
Abstract
Electrocatalytic CO2 reduction reaction (CO2RR) technology holds significant industrial potential. However, when faced with elevated-temperature environments caused by industrial-scale operations, the fundamental understanding of temperature-dependent CO2RR behavior in flow cells remains elusive. This study points out that elevated-temperature operation (>333 K) presents both challenges and opportunities for multi-carbon (C2+) production. While elevated temperature enhances reaction kinetics and reduces thermodynamic energy barriers, it simultaneously induces reconstruction of Cu-based catalyst, accelerates gas diffusion electrode flooding, and promotes *CO desorption together with hydrogen evolution reaction, collectively suppressing C2+ product formation and compromising long-term reactor stability. Through rational design of hydrophobic-enhanced Pd-Cu2O/polytetrafluoroethylene (PTFE)/Ag tandem electrodes, we overcome these challenges. Leveraging thermal reduced C-C coupling barriers, the optimized electrode achieves >70% Faradaic efficiency of C2+ across industrially relevant current densities (200–1000 mA cm−2) at 348 K. This strategy converts elevated temperature adversity into a kinetic and thermodynamic advantage, boosting C2+ cathodic energy efficiency by 1.3 times compared to ambient operation, establishing a promising paradigm for industrially viable CO2 electrolysis.
Similar content being viewed by others


Introduction
Electrocatalytic CO2 reduction to C2+ products offers a promising route for sustainable chemical synthesis and carbon emission mitigation1,2,3. Advances in electrocatalyst design, microenvironment engineering, and reactor optimization have enhanced C2+ production efficiency in ambient-temperature systems4,5,6. Nevertheless, when electrolyzers are scaled up and operated under industrial conditions, heat from Joule heating due to ohmic resistance, and limited heat dissipation collectively elevate electrode surface temperatures (Fig. 1a)7,8. Strikingly, scaling electrode areas from 4 cm2 to 25 cm2 rapidly elevates cell temperature to 353 K or induces violent boiling (Fig. 1b, Supplementary Fig. 1 and Supplementary Note 1), exceeding thermal limits of ion-exchange membranes and causing system failure. This demonstrates that elevated-temperature operation is unavoidable in industrial scaling. Understanding temperature impacts on CO2RR systems, including reactor integrity and reaction pathways, is essential for industrial implementation9,10.

a Schematic illustration of the source pathways of Ohm heat in a flow cell for industrial CO2RR applications. b Electrolyte temperature curve of large-area electrodes operating at high currents for 30 min in a flow cell (Room temperature: 298 K). c Schematic illustration of the effect of elevated temperature on CO2RR products. The black represents Cu catalyst, the brown represents Pd atom, and the combination of red, gray, and blue represents an OCCOH intermediate formed by C-C coupling. d A comparison of temperature and the FE of C2+ for CO2RR in previous reports (The highest FE and the FE at the highest temperature from these studies). e Breakdown of the plant-gate levelized cost per tonne of C2+ products with and without cooling, as calculated from a TEA. Source data are provided as a Source Data file.
Current understanding of temperature effects originates from H-cell studies, which provide preliminary insights into product selectivity and intermediate adsorption/desorption11,12,13. Koper et al. proposed that a slight increase in temperature promotes the formation of C2+ products, while excessive elevation accelerates the formation of the competing HER14. However, elevated temperature alters CO2 solubility, water vapor pressure, and electrolyte conductivity, thereby fundamentally reconstructing the reaction microenvironments15,16. Consequently, investigating temperature effects in H-cells is substantially hindered by the coupled influences of kinetic and mass transport changes, making it challenging to infer purely thermodynamic or intrinsic kinetic effects. Gas diffusion electrodes (GDEs) with stable gas–liquid–solid interfaces effectively mitigate mass transport limitations. Compared to membrane electrode assembly (MEA) electrolyzers, the flow-cell configuration offers a key advantage for fundamental research by enabling independent and precise thermal management and potential measurement of the cathode. Moreover, it avoids complications such as membrane degradation and water evaporation at elevated temperature, which are commonly encountered in MEA electrolyzers, thereby enabling a rigorous assessment of thermal impacts on reaction thermodynamics and kinetics17,18,19. Previous studies in flow cells demonstrate temperature-enhanced kinetics for C1 products, exemplified by reduced overpotentials for CO and H2 evolution on silver catalysts from 291 to 343 K20, while mechanistic work reveals that temperature-dependent *COOH/*H adsorption competition dictates CO/H2 selectivity21.
Despite these advances for C1 products, premature *CO desorption and accelerated hydrogen evolution reaction (HER) kinetics severely impede C-C coupling at elevated temperatures, limiting C2+ formation on Cu catalysts (Fig. 1c). Takanabe et al. demonstrate that temperatures exceeding 333 K significantly reduce the selectivity of C2+ products22. Han et al. Further confirms that lower operating temperature (270 K, −3 °C) favors C-C coupling and hydrogenation pathways of *CH-COH intermediates on Cu-based catalysts23, suggesting incompatibility between C2+ production conditions and industrial elevated-temperature environments. Additionally, elevated temperature may compromise the stability of both catalysts and GDEs in flow cells. Therefore, prior strategies have focused on circumventing, rather than addressing, the elevated-temperature challenge, for example, by lowering operating temperature via heat exchange and electrolyzers' cooling. Thus far, maintaining a high Faradaic efficiency (FE) for C2+ products in elevated-temperature flow cells remains a critical scientific challenge and a technological bottleneck urgently demanding resolution (Fig. 1d)13,14,16,23,24,25,26,27,28,29,30,31. Consequently, a comprehensive understanding of the adverse effects of elevated temperature on CO2RR in flow cells, together with the development of corresponding mitigation strategies, is essential for industrial implementation.
To bridge these gaps, we systematically investigate the effect of temperature (298–348 K) on CO2RR in flow cells. Experimental results reveal that elevated temperature induced dynamic restructuring of Cu-based catalysts, accelerated GDEs flooding due to increased water vapor pressure, and enhanced *CO desorption and HER kinetics. Therefore, elevated temperature inhibits the formation of C2+ products and indirectly promotes product loss in flow cells. To address these challenges, we engineered hydrophobic-enhanced Pd-Cu2O/PTFE/Ag tandem electrodes that achieve >70% FE of C2+ across 200–1000 mA cm−2 at 348 K and exhibit stable operation for extended periods (Fig. 1d). Operando Raman spectroscopy and density functional theory (DFT) calculations confirm that strengthened *CO adsorption and reduced C-C coupling energy barriers synergistically promote the formation of C2+ at elevated temperature. These strategies convert elevated-temperature challenges into opportunities, increasing cathodic energy efficiency (CEE) by 1.3 times compared to room-temperature operation. Preliminary techno-economic analysis (TEA) indicates that this strategy not only significantly reduces electricity consumption but also eliminates approximately 14.9% of cooling-related operating costs (Fig. 1e, Supplementary Fig. 2, Supplementary Note 2 and Supplementary Table 1 to 2). This work provides essential guidance for the scale-up and practical application of CO2 electrolysis technology.
Results
We systematically investigated the effect of temperature on CO2RR in a flow cell by simultaneously examining catalyst structure, three-phase interface stability, and key reaction intermediates and pathways, all of which collectively govern CO2RR activity. Given that ion exchange membranes are typically operated below 353 K, we investigated the impact of temperature on CO2RR within a safe range of 298-348 K32,33. To maintain a stable CO2RR interfacial temperature, both the flow cell and catholyte were heated to simulate the elevated temperature environment encountered in flow cell (Supplementary Fig. 3 and Supplementary Note 3). The flow cell temperature was monitored using two K-type thermocouples and infrared thermal imaging technology. Measurements commenced only after the temperature had reached the set-point and remained stable for 5 min.
The impact of temperature toward the catalyst structure
Cu is widely employed for C2+ production owing to its efficient C-C coupling capability, yet concerns persist regarding its structural vulnerability under industrial elevated-temperature industrial operation34,35. X-ray diffraction (XRD) analysis reveals distinct thermal oxidation pathways in magnetron-sputtered Cu electrodes (Fig. 2a). The as-synthesized electrodes exhibit characteristic Cu(111) and (200) diffraction peaks36,37. After immersion in 348 K 1.0 M KOH electrolyte, a new peak emerges at 36.5° corresponding to Cu2O(111), confirming surface oxidation38,39. Subsequent CO2RR operation restores the original Cu phase, demonstrating electrochemical reduction reversibility. X-ray photoelectron spectroscopy (XPS) verified this phenomenon (Supplementary Fig. 4a), i.e., Cu was oxidized in 348 K electrolyte, as evidenced by a distinct Cu(II) signal at 935 eV40. Cu LMM Auger electron spectroscopy (AES) confirms increased relative ratios of Cu(I) and Cu(II) signals at elevated temperature (Supplementary Fig. 4b)41. To eliminate environmental interference and probe dynamic redox behavior of the Cu electrode, electrochemical in situ X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements were conducted (Supplementary Fig. 5). Upon electrochemical reduction, the Cu K-edge XANES spectra (Fig. 2b and Supplementary Fig. 6a) confirm dominant Cu(0) states, matching the Cu foil references. However, once electrochemical control is removed, rapid surface oxidation occurs, as evidenced by a distinct Cu-O coordination peak at 1.6 Å in the EXAFS spectra (Fig. 2c, Supplementary Fig. 6b, and Supplementary Table 3)42. In addition to changes in chemical state, scanning electron microscope (SEM) images reveal that operating at the elevated temperature (348 K) caused slight morphological changes in Cu nanoparticles (Fig. 2d).

a XRD patterns of Cu electrode (2.5 cm×1.0 cm) under different conditions (I: Initial sample; II: After immersion in 298 K 1.0 M KOH electrolyte, pH = 13.7 ± 0.1; III: After immersion in 348 K 1.0 M KOH electrolyte; IV: After CO2RR in 348 K 1.0 M KOH electrolyte, pH = 13.7 ± 0.1. b Normalized Cu K-edge in situ XANES spectra and c Fourier transform k3-weighted EXAFS of Cu under different conditions in a 0.5 M KHCO3 electrolyte (pH = 6.7 ± 0.1, 6.8 ± 0.3 Ω). d SEM images of Cu under different conditions. e SEM images of Cu2O under different conditions. f Normalized Cu K-edge in situ XANES spectra and g Fourier transform k3-weignted EXAFS of Cu under different conditions in a 0.5 M KHCO3 electrolyte (pH = 6.7 ± 0.1, 6.8 ± 0.3 Ω). All reported potentials were not IR corrected. Source data are provided as a Source Data file.
Therefore, elevated-temperature (348 K) electrolyte immediately affects the structure of Cu catalysts, but continuous electrochemical stimulation masks the effects of elevated-temperature oxidation, so this dynamic oxidation-reduction process is easily overlooked during the CO2RR. Critically, elevated-temperature-induced oxidation alters CO2RR pathways. As the temperature increased from 298 K to 348 K, the FE of CH4 increased from 3.4% to 20.8%, while the FE of C2H4 decreased from 30.2% to 17.6% at −200 mA cm−2, concomitant with accelerated HER (Supplementary Fig. 7). During the 6-hour stability test (Supplementary Fig. 8), the FE of C2H4 and CH4 remained nearly constant at 298 K. Conversely, at 348 K, Cu oxidation and surface restructuring shifted selectivity toward CH4.
In contrast, the elevated-temperature-induced dynamic oxidation-reduction process shows a different pattern on Cu2O. Cu2O undergoes no further oxidation in 348 K electrolyte, and its morphology remains unchanged (Fig. 2e). XRD analysis confirms the absence of elevated-temperature-induced crystal structure alteration (Supplementary Fig. 9); the weak diffraction peaks at 44° and 51° are ascribed to electrochemically reduced Cu(0) (111) and (200) facets43. XPS shows a slight decrease in the Cu(I)/Cu(0) ratio after electrochemical reduction at 348 K (Supplementary Fig. 10). Notably, XANES spectra reveal that a metallic Cu phase forms during the prolonged electrochemical reduction reaction (Fig. 2f and Supplementary Fig. 11). When electrochemical control is removed, the metallic Cu is re-oxidized to Cu2O (Fig. 2g). This result confirms that the Cu2O crystal structure is unaffected by the elevated temperature, yet the metallic Cu generated during CO2RR is still oxidized, just like the Cu electrode mentioned above. In contrast, as the temperature increased from 298 K to 348 K, the Cu2O electrode exhibited no appreciable changes in the distribution of carbon products (Supplementary Fig. 12), apart from a drastic increase in the FE of H2. Operando surface-enhanced Raman spectroscopy (SERS) revealed the temperature dependence of product selectivity on Cu and Cu2O electrodes (Supplementary Figs. 13–17 and Supplementary Notes 4–7), that elevated‑temperature‑induced structural changes in Cu significantly promote the activation of interfacial water. Under the combined effects of low *CO coverage and excessive protonation, the product selectivity shifts from C2+ to CH4. In contrast to Cu, Cu2O maintains its structural integrity at 348 K and, crucially, preserves its favorable interfacial water structure. Therefore, in subsequent research, selecting Cu2O as the subject of study can effectively avoid the adverse effects arising from structural changes in the catalyst induced by elevated temperatures. While Cu2O nanocubes demonstrate superior structural stability relative to metallic Cu at elevated temperature, their CO2RR performance at 348 K remains severely compromised. This performance degradation points to additional elevated-temperature barriers beyond structural preservation.
The impact of temperature toward the GDE
Beyond catalyst instability, elevated temperatures compromise electrode integrity by disrupting the gas-liquid equilibrium. Product distribution analysis on the Cu2O electrodes shows a sharp rise in the FE of H2 and a simultaneous drop in the FE of CO and C2+ when the temperature increases from 298 K to 348 K (Supplementary Fig. 12). This anomalous enhancement in HER does not stem from catalyst degradation but from catastrophic flooding of the GDE, a direct consequence of elevated water vapor pressure that destabilizes the three-phase boundary (Fig. 3a). In the flow cell configuration, the three-phase interface separates the CO2 feed from the electrolyte via the catalyst layer. The short gas diffusion path greatly enhances mass transfer efficiency from the CO2 feed to the catalyst-electrolyte interface, enabling high current densities. However, this design also renders the cathode susceptible to flooding44. When the hydrophobic GDE structure is compromised during electroreduction, electrolyte infiltrates the catalyst layer, the GDE, and the gas channel, blocking CO2 access to active sites. At elevated temperature, the electrolyte’s saturated vapor pressure rises; water vapor formation accelerates flooding, further diminishing the population of active sites for CO2RR. The remaining sites in contact with the electrolyte continue to drive the hydrogen evolution reaction.

a Schematic representation of water flooding of cathode structures in a flow cell at elevated temperature. b FEs of H2 and c FEs of C2+ at different temperatures and current densities on Cu2O. d Double-layer charging current plotted against the CV scan rate at different temperatures on Cu2O and Cu2O/PTFE. e EIS of Cu2O and Cu2O/PTFE at different temperatures. f FEs of H2 and C2+ on Cu2O/PTFE at different current densities at 348 K. All error bars represent the standard deviation calculated from at least 3 data points. Source data are provided as a Source Data file.
The Cu layer formed by magnetron sputtering is highly dense and therefore hydrophobic, effectively preventing water vapor intrusion at elevated-temperature conditions (Supplementary Fig. 18a and Supplementary Note 8). In contrast, most Cu-based composite catalysts must be sprayed onto the GDE in the powder form; their high porosity compromises hydrophobicity and renders them more susceptible to flooding at elevated temperature (Supplementary Fig. 18b). The electrochemical in situ reconstruction of Cu2O is largely independent of H2 evolution; at 298 K the FE of H2 actually decreases when the current density is raised to −600 mA cm−2 (Fig. 3b). However, as the temperature rises further, the FE of H2 increases sharply, indicating that elevated temperatures disrupts CO2 transport and suppresses C2+ products formation (Fig. 3c and Supplementary Fig. 19). Damage to the three-phase interface also leads to product loss; the total FE falls to only 88.5%, of which H2 accounts for 74.9%. Thus, the electrode structure is more vulnerable at elevated temperatures and high current densities. Cyclic voltammetry (CV) in non-Faradaic regions shows that the Cdl value of Cu2O increases with temperature, giving a larger electrochemical active surface area (ECSA) at 348 K (Fig. 3d and Supplementary Table 4)45. In the flow cell, however, the ECSA encompasses both CO2RR and HER active sites; higher temperature therefore intensifies flooding and enlarges the HER-active area (Supplementary Fig. 20). After a 6-h stability test, the gas-solid-liquid interface is almost completely destroyed and H2 becomes the predominated reduction product (Supplementary Fig. 21).
Hydrophobic polytetrafluoroethylene (PTFE) particles were introduced into the catalyst layer to suppress water vapor intrusion (Supplementary Fig. 22)46. The Cdl value of Cu2O/PTFE remains nearly constant with increasing temperature, indicating that PTFE effectively stabilizes the three-phase interface at elevated temperature (Supplementary Fig. 23). Electrochemical impedance spectroscopy (EIS) shows that while elevated temperature lowers the interfacial charge transfer resistance, the insulating PTFE induces a slight increase in this value (Fig. 3e). The thickness (δ) of the CO2 diffusion layer on the catalyst was fitted at different temperatures (Supplementary Table 5). For Cu2O, δ decreased from 4.51 to 3.54 as temperature rose, consistent with water vapor instructions shortening reducing the CO2 diffusion path (Supplementary Fig. 24). In contrast, the almost unchanged δ for Cu2O/PTFE confirms that a stable three-phase interface that is insensitive to temperature. Consequently, PTFE effectively inhibits water flooding and limits the FE of H2 to only 13.0% at 348 K, even at −600 mA cm−2 (Fig. 3f). Thus, the shift of CO2RR products toward H2 on Cu2O at elevated temperature is mainly attributable to thermally accelerated water flooding that impedes CO2 diffusion. The PTFE not only mitigates flooding but also ensures robust GDE operation at elevated temperatures. Nevertheless, the FE of H2 on Cu2O/PTFE remains higher at 348 K than at 298 K (Supplementary Fig. 25), indicating that intrinsic kinetic limitations, beyond flooding, also contribute to suppressed C2+ formation.
The impact of temperature toward the C2+ product
The efficiency of C-C coupling is a key determinant of C2+ yield. Even with PTFE-stabilized GDE operation, elevated temperatures suppress C-C coupling, as evidenced by reduced C2+ FE and concomitant promotion of H2 and CO generation (Supplementary Fig. 25 and Fig. 4a). Investigating the temperature effect on C-C coupling therefore requires suppressing competitive hydrogen evolution and preventing *CO desorption. Although the Cu2O/PTFE electrode remains structurally stable at elevated temperatures, temperature rise still enhances its HER. The same trend was observed on magnetron-sputtered Cu electrodes. To further suppress H2 reduction, we coated an Ag layer onto the Cu2O/PTFE electrode, creating a spatially separated tandem architecture (Supplementary Fig. 26 and Supplementary Note 9)47. The Ag catalyst layer suppresses water dissociation and effectively reduces CO2 to CO, increasing the local *CO concentration while inhibiting *H adsorption on Cu2O surface (Fig. 4a and Supplementary Fig. 27)31,48 The CO2RR activity tests show that the Cu2O/PTFE/Ag tandem structure lowers the FE of H2 from 22.5% to 7.2% at 348 K and −200 mA cm−2 (Fig. 4b). In previous studies, Ag serves as a *CO generator, and higher *CO coverage can lower the C-C coupling barrier. However, Cu2O/PTFE/Ag exhibits a higher FE of CO than Cu2O/PTFE at both 298 K and 348 K (Supplementary Fig. 28 and Fig. 4b), indicating that the abundant *CO produced by Ag is not efficiently spilled over to Cu2O for coupling but is instead desorbed and vented. This suggests that the *CO-adsorption capability of Cu2O is equally critical; its capacity as a *CO receiver dictates the amount of *CO available for C-C coupling49. Because local *CO concentration and pH scale with current density, the *CO coverage at −200 mA cm−2 is lower than that at higher current densities, and the higher C-C coupling barriers yield lower FE of C2+. Moreover, Cu2O/PTFE/Ag shows a higher FE of CO at 348 K than at 298 K, confirming that elevated temperature not only fails to promote C-C coupling but also accelerates *CO desorption.

a Schematic diagram of elevated temperature promotion of CO2RR on the surface of Cu2O/PTFE, Cu2O/PTFE/Ag, and Pd-Cu2O/PTFE. b Corresponding FEs at different current densities on Cu2O/PTFE/Ag at 298 K and 348 K in 1.0 M KOH electrolyte (pH = 13.7 ± 0.1, 0.62 ± 0.03 Ω). c Product distributions and corresponding FEs at different current densities on Pd-Cu2O/PTFE at 348 K in 1.0 M KOH electrolyte. d Product distributions and corresponding FEs at different current densities on Pd-Cu2O/PTFE/Ag at 348 K in 1.0 M KOH electrolyte. e FEs and C2+/C1 ratio at −200 mA cm-2 on CP (Cu2O/PTFE), CPA (Cu2O/PTFE/Ag), PCP (Pd-Cu2O/PTFE), and PCPA (Pd-Cu2O/PTFE/Ag) at 298 K and 348 K. f Corresponding potentials and CEEs at different current densities on Pd-Cu2O/PTFE/Ag at 298 K and 348 K in 1.0 M KOH electrolyte. g Comparison of some typical results at −200 mA cm-2 at 348 K. All error bars represent the standard deviation calculated from at least 3 data points. All reported potentials were not IR corrected. Source data are provided as a Source Data file.
Recently, Xu et al. showed that alkali metal cations modulate both the activation enthalpy of reaction and the enthalpy of CO adsorption50. The CO2RR tests in electrolytes containing different alkali metal cations reveal that larger cations suppress water dissociation more effectively, with the FE of H2 following the order Li+> Na+> K+> Cs+ (Supplementary Fig. 29). Conversely, larger cation weakens *CO adsorption, so the FE of CO increases in the order Li+ <Na+ <K+ <Cs+. As the temperature rises, the FEs of H2 and CO increase in all alkali metal cation-containing electrolytes, confirming that elevated temperatures accelerate *CO desorption. Cs+, which exhibits the weakest *CO binding, gives the highest FE of CO. These results demonstrate that the interfacial *CO adsorption capacity governs C-C coupling efficiency at elevated temperature (Supplementary Note 10).
Therefore, converting the inherently adverse elevated temperature environment into a beneficial one hinges on suppressing *CO desorption. Because Pd possesses a markedly stronger *CO binding energy than Cu, highly dispersed Pd was deposited onto Cu2O via chemical reduction to stabilize *CO intermediates at elevated temperatures (Fig. 4a)51,52. Elemental maps confirm the uniform distribution of Pd across the Cu2O surface, and aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) further verifies its atomic-level dispersion (Supplementary Fig. 30). At 298 K, the CO2RR tests show that Pd incorporation lowered the FE of CO and promoted the C-C coupling, yielding 53.1% FE of C2+ at −200 mA cm−2 (Supplementary Fig. 31). However, Pd also strongly binds *H, enhancing water dissociation and increasing the FE of H253. When the temperature is raised to 348 K, H2 becomes the dominant side product (Fig. 4c). To balance these competing effects, Pd and Ag were co-incorporated into the Cu2O/PTFE electrode. In the resulting Pd-Cu2O/PTFE/Ag architecture, Pd selectively suppresses *CO desorption, while Ag inhibits HER and continuously supplies *CO. The presence of Pd and Ag is confirmed by XRD and XPS spectra (Supplementary Fig. 32), and SEM imaging reveals in the Pd-Cu2O/PTFE/Ag electrode (Supplementary Fig. 33). Leveraging this synergy, the Pd-Cu2O/PTFE/Ag electrode delivers a C2+ FE to 60.2% at −200 mA cm−2 and 298 K (Supplementary Fig. 34). Pd strengthens *CO adsorption on Cu2O sites fed by Ag, and the additional thermal energy at elevated temperature further increases the probability of C-C coupling. Consequently, the Pd-Cu2O/PTFE/Ag electrode achieves a C2+ FE of 72.6% at −200 mA cm−2 upon raising the electrolyte temperature to 348 K (Fig. 4d and Supplementary Fig. 35). Optimization of Pd loading reveals that 0.7 wt% Pd provided optimal *CO and *H adsorption, maximizing FE of C2+ products while minimizing FE of H2 (Supplementary Fig. S36). The low loading and high dispersion of Pd prevent its aggregation during prolonged elevated-temperature electroreduction, thereby maintaining the structural function (Supplementary Figs. 37 and 38). Notably, elevated temperature simultaneously suppresses CO/H2 generation and enhances C-C coupling at low current densities (Fig. 4e), overcoming the CO concentration limitation inherent to low current density operation. Benefiting from robust interfacial water management, the Pd-Cu2O/PTFE/Ag electrode remains stable during a 48-h test at 348 K (Supplementary Fig. 39). Furthermore, the elevated temperature accelerates CO2RR kinetics and lowers the required overpotential (Supplementary Fig. 40), thereby improving cathodic energy efficiency (CEE). Assisted by the elevated temperature, the Pd-Cu2O/PTFE/Ag electrode delivers a CEEC2+ of 48.9% at −200 mA cm−2 (Fig. 4f), 1.3-fold higher than at room temperature (37.7%) and 5.5-fold higher than the initial Cu2O/PTFE (8.9%)54. These results confirm that elevated temperature can promote C-C coupling under controlled conditions. Thus, by synergistically optimizing interfacial water management, strengthening *CO adsorption, and facilitating C-C coupling, the hydrophobic-enhanced Pd-Cu2O/PTFE/Ag electrode surmounts elevated-temperature limitations and achieves optimal C2+ production in flow cells at 348 K (Fig. 4g and Supplementary Table 6).
Mechanistic studies
Complementing our experimental observations of *CO desorption, Operando SERS provides direct evidence for interfacial intermediate dynamics in flow cells. Because the Ag tandem layer functions solely as a CO generator while C-C coupling occurs exclusively on Cu2O or Pd-Cu2O surfaces, the electrodes for Operando SERS measurements were fabricated without Ag layers to avoid interference from its strong surface-enhanced Raman scattering effect (Supplementary Fig. 41 and Supplementary Note 11). Raman spectra (Fig. 5a) reveal that at 298 K, when the Cu2O/PTFE electrode potential is lowered to -0.6 V (vs. RHE), characteristic peaks emerge at 2030-2090 cm−1, assigned to *CO atop stretching vibrations (*COatop)55. These signals indicate that *CO intermediate adsorbs predominantly atop Cu atoms and participates in C-C coupling. The concurrent appearance of a peak at 360 cm−1 (assigned to Cu-CO stretching) confirms strong potential-dependent Cu-CO interactions (Supplementary Fig. 42a). At 348 K, the disappearance of the *COatop peak signals (Fig. 5b) signifies suppressed C-C coupling56. The peak at 360 cm−1 now emerges only at higher potentials (Supplementary Fig. 42b), demonstrating that elevated temperature weakens Cu-CO binding and promotes *CO desorption. Collectively, these results underscore that catalyst-specific *CO adsorption strength is critical for C2+ formation. Depositing Pd onto Cu2O strengthens Cu-CO interactions; both the Cu-CO (360 cm−1) and *COatop peaks intensify at lower potentials at 298 K (Supplementary Fig. 42c and Fig. 5c), confirming Pd-enhanced *CO binding57. Importantly, Pd mitigates the elevated-temperature limitation; *COatop signals intensify at low potentials at 348 K (Fig. 5d), suggesting that elevated temperature accelerates localized *CO conversion, thereby facilitating low-potential C-C coupling. Additionally, the Pd-induced enhanced interfacial electric field strengthens the adsorption and activation of CO2−, thereby indirectly facilitating the protonation process of CO2−, evidenced by an enhanced CO2− peaks at 700 cm−1 and 1530–1560 cm−1 at low potentials (Supplementary Fig. 42d and Supplementary Note 12).

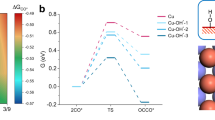
Operando SERS for CO2RR over the Cu2O/PTFE catalyst at different applied potentials in 1.0 M KOH electrolyte (pH = 13.7 ± 0.1, 1.2 ± 0.1 Ω) at a 298 K and b 348 K. Operando SERS for CO2RR over the Pd-Cu2O/PTFE catalyst at different applied potentials at c 298 K and d 348 K. e The *CO adsorption free energy and *H adsorption free energy on Cu2O and Cu2O/Pd at 298 K and 348 K. All reported potentials were not IR corrected. f Energy profiles of asymmetric coupling of *CO with *COH on Cu2O and Cu2O/Pd surfaces with different CO coverages. g Energy profiles of asymmetric CO-COH coupling on Cu2O and Cu2O/Pd surfaces at 298 K and 348 K. Source data are provided as a Source Data file.
To quantify the temperature-dependent adsorption energetics inferred from Raman spectra, DFT calculations were employed to model *CO binding on Cu2O and Pd-Cu2O. The Cu2O (100) surface and atomically dispersed Pd on Cu2O (100) (denoted Pd-Cu2O (100)) served as models (Supplementary Data 1). The adsorption energy calculations reveal that both *CO and *H binding weaken monotonically with temperature (Supplementary Fig. 43 and Fig. 5e), promoting *CO desorption and impeding subsequent C-C coupling, consistent with experimental results. Although *CO binding on Pd-Cu2O remains temperature-dependent, Pd-Cu2O exhibits stronger CO absorption than Cu2O across the entire temperature range. This enhancement arises from the increased density of states near the Fermi level in Pd 4 d orbitals, which upshifts the d-band center of Cu2O. Projected crystal orbital Hamiltonian population (pCOHP) analysis further reveals stronger Pd-C bonding, confirming the higher *CO adsorption energy (Supplementary Fig. 44). Given the critical role of surface *CO coverage in C-C coupling, we further examined the asymmetric *CO-COH coupling pathway under varying coverages58,59,60. Increasing *CO coverage on Cu2O from 1/18 ML to 5/18 ML (Supplementary Fig. 45 and Fig. 5f) progressively lowers the coupling barrier from 1.715 eV to 1.011 eV. This aligns with electrochemical observations. Higher current densities raise *CO coverage, reduce the C-C coupling barrier, and enhance the FE of C2+ products. At identical *CO coverage (5/18 ML), Pd-Cu2O exhibits a lower C-C coupling barrier than Cu2O (1.011 eV vs. 0.947 eV), rationalizing its superior C2+ FE at low current densities. Crucially, although elevated temperature promotes intermediate desorption, it simultaneously reduces C-C coupling barriers (Fig. 5g)61. Raising the temperature from 298 K to 348 K decreases the *OCCOH formation barrier by 0.124 eV for Cu2O and by 0.136 eV for Pd-Cu2O, underscoring the thermodynamic advantage of elevated temperature C-C coupling.
The influence of elevated temperature on C2+ production can be summarized as follows: (i) Elevated temperature accelerates *H and *CO desorption from the Cu2O surface, leading to substantial H2 and CO formation; (ii) Introducing an Ag tandem layer suppresses *H participation and continuously generates CO intermediates that migrate to the Cu2O surface. Although the locally high CO concentration facilitates the C-C coupling, weak *CO adsorption on Cu2O limits its ability to effectively capture and convert the migrated CO at elevated temperature; (iii) Incorporating Pd significantly strengthens the *CO adsorption on Cu2O, enabling localized accumulation of *CO and promoting the *OCCOH generation and subsequent C2+ formation. Additionally, Pd site mitigates *CO desorption at elevated temperature, leveraging the reduced C-C coupling barrier to further enhanced C2+ yields. Consequently, the Pd-Cu2O/PTFE/Ag electrode demonstrates significantly enhanced CO2RR activity at elevated temperature (Supplementary Note 13).
Discussion
In summary, this work fundamentally reframes the paradigm for elevated-temperature CO2 electrolysis. Rather than regarding elevated temperatures solely as a liability for C2+ production, we demonstrate that its intrinsic kinetic and thermodynamic advantages can be leveraged through rational electrode design. We first delineated the three primary challenges encountered under industrially relevant elevated-temperature (>333 K) operation in flow cells: (i) dynamic reconstruction of Cu catalysts, (ii) accelerated GDE flooding caused by increased vapor pressure, and (iii) detrimental *CO desorption coupled with enhanced HER. By transforming these challenges into opportunities, we engineered a hydrophobically stabilized tandem electrode (Pd-Cu2O/PTFE/Ag). This integrated architecture operates synergistically: Hydrophobic PTFE mitigates flooding to preserve the vital gas-liquid-solid interface; the Ag layer suppresses water dissociation while generating abundant *CO intermediates; atomically dispersed Pd significantly strengthens *CO adsorption on Cu2O, counteracting thermal desorption and enabling efficient C-C coupling by exploiting thermally lowered activation barriers. As a result, the optimized system achieves exceptional performance under industrially demanding conditions, achieving >70% FE of C2₊ products across a wide current density range (200–1000 mA cm−2) at 348 K. Most notably, this strategy of converting thermal energy into catalytic advantage boosts the CEE of C2+ by 1.3 times compared to ambient operation. This study provides a transformative perspective for CO2 electrolysis scale-up. By strategically managing interfacial processes at elevated temperatures, we unlock the dual benefits of accelerated reaction kinetics and enhanced thermodynamic favorability for C2+ production. TEA confirms that elevated-temperature C2+ production not only reduces electrolysis costs but also eliminates additional cooling energy consumption. This approach enables waste-heat utilization and represents a critical advance toward economically viable carbon-negative chemical manufacturing. Furthermore, the electrode design strategy proposed in this work may also provide valuable insights for other electrochemical synthesis fields, such as amine synthesis, biomass conversion, and fuel cells.
Methods
Chemicals and materials
Copper nitrate (Cu(NO3)2·3H2O, 98%), Sodium hydroxide (NaOH, 96%), L(+)-Ascorbic acid (99%), L-cysteine (98%), Sodium borohydride (NaBH4, 96%), and isopropanol (IPA) (C3H8O, 99%) were purchased from Sinopharm Chemical Reagent Co., Ltd. Potassium chloropalladite (K2PdCl4, 98%) was purchased from Shanghai Acmec Biochemical Technology Co., Ltd. Ag nanoparticles were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Polytetrafluoroethylene (PTFE, particle size: 200 nm) was purchased from Shanghai Macklin Biochemical Technology Co., Ltd. GDL (YLS-30T) and Nafion (5.0 wt%, DuPont 520) were purchased from Suzhou Sinero Technology Co., Ltd. All chemicals were analytical grade and used without additional purification. The ultrapure water used in the experiments was deionized water (DI) with a resistivity of 18.2 MΩ·cm.
Fabrication of the Cu2O catalyst
Cu2O nanocube particles were prepared by a wet chemical method. First, 120.0 mg Cu(NO3)2·3H2O was added to 90.0 mL DI and stirred for 5 min. Subsequently, 200.0 mg NaOH, which had been dissolved in 5.0 ml DI, was added under stirring conditions to form a blue precursor solution. Finally, L(+)-Ascorbic acid dissolved in 5.0 mL DI was slowly dripped into the precursor solution and stirred vigorously for 60 min until the solution turned orange. The product was collected, washed with DI water and ethanol three times by centrifugation, and dried in a vacuum oven at 60 °C for 12 h. The final product was Cu2O nanocube particles with a size of about 200 nm.
Fabrication of the Pd-Cu2O catalyst
The above-prepared 70.0 mg Cu2O was added to 70.0 mL ethanol and dispersed by sonication for 1 h. Subsequently, 1.2 mg of L-cysteine was added and sonication was continued for 30 min. A certain volume (2.0 mL, 4.0 mL, and 6.0 mL) of K2PdCl4 precursor solution (0.01 mol·L−1) was added to the mixed solution under high-speed stirring conditions. The amount of Pd added was varied by changing the volume. After 60 min of stirring, 1.42 mg NaBH4 dissolved in 1.25 mL DI was added to the mixed solution and stirred continuously for 60 min. The products were collected and washed by centrifugation with DI water and ethanol. The Pd-Cu2O composites were obtained after drying in a vacuum oven for 12 h. All samples labeled Pd-Cu2O represent samples of 4.0 mL of K2PdCl4 precursor solution added, and 0.7 wt% Pd was obtained by inductively coupled plasma optical emission spectrometer (ICP-OES) testing.
Preparation of Cu2O electrodes
All electrodes were prepared by the spraying method. 20.0 mg Cu2O was dispersed in 2.0 mL of isopropanol, and 50 μL of Nafion solution was added as a binder. Dispersing was carried out under ultrasonic conditions for 1 h to form a homogeneous ink. The GDE was placed on a heater at 70 °C and sprayed with a spray gun fitted with ink. The quality of the catalyst was determined by weighing the change in mass of the GDE before and after spraying. The loading of Cu2O was controlled to be 1.0 mg·cm−2 by spraying several times. Finally, the Cu2O electrode was cut to 2.5 cm × 1.0 cm for testing.
Preparation of Cu2O/PTFE electrodes
Cu2O/PTFE electrodes were prepared in a similar manner to that described above. 20.0mg of Cu2O and 50μL Nafion were added to 2.0 mL IPA to make solution A. 10.0 mg of polytetrafluoroethylene (PTFE) powder was added to 2.0 mL IPA to make solution B. Solution A and solution B, after ultrasonication for 60 min, were mixed, and ultrasonication was continued for another 30min to form ink. The steps for spraying were the same as described above.
Preparation of Cu2O/PTFE/Ag electrodes
A total of 10.0 mg of Ag and 50 μL Nafion were added to 2.0 mL IPA and sonicated for 30 min to form inks. Spraying of Ag was continued on Cu2O/PTFE electrodes to obtain Cu2O/PTFE/Ag electrodes. The loading of Ag was controlled to be 1.0 mg cm−2.
Preparation of Pd-Cu2O/PTFE/Ag electrodes
The Pd-Cu2O/PTFE/Ag electrode was prepared in the same way as the Cu2O/PTFE/Ag electrodes, but the Cu2O was replaced with Pd-Cu2O.
Preparation of Cu electrodes
Cu electrodes were prepared using a magnetron sputtering system (MSP-300B). A Cu layer with a thickness of about 400 nm was sputtered on carbon paper at a rate of 1 Å/sec.
Product analysis and performance evaluation for CO2RR
All electrocatalytic CO2 reduction reaction (CO2RR) measurements were tested on a CHI 760e electrochemical workstation with three electrodes. The flow cell reactor consisted of a cathodic gas runner plate, a cathodic flow chamber, and an anodic flow chamber. The volume of the cathode flow chamber is 0.7 cm3, and the reaction area is 1.0 cm2. The components are separated by PTFE gaskets. The cathode was a prepared gas diffusion electrode (2.5 cm × 1.0 cm), and the anode was IrOx/Ti (2.0 cm × 2.0 cm). Fresh 1.0 M KOH (pH = 13.7 ± 0.1) was used as the cathodic and anodic electrolyte. The electrolytes were all transported to the flow cell by a peristaltic pump at a flow rate of 13.0 mL min−1 and separated by an anion exchange membrane (3.0 cm × 3.0 cm, 130 μm). The anion exchange membrane was repeatedly flushed with pure water prior to use. CO2 (Messer gas, 99.99%) controlled by a mass flow controller (MFC) was delivered to the cathode gas flow channel plate. The gas flow rate at the outlet during the reaction was controlled to 30.0 mL·min−1 and monitored using a digital flowmeter. The product was collected after operating for 30 min in constant current mode. The gas phase products of CO2RR were analyzed using an online gas chromatograph. The chromatograph (PerkinElmer Clarus 680) was equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID). Ar (Messer gas, 99.999%) was used as carrier gas. The liquid phase products were analyzed using 1H nuclear magnetic resonance (1HNMR) spectroscopy on an Advance III 500-MHz Unity plus spectrometer (Bruker). A mixture of 500 μL of liquid phase product with 100 μL of DMSO was diluted to 100 ppm with D2O for testing. A saturated Ag/AgCl electrode was used as the reference electrode and was calibrated in H2‑saturated 0.5 M H2SO4 (pH = 0) prior to testing. A Pt electrode and a graphite rod were employed as the working electrode and counter electrode, respectively. Cyclic voltammetry (CV) was conducted at a scan rate of 1 mV s⁻¹, and the potential at zero current was taken as the calibrated EAg/AgCl. No IR compensation was employed for all electrochemical tests. All the potentials used were based on the reversible hydrogen electrode (RHE) using the following equation (Eq. 1):
Characterization
Scanning electron microscope (SEM) was performed on a HITACHI S-4800. X-ray photoelectron spectroscopy (XPS) measurements were conducted on a Thermo Scientific ESCALAB 250Xi instrument. Contact angle images were obtained by Bruker Theta Flex/DSA25. X-ray diffraction (XRD) patterns were obtained on a Rigaku Ultima IV diffractometer using Cu Kα radiation. High-resolution transmission electron microscope (HRTEM) measurements were carried out on a FEI Talos F200s electron microscope. Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) was captured using a JEOL GrandARM300F with double Cs correctors at an acceleration voltage of 300 kV. Inductively coupled plasma optical emission spectroscopy (ICP-OES) measurements were carried out by ThermoFisher iCap 7000. The electrochemical surface area (ECSA) of the electrode was quantified by measuring its double-layer capacitance (Cdl). CV was performed at various scan rates within a potential window where no Faradaic processes occur. The value of Cdl was then obtained by fitting the slope of the plot of the charging current against the CV scan rate. Electrochemical impedance spectra (EIS) measurements were carried out in the frequency range from 0.1 Hz to 100 kHz at 0 V (vs. RHE) with an amplitude of 10 mV. The resistance of the flow cell was measured using EIS under open circuit potentials (0.62 ± 0.03 Ω). The room temperature in the experiment was consistently maintained at 298 K (25 °C).
In situ X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopies were carried out using the Rapid XAFS HE (Anhui Absorption Spectroscopy Analysis Instrument Co., Ltd.) by transmission mode at 20 kV and 20 mA. A Si (553) spherically bent crystal analyzer (SBCA) with a radius of curvature of 500 mm served as the monochromator, thereby ensuring a diffraction geometry approaching a 90-degree backscatter angle at the absorption edge. After monochromatization, the X-rays pass through the sample and were collected using a high-energy-resolution silicon drift detector (SDD) to obtain the X-ray intensity. In measurements, a custom-made cell was utilized (The volume of the chamber is approximately 15.0 cm3), the samples were uniformly dispersed on carbon paper and immersed in a 0.5 M KHCO3 electrolyte (pH = 6.7 ± 0.1) saturated with CO2 gas. A saturated Ag/AgCl electrode and a Graphite rod electrode were used as the reference and counter electrodes, respectively. The XAFS data were acquired in transmission mode. During the XAFS measurements, the position of the absorption edge (E0) was calibrated using a standard Cu foil, and all data were collected within a single time period.
Operando surface-enhanced Raman spectroscopy (SERS) was carried out using a Horiba LabRAM HR Evolution Raman microscope in a modified flow cell (The configuration was identical to that of the flow cells used in electrochemical testing). The volume of the cathode flow chamber is 8.0 cm3, and the reaction area is 0.5 cm2. The electrolytes were all transported to the flow cell by a peristaltic pump at a flow rate of 13.0 mL min−1 and separated by an anion exchange membrane (3.0 cm × 1.5 cm, 130 μm). A saturated Ag/AgCl electrode and a IrOX/Ti electrode (2.0 cm ×1.0 cm) were used as the reference and counter electrodes, respectively. 1.0 M KOH (pH = 13.7 ± 0.1) was used as the electrolyte, and CO2 gas is continuously passed into the cathode. A 632.8 nm excitation laser was used. The signals were recorded at different applied potentials, using a 60-second integration and by averaging two scans.
Performance evaluation for CO2RR
The thickness of the diffusion layer δ can be determined as follows:
where Rd and Cd are the equivalent resistance and capacitance of the diffusion layer, and the values are obtained from the equivalent circuit fitting of EIS. D0 is the diffusion coefficient of CO2 in the electrolyte, which is affected by temperature changes62,63. D0 at different temperatures is calculated by the following equation:
where T is the thermodynamic temperature (298 K, 323 K, and 348 K), p is the pressure (Pa), MA and MB are the molecular weight of H2O and CO2, and VA and VB are the molar volumes of H2O and CO2. The D0 values at 298 K, 323 K, and 348 K are 0.1574, 0.1776, and 0.1986 cm2·s−1, respectively62.
FE for each product was calculated based on the following equation:
where zi is the number of electrons transferred for product, xi is the number of moles of the product, F is Faraday’s constant and Q is the total charge passed during electrolysis.
CEE for C2+ product was calculated based on the following equation:
where FEi is the FE for each C2+ product. Ei is the thermodynamic reduction potential of each C2+ product, Eethylene = 0.06 V vs RHE, Eethanol = 0.08 V vs. RHE, En-propanol = 0.10 V vs. RHE, and Eacetate = 0.12 V vs. RHE for the CO2RR64,65. Ecathode is the measured potential values in the experiment.
DFT calculations
In this work, all density functional theory (DFT) calculations were performed using the plane-wave code QUANTUM ESPRESSO66,67,68. The exchange-correlation potential was approximated with the Perdew−Burke−Ernzerhof functional69,70. The interaction between the cores and the valence electrons was represented with the projector augmented wave approach71, kinetic energy cutoff for wavefunctions and charge density were 50 and 500 Ry, respectively. The convergence criteria for electronic structure iterations and force on each atom during structure relaxation were set to 10−8 Ry and 5×10−4 Ry/Bohr, respectively. The 3×3 Cu2O (100) were modeled by two-layer slab with a 15 Å vacuum region to avoid interactions between periodic images. We sample the Brillouin zone with a 3×3×1 Γ-centered Monkhorst-Pack k-points for geometry optimizations. Gaussian smearing with a width of 10−3 Ry was used. For all the calculations, the van der Waals (vdW) interactions are described with the DFT-D3 method in Grimme’s scheme72. The transition states were determined using the climbing image nudged elastic band (CI-NEB) approach. The TS structure was optimized using quasi-Newton algorithm, and the TS were converged to a residual force smaller than 0.05 eV/Å. Additional, on-site Coulomb repulsion between the Cu 3 d orbitals were chosen as U = 6.0 eV was adopted. One of the surface Cu atom in the unit cell was substituted by Pd atom, which was used as the model of Pd1/Cu2O catalyst. Atomic coordinates of computational models for Cu2O (100) and Pd-Cu2O (100) are shown in Supplementary Data 1.
Data availability
The data generated in this study are provided in the Source Data file. Source data are provided with this paper.
References
Artz, J. et al. Sustainable conversion of carbon dioxide: an integrated review of catalysis and life cycle assessment. Chem. Rev. 118, 434–504 (2018).
Sullivan, I. et al. Coupling electrochemical CO2 conversion with CO2 capture. Nat. Catal. 4, 952–958 (2021).
Wakerley, D. et al. Gas diffusion electrodes, reactor designs and key metrics of low-temperature CO2 electrolysers. Nat. Energy 7, 130–143 (2022).
Chen, C., Li, Y. & Yang, P. Address the “alkalinity problem” in CO2 electrolysis with catalyst design and translation. Joule 5, 737–742 (2021).
Zhang, Z. et al. Additives-modified electrodeposition for synthesis of hydrophobic Cu/Cu2O with Ag single atoms to drive CO2 electroreduction. Adv. Mater. 37, 2411498 (2025).
Wang, Y. et al. Enhancing CO₂ electroreduction to multicarbon products by modulating the surface microenvironment of electrode with polyethylene glycol. Angew. Chem. Int. Ed. 64, 202420661 (2025).
Crandall, B. S. et al. Kilowatt-scale tandem CO2 electrolysis for enhanced acetate and ethylene production. Nat. Chem. Eng. 1, 421–429 (2024).
Chen, Q. et al. Electrocatalytic CO2 reduction to C2+ products in flow cells. Adv. Mater. 36, 2303902 (2024).
Sui, P.-F., Gao, M.-R., Wang, Y.-C. & Luo, J.-L. Recent progress in cathode material design for CO2 electrolysis: from room temperature to elevated temperatures. Acc. Mater. Res. 5, 699–711 (2024).
Pelzer, H. M., Kolobov, N., Vermaas, D. A. & Burdyny, T. Scaling and heating will drive low-temperature CO2 electrolysers to operate at higher temperatures. Nat. Energy 10, 549–556 (2025).
Li, K. et al. Superhydrophobicity-enabled efficient electrocatalytic CO2 reduction at a high temperature. ACS Catal 13, 9346–9351 (2023).
Proietto, F., Rinicella, R., Galia, A. & Scialdone, O. Electrochemical conversion of CO2 to formic acid using a Sn based cathode: combined effect of temperature and pressure. J. CO2 Util. 67, 102338 (2023).
Brandão, V. D. et al. Temperature effects on the surface CO population during CO2 electroreduction over copper. ACS Catal 15, 8979–8990 (2025).
Vos, R. E. et al. How temperature affects the selectivity of the electrochemical CO2 reduction on copper. ACS Catal 13, 8080–8091 (2023).
Kibria, M. G. et al. Electrochemical CO2 reduction into chemical feedstocks: from mechanistic electrocatalysis models to system design. Adv. Mater. 31, 1807166 (2019).
Ahn, S. T., Abu-Baker, I. & Palmore, G. T. R. Electroreduction of CO2 on polycrystalline copper: effect of temperature on product selectivity. Catal. Today 288, 24–29 (2017).
Hu, L. et al. Influence of environmental conditions on electrocatalytic CO2 reduction. ChemCatChem 16, e202301335 (2024).
Löwe, A. et al. Influence of temperature on the performance of gas diffusion electrodes in the CO2 reduction reaction. ChemElectroChem 6, 4497–4506 (2019).
Zhuansun, M. et al. Understanding the temperature effect on carbon-carbon coupling during CO and CO2 electroreduction in zero-gap electrolyzers. Chin. J. Chem. 42, 2705–2711 (2024).
Dufek, E. J., Lister, T. E. & McIlwain, M. E. Bench-scale electrochemical system for generation of CO and syn-gas. J. Appl. Electrochem. 41, 623–631 (2011).
Lin, L. et al. Temperature-dependent CO₂ electroreduction over Fe–N–C and Ni–N–C single-atom catalysts. Angew. Chem. Int. Ed. 60, 26582–26586 (2021).
Lu, X. et al. Selectivity control by modifying pressure, temperature, and ionomer decoration for CO2 electroreduction using gas-diffusion Cu electrodes. Chem Catal 4, 101030 (2024).
Liu, S. et al. Temperature-dependent pathways in carbon dioxide electroreduction. Sci. Bull. 70, 889–896 (2025).
Li, Y. et al. Perovskite-socketed sub-3 nm copper for enhanced CO2 electroreduction to C2+. Adv. Mater. 34, 2206002 (2022).
Yang, R. et al. In situ halogen-ion leaching regulates multiple sites on tandem catalysts for efficient CO2 electroreduction to C2+ products. Angew. Chem. Int. Ed. 61, 202116706 (2022).
Li, H. et al. High-rate CO2 electroreduction to C2+ products over a copper–copper iodide catalyst. Angew. Chem. Int. Ed. 60, 14329–14333 (2021).
Chen, R. et al. Atomically dispersed scandium in cuprous oxide weakens *CO adsorption to boost carbon dioxide electroreduction toward C2 Products. Adv. Funct. Mater. 35, 2415940 (2024).
Sang, J. et al. A reconstructed Cu2P2O7 catalyst for selective CO2 electroreduction to multicarbon products. Angew. Chem. Int. Ed. 61, 202114238 (2022).
Ma, L. et al. Cu supraparticles with enhanced mass transfer and abundant C–C coupling sites achieving ampere-level CO2-to-C2+ electrosynthesis. Nat. Commun. 16, 3421 (2025).
Chen, R. et al. Dynamically reconstructed triple-copper-vacancy associates confined in Cu nanowires enabling high-rate and selective CO2 electroreduction to C2+ products. Adv. Mater. 36, 2314209 (2024).
Vos, R. E., Smaak, J. P. & Koper, M. T. M. The temperature dependence of electrochemical CO2 reduction on Ag and CuAg alloys. J. Catal. 436, 115613 (2024).
Yang, R., Yesuraj, J. & Kim, K. Effect of flow channel shape and operating temperature on the performance of a proton exchange membrane electrolyzer cell. Energy Fuels 37, 12178–12191 (2023).
Zuo, P. et al. Ion exchange membranes: constructing and tuning ion transport channels. Adv. Funct. Mater. 32, 2207366 (2022).
Chen, X. et al. Multivalent Cu sites synergistically adjust carbonaceous intermediates adsorption for electrocatalytic ethanol production. Nat. Commun. 15, 7691 (2024).
Chen, C. et al. Asymmetrically coordinated Cu dual-atom-sites enables selective CO2 electroreduction to ethanol. Adv. Mater. 36, 2409797 (2024).
Xia, W. et al. Adjacent copper single atoms promote C–C coupling in electrochemical CO2 reduction for the efficient conversion of ethanol. J. Am. Chem. Soc. 145, 17253–17264 (2023).
Gutiérrez-Guerra, N. et al. Gas-phase electrocatalytic conversion of CO2 to chemicals on sputtered Cu and Cu–C catalysts electrodes. J. Energy Chem. 31, 46–53 (2019).
Chao, Y. et al. Integrating Cu+/Cu0 sites on porous nitrogen-doped carbon nanofibers for stable and efficient CO₂ electroreduction to multicarbon products. J. Energy Chem. 101, 453–462 (2025).
Xie, L. et al. Direct low concentration CO2 electroreduction to multicarbon products via rate-determining step tuning. Nat. Commun. 15, 10386 (2024).
He, Q. et al. Highly selective CO2 electroreduction to C2H4 using a dual-sites Cu(II) porphyrin framework coupled with Cu₂O nanoparticles via a synergetic-tandem strategy. Angew. Chem. Int. Ed. 3, 202407090 (2024).
Liu, W. et al. Electrochemical CO2 reduction to ethylene by ultrathin CuO nanoplate arrays. Nat. Commun. 13, 1877 (2022).
Wei, Z. et al. Constructing Ag/Cu2O interface for efficient neutral CO2 electroreduction to C₂H₄. Angew. Chem. Int. Ed. 64, 202417066 (2025).
Choi, W. et al. Exploring the influence of cell configurations on Cu catalyst reconstruction during CO2 electroreduction. Nat. Commun. 15, 8345 (2024).
O’Brien, C. P. et al. Scalability and stability in CO2 reduction via tomography-guided system design. Joule 8, 2903–2919 (2024).
Corral, D. et al. Bridging knowledge gaps in liquid- and vapor-fed CO2 electrolysis through active electrode area. Chem Catal 2, 3239–3253 (2022).
Xing, Z., Hu, X. & Feng, X. Tuning the microenvironment in gas-diffusion electrodes enables high-rate CO2 electrolysis to formate. ACS Energy Lett 6, 1694–1702 (2021).
Luan, P. et al. Selective electrosynthesis of ethanol via asymmetric C–C coupling in tandem CO2 reduction. ACS Catal 14, 8776–8785 (2024).
Cheng, Y. et al. Microenvironment tailoring for electrocatalytic CO2 reduction: effects of interfacial structure on controlling activity and selectivity. J. Am. Chem. Soc. 147, 12438–12448 (2025).
Ling, N. et al. Acidic media impedes tandem catalysis reaction pathways in electrochemical CO2 reduction. Angew. Chem. Int. Ed. 62, 202308782 (2023).
Xu, Y. et al. Cation effect on the elementary steps of the electrochemical CO reduction reaction on Cu. Nat. Catal. 7, 1120–1129 (2024).
Zhou, D. et al. Cooperation of different active sites to promote CO2 electroreduction to multi-carbon products at ampere-level. Angew. Chem. Int. Ed. 63, 202400439 (2024).
Nie, S., Wu, L., Liu, Q. & Wang, X. Entropy-derived synthesis of the CuPd sub-1nm alloy for CO2-to-acetate electroreduction. J. Am. Chem. Soc. 146, 29364–29372 (2024).
Greeley, J. et al. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 5, 909–913 (2006).
Wang, Y. et al. Enhanced local CO coverage on Cu quantum dots for boosting electrocatalytic CO2 reduction to ethylene. Adv. Funct. Mater. 35, 2417764 (2025).
Jiang, Y. et al. Structural reconstruction of Cu₂O superparticles toward electrocatalytic CO2 reduction with high C₂₊ products selectivity. Adv. Sci. 9, 2105292 (2022).
He, X. et al. Roles of copper(I) in water-promoted CO2 electrolysis to multi-carbon compounds. Nat. Commun. 15, 9923 (2024).
Du, Z.-Y. et al. Promoting water activation via molecular engineering enables efficient asymmetric C–C coupling during CO2 electroreduction. J. Am. Chem. Soc. 146, 32870–32879 (2024).
Xie, Y. et al. High carbon utilization in CO2 reduction to multi-carbon products in acidic media. Nat. Catal. 5, 564–570 (2022).
Chen, Y. et al. Efficient multicarbon formation in acidic CO2 reduction via tandem electrocatalysis. Nat. Nanotechnol. 19, 311–318 (2024).
Zhu, J. et al. Asymmetric triple-atom sites confined in ternary oxide enabling selective CO2 photothermal reduction to acetate. J. Am. Chem. Soc. 143, 18233–18241 (2021).
Li, L. et al. Neighboring nonmetal site as an intermediate modulator switching CO2 electroreduction pathway toward multicarbons. Joule 9, 101926 (2025).
Sheng, X., Ge, W., Jiang, H. & Li, C. Engineering the Ni-N-C catalyst microenvironment enabling CO2 electroreduction with nearly 100% CO selectivity in acid. Adv. Mater. 34, 2201295 (2022).
Zhang, W. et al. Investigations on the interfacial capacitance and the diffusion boundary layer thickness of ion exchange membrane using electrochemical impedance spectroscopy. J. Membr. Sci. 502, 37–47 (2016).
Lai, W., Qiao, Y., Zhang, J., Lin, Z. & Huang, H. Design strategies for markedly enhancing energy efficiency in the electrocatalytic CO2 reduction reaction. Energy Environ. Sci. 15, 3603–3629 (2022).
Chen, L. et al. Energy-efficient CO2 conversion to multicarbon products at high rates on CuGa bimetallic catalyst. Nat. Commun. 15, 7053 (2024).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Giannozzi, P. et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 29, 465901 (2017).
Giannozzi, P. et al. Quantum ESPRESSO toward the exascale. J. Chem. Phys. 152, 154105 (2020).
Dion, M., Rydberg, H., Schröder, E., Langreth, D. C. & Lundqvist, B. I. Van der Waals density functional for general geometries. Phys. Rev. Lett. 92, 246401 (2004).
Klimeš, J., Bowler, D. R. & Michaelides, A. Van der Waals density functionals applied to solids. Phys. Rev. B 83, 195131 (2011).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2023YFA1508000 to Z.C. and G.W.), the National Natural Science Foundation of China (22472139 to Z.C., U23A2087 to X.Y., U22A20392 to Z.C., 22227802 to Z.C., 22372137 to X.Y., 22102136 to Z.C., 22125205 to G.W., and U25A20622 to G.W.), the Fundamental Research Funds for the Central Universities (no. 20720250117 to Z.C.), the Natural Science Foundation of Shanghai (grant 25DZ3000400 to G.W.), the Liaoning Binhai Laboratory (LBLF-2023-02 to G.W.), and the Energy Revolution S&T Program of Yulin Innovation Institute of Clean Energy (E511020817 to G.W.).
Author information
Authors and Affiliations
Contributions
L.H. designed and performed the synthetic and electrochemical experiments, characterized the catalyst, and analyzed the data. Y.Y. performed the DFT calculations and analyzed the data. J.W. and D.Z. conducted Raman spectroscopic measurements. Y.Z. and J.H. assisted with the data analysis. X.Y., G.W., and Z.C. supervised the execution of the overall project. The results of the manuscript were discussed by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jun Gu, Masakazu Sugiyama and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, L., Yang, Y., Wang, J. et al. Synergistic electrode design for efficient CO2 electrolysis to multicarbon products at elevated temperatures. Nat Commun 17, 2684 (2026). https://doi.org/10.1038/s41467-026-69506-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69506-w
