
Abstract
The neurotrophic factor (NTF) family has recently expanded its role beyond neurological conditions, but its involvement in acute inflammatory lung diseases remains largely unclear. Using well-established acute lung injury (ALI) and sepsis models, we demonstrate that brain-derived neurotrophic factor (BDNF), a key NTF, is impaired in pulmonary epithelial cells and negatively correlates with the inflammatory response. Raising the BDNF level alleviates inflammatory lung injury, but these effects are absent in macrophage-deleted mice. Both in vivo and in vitro results show BDNF inhibits macrophage inflammation, and further proteomics analysis identifies macrophage TLR4 as a receptor that BDNF antagonizes via direct binding. The BDNF fragment (aa 104-115) is critical for BDNF-TLR4 interaction, and the corresponding synthetic BDNF-derived dodecapeptide (BDP-12) retains TLR4-antagonistic and anti-inflammatory effects both in vitro and in vivo, without pro-proliferative side effects. In conclusion, our findings reveal that epithelial-derived BDNF prevents macrophage inflammation by directly targeting TLR4 and highlights BDP-12 as a potential therapeutic agent for acute inflammatory diseases.
Similar content being viewed by others


Introduction
Neurotrophic factors (NTFs) are a class of proteins or peptides first identified in the nervous system, where they play crucial roles in regulating neuronal fate through binding to classic receptors such as TrkA, TrkB, TrkC, and p75NTR, respectively1. In recent years, it has been discovered that the NTFs family not only exerts essential functions in the neurological system but also plays significant roles in a variety of non-neurological pathophysiological processes. For instance, glial cell-derived neurotrophic factor (GDNF), derived from the bone tissues, promotes the proliferation and osteogenic differentiation of jaw bone marrow mesenchymal stem cells2. Ciliary neurotrophic factor (CNTF) activates AMPK pathways in skeletal muscle, helping to reverse obesity-induced insulin resistance3. Nerve growth factor (NGF) level has been found to be associated with the development and reproduction of primordial follicles4. Muscle-secreted neurturin (NRTN) enhances capillary density and oxidative capacity in myofibers, while favoring fatty acid metabolism over glycolysis through transcriptional reprogramming5. Additionally, neurotrophin-3 (NT-3), derived from cardiovascular progenitor cells, can protect cardiomyocytes from death during re-perfused myocardial infarction6. These findings highlight the functional diversity of the NTF family across different tissues. Therefore, a systematic exploration of the relationship and mechanisms between NTFs and certain diseases may provide new targets and strategies for their treatment.
Acute inflammation is a critical defense mechanism triggered by external factors such as infections, trauma, toxins, or other forms of injury7. Its primary role is to eliminate harmful substances and promote tissue repair. However, when the inflammatory response becomes excessive or prolonged, it can lead to the progression of acute inflammatory diseases. In the context of infections, pathogen-associated molecular patterns (PAMPs) stimulate tissue-resident macrophages via pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs)8,9. This process initiates the release of pro-inflammatory cytokines, which, when present in excessive amounts, induce apoptosis in parenchyma cells10. Concurrently, tissue-resident macrophages secrete adhesion molecules and chemokines, facilitating the infiltration of leukocytes, particularly neutrophils and monocytes, thereby exacerbating the inflammatory response. Furthermore, damage-associated molecular patterns (DAMPs) released by injured parenchyma cells act as additional pro-inflammatory stimuli, sustaining the inflammatory cascade, leading to rapid deterioration and potentially fatal outcomes11,12. Despite the sporadic reports, the connection and molecular mechanisms between NTFs and acute inflammatory diseases outside the brain are still largely unexplored. A comprehensive investigation into this area is of significant importance and may provide valuable insights and candidates for guiding the discovery of new therapeutic drugs for clinical anti-inflammatory treatment.
In this study, we utilized the well-established acute lung injury (ALI) and sepsis models to investigate the potential role of NTFs in regulating the progression of acute inflammatory diseases. Our findings revealed that brain-derived neurotrophic factor (BDNF), specifically expressed in the pulmonary epithelium, was significantly and negatively correlated with the progression of ALI and the severity of inflammation. Using high-throughput omics technologies, we further discovered that BDNF secreted by alveolar epithelial cells, or administered exogenously, can directly antagonize TLR4 in macrophages, thereby exerting anti-inflammatory effects and alleviating ALI and sepsis. Molecular studies confirmed that the BDNF aa 104-115 is the key segment responsible for binding to TLR4. Based on this fragment antagonizing TLR4, we synthesized the BDNF-derived dodecapeptide (BDP-12), which was ultimately identified as a therapeutic candidate for acute inflammation, without potential pro-proliferative side effects.
Results
Pulmonary epithelial-specific BDNF is negatively correlated with the progression of ALI
We performed RNA-seq using lung tissues of control and LPS-challenged mice to analyze the mRNA expression profile of 9 typical NTFs (Fig. 1A). In addition, we also checked two public RNA-seq datasets from P.aeruginosa-infected ALI model (GSE233206) and LPS-treated lungs (GSE214249) (Fig. 1A). Among the 9 typical NTFs, only BDNF mRNA (Bdnf) was found to be monotonically downregulated across all three datasets (Fig. 1B). Additional RNA-seq datasets from both murine models and human samples of pneumonitis also showed significantly decreased Bdnf expression compared to the control groups (Supplementary Fig. S1A–C). A rough time-dependent pattern of Bdnf transcripts was underscored in P. aeruginosa-(GSE204709) and hvKp-(GSE199546) infected lungs as well (Fig. 1C, D). Interestingly, the close relationship between BDNF and ALI was emphasized with the highest abundant and most significant decline of Bdnf expression in the lungs compared to other organs in an endotoxemia model (Supplementary Fig. S1D).

A Heatmap of typical NTKs (including Artn, Bdnf, Cntf, Gdnf, Ngf, Nrtn, Ntf3, Ntf5, and Pspn) from both self-performed sequencing data (PRJNA1183441 in SRA database) and publicly available datasets (GSE233206 and GSE214249 in the GEO database). B Veen diagram of differentially expressed genes in panel A. C The count value of Bdnf in GSE204709 across various infection time points (0–14 d). D The FPKM value of Bdnf in GSE199546 across various infection time points (0–60 h). E BDNF content in serum and BALF of non-ARDS and ARDS patients. F BDNF content in serum and BALF of mice treated with or without LPS. G tSNE plot of 13 cell clusters in mouse lungs. H Dot plot showing the expression of marker genes for the 13 cell clusters. I Feature plots illustrating the expression scores of Bdnf in combined and separate Sham and CLP groups. J Dot plot showing the expression of Bdnf in 13 cell clusters of the Sham and CLP group. K The protein level of BDNF in lung tissues from mice treated with or without LPS, and GAPDH was used as loading control. Corresponding relative BALF IL-6 levels for each group are shown on the right. L Protein level of BDNF in primary epithelial cells (ECs), primary fibroblasts (PFs), and primary macrophages (PMs) with or without 500 ng/mL LPS treatment for 24 h. Data are represented as mean ± SEM by unpaired 2-tailed Student’s t-test (E, F, K). Source data are provided as a Source Data file.
BDNF protein, as a secreted cytokine cleaved from its precursor proBDNF and exhibiting 100% homology across various species (Supplementary Fig. S2A, B), was measured in both human and mouse samples. Notably, a significant decrease in BDNF levels was observed exclusively in BALF from both ALI mice and ARDS patients, without corresponding changes in blood, suggesting that the ALI progression is associated with the change of local production of BDNF in lung, rather than the systemic or central BDNF secretion (Fig. 1E, F). To further investigate the cellular source of BDNF in lungs, 13 distinct lung cell types were identified in cecal ligation and puncture (CLP)-induced and sham-operated mice using 26 specific cell markers (Fig. 1G, H). Mapping Bdnf transcripts revealed an overall decrease, with predominant expression and reduction observed in lung epithelial cells following CLP modeling (Fig. 1I, J). These findings were corroborated by additional scRNA-seq datasets (Supplementary Fig. S1E, F). Parallelly, significant reductions in BDNF protein level were detected in LPS-treated mouse lungs and primary epithelial cells, but not in LPS-treated primary fibroblasts or macrophages (Fig. 1K, L). Together, these results highlight a critical association between reduced BDNF expression in lung epithelial cells and the pathogenesis of ALI.
Epithelial BDNF expression is regulated by GATA2 during ALI
We then tried to elucidate the regulatory mechanisms behind the decreased expression of BDNF in lung epithelial cells during ALI. We employed five databases for transcription factor prediction, identifying a total of 11 high-potential common transcription factors (Supplementary Fig. S3A). We then assessed the expression changes of these transcription factors using transcriptomic data, revealing that seven of them exhibited a consistent downregulation trend (Supplementary Fig. S3B, C). Further single-cell transcriptomic analysis indicated that only GATA2 and E2F1 displayed decreased expression levels in damaged lung epithelial cells (Supplementary Fig. S3D). Protein-level analysis corroborated these findings, confirming the reduction of GATA2 and E2F1 in lung tissues of ALI mice (Supplementary Fig. S3E, F). We then overexpressed GATA2 and E2F1 in MLE-12 cells and found that overexpression of GATA2 resulted in a significant increase in Bdnf transcription (Supplementary Fig. S3G). We then further applied ChIP-qPCR to verify the ability of GATA2 to directly bind and regulate the Bdnf gene promoter. Based on our lung RNA-seq dataset, we identified two BDNF transcripts with high basal expression that were markedly downregulated after LPS stimulation (Supplementary Fig. S3H). Four primer pairs were designed to cover the promoter regions corresponding to these transcripts. ChIP-qPCR using primer set 2 demonstrated that GATA2 directly binds to the Bdnf promoter, and this binding was substantially reduced in LPS-treated epithelial cells (Supplementary Fig. S3I). Together, these findings indicate that GATA2 is a key transcriptional regulator of Bdnf in lung epithelial cells, and that its downregulation during ALI contributes to the reduced BDNF expression observed in injured lungs.
Epithelial-specific BDNF overexpression and exogenous BDNF administration alleviate LPS-induced ALI in mice
Then, the lung epithelial-specific BDNF-overexpressed mice (Ad-BDNF) were successfully constructed via injecting AAV6 encoding mature BDNF and the lung epithelial-specific promoter, and then modeled to ALI by LPS installation (Fig. 2A; and Supplementary Fig. S4A, B). From the results of wet-to-dry ratio, protein concentration in BALF, and histological staining, we found distinct indications of pulmonary edema, protein leakage, and increased thickness of the alveolar wall in ALI mice. However, these pathological features were ameliorated in LPS-challenged Ad-BDNF mice (Fig. 2B–E). Furthermore, the heightened total cell counts in BALF pointed to immunocyte infiltration in LPS-exposed lungs, while BDNF overexpression effectively blocked this infiltration and reduced the number of neutrophils, as evidenced by diminished MPO activity and cell nuclear staining (Fig. 2F–H). This effect was consistent with the decreased expression of the adhesion molecules Icam1 and Vcam1, which are critical for immune cell infiltration and localization (Fig. 2I, J). Flow cytometric analysis of BALF samples also demonstrated the attenuation of both neutrophil and macrophage infiltration in LPS-challenged Ad-BDNF mice (Fig. 2K). In addition, BDNF overexpression significantly increased survival rates in mice with E.coli-induced septic mice (Fig. 2L).

A Schematic illustration of mice treated with AAV6 and modeled using LPS. B Lung wet/dry weight ratio measured in mice from panel (A). C Total protein levels in BALF samples collected from mice in (A). D Representative H&E-stained images of lung tissues collected from mice in panel (A). E Quantification of lung injury scores for (D). F Total cell counts in BALF samples collected from mice in (A). G MPO activity levels in lung lysates from mice in panel (A). H Neutrophil counts assessed by Wright-Giemsa staining in BALF samples from mice in (A). I, J mRNA levels of Icam1 (I) and Vcam1 (J) detected by RT-qPCR in lung tissues from mice in panel (A). K Flow cytometric detection of infiltrating cell populations in BALF samples from mice in (A). L Survival curve of AAV6-infected mice in an E.coli-induced sepsis model. M Schematic illustration of mice modeled using LPS and treated with rBDNF. N Lung wet/dry weight ratio measured in mice from panel (M). O Total protein levels in BALF samples collected from mice in (M). P Representative H&E-stained images of lung tissues collected from mice in panel (M). Q Quantification of lung injury scores for (P). R Total cell counts in BALF samples collected from mice in panel (M). S MPO activity levels in lung lysates from mice in (M). T Neutrophil counts assessed by Wright-Giemsa staining in BALF samples from mice in (M). U, V mRNA levels of Icam1 (U) and Vcam1 (V) detected by RT-qPCR in lung tissues from mice in (M). W Flow cytometric detection of infiltrating cell populations in BALF samples from mice in (M). X Survival curve of rBDNF-treated mice in an E.coli-induced sepsis model. Data are represented as mean ± SEM by one-way ANOVA with Tukey’s test. n = 6 per group. Source data are provided as a Source Data file.
Recombinant BDNF protein (rBDNF) was also administered intravenously to evaluate the therapeutic effect of exogenous BDNF against ALI in mice (Fig. 2M). The mice treated with rBDNF showed increased BNDF protein levels in serum and lung tissues (Supplementary Fig. S4C, D). As expected, exogenous rBDNF administration alleviated wet-to-dry ratio, protein concentration in BALF, and histopathological injury in LPS-treated mice (Fig. 2N–Q). Immunocyte infiltration was also inhibited by rBDNF in ALI mice, as indicated by total cell counts in BALF, MPO activity, neutrophil staining, qPCR analysis of lung Icam1 and Vcam1, and flow cytometry assay (Fig. 2R–W). Also, rBDNF administration significantly reduced septic death in E.coli-injected mice (Fig. 2X). In summary, these data show that either epithelial-specific BDNF overexpression or exogenous rBDNF treatment alleviates LPS-induced ALI in mice.
BDNF alleviates ALI through inhibiting inflammation in macrophages
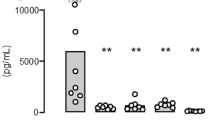
Given that macrophage inflammation is a key driver of ALI, we further investigated whether BDNF is directly associated with the inflammatory response in ALI. Our results revealed a negative correlation between Bdnf transcript levels and Il6, a gene encoding the pro-inflammatory cytokine IL-6 in RNA-seq datasets of ALI mice (Fig. 3A). Similarly, secreted BDNF protein content was negatively correlated with both IL-6 and TNF-α concentrations in both mice and human BALF (Fig. 3B, C). These data suggest that BDNF negatively regulates the inflammatory response during ALI progression.

A Correlation analysis between count values and FPKM values of Bdnf and Il6 in GSE204709 and GSE199546, respectively. B Correlation analysis between BDNF levels in BALF and TNF-α and IL-6 in mice. C Correlation analysis between BDNF levels in human BALF and TNF-α and IL-6. D Primary macrophages were pre-treated with or without 10 nM BDNF for 1 h and then exposed to 500 ng/mL LPS for 6 h. The heatmap shows the transcription levels of more than 40 inflammatory factors detected by PCR array. E Heatmap showing mRNA levels of pro-inflammatory factors Tnf, Il6, Il1b, Ccl2, Ifna1, Ifnb1, and Ifng in lung tissues of mice described in Fig. 2M. F, G Serum (F) and BALF (G) levels of TNF-α, IL-6, and IFN-α in the ALI mouse model described in Fig. 2M. H Schematic illustration of mice undergoing macrophage deletion, ALI modeling, and rBDNF injection. I Representative H&E-stained images of lung tissues. J Quantification of lung injury scores for (I). K Lung wet/dry weight ratio. L Total protein levels in BALF samples. M Total cell counts in BALF samples. N Survival curve of mice with or without macrophage deletion and rBDNF injection in an E.coli-induced sepsis model. Data are represented as mean ± SEM by one-way ANOVA with Tukey’s test. n = 6 per group (E–G, J–M). Source data are provided as a Source Data file.
An inflammatory gene PCR array further demonstrated the anti-inflammatory effects of rBDNF in LPS-challenged macrophages, showing that rBDNF suppresses LPS-induced expression of over 40 key inflammatory genes, including chemokines, interleukins, and interferons (Fig. 3D). ALI mice treated with rBDNF also showed decreased inflammatory cytokines transcription, accompanied by reduced secretion of TNF-α, IL-6, and IFN-α in both serum and BALF (Fig. 3E–G). Subsequently, to determine the role of macrophages in the anti-ALI effects of rBDNF in mice, we used clodronate liposomes (Clo-lip) to delete macrophages in mice (Fig. 3H). Macrophage deletion led to increased tolerance to LPS challenge, but when rBDNF was administered after macrophage depletion, the therapeutic effects of rBDNF were lost, as indicated by no further reduction in histopathological injury, wet-to-dry ratio, BALF protein concentration, immune cell infiltration, and sepsis-induced mouse mortality (Fig. 3I-N). These data indicate that macrophages are the primary effector cells targeted by BDNF. Epithelial cell-secreted BDNF establishes a new cell-cell communication pathway to mitigate macrophage-driven inflammation during ALI.
Identification of BDNF as a antagonistic ligand of TLR4 in macrophages
It is unclear which receptor in macrophages recognizes the extracellular BDNF and mediates its anti-inflammatory effect. Initially, we hypothesized that tyrosine receptor kinase B (TrkB), the canonical and previously reported receptor of BDNF, was responsible for these effects in macrophages. However, when Ntrk2 (encoding TrkB) was silenced, the anti-inflammatory effects of BDNF in macrophages remained, indicating that TrkB was not the primary mediator (Supplementary Fig. S5A, B).
Then, dual-proteomics and bioinformatics approaches were employed to uncover the actual but unknown receptor governing the BDNF’s anti-inflammatory ability (Fig. 4A). These analyses highlighted TLR4 and CCR1 as high-potential BDNF-interacting candidates (Fig. 4B). Subsequent co-immunoprecipitation assays demonstrated that BDNF interacted with TLR4, but not CCR1 (Fig. 4C–D). Surface plasmon resonance analysis further confirmed a direct and high-affinity binding between recombinant BDNF and TLR4 proteins (Fig. 4E). Consistently, Fig. 4D also showed that BDNF did not interact with the TLR4 accessory protein MD2, supporting TLR4 as the primary binding partner. Treatment of macrophages with rBDNF increased BDNF-TLR4 complex content, inversely correlated with MD2-TLR4 complex, suggesting that BDNF competitively binds to TLR4 to prevent MD2-TLR4 interaction in LPS-challenged macrophages (Fig. 4F–H; and Supplementary Fig. S5C). After activation, TLR4 recruits two adaptor proteins (MyD88 and TRIF) to activate downstream kinases (TAK1, MAPKs, TBK1, and IRF3) and transcriptional factors such as NF-κB, AP-1, and IRF, respectively, inducing inflammatory gene transcription and cytokine expression13. We then examined the downstream signaling and response in the TLR4 inflammatory cascade in primary macrophages. As shown in the following results, we found that rBDNF treatment dose-dependently inhibited LPS-induced phosphorylation of TAK1, MAPKs, TBK1, and IRF3 (Fig. 4I; and Supplementary Fig. S5D), as well as activation of transcriptional factors (including NF-κB, AP-1, and IRF) (Fig. 4J), corresponding inflammatory genes transcription (including Tnf, Il6, Il1b, Ccl2, Ifna1, Ifnb1, and Ifng) (Fig. 4K), and cytokine production (including TNF-α, IL-6, and IFN-α) (Fig. 4L) in primary macrophages. We also examined the TLR4 inflammatory cascade in rBDNF-treated mice. As shown in Supplementary Fig. S6A, rBDNF administration increased BDNF-TLR4 interaction in mouse lung tissues. LPS challenges induced MD2-TLR4 interaction, phosphorylation of TAK1, MAPKs, TBK1, and IRF3, and NF-κB activation, while rBDNF treatment reversed these changes in mouse lung tissues (Supplementary Fig. S6B–G).

A Schematic illustration of the workflow integrating protein microarray screening, IP-MS analysis, and subcellular localization filtering to determine the potential receptor for BDNF. B Venn diagram showing TLR4 and CCR1 as the two membrane proteins with potential BDNF-binding capacity. C Co-immunoprecipitation analysis in 293 T cells transfected with or without CCR1 and BDNF plasmids, demonstrating no detectable interaction between CCR1 and BDNF. D 293 T cells were transfected with or without TLR4, MD2, and BDNF plasmids. Co-immunoprecipitation revealed an interaction between BDNF and TLR4, but not MD2. E Surface plasmon resonance analysis of direct affinity between BDNF and TLR4. (F-H) Primary macrophages were pre-treated with or without 5, 10 nM rBDNF for 1 h and then exposed to 500 ng/mL LPS for 0.5 h. F The binding level of BDNF with TLR4 in protein lysates was determined by ELISA assay. G TLR4 was immunoprecipitated (IP) and the associated MD2 level was detected using immunoblotting (IB). H The binding level of MD2 with TLR4 in protein lysates was determined by ELISA assay. I Primary macrophages were pre-treated with or without rBDNF for 1 h and then exposed to 500 ng/mL LPS for 0.5 h. Activation of the MyD88-dependent pathway was assessed by probing for IκB-α, phosphorylated TAK1, and phosphorylated MAPK (ERK, JNK, and p38) proteins. Activation of the TRIF-dependent pathway was assessed by probing for phosphorylated TBK1 and IRF3. Unphosphorylated proteins were used as the controls. J Wild-type macrophages transfected with luciferase reporter plasmids were pre-treated with rBDNF and exposed to LPS for 24 h. Relative luciferase activity of NF-κB p65, AP-1, and IRFs was measured. K Primary macrophages were pre-treated with rBDNF and then exposed to LPS for 6 h. Relative mRNA levels of pro-inflammatory factors Tnf, Il6, Il1b, Ccl2, Ifna1, Ifnb1, and Ifng were determined. L Primary macrophages were pre-treated with rBDNF and then exposed to LPS for 24 h. Cytokine levels of TNF-α, IL-6, and IFN-α in cultured medium were measured. M Schematic illustration of mice undergoing bone marrow transplantation, ALI modeling, and rBDNF injection. N Representative H&E-stained images of lung tissues. O Quantification of lung injury scores for (N). P Total protein levels in BALF samples. Q Lung wet/dry weight ratio. R Total cell counts in BALF samples. S Survival curve of born marrow transplanted mice with or without rBDNF injection in an E.coli-induced sepsis model. Data are represented as mean ± SEM by one-way ANOVA with Tukey’s test. n = 3 (H, J), or n = 4 (F, L, and K), or n = 6 (O–R) per group (E–G, J–M). Source data are provided as a Source Data file.
We then examined if TLR4 mediates the anti-inflammatory effects of BDNF in vivo. We constructed macrophage-specific Tlr4-deficient mice which were generated through bone marrow transplantation, and then exposed these mice to LPS or E.coli challenges (Fig. 4M). As shown in Fig. 4N–S, rBDNF treatment failed to further protect mouse lung tissues and septic death in macrophage-specific Tlr4-/- mice challenged with LPS or E.coli.
BDNF has been reported to suppress the inflammatory response of microglia through TrkB14. However, paradoxical evidence demonstrates the absence of TrkB in microglia, leaving the true receptor unclear15. Based on the high abundance of TLR4 in microglia, we hypothesize that BDNF inhibits neuroinflammation by targeting TLR4. To test this, we knocked down Tlr4 in microglia and found that BDNF’s anti-inflammatory effects were markedly reduced upon LPS stimulation (Supplementary Fig. S7A, B). Taken together, our data identified TLR4 as a new receptor for BDNF and revealed that BDNF inhibits LPS-induced inflammation in macrophages by antagonizing TLR4.
The aa 104-115 peptide segment in BDNF binds to the extracellular LRR domain of TLR4
We try to unravel the BDNF-TLR4 binding mechanism. TLR4 protein consists of the extracellular LRR domain, intracellular TIR domain, and the transmembrane sequence (Fig. 5A). Our immunoprecipitation assay showed that TLR4 lacking LRR domain failed to form complex with BDNF, indicating that BDNF binds to the extracellular LRR domain of TLR4 (Fig. 5B). Immunoprecipitation assay further validated that rBDNF protein could interact with the recombinant protein of LRR domain (rLRR) (Fig. 5C). Subsequently, rBDNF was enzymatically digested to generate BDNF-derived short peptides (BDPs) to identify the TLR4-binding segment in BDNF (Fig. 5D; and Supplementary Table S1). Among these BDPs, a dodecapeptide segment of BDNF aa 104-115 (BDP-12, IDTSCVCTLTIK) was identified as the key fragment responsible for rBDNF-rLRR interaction by using immunoprecipitation-MALDI-TOF analysis (Fig. 5D–F). To reinforce this discovery, we synthesized two random BDPs (BDP-18 and BDP-15), which are far away from the binding segment BDP-12, as the negative comparison (Fig. 5E). We mixed BDP-12, BDP-18, and BDP-15 with rLRR, respectively, and then performed the immunoprecipitation-MALDI-TOF analysis. As expected, only BDP-12 showed the interaction with rLRR protein (Fig. 5G). Circular dichroism analysis also showed BDP-12 could change the spectrum of rLRR protein in 210-240 nm, indicating BDP-12 affects the secondary structure of rLRR, while BDP-18 and BDP-15 failed (Fig. 5H). Further normal CETSA or ITDRF-CETSA studies highlighted the binding ability of BDP-12 with TLR4 in macrophages (Fig. 5I, J). As negative comparisons, BDP-18 and BDP-15 failed to interact with TLR4, and BDP-12 did not bind to MD2 (Fig. 5I, J; and Supplementary Fig. S8). Finally, we constructed a plasmid encoding BDNF lacking aa 104-115 (BDNFΔ) and the immunoprecipitation results showed that BDNFΔ lost its binding ability with the extracellular domain of TLR4 (Fig. 5K). Taken together, the extracellular LRR of TLR4 and BDNF aa 104-115 cooperate to mediate the binding between BDNF and TLR4.

A Schematic illustration of the domain composition in rTLR4, TLR4ΔLRR, and TLR4ΔTIR. B 293 T cells were transfected with or without BDNF, TLR4ΔLRR, and TLR4ΔTIR plasmids. The binding ability between BDNF and TLR4ΔTIR or TLR4ΔLRR was assessed by Co-IP. C rTLR4(LRR domain) was mixed with or without rBDNF, followed by immunoprecipitation with a BDNF antibody and then the associated rTLR4 level was detected using immunoblotting. D Schematic illustration of tryptic digestion and immunoprecipitation combined with mass spectral analysis. E Schematic showing tryptic cleavage sites in BDNF, with BDNF fragments aa 8-25, aa 27-41, and aa105-116 defined as BDP-18, BDP-15, and BDP-12, respectively. F MALDI-TOF spectrogram showing a prominent BDP-12 peak after immunoprecipitating with a TLR4 antibody. The corresponding position in BDNF and sequence are shown below. G MALDI-TOF spectrograms of BDPs standards-rTLR4 mixtures immunoprecipitated with TLR4 antibody. H Circular dichroism analysis of rTLR4 after mixing with gradient concentrations of BDPs standards. I Primary macrophages were treated with BDPs (2 μM) or solvent control for 24 h, followed by CETSA analysis of TLR4 and GAPDH across a temperature range from 40 to 75 °C. Densitometric quantification is shown below. J Primary macrophages were treated with gradient BDPs (0–5 μM) or solvent control for 24 h. ITDRF-CETSA analysis of TLR4 and GAPDH was assessed at 61 °C. Densitometric quantification is shown below. K BDNF mutation with aa 105–119 deletion (BDNFΔ) was constructed as shown above. Following transfection of 293 T cells with wild-type BDNF or BDNFΔ, Co-IP was performed to detect the associated TLR4ΔTIR level. Data are represented as mean ± SEM; n = 3. Source data are provided as a Source Data file.
Identification of key residues involved in BDNF-TLR4 interaction
Molecular docking was employed to further investigate the residues critical for the interaction between BDNF aa 104–115 and TLR4 LRR domain (Fig. 6A). Residue-binding free energy analysis pointed out the significance of T107 and L113 in BDNF, along with L444, F463, and F487 in TLR4 (Supplementary Table S2, S3). Immunoprecipitation assay revealed that BDNF mutation at either T107A or L113A resulted in reduced affinity to TLR4, and the affinity almost disappeared when both mutations were present at T107A/L113A (Fig. 6B). TLR4 mutations including TLR4F463A and TLR4F487A also exhibited impaired or diminished interaction with BDNF (Fig. 6C). Besides, BDNF variants were overexpressed in 293 T cells and secreted into the culture medium, which was then applied to stimulate macrophages (Fig. 6D). The concentrations of the various BDNF variants were consistent across the media (Fig. 6E). Analysis of mRNA levels of the inflammatory markers revealed that BDNFT107A and BDNFL113A displayed a partial reversal, while BDNFT107A/L113A completely abolished the anti-inflammatory effect in LPS-challenged macrophages (Fig. 6F). Taken together, these data underscore the critical roles of T107 and L113 in BDNF and F463 and F487 in TLR4, which jointly contribute to the BDNF-TLR4 interaction.

A Optimal conformation of the BDNF-TLR4 complex. B The binding ability between BDNF and its variants with TLR4 was determined by Co-IP. C The binding ability between TLR4 and its variants with BDNF was determined by Co-IP. D The schematic illustration of the preparation of medium containing BDNF or its variants, followed by treatment of macrophages with or without LPS exposure. E The protein level of BDNF and its variants in the collected medium was assessed by immunoblotting. F mRNA level of Tnf, Il6, and Il1b in macrophages treated as described in (D). Data are represented as mean ± SEM by one-way ANOVA with Tukey’s test; n = 4. Source data are provided as a Source Data file.
BDP-12 shows strong anti-inflammatory effects without pro-proliferative activity
We tried to examine whether the BDNF-derived BDP-12 could be developed as a peptide drug for treating inflammatory lung injury. BDNF, known for its pro-proliferative effects via activating TrkB, is overexpressed in several cancers and is considered as a potential proto-oncogene16,17,18. Since BDP-12 differs from the aa 93-96 within the β-turn of BDNF loop 4, which is the segment known to mediate TrkB activation and promote cell proliferation19, we guess that BDP-12 may avoid the pro-proliferative side effect, a key advantage for its development as an anti-inflammatory drug candidate. We administered rBDNF or BDP-12 to mice for three consecutive days and then assessed PCNA-positive cells in lung tissues. rBDNF treatment induced a marked increase in proliferating (PCNA+) cells, particularly within the epithelial compartment (ECAD+-PCNA+), while macrophages (F4/80+-PCNA+) showed no apparent proliferative response. In contrast, BDP-12 did not trigger any detectable increase in cell proliferation in the lungs (Fig. 7A; and Supplementary Fig. S9A, B). Similarly, in vitro experiments using alveolar epithelial cells revealed that, unlike rBDNF, BDP-12 did not induce TrkB phosphorylation and cell proliferation (Fig. 7B, C; Supplementary Fig. S9C). These data show the safety profile of BDP-12 over BDNF, without potential oncogenic side-effects.

A Mice treated with rBDNF or BDP-12 for 3 d underwent immunohistochemistry staining for PCNA in lung tissues. Quantification of PCNA+ cells was shown in the right. B MLE-12 cells were treated with 10 nM rBDNF or 2 μM BDP-12 for 0.5 h. Then the phosphorylated TrkB, total TrkB, and GAPDH were assessed by immunoblotting. C MLE-12 cells were treated with gradient rBDNF (0–20 nM) or BDP-12 (0–5 μM) for 24 h. Relative cell amounts were quantified using the CCK8 assay (n = 4). D Primary macrophages were pre-treated with 2, 4 μM BDP-12 and then exposed to LPS for 0.5 h. Then the TLR4 was immunoprecipitated and the associated MD2 level was detected using immunoblotting. E–G WT and Tlr4-/- macrophages were pre-treated with BDP-12 (2, 4 μM) and then exposed to LPS for 6 h. Relative mRNA levels of Tnf (E), Il6 (F), and Il1b (G) were measured. H Schematic illustration of mice undergoing ALI modeling and BDP-12 injection. I Representative H&E-stained images of lung tissues. J Quantification of lung injury scores for (I). K Total protein levels in BALF samples. L Lung wet/dry weight ratio. M Total cell counts in BALF samples. N Survival curve of mice with or without BDP-12 injection in an E.coli-induced sepsis model. O The level of MD2-TLR4 complex formation in lung tissues was assessed by Co-IP. P Heatmap showing mRNA levels of pro-inflammatory factors Tnf, Il6, Il1b, Ccl2, Ifna1, Ifnb1, and Ifng in lung tissues. Q, R Serum (Q) and BALF (R) levels of TNF-α, IL-6, and IFN-α in the ALI mouse model. Data are represented as mean ± SEM by one-way ANOVA with Tukey’s test. n = 4 (C, E–G), or n = 6 (A, J–M, P–R) per group. Source data are provided as a Source Data file.
Subsequently, we tested the anti-inflammatory activity of BDP-12 both in vitro and in vivo. BDP-12 treatment dose-dependently disrupted the formation of the MD2-TLR4 complex (Fig. 7D; and Supplementary Fig. S9D) and significantly inhibited the transcription of inflammatory genes (Fig. 7E–G). In contrast, the two negative controls, BDP-15 and BDP-18, failed to suppress inflammatory gene transcription in LPS-challenged macrophages (Supplementary Fig. S9E). Notably, comparison of the dose-response curves revealed that the maximal anti-inflammatory efficacy of rBDNF reached ~60%, whereas BDP-12 achieved over 85%, underscoring the superior therapeutic potential of BDP-12 (Supplementary Fig. S9F). As expected, LPS induced only a weak inflammatory response in Tlr4-deficient macrophages, which may be mediated by alternative inflammatory pathways independent of TLR4. However, BDP-12 treatment showed no effect on this residual inflammatory response (Fig. 7E–G), confirming that BDP-12 exerts its anti-inflammatory effects specifically by targeting TLR4.
Given that neutrophils also express high levels of TLR4, we isolated primary neutrophils to assess whether BDP-12 exerts similar effects in this population. Interestingly, BDP-12 failed to suppress LPS-induced inflammatory responses in neutrophils (Supplementary Fig. S9G), which may be attributable to the ability of neutrophils to phagocytose LPS and activate intracellular alternative pro-inflammatory pathways20. These findings collectively demonstrate that BDP-12 potently and specifically targets macrophage TLR4 to exert its anti-inflammatory effects.
BDP-12 alleviates ALI through inhibiting MD2-TLR4-mediated inflammation
The protective effect of BDP-12 was further explored in LPS-induced ALI mice (Fig. 7H). Before assessing the in vivo efficacy of BDP-12, we evaluated its pharmacokinetic characteristics and found that BDP-12 showed high and sustained pulmonary concentration in mice during the experimental time (Supplementary Fig. S9H). Histological analysis revealed that BDP-12 dose-dependently ameliorated hyaline membrane formation and reduced alveolar wall thickness (Fig. 7I, J). Trends in LPS-induced pulmonary edema and protein leakage aligned with H&E results (Fig. 7K, L). Measurements of BALF total cell count revealed a significant reduction in immunocyte infiltration in ALI mice with BDP-12 injection (Fig. 7M). Moreover, BDP-12 treatment resulted in improved survival rates in LPS-induced septic mice (Fig. 7N). Mechanistically, BDP-12 dose-dependently blocked MD2-TLR4 interaction (Fig. 7O; Supplementary Fig. S9I) and suppressed the production of inflammatory cytokines in LPS-challenged mouse lung tissues, serum, and BALF (Fig. 7P–R).
In addition, we noted that both AAV-BDNF (preventive-like administration) and exogenous rBDNF protein (therapeutic-like administration) exerted protective effects in the ALI model. This prompted us to investigate whether BDP-12 would exhibit similar benefits when administered at different time points. Therefore, BDP-12 was delivered either 6 h before LPS challenge (preventive) or 0.5 h after challenge (therapeutic) (Supplementary Fig. S10A). Remarkably, both preventive and therapeutic BDP-12 administration significantly ameliorated lung histopathological injury, restored membrane barrier integrity, reduced inflammatory cell infiltration, and suppressed inflammatory responses (Supplementary Fig. S10B–H). Notably, the preventive regimen showed a modestly greater protective effect, likely attributable to the slow clearance and prolonged retention of BDP-12 in lung tissue.
We further examined the pharmacological effects of BDP-12 in the cecal ligation and puncture (CLP)-induced polymicrobial septic mouse model, which is recognized as the gold standard for mimicking the clinical features of sepsis in patients (Fig. 8A). As expected, BDP-12 injection exhibited systemic protective effects in a dose-dependent manner, as evidenced by the reductions in pathological damage not only in the lungs but also in the kidneys and hearts (Fig. 8B–F). Focusing on the lungs, BDP-12 significantly alleviated lung edema, restored permeability, and reduced immune cell infiltration in CLP-challenged mouse lung tissues (Fig. 8G–N). Mechanistically, BDP-12 also prevented the MD2-TLR4 interaction in lung tissue (Fig. 8O, P) and inflammatory gene expression in lung tissue, BALF, and serum (Fig. 8Q–S). Collectively, these data indicate the substantial translational potential of BDP-12 as a therapeutic agent for ALI, sepsis, and systemic inflammation, without oncogenic side effects.

A Schematic illustration of mice undergoing CLP modeling and BDP-12 injection. B Representative H&E-stained images of lung tissues. C Quantification of lung injury scores for (B). D Representative H&E-stained images of heart and kidney tissues. E Quantification of the tubular injury scores for panel (D). (F) Quantification of the cardiac injury scores for (D). G Total protein levels in BALF samples. H Lung wet/dry weight ratio. I Total cell counts in BALF samples. J MPO activity levels in lung lysates. K Neutrophil counts assessed by Wright-Giemsa staining in BALF samples. L Immunohistochemistry staining of F4/80 in lung tissues. M Quantification of F4/80 immunoreactivity corresponding to panel (L). N mRNA levels of Icam1 and Vcam1 detected by RT-qPCR in lung tissues. O The level of MD2-TLR4 complex formation in lung tissues was assessed by Co-IP. P Densitometric quantification of (O). Q Heatmap showing mRNA levels of pro-inflammatory factors Tnf, Il6, Il1b, Ccl2, Ifna1, Ifnb1, and Ifng in lung tissues. R, S BALF (R) and serum (S) levels of TNF-α, IL-6, and IFN-α in the CLP-induced sepsis model. Data are represented as mean ± SEM by one-way ANOVA with Tukey’s test. n = 6 per group. Source data are provided as a Source Data file.
Discussion
Here, we illustrate the role of BDNF in acute inflammatory lung diseases. Three key findings of this study inlcude: (i) the local expression of BDNF in pulmonary epithelial cells is significantly negatively correlated with the progression of ALI; (ii) BDNF secreted by lung epithelial cells exerts anti-inflammatory effects by directly targeting and antagonizing TLR4 in macrophages; (iii) BDP-12, a BDNF-derived peptide designed from BDNF-TLR4 interaction, showed excellent anti-inflammatory activity both in vitro and in vivo without the proliferative side-effects. In summary, the interaction between alveolar epithelial cell-derived BDNF and macrophage TLR4 represents a intercellular communication in the regulation of lung inflammation. The translated peptide, BDP-12, is a promising anti-inflammatory candidate for the treatment of ALI/sepsis.
As a well-known NTF family member, BDNF is essential for survival, growth, and differentiation of neurons, and BDNF deficiency is lethal21. The classic functions of BDNF primarily involve its interaction with its high-affinity receptor TrkB, activating downstream signaling pathways including PI3K-AKT, Jak-STAT, PLCγ-PKC, and Ras-MAPK. Through these pathways, BDNF plays a therapeutic role in brain development, neuroprotection, and resistance to neurodegenerative changes22. Interestingly, recent studies have shown that BDNF also plays significant roles outside the nervous system: loss of BDNF in cardiomyocytes leads to myocardial degeneration and heart failure23; BDNF-knockdown mice spontaneously develop non-alcoholic steatohepatitis24; and BDNF expression in the renal collecting ducts may be associated with chronic kidney disease25. In this study, we not only identified the lung as one of the organs with significantly high BDNF expression but also clarified the role of pulmonary BDNF in the progression of LPS-induced pneumonia by targeting TLR4. At the molecular level, BDNF and the MD2, a co-receptor of TLR4, spatially overlap in their competition for TLR4 interaction. MD2 primarily forms hydrophobic interactions with TLR4’s residues F440, L444, and F463, as well as hydrogen bonds with S416, N417, and E43926. Docking studies indicate that BDNF interacts with multiple residues in the TLR4 aa 419-487 region, including L444, F463, and E439. The similar binding modes suggest that the competition between BDNF and MD2 influences the balance of TLR4’s inactive and active states. The precise mode needs to be illustrated using generating a co-crystal structure of the BDNF-TLR4 complex. Overall, our study for the first time demonstrates BDNF’s new role as an antagonist of TLR4 and an anti-inflammatory protein against ALI/sepsis.
We identified GATA2 as a key transcriptional activator of BDNF in lung epithelial cells. Its LPS-induced downregulation directly explains the concomitant drop in BDNF. This loss is likely mediated by promoter hypermethylation of Gata2, as reported in an ALI rat model27. Furthermore, the BDNF-TLR4 axis is compromised during ALI through a self-amplifying inflammatory loop: TLR4 upregulation enhances sensitivity to inflammatory stimuli28, while NF-κB activation by the ensuing cytokine storm may further suppresses BDNF transcription29. Accordingly, we regarded restoration of BDNF levels as a promising strategy to limit ALI progression and initially considered recombinant BDNF protein as a potential therapeutic agent. However, numerous studies have reported that the BDNF-TrkB axis was highly activated in cancers, suggesting it may act as a potential oncogenic factor and risk30,31,32. As a result, we shifted our focus to developing BDNF derivatives. In fact, several molecules known as BDNF mimetics have already been developed, including BDNF loop dipeptide mimetics such as GTS-201, GSB-106, and GSB-214, as well as the natural product 7,8-dihydroxyflavone33,34,35,36,37. These molecules are defined as BDNF mimetics based on their ability to activate TrkB signaling. Here, we employed proteolysis to identify active BDNF fragments targeting TLR4 and designed BDP-12 as an antagonist of TLR4. We confirmed that BDP-12 showed no pro-proliferative side effects but excellent anti-inflammatory activity. We also acknowledge that pro-proliferative activity of BDNF is not entirely detrimental; a certain degree of proliferative activity is beneficial for lung tissue repair following injury, as demonstrated by the BDNF-TrkB axis in devastating lung diseases38. However, excessive or ectopic proliferation can lead to maladaptive repair, fibrotic remodeling, or heightened oncogenic risk. Therefore, we believe that the specificity of BDP-12 offers a more flexible therapeutic option. While BDP-12 can be administered alone when strong anti-inflammatory action is paramount, it can also be strategically used in combination with other regenerative therapies to support epithelial repair when deemed appropriate.
Furthermore, strategic molecular modifications to BDP-12, such as truncation, amino acid substitution, or targeted chemical modification, should be explored to optimize several critical drug properties. These optimization goals include enhancing its binding affinity for TLR4, improving its solubility, stability, targeted tissue distribution, and pharmacokinetic parameters, and minimizing potential immunogenicity. Pursuing these avenues will be an essential step toward the successful drug development and clinical translation of BDP-12.
TLR4 has been considered as a therapeutic target for inflammatory diseases. Eritoran, a lipid A analog, indirectly inhibits TLR4 activation by targeting MD2. However, it failed to improve patient survival rates in clinical trials39. TAK-242, a covalent inhibitor that targets the intracellular TIR domain of TLR4, also failed in Phase 3 trials due to insufficient anti-inflammatory effects in patients, likely due to a suboptimal pharmacokinetic profile and the complex metabolic processes after cellular entry40. These setbacks suggest that a more direct way of targeting the extracellular domain of TLR4, rather than its co-receptors or the intracellular TIR domain, may offer better efficacy and therapeutic potential. In the current study, we first identified BDNF, as a endogenous antagonist targeting the extracellular LRR domain of TLR4. Based on BDNF, we developed BDP-12, a linear and druggable peptide candidate. BDP-12 shows strong anti-inflammatory effects both in vitro and in vivo, without pro-proliferative activity. We believe that BDP-12, with its distinct TLR4-targeting strategy and its natural property, may be better accepted by the body, thus holding greater potential for clinical translation and drug development.
Although not predominant, the single-cell sequencing also revealed a considerable expression level of BDNF in vascular smooth muscle cells. This finding aligns with the previously reported role of BDNF as an exerkine41. Previous studies have shown that exercise enhances BDNF expression and secretion in muscle cells, promoting metabolic reprogramming42. The muscle-derived BDNF also limits the progression of related chronic diseases, while the underlying mechanism remains unclear43,44,45. Therefore, we believe that changes in muscle-derived BDNF in other organs, such as the cardiovascular system, may mediate the onset and progression of related diseases. Besides, our research group has previously reported that the MD2-TLR4 complex not only participates in acute inflammatory responses but also recognizes various metabolism-associated molecular patterns (MAMPs) such as palmitic acid, advanced glycation end products, and oxLDL, thereby mediating chronic inflammation in metabolic diseases46,47,48,49 Thus, we may guess that BDNF attenuates some metabolic diseases through antagonizing TLR4 and then reducing TLR4-driven chronic inflammation. This also suggests that BDP-12 may have potential therapeutic effects in chronic inflammatory diseases, warranting further investigation.
Methods
Antibodies and general reagents
The main antibodies and reagents used in this study were provided in Supplementary Table S4.
Cell isolation and culture
Mouse primary fibroblasts were isolated according to a previously established protocol and cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin50. Briefly, fresh lung tissue is obtained from euthanized mice and rinsed thoroughly to remove excess blood. Under sterile conditions, the lung tissue is minced into small pieces and evenly distributed across a 10 cm culture dish. Complete culture medium is added, and the dish is incubated at 37 °C with 5% CO₂ for 24 h. The medium is then carefully removed and replaced with fresh complete culture medium, with subsequent medium changes performed every 3 d. The outgrowth of fibroblasts is monitored under a microscope. Once the cells approach confluence, all remaining tissue fragments are removed, and the fibroblasts are further cultured as required for experimental purposes.
Mouse primary macrophages were prepared as described previously and maintained in RPMI-1640 medium with 10% FBS and 1% penicillin-streptomycin13. Briefly, mice were intraperitoneally injected with 2 mL of 6% thioglycolate solution (prepared with 0.3 g beef extract, 1 g tryptone, and 0.5 g sodium chloride in 100 mL double-distilled water, filtered through a 0.22 μm filter) and maintained under pathogen-free conditions for 3 days. Peritoneal macrophages were isolated by washing the peritoneal cavity with 8 mL RPMI-1640 medium. Collected cells were centrifuged and resuspended in complete RPMI-1640 medium. Primary cultures were seeded at 700,000 cells per 35 mm well, with non-adherent cells removed by medium washing 4 h after seeding. Adherent macrophages were used for experiments 24 h post-plating.
Mouse primary neutrophils were isolated from mouse bone marrow using the Mouse Neutrophil Negative Selection Kit (MCE; NJ, USA) according to the manufacturer’s instructions. Briefly, femurs and tibias were flushed with ice-cold PBS containing 2% FBS to obtain single-cell suspensions, which were subsequently filtered through a 40-µm strainer. Red blood cells were lysed using ACK lysis buffer. The cell suspension was then incubated with a cocktail of biotin-conjugated antibodies targeting non-neutrophil populations, followed by magnetic beads. Labeled cells were depleted using a magnetic separation column, and the untouched neutrophils were collected in the flow-through fraction.
Mouse primary alveolar epithelial cells were obtained from Procell (CP-M330; Wuhan, China) and cultured in their specified medium (Procell: CM-M003). The mouse pulmonary epithelial cell line MLE-12 was purchased from Cellverse (iCell-m036; Shanghai, China) and cultured in a specified medium (Cellverse: iCell-m036-001b). HEK-293T cell line and BV-2 microglial cell line (Procell: CL-0493) were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin.
Mice
All animal care and experimental procedures were approved by the Animal Policy and Welfare Committee of the Wenzhou Medical University (Wenzhou, Zhejiang, China; approval number: wydw2020-0012). Six-week-old male C57BL/6JGpt (Wild-type) mice and Male Tlr4−/− mice (strain number: T051714) were purchased from Gempharmatech (Jiangsu, China). All animals were housed in a pathogen-free room at 22 °C ± 2 °C, 50%–60% humidity, 12 h: 12 h light/dark cycle and fed with a standard rodent diet and water that was available ad libitum. The animals were acclimatized to the laboratory for 2 weeks before initiating the studies. All animal experiments were performed and analyzed by blinded researchers. The mice were divided randomly into groups and housed in the microisolator cages with 6 mice per cage. Each mouse was assigned a temporary random number within the weight range and then given a permanent numerical designation in the cages. For each group, one cage was randomly selected from the pool of all cages. Our study examined male mice because male animals exhibited less variability in phenotype.
Human serum and BALF samples
Blood samples were collected from ARDS patients (n = 12) and non-ARDS patients (n = 5), along with bronchoalveolar lavage fluid (BALF) samples from ARDS patients (n = 10) and non-ARDS patients (n = 13) at the Affiliated Cangnan Hospital of Wenzhou Medical University (Approval no. 2024-004). Following collection, the samples were centrifuged at 3000 × g, and the resulting supernatants and serum were carefully separated and stored for subsequent analysis.
Adeno-associated virus infection
Adeno-associated virus serotype 6 (AAV6) encoding mature BDNF with an N-terminal fusion albumin signal peptide sequence under the control of the epithelial-specific surfactant protein C (SPC) promoter (AAV6-SPC-BDNF) and negative control (AAV6-SPC-NC) were obtained from Genechem (Shanghai, China). Mice were anesthetized with 2% sodium pentobarbital (80 mg/kg, i.p.), with heart rate, body temperature, and toe pinch reflex monitored to ensure adequate depth of anesthesia. AAV6 was then administered via intratracheal injection at a dose of 5 × 1010 vg per mouse. The mice were subsequently maintained for 2 weeks to achieve optimal infection efficiency.
Macrophages deletion
Eight-week-old mice were intranasally challenged with 50 μL clodronate liposomes combined with an intravenous injection of 150 μL clodronate liposomes. PBS-containing liposomes were used as a control. The mice were subsequently maintained for 2 days to achieve optimal clearance efficiency.
Bone marrow transplantation
Eight-week-old male recipient mice were housed in filter-top cages and provided with sterile water containing 1000 U/L polymyxin B and 1.1 mg/L neomycin for one week prior to receiving total body irradiation (6 Gy). 12 h post-irradiation, femurs, and tibias were collected from donor mice, and bone marrow cells were flushed out using a 24-gauge syringe. Following red blood cell lysis, the cell suspension was filtered through a 100-μm nylon mesh to create a single-cell suspension. Each irradiated recipient mouse then received an intravenous injection of 2 × 106 bone marrow cells. The mice were allowed a two-week recovery period before proceeding with subsequent experiments.
LPS-induced ALI model and endotoxemia model
Mice were adequately anesthetized with 2% sodium pentobarbital (80 mg/kg, i.p.). Following anesthesia, mice were challenged via intratracheal injection with LPS (5 mg/kg, dissolved in 0.9% saline) or 0.9% saline alone. Recombinant BDNF (300 ng), BDNF-derived dodecapeptide (BDP-12; 4, 10 mg/kg), or a solvent control were injected intravenously or intraperitoneally at different time point according to the experimental design. After 6 h, the mice were sacrificed, and BALF was collected by perfusing the lungs with 2 × 200 μL of PBS solution through a tracheal cannula. Blood and lung tissue samples were also collected for subsequent analyses.
The collected BALF samples were centrifuged at 3000 × g for 5 min. The supernatant was analyzed for total protein levels and cytokine concentrations using ELISA. Cell pellets were resuspended in 100 μL of 0.9% saline, and total cell counts were determined using a hemocytometer. Neutrophil counts were obtained by counting 200 cells on a smear prepared with Wright–Giemsa staining. Lung wet-to-dry weight ratios were assessed as an index of pulmonary edema. Right middle lobes were collected, and wet weights were recorded. Tissues were then dried in a thermostatically controlled oven at 65 °C for 24 h, after which dry weights were measured.
Additionally, to induce an endotoxemia model, mice were intravenously injected with 5 mg/kg LPS and sacrificed 12 h later. Various visceral organs were then collected for further analysis.
Survival analysis in E.coli-induced sepsis model
E.coli strain DH5α was grown in Luria Broth (LB) media and the density was determined at 600 nm (OD600) using NanoDrop2000. The corresponding colony-forming units (CFUs) were determined on LB agar plates. Viable E. coli DH5α (1 × 109 CFU per mice) in 0.5 mL PBS was injected into the peritoneal cavity (i.p.). Survival of the mice was monitored every 6 h for 3 days.
Cecal ligation and puncture-induced polymicrobial sepsis model
Cecal ligation and puncture (CLP) was performed to induce polymicrobial sepsis in mice. Mice were first anesthetized with 2% sodium pentobarbital (80 mg/kg, i.p.), after which the abdomen was shaved and sterilized with ethanol. A midline incision was made to expose the cecum, which was ligated ~40% from the tip using sterile sutures. A single puncture was created in the ligated segment with a 21-gauge needle to allow a small amount of fecal material to extrude. The cecum was then gently returned to the abdominal cavity, and the incision was closed in layers using absorbable sutures. Postoperative care included fluid supplementation and maintaining a warm environment to promote recovery. 1 h later, BDP-12 (4, 10 mg/kg) or a solvent control was injected intraperitoneally according to the experimental design. 12 h post-CLP, the mice were sacrificed, and blood, BALF, and major visceral tissues were collected for subsequent analyses.
Histological analysis
Tissues were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned to a thickness of 5 μm. For routine histology, the sections were stained with hematoxylin and eosin. The lung injury scores, tubular injury scores, and cardiac injury scores were blindly determined by pathologists using the criteria in previous reports13,51,52.
Immunohistochemistry (IHC) and immunofluorescence (IF) staining were performed on deparaffinized and rehydrated lung sections. For both procedures, antigen retrieval was carried out by heating sections in sodium citrate buffer (pH 6.0). After cooling, sections were blocked with 1% bovine serum albumin (BSA; Sigma-Aldrich) in PBS, and permeabilized with 0.3% Triton X-100 when required (IF). For IHC, sections were incubated with primary antibodies (1:100) overnight at 4 °C, followed by horseradish peroxidase (HRP)–conjugated secondary antibodies for 1 h at 37 °C. Immunoreactivity was visualized using diaminobenzidine (DAB) and counterstained with hematoxylin. For IF, sections were incubated with mixed primary antibodies raised in different species (1:100 each) overnight at 4 °C, followed by species-specific fluorophore-conjugated secondary antibodies for 1 h at room temperature in the dark; nuclei were counterstained with DAPI. Images were captured using brightfield (IHC) or fluorescence (IF) microscopy. Quantification of staining intensity and colocalization was performed using ImageJ software (NIH, Bethesda, MD, USA).
Tissue distribution of BDP-12
Mice were intraperitoneally injected with BDP-12 at 10 mg kg−1 and sacrificed under anesthesia at 4, 8, and 12 h. Organs including heart, liver, spleen, lung and kidney were collected, weighed and homogenized with 0.9% saline solution (1:2 w/v). The homogenate was centrifuged at 12000 × g, 4 °C for 10 min; an aliquot of the supernatant was mixed with trifluoroacetic acid containing loratadine (100 ng mL−1) as internal standard, vortexed for 3 min, and re-centrifuged at 12000 × g for 10 min. The final supernatant was transferred to an autosampler vial and BDP12 concentration was quantified by UPLC-MS/MS.
Myeloperoxidase activity assay
Neutrophil tissue infiltration was evaluated in terms of myeloperoxidase (MPO) activity in lung tissue samples using an MPO Activity Assay Kit (ThermoFisher). Lung tissues were homogenized in 1 ml of 50 mM potassium PBS (pH 6.0) containing 0.5% hexadecyltrimethylammonium hydroxide and centrifuged at 15000 × g, 4 for 20 min. Ten microliters of the supernatant were transferred into PBS (pH 6.0) containing 0.17 mg/ml 3,3’-dimethoxybenzidine and 0.0005% H2O2. MPO activity in the supernatant was determined by measuring the absorbance at 460 nm. Total protein levels were measured using a Pierce BCA Protein Assay Kit (ThermoFisher). Data are presented as U/g tissue.
Western blot
Proteins were isolated using RIPA lysis and extraction buffer, and protein concentrations were measured using the Pierce BCA Protein Assay Kit. Protein lysates were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. The membranes were blocked with skim milk for 1 h at room temperature, and then incubated with primary antibodies, at 4 °C overnight. Secondary antibodies were applied for 1 h at room temperature. Immunoreactivity was visualized using an enhanced chemiluminescence reagent (Bio-Rad; Hercules, CA, USA). Protein-protein interactions were evaluated using co-immunoprecipitation. Protein lysates were incubated with specific precipitating antibodies at 4 °C overnight, precipitated with protein A/G agarose beads, and used for the detection of interacting proteins by means of immunoblotting. ImageJ analysis software was used for densitometric quantification of the blots. Densitometric quantification data were normalized to the total protein levels or loading controls.
Transcriptome sequencing
Total RNA from the tissues was isolated and purified using TRIzol reagent following the manufacturer’s procedure. The RNA amount and purity of each lung sample were quantified using NanoDrop ND-1000 (NanoDrop; DE, USA). The RNA integrity was determined using Bioanalyzer 2100 (Agilent; CA, USA), and confirmed by electrophoresis with denaturing agarose gel. Poly (A). RNA is purified from 1 μg total RNA using Dynabeads Oligo (dT)25 (ThermoFisher). Then the poly(A) RNA was fragmented into small pieces using a Magnesium RNA Fragmentation Module (NEB; MA, USA). The cleaved RNA fragments were then reverse transcribed into cDNA by SuperScript™ II Reverse Transcriptase (ThermoFisher). Finally, the sequence analysis was performed on an Illumina Novaseq™ 6000 following the manufacturer’s instructions. After generating the final transcriptome, the expression levels of all transcripts were estimated. The differentially expressed mRNAs were selected with adj. p value < 0.05.
ELISA assay
Cytokines and BDNF levels were quantified in mouse serum, BALF, various tissues, cell culture media, and human samples. Mouse TNF-α, IL-6, IFN-α, and BDNF levels were measured using ELISA kits according to the manufacturer’s instructions, as were human TNF-α, IL-6, and BDNF levels. Additionally, we constructed custom ELISAs to detect the BDNF-TLR4 and MD2-TLR4 complexes. Briefly, 96-well plates were coated with TLR4 antibody and blocked with BSA. Protein lysates from cells subjected to different treatments were then added, followed by incubation with either MD2 or BDNF antibody. After washing, HRP-conjugated secondary antibody against MD2 or BDNF antibody was added, and the reaction was developed using TMB solution. Absorbance was measured at 450 nm using a SpectraMax® M5 Multi-Mode Microplate Reader (Molecular Devices; San Jose, CA, USA) for all ELISA assays.
Flow cytometry
Cell pellets obtained from BALF were resuspended in 100 μL of FACS buffer (PBS with 2% FBS and 1 mM EDTA). The cells were stained with FITC-anti-CD45, PE-anti-F4/80, and PerCP-anti-Ly6G for 30 min on ice. Following staining, the cells were washed three times with FACS buffer and analyzed using a BD Accuri™ C6 Cytometer (BD Biosciences; Franklin Lakes, NJ, USA). Total viable single cells were initially gated for CD45-positive immune cells and subsequently analyzed for F4/80 and Ly6G expression. Neutrophils were identified as Ly6G+, while macrophages were identified as F4/80+.
Real-time qPCR
Total RNA was extracted from tissues using Trizol Reagent. RNA was reverse-transcribed using PrimeScriptTM RT reagent (Takara: RR047A). Real-time PCR was conducted using TB Green® Premix Ex TaqTM II (Takara: RR820A) on CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Relative expression was normalized to Actb. Target gene sequences are listed in Supplementary Table S5. For some studies, we utilized a pre-coated inflammatory factor qPCR Array (CT Bioscience; Changzhou, Jiangsu, China). Data from pre-loaded arrays was normalized to Hprt1.
Chromatin immunoprecipitation (ChIP)-PCR
ChIP assays were performed in MLE-12 cells to assess whether GATA2 binds to the Bdnf promoter. Cells were crosslinked with 1% formaldehyde, quenched with glycine, and lysed according to standard procedures. Chromatin was fragmented using micrococcal nuclease (MNase) digestion to yield nucleosomal DNA fragments (150-500 bp). The digested chromatin was incubated overnight at 4 °C with anti-GATA2 antibody or normal IgG control, and immune complexes were captured with Protein A/G magnetic beads. Following a series of stringent washes, crosslinks were reversed and DNA was purified using a column-based method. Four primer pairs (Supplementary Table S6) were designed to span four contiguous segments of the Bdnf promoter, enrichment at each segment was assessed by qPCR.
Cell transfections
To silence Ntrk2 (encoding TrkB) in macrophages, we purchased the corresponding Ntrk2 targeted siRNA (MCE: HY-RS09632) and transfected it into the cells using Lipofectamine RNAiMAX (ThermoFisher: 13778150) according to the manufacturer’s instructions. To knockout Tlr4 (encoding TrkB) in microglial, we constructed the corresponding targeted All-in-one CRISPR/Cas9 plasmid through GeneScript and transfected it into the cells using Lipofectamine 2000 (ThermoFisher: 11668019) according to the manufacturer’s instructions. For the overexpression of BDNF, TLR4, CCR1, and MD2 in HEK-293T cells, we utilized pcDNA3.1 plasmids containing the coding sequences of wild-type or mutated genes, synthesized by GeneScript, and transfected the cells using Lipofectamine 2000. In the luciferase reporter assay, we transfected the p65 reporter plasmid (Beyotime: D2206), AP-1 reporter plasmid (Beyotime: D2108), and ISRE reporter plasmid (Beyotime: D2179) into the cells using Lipofectamine 2000, following the manufacturer’s guidelines for all transfections.
Proteome microarray assays
HuProt™ v4.0 Human Proteome Microarrays (CDI Laboratories; Baltimore, MD, USA), containing over 21,000 unique human proteins, were used to identify BDNF-interacting proteins. The microarrays were blocked with a blocking buffer, and either biotinylated BDNF or biotinylated BSA was added. Following washing, Cy5-streptavidin was incubated. Scanning was performed using the GenePix 4200 A (Axon Instruments; Sunnyvale, CA, USA), and the raw data were analyzed with GenePix Pro v6.0 software. The signal-to-noise ratio (SNR) was defined as the ratio of the median foreground signal to the median background signal for each protein. The SNR for each protein was averaged from two duplicate spots on each microarray. Proteins with an SNR in the BDNF-treated group greater than both the BSA-treated group and 1.5 were considered potential interacting proteins.
Immunoprecipitation-coupled mass spectrometry (IP-MS)
Primary macrophages were stimulated with recombinant BDNF for 1 h and subsequently lysed in NP-40 lysis buffer containing protease and phosphatase inhibitors. After clarification by centrifugation, the supernatants were incubated overnight at 4 °C with anti-BDNF antibody (2 μg per mg protein) pre-bound to magnetic Protein A/G beads (Thermo Fisher). Following extensive washing to eliminate nonspecific interactors, bound proteins were eluted by heating in denaturing buffer. Samples were subjected to in-solution tryptic digestion after reduction (10 mM DTT) and alkylation (55 mM iodoacetamide), and the resulting peptides were desalted on C18 cartridges. Peptide mixtures were analyzed by DIA-based LC–MS/MS on a high-resolution mass spectrometer. Data processing was performed against the Mus musculus UniProt database, using a 1% false discovery rate at both peptide and protein levels.
Surface plasmon resonance assay
The binding affinity of BDNF to rTLR4 was determined using a Biacore T200 instrument (Cytiva; Marlborough, MA, USA) with a CM5 sensor chip (Cytiva). Briefly, rTLR4 was loaded onto the sensors using an Amine Coupling Kit (Cytiva). rBDNF samples (at concentrations of 0, 3.13, 6.25, 12.5, 25, 50, or 100 nM) were prepared in a running buffer (PBS, 0.5% P20). The sensor and sample plates were placed on the instrument and the rBDNF samples flowed over the black and target sensors. Seven concentrations were injected successively, at a flow rate of 30 μL·min-1, for a 200-s association phase, which was followed by a 200-s dissociation phase at 25 °C. The final graphs were obtained by subtracting the blank sensorgrams and blank samples from the duplex. The data were analyzed using Biacore T200 software EV (Cytiva). The dissociation constant (KD) was calculated by global fitting of kinetic data from various concentrations of rBDNF using a 1:1 Langmuir binding model.
Tryptic digestion and immunoprecipitation
Immobilized TPCK trypsin (30 μL) was used to digest 30 μg of human BDNF at 37 °C for 1 hour. The cleavage sites and resulting peptides were analyzed using PeptideCutter (https://web.expasy.org/peptide_cutter/), and fragments containing 10-20 residues were synthesized as standard polypeptides53. The BDNF fragments or standard polypeptides were aliquoted and incubated with 30 μg of human TLR4 or a solvent control overnight on ice. Following this, protein A/G agarose beads and a TLR4 antibody were added to immunoprecipitated TLR4 and its interacting molecules. After centrifugation, the beads were collected, denatured in 0.1 M Glycine-HCl (pH 2.7), and heated at 95 °C for 5 min. Finally, the beads were removed by centrifuge again and the supernatant was used for subsequent Mass spectrometry identification.
Mass spectrometry
The samples derived from tryptic digestion and immunoprecipitation were loaded onto ground steel (Bruker: 8280784; Billerica, MA, USA) using the dried droplet method. Briefly, α-Cyano-4-hydroxycinnamic acid (HCCA) was dissolved in TA30 (30:70 v/v Acetonitrile: 0.1% TFA in water) to create a saturated solution. Equal volumes of the samples and HCCA solution were mixed and spotted onto the ground steel to air dry. Finally, the ground steel was analyzed using the Autoflex Max MALDI-TOF mass spectrometer (Bruker).
Circular dichroism spectroscopy
Polypeptide standards synthesized based on the amino acid sequence fragments of BDNF were mixed with 16 µM recombinant TLR4 at concentrations of 0, 8, 16, and 32 µM, respectively. Subsequently, changes in ellipticity were immediately measured using a ChiraScan circular dichroism spectrometer (Applied Photophysics; Leatherhead, Surrey, UK) over the wavelength range of 200–280 nm.
Cellular thermal shift assay
Cellular thermal shift assay (CETSA) experiments were conducted following a standard CETSA protocol54. Briefly, cultured primary macrophages were treated with standard polypeptides for 24 h. The cells were then harvested and resuspended in phosphate-buffered saline. Treated samples were aliquoted and heated at different temperatures for 3 min in a PCR plate (ThermoFisher). Following heating, a protease inhibitor cocktail was added, and the cells were lysed using three freeze-thaw cycles with liquid nitrogen and a heat block. The cell lysates were centrifuged at 20,000 × g for 20 min. Supernatants were analyzed by Western blot.
Additionally, isothermal dose-response fingerprints (ITDRF)-CETSA were performed on the same cells. Briefly, cultured primary macrophages were treated with gradient concentrations of standard polypeptides for 24 h. The cells were harvested and resuspended as before, then heated at 61 °C for 3 min. The cells were subsequently lysed and analyzed using the same procedures.
Molecular docking
The crystal structures of TLR4 (PDB code: 3FXI) and BDNF (PDB code: 1B8M) were obtained from the Protein Data Bank. Molecular docking between TLR4 and BDNF was performed using Hawkdock (http://cadd.zju.edu.cn/hawkdock/)55. Among the top 10 results, only the conformation with interactions involving BDNF amino acids 104–116 was selected for MM/GBSA analysis. The top 10 key residues responsible for protein-protein interactions were identified based on per-residue decomposition energy calculations.
Proliferation and viability assay
The effects of rBDNF and BDP-12 on cell proliferation and viability were assessed using a CCK-8 kit according to the manufacturer’s instructions. Additionally, mice were administered rBDNF (300 ng) and BDP-12 (4, 10 mg/kg) once daily for 3 consecutive days, after which they were sacrificed, and lung tissues were collected for immunohistochemical staining of PCNA, as well as dual-immunofluorescence co-staining for ECAD + PCNA and F4/80 + PCNA to determine in vivo proliferative activity.
RNA-seq data processing
RNA sequencing was performed using the Illumina NovaSeq™ 6000 platform to generate paired-end reads. Raw sequencing data in FASTQ format were first processed using FastQC to assess the quality of the reads. Adapter sequences and low-quality bases were removed using Trimmomatic to ensure high-quality data. Clean reads were then aligned to the reference genome using HISAT2. The resulting aligned reads in BAM format were sorted and indexed using SAMtools. Gene-level quantification was performed using featureCounts from the Subread package to obtain raw read counts. Differential gene expression analysis was conducted using DESeq2 in R. Genes with an adj. p value < 0.05 and |log2(fold change)| > 1 were considered significantly differentially expressed.
Publicly available single-cell and bulk transcriptomic data
Public single-cell RNA sequencing data for CLP and sham lung samples were obtained from the GEO database (accession number: GSE207651)56. Subsequent analyses and visualizations were conducted using the Majorbio cloud platform (https://www.majorbio.com/)57. More single-cell expression results of liver Bdnf were obtained from public databases including The Human Protein Atlas (https://www.proteinatlas.org/) and Single Cell Expression Atlas (https://www.ebi.ac.uk/gxa/sc/home)58,59. Additionally, several bulk RNA sequencing datasets from both human and mouse lungs, with or without acute pneumonia, were sourced from the GEO database, including GSE233206, GSE214249, GSE104214, GSE199546, GSE38092, GSE64027, GSE150910, and GSE21369. Differentially expressed gene lists were extracted directly from the supplementary files of each dataset or analyzed using the GEO2R web tool when applicable. For datasets providing only raw counts or FPKM values, expression matrices were analyzed using the ExpressAnalyst web tool (https://www.expressanalyst.ca/)60.
Statistics & reproducibility
All data are reported as Mean ± SEM. Statistical analysis was performed with GraphPad Prism 8.0 software (San Diego, CA, USA). Sample sizes were not predetermined using statistical methods but were consistent with those commonly used in the field. All assays were performed in at least three independent experiments. Each dot point represents an individual animal or independent experiment. The Shapiro–Wilk test was applied to assess normality. For comparisons between two groups, unpaired Student’s t-tests were used, or the Mann–Whitney U-test if applicable. For multiple group comparisons, two-sided one-way ANOVA followed by Tukey’s post hoc test was conducted. Statistical tests were selected based on data distribution, with statistical significance defined as p < 0.05. All data collection and analysis were conducted in a blinded manner, and no animals were excluded.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw data generated from this study including RNA-Seq has been deposited in SRA (accession number: PRJNA1183441, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1183441). Source data are provided with this paper.
References
Wei, M., Wu, T. & Chen, N. Bridging neurotrophic factors and bioactive peptides to Alzheimer’s disease. Ageing Res Rev. 94, 102177 (2024).
Wang, Y. et al. GDNF promotes the proliferation and osteogenic differentiation of jaw bone marrow mesenchymal stem cells via the Nr4a1/PI3K/Akt pathway. Cell Signal 108, 110721 (2023).
Watt, M. J. et al. CNTF reverses obesity-induced insulin resistance by activating skeletal muscle AMPK. Nat. Med 12, 541–548 (2006).
Zhang, T. et al. HDAC6-dependent deacetylation of NGF dictates its ubiquitination and maintains primordial follicle dormancy. Theranostics 14, 2345–2366 (2024).
Correia, J. C. et al. Muscle-secreted neurturin couples myofiber oxidative metabolism and slow motor neuron identity. Cell Metab. 33, 2215–2230.e2218 (2021).
Bi, W. et al. Neurotrophin-3 contributes to benefits of human embryonic stem cell-derived cardiovascular progenitor cells against reperfused myocardial infarction. Stem Cells Transl. Med 10, 756–772 (2021).
Soliman, A.M., Barreda, D.R. Acute inflammation in tissue healing. Int. J. Mol. Sci. 24, 641 (2022).
Wang, Z., Li, S. & Huang, B. Alveolar macrophages: Achilles’ heel of SARS-CoV-2 infection. Signal Transduct. Target Ther. 7, 242 (2022).
Zhang, H. et al. The role of innate immunity in pulmonary infections. Biomed. Res Int 2021, 6646071 (2021).
Bezerra FS. et al. Oxidative stress and inflammation in acute and chronic lung injuries. Antioxidants (Basel) 12, 548 (2023).
Duan, J. X. et al. Extracellular citrate serves as a DAMP to activate macrophages and promote LPS-induced lung injury in mice. Int Immunopharmacol. 101, 108372 (2021).
Peng, W. et al. Autophagy alleviates mitochondrial DAMP-induced acute lung injury by inhibiting NLRP3 inflammasome. Life Sci. 265, 118833 (2021).
Zhu, W. et al. Licochalcone A protects against LPS-induced inflammation and acute lung injury by directly binding with myeloid differentiation factor 2 (MD2). Br. J. Pharm. 180, 1114–1131 (2023).
Wu, S. Y. et al. BDNF reverses aging-related microglial activation. J. Neuroinflammation 17, 210 (2020).
Niu, C. et al. Genetic dissection of BDNF and TrkB expression in glial cells. Biomolecules 14, 91 (2024).
Xiong, X. et al. Serum brain-derived neurotrophic factor (BDNF) as predictors of childhood neuroblastoma relapse. BMC Cancer 23, 670 (2023).
Meng, L. et al. Targeting the BDNF/TrkB pathway for the treatment of tumors. Oncol. Lett. 17, 2031–2039 (2019).
Radin, D. P. & Patel, P. BDNF: an oncogene or tumor suppressor? Anticancer Res 37, 3983–3990 (2017).
Zainullina, L. F., Vakhitova, Y. V., Lusta, A. Y., Gudasheva, T. A. & Seredenin, S. B. Dimeric mimetic of BDNF loop 4 promotes survival of serum-deprived cell through TrkB-dependent apoptosis suppression. Sci. Rep. 11, 7781 (2021).
Tourneur, L. & Witko-Sarsat, V. Inflammasome activation: Neutrophils go their own way. J. Leukoc. Biol. 105, 433–436 (2019).
Voigt, M.W., Schepers, J., Haas, J., von Bohlen Und Halbach O. Reduced levels of brain-derived neurotrophic factor affect body weight, brain weight and behavior. Biology (Basel) 13, 159 (2024).
Kowianski, P. et al. BDNF: a key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol. Neurobiol. 38, 579–593 (2018).
Li, L. et al. Ablation of cardiomyocyte-derived BDNF during development causes myocardial degeneration and heart failure in the adult mouse heart. Front Cardiovasc Med 9, 967463 (2022).
Ichimura-Shimizu, M. et al. Brain-derived neurotrophic factor knock-out mice develop non-alcoholic steatohepatitis. J. Pathol. 261, 465–476 (2023).
Tao, Y. S. et al. Expression of brain-derived neurotrophic factor in kidneys from normal and cyclosporine-treated rats. BMC Nephrol. 19, 63 (2018).
Park, B. S. et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195 (2009).
Zhang, X. Q. et al. Genome‑wide analysis of DNA methylation in rat lungs with lipopolysaccharide‑induced acute lung injury. Mol. Med Rep. 7, 1417–1424 (2013).
Yu, B., Li, Q. & Zhou, M. LPS‑induced upregulation of the TLR4 signaling pathway inhibits osteogenic differentiation of human periodontal ligament stem cells under inflammatory conditions. Int J. Mol. Med 43, 2341–2351 (2019).
Maqbool, A., Lattke, M., Wirth, T. & Baumann, B. Sustained, neuron-specific IKK/NF-kappaB activation generates a selective neuroinflammatory response promoting local neurodegeneration with aging. Mol. Neurodegener. 8, 40 (2013).
Yu, Q. et al. BDNF is a prognostic biomarker involved in immune infiltration of lung adenocarcinoma and is associated with brain metastasis. Immunology 168, 320–330 (2023).
Malekan, M. et al. BDNF and its signaling in cancer. J. Cancer Res Clin. Oncol. 149, 2621–2636 (2023).
Silverman, D. A. et al. Cancer-associated neurogenesis and nerve-cancer cross-talk. Cancer Res 81, 1431–1440 (2021).
Markin, P. A. et al. LC-MS/MS determination of GTS-201, a dipeptide mimetic of the brain-derived neurotrophic factor, and neurotransmitter metabolites with application to a pharmacokinetic study in rats. J. Pharm. Biomed. Anal. 223, 115125 (2023).
Povarnina, P. Y., Antipova, T. A., Logvinov, I. O., Gudasheva, T. A. & Seredenin, S. B. capital ES, cyrillichronically administered BDNF dipeptide mimetic GSB-106 prevents the depressive-like behavior and memory impairments after transient middle cerebral artery occlusion in rats. Curr. Pharm. Des. 29, 126–132 (2023).
Povarnina, P. Y. et al. A low-molecular-weight BDNF mimetic, dipeptide GSB-214, prevents memory impairment in rat models of Alzheimer’s disease. Acta Nat. 14, 94–100 (2022).
Hang, P. Z. et al. BDNF mimetic 7,8-dihydroxyflavone rescues rotenone-induced cytotoxicity in cardiomyocytes by ameliorating mitochondrial dysfunction. Free Radic. Biol. Med 198, 83–91 (2023).
Korkmaz, O. T. Can Brain-derived Neurotrophic Factor (BDNF) Mimetics be a Way Out for Neurodegenerative Diseases? Curr. Pharm. Des. 29, 246–250 (2023).
Paris, A. J. et al. STAT3-BDNF-TrkB signalling promotes alveolar epithelial regeneration after lung injury. Nat. Cell Biol. 22, 1197–1210 (2020).
Sohn, J. W. Critical care paper review 2012. Tuberc. Respir. Dis. (Seoul.) 73, 1–10 (2012).
Samarpita, S., Kim, J. Y., Rasool, M. K. & Kim, K. S. Investigation of toll-like receptor (TLR) 4 inhibitor TAK-242 as a new potential anti-rheumatoid arthritis drug. Arthritis Res Ther. 22, 16 (2020).
Chow, L. S. et al. Exerkines in health, resilience and disease. Nat. Rev. Endocrinol. 18, 273–289 (2022).
Chan, W. S. et al. Exercise-induced BDNF promotes PPARdelta-dependent reprogramming of lipid metabolism in skeletal muscle during exercise recovery. Sci. Signal 17, eadh2783 (2024).
Yang X. et al. Muscle-generated BDNF is a sexually dimorphic myokine that controls metabolic flexibility. Sci. Signal 12, eaau1468 (2019).
Halievski, K. et al. Muscle BDNF improves synaptic and contractile muscle strength in Kennedy’s disease mice in a muscle-type specific manner. J. Physiol. 598, 2719–2739 (2020).
Delezie, J. et al. BDNF is a mediator of glycolytic fiber-type specification in mouse skeletal muscle. Proc. Natl. Acad. Sci. USA 116, 16111–16120 (2019).
Wang, X., Wang, Y., Antony, V., Sun, H. & Liang, G. Metabolism-associated molecular patterns (MAMPs). Trends Endocrinol. Metab. 31, 712–724 (2020).
Wang, Y. et al. Saturated palmitic acid induces myocardial inflammatory injuries through direct binding to TLR4 accessory protein MD2. Nat. Commun. 8, 13997 (2017).
Wang, Y. et al. MD2 activation by direct AGE interaction drives inflammatory diabetic cardiomyopathy. Nat. Commun. 11, 2148 (2020).
Chen, T. et al. Macrophage-derived myeloid differentiation protein 2 plays an essential role in ox-LDL-induced inflammation and atherosclerosis. EBioMedicine 53, 102706 (2020).
Edelman, B. L. & Redente, E. F. Isolation and characterization of mouse fibroblasts. Methods Mol. Biol. 1809, 59–67 (2018).
Xiong, J. et al. DUSP2-mediated inhibition of tubular epithelial cell pyroptosis confers nephroprotection in acute kidney injury. Theranostics 12, 5069–5085 (2022).
Yuan, Y. et al. Protective effect of excretory-secretory proteins from Trichinella spiralis muscle larvae against myocardial injury in septic mice. Nan Fang. Yi Ke Da Xue Xue Bao 42, 824–831 (2022).
Wu, N., Li, P., Shuang, Q. & Wuhanqimuge Screening and molecular dynamics simulation of ACE inhibitory tripeptides derived from milk fermented with Lactobacillus delbrueckii QS306. Food Funct. 15, 2655–2667 (2024).
Jafari, R. et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 9, 2100–2122 (2014).
Weng, G. et al. HawkDock: a web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res 47, W322–W330 (2019).
Yang, R., Zheng, T., Xiang, H., Liu, M. & Hu, K. Lung single-cell RNA profiling reveals response of pulmonary capillary to sepsis-induced acute lung injury. Front Immunol. 15, 1308915 (2024).
Ren, Y. et al. Majorbio cloud: a one-stop, comprehensive bioinformatic platform for multiomics analyses. Imeta 1, e12 (2022).
Karlsson M. et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 7, eabh2169 (2021).
George, N. et al. Expression Atlas update: insights from sequencing data at both bulk and single cell level. Nucleic Acids Res 52, D107–D114 (2024).
Liu, P. et al. ExpressAnalyst: a unified platform for RNA-sequencing analysis in non-model species. Nat. Commun. 14, 2995 (2023).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (82404637 to W.Z.), Postdoctoral Fellowship Program of CPSF (GZC20231957 to W.Z.), China Postdoctoral Science Foundation (2024M752443 to W.Z.), Zhejiang Provincial Health Science and Technology Plan (2025KY322 to W.Z.). We thank Scientific Research Center of Wenzhou Medical University for consultation and instrument availability that supported this work.
Author information
Authors and Affiliations
Contributions
Conceptualization: W.L. and G.L.; data curation: W.Z., L.J., Q.Z., Y.C., Y.Z., P.C., J.Y., Y.X., M.L., Y.Z., R.W., and Y.L.; formal analysis: W.Z., L.J., Q.Z., and Y.C.; investigation: W.Z., L.J., Q.Z., Y.C., J.Y., Y.X., M.Lin, and Y.Z.; supervision: G.L. and J.Y.H.; writing–original draft: W.Z.; writing–review and editing: G.L., W.L., Z.Z.; funding acquisition: W.Z. The order of the first authors’ names was determined on the basis of their contributions to the work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ioannis Charalampopoulos, who co-reviewed with Konstantina Chanoumidou, Atsushi Murao, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, W., Jin, L., Zhang, Q. et al. Brain-derived neurotrophic factor and the derived dodecapeptide function as Toll-like receptor 4 antagonists in acute lung injury. Nat Commun 17, 2786 (2026). https://doi.org/10.1038/s41467-026-69541-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69541-7
