
Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD) is a global health concern that affects nearly one-quarter of the world’s population. General control non-repressed protein 5 (GCN5), a histone acetyltransferase (HAT), has been implicated in the progression of several diseases, but its role in MASLD remains unclear. Here, we provide the experimental evidence that progressive human and male murine MASLD is driven by GCN5, but not by p300/CREB binding protein associated factor (PCAF) activation. Hepatocyte-specific GCN5 overexpression accelerates MASLD progression, whereas its ablation alleviates disease severity. Moreover, pharmacological inhibition of GCN5 with CPTH2 protects against MASLD. Metabolomics and RNA-seq analyses demonstrate that GCN5 promotes de novo lipogenesis (DNL) by upregulating SREBP1c-mediated transcription of lipogenic genes. Mechanistically, GCN5 acetylates histone H3 at the SREBP1c promoter, enhancing transcription through its intrinsic acetyltransferase activity. Our findings further identify GCN5 as a key regulator of LXRα-induced SREBP1c expression, suggesting that targeting GCN5 may selectively inhibit SREBP1c-driven DNL without impairing LXRα-mediated reverse cholesterol transport (RCT). Notably, combined treatment with the Liver X Receptor (LXR) agonist T0901317 and CPTH2 synergistically reduced lipid accumulation in vitro and in vivo, highlighting a promising therapeutic strategy for MASLD.
Similar content being viewed by others

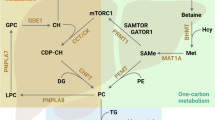
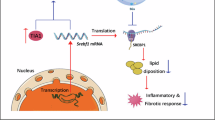
Introduction
Non-alcoholic fatty liver disease (NAFLD), now known as metabolic dysfunction-associated steatotic liver disease (MASLD), is a global health concern, that affects nearly one-quarter of the world’s population1. MASLD encompasses a spectrum of conditions, ranging from simple steatosis to metabolic dysfunction-associated steatohepatitis (MASH), which markedly increases the risk of cirrhosis, hepatocellular carcinoma (HCC), and systemic metabolic disorders2. In 2024, the U.S. Food and Drug Administration (FDA) approved Resmetirom, an oral selective agonist of thyroid hormone receptor (THR)-β, for the treatment of MASH with liver fibrosis. This approval marked the introduction of the first targeted drug therapy for MASH3. However, further validation of its clinical efficacy is warranted. Thus, there is an urgent need to elucidate the mechanisms underlying the onset and progression of MASLD to enable the development of effective therapeutic interventions4.
MASLD arises from a complex interplay of various environmental and genetic factors. According to the double-hit theory of pathogenesis, the first hit is the accumulation of triglycerides in hepatocytes, followed by a second hit characterized by the activation of inflammatory mediators leading to hepatocellular damage, inflammation, and fibrosis5,6. More recent models propose that multiple concurrent factors contribute to MASLD development. Fatty acids and their metabolites have been implicated in the transition from simple steatosis to MASH. Insulin resistance (IR) promotes hepatic accumulation of free fatty acids, resulting in endoplasmic reticulum (ER) stress, oxidative stress, and ultimately hepatocyte apoptosis. In turn, fatty acids exacerbate IR in the liver by promoting lipid storage7. Despite these insights, the precise mechanisms driving MASLD pathogenesis remain unclear.
Epigenetics provides a compelling framework for understanding how lifestyle, environmental exposures, and other risk factors influence MASLD development8. Histone acetylation, a post-translational modification that occurs mainly at specific lysine residues in the N-terminal tails of histones H3 and H4, regulates chromatin accessibility and gene expression9. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) maintain histone acetylation in a dynamic equilibrium. Evidence indicates that histone acetylation patterns are altered in MASLD. For example, studies in Japanese macaques have demonstrated increased acetylation at H3K14 in fetal hepatic tissue, as well as elevated acetylation at H3K9 and H3K18. In addition, a maternal high-fat diet (HFD) reduced HDAC1 protein levels in the fetal liver. These findings suggest that maternal obesity induced by a HFD alters fetal chromatin structure through histone modifications10,11,12,13.
General control non-repressed protein 5 (GCN5) was the first HAT to be cloned and identified in yeast and belongs to the N-acetyltransferase superfamily14. GCN5 is widely conserved across eukaryotes and contains a C-terminal bromodomain (BRD) that recognizes acetylated histones, a catalytically active and relatively conserved HAT domain, and a PCAF homology domain that recognizes the N-terminus of nucleosomes15. GCN5 is primarily expressed in the liver, brain, thyroid, kidney, and spleen, and is mainly localized in the nucleus and associated with the microtubule cytoskeleton. In mammals, GCN5 shares 73% sequence homology with p300/CREB binding protein–associated factor (PCAF); both possess histone acetyltransferase and lysine acetyltransferase activity16. GCN5 acetylates histone H3 at lysine residues K9 and K14, and it also acetylates non-histone proteins. Functionally, GCN5 regulates cell proliferation, metabolism, and inflammation, and its dysregulation has been linked to the progression of cancer, metabolic, autoimmune, and neurological disorders17. Recently, GCN5 has emerged as a promising therapeutic target, and high-throughput screening approaches have been established to identify selective inhibitors. To date, more than 20 small-molecule inhibitors with distinct structural scaffolds have been reported18,19.
Studies indicate that GCN5 regulates hepatic gluconeogenesis and is closely associated with diabetes20,21,22,23, but its role in MASLD has not been elucidated. Based on clinical and experimental evidence, we investigated the involvement of hepatocyte GCN5 in MASLD progression. We demonstrate that GCN5 promotes hepatic de novo lipogenesis (DNL) by regulating the transcriptional activity of LXRα/RXRα-mediated SREBP1c.
Results
GCN5 expression is triggered in patients and mice with MASLD
We first examined the dynamics of GCN5 (KAT2A) and PCAF (KAT2B) expression in liver tissue from individuals with healthy livers (control group, n = 11) and patients with progressive MASLD (mild to severe steatosis, n = 117) (Supplementary Data 1). Western blotting and immunohistochemistry analyses showed that GCN5, but not PCAF protein levels, were markedly higher in liver tissue from patients with MASLD than in healthy controls (Fig. 1A–D). Consistently, qRT-PCR analysis revealed significantly higher KAT2A, but not KAT2B, mRNA expression in liver tissue from patients with MASLD (Fig. 1E). Both protein and mRNA levels of hepatic GCN5, but not PCAF, positively correlated with MASLD progression (Fig. 1A–E). Supporting the role of GCN5 in disease acceleration, we observed a positive correlation of intrahepatic GCN5 expression with MASLD progression determined by the MASLD activity score (MAS), and the levels of AST, ALT, TG, TC, LDL-c, and bile acid in blood samples from patients with MASLD (Fig. 1F–L). The blood levels of HDL-c, insulin, and glucose did not correlate with the protein levels of GCN5 or PCAF (Supplementary Fig. S1A–C).

A–E GCN5 and PCAF expression levels in human liver samples from individuals without hepatic steatosis (non-steatosis, n = 11 patients) or patients with hepatic steatosis (mild steatosis, n = 30 patients; moderate steatosis, n = 61 patients; severe steatosis, n = 26 patients) were determined by western blot (A, B) immunohistochemistry (C, D) and qRT-PCR (E). F–L Spearman correlation analysis revealed significant associations of hepatic GCN5 and PCAF expression levels with MAS, AST, ALT, serum TG, TC, LDL-c, and bile acid levels. M Schematic diagram of HFD-induced MASLD in mice for 8, 16, and 32 weeks. N–R Male C57BL/6 J mice (n = 3 independent mice per group) were sacrificed, and liver sections were analyzed by western blot (N, O) immunohistochemistry (P, Q) and qRT-PCR (R). Scale bar: 50 μm. Data are presented as mean ± SEM. Statistical analyses were performed using two-tailed Student’s t test (O–R) and one-way ANOVA with Dunnett’s post-test (B–E). Exact p-values are provided in the figure. Source data are available in the Source Data file.
Based on these findings, we further evaluated GCN5 and PCAF expression during MASLD progression and investigated GCN5 induction and nuclear localization in a murine model. Mice were fed an HFD for 8, 16, or 32 weeks (Fig. 1M). This model is well established for studying sequential stages of MASLD progression. Consistent with earlier studies, HFD-fed mice developed hepatic steatosis with ballooned hepatocytes and varying degrees of lobular inflammation24. Moreover, in line with our findings in human MASLD, western blotting and immunohistochemistry confirmed strong induction and nuclear accumulation of GCN5, but not PCAF, in hepatocytes during MASLD progression (Fig. 1N–Q). Hepatic Kat2a, but not Kat2b, mRNA levels were also elevated with disease progression (Fig. 1R). We next examined GCN5 expression across different liver cell types. GCN5 protein and mRNA levels were significantly increased in HL-7702 and HepG2 cells, as well as in mouse primary hepatocytes treated with PAOA (Supplementary Fig. S1D–L).
To investigate the mechanism underlying lipotoxicity-induced GCN5 expression, we used JASPAR analysis to predict transcription factors (TFs) potentially binding to the KAT2A promoter. This analysis identified four candidates: KLF5, KLF15, KLF16, and ZNF384 (Supplementary Fig. S1M). Functional validation was performed using luciferase reporter constructs containing either the full-length (– 2000 to + 200 bp) or truncated versions of the KAT2A promoter. Dual-luciferase assays showed that only KLF15 significantly activated the full-length promoter (Supplementary Fig. S1N). This result was corroborated in PAOA-treated HL-7702 hepatocytes, where KLF15 overexpression specifically increased KAT2A transcription (Supplementary Fig. S1O). To further characterize this interaction, we performed chromatin immunoprecipitation (ChIP) assays with primers targeting the predicted KLF15 binding sites identified by JASPAR. The results confirmed direct binding of KLF15 to the KAT2A promoter region spanning –1330 to –1180 bp (Supplementary Fig. S1P). Importantly, site-directed mutagenesis of the core binding motif within this region abolished KLF15-mediated promoter activation (Supplementary Fig. S1Q), providing mechanistic evidence that KLF15 regulates KAT2A expression through this cis-element. Together, these experiments in progressive human and murine MASLD establish the first experimental evidence that GCN5 activation contributes to disease acceleration.
Hepatocellular GCN5 overexpression exacerbates MASLD progression in vivo and increases lipid accumulation in hepatocytes in vitro
To confirm the association between hepatic GCN5 expression and MASLD progression, we generated a wild-type (WT) KAT2A expression vector using adeno-associated virus serotype 8 (AAV-Kat2a WT) (Fig. 2A). Western blotting confirmed that tail vein injection of AAV-Kat2a WT selectively increased GCN5 expression in the liver, but not in other tissues (Fig. 2B, and Supplementary Fig. S2A, B). Compared with AAV-LacZ controls, Kat2a WT overexpression in the liver did not alter body weight or food intake (Supplementary Fig. S2C, D). However, Kat2a WT overexpression increased liver weight (LW) and the liver-to-body weight ratio (LW/BW) in high-fat diet (HFD)-fed mice (Fig. 2C). Hematoxylin and eosin (H&E) and Oil-Red O staining further showed that Kat2a WT markedly promoted hepatic lipid accumulation in HFD-fed mice (Fig. 2D, and Supplementary Fig. S2E), consistent with elevated liver and serum TG, TC, HDL-c, and LDL-c levels (Fig. 2E, F). In addition, hepatocyte ballooning was more pronounced in HFD-fed mice overexpressing Kat2a WT (Fig. 2D). Serum AST and ALT levels were also significantly elevated in Kat2a WT-overexpressing HFD mice compared with control-HFD mice (Fig. 2G). As a negative control, we expressed an acetyltransferase-defective mutant (GCN5-E575Q, AAV-Kat2a MUT) (Fig. 2H). Tail vein injection of AAV-Kat2a MUT successfully increased hepatic GCN5 expression without affecting other tissues (Fig. 2I and Supplementary Fig. S2F, G). Like AAV-LacZ controls, Kat2a MUT overexpression did not affect body weight or food intake (Supplementary Fig. S2H, I). Unlike Kat2a WT, however, Kat2a MUT did not promote abnormal lipid accumulation in the liver or blood of HFD-fed mice (Fig. 2J–N, and Supplementary Fig. S2J).

A Scheme for the generation of hepatocyte-specific Kat2a-overexpressing mice injected with AAV-Kat2a WT or empty AAV-LacZ via tail vein. B Successful overexpression of GCN5 protein in the liver was verified by western blot (n = 3 independent mice). C Liver weight (LW) and LW-to-body weight ratio (LW/BW) of AAV-Kat2a WT, and AAV-LacZ mice (n = 6 independent mice). D Representative H&E and Oil Red O staining of the liver. Scale bar: 50 μm. E The levels of serum TG, TC, HDL-c, and LDL-c (n = 6 independent mice). F The contents of liver TG and TC (n = 6 independent mice). G The levels of serum AST and ALT (n = 6 independent mice). H Scheme for the generation of hepatocyte-specific Kat2a-overexpressing mice injected with AAV-Kat2a Mutant (E575Q) or empty AAV-LacZ via the tail vein. I Successful overexpression of GCN5 protein in the liver was verified by western blot (n = 3 independent mice). J Liver weight (LW) and LW-to-body weight ratio (LW/BW) of AAV-Kat2a Mutant (E575Q) and AAV-LacZ mice (n = 10 independent mice). K Representative H&E and Oil Red O staining of the liver (n = 10 independent mice). Scale bar: 50 μm. L The levels of serum TG, TC, HDL-c, and LDL-c (n = 10 independent mice). M The contents of liver TG and TC (n = 10 independent mice). N The levels of serum AST and ALT (n = 10 independent mice). O–T Hepatocytes were transfected with KAT2A WT and KAT2A Mutant (E575Q) plasmids for 12 h, and then the cells were incubated with PA (200 μM) and OA (400 μM) for 24 h. The cellular TG and TC levels were detected in HL-7702 (O) HepG2 (Q) and mouse primary hepatocytes (S) (n = 3 independent cultures). The lipids were stained with Nile Red and BODIPY 493/503 (Bodipy) in HL-7702 (P) HepG2 (R) and mouse primary hepatocytes (T) (n = 6 independent cultures), scale bar: 20 μm. Error bars are represented as mean ± SEM. Statistical analysis was done using the two-tailed Student’s t test (A–N) and one-way ANOVA with Dunnett’s post-test (O–T). Source data are provided as a Source Data file.
To further assess the effects of GCN5 on hepatocellular lipid metabolism, WT and acetyltransferase-defective (E575Q) KAT2A constructs were transfected into HepG2, HL-7702, and primary mouse hepatocytes (Supplementary Fig. S2K–M). Overexpression of KAT2A WT, but not KAT2A MUT, significantly increased cellular TG and TC levels (Fig. 2O, Q, S). Consistently, staining with the neutral lipid dyes Nile Red and BODIPY 493/503 (Bodipy) revealed that lipid droplet size and number were significantly higher in hepatocytes overexpressing KAT2A WT, but not KAT2A MUT (Fig. 2P, R, T). Together, these results demonstrate that hepatocellular GCN5 overexpression exacerbates MASLD progression in vivo and promotes lipid accumulation in hepatocytes in vitro.
Hepatocyte-specific GCN5 deletion ameliorates MASLD/MASH
To investigate the role of GCN5 in MASLD pathogenesis, hepatocyte-specific Kat2a-knockout (Kat2aHKO) mice were generated by crossing Kat2afl/fl mice with Alb-Cre mice and feeding them an HFD (Fig. 3A and Supplementary Fig. S3A, B). qRT-PCR and western blotting confirmed successful knockout of Kat2a in hepatocytes (Fig. 3B and Supplementary Fig. S3C–D). Hepatocyte-specific Kat2a deletion did not affect body weight or food intake under HFD conditions (Supplementary Fig. S3E, F). Importantly, compared with WT littermates, Kat2aHKO mice had lower LW and LW/BW (Fig. 3C), as well as less severe hepatic steatosis after 10 weeks of HFD feeding, as shown by H&E and Oil Red O staining (Fig. 3D and Supplementary Fig. S3G). Kat2aHKO mice also developed less severe hypertriglyceridemia and hypercholesterolemia than Kat2afl/fl mice (Fig. 3E). Hepatic TG and TC contents were markedly reduced in Kat2aHKO mice compared with Kat2afl/fl mice (Fig. 3F). Hepatocyte ballooning was also attenuated in Kat2aHKO mice (Fig. 3D). Serum AST and ALT levels were significantly lower in Kat2aHKO mice than in Kat2afl/fl mice (Fig. 3G). To further assess the role of GCN5 in MASLD progression, mice were fed a high-fructose diet (HFrD) for 6 weeks (Supplementary Fig. S4A–K) or an Amylin liver NASH (AMLN) diet for 16 weeks (Supplementary Fig. S5A–K). Results from both models were consistent with those obtained in HFD-fed mice.

A Schematic diagram of HFD diet-induced MASLD models in Kat2aHKO and Kat2afl/fl mice. B Successful depletion of GCN5 protein in the liver was verified by western blot (n = 3 independent mice). C LW and LW/BW of Kat2aHKO and Kat2afl/fl mice (n = 6 independent mice). D Representative H&E and Oil Red O staining of the liver (n = 6 independent mice). E The levels of serum TG, TC, HDL-c, and LDL-c (n = 6 independent mice). F The contents of liver TG and TC (n = 6 independent mice). G The levels of serum AST and ALT (n = 6 independent mice). H Schematic diagram of FPC diet-induced MASLD and MASH models in Kat2aHKO and Kat2afl/fl mice. I Successful depletion of GCN5 protein in the liver was verified by western blot (n = 3 independent mice). J LW and LW/BW of Kat2aHKO and Kat2afl/fl mice (n = 6 independent mice). K The levels of serum TG, TC, HDL-c, and LDL-c (n = 6 independent mice). L The contents of liver TG and TC (n = 6 independent mice). M, N Representative H&E (M) and Oil Red (N) of the liver (n = 6 independent mice). O The levels of serum AST and ALT (n = 6 independent mice). P Representative Sirius red and F4/80 staining of the liver (n = 6 independent mice). Q The mRNA expression in liver tissue was detected by qRT-PCR (n = 6 independent mice). R Schematic diagram of HFMRCD diet-induced MASH models in Kat2aHKO and Kat2afl/fl mice. S Successful depletion of GCN5 protein in the liver was verified by western blot (n = 3 independent mice). T LW and LW/BW of Kat2aHKO and Kat2afl/fl mice (n = 6 independent mice). U The levels of serum TG, TC, HDL-c, and LDL-c (n = 6 independent mice). V The contents of liver TG and TC (n = 6 independent mice). W Representative H&E, Oil Red O, Sirius red, and F4/80 staining of the liver (n = 6 independent mice). X The levels of serum AST and ALT (n = 6 independent mice). Y, Z The mRNA expression in liver tissue was detected by qRT-PCR (n = 4 independent mice). Scale bar: 50 μm. Error bars are represented as mean ± SEM. Statistical analysis was done using the two-tailed Student’s t test (C–Z). Exact p-values are indicated in the figure. Source data are provided as a Source Data file.
We next fed Kat2afl/fl and Kat2aHKO mice a fructose–palmitate–cholesterol–trans-fat (FPC) diet for 16 or 24 weeks to investigate the involvement of GCN5 in MASLD to MASH (Fig. 3H, I, and Supplementary Fig. S6A). Body weight and food intake did not differ significantly between Kat2aHKO and Kat2afl/fl mice (Supplementary Fig. S6B, C). After 16 and 24 weeks of FPC feeding, Kat2aHKO mice showed significantly reduced LW and LW/BW (Fig. 3J), lower fasting serum TG, TC, and LDL-c levels, and higher HDL-c levels (Fig. 3K). H&E and Oil Red O staining and hepatic TG and TC measurements confirmed reduced hepatic steatosis in Kat2aHKO mice compared with Kat2afl/fl mice (Fig. 3L–N and Supplementary Fig. S6D, E). In addition, Kat2aHKO ameliorated FPC diet-induced liver injury, as indicated by decreased plasma AST and ALT levels (Fig. 3O). Furthermore, Kat2aHKO mice showed lower degrees of inflammation and fibrosis than Kat2afl/fl mice, as evidenced by F4/80, and Sirius Red staining (Fig. 3P and Supplementary Fig. S6F, G). These phenomena were accompanied by reduced expression of MASH-related inflammatory factors, such as IL6, IL16, Cxcl1, Cxcl5, and Cxcl10, as well as lower expression of profibrotic and MASH-related profibrotic genes, including Acta2, α-Sma, ctgf, Tgfβ, Col3a1, and Col1a1 (Fig. 3Q). To investigate the role of GCN5 deficiency in a specific MASH model, we used a mouse model of a high-fat and methionine-choline-deficient refined diet (HFMRCD)-induced MASH, in which the animals exhibited more profound inflammatory responses and fibrosis symptoms (Fig. 3R, S and Supplementary Fig. S6H). Body weight and food intake were not significantly different between Kat2aHKO and Kat2afl/fl mice (Supplementary Fig. S6I, J). However, the LW and LW/BW of Kat2aHKO mice were significantly decreased (Fig. 3T). Kat2aHKO mice had lower fasting blood lipid levels, including TG, TC, and LDL-c, and higher HDL-c levels (Fig. 3U). Furthermore, H&E staining, Oil Red O staining, and hepatic TG and TC concentrations showed that Kat2aHKO mice had reduced hepatic steatosis in response to the HFMRCD diet (Fig. 3V, W and Supplementary Fig. S6K). In addition, Kat2aHKO mitigated HFMRCD-induced liver injury, as indicated by decreased plasma AST and ALT levels (Fig. 3X). Kat2aHKO mice also exhibited lower degrees of inflammation and fibrosis than Kat2afl/fl mice, as evidenced by F4/80 and Sirius Red staining (Fig. 3W and Supplementary Fig. S6L, M). These effects were accompanied by reduced expression of MASH-related inflammatory factors, such as IL6, IL16, Cxcl1, Cxcl5, and Cxcl10 (Fig. 3Y), and lower expression of profibrotic and MASH-related profibrotic genes, including Acta2, α-Sma, ctgf, Tgfβ, Col3a1, and Col1a1 (Fig. 3Z). Moreover, hepatic GCN5 deficiency markedly decreased HFMRCD-induced overactivation of signaling pathways involving the transcription factor NF-κB by promoting the phosphorylation of the NF-κB inhibitor α (Iκbα) and the p65 NF-κB subunit (Supplementary Fig. S6N). The TGF-β1/Smad signaling pathway is of great importance in the development of liver fibrosis25. As expected, the marked increase in phosphorylated SMAD2/3 protein levels was reduced in Kat2aHKO mice (Supplementary Fig. S6N). Meanwhile, protein expression of Collagen I and α-SMA was also reduced in Kat2aHKO mice (Supplementary Fig. S6N). Furthermore, we generated a KAT2A knockout HL-7702 cell line (KAT2A KO) using the CRISPR-Cas9 system to investigate the function of GCN5 (Supplementary Fig. S7A, B). Knockout of KAT2A reduced cellular TG by 21% and TC by 33% (Supplementary Fig. S7C). Nile Red and Bodipy staining of hepatocytes showed that lipid droplet accumulation, including droplet size and number, was significantly decreased in KAT2A KO cells (Supplementary Fig. S7D–F). Moreover, cellular TG and TC levels were not significantly decreased in KAT2A KO cells replenished with KAT2A WT, but not with KAT2A MUT (E575Q) (Supplementary Fig. S7C). Nile Red and Bodipy staining yielded similar results (Supplementary Fig. S7D–F). Collectively, these data demonstrate that hepatocyte-specific GCN5 deficiency protects against diet-induced hepatic steatosis and subsequent inflammation and fibrosis, suggesting that GCN5 may act as a positive regulator of MASLD progression.
CPTH2, a potent GCN5 inhibitor, protects against MASLD with good safety
Pharmacotherapies targeting MASLD remain an unmet medical need. We used CPTH2, a potent inhibitor of GCN5, to evaluate whether GCN5 represents a viable therapeutic target for MASLD. First, we assessed the pharmacokinetics of CPTH2. As shown in Supplementary Fig. S8A, CPTH2 concentrations peaked at 33.17 ± 9.67 ng/ml within approximately 0.875 h after intragastric administration. The plasma area under the concentration-time curve (AUC) was 105.45 ± 8.58 ng·h/mL (Supplementary Fig. S8A). One hour after administration, CPTH2 was most concentrated in the liver, with a concentration of 2172.3 ng/g, which was markedly higher than that in other tissues (Supplementary Fig. S8B). These findings suggest that CPTH2 is predominantly enriched in the liver.
MASLD was established in mice fed an HFD for 4 weeks, after which the mice were orally administered CPTH2 (20 mg/kg/day) while continuing the HFD for an additional 6 weeks (Fig. 4A, B and Supplementary Fig. S8C). Throughout the experiment, body weights did not differ between CPTH2-treated and HFD-fed mice (Supplementary Fig. S8D), and food intake remained unchanged (Supplementary Fig. S8E). The HFD-induced increases in LW and LW/BW were reduced by CPTH2 treatment (Fig. 4C). Moreover, H&E and Oil Red O staining showed that CPTH2 markedly decreased lipid accumulation in HFD-fed mice (Fig. 4D and Supplementary Fig. S8F), consistent with reduced liver and serum TG, TC, HDL-c, and LDL-c levels (Fig. 4E, F). In addition, hepatocyte ballooning was diminished in CPTH2-treated HFD-fed mice (Fig. 4D). Serum AST and ALT levels were significantly lower in CPTH2-treated obese mice compared with control HFD-fed mice (Fig. 4G).

A–G Male C57BL/6 J mice at 8 weeks of age were randomly grouped. Mice were fed an HFD for 4 weeks, and then vehicle or CPTH2 (20 mg/kg/day) was administrated to mice by gastric irrigation daily. After 6 weeks of treatment, the mice were sacrificed and subjected to a series of analyses as indicated below. A Schematic diagram of HFD diet-induced MASLD models treated with CPTH2. B The protein levels of GCN5 and PCAF in the liver were verified by western blot (n = 3 independent mice). C LW, and LW/BW (n = 5 independent mice). D Oil red O staining and histological analysis of the liver (n = 5 independent mice). Scale bar: 50 μm. E The serum TG, TC, HDL-c, and LDL-c levels (n = 5 independent mice). F The liver TG and TC levels (n = 5 independent mice). G The serum AST, and ALT levels (n = 5 independent mice). H–N Male C57BL/6 J mice at 8 weeks of age were randomly grouped. Mice were fed the FPC diet for 16 weeks, and then vehicle or CPTH2 (20 mg/kg/day) was administrated to mice by gastric irrigation daily. After 6 weeks of treatment, the mice were sacrificed and subjected to a series of analyses as indicated below. H Schematic diagram of FPC diet-induced MASLD models treated with CPTH2. I The protein levels of GCN5 and PCAF in the liver were verified by western blot (n = 3 independent mice). J LW, and LW/BW (n = 6 independent mice). K Oil red O staining and histological analysis of the liver (n = 6 independent mice). Scale bar: 50 μm. L The serum TG, TC, HDL-c, and LDL-c levels (n = 6 independent mice). M The liver TG and TC levels (n = 6 independent mice). N The serum AST, and ALT levels (n = 6 independent mice). O–Q KAT2A KO cells were transfected with indicated plasmids for 12 h, incubated with PAOA with CPTH2 (50 μM) for 24 h, then stained with Nile Red and Bodipy (O), scale bar: 20 μm. Quantification of the cellular lipids was analyzed by ImageJ (P) (n = 6 independent cultures). The cellular TG and TC levels (Q) (n = 3 independent cultures). Error bars are represented as mean ± SEM. Statistical analysis was done using the two-tailed Student’s t test (C–G, J–N) and one-way ANOVA with Dunnett’s post-test (P–Q). Exact p-values are indicated in the figure. Source data are provided as a Source Data file.
MASLD was also established in mice fed an FPC diet for 10 weeks, after which the mice were orally administered CPTH2 (20 mg/kg/day) while continuing the FPC diet for an additional 6 weeks (Fig. 4H, I). During the experiment, body weight did not differ between CPTH2-treated and FPC diet-fed mice (Supplementary Fig. S8G), and food intake remained unchanged (Supplementary Fig. S8H). The FPC diet-induced increases in LW and LW/BW were reduced by CPTH2 treatment (Fig. 4J). Moreover, H&E and Oil Red O staining showed that CPTH2 markedly decreased lipid accumulation in FPC diet-fed mice (Fig. 4K and Supplementary Fig. S8I), consistent with reductions in liver and serum TG, TC, HDL-c, and LDL-c levels (Fig. 4L, M). In addition, hepatocyte ballooning was reduced in CPTH2-treated FPC diet-fed mice (Fig. 4K). Serum AST and ALT levels were significantly lower in CPTH2-treated mice compared with control FPC diet-fed mice (Fig. 4N).
Human HL-7702 normal liver cells were treated with CPTH2 (25 and 50 μM) in the presence of PAOA for 16 h without significant cytotoxicity (Supplementary Fig. S9A). Using the neutral lipid dyes Nile Red and Bodipy to stain lipid droplets, CPTH2 significantly reduced cellular lipid droplet accumulation, including both size and number (Supplementary Fig. S9D, E). The lipid-lowering effect of CPTH2 was further confirmed by reduced cellular TG and TC levels (Supplementary Fig. S9F). Similar results were observed in HepG2 cells and mouse primary hepatocytes under the same conditions, with CPTH2 significantly decreasing lipid droplet size and number (Supplementary Fig. S9G, H, J, K) as well as TG and TC levels (Supplementary Fig. S9I, L), again without cytotoxicity (Supplementary Fig. S9B, C).
To confirm that the lipid-lowering effects of CPTH2 are specifically dependent on GCN5 rather than off-target effects, we performed validation experiments in KAT2A KO cells. As shown in Fig. 4O, P, CPTH2 failed to reduce PAOA-induced lipid accumulation in KAT2A KO HL-7702 cells, in which TG and TC levels remained unchanged (Fig. 4Q). However, when KAT2A KO HL-7702 cells were replenished with KAT2A WT, CPTH2 significantly decreased PAOA-induced lipid accumulation as well as TG and TC levels (Fig. 4O–Q). In contrast, replenishment with KAT2A MUT in KAT2A KO HL-7702 cells did not restore the lipid-lowering activity of CPTH2 (Fig. 4O–Q). These findings indicate that the protective effect of CPTH2 against MASLD depends on GCN5, supporting its potential as a safe therapeutic target.
Loss of GCN5 in the liver suppresses DNL
To investigate the mechanism by which GCN5 regulates MASLD progression, liquid chromatography–mass spectrometry (LC-MS) profiling of hepatic free fatty acids (FFA) and total esterified lipids (TG, cholesterol ester, and phospholipid) revealed an overall reduction of short-chain and very short-chain fatty acids in the livers of Kat2aHKO mice (Fig. 5A–C). Interestingly, Kat2a depletion did not affect the levels of the essential polyunsaturated fatty acids (PUFAs) C18:2n6 (linoleic acid) and C18:3n3 (α-linolenic acid), which cannot be synthesized de novo and are derived from diet or adipose tissue (Fig. 5B, C). Similarly, the n-3 and n-6 families of highly unsaturated fatty acids with 20–22 carbons, synthesized from dietary linoleic acid by the desaturases FADS1 and FADS2, were not altered in Kat2aHKO livers (Fig. 5B, C). Kat2a KO hepatocytes exhibited a reduced de novo lipogenesis (DNL) index, calculated as the ratio of C16:0 to C18:2n6 (Fig. 5D), suggesting that Kat2a depletion inhibits hepatic fat accumulation primarily by limiting endogenous biosynthesis. Accordingly, LC-MS quantification of the lipogenic precursors acetyl-CoA and malonyl-CoA, and gas chromatography–mass spectrometry (GC-MS) analysis of other lipogenic intermediates, including citric acid, dihydroxyacetone phosphate (DHAP), glycerol, and glycerol-3P, demonstrated significant reductions in Kat2aHKO livers (Fig. 5E, F and Supplementary Fig. S10A).

A Schematic representation of the liver of Kat2aHKO mice and Kat2aflf mice metabolomics. B, C Relative levels of non-esterified (NEFA) (B) and esterified FA (EFA) (C) in the liver of Kat2aHKO mice and Kat2aflf mice (n = 6 independent mice). D Relative DNL index is measured as the ratio of hepatic C16:0 content to C18:2n6 content (n = 6 independent mice). E, F Relative abundance of selected metabolites involved in glucose metabolism and lipogenesis illustrated in Supplementary Fig. S5A (n = 3 independent mice). G–I RNA sequencing was performed on Kat2aHKO and Kat2afl/fl mice fed an HFD for 10 weeks (n = 3 independent mice). G Volcano plot representation of significantly up- and down-regulated genes. H Gene Ontology analysis. I Gene Set Enrichment Analysis plot of enrichment. J–O The indicated gene expression in liver tissues of Kat2aHKO and Kat2afl/fl mice (J–L), or AAV Kat2a and AAV LacZ mice (M–O) (n = 6 independent mice). P, Q Kat2aHKO and Kat2afl/fl mice were fed an HFD diet for 11 days, fasted from 9 a.m. to 7 p.m., refed for 2 h, and force-fed a bolus of 13C-fructose and 12C-glucose. The mice were fed overnight and killed the next morning. LC-MS was performed to examine the amount of 13C label incorporation into hepatic fatty acids (n = 6 independent mice). R, S Kat2aHKO and Kat2afl/fl mice were fed an HFD diet regimen for 11 days and then injected intraperitoneally (i.p.) with deuterium oxide (2H2O) at ~ 7 p.m. Five hours later, the mice were killed, and their livers harvested. LC-MS was performed to examine the amount of deuterium label incorporation into hepatic fatty acids (n = 6 independent mice). Error bars are represented as mean ± SEM. Statistical analysis was done with a two-tailed Student’s t test. Exact p-values are indicated in the figure. Source data are provided as a Source Data file.
To further elucidate the role of GCN5 in hepatic lipid metabolism, RNA sequencing was performed on livers from Kat2aHKO and Kat2afl/fl mice after 10 weeks of HFD feeding. A total of 163 genes were upregulated, and 516 genes were downregulated in Kat2aHKO mice compared with Kat2afl/fl mice (Fig. 5G). Transcriptomic profiling revealed that GCN5 ablation downregulated pathways enriched in fatty acid metabolism, while genes involved in triglyceride uptake and oxidation were not significantly affected (Fig. 5H, I). Consistently, transcriptomic analysis of HL-7702 hepatocytes yielded similar results (Supplementary Fig. S10B–D), indicating conservation of GCN5 function in liver lipid metabolism. At the molecular level, genes involved in fatty acid synthesis, elongation, and desaturation were broadly downregulated in KAT2A KO hepatocytes, without significant changes in the expression of genes related to lipid transport or fatty acid oxidation (Fig. 5J–L). Conversely, DNL-related genes were significantly upregulated in the AAV-Kat2a group compared with the AAV-LacZ group (Fig. 5M–O). Collectively, these findings demonstrate that loss of GCN5 suppresses the hepatic DNL program.
To directly assess DNL, mice were administered 13C-fructose, a rich carbon source for hepatic DNL, and 13C incorporation into liver fatty acids was quantified. Kat2aHKO mice fed a short-term HFD or FPC diet exhibited an approximately 50% reduction in DNL, without changes in body weight (Fig. 5P, Q, Supplementary Fig. S10E, F, I, J). Comparable results were obtained in mice injected with deuterium oxide, where 2H incorporation into liver fatty acids provided an alternative measure of DNL (Fig. 5R, S and Supplementary Fig. S10G, H, K, L). In vitro, Kat2a knockout markedly inhibited fatty acid de novo synthesis, with minimal effects on fatty acid oxidation, lipid transport, and re-esterification (Supplementary Fig. S10M–P). Taken together, these findings demonstrate that GCN5 depletion robustly suppresses anabolic DNL.
Loss of GCN5 suppresses hepatic DNL by inhibiting the SREBP1c pathway
Transcription of DNL genes is largely mediated by sterol regulatory element-binding proteins (SREBPs), carbohydrate response element-binding protein (ChREBP), and upstream stimulatory factors (USF)25. To investigate GCN5-dependent pathways in hepatic DNL regulation, we examined the effects of GCN5 on the activities of these transcription factors. As shown in Supplementary Fig. S11A, KAT2A WT significantly increased the transcriptional activity of SREBP, but not that of ChREBP or USF. In contrast, KAT2A MUT overexpression did not alter SREBP activity. In mammalian cells, SREBPs—comprising three isoforms (SREBP1a, SREBP1c, and SREBP2)—act as master regulators of lipid metabolism. SREBP2 preferentially activates genes involved in cholesterol synthesis, whereas SREBP1 promotes triglyceride synthesis26. Given that GCN5 regulates triglyceride biosynthesis, we focused on SREBP1 as its downstream effector. SREBP1, embedded in the endoplasmic reticulum (ER) membrane, is activated by proteolytic cleavage of its precursor form (p-SREBP1) into its soluble mature form (m-SREBP1)26. Overexpression of KAT2A WT in hepatocytes significantly increased the protein levels of p-SREBP1 and m-SREBP1 (Supplementary Fig. S11B), and SREBP1 target genes, such as SREBF1, ACC, FASN, SCD1, SCD2, and FADS2 (Supplementary Fig. S11C). However, there were no significant changes in SREBP transcriptional activity, SREBP1 protein expression, and downstream target gene levels in liver cells overexpressing KAT2A MUT (Supplementary Fig. S11A–C). KAT2A WT, but not KAT2A MUT hepatic overexpression mice showed markedly increased protein levels of SREBP1 (p-SREBP1, and m-SREBP1) and expression of its target genes, including Srebf1c, Acc, Fasn, Scd1, Scd2, and Fads2 (Fig. 6A, D). In contrast, KAT2A KO hepatocytes and Kat2aHKO mice displayed significantly suppressed p-SREBP1 and m-SREBP1 protein production, accompanied by reduced expression of downstream target genes (Fig. 6B, E and Supplementary Fig. S11D–F). Pharmacological inhibition of GCN5 with CPTH2 in hepatocytes and mice similarly decreased SREBP transcriptional activity and downregulated SREBP1 and its target genes (Fig. 6C, F and Supplementary S11G–I). Notably, CPTH2 did not inhibit the SREBP pathway in KAT2A KO hepatocytes (Supplementary Fig. S11J–L). However, reintroduction of KAT2A WT, but not KAT2A MUT, restored CPTH2 responsiveness, confirming that CPTH2 regulates the SREBP pathway in a GCN5-dependent manner (Supplementary Fig. S11J–L). Finally, the association between elevated hepatic GCN5 and SREBP1 protein levels was validated in MASLD patient samples (Supplementary Fig. S11M).

A–C The hepatic protein levels of SREBP1 in the AAV LacZ, AAV Kat2a WT, and AAV Kat2a MUT (E575Q) mice (A) Kat2aHKO and Kat2afl/fl mice (B) and CPTH2-treated mice (C) (n = 3 independent mice). D–F The hepatic mRNA levels of indicated genes in the AAV-LacZ and AAV-Kat2a WT (n = 6 independent mice), AAV-LacZ and AAV-Kat2a MUT (E575Q) mice (n = 5) (D) Kat2aHKO and Kat2afl/fl mice (E) (n = 6 independent mice), and CPTH2-treated mice (F) (n = 3 independent mice) were detected by qRT-PCR. G Schematic diagram of HFD-induced MASLD in Kat2aHKO and Kat2afl/fl mice injected with a titer of 2.5 × 1011 adenovirus overexpressing mSREBP1c. H Successful overexpression of m-SREBP1 protein in the liver was verified by western blot (n = 3 independent mice). I LW and LW/BW (n = 6). J Representative H&E and Oil Red O staining of the liver. K The contents of liver TG and TC (n = 6 independent mice). L The levels of serum TG, TC, HDL-c, and LDL-c (n = 6 independent mice). M The levels of serum AST and ALT (n = 6 independent mice). N The hepatic mRNA levels of indicated genes were detected by qRT-PCR (n = 4 independent mice). O Schematic diagram of FPC-induced MASLD in Kat2aHKO and Kat2afl/fl mice injected with a titer of 2.5 × 1011 adenovirus overexpressing mSREBP1c. P LW and LW/BW (n = 6 independent mice). Q The contents of liver TG and TC (n = 6 independent mice). R Representative H&E and Oil Red staining of the liver. S The levels of serum TG, TC, HDL-c, and LDL-c (n = 6 independent mice). T The levels of serum AST and ALT (n = 6 independent mice). Scale bar: 50 μm. Error bars are represented as mean ± SEM. Statistical analysis was done using the two-tailed Student’s t test (D–F) and one-way ANOVA with Dunnett’s post-test (I–T). Exact p-values are indicated in the figure. Source data are provided as a Source Data file.
To confirm that the protective effect of GCN5 deficiency is mediated through the SREBP1 pathway during MASLD progression, we re-expressed p-SREBP1c or m-SREBP1c in KAT2A KO hepatocytes and performed in vitro PAOA-induced lipid accumulation assays (Supplementary Fig. S12A–C). SREBP1c rescue markedly increased the expression of DNL-related genes (Supplementary Fig. S12D) and restored cellular TG and TC contents in KAT2A KO hepatocytes (Supplementary Fig. S12E). In addition, overexpression of p-SREBP1 and m-SREBP1 reversed the inhibitory effect of CPTH2 on the SREBP pathway in HepG2 cells and primary hepatocytes (Supplementary Fig. S12F–M).
To determine whether the anti-MASLD effect of GCN5 deficiency is dependent on SREBP1 in vivo, m-SREBP1c was specifically overexpressed in the liver via tail vein injection of adeno-associated virus (AAV)-m-SREBP1c or AAV-LacZ after 4 weeks of HFD feeding (Fig. 6G, H). Weight gain and food intake did not significantly differ between groups (Supplementary Fig. S13A, B). Liver-specific overexpression of SREBP1c disrupted the reduction in LW and LW/BW ratios observed in Kat2a-deficient mice (Fig. 6I). H&E and Oil Red O staining revealed that the decrease in hepatic lipid accumulation in Kat2aHKO mice was reversed by SREBP1c overexpression (Fig. 6J), consistent with liver and serum TG, TC, LDL-c, and HDL-c levels (Fig. 6K, L). Consequently, the decrease in serum AST and ALT levels in liver-specific Kat2a-deficient mice was significantly attenuated by SREBP1c overexpression (Fig. 6M). At the molecular level, Kat2a knockout significantly downregulated SREBP1c target genes, whereas SREBP1c overexpression restored their expression (Fig. 6N). To further validate these findings, we applied an FPC diet-induced MASLD model. Similar to the HFD model, liver-specific m-SREBP1c overexpression in Kat2aHKO mice significantly reversed the lipid-lowering effect of GCN5 deficiency (Fig. 6O–T and Supplementary Fig. S13C, D). Collectively, these data demonstrate that SREBP1c is essential for the regulation of MASLD progression by GCN5.
GCN5 epigenetically promotes SREBP1c transcriptional expression
SREBP1c activity is regulated at both the mRNA and protein levels26. In addition to its canonical function as a histone acetyltransferase, GCN5 acetylates cytoplasmic proteins and can regulate protein stability17. Therefore, GCN5 may influence DNL gene expression by modulating the SREBP1c pathway at one or more regulatory levels. Although GCN5 depletion downregulates SREBF1c mRNA and precursor protein levels, SREBF1c is also an autoregulatory target of SREBP1c, making it unclear whether GCN5 regulates SREBP1c expression via epigenetic or post-translational mechanisms.
To investigate this, Flag-tagged SREBP1c was immunoprecipitated under stringent conditions using SDS-containing buffers, and acetylated SREBP1c levels were detected by western blotting with an acetyl-Lys antibody. CPTH2 treatment did not alter SREBP1c acetylation (Supplementary Fig. S14A), and GCN5 was not found to interact with SREBP1c (Supplementary Fig. S14A). In the presence of the translation inhibitor cycloheximide, p-SREBP1 and m-SREBP1 levels gradually declined, but CPTH2 did not accelerate their degradation (Supplementary Fig. S14B, C). These results indicate that GCN5 inhibition does not promote p-SREBP1 or m-SREBP1 degradation. We therefore hypothesized that GCN5 regulates the SREBP1 pathway through epigenetic mechanisms. As a histone acetyltransferase, GCN5 modifies histone 3 at lysine 9 and lysine 1417. Consistently, H3K9ac and H3K14ac levels were elevated in MASLD patients with high GCN5 expression and correlated positively with GCN5 levels (Supplementary Fig. S14D, E). Similar results were observed in HFD-induced MASLD and ob/ob mouse models (Supplementary Fig. S14F, G), as well as in PAOA-treated hepatocytes in vitro (Supplementary Fig. S14H).
Subsequently, H3K9ac and H3K14ac levels were markedly reduced in KAT2A knockout HL-7702 cells and liver-specific Kat2a knockout mice (Fig. 7A and Supplementary Fig. S14I). Similarly, inhibition of GCN5 activity by CPTH2 in hepatocytes or mice significantly decreased H3K9ac and H3K14ac protein levels in both the cells and liver tissue (Fig. 7B and Supplementary Fig. S14J). In contrast, overexpression of KAT2A significantly elevated H3K9ac and H3K14ac levels in HL-7702 cells and the liver (Fig. 7C and Supplementary Fig. S14K).

A–C The hepatic protein levels of H3K9ac and H3K14ac in the Kat2aHKO and Kat2afl/fl mice (A), CPTH2-treated mice (B) and AAV-LacZ, AAV-Kat2a WT, and AAV-Kat2a Mutant (E575Q) mice (C) (n = 3 independent mice). D–G Genome-wide distribution of GCN5 and H3K9ac was analyzed by ChIP-seq obtained from the GEO database (GCN5 ChIP-seq: GSM1003804 and GSE94229; H3K9ac ChIP-seq: GSM1000141 and GSM918712). D The heat map shows the distribution of GCN5 and H3K9ac at transcription start sites. E Pie chart showing genomic annotations of GCN5 ChIP-seq peaks. F Venn diagram showing the overlap (571 genes) of GCN5 (615 genes) and H3K9ac (7863 genes) bound genes. G Genome tracks showing GCN5 and H3K9ac ChIP-seq occupancy at SREBF1 promoter. H, I HL-7702 cells were treated with CPTH2 (50 μM) for 24 h. H3K9ac and GCN5 recruitment to the promoter of the SREBF1 gene were detected by ChIP-qPCR (n = 3 independent cultures). J, K HFD diet-induced MASLD models treated with CPTH2. The liver H3K9ac and GCN5 recruitment to the promoter of the SREBF1 gene were detected by ChIP-qPCR (n = 4 independent mice). L, M H3K9ac recruitment to the promoter of the SREBF1c gene in the KAT2A KO HL-7702 cells (L) and liver-specific Kat2a knockout mice (M) detected by ChIP-qPCR (n = 3 independent cultures/mice). N, O Overexpression of KAT2A WT or acetyltransferase-defective KAT2A MUT in HL-7702 cells (N) and MASLD liver tissue (O) H3K9ac recruitment to the promoter of the SREBF1c gene were detected by ChIP-qPCR (n = 3 independent cultures/mice). Error bars are represented as mean ± SEM. Statistical analysis was done using one-way ANOVA with Dunnett’s post-test. Exact p-values are indicated in the figure. Source data are provided as a Source Data file.
Next, the genome-wide distributions of GCN5 and H3K9ac were analyzed using ChIP-seq. Enrichment of GCN5 and H3K9ac was observed at transcription start sites (TSSs), showing highly overlapping gene read distributions (Fig. 7D). Specifically, GCN5 was predominantly enriched at promoter and 5’UTR regions (Fig. 7E), and substantial overlap was observed between GCN5 and H3K9ac binding sites, including at SREBF1c (Fig. 7F). ChIP-seq data indicated that GCN5 and H3K9ac co-occupy the SREBF1c promoter (Fig. 7G), suggesting that GCN5 is recruited to this region to regulate transcription. ChIP assays confirmed that GCN5 and H3K9ac are recruited to the SREBF1c promoter in HL-7702 cells and MASLD liver tissue (Fig. 7H–K). Inhibition of GCN5 activity by CPTH2 significantly reduced the recruitment of GCN5 and H3K9ac to the promoters (Fig. 7H–K). Similarly, in KAT2A knockout hepatocytes and liver-specific Kat2a knockout mice, H3K9ac enrichment at the SREBF1c promoter markedly decreased compared with wild-type controls (Fig. 7L, M). Conversely, overexpression of KAT2A WT, but not the acetyltransferase-defective KAT2A MUT, enhanced H3K9ac association with the SREBF1c promoter in HL-7702 cells and MASLD liver tissue (Fig. 7N, O). Together, these results demonstrate that GCN5 promotes SREBF1c transcription by modulating histone H3 acetylation at its promoter via its acetyltransferase activity.
GCN5 promotes the transcriptional expression and activity of LXRα/RXRα-mediated SREBP1c, but not ABCA1
Previous studies have suggested that liver X receptor (LXR) functions as a transcriptional activator of SREBP1c27. To determine whether GCN5 cooperates with LXRα/RXRα to regulate SREBP1c expression, we performed cotransfection and luciferase reporter assays. KAT2A WT, but not KAT2A MUT, enhanced the transcriptional activity of LXRα on a synthetic LXRE promoter in a ligand (T0901317)-dependent manner (Fig. 8A). We further examined their coactivator function on two physiologically relevant natural promoters containing LXREs, specifically SREBF1c and ABCA1. KAT2A WT increased transcriptional activity of the SREBF1c promoter but had no effect on the ABCA1 promoter (Fig. 8B and Supplementary Fig. S15A, B), whereas KAT2A MUT did not enhance transcription of either promoter (Fig. 8B and Supplementary Fig. S15A, B). Importantly, neither KAT2A WT nor KAT2A MUT stimulated transcription from SREBF1c or ABCA1 promoters containing LXRE mutations (Fig. 8C and Supplementary Fig. S15C), indicating that GCN5’s coactivator function depends specifically on LXR recruitment to LXREs. To validate target gene-specific activity, we also tested GW3965, a structurally distinct LXR ligand28. Consistently, GCN5 selectively activated the SREBF1c promoter in an LXR-dependent manner, whereas ABCA1 promoter activity remained unaffected (Fig. 8B, C, Supplementary Fig. S15B, C).

A–C HL-7702 cells were transfected with LXRα/RXRα/GCN5, and a luciferase reporter plasmid under the control of synthetic LXRE (A) SREBF1c WT promoter (B) SREBF1c Mutant promoter (C), incubated with T0901317 (T09) and GW3965 (GW) (n = 3 independent cultures). D, E HL-7702 cells were transfected with Myc-tag-LXRα and Flag-tag-GCN5 plasmids for 24 h and treated with CPTH2 for 24 h. The interaction of GCN5 and LXRα was detected by IP (representative of 3 independent experiments with similar results). F The schematic diagram of GCN5 and its truncations. G HL-7702 cells transfected with Myc-tag-LXRα and the indicated Flag-tag-GCN5 constructs for 24 h and were subjected to immunoprecipitation (representative of 3 independent experiments with similar results). H The schematic diagram of LXRα and its truncations. Scale bar: 20 μm. I HL-7702 cells transfected with Flag-tag-GCN5 and indicated Myc-tag-LXRα-truncated constructs for 24 h and were subjected to immunoprecipitation (representative of 3 independent experiments with similar results). J Co-localization of GCN5 and LXRα by immunofluorescence (representative of 3 independent experiments with similar results), scale bar: 10 μm. K Schematic diagram of primer locations used for the ChIP assay across the SREBF1c genomic structure. L, M HL-7702 cells were transfected with siRNAs for 48 h. The cells were incubated with T0901317 and GW3965 for 24 h. ChIP assays were performed with anti-GCN5 (L) or anti-H3K9ac (M) antibodies. Recruitment of GCN5 and H3K9ac to the LXREs of the SREBF1c gene was determined by qRT-PCR (n = 3 independent cultures). N, O KAT2A KO HL-7702 cells were treated with T0901317 and GW3965 for 24 h. ChIP assays were performed with anti-LXRα (N) or anti-H3K9ac (O) antibodies. Recruitment of LXRα and H3K9ac to the LXREs of the SREBF1c gene was determined by qRT-PCR (n = 3 independent cultures). P–R KAT2A KO HL-7702 cells were treated with T0901317 and GW3965 for 16 h (n = 3 independent cultures). P The protein levels of SREBP1 were detected by western blot. Q The SRE-Luc activity was detected by luciferase assay. R The indicated gene expression was detected by qRT-PCR. Error bars are represented as mean ± SEM. Statistical analysis was done using one-way ANOVA with Dunnett’s post-test. Exact p-values are indicated in the figure. Source data are provided as a Source Data file.
The above experiments confirmed that GCN5 and LXRα synergistically regulate SREBF1c expression. To further elucidate their molecular relationship, we performed co-immunoprecipitation (co-IP) assays in HL-7702 cells co-transfected with Flag-tagged GCN5 and Myc-tagged LXRα. The results revealed a robust physical interaction between GCN5 and LXRα (Fig. 8D). Reciprocal co-IP experiments consistently showed that LXRα efficiently precipitated GCN5, confirming their strong mutual association (Fig. 8E). Importantly, treatment with CPTH2 significantly decreased the interaction between GCN5 and LXRα (Fig. 8D, E). To map the interaction domains, we generated full-length (FL) GCN5 and a series of truncation mutants co-transfected with LXRα into HL-7702 cells. Co-IP indicated that the PCAF-N domain of GCN5 (1–407 aa) is both necessary and sufficient for interaction with LXRα (Fig. 8F, G). Conversely, to identify the LXRα domains mediating the interaction, we constructed several LXRα deletion mutants, including the N-terminal and DNA-binding domain (1–163 aa, unable to bind MEIS proteins), ligand-binding domain (164–326 aa), and C-terminal (327–447 aa, impaired DNA binding) (Fig. 8H). Co-IP experiments revealed that the N-terminal and DNA-binding domain is required for LXRα to interact with GCN5 (Fig. 8I). Immunofluorescence co-localization experiments in native human hepatocytes further confirmed the interaction between GCN5 and LXRα (Fig. 8J). In silico AlphaFold3 predictions also supported a direct protein-protein interaction, identifying hydrogen bond pairs between GCN5 and LXRα, including LYS124:ASP11, ARG206:ASP11, ARG206:ILE12, LYS203:PRO10, and LYS203:SER16 (Supplementary Fig. S15D).
To investigate the recruitment of GCN5 to endogenous chromatin, we performed ChIP analysis of the SREBF1c and ABCA1 promoter regions. GCN5 and H3K9ac were recruited to the LXRE of the endogenous SREBF1c promoter in response to treatment with T0901317 or GW3965 (Fig. 8K–M). In contrast, GCN5 and H3K9ac were not recruited to the LXRE of the ABCA1 promoter (Supplementary Fig. S15E, F). To determine whether this recruitment is LXRα-dependent, we knocked down LXRα in hepatocytes using siRNA, which was validated by qRT-PCR (Supplementary Fig. S15G). Under LXRα-knockdown conditions, ligand treatment no longer stimulated the recruitment of GCN5 and H3K9ac to the SREBF1c LXRE (Fig. 8L, M), whereas recruitment to the ABCA1 LXRE remained absent regardless of LXRα expression (Fig. S15E, F). These findings indicate that GCN5 is selectively recruited to the SREBF1c promoter in a ligand- and LXRα-dependent manner.
To further examine the role of GCN5 in promoter activation, ChIP assays were performed using antibodies against LXRα and H3K9ac. Depletion of GCN5 abolished T0901317-dependent recruitment of LXRα and H3K9ac to the SREBF1c LXRE (Fig. 8N, O), while recruitment to the ABCA1 LXRE was not significantly altered (Supplementary Fig. S15H, I). Similarly, GCN5 depletion reduced GW3965-dependent recruitment of LXRα and H3K9ac to the SREBF1c LXRE but not to the ABCA1 LXRE (Fig. 8N, O), demonstrating that GCN5 selectively stimulates SREBF1c expression via chromatin remodeling.
To assess the functional consequences, KAT2A knockout (KO) cells were used to measure SREBF1c and ABCA1 transcript levels by qRT-PCR. Both GW3965 and T0901317 significantly increased SREBF1c mRNA levels (Fig. 8P); however, KAT2A depletion markedly suppressed these ligand-dependent increases (Fig. 8R). In addition, the ability of these ligands to elevate SREBP1 protein levels and transcriptional activity was significantly diminished in KAT2A KO hepatocytes (Fig. 8P–Q), resulting in reduced expression of downstream target genes, including ACC, FASN, SCD1, SCD2, and FADS2 (Fig. 8R). These results demonstrate that GCN5 specifically promotes LXRα/RXRα-mediated transcriptional expression and activity of SREBP1c, but not ABCA1.
Synergistic reduction of lipid accumulation by T0901317 and CPTH2
LXRα agonizts are used to treat atherosclerosis by promoting the expression of genes such as ABCA1 and enhancing reverse cholesterol transport (RCT)29. However, they also increase DNL by upregulating SREBP1c, which raises the risk of fatty liver30. Based on the selective regulatory effect of GCN5 on SREBP1c versus ABCA1, we hypothesized that CPTH2 could simultaneously inhibit LXRα-induced DNL while preserving RCT. In vitro, T0901317 and GW3965 increased the mRNA levels of Abca1, Abcg5, and Abcg8, promoting RCT and reducing cholesterol levels in RAW264.7 cells (Fig. 9A–C). In hepatocytes, these ligands increased SREBP1c expression and its downstream targets, enhanced DNL, and elevated TG levels (Fig. 9D–G). Neutral lipid staining with Nile Red and Bodipy revealed that T0901317 and GW3965 significantly increased lipid droplet accumulation (Fig. 9H and Supplementary Fig. S16A–C).

A–G Cells were incubated with PAOA and the indicated compounds (T0901317, GW3965, or CPTH2) for 24 h (n = 3 independent cultures). A, E The indicated gene expression was detected by qRT-PCR. B The RCT level was measured. C The cellular TC levels. D The SREBP1 protein levels were detected by western blot. F De novo lipogenesis was measured in the HL-7702 cells. G The cellular TG levels. H The treated cells were stained with Nile Red and Bodipy (representative of 6 independent experiments with similar results). Scale bar: 20 μm. I–R Male C57BL/6 J mice at 8 weeks of age were randomly grouped (n = 6 independent mice). Mice were allowed ad libitum access to water and an HFD diet. The indicated compounds were administered to mice by gastric irrigation every day. After 2 weeks of treatment, the mice were sacrificed and subjected to a series of analyses as indicated below. I Schematic diagram of HFD diet-induced MASLD models treated with indicated compounds. J LW and LW/BW. K The liver TG and TC levels (n = 6 independent mice). L The serum TG, TC, HDL-c, and LDL-c levels (n = 6 independent mice). M Representative Oil red O staining in the liver and histological analysis of the liver. Scale bar: 50 μm. N Quantification of hepatic lipid droplets was performed using ImageJ (n = 6 independent mice). O The serum ALT, and AST levels (n = 6 independent mice). P The SREBP1 protein levels were detected by western blot. Q, R The indicated gene expression was detected by qRT-PCR (n = 3 independent mice). Error bars are represented as mean ± SEM. Statistical analysis was done using one-way ANOVA with Dunnett’s post-test. Exact p-values are indicated in the figure. Source data are provided as a Source Data file.
Treatment with CPTH2 did not impair RCT or the expression of RCT-related genes but significantly reduced T0901317- and GW3965-induced SREBP1c expression and downstream DNL genes, including ACC, FASN, SCD1, SCD2, and FADS2 (Fig. 9D, E). Consequently, hepatic TG accumulation and lipid droplet content were markedly decreased in HL-7702 cells and mouse primary hepatocytes (Fig. 9F–H and Supplementary Fig. S16A–C). In vivo, HFD-fed mice were gavaged with T0901317 (5 mg/kg), CPTH2 (20 mg/kg), or their combination for 2 weeks (Fig. 9I). Food intake and body weight were not significantly altered (Supplementary Fig. S16D, E). T0901317-induced increases in LW and LW/BW ratios were attenuated by CPTH2 (Fig. 9J). The serum TC, LDL-c, TG, and hepatic TC levels in T0901317-treated mice were significantly lower than those of the vehicle-treated mice (Fig. 9K, L). However, the liver TG levels were largely increased in HFD-fed mice treated with T0901317 (Fig. 9K), with hepatic lipids accumulation (Fig. 9M, N). Interestingly, co-administration with CPTH2 significantly reduced T0901317-induced liver TG levels, and the combination treatment also further lowered serum TC, LDL-c, and hepatic TC compared with either treatment alone (Fig. 9K, L). Histological analyses using H&E and Oil Red O staining confirmed that lipid accumulation was substantially decreased by co-administration (Fig. 9M), and serum ALT and AST levels were also significantly reduced (Fig. 9O). At the molecular level, CPTH2 repressed T0901317-induced hepatic SREBP1c expression and its target genes without affecting RCT-related gene transcription (Fig. 9P–R). These findings indicate that combined administration of T0901317 and CPTH2 exerts synergistic lipid-lowering effects in vitro and in vivo, inhibiting DNL while preserving RCT.
Discussion
GCN5 acts as an epigenetic regulator, modifying histones, as individual regulators and non-histone proteins14. This makes it a key player in human diseases such as cancer, diabetes, osteoporosis, and malaria. Therefore, GCN5 is a promising therapeutic target with considerable potential. However, the 73% homology between GCN5 and PCAF has been shown to have the same function in multiple diseases15,17,31,32. In this study, we found that GCN5 was highly expressed, but PCAF was not in the livers of patients with MASLD and mice. Using this database and our clinical data for correlation analysis, we found that the expression of GCN5, but not that of PCAF, was positively correlated with MASLD progression. These results suggested that GCN5 and PCAF may not play the same role in MASLD progression. We generated a liver-specific overexpression of KAT2A WT and an acetyltransferase-defective GCN5-E575Q mutant of KAT2A in NALFD mice. KAT2A WT and KAT2A MUT were transfected in HL-7702, HepG2 cells, and mouse primary hepatocytes. Overexpression of KAT2A WT, but not KAT2A MUT, exacerbated MASLD progression in vivo and increased lipid accumulation in hepatocytes in vitro. To explore the role of GCN5 in NAFL pathogenesis, hepatocyte-specific Kat2a-knockout (Kat2aHKO) was generated by crossing Kat2a flox/flox (Kat2afl/fl) mice with Alb-Cre mice. We generated a KAT2A knockout HL-7702 cell line (KAT2A KO) using the CRISPR-Cas9 system to investigate the function of KAT2A. We found that hepatocyte-specific Kat2a deficiency protected against HFD/HFrD/HFMRCD/FCP/AMLN-induced hepatic steatosis and subsequent inflammation and fibrosis, suggesting that GCN5 may act as a positive regulator of MASLD progression. Moreover, we used CPTH2, a GCN5 potent inhibitor, that protects against MASLD.
In 2006, Lerin et al. demonstrated that under fasting conditions, GCN5 inhibits gluconeogenesis in the liver through the acetylation of PGC-1α, thereby lowering blood glucose levels21. This mechanistic framework was substantially expanded by Sakai et al. in 2016, who elucidated the molecular basis of the nutritional state-dependent regulation of GCN5 activity. During fed states, GCN5 primarily acetylates PGC-1α to repress the expression of gluconeogenic genes, whereas fasting or diabetic conditions trigger glucagon-cAMP signaling-induced phosphorylation of GCN5 at Ser275, switching its substrate preference from PGC-1α to histone H3 and thereby activating the gluconeogenesis23. The regulation of gluconeogenesis mediated by GCN5 shows a significant dependence on specific physiological or pathological conditions. In this study, we found that the blood levels of insulin and glucose were not correlated with the protein levels of GCN5 and PCAF, while Sakai et al.‘s findings are based solely on mouse models and primary hepatocytes, with no human data or correlation analysis provided.MASLD. In our current investigation employing MASLD or MASH models, the metabolic profile was predominantly characterized by hyperlipidemia and mild hyperglycemia, with minimal activation of the glucagon-cAMP/PKA signaling axis24,33,34.
FA metabolism disorder is not only the basis of MASLD occurrence and development, but also one of the most critical links. FA and TG production of FA and TG is mainly regulated at the transcriptional level by various nutritional metabolic states in the liver. SREBP1c is a key transcription factor in liver DNL that directly regulates the gene expression of key enzymes involved in FA and TG synthesis, such as ACLY, ACC, FAS, and SCD135. Abnormally high expression of the SREBP1c gene and protein has been found in a variety of mouse and rat models of MASLD and in the livers of patients. Overexpression of SREBP1c induces hepatic DNL, lipid accumulation, endoplasmic reticulum stress (ER stress), and the unfolded protein response (UPR). Long-term ER stress and the UPR reaction further activate SREBP1c, thus aggravating hepatic steatosis, cellular stress, and inflammation. Chronic inflammation increases the risk of liver fibrosis. The SREBP1 pathway is involved in the first and second steps of MASLD formation36. Exogenous SREBP1c overexpression leads to hepatic lipid accumulation and induce MASLD37. Systemic SREBP1c knockout reversed hepatic lipid accumulation in obese mice38. SREBP1c is originally synthesized as an inactive precursor anchored to the ER membrane and forms a complex with the SREBP cleavage-activating protein (SCAP). To achieve activation, SREBP exits the ER and travels to the Golgi apparatus, where it is sequentially hydrolyzed by site-1 and site-2 protease. The release of transcriptionally active NH2-terminal domains that can migrate to the nucleus to regulate the transcription of their target genes by binding to sterol regulatory elements (SREs) in their promoter region36. To date, a series of SREBP inhibitors have been reported, including oxysterols, such as 25-hydroxycholesterol. As these compounds abrogate the movement of the SCAP-SREBPs complex to the Golgi apparatus, SREBPs activity is inhibited, alleviating the symptoms of metabolic diseases, such as obesity, hyperlipidemia, and insulin resistance39. However, free cholesterol accumulation in the ER activates the accumulation of unfolded/misfolded protein, thereby increasing ER stress and apoptosis. Prolonged ER stress induces cardiac myocyte apoptosis, fat accumulation, insulin resistance, and non-alcoholic steatohepatitis40. Therefore, the discovery of new mechanisms regulating the SREBP1c pathway and the development of related inhibitors to avoid these adverse reactions are the strategies for the treatment of MASLD41. In this study, we found that the loss of GCN5 suppressed hepatic DNL by inhibiting the SREBP1c pathway. An in-depth mechanistic study found that GCN5 promotes the gene expression of SREBF1c by altering the acetylation state of histone 3 in the promoter region of SREBF1c through its inherent acetyltransferase activity. Our data demonstrates that GCN5 deletion suppresses SREBP-1c activity, leading to downregulation of mRNA expression, including ACLY and ACC. Thus, reduced cytosolic acetyl-CoA and malonyl-CoA pools are direct consequences of impaired SREBP-1c-mediated DNL. Therefore, we found that CPTH2 did not cause side effects such as ER stress or the UPR reaction during MASLD treatment.
These research findings suggest that targeting LXR may offer a promising therapeutic approach for combating atherosclerosis through the modulation of macrophage-driven inflammation and promotion of ABCA1-dependent RCT42. The RCT is a vital pathway for eliminating surplus cellular cholesterol from peripheral tissues to the liver for excretion in the bile and feces, thereby reducing the progression of atherosclerosis42,43. Administration of the LXR ligands T0901317 or GW3965 has been shown to enhance RCT facilitated by ABCA1, resulting in increased serum HDL levels and decreased atherosclerosis. Furthermore, LXR exerts cardioprotective effects by inhibiting inflammatory gene expression in macrophages. The potential therapeutic application of LXR agonizts in patients with atherosclerosis are promising29,44. However, LXR activation has been associated with an increase in SREBF1c mRNA, a crucial regulator of DNL, leading to elevated triglyceride levels in both the serum and liver. To optimize the therapeutic efficacy of LXR agonizts, it is imperative to develop strategies that counteract their negative effects on fatty acid metabolism45,46,47. Our study demonstrates the involvement of GCN5 in the transcriptional regulation of SREBP1c by LXRα. Through nuclear extract-binding experiments, we observed that GCN5 interacts with the LXRα ligand-binding domain in a ligand-dependent manner. Interestingly, GCN5 selectively activated SREBF1c, but not ABCA1, despite both being LXR target genes. This specificity was attributed to GCN5’s preferential recruitment to the SREBF1c promoter over the ABCA1 promoter. Furthermore, the recruitment of GCN5 to the LXRE of SREBF1c in response to ligand stimulation was found to be entirely dependent on LXRα. A ChIP assay utilizing anti-H3K9ac antibodies revealed that GCN5 can promote chromatin remodeling by acetylating H3 close to the LXRE of the SREBF1c gene. This implies that GCN5 may enhance SREBP1c transcription by increasing histone acetylation levels. As a result, the reduction of GCN5 specifically attenuated the expression of genes involved in DNL, while leaving the expression of genes related to the RCT of the LXR ligand unaffected.
In the context of RCT, we assessed the expression of ABC transporters (specifically ABCA1). RCT is a complex biological process that involves multiple tissues, such as macrophages, the liver, and the intestine42. In addition, ABC transporters play a crucial role in facilitating RCT throughout the entire pathway, and not solely in the efflux of cholesterol from macrophages48. Therefore, we focused on the final stages of RCT to examine the impact of GCN5 on LXR-induced RCT. This study proposes that the manipulation of GCN5 may serve as a mechanism to selectively inhibit SREBP1c-mediated DNL while maintaining RCT stimulation by LXRα. Therefore, GCN5 may be a potential target for the treatment of fatty liver and for the development of LXR agonizts that can effectively prevent atherosclerosis. In addition, we found that the combined administration of T0901317 and CPTH2 might have a synergistic lipid-lowering effect in vitro and in vivo.
Methods
Human study and approval
The MASLD patients recruited from Changzhou No. 2 People’s Hospital were diagnosed by ultrasonography and serologic testing. Inclusion criteria were as follows: (1) age between 18 and 65 years; (2) clinical diagnosis of non-alcoholic fatty liver (NAFL) or non-alcoholic steatohepatitis (NASH); (3) provision of written informed consent. Exclusion criteria included: (1) severe comorbidities affecting major organs (e.g., heart, liver, or kidney); (2) history of malignant tumors; (3) pregnancy or lactation; (4) psychiatric disorders. The liver tissue samples of patients were obtained intraoperatively from bariatric surgery, and subjects with excessive alcohol intake or other liver diseases (such as viral hepatitis and drug damage et al.) were excluded. Adjacent non-cancer normal liver tissues were collected from liver cancer surgical resection as normal control liver specimens, except those with metabolic abnormalities. The study was approved by the Ethics Committee of the Changzhou No. 2 People’s Hospital and followed the ethical guideline Declaration of Helsinki. Written informed consent was provided by each subject ([2023]KY124-01).
Animal
Association for Assessment and Accreditation of Laboratory Animal Care International has accredited the animal experimental center. Animals received human care following the National Institutes of Health’s Guide to the Care and Use of Laboratory Animals. All animal experiments and care were approved by the Animal Ethics Committee of China Pharmaceutical University (2024-06-017). Male C57BL/6 J wild-type mice, ob/ob mice, and littermate controls (SPF grade, 6-7 weeks old, 20 to 22 g) were obtained from GemPharmatech Co. Ltd (Nanjing, China). Animals were maintained on a 12-h light-dark cycle at 22–24 °C, relative humidity kept at 50–60% and had free access to water and Normal chow (Jiangsu Synergetic Biology Co., Ltd, 1010039) unless otherwise stated.
Diet-induced mouse MASLD models
High-fat diet-induced mouse MASLD models were constructed by feeding mice an HFD diet (60 kcal% fat, 20.6 kcal% carbohydrate,19.4 kcal% protein) for 8, 16, or 32 weeks. FPC diet-induced mouse MASLD models were constructed by feeding mice an FPC diet (52 kcal% fat, 34.5 kcal% sucrose, and 1.25% cholesterol; Teklad, TD190142) combined with 42 g/L sugar solution (55% glucose + 45% fructose) in drinking water for 16 weeks. HFrD diet-induced mouse MASLD models were constructed by feeding mice an HFrD diet (10 kcal% fat and 60 kcal% fructose; Research Diets, Inc. D02022704K) for 6 weeks. AMLN diet-induced mouse MASLD models were constructed by feeding mice an AMLN diet (40 kcal% fat, 20 kcal% fructose and 2% cholesterol; Research Diets, D09100310) for 16 weeks.
Diet-induced mouse MASH models
A mouse MASH model was built by feeding mice an HFMRCD diet (60 kcal% fat with low methionine and no Choline; Xietong Shengwu, XTMRCD-60) for 6 weeks. Another mouse MASH model was built by feeding mice an FPC diet (52 kcal% fat, 34.5 kcal% sucrose, and 1.25% cholesterol; Teklad, TD190142) combined with 42 g/L sugar solution (55% glucose + 45% fructose) in drinking water for 24 weeks.
AAV8-mediated gene overexpression
For AAV8 transduction, mice were fed 4–7 weeks of HFD and then injected with 2.5e11 VG of AAV-LacZ/AAV-KAT2A WT/AAV-KAT2A Mut or AAV-m-SREBP1c through the tail vein. The thyroxine-binding globulin (TBG) promoter was adopted for hepatocyte-specific expression.
Generation of Kat2a HKO mice
Hepatocyte-specific Kat2a knockout (Kat2aHKO) mice were established with the CRISPR-Cas9 system and Cre-loxP-mediated recombination technology. Kat2afl/fl mice and Albumin-Cre (Alb-Cre) mice were obtained from GemPharmatech Co., Ltd (Nanjing, China). All the mice were C57BL/6 J background. Kat2aHKO mice were generated by crossing homozygous Kat2afl/fl mice with Alb-Cre mice. Exon 7–11 of Kat2a in hepatocytes were depleted from Kat2afl/fl mice. PCR was applied for genotyping using DNA extracted from mouse tails. The genotyping primers were as follows:
Kat2a-F, 5’-AAGTGAAGCCATTACCTATCTCTGTGCC-3’;
Kat2a-R, 5’-GCCAGGATAGAGACTGGAGACCCAA-3’;
Cre-F, 5’-GGGCAGTCTGGTACTTCCAAGCT-3’;
Cre-R, 5’-TAGCTACCTATGCGATCCAAACAAC-3’;
WT-F, 5’-CAGCAAAACCTGGCTGTGGATC-3’;
WT-R, 5’-ATGAGCCACCATGTGGGTGTC-3’.
The primer pairs Kat2a-F and Kat2a-R were used to detect the flox allele (wildtype allele = 257 bp, flox allele = 359 bp). The primer pairs Cre-F and Cre-R were used to detect the Alb-Cre allele (Alb-Cre allele = 340 bp; wildtype allele = None). The primer pairs WT-F and WT-R were used to detect whether Alb-Cre mice were homozygous or heterozygous (homozygote = None; heterozygote = 412 bp).
Mouse primary hepatocyte isolation
Male mice were first anesthetized using isoflurane (1.5% in O₂). A catheter was then inserted into the vena cava, and the portal vein was cut. The liver was subsequently perfused via the catheter with oxygenated, 37 °C Buffer A (1 × PBS, 5 mM EGTA). This was followed by perfusion with oxygenated, 37 °C Buffer B (1 × PBS, 1 mM CaCl₂, collagenase type IV). Finally, the liver was transferred to a Petri dish containing Buffer C (1 × PBS, 2 mM CaCl₂, 0.6% BSA) and gently dissociated using forceps. Liver perfusions were centrifuged at 48 g for 5 min after digestion using sterile gauze. Next, primary hepatocytes were isolated from mice using sterile gauze. A pellet of hepatocytes was resuspended in Medium B, and the supernatant was discarded after three washes. In collagen-coated plates, hepatocytes were plated, and cell viability was confirmed via the trypan blue exclusion test. Cell viability greater than 70% was considered acceptable for moving forward with the research.
Prediction of transcription factors binding to the KAT2A promoter
Candidate transcription factors (TFs) capable of binding to the KAT2A promoter were predicted using the JASPAR database (https://jaspar.genereg.net/; release 2022). The analyzed promoter region was defined as 2,000 bp upstream of the transcription start site (TSS) to 200 bp downstream for both the human (GRCh38/hg38: chr17:40,100,000-40,102,200) and mouse (GRCm39/mm39: chr11: 69,900,000-69,902,200) KAT2A genes. The search was performed against the JASPAR CORE vertebrate collection of transcription factor binding profiles. Scanning was under the screening condition of p-value < 10−5, all predicted TFs meeting this threshold in either species were recorded. To prioritize TFs with potential conserved regulatory roles, we intersected the prediction results from the human and mouse KAT2A promoter sequences. This analysis yielded six candidate TFs. Subsequently, we reviewed the existing literature for these six TFs and selected four that have previously documented roles in lipid metabolism for further experimental validation.
Viability assay
Cell viability was assessed in HepG2, HL-7702, and primary hepatocytes using the MTT assay. Briefly, cells were plated in 96-well plates at a density of 1 × 10⁴ cells per well. After 24 h, the cells were treated with the specified concentrations of CPTH2 for another 24 h. Then, 10 μl of MTT solution (5 mg/ml) was added to each well, and the plates were incubated at 37 °C for 4 h. Subsequently, 100 μl of formazan solution was added to each well, followed by incubation at 37 °C for 3–4 h. Absorbance was measured at 570 nm using a microplate reader (BMG POLARstar Omega).
BODIPY493/503 and Nile Red staining
Cells were fixed with 4% paraformaldehyde and then stained with 1 μM Nile Red or 5 μM BODIPY493/503 (Bodipy) for 30 min at room temperature in the dark. After being stained with DAPI, excess dye was washed away with PBS three times. The fluorescence of intracellular lipid droplets was photographed by a laser confocal microscope (Olympus FV3000). ImageJ (Version 1.5a) was used to quantify intracellular neutral lipids.
Western blotting
After collection, cells were solubilized in lysis buffer supplemented with SDS, loading buffer, protease inhibitors, and phosphatase inhibitors. Samples were then thermally denatured at 95 °C for 10 min and subjected to electrophoretic separation on 8–12% SDS-polyacrylamide gels. Subsequently, proteins were transferred onto nitrocellulose membranes via electroblotting. Membranes were blocked for 1 h at room temperature with 5% non-fat dry milk and then incubated with primary antibodies at 4 °C overnight. Following thorough washing, membranes were exposed to HRP-conjugated secondary antibodies for 1 h at room temperature. Detection was carried out using an enhanced chemiluminescence substrate, and blot images were captured with a Tanon 5200 imaging system (Tanon, China). Quantitative analysis of band intensity was conducted using ImageJ software (Version 1.5a). Uncropped blots are available in the Source Data file.
Cellular, liver, and serum lipid determination
A subset of cells was reserved for protein quantification via BCA assay, while the remainder underwent lipid extraction. Cellular total cholesterol (TC) and triacylglycerol (TG) were extracted in chloroform/methanol (2:1, v/v) at room temperature for 3 h. After adding 500 μl of 0.1 M NaCl, samples were vortexed, centrifuged, and the lower organic phase collected and dried under nitrogen. Dried lipids were redissolved in 50 μl ethanol with 1% Triton X-100 (Sigma-Aldrich, T9284). TG and TC levels were determined using commercial assay kits and normalized to protein content.
For hepatic TG and TC measurement, 50 mg liver tissue was homogenized in PBS. Part of the homogenate was used for the BCA protein assay; 0.4 ml was mixed with 1.6 ml chloroform/methanol for extraction as above.
Serum TG, TC, HDL-c, and LDL-c were measured using corresponding kits per manufacturer’s protocols.
Measurement of de novo fatty acid synthesis
KAT2A knockout and wild-type HL-7702 cells were treated under specified conditions in Medium B for 16 h. The de novo fatty acid synthesis rate was assessed by incubation with a 12 µCi/ 60 mm dish of [14C] acetate for 2 h. After washing, cells were lysed in 0.1 N NaOH and saponified by autoclaving. Nonpolar lipids (cholesterol) were extracted using petroleum ether and evaporated under nitrogen. Following acidification with concentrated HCl, polar lipids (fatty acids) were similarly extracted, dried, and dissolved in 5 ml scintillation cocktail for DPM measurement.
Oxygen consumption rate (OCR)
HL-7702 cells were plated in Seahorse XF96 microplates (Agilent Technologies, USA) at 5 × 104 cells/well and treated with OA (200 mM) and PA (100 mM). Prior to OCR assessment, cells were incubated for 1 hour at 37 °C in a CO₂-free incubator using XF base minimal DMEM supplemented with 10 mM glucose, 0.5 mM L-carnitine, and 1 mM L-glutamine. OCR was monitored using the Seahorse XFe96 extracellular flux assay kit under calibrant conditions. Sequential injections of 2 μM etomoxir, 2.5 μM oligomycin, 1 μM FCCP, and 0.5 μM rotenone/antimycin A were performed during the assay. In addition, 30 μl of BSA-control or BSA-palmitic acid (BSA-PA) was introduced into each well before measurement. Data were acquired on a Seahorse XFe96 Analyzer and analyzed with Wave 2.6.1 software.
FFA uptake
The uptake of free fatty acids (FFA) was quantified utilizing a fluorometric assay kit (Abcam, ab176768). HL-7702 cells were seeded at 1 × 105 cells per well, resuspended in PBS, and maintained at 37 °C in a CO₂ incubator for 30 min. Subsequently, a fluorescent FFA probe was added. Following an additional incubation period of 1 h, fluorescence intensity was detected in bottom-read mode with excitation/emission set at 485/515 nm (or FITC channel) on a microplate reader.
Cholesterol efflux test
RAW264.7 cells were inoculated at a density of about 1 × 106 cells/well in a 6-well plate. The next day, the medium was removed and treated for 24 h with drugs or other means. Then, 1.5 μM 22-NBD cholesterol was added to a medium containing 2.5% FBS for 1 h. The cells were washed twice with Puck buffer and placed in a serum-free medium. Efflux was initiated by adding HDL (10 μg/ml). Following 4 h of incubation, cells were harvested and resuspended in FACS buffer. Fluorescence (FL-1) was quantified via flow cytometry, and data were analyzed using Cell Quest Pro software. HDL-dependent cholesterol efflux was calculated as: (fluorescence without HDL − fluorescence with HDL) / fluorescence without HDL × 100%.
Reporter gene assay
Promoter-reporter constructs (SREBP-1c-Luc, SREBP-1c mtLXRE-Luc, ABCA1-Luc, and ABCA1 mtLXRE-Luc) were generated by PCR-based cloning, with primer sequences provided in Table 1. HL-7702 cells were seeded in 96-well plates, transfected with the corresponding luciferase reporter plasmids, and treated as indicated. After treatment, cells were lysed using reporter gene lysis buffer (Beyotime, RG0036) for 30 minutes at room temperature. Lysates from each well were transferred to a white 96-well plate. Protected from light, luciferase substrate (Vazyme, DL101-01) was added, and luminescence was immediately measured using a BMG POLARstar Omega microplate reader. Luminescence values were normalized to the protein concentration of each sample.
Structure prediction
The amino acid sequences for human GCN5 and LXRα were retrieved from UniProt, with the respective Protein IDs: GCN5 (Q92830, https://www.uniprot.org/uniprotkb/Q92830/entry), LXRα (Q13133, https://www.uniprot.org/uniprotkb/Q13133/entry). 3D complex structures of GCN5 and LXRα were predicted using AlphaFold3 (https://www.alphafoldserver.com). Resulting models were visualized and analyzed with PyMOL.
qRT-PCR
Total RNAs were extracted by RNA-easy isolated reagents. cDNA was reverse transcribed with the HiScript Reverse Transcriptase kit. qRT-PCR was carried out on a LightCycler 96 system (Roche) with SYBR Green Master Mix. The relative expression of genes was calculated by the ΔΔCt method. Primer sequences are provided in Table 2.
Knockout of KAT2A by CRISPR-Cas9
A CRISPR-Cas9 genome editing system was applied to establish the KAT2A KO cell. Briefly, sgRNA sequence (5’-ATGGGGCAAACTCTCCAATC-3’) was designed with the Broad Institute CRISPick tool (https://portals.broadinstitute.org/gppx/crispick/public) and cloned into the pUC-CBh-gRNA vector named pUC-CBh-KAT2A-gRNA. HL-7702 cells were transfected with the donor construct EF1A-GFP-T2A-Puro (conferring puromycin resistance) using Lipofectamine 3000. After 48 h, transfected cells were selected with puromycin (2 μg/ml) for 72 h. Single colonies were isolated, and the KAT2A knockout was confirmed by western blot.
Chromatin immunoprecipitation (ChIP) and ChIP-seq analysis
The ChIP assay was carried out using the Simple ChIP® Plus Sonication Chromatin IP Kit (Cell Signaling Technology, 56383S). Following treatment, cells were cross-linked with 1% formaldehyde for 10 min at room temperature and quenched with 0.125 M glycine for 5 min. For liver tissues, approximately 100 mg was minced into 1 mm³ pieces and incubated in PBS containing protease inhibitors before fixation. Cells were washed twice with cold PBS, collected in PBS with protease inhibitors, and centrifuged at 1000 × g for 5 min at 4 °C. The pellet was resuspended in Cell Lysis Buffer and lysed on ice for 10 min twice. Nuclei were lysed with Nuclear Lysis Buffer, and chromatin was sheared using a Branson SFX250 Sonifier (50% amplitude, 6 min cycle, 1 s on/off). After centrifugation at 21,000 × g for 10 min at 4 °C, the supernatant was collected. Each 10 μg of chromatin was immunoprecipitated with 2 μg of specific antibody or control IgG overnight at 4 °C, followed by incubation with Protein G beads for 2 h at 4 °C. Beads were washed, and chromatin was eluted and reverse cross-linked. DNA was purified and quantified by qPCR. Primer sequences are listed in Table 3.
ChIP-seq data were obtained from the GEO database (GCN5 ChIP-seq: GSM1003804 and GSE94229; H3K9ac ChIP-seq: GSM1000141 and GSM918712). Raw sequencing reads were trimmed to remove adapter sequences and low-quality fragments using Skewer (v0.2.2). Quality control was performed with FastQC (v0.11.5). Cleaned reads were aligned with the mm9 mouse genome by Bowtie2 (version 2.5.2). Peak calling for tissues was performed using MACS2 (version 2.1.1) to obtain protein and DNA interaction binding sites. ChIP-seq peaks were visualized with IGV (version 2.17.2). Heatmaps were created using R 3.6.1 and the pheatmap package (1.0.12).
RNA-seq analysis
KAT2A knockout and wild-type HL-7702 cells were separately seeded into 6-well plates and treated with PAOA for 24 h, and the livers of Kat2aHKO mice and Kat2aflf mice which fed with 10 weeks HFD diet, then total RNA was extracted using RNA-easy isolated reagent. RNA quantification was detected using NanoPhotometer (Thermo Fisher), followed by examination of RNA integrity and concentration with Agilent 2100 RNA Nano 6000 Assay Kit (Agilent Technologies). mRNA was enriched with Oligo d(T) Magnetic Beads, fragmented, and converted into double-stranded cDNA. The cDNA underwent end repair, adenylation, adapter ligation, and size selection (~ 350 bp). After PCR amplification, the resulting library was quantified with Qubit 3.0 (Thermo Fisher) and quality-checked using the Agilent 2100 Bioanalyzer (Agilent Technologies). Sequencing was performed on an MGI DNBSEQ-T7 platform. Raw reads were processed with Fqtools Plus to remove adapters and low-quality sequences. Clean reads were aligned to the hg38 reference genome using HISAT2 (v2.2.1). Differential gene expression analysis was conducted with DESeq2 (v23.1.0) under thresholds of |log₂FC | ≥ 1 and p-value ≤ 0.05. Gene Ontology enrichment was performed using cluster Profiler (v3.5.1; p-value < 0.05). Visualization was carried out with GraphPad Prism (v8), including heatmaps and volcano plots.
Metabolomics
Sample extraction
Liver tissues from Kat2aHKO mice and Kat2afl/fl mice were snap-frozen, pulverized, and aliquoted. Metabolites were extracted using a two-phase system. Powdered tissue was homogenized in 50% methanol and acetonitrile. Phase separation was induced by adding methylene chloride and water, followed by vortexing and centrifugation (4 °C, 20 min). The lower organic phase was collected and dried under N₂ for LC-MS analysis of fatty acids. The upper aqueous phase was processed for two analyses: one aliquot was dried, reconstituted in water, and analyzed by LC-MS for CoA species; the remainder was dried, derivatized, and subjected to GC-MS for polar metabolites (DHAP, Glycerol, Glycerol-3P, and Citric acid).
Free and esterified fatty acids analysis by LC-MS
The dried organic phase was reconstituted in a methanolic KOH solution containing an internal standard (rD27-myristic acid) for saponification. After vortexing, sonication, and incubation, methylene chloride was added, and the mixture was recentrifuged. The aqueous layer was acidified, and free fatty acids were extracted with hexane. The organic layer was dried, reconstituted in methyl acetate, and derivatized with tetramethylammonium hydroxide to generate fatty acid methyl esters (FAMEs). Chromatographic separation was carried out using an Acquity UPLC BEH C18 column (2.1 × 100 mm, 1.7 μm) maintained at 55 °C with a flow rate of 0.3 ml/min. Mobile phase A consisted of water-acetonitrile (40:60, v/v), and mobile phase B was isopropanol-acetonitrile (90:10, v/v), both containing 5 mM ammonium formate and 0.1% formic acid. A 35 min gradient was used. Mass spectrometry was conducted on a SCIEX X500B QTOF instrument using both positive and negative ESI modes. Data were acquired in data-independent mode over m/z 100–1250. Quality control samples were analyzed throughout the sequence to ensure instrument stability. Signal intensities were normalized to protein concentration, and data were processed using SCIEX OS software (v. 3.0).
Acyl-CoA analysis by LC-MS/MS
Acyl-CoA species were quantified using HILIC-MS/MS with an Agilent 6470 QQQ mass spectrometer. Chromatographic separation was achieved on a Waters XBridge BEH Amide column (2.1 × 150 mm, 2.5 μm) maintained at 40 °C. The mobile phase comprised (A) 10 mM ammonium acetate (pH 9.0) and (B) acetonitrile. The gradient elution program was set as follows: 90% to 60% B over 10 min at a flow rate of 0.2 ml/min, followed by a 7 min re-equilibration period. The injection volume ranged from 2 to 5 μL. MS detection used positive ESI mode with MRM acquisition. Source conditions were set as follows: gas temperature 300 °C, gas flow 5 L/min, nebulizer pressure 45 psi, and capillary voltage 3500 V. Quantitation was achieved using a stable isotope-labeled internal standard. Quality control samples were regularly interspersed throughout the analytical sequence. Data were processed with MassHunter Quant software.
Polar metabolites analysis by GC-MS
Following acyl-CoA analysis, samples underwent vacuum centrifugation to complete dryness. Derivatization was initiated by reconstituting residues in methoxylamine hydrochloride solution (10 mg/ml) with a 30 min incubation at room temperature. Subsequently, N-methyl-N-(tert-butyldimethylsilyl) trifluoroacetamide (MTBSTFA) was added, and samples were heated at 70 °C for 1 h. Chromatographic separation was achieved using an Agilent 5975 C GC-MS system equipped with a DB-5MS + DG capillary column (30 m × 250 μm × 0.25 μm). The injection port was maintained at 280 °C and operated in splitless mode with 1 μL injections. The temperature program commenced at 60 °C (1 min hold), followed by a 10 °C/min ramp to 320 °C, with a final 10 min hold. Helium carrier gas flow was optimized for metabolite separation. The mass spectrometer interface and quadrupole were maintained at 285 °C and 150 °C, respectively, with electron impact ionization at 70 eV. Mass spectrometric detection incorporated both full scan (50–700 m/z) and selected ion monitoring modes. Quantification was based on characteristic M-57 fragment ions using Agilent MassHunter Quantitation software, with metabolite identification verified against authentic standards through retention time and mass spectral matching.
Immunofluorescence
After washing with precooled PBS, cells were fixed in 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. They were then blocked with fetal bovine serum. Following removal of the blocking solution, cells were incubated with fluorescent-conjugated primary antibody overnight at 4 °C. The next day, cells were washed with PBS containing 0.1% Tween 20 and PBS, then incubated with secondary antibody for 1 h. Nuclei were stained with DAPI, and images were acquired using a laser scanning confocal microscope (Olympus FV3000).
Co-immunoprecipitation
For co-IP analysis, cells were lysed in 500 µl of lysis buffer (50 mM Tris-Cl, pH 7.4, 1 mM EDTA, 0.2% Triton X-100, protease inhibitor) on ice for 30 min. After centrifugation, 50 µl of the supernatant was kept as input for the western blot. The rest lysate was incubated with the corresponding antibody at 4 °C overnight, followed by incubation with protein A/G agarose beads (Santa Cruz Biotech., sc-2003) for 3 h. Then, the beads/protein complex was washed 5 times with cold lysis buffer and boiled with 40 µl of SDS loading buffer, and analyzed by western blot.
Measurement of de novo lipogenesis by 13C fructose gavage
To measure lipogenesis with 13C-fructose gavage, Kat2aHKO and Kat2afl/fl mice were fed an HFD diet for 11 days or an FPC diet for 9 days. After a 10 h fast, mice were refed for 2 hours, and received oral gavage (2 g/kg) with a 1:1 mixture of 12C D-glucose and 13C fructose. Mice were sacrificed, and livers were collected and snap-frozen in liquid nitrogen in the following day. Lipids were extracted from 20 mg of liver tissue through saponification and analyzed via LC-MS as previously described. Signal intensities were obtained for each fatty acid isotopomer (M + i), where M denotes the unlabeled parent ion and i represents the number of 13C atoms incorporated (ranging from 0 to n, n being the total carbon number of the fatty acid). The fractional enrichment was calculated as follows: for each fatty acid, the intensity of each isotopomer was multiplied by i, summed across all isotopomers, divided by n and the total ion count, and multiplied by 100 to obtain percentage enrichment.
Measurement of de novo lipogenesis by 2H2O injection
To measure lipogenesis with 13C-fructose gavage, Kat2aHKO and Kat2afl/fl mice were fed an HFD diet for 11 days or an FPC diet for 9 days. Following the feeding period, mice received an intraperitoneal injection (30 µL/g) of 99.9% ²H₂O (Sigma-Aldrich, 151882) in 0.9% NaCl. Five hours post-injection, the mice were euthanized, and the livers were collected and snap-frozen in liquid nitrogen. Lipids were extracted from 20 mg of frozen liver tissue via saponification and analyzed by LC-MS as described. Ion counts were acquired for each fatty acid isotopomer (M + i), where M represents the unlabeled parent ion and i corresponds to the number of incorporated deuterium atoms (ranging from 0 to n, n being the total number of hydrogen atoms in the fatty acid eligible for labeling). Deuterium enrichment was calculated as follows: for each fatty acid, the intensity of each isotopomer was multiplied by i, summed over all isotopomers, divided by n and the total ion count, and multiplied by 100 to obtain percentage enrichment.
CPTH2 treatment
Male C57BL/6 J wild-type mice (8 weeks old) were fed 4 weeks of HFD diet or 10 weeks of FPC diet. Then mice were treated with vehicle A (10% DMSO, 40% PEG3000, 5% Tween-80, 45% saline, v/v) or CPTH2 (20 mg/kg) in vehicle A by intragastric gavage every day, along with HFD fed. After another 6 weeks, mice were euthanized for analysis.
CPTH2 and T0901317 treatment
Male C57BL/6 J wild-type mice (8 weeks old) were fed 6 weeks of HFD. Then mice were treated with vehicle A/ T0901317 (0.5 mg/kg)/ CPTH2 (20 mg/kg) or T0901317 and CPTH2 in the vehicle by intragastric gavage every day, along with HFD fed. After another 2 weeks, mice were euthanized for analysis.
Histological analysis of liver
Livers were fixed in 4% paraformaldehyde at 4 °C overnight, embedded in paraffin wax, and sectioned into 5 μm thickness slices. Liver sections were stained with hematoxylin and eosin (H&E) staining to estimate morphology. Liver sections were stained by oil red O and counterstained with hematoxylin to evaluate lipid accumulation. Liver sections were stained by Sirius Red to assess liver fibrosis. The indicated antibodies were applied for Immunohistochemistry (IHC) staining. The sections were observed and captured using an upright fluorescence microscope (Olympus, BX53, Tokyo, Japan). The lipid drops were quantified by ImageJ (Version 1.5a).
Materials
Lipofectamine 3000 (L3000008) and BODIPY493/503 (D3922) were purchased from Thermo Fisher Scientific. Nile Red (SS1956) was purchased from BIOFUNT. Bovine Serum Albumin (BSA) (B2064), palmitic acid (PA) (P5585), oleic acid (OA) (O1008), puromycin (P7255), compactin (1443216), 2H2O (151882), Oil red O (O0625), and Triton X-100 (T8787) were purchased from Sigma-Aldrich. Simple ChIP Plus Sonication Chromatin IP Kit (56383S) was purchased from Cell Signaling Technology. T0901317 (HY-10626), GW3965 (HY-10627), and CPTH2 (HY-W013274) were purchased from MedChemExpress. Anti-mouse horseradish peroxidase (HRP) (A0216), anti-rabbit horseradish peroxidase (HRP) (A0208), BCA protein concentration assay kit (P0010), MTT (C0009S), reporter gene cell lysis buffer (RG126M), SDS (ST626), SDS-PAGE Sample Loading Buffer (5X) (P0015L) were purchased from Beyotime. Protease inhibitors (04693124001) and Phosphatase inhibitors (4906845001) were purchased from Roche. Nitrocellulose (NC) membrane (HATF00010) was purchased from Millipore. Tris-HCl buffer (T301502) and mevalonate (D304342) were purchased from Aladdin. 4% paraformaldehyde (PFA) (BL539A) was purchased from Biosharp. TG (A110-1-1), TC (A111-1-1), HDL-c (A112-1-1), and LDL-c (A113-1-1) assay kits were purchased from Nanjing Jiancheng Bioengineering Institute. HFD diets (D12492), HFrD diets (D02022704K) and AMLN diets (D09100310) were purchased from Research Diets. FPC diets (TD190142) were purchased from Teklad. HFMRCD diets (XTMRCD-60) and normal diets (1010039) were purchased from Jiangsu Synergetic Biology Co., Ltd. RNA-easy isolated reagent (R701), HiScript Reverse Transcriptase kit (R123-01), Dual Luciferase Reporter Assay Kit (DL101-01), and qPCR SYBR Green Master Mix (Q111-02) were purchased from Vazyme. Protein A/G agarose bead (sc-2003) was purchased from Santa Cruz Biotech. The mixture of 12C D-glucose and 13C fructose (FRU-011) was purchased from Omicron.
Primary antibodies
Anti-Flag (ab205606, diluted at 1:5000 for WB, IP: 0.5-4.0 μg for 1.0-3.0 mg of total protein lysate), anti-LXRα (ab41902, diluted at 1:1000 for WB, diluted at 1:100 for IF), and anti-SREBP1 (ab3259, diluted at 1:1000 for WB) antibodies were purchased from Abcam. Anti-GAPDH (AF0006, diluted at 1:10000 for WB) antibody was purchased from Beyotime. Anti-H3K14ac (7627 T, diluted at 1:1000 for WB) and anti-H3K9ac (9649S, diluted at 1:1000 for WB) antibodies were purchased from Cell Signaling Technology. Anti-PCAF (A22719, diluted at 1:1000 for WB), anti-GCN5 (A2224, diluted at 1:2000 for WB), and anti-Myc (AE010, diluted at 1:2000 for WB, IP: 1.0-4.0 μg for 1.0-3.0 mg of total protein lysate) antibodies were purchased from Abclonal. Anti-p-IκBα (WL02495, diluted at 1:500 for WB), anti-IκBα (WL01936, diluted at 1:1000 for WB), anti-p-p65 (WL02169, diluted at 1:1000 for WB), anti-p65 (WL01273b), anti-p-SMAD2/3 (WL02305, diluted at 1:500 for WB), anti-SMAD2/3 (WL01520, diluted at 1:500 for WB), anti-Collagen I (WL0088, diluted at 1:500 for WB), and anti-α-SMA (WL02510, diluted at 1:1000 for WB) antibodies were purchased from Wanleibio. Alexa Fluor® 488 AffiniPure® Alpaca Anti-Rabbit IgG (H + L) (611-545-215, diluted at 1:500 for IF) and Alexa Fluor® 594 AffiniPure® Alpaca Anti-Mouse IgG (H + L) (615-585-214, diluted at 1:500 for IF) antibodies were purchased from Jackson ImmunoResearch.
Plasmids
pCMV3-C-Flag-p-SREBP1c, pCMV3-C-Flag-m-SREBP1c, pCMV3-C-HA-RXRα, pCMV3-C-Flag-GCN5, pCMV3-C-Myc-GCN5, pCMV3-C-Flag-GCN5 (E575Q), pCMV3-C-Myc-LXRα, pCMV3-C-Myc-LXRα (1-163 aa), pCMV3-C-Myc-LXRα (164-326 aa), pCMV3-C-Myc-LXRα (327-447 aa), pCMV3-C-Flag-GCN5 (1-407 aa), pCMV3-C-Flag-GCN5 (408-656 aa), pCMV3-C-Flag-GCN5 (657-837 aa) plasmids were constructed by gene synthesis and site-directed mutagenesis.
Culture medium
Medium A was prepared using DMEM (Keygen, KGL1206) supplemented with 10% fetal bovine serum (Gibco, A5670701), 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate. Medium B consisted of an equal-volume mixture of Ham’s F-12K (Gibco, 21127030) and DMEM, supplemented with 5% LPDS (Kalen Biomedical, 880100), 10 μM compactin (Sigma-Aldrich, 1443216), and 50 μM mevalonate (Aladdin, D304342).
Quantification and statistical analysis
Data were analyzed using GraphPad Prism (Version 8). All data are represented as mean ± SEM. Statistical significance was analyzed by the Student’s t test between the two groups. When comparison between three or more groups, one-way ANOVA or two-way ANOVA was used. The D’Agostino-Pearson normal test was used to determine whether the data conformed to normal distribution.
Ethics statement
The human study was approved by the Ethics Committee of the Changzhou No. 2 People’s Hospital and followed the ethical guideline Declaration of Helsinki. Written informed consent was provided by each subject ([2023]KY124-01). Informed written consent was obtained from all participants involved in the research. All animal experiments and care were approved by the Animal Ethics Committee of China Pharmaceutical University (2024-06-017).
Data availability
The RNA-seq data of WT and KAT2A-deficient hepatocytes generated in this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) under accession code GSE279791. RNA-seq data of Kat2aHKO and Kat2afl/fl mouse livers have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2025) at the National Genomics Data Center (Nucleic Acids Res 2025), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences, under accession code CRA033103. ChIP-seq datasets used in this study are available from the GEO database (GCN5 ChIP-seq: GSM1003804, GSE94229; H3K9ac ChIP-seq: GSM1000141, and GSM918712. This paper does not report original code. Source data values, including uncropped immunoblots and gels, are provided with this paper as Source Data files. All other data supporting the findings of this study are available within the paper and its Supplementary Information. Source data are provided in this paper.
Code availability
No custom code was used in this study.
References
Hsu, C. L. & Loomba, R. From NAFLD to MASLD: implications of the new nomenclature for preclinical and clinical research. Nat. Metab. 6, 600–602 (2024).
Riazi, K. et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 7, 851–861 (2022).
Harris, E. FDA Okays first drug for scarring from fatty liver disease. JAMA 331, 1439 (2024).
Wei, S., Wang, L., Evans, P. C. & Xu, S. NAFLD and NASH: etiology, targets and emerging therapies. Drug Discov. Today 29, 103910 (2024).
Farrell, G. C. & Larter, C. Z. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 43, S99–S112 (2006).
Haider, D. G. et al. Increased plasma visfatin concentrations in morbidly obese subjects are reduced after gastric banding. J. Clin. Endocrinol. Metab. 91, 1578–1581 (2006).
Buzzetti, E., Pinzani, M. & Tsochatzis, E. A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 65, 1038–1048 (2016).
Anstee, Q. M. & Day, C. P. The genetics of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 10, 645–655 (2013).
Sodum, N., Kumar, G., Bojja, S. L., Kumar, N. & Rao, C. M. Epigenetics in NAFLD/NASH: Targets and therapy. Pharmacol. Res. 167, 105484 (2021).
Eslam, M., Valenti, L. & Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 68, 268–279 (2018).
Jonas, W. & Schurmann, A. Genetic and epigenetic factors determining NAFLD risk. Mol. Metab. 50, 101111 (2021).
Loomba, R., Friedman, S. L. & Shulman, G. I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184, 2537–2564 (2021).
Aagaard-Tillery, K. M. et al. Developmental origins of disease and determinants of chromatin structure: maternal diet modifies the primate fetal epigenome. J. Mol. Endocrinol. 41, 91–102 (2008).
Brownell, J. E. & Allis, C. D. Special HATs for special occasions: linking histone acetylation to chromatin assembly and gene activation. Curr. Opin. Genet. Dev. 6, 176–184 (1996).
Haque, M. E. et al. The GCN5: its biological functions and therapeutic potentials. Clin. Sci. 135, 231–257 (2021).
Jin, Q. et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 30, 249–262 (2011).
Xiao, H. T., Jin, J. & Zheng, Z. G. Emerging role of GCN5 in human diseases and its therapeutic potential. Biomed. Pharmacother. 165, 114835 (2023).
Kumar, A. et al. Designing novel inhibitors against histone acetyltransferase (HAT: GCN5) of Plasmodium falciparum. Eur. J. Med. Chem. 138, 26–37 (2017).
Xiong, H. et al. Discovery of 1,8-acridinedione derivatives as novel GCN5 inhibitors via high throughput screening. Eur. J. Med. Chem. 151, 740–751 (2018).
Dominy, J. E. Jr. et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol. Cell 48, 900–913 (2012).
Lerin, C. et al. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 3, 429–438 (2006).
Sakai, M. et al. The GCN5-CITED2-PKA signalling module controls hepatic glucose metabolism through a cAMP-induced substrate switch. Nat. Commun. 7, 13147 (2016).
Tavares, C. D. et al. The methionine transamination pathway controls hepatic glucose metabolism through regulation of the GCN5 acetyltransferase and the PGC-1alpha transcriptional coactivator. J. Biol. Chem. 291, 10635–10645 (2016).
Im, Y. R. et al. A systematic review of animal models of NAFLD finds high-fat, high-fructose diets most closely resemble human NAFLD. Hepatology 74, 1884–1901 (2021).
Wang, Y., Viscarra, J., Kim, S. J. & Sul, H. S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 16, 678–689 (2015).
Ferre, P., Phan, F. & Foufelle, F. SREBP-1c and lipogenesis in the liver: an update1. Biochem. J. 478, 3723–3739 (2021).
Repa, J. J. et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 14, 2819–2830 (2000).
Leik, C. E. et al. GW3965, a synthetic liver X receptor (LXR) agonist, reduces angiotensin II-mediated pressor responses in Sprague-Dawley rats. Br. J. Pharmacol. 151, 450–456 (2007).
Joseph, S. B. et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl. Acad. Sci. USA 99, 7604–7609 (2002).
Kirchgessner, T. G. et al. Beneficial and adverse effects of an LXR agonist on human lipid and lipoprotein metabolism and circulating neutrophils. Cell Metab. 24, 223–233 (2016).
Aseem, S. O. et al. Epigenomic evaluation of cholangiocyte transforming growth factor-beta signaling identifies a selective role for histone 3 lysine 9 acetylation in biliary fibrosis. Gastroenterology 160, 889–905.e810 (2021).
Fournier, M. et al. KAT2A/KAT2B-targeted acetylome reveals a role for PLK4 acetylation in preventing centrosome amplification. Nat. Commun. 7, 13227 (2016).
Gallage, S. et al. A researcher’s guide to preclinical mouse NASH models. Nat. Metab. 4, 1632–1649 (2022).
Hebbard, L. & George, J. Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 8, 35–44 (2011).
Moslehi, A. & Hamidi-Zad, Z. Role of SREBPs in liver diseases: a mini-review. J. Clin. Transl. Hepatol. 6, 332–338 (2018).
Shimano, H. & Sato, R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat. Rev. Endocrinol. 13, 710–730 (2017).
Shimomura, I., Bashmakov, Y. & Horton, J. D. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J. Biol. Chem. 274, 30028–30032 (1999).
Moon, Y. A. et al. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 15, 240–246 (2012).
Adams, C. M. et al. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J. Biol. Chem. 279, 52772–52780 (2004).
Tang, J. J. et al. Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab. 13, 44–56 (2011).
Zheng, Z. G. et al. Discovery of a potent SCAP degrader that ameliorates HFD-induced obesity, hyperlipidemia and insulin resistance via an autophagy-independent lysosomal pathway. Autophagy 17, 1592–1613 (2021).
Im, S. S. & Osborne, T. F. Liver x receptors in atherosclerosis and inflammation. Circ. Res. 108, 996–1001 (2011).
Zhang, Y. et al. Liver LXRalpha expression is crucial for whole body cholesterol homeostasis and reverse cholesterol transport in mice. J. Clin. Invest. 122, 1688–1699 (2012).
Joseph, S. B., Castrillo, A., Laffitte, B. A., Mangelsdorf, D. J. & Tontonoz, P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 9, 213–219 (2003).
Higuchi, N. et al. Liver X receptor in cooperation with SREBP-1c is a major lipid synthesis regulator in nonalcoholic fatty liver disease. Hepatol. Res. 38, 1122–1129 (2008).
Liang, G. et al. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J. Biol. Chem. 277, 9520–9528 (2002).
Wang, B. & Tontonoz, P. Liver X receptors in lipid signalling and membrane homeostasis. Nat. Rev. Endocrinol. 14, 452–463 (2018).
Kim, G. H. et al. Hepatic TRAP80 selectively regulates lipogenic activity of liver X receptor. J. Clin. Invest. 125, 183–193 (2015).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82574477), the Natural Science Foundation of Jiangsu Province (BK20250204), the Jiangsu Provincial Traditional Chinese Medicine Science and Technology Development Plan (QN202426), Jiangsu Province “333 High-level Talents Training Project” [(2024)3-0189], Youth Talent Support Project of the Jiangsu Association for Science and Technology (TJ-2023-053) to Z.-G.Z., the Fundamental Research Program of Shanxi Province (202403021221211), the Research Project Supported by the Shanxi Scholarship Council of China (2023-158) to L.-N.L., and the Chief Scientist Research Project of Hubei Shizhen Laboratory (HSL2024SX0005) to H.Y.
Author information
Authors and Affiliations
Contributions
Study design: Z.-G.Z., L.-N. L., H.Y., and P.L. Writing—original draft: Z.-G.Z., H.-T.X., P.S., and J.J. Writing—review and editing: Z.-G.Z., H.-T.X., P.S., and J.J. Investigation: Z.-G.Z., H.-T.X., J.J., P.S., Y.-D. W., Y.-Y.X., H.W., X.-X.S., Q.-F.G., J.H., Y.-R.W., J.Z., B.-Y.C., and P.Z. Validation: Z.-G.Z., H.-T.X., P.S., J.J., Y.-D.W., Y.-Y.X., H.W., X.-X.S., Q.-F.G., J.H., Y.-R.W., J.Z., B.-Y.C., and P.Z. Methodology: Z.-G.Z., H.-T.X., P.S., J.J., Y.-D.W., Y.-Y.X., X.-X.S., H.W., J.H., and Y.-R.W. Data interpretation: Z.-G.Z., L.-N.L., and P.L. Supervision: Z.-G.Z.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jianhong Ching, who co-reviewed with Ming Shen Tham and the other anonymous reviewer(s) for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiao, HT., Song, P., Jin, J. et al. GCN5 drives MASLD progression through LXRα/SREBP1c signaling pathway–mediated de novo lipogenesis. Nat Commun 17, 2821 (2026). https://doi.org/10.1038/s41467-026-69736-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69736-y
