neutralization assays: G.W., C.D., Y.-N.Z., and L.H.; flow cytometry: Y.-N.Z; manuscript writing: L.H., Y.-Z.L., Y.-N.Z., M.N., B.M.J., G.O., M.C., and J.Z. All authors review and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
J.Z. is the Co-Founder and a Scientific Advisory Board member of Uvax Bio, LLC, and holds associated financial interests. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
He, L., Lee, YZ., Zhang, YN. et al. Native-like soluble E1E2 glycoprotein heterodimers on self-assembling protein nanoparticles for hepatitis C virus vaccine design. Nat Commun 17, 2633 (2026). https://doi.org/10.1038/s41467-026-69418-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69418-9
