
Abstract
Allogeneic stem cell transplantation (allo-HSCT) has recently been approved as standard therapy for transfusion-dependent thalassemia (TDT) but remains limited to the use of HLA-matched sibling donors (MSDs), due to a lack of large-scale prospective studies evaluating the use of grafts from alternative donors. Here, we report the results of a non-randomised, interventional, phase 4 clinical trial evaluating allo-HSCT from alternative donors for the treatment of TDT. A total of 823 patients with TDT were transplanted with grafts from MSDs (n = 331) or alternative donors, including matched unrelated donors (MUDs; n = 352) and haploidentical related donors (Haplos; n = 140). Conditioning was with busulfan, cyclophosphamide, fludarabine and anti-thymocyte globulin. Graft-versus-host disease (GvHD) prophylaxis was cyclosporine, methotrexate (MTX) and mycophenolate mofetil (MMF) for recipients of MSDs and tacrolimus, MTX, MMF for others. The primary endpoints were 2-year overall survival (OS) and event-free survival (EFS). Two-year OS for MSDs, MUDs, Haplos was 97.2% (95% CI, 95.4-99.0), 93.1% (90.5-95.9) and 95.4% (91.9-99.1); EFS was 97.2% (95.4-99.0), 92.9% (90.1-95.7) and 94.7% (90.9-98.6); GvHD-free, relapse-free survival (GRFS) was 91.4% (88.4-94.6), 77.0% (72.6-82.7) and 75.6% (68.5-83.4) respectively. Two-year OS and EFS for MUDs were lower than those for MSDs (both P < 0.05) and were not significantly different with those for Haplos (both P > 0.05). Transplant-related mortality was 4.4% (3.0-5.9) and graft failure rate was 0.5%. The incidence of grades 2-4 acute GvHD and moderate-severe chronic GvHD from alternative donors were higher than those from MSDs (28.9% [24.7-33.2] vs 7.5% [4.9-10.7], P < 0.001; 12.3% [9.2-15.7] vs 5.0% [2.9-7.9], P < 0.01). In summary, these findings may help expand the donor pool for patients with TDT lacking MSDs (ClinicalTrials.gov: NCT04009525).
Similar content being viewed by others

Introduction
The hemoglobinopathies thalassemia and sickle cell disease (SCD) are common genetic diseases. Approximately 300,000 SCD infants and 40,000 β-thalassemia infants are born annually1,2 and the severe variants consume huge health-care resources, related to life-long iron chelation and blood transfusions. Over 80% of these patients live in low or middle-income countries, and less than 15% of patients with transfusion-dependent thalassemia (TDT) live beyond 35 years due to inadequate treatments3. Thus, curative strategies, i.e., allogeneic hemopoietic stem cell transplantation (allo-HSCT) and/or gene therapy are needed.
Allo-HSCT with HLA-matched sibling donors (MSDs) has been standard therapy for TDT patients, event-free survival (EFS) reaching 80–90%4. Unfortunately, MSDs could be found for only 25–30% of TDT patients. Alternative donors (matched unrelated donors [MUDs] and haploidentical related donors [Haplos]) have been explored5,6, but allo-HSCT with alternative donors is characterized by high rates of graft failure (GF), and graft-versus-host disease (GvHD), yielding inferior overall survival (OS) and EFS compared with MSDs7,8. Although alternative donor outcomes have been comparable with MSDs in hematological malignancies9, their use for TDT is still investigational, and a lack of large-scale prospective studies prevents the application of alternative donor allo-HSCT for TDT.
We previously reported a 3-year EFS of 97% after allo-HSCT for 184 TDT patients with MSDs, using the protocol GX-07-TM10. With support from the Chinese Red Cross and the Guangxi local government, we launched the project Severe Thalassemia Patients Treatment Plan in 2019, aiming to promote allo-HSCT for TDT and transplant 800 patients within three years. We designed this prospective, multi-center trial to confirm our previous results and to assess outcomes using alternative donors.
Results
Patients
Between July 5, 2019, and July 31, 2023, 841 patients from 16 Chinese centers were screened (Fig. 1, Supplementary Table 1). Eighteen patients met the exit criteria, and 823 patients were eligible for the final analysis, including 787 with β-TDT and 36 with α-TDT. Among them, 331 received transplants from MSDs and 492 from alternative donors, comprising 352 MUDs and 140 Haplos. Median follow-up after transplantation was 23 months (IQR 13–36, range 3–52). The last follow-up was October 31, 2023. Patients’ baseline characteristics and outcomes are listed in Table 1, Supplementary Table 2 and Table 2.

Eight hundred and forty-one patients were screened. Eighteen patients met the exit criteria, and 823 patients were eligible for the final analysis.
Engraftment and graft failure
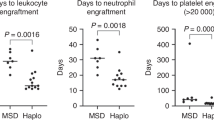
The median time to neutrophil engraftment was 12 days (IQR 11–14), 12days (IQR 11–13) and 12 days (IQR 11–14) in MSDs, MUDs and Haplos, respectively. The median time to platelet engraftment was 14 days (IQR 12–20), 12 days (IQR 11–15) and 14 days (IQR 12–17) in the three type donors (Table 2). Neutrophil engraftment did not differ between MSDs, MUDs and Haplos. Compared with MSDs and Haplos, the median time to platelet engraftment was less in MUDs (p < 0.01) (Table 2). Primary engraftment was achieved in 819 patients, 4 patients experienced GF (GF was 0.5%; 1 primary GF, 3 secondary GF, 2 died and 2 became transfusion dependent).
Primary endpoints
The primary endpoint was achieved. The observed 2-year OS was 95.2%, significantly exceeding the predefined reference value of 91.5% (P < 0.001). Two-year OS and EFS were 95.2% (95% confidence interval (CI), 93.7–96.7), and 94.9% (93.4–96.5) for all patients, with both 97.2% (95.4–99.0) for MSDs, 93.1% (90.5–95.9) and 92.9% (90.1–95.7) for MUDs, 95.4% (91.9–99.1) and 94.7% (90.9–98.6) for Haplos. The 2-year GvHD-free, relapse-free survival (GRFS) was 82.7% (80.0–85.4) for all patients, 91.4% (88.4–94.6) for MSDs, and 77.0% (72.6–81.7) for MUDs and 75.6% (68.5–83.4) for Haplos (Table 2, Fig. 2). In a post hoc analysis, patients with MUDs and Haplos had slightly lower OS and EFS, and higher transplant-related mortality (TRM) compared with MSDs recipients (Table 2). There was no center-dependent effect observed for these results.

Kaplan–Meier curves for overall survival (A), event-free survival (B), GvHD and relapse-free survival (C) and transplant related mortality (D). Time-to-event rates were estimated using the Kaplan–Meier method, and hazard ratios were estimated using the two-sided Cox proportional hazards model with centers as a random-effect. OS Overall survival, EFS Event free survival, GRFS Graft versus host disease and relapse free survival, TRM Transplant related mortality.
In univariate analysis, OS and EFS were significantly lower for MUDs compared to MSDs (hazard ratio (HR) 2.74, 95% CI 1.25–5.99; P = 0.012 and HR 2.90, 95% CI 1.33–6.32; P = 0.007, respectively), for splenectomy group compared to non-splenectomy (3.35, 1.66–6.76; P = 0.001 and 3.12, 1.56–6.24; P = 0.001), for patients older than 15 years (4.86, 1.91–12.36; P = 0.001 and 4.48, 1.79–11.23; P = 0.001), and for patients with grades 2–4 and 3–4 acute GvHD (aGvHD) compared to those without aGvHD (5.59, 2.91–10.72; P < 0.001 and 5.08, 2.69–9.59; P < 0.001 for 2–4 aGvHD, respectively; 10.39, 5.39–20.04; P < 0.001 and 9.59, 5.00–118.16; P < 0.001 for 3–4 aGvHD, respectively). OS and EFS were significantly lower in patients with infections (13.30, 1.73–101.93; P = 0.013 and 7.68, 1.70–34.68; P = 0.008, respectively), cytomegalovirus (CMV) reactivation (2.45, 1.27–4.74; P = 0.008 and 2.54, 1.32–4.87; P = 0.005, respectively) and hemorrhagic cystitis (HC) (2.46, 1.29–4.69; P = 0.006 and 2.85, 1.52–5.35; P = 0.001, respectively), compared to unaffected patients. Additionally, EFS was also lower in patients with veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS) (3.62, 1.29–10.18; P = 0.015) compared to those without VOD/SOS (Supplementary Table 3). The total number of CD34+ cells infused showed a protective effect; thus, OS were improved with higher CD34+ cell doses infused (0.89, 0.82–0.98; P = 0.013, Supplementary Table 3). Factors that have a negative impact on OS and EFS are found in Supplementary Table 3. Post hoc multivariable analysis of risk factors for OS and EFS showed that grades 2–4 aGvHD (HR 5.24, 95% CI 2.70–10.20 and 4.56, 2.40–8.69), age >15 years (3.07, 1.06–8.88 and 2.88, 1.01–8.23), and previous splenectomy (3.36, 1.49–7.54 and 3.15, 1.41–7.02) were significant negative predictors for OS and EFS compared to patients without these risk factors (Fig. 3).

Statistical significance was assessed using two-sided multivariate COX regression analysis. The error bars show hazard ratios with 95% confidence interval. OS Overall survival, HSCT hematopoietic stem cell transplantation, aGVHD acute graft versus host disease, EFS Event free survival, G Granulocyte colony-stimulating factor, BM Bone marrow, PBSCs Peripheral blood stem cells, CB Cord blood, cGVHD chronic graft versus host disease. a Forty-eight patients received stem cell type included 35 cases of G-BM, 10 cases of G-BM & PBSCs & CB, 2 cases of G-PBSCs & CB and 1case of CB. b Ninety patients in total, including 42 cases of BM & CB, 35 cases of G-BM, 10 cases of G-BM & PBSCs & CB, 2 cases of G-PBSCs & CB and 1 case of CB.
Secondary endpoints
The cumulative incidence of grades 2–4, and 3–4 aGvHD was 7.5% (95% CI, 4.9–10.7) and 1.9% (0.8–3.9) in patients for MSDs, 27.6% (22.8–32.6) and 14.6% (10.8–18.9) for MUDs and 32.3% (24.2–40.6) and 14.7% (8.9–21.9) for Haplos. Two-year cumulative incidence of chronic GvHD (cGvHD) and moderate-severe cGvHD was 8.5% (5.7–12.0) and 5.0% (2.9–7.9) for MSDs, 19.1% (14.8–23.9) and 11.2% (7.9–15.2) for MUDs, and 22.6% (15.2–30.8) and 15.1% (8.9–22.9) for Haplos. Among the 668 patients with follow-up longer than one year, cGvHD occurred in 8.9% (5.9–12.5) of MSDs and 18.8% (14.3–23.8) for MUDs and 23.1% (15.5–31.7) for Haplos; moderate–severe cGvHD occurred in 5.4% (3.2–8.5) for MSDs, 10.7% (7.3–14.8) for MUDs and 14.7% (8.4–22.7) for Haplos, respectively (Supplementary Table 4). These analyses show a consistent pattern of higher cGvHD rates after alternative donors (MUDs and Haplos) than after MSDs (Table 2, Supplementary Fig. 1). Of the 112 cases of cGvHD were recorded, 56% had prior aGvHD. The organ scores of cGvHD were shown in Supplementary Table 5.
In univariable analysis, MUDs (HR 4.09, 95% CI 2.62–6.37; HR 7.00, 95% CI 3.15–15.60) and Haplos (5.03, 3.06–8.27; 7.35, 3.05–17.70) increased risk for grades 2–4 and 3–4 aGvHD compared with MSDs. For moderate-severe cGvHD, the risks were significantly higher for patients with MUDs (2.21, 1.24–3.94) and Haplos (2.58, 1.30–5.10), compared to MSDs. History of aGvHD markedly influenced cGvHD risk (4.42, 2.74–7.14 for grades 2–4 aGvHD; 8.03, 4.74–13.60 for grades 3–4 aGvHD) (Supplementary Tables 6, 7). The proportion of patients with moderate-severe cGvHD at 2 years is shown in Table 2.
Post hoc multivariable analysis revealed that the risk of grades 2–4 and 3–4 aGvHD was significantly higher in patients with MUDs (HR 3.38, 95% CI 1.74–6.58 for grades 2–4; 7.37, 2.12–25.56 for grades 3–4) and Haplos (4.89, 2.88–8.30 for grades 2–4; 7.59, 3.07–18.79 for grades 3–4), compared to MSDs. Additionally, grades 2–4 aGvHD were associated with an increased risk of developing moderate-severe cGvHD (3.80, 2.29–6.32) (Fig. 3).
Safety
Infectious complications exhibited notable variations among donor types. Haplo-patients had higher incidences of pneumonia (48.6%), CMV (52.1%) and Epstein-Barr virus (EBV) (32.9%) reactivation compared with those having MSDs and MUDs. Moreover, the rates of bacterial and fungal infections, septicemia, and posterior reversible encephalopathy syndrome (PRES) were similar between groups. VOD/SOS, diagnosed in 27 patients (3.3%), without difference between the subgroups (Table 2). Hemorrhagic cystitis was observed in 190 (23.0%) patients, 48 (14.5%) were MSDs, 99 (28.1%) MUDs, and 43 (30.7%) Haplos, respectively, for all SAEs see Supplementary Table 8.
The overall 2-year TRM was 4.4% (95% CI, 3.0–5.9) for all patients, 2.5% (0.8–4.2) for MSDs, 6.3% (3.7–9.9) for MUDs, and 4.6% (0.9–8.1%) for Haplos (Table 2, Fig. 2D). The main causes of death in 38 cases were infections (two patients died from the COVID-19 infection), GvHD and intracranial hemorrhage (Supplementary Fig. 2). Fifty-four (6.6%) patients developed mixed chimerism (MC) at a median of 133 (range 15–574) days. The 12.4% MC in patients with MSDs was higher than 3.1% in MUDs and 1.4% in Haplos (Table 2 and Supplementary Table 9).
There was no significant difference in outcome for patients ages 2–7 and 7–15 years, but patients over 15 years exhibited lower two-year OS, EFS, and higher TRM than the younger cohort(s) (Supplementary Fig. 3). After adjustment for age and hepatomegaly (>5 cm), the 92 splenectomised patients still had inferior OS, EFS, and increased TRM, although they had faster neutrophil and platelet engraftment compared with non-splenectomised patients. The infection-related mortality in splenectomised group was higher than that in non-splenectomised patients (7.6% vs 2.5%, P = 0.010). (Supplementary Table 10).
Discussion
This is to date the largest prospective, multicenter allo-HSCT trial for TDT, using both MSDs and alternative donors. The OS and EFS in 331 patients with MSDs were both 97%, consistent with our previous study10. To our knowledge, these results are among the best outcomes reported for allo-HSCT in TDT. It confirms that the GX-07-TM protocol is a valid option for TDT patients using MSDs. Moreover, the OS and EFS in 492 patients with alternative donors were 94% and 93%, respectively. Although the 2-year OS and EFS of patients with alternative donors were statistically lower than those with MSDs, this small difference is still encouraging compared with previous reports8,11,12. In most previous studies, the EFS was about 70–89%8,11,12. Recently, some small-scale studies reported the EFS of patients with Haplos reached 95–96%, using published protocols5,6. Based on these results, we propose that alternative donors can be used as a frontline option for TDT patients at centers with expertise in alternative donor transplantation when lacking access to MSDs.
In this trial, the 2-year OS and EFS were both 93% for patients with MUDs, and 95% with Haplos. The 2-year OS, EFS, GRFS, TRM, GF rate and incidences of GvHD were not significantly different between the patients with MUDs and Haplos. The results showed similar outcomes between these two groups. Our data suggest that Haplos can provide a suitable alternative to MUDs. Also, for all 823 patients, 2-year TRM was only 4.4% and the GF rate was 0.5%, with 2-year OS and EFS being 95%. The trial confirmed that allo-HSCT is a safe, curative therapy for TDT patients.
Graft failure is a known problem after allo-HSCT for hemoglobinopathies, especially when using alternative donors. It may be partly due to allo-immunization following long-term transfusions before transplantation, resulting in high titers of donor specific anti-HLA-antibodies. The GF rate in TDT patients has been reported to be as high as 6.2–8.6% with MSDs, and 7.3–21.8% with alternative donors, respectively7,8, contrasting our GF rate of 0.5%. The following factors may account for this: ⑴ The GX-07-TM protocol is an intensive myeloablative/immunosuppressive regimen administering busulfan (Bu), cyclophosphamide (Cy), fludarabine (Flu) and anti-thymocyte globulin (ATG). In our previous study, the GF rate was 0.6%10. GF is likely associated with residual host T-cells. Thus, a reduced-intensity conditioning regimen will be associated with a higher GF rate13, an intensified conditioning regimen ensures the myeloablative/immunosuppressive properties and assist in engraftment, lowering the GF risk14. ⑵ ATG favored a reduced GF risk and is accepted as part of standard conditioning regimens for TDT15,16. The use of T-replete grafts with ATG and granulocyte colony stimulating factor (G-CSF) in the Beijing Protocol improved the outcomes of allo-HSCT from Haplos17,18. ⑶ High CD34+ cell doses and mixed G-CSF mobilized bone marrow and peripheral blood stem cells (G-BM& PBSCs) and G-PBSCs as stem cell sources may assist in reducing the GF risk10,15. A low stem cell dose has been associated with a high GF risk, a high CD34+ cell dose was associated with earlier engraftment, lower GF risk15. The GF rate in patients received CD34 + stem cell dose <3 × 106/kg was 12%,which was significantly higher than the 1–7% seen in patients who received higher cell doses(>3 × 106/kg)15. However, a high CD34+ cell dose may increase the GvHD risk. This needs to be balanced against GF risk. The median CD34+ cell dose in this trial was 9 × 106/kg. We recommend a CD34+ cell dose in allo-HSCT for TDT patients in the range of 6–12 × 106/kg. ⑷ Close monitoring of chimerism and early intervention for mixed/decreasing chimerism may also reduce secondary GF10. The measures described appear to overcome the GF risk after allo-HSCT for TDT, especially benefiting patients with alternative donors. In addition, late graft failure in TDT often reflects autologous reconstitution from insufficient myeloablation rather than immune rejection. Our regimen’s strong myeloablative component, together with high CD34+ doses and early chimerism-guided interventions, likely minimized this risk and contributed to the exceptionally low GF rate.
The incidence of grades 2–4 and 3–4 aGvHD from MSDs were only 7.5% and 1.9%, respectively. The incidence of grade 2–4 aGvHD in previous reports were about 11.8–20%7,8,11. The incidences of cGvHD and moderate-severe cGvHD from MSDs were 8.5% and 5.0%, respectively, lower than previously reported7,8,11. It might be attributed to our GvHD prophylaxis regimen and to our use of post-transplant immunosuppression for over one year. Using the same GvHD prophylaxis regimen, a study reported the incidence of grades 2–4 and 3–4 aGvHD to be only 4.5% and 1.9% in aplastic anemia patients from MSDs19. In our opinion, the GvHD prophylaxis regimen of CsA, MTX, MMF and ATG or other T-cell depleting serotherapy could replace a more traditional GvHD prophylaxis regimen for patients with non-malignant disorders from MSDs, but it would probably be best to use a personalized approach, based on the risk for GvHD, GF, disease relapse, and the risk of infection.
Previous studies showed tacrolimus (Tac) as more effective than CsA for GvHD prophylaxis20,21. Thus, we used Tac instead of CsA in patients with alternative donors. The incidences of grades 2-4 aGvHD, cGvHD were 27.6%, 19.1% from MUDs, and 32.3%, 22.6% from Haplos, respectively. These results were similar to previous reports5,7,8, and higher than those with MSDs. In previous reports, the incidence of grade 2–4 aGvHD and cGvHD from alternative donors were about 23.5–42% and 8.3–45%, respectively5,7,8. Interestingly, both the incidence of aGvHD and cGvHD were similar between patients with MUDs and Haplos in this trial. We propose that the different stem cell sources may have balanced the HLA disparity between these two groups. In this trial, aGvHD was one of the main causes of death and a risk factor for cGvHD. The 2-year GRFS of patients with alternative donors was lower than patients with MSDs. Therefore, the GvHD prophylaxis regimen of Tac, MTX, MMF and ATG is still suboptimal in this subgroup. Optimizing GvHD prophylaxis regimens for patients receiving transplants from alternative donors remains essential to further improve outcomes. Recent studies have reported that could reduce the incidence of aGvHD and cGvHD and improve the outcomes of patients transplanted with Haplos, using the novel GvHD prophylaxis such as the PTCy combination with ATG or TCRα/β+/CD19+–T cell depletion6,22,23. In our center, we previously reported that adding CD25 antibody to our prophylaxis protocols (Tac, MTX, MMF and ATG) could reduce the incidence of aGvHD and cGvHD and increase the GRFS of patients with TDT from alternative donors24. A prospective multicenter randomized controlled trial to confirm these results is underway (NCT06657391).
The threshold age for optimal transplant outcomes in TDT patients has been reported to be around 14–15 years7,8,11. Our trial confirmed that age >15 years is an adverse factor. Some investigators reported an optimal age for allo-HSCT in TDT to be <7 years, younger patients having better outcomes8,11. However, the 2-year OS, EFS and TRM in this trial were similar for all patients up to 15 years old. The improved transplant protocol combined with optimization of the patient’s clinical condition before transplantation might raise the safe transplant age. Gene therapy is rapidly emerging as a potentially curative treatment option for patients with TDT and SCD25,26,27. Gene therapy may ultimately be preferred for older children or adults lacking MSDs. Recently, two gene therapeutic drugs, Zynteglo and Casgevy were approved by the US FDA for treating TDT patients. However, larger clinical trials are still needed before such treatment can be ascertained as safe and effective on a routine basis.
Splenomegaly and splenectomy in TDT patients are still common in resource-poor countries. Few studies, with small patient numbers, have reported on splenectomy before allo-HSCT, and the results are controversial28,29. Our findings indicate that splenectomy was associated with inferior overall outcomes, although engraftment of neutrophils and platelets occurred more rapidly in splenectomised patients. Infection-related mortality was significantly higher in the splenectomised group compared with non-splenectomised patients. In our experience, splenectomy before transplantation is best avoided. We recommend improving pre-HSCT management, such that regular blood transfusions are routinely combined with iron chelation, and implementing allo-HSCT earlier in the disease course.
Although an intensive conditioning regimen was used, no serious organ toxicities occurred. All patients were children or adolescents, and patients with preexisting severe organ damage were excluded from the trial. The incidence of VOD/SOS in this study (3.3%) was lower than that of our previous report (10.4%), which may be attributed to the use of intravenous Bu30 and our experience of identifying early symptoms of VOD/SOS and management of adverse drug reactions such as those with calcineurin inhibitors and azoles. We previously reported the quality of life for TDT patients after allo-HSCT using the GX-07-TM protocol. The results show that cGvHD was independently associated with lower health-related quality-of-life scores, and more than 4 years after transplantation, patients achieved a health-related quality of life equal to that of their healthy peers31. These results suggest the GX-07-TM protocol is well-tolerated and safe. In addition, measures including hydroxyurea and intensive iron chelation before transplantation, early diagnosis and management of transplant complications including GvHD, VOD/SOS and PRES, and potentially also early post-transplant initiation of iron chelation were valuable to improve outcomes. The experience of the Guangxi allo-HSCT thalassemia project can quickly increase the number of transplanted patients, which is important in limited-resource settings.
Limitations of the study include the non-randomized design, relatively short follow-up, which precludes full assessment of long-term engraftment, late complications, and secondary malignancies, though ongoing surveillance continues. Another limitation is that we did not monitor the intravenous Bu levels for most patients, as this was unavailable at most Chinese HSCT centers, although the Bu pharmacokinetic data in small number TDT patients in our center showed plasma concentrations within the effective myeloablative range. Previous studies indicate that intravenous Bu exposure is lower in Chinese patients32. Thus, the GX-07-TM protocol may require dose adjustment for different ethnic populations to ensure optimal exposure and safety. Despite these limitations, the prospective multi-center design and standardized treatment approach provide valuable real-world evidence on allo-HSCT outcomes in TDT across multiple donor types.
In conclusion, we good excellent 2-year OS and EFS after allo-HSCT in patients with TDT using MSDs or alternative donors. The outcomes were similar between patients with MUDs and Haplos. Allo-HSCT with alternative donors can be used as a frontline option for TDT patients lacking MSDs. An intensive myeloablative regimen and targeting a high CD34+ cell dose ameliorated the GF problem. GvHD prophylaxis with CsA, MTX, MMF and ATG effectively prevented GvHD in patients with MSDs, while patients receiving alternative donors would benefit from an improved GvHD-prophylaxis strategy. Age above 15 years, grades 2–4 aGvHD and splenectomy were identified as adverse factors. The GX-07-TM protocol is well-tolerated, cost-effective. Promoting allo-HSCT in countries with limited resources may help save the lives of patients with severe haemoglobinopathies and improve their quality of life.
Methods
Study design and participants
This was a prospective, multicenter clinical trial at sixteen hospitals in China (Supplementary Table 1). Inclusion criteria were: (1) diagnosis of TDT by hemoglobin electrophoresis, DNA analysis, and blood transfusion dependence; (2) age 2–20 years; (3) availability of matched sibling- or alternative donor; and (4) performance status (0–2, Karnofsky score >70%, or Lansky score ≥60% for children). Exclusion criteria: (1) cardiac ejection fraction <50%, severe cardiovascular/pulmonary disorders (PaO2 ≤ 60 mm Hg at rest, room air); (2) liver dysfunction (ALT or AST ≥ 4 times the upper normal limit [ULN] of institutional reference)3. creatinine ≥2 times the ULN or creatinine clearance <30 mL/min; (4) uncontrolled active infection; (5) CMV or EBV > 400 copies/mL in blood by quantitative PCR; (6) hepatitis C or -B positivity; (7) HIV positivity; (8) previous transplant; and (9) any condition deemed by the investigator to make the patient unsuitable for trial.
The medical ethics committee at each hospital approved the trial, which was conducted according to the Declaration of Helsinki. All patients and donors provided written informed consent/assent. Consent was obtained from the parents or legal guardians of all participating children prior to enrollment. For adolescents capable of providing assent, age-appropriate information was provided, and their assent was also obtained whenever applicable. The ethics committee reference number was [Medicine Ethics] 2019 No. (009). The study protocol and statistical analysis plan are included in Supplementary Data 1.
Procedures
Patients were assigned to groups with MSDs (10/10 or 9/10 at HLA-A, -B, -C, -DR, -DQ) and alternative donors (MUDs [10/10 or 9/10 matches] or Haplos) based on donor availability. MSDs were preferred, followed by MUDs. In the absence of these, Haplos were considered17.
All patients received the same conditioning regimen: IV busulfan (BUSULFEX™, 1 mg/kg i.v. 4× daily on days −9 to −6), cyclophosphamide (50 mg/kg i.v. once daily, days −5 to −2), fludarabine (50 mg/m² i.v. once daily from days −12 to −10), and anti-thymocyte globulin (Thymoglobulin™, 2.5 mg/kg i.v. once daily, days −4 to −1) (Supplementary Fig. 4). Everyone received 20–30 mg/kg hydroxyurea orally daily for 2–3 months before transplantation. Patients with MSDs and Haplos received G-BM & PBSCs, or BM and cord blood (CB) grafts33. Patients with MUDs received G-CSF mobilized PBSCs. For GvHD prophylaxis, patients with MSDs received CsA, MTX, and MMF10. Patients with alternative donors received Tac, 0.03 mg/kg/day), MTX10, and MMF (250 mg/day for 90 days).
Supportive care routines are outlined in a published protocol10. Laboratory tests, and clinical assessments, including vital signs, were performed before transplantation, every 1–2 days while in-patient, after discharge weekly for the first 3 months post-transplantation, biweekly from the fourth to sixth months, and then every 4 weeks until the study was completed.
Outcomes
Primary endpoints were 2-year OS and EFS, OS defined as time from transplant to death from any cause, or censored at time of last follow-up. EFS was defined as the time from transplantation to death from any cause, graft failure, or recurrence of transfusion dependence. The FAS included all patients who underwent transplantation and received at least one dose of the conditioning regimen, GF or recurrent TDT, similarly censored for patients alive. Secondary endpoints were aGvHD and cGvHD, neutrophil and platelet recovery, engraftment of donor cells, TRM, GF incidence, incidence of serious infections, reactivations of CMV, adenovirus, EBV, and MC. GRFS at 2 years was defined as free events including grades 3–4 aGvHD, cGvHD requiring systemic treatment, thalassemia relapse, or death from any cause34. GvHD was classified by the Glucksberg and NIH classifications. Neutrophil and Platelet engraftment, TRM, GF, MC were defined as in prior studies10,15. The diagnosis of VOD/SOS was according to McDonald35. The number of adverse events and organism group (bacteria, virus, or fungus) were provided for infections.
Statistical analysis
Two-year OS after allo-HSCT was ~91.5% without distinguishing between donor types according to previous reports36. Assuming a 2-year OS of at least 94%10,37 to be significantly different from the null targeted value of 91.5% for a two-sided α-level of 5%, power of 90% and a dropout rate of 15%, with recruitment time of 36 months and follow-up time of 12 months, ~800 patients were required.
We summarized data using medians and interquartile ranges, or proportions, and made comparisons with Kruskal–Wallis test, Wilcoxon rank-sum test, Chi-square test or Fisher Exact test, as appropriate. For the OS, EFS, GRFS, and TRM, time-to-event rates were estimated using the Kaplan-Meier method, and hazard ratios were estimated using the Cox proportional hazards model with centers as a random-effect. For complications such as MC, VOD/SOS, HC, PRES and infections, the odds ratio was estimated using the Logistic model. In the presence of competing risks, the Fine and Gray model was used for GvHD. The risk factors included in the multivariate analysis were predetermined according to their importance and presumed relevance to outcome. SPSS v23.0 and R v4.2.1 were used for analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are included in this article and its supplementary information file. The study protocol and statistical analysis plan are included in Supplementary Data 1. Source data are provided with this paper.
References
Kato, G. J. et al. Sickle cell disease. Nat. Rev. Dis. Primers 4, 18010 (2018).
Modell, B. Global epidemiology of haemoglobin disorders and derived service indicators. Bull. World Health Organ. 2008, 480–487 (2008).
Kattamis, A., Kwiatkowski, J. L. & Aydinok, Y. Thalassaemia. Lancet 399, 2310–2324 (2022).
Leonard, A. et al. Curative therapy for hemoglobinopathies: an International Society for Cell & Gene Therapy Stem Cell Engineering Committee review comparing outcomes, accessibility and cost of ex vivo stem cell gene therapy versus allogeneic hematopoietic stem cell transplantation. Cytotherapy 24, 249–261 (2022).
Anurathapan, U. et al. Hematopoietic stem cell transplantation for severe thalassemia patients from haploidentical donors using a novel conditioning regimen. Biol. Blood Marrow Transpl. 26, 1106–1112 (2020).
Wang, X. et al. Co-transplantation of haploidentical stem cells and a dose of unrelated cord blood in pediatric patients with thalassemia major. Cell Transpl. 30, 963689721994808 (2021).
Swaminathan, V. V. et al. Matched family versus alternative donor hematopoietic stem cell transplantation for patients with thalassemia major: experience from a tertiary referral center in South India. Biol. Blood Marrow Transpl. 26, 1326–1331 (2020).
Li, C. et al. Related and unrelated donor transplantation for β-thalassemia major: results of an international survey. Blood Adv. 3, 2562–2570 (2019).
Nagler, A. et al. Matched related versus unrelated versus haploidentical donors for allogeneic transplantation in AML patients achieving first complete remission after two induction courses: a study from the ALWP/EBMT. Bone Marrow Transpl. 58, 791–800 (2023).
Li, Q. et al. G-CSF-mobilized blood and bone marrow grafts as the source of stem cells for HLA-identical sibling transplantation in patients with thalassemia major. Biol. Blood Marrow Transpl. 25, 2040–2044 (2019).
Baronciani, D. et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry, 2000-2010. Bone Marrow Transpl. 51, 536–541 (2016).
Yesilipek, M. A. et al. Thalassemia-free and graft-versus-host-free survival: outcomes of hematopoietic stem cell transplantation for thalassemia major, Turkish experience. Bone Marrow Transpl. 57, 760–767 (2022).
Masouridi-Levrat, S., Simonetta, F. & Chalandon, Y. Immunological basis of bone marrow failure after allogeneic hematopoietic stem cell transplantation. Front. Immunol. 7, 362 (2016).
Olsson, R. F. et al. Primary graft failure after myeloablative allogeneic hematopoietic cell transplantation for hematologic malignancies. Leukemia 29, 1754–1762 (2015).
Olsson, R. et al. Graft failure in the modern era of allogeneic hematopoietic SCT. Bone Marrow Transpl. 48, 537–543 (2013).
Goussetis, E. et al. HLA-matched sibling stem cell transplantation in children with β-thalassemia with anti-thymocyte globulin as part of the preparative regimen: the Greek experience. Bone Marrow Transpl. 47, 1061–1066 (2012).
Zhang, X. H. et al. The consensus from The Chinese Society of Hematology on indications, conditioning regimens and donor selection for allogeneic hematopoietic stem cell transplantation: 2021 update. J. Hematol. Oncol. 14, 145 (2021).
Chang, Y. J. et al. Controlled, randomized, open-label trial of risk-stratified corticosteroid prevention of acute graft-versus-host disease after haploidentical transplantation. J. Clin. Oncol. 34, 1855–1863 (2016).
Xu, Z. L. et al. Comparable long-term outcomes between upfront haploidentical and identical sibling donor transplant in aplastic anemia: a national registry-based study. Haematologica 107, 2918–2927 (2022).
Ratanatharathorn, V. et al. Phase III study comparing methotrexate and tacrolimus (prograf, FK506) with methotrexate and cyclosporine for graft-versus-host disease prophylaxis after HLA-identical sibling bone marrow transplantation. Blood 92, 2303–2314 (1998).
Nash, R. A. et al. Phase 3 study comparing methotrexate and tacrolimus with methotrexate and cyclosporine for prophylaxis of acute graft-versus-host disease after marrow transplantation from unrelated donors. Blood 96, 2062–2068 (2000).
Anurathapan, U. et al. Haploidentical hematopoietic stem cell transplantation in thalassemia. Hemoglobin 46, 2–6 (2022).
Gaziev, J. et al. Haploidentical HSCT for hemoglobinopathies: improved outcomes with TCRαβ(+)/CD19(+)-depleted grafts. Blood Adv. 2, 263–270 (2018).
Liu, R. R. et al. Basiliximab in the prevention of aGVHD for unrelated donor hematopoietic stem cell transplantation in patients with thalassemia major: a prospective, multicenter, open-label, randomized controlled study. Blood 140, 1383–1384 (2022).
Thompson, A. A. et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N. Engl. J. Med. 378, 1479–1493 (2018).
Locatelli, F. et al. Betibeglogene autotemcel gene therapy for non-β(0)/β(0) genotype β-thalassemia. N. Engl. J. Med. 386, 415–427 (2022).
Frangoul, H. et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 384, 252–260 (2021).
Chen, L. et al. Effects of splenectomy before transplantation on β-thalassaemia major with matched sibling and unrelated hematopoietic stem cell transplantation. Blood 132, 5782–5782 (2018).
Sanpakit, K. et al. Impact of splenectomy on outcomes of hematopoietic stem cell transplantation in pediatric patients with transfusion-dependent thalassemia. Pediatr. Blood Cancer 67, e28483 (2020).
Lai, X. et al. Hepatic veno-occlusive disease/sinusoidal obstruction syndrome after hematopoietic stem cell transplantation for thalassemia major: incidence, management, and outcome. Bone Marrow Transpl. 56, 1635–1641 (2021).
Liang, H. et al. Health-related quality of life in pediatric patients with β-thalassemia major after hematopoietic stem cell transplantation. Bone Marrow Transpl. 57, 1108–1115 (2022).
Huang, J. J., Chen, B., Hu, J. & Yang, W. H. Limited sampling strategy for predicting busulfan exposure in hematopoietic stem cell transplantation recipients. Int. J. Clin. Pharm. 39, 662–668 (2017).
Tucunduva, L. et al. Combined cord blood and bone marrow transplantation from the same human leucocyte antigen-identical sibling donor for children with malignant and non-malignant diseases. Br. J. Haematol. 169, 103–110 (2015).
Holtan, S. G. et al. Composite end point of graft-versus-host disease-free, relapse-free survival after allogeneic hematopoietic cell transplantation. Blood 125, 1333–1338 (2015).
McDonald, G. B., Sharma, P., Matthews, D. E., Shulman, H. M. & Thomas, E. D. Venocclusive disease of the liver after bone marrow transplantation: diagnosis, incidence, and predisposing factors. Hepatology 4, 116–122 (1984).
Li, C. et al. A novel conditioning regimen improves outcomes in β-thalassemia major patients using unrelated donor peripheral blood stem cell transplantation. Blood 120, 3875–3881 (2012).
Li, Q., Wu, M., Zhang, Z., Liu, L. & Lai, Y. Unrelated donor peripheral blood stem cell transplantation for thalassaemia: a single institution experience of 53 patients. Blood 130, 357 (2017).
Acknowledgements
Some patients enrolled in this study received support from Angel Program Caring for Life Thalassemia Rescue Project from Red Cross Society of China Guangxi Branch. Robert Peter Gale, Qifa Liu, Yu Wang, Ming Wan, Peiyan Kong, Lanping Xu, Yang Liang all contributed to the draft of the manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed substantially to the manuscript. R.R.L., H.W.X., C.J.Q., J.H., J.M.L., H.C., X.Z., X.L.Y., G.Y.N., G.Y.W., Z.L., W.T., H.Z., X.T.W., M.L., C.D.L., T.H.Y., S.B.W., J.X.H., Y.Y.H., L.W., Z.M.Z., L.J.L. contributed to data acquisition. R.R.L., Q.Z., H.W.X., Z.B.W., M.Q.W., L.L.S., Y.R.L. contributed to study design, data analysis, and data interpretation. R.R.L., Y.R.L. wrote the first draft of the manuscript. B.S.A., X.J.H. contributed to study design and data interpretation. All authors approved the final version to be published and agree to be accountable for all aspects of the work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Diana Giannarelli, Ashutosh Lal and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, R., Xiao, H., Qin, C. et al. A multi-center clinical trial of allogeneic hematopoietic stem cell transplantation in transfusion-dependent thalassemia. Nat Commun 17, 3083 (2026). https://doi.org/10.1038/s41467-026-69756-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69756-8
