
Abstract
Progress in extending the cycle life of lithium metal batteries has primarily been achieved through strategies focused on slow charging and fast discharging conditions, with limited practical impact. However, accelerated performance degradation under the opposite operation protocols—fast charging and slow discharging—remains a challenge. Here, we introduce (difluoromethyl)trimethylsilane with a large electrostatic potential difference as a diluent for localized high-concentration electrolytes. The presence of this diluent limits the size of ion clusters and promotes the transfer of Li ions through the swollen solid-electrolyte-interphase layer for robust fast charging. The diluent also increases electrode polarization, which leads to spatially uniform Li stripping sites and allows reliable discharging even at low current densities. As a result, the electrolyte achieves an average Coulombic efficiency of 98.12% even at 12 mA cm−2 in Li | |Cu asymmetric cells. In full cells, a state-of-charge of 77.3% is achieved within 6 minutes while retaining 81.3% capacity after 200 cycles at a 10 C charging rate, demonstrating their stability under fast-charging and slow-discharging protocols.
Similar content being viewed by others


Introduction
Lithium metal batteries (LMBs) represent a paradigm shift in energy storage technology and are ushering in a new era of high-performance power sources. The utilization of metallic lithium as a negative electrode in LMBs has unprecedented advantages, most notably its unparalleled capacity of 3860 mAh g−1 and the lowest redox potential of −3.04 V versus the standard hydrogen electrode. LMBs are therefore poised to transcend the energy density limitations inherent to current intercalation-based lithium-ion batteries (LIBs). This quantum leap in performance promises not just to extend the driving range but also to redefine the auxiliary boundaries of rechargeable batteries in powering emerging vehicle functions, such as autonomous driving and infotainment systems1,2,3.
The viability of LMBs relies on the reversibility of Li (de)plating. Dendritic growth and uneven stripping are both responsible for the formation of isolated Li plates (dead Li) and the parallel acceleration of electrolyte decomposition to impair the reversibility of individual cycles4,5,6. This situation worsens at accelerated plating (fast-charging) and prolonged stripping (slow-discharging) rates7,8,9. The battery community has developed numerous strategies such as surface modification10, installation of hosts11,12, and electrolyte engineering13,14 to address these issues. Yet, the majority of these approaches are effective under slow-charging and fast-discharging conditions. In particular, electrolyte engineering has elucidated the emerging opportunities related to tuning the solvation structure near the Li ion, and localized high concentration electrolytes (LHCEs) involving diluents are the most representative in this regard15,16,17. These LHCEs have ion-pair configurations in which the anions are involved in the solvation structure to form either contact ion pairs (CIPs) or aggregates (AGGs), including smaller AGG1 and larger AGG2, to build an inorganic-rich solid-electrolyte interphase (SEI) layer. This inorganic-rich SEI supports reversible Li plating and stripping owing to its strengthened physical properties18. Nevertheless, these remedies lose their effectiveness when the cell operates under more practically useful conditions: rapid energy storage paired with prolonged utilization before requiring recharging, in other words, fast-charging and slow-discharging, respectively.
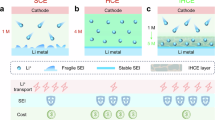
Unfortunately, warranting the reversibility of LMBs under these preferred operational conditions is nontrivial. Even if the anion-derived SEI layer induces densely packed Li plates, the electrode would still be vulnerable to local dendritic growth once the region near the electrode surface becomes depleted of Li ions at higher charging rates16,19,20. The community has often explained this dependence on the plating current using concepts such as Sand’s time, deionization shock, and the tip effect, without fully considering the interphase on the surface19,20,21,22,23,24. In the Li metal system, however, consideration of the SEI layer is essential for understanding the key performance factors. Since the discovery of the existence of the SEI layer, advanced analytical techniques have revealed new characteristics of this layer, such as its self-standing nature during reversible Li (de)plating25,26, swollenness due to penetration of electrolyte components rather than complete blockage6,13,27, and the significantly restricted Li ion transport therein compared to the bulk electrolyte4,28,29,30,31. Considering these aspects, it can be deduced that facilitating de-solvation and transfer of Li ions through narrow pathways within the SEI layer is crucial to alleviate Li ion depletion near the electrode surface and thus achieve fast charging capability (Fig. 1, left).

Descrition of Li+ transport through a swollen SEI layer (Left). Reaction uniformity scheme on lithium metal under electrode polarization during plating and stripping (Right).
On the other hand, stripping at a decelerated rate was found to be detrimental to reversibility due to the limited availability of stripping sites32,33. The electrode surface inevitably contains structurally defective sites, such as point defects and grain boundaries, with low activation barriers for electrochemical reactions that cause undesired localized charge transfer33,34,35,36. Even within a single Li deposit, imbalanced stripping can lead to the formation of isolated deposited Li, referred to as dead Li4,29,37. In conventional electrochemistry, a highly polarized electrode, indicative of increased overpotential at a given current density, has been proven to mitigate the reaction heterogeneity33,38,39. In this line, the beneficial effect of an increased overpotential lies in its ability to overcome reaction barriers and extensively activate reaction sites to ultimately realize uniform Li (de)plating (Fig. 1, right). Additionally, according to classical nucleation theory, increasing the overpotential of an electrode increases the number of deposition sites while reducing their size due to a lowered nucleation barrier, as quantified by the nucleation overpotential (|\({\eta }_{{{\rm{nuc}}}}|\))38,39,40.
Considering these aspects, herein, we identify the determinants for the reversibility of Li (de)plating under fast-charging and prolonged-discharging conditions and propose an LHCE design strategy based on electrostatic potential control. A rigid SEI, which can be dictated by electrolyte chemistries, is preferred to reach a certain level of reversibility, but may not be sufficient to avoid the dendritic growth associated with fast Li plating unless the constrained Li+ transport through the SEI layer is resolved. In LHCEs, Li ion transport kinetics is affected by the size of ion clusters, which are highly dependent on the molecular structure of the diluent, as well as the SEI properties. In this regard, we aimed to reduce the size of the ion clusters to facilitate the Li ion transport through the SEI layer while maintaining the high-concentration effect, which induces an inorganic-rich SEI. This electrolyte design perspective led us to pursue an electrolyte with a relatively high content of small ion clusters. The preference of small clusters guided us to consider the electrostatic potential difference (ΔESP) of a diluent molecule as a key metric because the polarized diluent can shield cation-anion electrostatic interaction more effectively and thus allay the tendency to aggregate ions, even without direct coordination. Along this line, a LHCE containing a diluent, (difluoromethyl)trimethylsilane (DFTMS), with a large ΔESP exhibited 74.8% and 22.1% higher proportions of ion pairs of suitable sizes under CIP and AGG1, respectively, compared to its trifluoromethyl-substituted counterpart, namely, (trifluoromethyl)trimethylsilane (TFTMS). The increased content of small clusters promotes the kinetics of Li ion transport through the SEI layer for stable Li plating at fast-charging modes. On the other hand, the addition of DFTMS improves the uniformity of Li stripping at slow discharging rates by effectively raising the electrode polarization and therefore increasing the availability of electrochemical reaction sites. As a result, the electrolyte 2 M lithium bis(fluorosulfonyl)imide (LiFSI) 1,2-dimethoxyethane (DME)-DFTMS (1/2 = v/v) exhibited stable cycling with an average Coulombic efficiency (CE) of 98.12% at a high current density of 12 mA cm−2 in a Li | |Cu asymmetric cell configuration. When examined in a full-cell with a limited N/P ratio (the capacity ratio between the negative and positive electrodes) of 2.5, 78.5% of the original capacity was retained after 300 cycles, even with fast charging that reached a state-of-charge (SOC) of 87.0% within 15 min during each cycle. Moreover, 81.3% of the capacity was retained after 200 cycles, even with fast charging that reached an SOC of 77.3% in 6 min, which corresponds to a 10 C rate (16 mA cm−2). The current study presents the ΔESP and electrode polarization as key parameters for designing LHCEs for LMBs capable of operating under the desired fast charging and prolonged discharging conditions. Furthermore, we propose a transport scheme of Li-ion clusters in the SEI layer, characterized by kinetically hindered ion-cluster transport in the upper swollen SEI, followed by gradual de-solvation as the clusters approach the electrode surface.
Results
Factors affecting reversibility in fast charging and slow discharging
As a variety of interrelated interfacial factors can complicate the electrochemical behavior, establishing ideal interfacial conditions is not trivial. To identify the determinants for fast charging coupled with prolonged discharging, we conducted preliminary tests using an electrolyte that is widely used in LMBs: LiFSI in DME. Higher concentrations of the LiFSI salt alter the bulk ionic conductivity, yet also change the interfacial properties, which would complicate the interpretation of the overall phenomena41,42,43. To exclude the effect of the interface, apart from preparing cells with a single polyethylene (PE) separator and 2 M and 4 M LiFSI DME as the electrolyte, we additionally prepared a cell with the identical 2 M LiFSI DME electrolyte but installed three separators. This cell, with the three separators and 2 M LiFSI DME, exhibited even higher ohmic resistance than that with one separator and 4 M LiFSI DME, but its interfacial resistance was similar to that with one separator and 2 M LiFSI DME, as indicated by electrochemical impedance spectroscopy (EIS) analysis (Supplementary Fig. S1). The average Coulombic efficiencies (CEs) of these cells were assessed by Adams’ method (Supplementary Fig. S2). Notably, despite the similar interfacial resistances, the addition of separators increased the average CE from 96.83% to 98.02% for the same 2 M LiFSI DME electrolyte. In a separate experiment, 4 M LiFSI DME with one separator yielded the highest CE of 99.2%, confirming that the high-concentration strategy remains effective for improving the reversibility of Li (de)plating at a slow rate of 1 mA cm−2 through the formation of an inorganic-rich SEI41,42. Nonetheless, this high-concentration strategy alone did not warrant reliable cycling at higher rates above 12 mA cm−2, as sudden potential drops indicative of soft short-circuits were observed (Supplementary Fig. S3). The much lower Li+ diffusivity through the SEI layer (approximately 10−9–10−12 cm2 s−1) than that in the bulk electrolyte (~10−6 cm2 s−1)28,29,31,44,45 suggests that dendritic growth induced by depletion during fast Li plating is mainly related to limited mass transfer of Li ions in the SEI layer5,46,47. This perspective aligns well with recent reports48,49,50,51 indicating that the reversibility of Li deposition during fast charging highly depends on Li ion transport through SEI, rather than bulk ionic conductivity.
To clarify the improved reversibility in the cell with three PE separators despite using the same electrolyte, we examined the potential profiles (Supplementary Fig. S4). The three-separator cell exhibited a higher overpotential of 20.9 mV compared to that of the single-separator cell of 10.9 mV due to the increased ohmic resistance. The depth of the dips at the beginning of the potential profiles during Li plating indicated that \(|{\eta }_{{{\rm{nuc}}}}|\) decreased from 40.0 mV (one separator) to 23.4 mV (three separators). Hence, based on classical nucleation theory, the lower \(|{\eta }_{{{\rm{nuc}}}}|\) of the three-separator cell rationalizes its more extensive coverage in terms of Li deposits observed through scanning electron microscopy (SEM) analysis (Supplementary Fig. S5). Notably, the distribution and size of the stripping sites need to be interpreted within a different framework, as classical nucleation theory primarily pertains to deposition. For the one-separator cell, larger pits were observed at localized sites, indicating the uneven spatial and dimensional distributions of Li stripping reactions. Conversely, in the three-separator cell, the pits were shallower and more uniformly distributed across the area of the electrode. This uniform distribution of reaction sites for Li stripping suggests that an appropriate increase in the polarization can help maintain the reaction uniformity, which is beneficial for prolonged discharge processes.
Molecular structure design of diluent for small ion clusters
LHCEs have proven to be an effective electrolyte design for reversible Li (de)plating under slow plating conditions through the formation of a rigid SEI layer. Apart from this, facilitating the transport of Li ions through the SEI layer is also crucial for suppressing depletion-induced dendritic growth during fast charging. Increasing the CIP/AGG ratio by augmenting the portion of CIPs or reducing the size of ion clusters reportedly promotes rapid mass transfer kinetics16,52,53.
The proportions of solvent-separated ion pairs (SSIPs), CIPs, and AGGs vary depending on the electrolyte composition. The electrostatic potential (ESP) of a molecule, determined by the electronic structures of its constituent atoms and their intramolecular interactions, dictates the solvation environment and the resulting ion-pair states in the electrolyte54,55. In response to the electric field created by the ions in an electrolyte, the ESP of a molecule can adjust its orientation to electrostatically counteract the field: the site with the minimum ESP (ESPmin) aligns toward neighboring cations, while the site with the maximum ESP (ESPmax) orients toward neighboring anions (Fig. 2a). Based on a similar logic, a molecule with the most negative ESPmin would have a strong tendency to coordinate with Li ions. As the absolute value of ESPmin decreases, the molecule becomes a weakly solvating solvent or even a diluent that barely participates in the primary solvation shell. On the other hand, the difference between ESPmin and ESPmax (ΔESP) of a diluent can still influence the ion-pair states outside the solvation shell through its charge-compensation ability56,57. That is, a wide ΔESP in a diluent can buffer the attractive force between cations and anions, thereby effectively disaggregating large ion clusters (e.g., AGG2) into smaller ones (e.g., AGG1 and CIP).

a Relationship between ΔESP of the diluent and the cluster size of an ion-pair. b Electrostatic potential map of various diluents of interest. Atom types: carbon (dark gray), hydrogen (white), silicon (blue-gray), fluorine (light blue). c Relative compositions of ion-pair states in the electrolytes. d 7Li nuclear magnetic resonance (NMR) spectra of the electrolytes. e Small-angle X-ray scattering (SAXS) spectra of the electrolytes. f Ionic conductivities of the electrolytes.
With this effect of ΔESP in mind, we screened diluents to identify those with a molecular structure that would support the creation of highly dispersed ion pairs with smaller sizes. This approach was intended to increase ΔESP without significantly raising the absolute value of ESPmin, as a large negative ESPmin could lead to an unfavorable SSIP state. Also, we note that it is crucial to compare diluents with reasonably similar molecular frameworks, as large structural differences or coordinating functionalities could obscure and confound the contribution of ΔESP. From the perspective of the atomic arrangement, replacing the central atom with a larger one can increase ΔESP because the positive and negative charges within the molecule become less shielded. As a shred of direct evidence, the replacement of the central carbon atom in neopentane by silicon (Si) to form tetramethylsilane (TMS) increases ΔESP from 29.86 to 50.97 kJ mol−1 as the distance between the central atom and the methyl groups increases from 1.54 to 1.89 Å (Fig. 2b and Supplementary Data 1). Beyond the atomic size, asymmetry in the molecular structure can further increase ΔESP. Along this direction, the incorporation of fluorine (F) is particularly notable because its high electronegativity polarizes the electron distribution within the molecule. However, the degree of fluorination needs to be adjusted to minimize ESPmin and thus optimize ΔESP (Supplementary Fig. S6). In this regard, the trifluoromethyl group (−CF3) may be less effective than its mono- and di-fluoromethyl counterparts because the three fluorine atoms can offset the electron withdrawing effect of one another to yield a lower net |ESPmin | 58,59. Our comparison of two TMS-derived diluents, DFTMS and TFTMS, revealed that ESPmin (−94.89 kJ mol−1) of DFTMS is clearly lower than that of TFTMS (−73.29 kJ mol−1) (Fig. 2b). However, because their ESPmax values do not differ much (81.82 and 88.88 kJ mol−1, respectively), their ΔESP values differ considerably: DFTMS (176.71 kJ mol−1) and TFTMS (162.17 kJ mol−1). We note that this study does not intend to determine an absolute ΔESP threshold for effective ion-pair disaggregation. Rather, we consider ΔESP to be a key parameter for understanding the differences in ion-cluster size and fast-charging capability among LHCEs.
In line with the ESPmin results, density functional theory (DFT) calculations indicate that DFTMS has a stronger affinity for binding Li ions than TFTMS (Supplementary Fig. S7). However, both affinities are still significantly lower than that of DME, supporting the classification of DFTMS and TFTMS as diluents. Although the highest occupied molecular orbital (HOMO) levels of both diluents are relatively low (Supplementary Fig. S8) and they are thus expected to have decent oxidative stabilities, the concentration of LiFSI salt is required to be at least 2 M in LHCEs to avoid free DME molecules and FSI anions, as indicated by the stepped chronoamperometry results of 1.5 M and 2 M samples up to 5.0 V vs. Li+/Li (Supplementary Fig. S9). Therefore, a salt concentration of 2 M was employed. Our assessment of the diluent contents for LHCEs at LiFSI of 2 M showed that TFTMS could not be added at a volume ratio greater than 2 relative to DME (1:2 v) due to its limited polarity, closely linked to small ΔESP or higher ESPmax of TFTMS. For example, 2 M LiFSI DME-TFTMS at the 1:2.5 v ratio had poor homogeneity (Supplementary Fig. S10). Therefore, we set the volume ratio to 1:2 v to ensure oxidative stability (Supplementary Fig. S11) without any dispersion issue: 2 M LiFSI DME-DFTMS 1:2 v and 2 M LiFSI DME-TFTMS 1:2 v.
Raman spectroscopy was used to determine the ion-pair compositions of the electrolytes (Fig. 2c and Supplementary Fig. S12). In the 2 M LiFSI DME, only 47.74% of the total ion-pairs were CIP and AGGs (the sum of CIP, AGG1, and AGG2), whereas 2 M LiFSI DME-DFTMS and 2 M LiFSI DME-TFTMS both contained over 96% under these compositions. Notably, the DFTMS-based LHCE had distinctly higher portions of small ion clusters (CIP and AGG1: 16.54% and 48.78%, respectively) yet much less larger ones of AGG2 (31.36%) compared to those in the TFTMS-based LHCE (CIP, AGG1, and AGG2: 9.46%, 39.94%, and 47.13%, respectively). Both molecular dynamic (MD) simulation (Supplementary Fig. S13 and Supplementary Data 2) and 7Li NMR chemical shift (Fig. 2d) results indicate that the coordination of the two diluents with Li+ is insignificant, informing that the solvating components in the primary solvation shell had a negligible effect on the distinct ion-pair distributions. Therefore, the prominently higher proportions of CIP and AGG1 in the DFTMS-based LHCE, increased by 74.8% and 22.1%, respectively, compared to its TFTMS-based counterpart, are anticipated to result from the electrostatic shielding effect of DFTMS outside the primary solvation shell.
Small-angle X-ray scattering (SAXS) and wide-angle X-ray scattering (WAXS) spectroscopy supported the enhanced formation of small ion clusters in 2 M LiFSI DME-DFTMS 1:2 v (Fig. 2e and Supplementary Fig. S14)16. SAXS analysis (Fig. 2e) revealed an enhanced intensity, and hence greater prevalence, of ion-pairs in the two LHCEs compared to 2 M LiFSI DME over the vector (q) range of 0.02 − 0.5 Å. Between the two LHCEs, the intensity of the DFTMS-based one was lower in the lower q regime (q < 0.1 Å), but the trend reversed thereafter, inferring the decreased average dimensions of ion clusters in 2 M LiFSI DME-DFTMS compared to 2 M LiFSI DME-TFTMS. These results are well aligned with the greater abundance of CIP and AGG1 in 2 M LiFSI DME-DFTMS, indicated by the Raman spectra (Fig. 2c). The bulk ionic conductivity can also be associated with the size of charged clusters16. Particularly, 2 M LiFSI DME with the highest portion of SSIPs had the highest ionic conductivity of 17.78 mS m−1 among the electrolytes (Fig. 2f). Between the two LHCEs, the ionic conductivity of 2 M LiFSI DME-DFTMS 1:2 v (5.88 mS cm−1) was higher than that of 2 M LiFSI DME-TFTMS 1:2 v (4.57 mS cm−1) due to the increased portion of small ion clusters in the 2 M LiFSI DME-DFTMS 1:2 v. Overall, the larger ΔESP originating from the optimal level of fluorination of DFTMS (compared with TFTMS) induces the formation of more disintegrated ion clusters in its LHCE solution, which is beneficial for facile de-solvation and ionic transport through the SEI layer and thus fast charging LMBs. In previous studies10,60, TFTMS was introduced as an additive for interface modification in LMBs and lithium-sulfur batteries. However, the ion-pair state and relevant cluster sizes in relation to its ESP distribution were not considered, nor was its partially fluorinated version investigated.
Evaluation of reversibility at slow-charging rates
Evaluation of the reversibility of Li (de)plating at a relatively slow rate of 1 mA cm−2 indicated that the average size of ion clusters appeared to have an insignificant effect. At a current density of 1 mA cm−2 with a deposition capacity of 1 mAh cm−2, a Li | |Cu asymmetric cell with 2 M LiFSI DME exhibited the disappointing average Coulombic efficiency (CE) of 91.57% (from the 15th to the 300th cycle), with abrupt degradation after 120 cycles (Fig. 3a). Alternatively, the LHCE cells of 2 M LiFSI DME-DFTMS 1:2 v and 2 M LiFSI DME-TFTMS 1:2 v demonstrated stable reversibility, delivering higher average CEs of 99.07% and 98.94% for 300 cycles, respectively. Analogously, Adams’ measurements at a low current density of 0.5 mA cm−2 also revealed high CEs of 99.38 and 99.30% for both LHCEs, respectively, whereas the cell with 2 M LiFSI DME had a distinctly lower CE of 97.66% (Fig. 3b). These CE results are consistent with the extent of dendritic deposition on the Li electrodes in the SEM analysis (Fig. 3c); dendrites were observed in the 2 M LiFSI DME after 20 cycles at a current density of 1 mA cm−2 with a deposition capacity of 1 mAh cm−2, whereas the deposits in both LHCEs were comparatively planar after the same 20 cycles.

a Comparison of Coulombic efficiencies for Li | |Cu asymmetric cells when evalauted at a current density of 1 mA cm−2 with a deposition capacity of 1 mAh cm−2. b Li plating/stripping reversibility assessed via the Adams’ measurements in Li | |Cu asymmetric cells. c Li deposition morphology after Li deposition in the 21st cycle, characterized SEM at a current density of 1 mA cm−2 with a deposition capacity of 1 mAh cm−2. d X-ray photoelectron spectroscopy (XPS) profiles of the deposited Li after 5 cycles at 1 mA cm−2 with a deposition capacity of 1 mAh cm−2. e Cycling performance of Li | |NCM811 full-cells and f their potential profiles at the 5th and 200th cycles at 1 C rate (1 C = 200 mA g−1).
The reversibility of Li (de)plating at moderate plating rates reportedly depends on the physicochemical properties of the SEI layer. X-ray photoelectron spectroscopy (XPS) revealed that the SEI layer formed in 2 M LiFSI DME was rich in organic components derived from the decomposition of the primarily solvating DME (Fig. 3d). In contrast, the SEI layers of both LHCEs were characterized by abundant inorganic components, with a minor chemical difference that manifested in their similar XPS peaks. This similarity is reflected in their comparable linear sweep voltammetry (LSV) profiles related to electrolyte reduction (Supplementary Fig. S15). Interestingly, despite the presence of Si in the molecular structures of DFTMS and TFTMS, the minor presence of Si components in the SEI layers was consistent with previous studies in which silane was used as a solvent or diluent in the electrolyte61,62,63,64. The insignificant Si signature implies that the inorganic components in the SEI layers mainly originate from the decomposition of the anion rather than the diluents.
The cyclability of the full-cell in conventional constant current (CC) mode at 1 C for both charge and discharge was evaluated by preparing Li (20 μm)||NCM811 (8 mg cm−2) full-cells with a low N/P ratio of 2.5 (Fig. 3e, f). For the 2 M LiFSI DME full-cell, the capacity progressively declined from the beginning of cycling with severe degradation after 150 cycles, reflecting the aforementioned oxidative instability and irreversible Li (de)plating for this electrolyte. In contrast, the 2 M LiFSI DME-DFTMS 1:2 v full-cell cycled stably with capacity retention of 90.0% and 81.9% after 238 and 350 cycles, respectively. Consistent with the similar reversibility observed in the Li | |Cu asymmetric cell tests, the 2 M LiFSI DME-TFTMS 1:2 v full-cell retained 90.1% and 71.3% of its capacity after 195 and 350 cycles, respectively. These results are comparable to those of its DFTMS-based LHCE counterpart, suggesting that the size of ion clusters was not the main factor governing the cyclability at slow plating rates.
Evaluation of reversibility at fast-charging conditions
The disparity in the reversibility between the two LHCEs became more pronounced at higher current densities. At the intermediate current density of 4 mA cm−2, Li | |Cu asymmetric cells with both LHCEs demonstrated similar average CEs of 98.74% and 98.64% from the 15th to the 250th cycles, respectively (Fig. 4a). However, at the significantly higher current density of 12 mA cm−2 with a fixed capacity of 1 mAh cm−2, the average CE for the 2 M LiFSI DME-TFTMS 1:2 v cell of 85.00% was considerably lower such that it was comparable to that of the 2 M LiFSI DME cell of 83.59% (Fig. 4b). In stark contrast, the 2 M LiFSI DME-DFTMS 1:2 v cell maintained its reversibility with an average CE of 98.12%, clearly succeeding in avoiding the degradation in reversibility caused by depletion-induced dendrite formation.

Coulombic efficiencies of Li | |Cu asymmetric cells at a 4 mA cm−2 and b 12 mA cm−2 for both Li plating and stripping with a deposition capacity of 1 mAh cm−2. c Coulombic efficiencies of re-assembled Li | |Cu asymmetric cells with different LHCEs at 12 mA cm−2 with a deposition capacity of 1 mAh cm−2. d SEM analysis of the 1st Li deposition at 1 mA cm−2 and 12 mA cm−2 with a fixed capacity 3 mAh cm−2. The scale bars in the inset images are 20 μm. e Current and potential profiles of three-electrode cells obtained through galvanostatic intermittent titration technique (GITT). f Potential relaxation of the working electrodes during the rest periods after plating at 1, 3, and 6 mA cm−2 in the GITT analysis.
In a further investigation of the determinants of the rate capability, we re-evaluated the CEs at 12 mA cm−2 by re-assembling the electrodes with pre-formed SEI layers in the individual electrolytes. The SEI layer on the Cu electrode was pre-formed by operating the cells for five cycles with each LHCE at 1 mA cm−2 with 1 mAh cm−2. These Cu electrodes were then carefully washed with DME to remove residual LHCEs without damaging the pre-formed layer. Subsequently, the electrodes were re-assembled into new cells with fresh Li counter electrodes and each LHCE to assess the impact of the pre-formed SEI layers on the CEs at 12 mA cm−2 with 1 mAh cm−2 (Fig. 4c). Notably, regardless of the pre-formed SEI layer, the CE stability was determined mainly by the electrolyte; the re-assembled cells with 2 M LiFSI DME-DFTMS 1:2 v exhibited stable CEs (red and orange, in Fig. 4c) whereas the CEs of those with 2 M LiFSI DME-TFTMS 1:2 v were highly fluctuating (green and blue, in Fig. 4c). Furthermore, we identified insignificant differences in SEI thickness (Supplementary Fig. S16) and chemical compositions (Supplementary Fig. S17) between the two LHCEs, in agreement with the XPS results. Even when the ohmic resistance of the 2 M LiFSI DME-DFTMS 1:2 v cell was increased by incorporating three PE separators, its reversibility at 12 mA cm−2 remained high (Supplementary Fig. S18). All of these results indicate that neither the SEI structure nor the bulk ionic conductivity is the key determinant of the reversibility at high rates.
The morphology of the Li deposits was inspected using SEM to assess the dependence thereof on the current density and electrolyte (Fig. 4d and Supplementary Fig. S19). At a low current density of 1 mA cm−2, morphologies in the reference 2 M LiFSI DME electrolyte revealed localization of deposits with particulate and dendritic structures, whereas the deposits in both LHCEs were homogeneous with full coverage. Notably, at the high current density of 12 mA cm−2, the morphologies of the deposits in the 2 M LiFSI DME and 2 M LiFSI DME-TFTMS 1:2 v evolved into thin needle-like features, which are often observed with poor SEI-forming electrolytes such as carbonates or under mass transfer-limiting conditions. In contrast, the deposits in the 2 M LiFSI DME-DFTMS 1:2 v maintained their planarity. Interestingly, when the ion-cluster aggregation was mitigated by reducing the salt concentration to 1 M (Supplementary Fig. S20), a more blunted deposition appeared in the 1 M LiFSI DME-TFTMS 1:2 v, instead of the needle-like morphology (Supplementary Fig. S21). This morphological improvement was accompanied by enhanced Coulombic efficiency during the initial 10 cycles (Supplementary Fig. S22). Nevertheless, the compromised robustness of the SEI layer stemming from less anion-decomposition could not ensure the long-term cyclability. Taken together, such a significant morphological difference between the two 2 M based-LHCEs at high current densities is closely linked to their distinct capabilities in overcoming the mass transfer limitations along the SEI layers.
The reversibility was also evaluated for the Li | |NCM811 full-cells at high rates by imposing a charging current of 3 C (Supplementary Fig. S23). Unlike the cyclability at the 1 C rate shown in Fig. 3e, the CEs of the 2 M LiFSI DME full-cell decreased to below 90% at the beginning of cycling, despite its decent stability during the formation cycles at 0.2 C (Supplementary Fig. S24). The excessive charge capacities, along with the normal discharge profiles, suggest that the inferior CEs were caused by oxidative instability. In contrast, due to their enhanced oxidative stability, the CEs of both LHCE full-cells remained stable even at the 3 C rate. However, the cyclability of the cell with 2 M LiFSI DME-TFTMS 1:2 v decayed faster compared to that of its 2 M LiFSI DME-DFTMS 1:2 v counterpart, which retained 78.7% capacity after 200 cycles even at this high C-rate.
Interfacial phenomena during plating predominantly involve two critical processes: the transport of Li+ clusters through the SEI layer and the charge transfer of the clusters accompanied with the de-solvation. To elucidate the primary factor underlying the stability of fast plating performance of 2 M LiFSI DME-DFTMS 1:2 v, we conducted comprehensive electrochemical analyses. Tafel plots constructed through cyclic voltammetry (CV) revealed marginal differences between the two LHCEs (Supplementary Fig. S25). Furthermore, charge transfer resistances (\({R}_{{{\rm{CT}}}}\)) were evaluated across a temperature range of −10 °C–25 °C using electrochemical impedance spectroscopy (EIS) to construct Arrhenius plots (Supplementary Fig. S26 and Table S1). Like the Tafel plots, the activation energies of the two LHCEs related to \({R}_{{\mathrm{CT}}}\) showed minimal difference. These findings point to the fact that neither the exchange current density nor the charge transfer resistance, representing de-solvation and electron transfer kinetics, are the primary determinants in the observed disparity in fast charging capabilities.
To investigate the transport kinetics of Li ion clusters through the SEI layer, we conducted a modified galvanostatic intermittent titration technique (GITT) analysis (Fig. 4e). To this end, we employed homemade three-electrode cells to exclusively monitor the potential changes of the working electrode (Supplementary Fig. S27, top). When Li+ transport within the SEI layer is limited, relatively rapid Li+ consumption through plating process leads to depletion of the Li+ concentration at the Li metal surface (\({C}_{{{{\rm{Li}}}}^{+}}^{{{\rm{s}}}}\)) (Supplementary Fig. S27, bottom). The more restricted transport in the TFTMS-based LHCE with larger ion clusters amplifies the concentration gradient across the SEI layer, making it more susceptible to short-circuiting compared to its DFTMS-based counterpart (Supplementary Fig. S28). When the cell rests right after plating, the Li+ concentration gradient is relieved as Li ions diffuse into the SEI layer. Accordingly, the working electrode potentials gradually relax toward 0 V vs. Li+/Li. In this context, the relaxation of potentials is prolonged as the plating rate increases or the clusters’ transport through the SEI layer becomes more restricted. Noticeably, the TFTMS-based LHCE exhibited more pronounced retardation in the relaxation, whereas facile diffusion of Li+ clusters in the DFTMS-based LHCE afford faster relaxation (Fig. 4f). These observations suggest that the enhanced capability of fast charging in the 2 M LiFSI DME-DFTMS 1:2 v is greatly associated with more efficient Li+ transport through the SEI layer.
Electrochemical performance in fast charging and slow discharging
Numerous studies4,33,46 have highlighted the challenge of maintaining the cyclability under prolonged discharge conditions, and have emphasized the difficulty in achieving uniformly distributed stripping sites. To evaluate the uniformity of the stripped sites after the 1st discharge, we used SEM to inspect the stripped Li electrodes (Fig. 5a and Supplementary Fig. S29). At the highest stripping rate of 12 mA cm−2, the stripping morphologies were uniformly distributed across the entire surfaces of the electrodes for all the electrolytes. At lower stripping rates, however, the gradual localization of these stripping sites into specific regions provided clear evidence of non-uniform stripping at prolonged discharge rates, as was also previously noted7,8,9,32,65. Notably, contrary to the 2 M LiFSI DME, which developed large and deep pits at 1 mA cm−2, the two LHCEs displayed spatially extensive and shallow pits at this stripping rate, indicative of better reversibility during prolonged discharge.

a SEM images of the Li electrodes after the 1st stripping at 1 mA cm−2 and 12 mA cm−2 with a capacity of 3 mAh cm−2. b Average overpotentials of the Li | |Li symmetric cells at incrementally increasing current densities. The plating and stripping current densities were identical. c Potential profiles of the 3rd cycle in Li | |Cu asymmetric cells at 1 mA cm−2 with a capacity of 1 mAh cm−2. d Electrochemical impedance spectroscopy (EIS) profiles of Li | |Li symmetric cells after 5 cycles at 1 mA cm−2 with a capacity of 1 mAh cm−2. Inset: RCT extracted from EIS profiles. e Constant current/constant voltage (CC/CV) profiles of Li | |NCM811 full-cells and f their cycling performance when charged at 4 C (1 C = 200 mA g−1) in CC/CV mode and discharged at nC in CC mode in each cycle. g Cyclability evaluations of Li | |NCM811 full-cells at an fast-charging rate of 10 C with a 6-min time cutoff and 4 C discharge and h their potential profiles at the 10th, 100th, and 200th cycles.
The electrochemical responses were analyzed to further probe the dependence of the stripping behavior on the current density and choice of electrolyte. The average overpotentials increased with increasing current densities for all the electrolytes (Fig. 5b). Intriguingly, the overpotentials in the two LHCEs were considerably higher than those in the 2 M LiFSI DME even at low current densities such as 1 mA cm−2. From the perspective of plating, the higher overpotentials in the LHCEs lowered the nucleation barriers, as indicated by the decrease in |\({\eta }_{{{\rm{nuc}}}}|\) on the potential profiles (Fig. 5c). These elevated overpotentials in the LHCEs also play a central role in promoting spatially uniform and extended stripping reactions by helping to overcome the activation barrier, as demonstrated by the experimental results with varying the number of separators (Fig. 1). In addition, as is evident from the EIS measurements, these high overpotentials for the LHCEs are associated with relatively greater \({R}_{{{\rm{CT}}}}\) (Fig. 5d and Supplementary Table S2). This increase in \({R}_{{{\rm{CT}}}}\) translates into large overpotentials for the LHCEs even at low rates, and results in the uniform distribution of Li plating and stripping sites. The beneficial effect of high \({R}_{{{\rm{CT}}}}\) on the reaction uniformity aligns with previous studies66,67,68, in which the interfacial resistance was proposed as a key metric and the results were rationalized using the Damkohler (Da) and Wagner (Wa) numbers. Considering the detection of negligible Si components in the SEI layer (Fig. 3d) and the insignificant increases in the overpotentials of electrolytes containing only 10 vol% DFTMS or TFTMS (Supplementary Fig. S30), the observed high \({R}_{{{\rm{CT}}}}\) is likely attributable to the solvation environments induced by the DFTMS and TFTMS diluents rather than the SEI characteristics. Considering the high \({R}_{{{\rm{CT}}}}\) consistently observed in silane-based electrolytes,61,63,64 the silicon atom in the two diluents may effectuate the rise in \({R}_{{{\rm{CT}}}}\).
Benchmarking the extreme fast-charge standard for commercial LIBs (achieving 80% SOC within 15 min)69, we evaluated the fast-charging performance of full-cells using a constant current and constant voltage (CC/CV) charging protocol at 4 C, with a time cutoff of 15 min for the combined CC and CV phases (Fig. 5e). To evaluate the reversibility at prolonged discharge coupled with fast charge, the discharge rate (nC) was varied for each full-cell to include a prolonged rate (0.3 C), an intermediate rate (1 C), and a fast rate (4 C). Consistent with the SEM images of the Li | |Li symmetric cell tests (Fig. 5a), the cyclability of the 2 M LiFSI DME full-cells deteriorated significantly at lower discharge rates due to the formation of unevenly localized stripping sites (Fig. 5f). For example, abrupt capacity decay was observed around the 37th cycle at the 0.3 C discharge rate. In stark contrast, full-cells with 2 M LiFSI DME-DFTMS 1:2 v (Fig. 5f) and 2 M LiFSI DME-TFTMS 1:2 v (Supplementary Fig. S31) retained 93.5% and 88.2% of their original capacities for 100 cycles at the same 0.3 C discharge rate. The 2 M LiFSI DME-DFTMS 1:2 v full-cell reached a high SOC of 87.0% at 4 C charge within the 15-min cutoff (compared to the formation capacity at 0.2 C) and stable capacity retention of 78.5% for 300 cycles at a 4 C discharge rate. Notably, even at a fast charging rate of 10 C (16 mA cm−2) with a 6-min time cutoff, this cell demonstrated improved rate capability: an SOC of 77.3% and capacity retention of 81.3% for 200 cycles (Fig. 5g, h). By contrast, the 2 M LiFSI DME-TFTMS 1:2 v full-cell performed less effectively due to its larger ion clusters. Under the same 10 C charge and 6 min cutoff conditions, it reached an SOC of 74.0% and maintained only 32.2% after 200 cycles.
The effect of electrolyte design on Li ion-cluster transport through the SEI layer is graphically illustrated in Fig. 6. Although enriching inorganic components in the SEI layer via ion-pair clustering is an effective approach under the slow charging condition, reducing the size of Li-ion clusters is preferred for their facile transport through the SEI layer and thus avoiding the depletion-induced dendritic growth during fast-charging.

The top panel illustrates the molecular design strategies to enhance the electrostatic potential difference of the diluent. The bottom panel provides a comparative schematic of the resulting Li-ion transport behaviors within the swollen SEI layer.
Indeed, prior studies have well established that ion-cluster disaggregation can be controlled by factors within the primary solvation shell, such as coordination affinity and steric hindrance58,59. In contrast, this study highlights the role of ΔESP of non-coordinating diluents. Even without direct solvation, a diluent with a larger ΔESP effectively disassembles the ion clusters by electrostatically weakening the interactions between charged clusters. Although the dielectric constant is a well-established macroscopic descriptor for accessing Coulombic shielding in individual media, at the molecular level, we propose ΔESP to target diluents in LHCEs with reasonably similar molecular structures, particularly for estimating their ion cluster sizes and fast-charging performance. Our rationale was further supported by comparisons with other LHCE systems, where diluents with higher ΔESP values exhibit more pronounced ion-cluster disaggregation and improved fast-charging performance (Supplementary Figs. S32−S41). Nonetheless, we note that diluents with substantial structural differences may show varying degrees of correlation between ΔESP and ion-cluster disaggregation.
Furthermore, this transport phenomena in the SEI layer must be mutually affected by the properties of the SEI matrix. Based on the recently proposed swollen SEI model27, we establish a scenario for Li-ion transport in the SEI layer: Li ion clusters enter the relatively loose upper portion of the swollen SEI layer (with limited kinetics) and gradually become de-solvated as they approach the electrode surface. Notably, as indicated by the similar \({R}_{{\mathrm{CT}}}\) between the two LHCEs (Fig. 5d), the de-solvation process near the electrode surface was found to have minimal impact on the charging kinetics.
Discussion
Fast charging combined with prolonged discharging has long been challenging to maintain over a significant number of cycles in LMBs due to the Li (de)plating characteristics that impede reversibility. During charging, ion depletion near the electrode surface, which becomes more pronounced with increasing current density, is the main obstacle that hinders fast charging ability, whereas the localized, nonuniform formation of stripping sites makes prolonged discharging challenging. To overcome these fundamental issues, this study identified two key parameters associated with Li-ion transport and reactions on the Li metal: the size of Li-ion clusters for facile Li ion transport through the SEI layer and electrode polarization (or overpotential). DFTMS was identified as a suitable diluent to address these problems. The enlarged molecular size arising from the incorporation of Si and the partial fluorination jointly heighten its ΔESP, allowing ion pairs in the electrolyte to be electrostatically shielded, thus reducing the sizes of Li-ion clusters. The reduced cluster sizes facilitate Li ion transport through the SEI layer, enabling robust fast charging without severe dendritic growth. Moreover, the minimal difference in the de-solvation energy between the two LHCEs, together with the aforementioned cluster size-dependent kinetics, led us to propose a picture on the SEI conformation and Li-ion transport therein: Li-ion clusters diffuse through the upper portion of the loosened SEI layer with limited kinetics and are gradually de-solvated as they approach the electrode surface. We anticipate that the elucidation of the SEI layer in terms of its internal ion transport mechanisms will play a crucial role in developing more advanced electrolytes. On the other hand, the suitably increased overpotential resulting from the addition of DFTMS promotes spatially uniform stripping sites for stable discharge even at moderate stripping rates. This study highlights the potential for realizing the desired combination of fast charging and prolonged discharging in LMBs through optimizing the molecular design of diluents and a deeper understanding of the SEI properties on Li metal electrodes.
Methods
Materials
1,2-dimethoxyethane (DME, anhydrous, 99.5% inhibitor free), (trifluoromethyl)trimethylsilane (TFTMS, 99%), 1,1,2,2-tetrafluoroethyl 2,2,3,3-tetrafluoropropylether (TTE, >99%), and methyl trifluoroacetate (MTFACO, 99%) were purchased from Sigma-Aldrich (USA). Lithium bis(fluorosulfonyl)imide (LiFSI, >98%), (difluoromethyl)trimethylsilane (DFTMS, >97%), bis(2,2,2-trifluoroethyl) ether (BTFE, >99%), and methyl difluoroacetate (MDFACO, >97%) were purchased from TCI Chemicals (Japan). Li (20 μm thick on Cu foil), Cu foil, Al foil, LiNi0.8Co0.1Mn0.1 (NCM811), poly(vinylidene difluoride) (PVDF, HSV900) binder, and the polyethylene (PE) separator were obtained from Wellcos Corporation (South Korea). Super P was purchased from MTI Corporation (South Korea). N-methyl-2-pyrrolidone (NMP) was purchased from Junsei (Japan).
Electrochemical measurements
All coin cells and electrolytes were prepared and handled in an argon-filled glovebox. All electrochemical measurements were conducted by fabricating CR2032-type coin cells, into which 60 μL electrolyte was injected. The Li foil electrodes were 12 mm in diameter and 300 μm in thickness for the Li | |Cu asymmetric cells and Li | |Li symmetric cells, whereas the dimensions of the thin lithium foil were 10 mm and 20 μm (0.0079 g including Cu foil), respectively, for testing full-cells with a low N/P ratio of 2.5. A polyethylene separator (18 mm diameter; 0.02751 g) and a 1 T stainless steel spacer (1.6019 g) were used. The internal components included a case (0.9090 g), cap (0.7946 g), spring (0.1664 g), and gasket (0.0986 g). Prior to conducting measurements, all cells were allowed to rest for 6 hours to ensure sufficient infiltration of the electrolytes through the electrodes. For the GITT analysis, homemade three-electrode cells were used. In these cells, working electrode (WE) and counter electrode (CE) were Li foil, while reference electrode (RE) was Li-deposited Cu mesh. Throughout the GITT measurement, data points were acquired at a frequency of 1 Hz (one point per second) during rest periods. To evaluate the electrochemical characteristics, galvanostatic cycling, LSV, and chronoamperometry were conducted using a WonATech WBCS3000 battery testing system (South Korea). The interfacial properties were investigated via potentiostatic electrochemical impedance spectroscopy (PEIS) on a Bio-Logic VSP workstation. The PEIS spectra were recorded within a frequency spectrum of 1 MHz to 50mHz with a sinusoidal potential amplitude of 10 mV. The sampling density was 20 points per decade. Before each measurement, the cells were allowed to rest at open-circuit voltage (OCV) for 1 h to reach a quasi-stationary state. To ensure reproducibility, all electrochemical tests were performed in duplicate (n = 2). The EIS spectra were fitted using an equivalent circuit model of Rs + (RSEI/CPESEI) + (RCT/CPECT) + (Rint/CPEint), where Rs, RSEI, RCT, and Rint represent the series resistance, SEI layer resistance, charge-transfer resistance, and additional interfacial resistance, respectively. Constant phase elements (CPE) were used instead of pure capacitors to account for the non-ideal capacitive behavior of the electrodes.
To prepare the positive electrodes for full-cell evaluation, a homogenized paste was formulated by dispersing NCM811, Super P, and PVDF binder (weight ratio = 94:3:3) within N-methyl-2-pyrrolidone (NMP). This mixture was subsequently applied to an aluminum foil current collector through a doctor-blade coating process. The resulting electrodes were subjected to a two-step drying protocol: an initial treatment at 80 °C under ambient pressure for 6 h, followed by overnight storage in a vacuum oven at 60 °C. Afterwards, the single-side coated NCM811 electrodes were pressed using a roll press to reach a density of 3 g cm−3. The loading of the NCM811 active material was 8 mg cm−2, which corresponds to 1.6 mAh cm−2. The positive electrodes were then cut into circular discs with a diameter of 10 mm (0.010915 g of active material on 0.004294 g of Al current collector). All the full-cells were activated by operating them for three formation cycles at 0.2 C (1 C = 200 mA g−1) in CC mode for both charge and discharge in the potential range of 2.7 − 4.4 V. All electrochemical experiments were carried out in a climatic chamber at 25 \(\pm\)1 °C. The capacity retention was calculated relative to the maximum discharge capacity achieved immediately after the formation process (three cycles at 0.2 C).
Characterization of materials
Field emission scanning electron microscopy (FE-SEM) images were captured using a JSM-7800F Prime instrument (JEOL, Japan) at an accelerating voltage of 15 kV. X-ray photoelectron spectroscopy (XPS) was conducted on an AXIS-His (KRATOS, U.K.), with the XPS peak positions calibrated against the C 1 s peak at 284.8 eV. Raman vibrational signatures were captured via a 785 nm laser excitation using a Thermo Fisher DXR2xi spectrometer (USA). To investigate the chemical properties of the electrolytes, 7Li nuclear magnetic resonance (NMR) spectra were collected on a 500 MHz Bruker Avance III system (USA). 1 M LiCl in D2O was used as a reference within a coaxial NMR tube setup. Furthermore, an InLab 710 probe (METTLER TOLEDO, USA) was employed to evaluate the ionic conductivity at 25 °C. These analyses, along with FE-SEM characterizations, were carried out at the National Centre for Inter-university Research Facilities (NCIRF) at Seoul National University. For ex situ characterizations (SEM, XPS, and Cryo-TEM), the cycled cells were disassembled in an argon-filled glovebox (H2O < 0.1 ppm, O2 < 0.1 ppm). The harvested lithium electrodes were gently rinsed with anhydrous DME to remove residual electrolyte and salts, followed by drying. DFT calculations were carried out using the Gaussian 09 software package (Frisch, M.J. et al. Gaussian 09, Revision A.02; Gaussian, Inc., Wallingford, CT, 2016), supported by the Korea Institute of Science and Technology Information (KISTI). Geometry optimizations and molecular orbital calculations were performed without imposing symmetry restrictions, using the B3LYP hybrid density functional and the 6-311 G + + (d, p) basis sets. MD simulations were performed using GROMACS version 2020.6. The parameter values for each molecule were derived from the optimized potentials for liquid simulation all-atom (OPLS-AA) force field. For Li+/FSI−, previously reported values70 with a charge scaled by 0.8 were employed. The parameters of solvents and diluents, including topology files, bonded parameters, and Lennard–Jones parameters, were obtained from the LigParGen server. For each system (77 LiFSI, 124 DME, 175 TFTMS (189 DFTMS) for 2 M LHCE electrolytes), energy minimization was first performed using the steepest descent method. The system was then equilibrated at 1 bar and 300 K for 1 ns using the V-rescale thermostat and Berendsen barostat. A subsequent equilibrium run was carried out for 50 ns using the Nose-Hoover thermostat and Parrinello-Rahman barostat, with a time step of 1 fs. The final production simulation was conducted in the NVT ensemble at 300 K for 10 ns with the Nose-Hoover thermostat at the same time step. The cutoff distance for Lennard-Jones and electrostatic interactions was set to 1.0 nm. Long-range electrostatics were calculated using the particle mesh Ewald (PME) method. Holonomic constraints on hydrogen atoms were applied using the LINCS algorithm to maintain stability. Small- and wide-angle X-ray scattering (SAXS/WAXS) data were obtained at the 9 A U-SAXS beamline and 4 C SAXS II beamline at Pohang Accelerator Laboratory (Pohang, Republic of Korea). Cryogenic transmission electron microscope (Cryo-TEM) analysis was conducted at 120 kV with a Talos L120C (FEI company, Czech Rep.) at Seoul National University National Instrumentation Center for Environmental Management (SNU NICEM). Time-of-flight secondary ion mass spectrometry (TOF-SIMS) analysis was conducted using an M6 instrument (ION-TOF, Münster, Germany) at the Advanced Analysis Center of the Korea Institute of Science and Technology (KIST). Bi3+ ions (30 keV, 0.2 pA) were used as the primary ion beam for analysis. All measurements were performed in negative ion mode.
Data availability
The data generated in this study are provided in the Supplementary Information and Source Data file. Source data are provided with this paper.
References
Kim, S. et al. Lithium-metal batteries: from fundamental research to industrialization. Adv. Mater. 35, 2206625 (2023).
Louli, A. et al. Diagnosing and correcting anode-free cell failure via electrolyte and morphological analysis. Nat. Energy 5, 693–702 (2020).
Jie, Y. et al. Towards long-life 500 Wh kg−1 lithium metal pouch cells via compact ion-pair aggregate electrolytes. Nat. Energy 9, 987–998 (2024).
Li, Y. et al. Correlating structure and function of battery interphases at atomic resolution using cryoelectron. Microsc. Joule 2, 2167–2177 (2018).
He, Y. et al. Origin of lithium whisker formation and growth under stress. Nat. Nanotech. 14, 1042–1047 (2019).
Kwon, H. et al. Borate–pyran lean electrolyte-based Li-metal batteries with minimal Li corrosion. Nat. Energy 9, 57–69 (2024).
Zheng, J. et al. Highly stable operation of lithium metal batteries enabled by the formation of a transient high-concentration electrolyte layer. Adv. Energy Mater. 6, 1502151 (2016).
Zheng, J. et al. Electrolyte additive enabled fast charging and stable cycling lithium metal batteries. Nat. Energy 2, 17012 (2017).
Qian, J. et al. Anode-free rechargeable lithium metal batteries. Adv. Funct. Mater. 26, 7094–7102 (2016).
Sun, Z. et al. Directing (110) Oriented lithium deposition through high-flux solid electrolyte interphase for dendrite-free lithium metal batteries. Angew. Chem. Int. Ed. 62, e202309622 (2023).
Wu, Z. et al. Growing single-crystalline seeds on lithiophobic substrates to enable fast-charging lithium-metal batteries. Nat. Energy 8, 340–350 (2023).
Yan, K. et al. Selective deposition and stable encapsulation of lithium through heterogeneous seeded growth. Nat. Energy 1, 1–8 (2016).
Kim, M. S. et al. Revealing the multifunctions of Li3N in the suspension electrolyte for lithium metal batteries. ACS nano 17, 3168–3180 (2023).
Ma, B. et al. Molecular-docking electrolytes enable high-voltage lithium battery chemistries. Nat. Chem. 16, 1427–1435 (2024).
Zhao, Y. et al. Fluorinated ether electrolyte with controlled solvation structure for high voltage lithium metal batteries. Nat. Commun. 13, 2575 (2022).
Kim, S. C. et al. High-entropy electrolytes for practical lithium metal batteries. Nat. Energy 8, 814–826 (2023).
Efaw, C. M. et al. Localized high-concentration electrolytes get more localized through micelle-like structures. Nat. Mater. 22, 1531–1539 (2023).
Hobold, G. M., Wang, C., Steinberg, K., Li, Y. & Gallant, B. M. High lithium oxide prevalence in the lithium solid–electrolyte interphase for high Coulombic efficiency. Nat. Energy 9, 580–591 (2024).
Maraschky, A. & Akolkar, R. Mechanism explaining the onset time of dendritic lithium electrodeposition via considerations of the Li+ transport within the solid electrolyte interphase. J. Electrochem. Soc. 165, D696 (2018).
Mani, A. & Bazant, M. Z. Deionization shocks in microstructures. Phys. Rev. E 84, 061504 (2011).
Santos, E. & Schmickler, W. The crucial role of local excess charges in dendrite growth on lithium electrodes. Angew. Chem. Int. Ed. 60, 5876–5881 (2021).
Rosso, M. Electrodeposition from a binary electrolyte: new developments and applications. Electrochim. Acta 53, 250–256 (2007).
Chazalviel, J.-N. Electrochemical aspects of the generation of ramified metallic electrodeposits. Phys. Rev. A 42, 7355 (1990).
Kim, M. et al. Cationic additive with a rigid solvation shell for high-performance zinc ion batteries. Angew. Chem. Int. Ed. 61, e202211589 (2022).
Cao, X. et al. Stability of solid electrolyte interphases and calendar life of lithium metal batteries. Energy Environ. Sci. 16, 1548–1559 (2023).
Han, B. et al. Cryo-electron tomography of highly deformable and adherent solid-electrolyte interphase xxoskeleton in Li-metal batteries with ether-based electrolyte. Adv. Mater. 34, 2108252 (2022).
Zhang, Z. et al. Capturing the swelling of solid-electrolyte interphase in lithium metal batteries. Science 375, 66–70 (2022).
Boyle, D. T. et al. Resolving current-dependent regimes of electroplating mechanisms for fast charging lithium metal anodes. Nano Lett. 22, 8224–8232 (2022).
Kushima, A. et al. Liquid cell transmission electron microscopy observation of lithium metal growth and dissolution: Root growth, dead lithium and lithium flotsams. Nano Energy 32, 271–279 (2017).
Park, K., Song, J. & Lee, K. T. Fast charging lithium metal batteries with liquid and solid-state electrolytes. Batter. Supercap. 6, e202300344 (2023).
Yuan, X., Liu, B., Mecklenburg, M. & Li, Y. Ultrafast deposition of faceted lithium polyhedra by outpacing SEI formation. Nature 620, 86–91 (2023).
Gireaud, L., Grugeon, S., Laruelle, S., Yrieix, B. & Tarascon, J.-M. Lithium metal stripping/plating mechanisms studies: a metallurgical approach. Electrochem. Commun. 8, 1639–1649 (2006).
Shi, F. et al. Lithium metal stripping beneath the solid electrolyte interphase. Proc. Natl. Acad. Sci. USA 115, 8529–8534 (2018).
Xu, Y. et al. Sweeping potential regulated structural and chemical evolution of solid-electrolyte interphase on Cu and Li as revealed by cryo-TEM. Nano Energy 76, 105040 (2020).
Nagy, K. S., Kazemiabnavi, S., Thornton, K. & Siegel, D. J. Thermodynamic overpotentials and nucleation rates for electrodeposition on metal anodes. ACS Appl. Mater. Interfaces 11, 7954–7964 (2019).
Golozar, M. et al. In situ observation of solid electrolyte interphase evolution in a lithium metal battery. Commun. Chem. 2, 131 (2019).
Wang, C. et al. Tension-induced cavitation in Li-metal stripping. Adv. Mater. 35, 2209091 (2023).
Pei, A., Zheng, G., Shi, F., Li, Y. & Cui, Y. Nanoscale nucleation and growth of electrodeposited lithium metal. Nano Lett. 17, 1132–1139 (2017).
Kim, M. et al. Surface overpotential as a key metric for the discharge–charge reversibility of aqueous zinc-ion batteries. J. Am. Chem. Soc. 145, 15776–15787 (2023).
Yan, K. et al. Temperature-dependent nucleation and growth of dendrite-free lithium metal anodes. Angew. Chem. Int. Ed. 58, 11364–11368 (2019).
Ren, X. et al. High-concentration ether electrolytes for stable high-voltage lithium metal batteries. ACS Energy Lett. 4, 896–902 (2019).
Qian, J. et al. High rate and stable cycling of lithium metal anode. Nat. Commun. 6, 6362 (2015).
Philip, M. A. et al. Enabling high capacity and coulombic efficiency for Li-NCM811 cells using a highly concentrated electrolyte. Batter. Supercap. 4, 294–303 (2021).
Hobold, G. M., Kim, K.-H. & Gallant, B. M. Beneficial vs. inhibiting passivation by the native lithium solid electrolyte interphase revealed by electrochemical Li+ exchange. Energy Environ. Sci. 16, 2247–2261 (2023).
Tao, R. et al. Kinetics tuning the electrochemistry of lithium dendrites formation in lithium batteries through electrolytes. ACS Appl. Mater. Interfaces 9, 7003–7008 (2017).
Wood, K. N. et al. Dendrites and pits: untangling the complex behavior of lithium metal anodes through operando video microscopy. ACS Cent. Sci. 2, 790–801 (2016).
Jagger, B. & Pasta, M. Solid electrolyte interphases in lithium metal batteries. Joule 7, 2228–2244 (2023).
Xia, Y. et al. Designing an asymmetric ether-like lithium salt to enable fast-cycling high-energy lithium metal batteries. Nat. Energy 8, 934–945 (2023).
Weiss, M. et al. Fast charging of lithium-ion batteries: a review of materials aspects. Adv. Energy Mater. 11, 2101126 (2021).
Lv, S. et al. Operando monitoring the lithium spatial distribution of lithium metal anodes. Nat. Commun. 9, 2152 (2018).
Xu, K. Navigating the minefield of battery literature. Commun. Mater. 3, 31 (2022).
Holoubek, J. et al. Tailoring electrolyte solvation for Li metal batteries cycled at ultra-low temperature. Nat. Energy 6, 303–313 (2021).
Ruan, D. et al. Solvent versus anion chemistry: unveiling the structure-dependent reactivity in tailoring electrochemical interphases for lithium-metal batteries. Jacs Au 3, 953–963 (2023).
Zhang, H. et al. Simultaneous stabilization of lithium anode and cathode using hyperconjugative electrolytes for high-voltage lithium metal batteries. Angew. Chem. Int. Ed. 62, e202218970 (2023).
Murray, J. S. & Politzer, P. The electrostatic potential: an overview. Wiley Interdiscip. Rev. Comput. Mol. Sci. 1, 153–163 (2011).
Wang, J. et al. A wide bandgap acceptor with large dielectric constant and high electrostatic potential values for efficient organic photovoltaic cells. J. Am. Chem. Soc. 145, 13686–13695 (2023).
Wang, H. et al. Correlating Li-ion solvation structures and electrode potential temperature coefficients. J. Am. Chem. Soc. 143, 2264–2271 (2021).
Zhang, G. et al. A monofluoride ether-based electrolyte solution for fast-charging and low-temperature non-aqueous lithium metal batteries. Nat. Commun. 14, 1081 (2023).
Wang, Y. et al. Difluoroester solvent toward fast-rate anion-intercalation lithium metal batteries under extreme conditions. Nat. Commun. 15, 5408 (2024).
Liu, T. et al. Low-density fluorinated silane solvent enhancing deep cycle lithium–sulfur batteries’ lifetime. Adv. Mater. 33, 2102034 (2021).
Li, Y. et al. Single-solvent-based electrolyte enabling a high-voltage lithium-metal battery with long cycle life. Adv. Energy Mater. 13, 2300918 (2023).
Chen, L. et al. Dynamic shielding of electrified interface enables high-voltage lithium batteries. Chem 10, 1196–1212 (2024).
Huang, Y. et al. Eco-friendly electrolytes via a robust bond design for high-energy Li metal batteries. Energy Environ. Sci. 15, 4349–4361 (2022).
Li, M. et al. Two birds with one stone: engineering siloxane-based electrolytes for high-performance lithium–sulfur polyacrylonitrile batteries. J. Mater. Chem. A 11, 11721–11729 (2023).
Adams, B. D., Zheng, J., Ren, X., Xu, W. & Zhang, J. G. Accurate determination of coulombic efficiency for lithium metal anodes and lithium metal batteries. Adv. Energy Mater. 8, 1702097 (2018).
Liu, Y. et al. Insight into the critical role of exchange current density on electrodeposition behavior of lithium metal. Adv. Sci. 8, 2003301 (2021).
Jo, S. et al. The roles of nucleation and growth kinetics in determining Li metal morphology for Li metal batteries: columnar versus spherical growth. J. Mater. Chem. A 10, 5520–5529 (2022).
Hao, F., Verma, A. & Mukherjee, P. P. Mesoscale complexations in lithium electrodeposition. ACS Appl. Mater. Interfaces 10, 26320–26327 (2018).
Liu, Y., Zhu, Y. & Cui, Y. Challenges and opportunities towards fast-charging battery materials. Nat. Energy 4, 540–550 (2019).
Gouveia, A. S. et al. Ionic liquids with anions based on fluorosulfonyl derivatives: from asymmetrical substitutions to a consistent force field model. Phys. Chem. Chem. Phys. 19, 29617–29624 (2017).
Acknowledgements
J.W.C. acknowledges financial support from the National Research Foundation of Korea (RS-2024-00335274 and RS-2023-00261543) and generous support from the Institute of Engineering Research (IOER), the Institute for Battery Research Innovation (IBRI), and the Research Institute of Advanced Materials (RIAM) at Seoul National University. This work was also supported by the Swiss National Science Foundation (SNF) (Grant No. Sinergia CRSII5_202296).
Author information
Authors and Affiliations
Contributions
M.K. and J.K. contributed equally to this work. M.K., J.K., and J.W.C. conceived the idea. M.K. and J.K. performed the electrochemical measurements and physicochemical characterizations. M.K. conducted the DFT calculations and X-ray scattering analyses. J.K. conducted the MD simulations. M.B. assisted in conceptualizing the research. J.O. and J.L. contributed to the electrochemical measurements. M.K., J.K., A.C., and J.W.C. wrote the manuscript. A.C. and J.W.C. supervised the project. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zehao Cui, Tao Li, Qisheng Wu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, M., Kim, J., Baek, M. et al. Electrolyte diluent with large electrostatic potential difference for fast charging and slow discharging lithium metal batteries. Nat Commun 17, 3183 (2026). https://doi.org/10.1038/s41467-026-69870-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69870-7
