
Abstract
A plausible hypothesis for the origin of biological homochirality invokes chiral symmetry breaking, transfer, and amplification driven by autocatalysis. Here we experimentally demonstrate that crystallization-driven template autocatalysis (CDTA) induces mirror symmetry breaking and amplification in helices. CDTA enables the reductive cyclotetramerization of hydrogen-bonded naphthalonitrile precursors into crystalline fibers of naphthalocyanine derivatives. In achiral or racemic systems, a kinetically controlled right-handed helical bias emerges during secondary nucleation and develops into P-helical dominance as the fibers elongate, achieving mirror symmetry breaking. CDTA also transfers the single handedness of chiral seeds formed from enantiopure analogs to achiral naphthalocyanines through template-assisted replication, resulting in chiral amplification. A key mechanistic step involves the preorganization of naphthalonitrile molecules in a counterclockwise direction at the termini of P-helical fibers via J-type π–π stacking and hydrogen-bonding interactions for autocatalytic transformation. Thus, once mirror symmetry is broken, the resulting chiral imbalance is amplified in a self-replicating manner.
Similar content being viewed by others

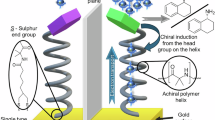
Introduction
The emergence of biological homochirality represents one of the most perplexing mysteries in science, captivating generations of researchers1,2. In 1953, Frank proposed a model of asymmetric autocatalysis, wherein an enantiomeric product acts as a chiral catalyst for its own asymmetric synthesis3. This conceptual model postulated that an initial minute enantiomeric excess breaks chiral symmetry, followed by enantioenrichment towards homochirality driven by asymmetric autocatalysis4,5. The first experimental validation of this principle came in 1995 with the Soai reaction, which demonstrated the stochastic generation of enantiomerically enriched compounds from achiral reactants6. This autocatalytic reaction constitutes a spontaneously symmetry-breaking and chirality-amplifying process capable of achieving molecular homochirality7,8,9,10. Over the past decades, diverse strategies, including other autocatalytic reactions11,12,13,14, templated polymerization15,16, conglomerate deracemization17,18,19, and kinetic control20,21 have been extensively studied to achieve symmetry breaking and chiral amplification at the molecular level.
Besides, supramolecular homochirality exists in nature as well. For instance, the double-helix architecture of DNA molecules is predominantly right-handed22. Molecular self-assembly has serendipitously revealed that some achiral building blocks form chiral supramolecular structures with enantiomeric imbalance23,24. Early reports of such anomalous symmetry breaking focused primarily on liquid crystalline systems25,26,27. Recent research interest has shifted toward solution-phase processes for generating supramolecular chirality from achiral molecules, particularly within asymmetric environments such as mechanical vortices28,29,30, external polarized fields31,32, spiral confinements33, and chiral matrices34,35. However, the most intriguing scenario involves spontaneous symmetry breaking without discernible asymmetric influences, where asymmetry originates solely from statistical fluctuations36,37,38. In parallel, two key chemical principles of the majority rule39,40,41 and sergeants-and-soldiers (S&S) effect42,43 have been established for amplifying supramolecular chirality. Both symmetry breaking and chiral amplification have thus been rationalized at the supramolecular level.
The question of how homochirality emerged within primordial self-replication events remains profoundly elusive. This is partly because the chirality of artificial self-replicators induced by the interplay of autocatalysis and self-organization has received little attention44. Otto and coworkers developed enantioselective self-replicators capable of chirality-amplified growth exclusively from enantiopure precursors matching their own handedness45. However, their system did not address the first step of the mirror symmetry breaking. Recently, Aida and coworkers reported solvent-free autocatalytic supramolecular polymerization, enabling high-yielded synthesis of single-crystalline phthalocyanine derivatives46. Our further study identified the trans-amide conformation as playing a key role in this crystallization-driven template autocatalysis47 (CDTA).
In this study, we extend CDTA to the efficient synthesis of naphthalocyanine (NC) analogs using fan-shaped, hydrogen-bonding (H-bonding) naphthalonitrile (NN) derivatives as precursors (Fig. 1a). Investigation of the chirality effects on CDTA revealed an unexpected outcome that achiral or racemic NC products consistently organize into columnar helices with predominance of right-handed (P-type) over left-handed (M-type) helicity. However, CDTA using enantiopure NN precursors exclusively yields single-handed helices formed by the corresponding NC derivatives (Fig. 1b). A key step involves the NC templates at the termini of P- or M-helical fibers, which likely preorganize incoming NN molecules in a counterclockwise (CCW) or clockwise (CW) orientation, respectively. This preorganization, mediated by J-type π–π stacking and H-bonding interactions, facilitates their cyclotetramerization for autocatalytic transformation (Fig. 1b). Owing to this template-assisted replication mechanism, any helical bias emerging during nucleation is significantly amplified, ultimately affording enrichment in defined helicity.

a CDTA reaction of naphthalonitrile (NN) precursors into the corresponding naphthalocyanine (NC) derivatives in the presence of a thiol compound (R’SH). b Schematic of the mirror symmetry breaking and amplification in columnar helices induced by CDTA. The target product, if any is formed, nucleates and elongates via noncovalent interactions, affording crystalline helical fibers. The termini of P- or M-helical fibers may certainly preorganize incoming precursors in a counterclockwise or clockwise orientation, respectively, and efficiently promote their chemical transformation in an autocatalytic manner.
Results
CDTA reactions of NN precursors
Motivated by molecular engineering of CDTA (refs. 46,47,48), we designed and synthesized a fan-shaped NNA featuring two amide groups and peripheral achiral n-butyl ether chains (Fig. 1a). To study chirality effects, two enantiopure NNR and NNS were prepared as references by partial substitution with (R)- or (S)−2-methylbutyl ether chains as chiral centers, respectively (Fig. 1a). All NN precursors were thoroughly characterized by nuclear magnetic resonance spectroscopy (NMR; Supplementary Figs. 1–3) and high-resolution mass spectrometry (HR-MS; Supplementary Figs. 4 and 5).
Following a well-established CDTA protocol (see Methods), we subjected the reaction mixtures of NN precursors and 1-dodecanethiol (DCTH, 25 mol%), acting as both an essential proton/electron donor and a solvent for NC synthesis, to heating at 185 °C for 15 h (Fig. 2a). In situ optical microscopy (OM) images (Fig. 2b) illustrated that after ~3 h of heating, numerous yellow-green colored twisted fibers nucleated abruptly and subsequently underwent rapid growth within 7 h (Supplementary Movie 1). Polarized optical microscopy (POM; Fig. 2c) confirmed the crystalline nature of the as-formed fibers with distinct birefringence. Isolation of these crystalline fibers by simply washing with methanol afforded analytically pure naphthalocyanine NCA (Supplementary Fig. 6). Further spectroscopic analysis revealed that the residual NNA precursors were completely dissolved in the wash solvent, with only trace amounts of side products detectable (Supplementary Fig. 7), demonstrating the high efficiency and selectivity of the CDTA reaction. We likewise heated enantiopure NNR (Supplementary Movie 2) and NNS (Supplementary Movie 3) to yield crystalline fibers identified spectroscopically as enantiopure NCR and NCS, respectively (Fig. 2d, e, Supplementary Figs. 8 and 9). Time-dependent absorbance on the characteristic Q-bands of all NC products displayed pseudo-sigmoidal kinetic profiles with autocatalytic feature (Fig. 2f).

a Schematic illustration of in situ optical microscopy (OM) observation on CDTA reactions on a hot stage. b Real-time OM images of the reaction mixture obtained by CDTA of NNA with 1-dodecanethiol (DCTH) upon heating at 185 °C. Scale bars, 50 μm. OM (left) and polarized optical microscopy (POM, right) images of the reaction mixtures obtained by CDTA of NNA (c), NNR (d), and NNS (e) with DCTH after heating at 185 °C for 15 h. White arrows represent transmission axes of the polarizer (P) and analyzer (A). Scale bars, 50 μm. f, Time-dependent profiles of absorbance (Abs.) at 810 nm of the reaction mixtures obtained by NNA (green), NNR (red), and NNS (blue) with DCTH upon heating at 185 °C. g Phase-transition diagram of NN precursors and the corresponding NC products. Cr. and Iso. denoted as crystalline and isotropic phases, respectively. h PXRD patterns of as-formed [NCA]CF (green), [NCR]CF (blue), and [NCS]CF (red) after washing with methanol and schematic illustration of its columnar order with a 2D rectangular geometry of C2/m.
A systematic study revealed that CDTA using NN precursors is considerably affected by the reaction temperature. Elevation of the heating temperature from 175 to 195 °C increasingly produced the more distorted fibers (Supplementary Figs. 10 and 11). As established previously46,47, CDTA requires reactive precursors to keep melting while template products maintain crystalline fibers with columnar packing. Differential scanning calorimetry (DSC) analysis (Supplementary Fig. 12) of NN precursors and corresponding NC derivatives confirmed that all NC products exhibit higher melting points and superior thermal stability compared to their NN precursors (Fig. 2g). As a consequence, the as-formed crystalline fibers of NC products, denoted hereafter as [NC]CF, remained structurally intact within the molten NN matrices, enabling efficient CDTA reaction within an optimal temperature window. Notably, the overheating-induced melting of [NC]CF permanently eliminated their fibrous architecture and thermal history (Supplementary Fig. 12), suggesting that [NC]CF obtained via CDTA might be in a non-equilibrium stationary state.
Further structural characterization identified that all NCs adopt nearly identical crystalline packing irrespective of the chirality. As shown in Fig. 2h, powder X-ray diffraction (PXRD) patterns of the isolated [NC]CF exhibited intense peaks indexable to a rectangular columnar lattice in a space group of C2/m (Supplementary Tables 1–3). Evaluation of unit cell parameters indicated that each column comprises J-aggregated NC stacks with a tilt angle of ~20° (Fig. 2h), consistent with the distinct red-shifted absorption observed in Q-bands (Supplementary Fig. 6e). Polarized Fourier transform infrared (FT-IR) spectroscopy corroborated this molecular arrangement, where the characteristic amide C=O and N–H stretches exhibited maximum absorbance when the polarized vector was parallel to the fiber axis (Supplementary Fig. 13). This dichroism implied the long-range alignment of H-bonded amide groups along the long axis of a fiber, which was subsequently supported by theoretical calculations discussed later.
CDTA-induced mirror symmetry breaking
One aim of this study was to explore the chiral properties of [NC]CF obtained by CDTA. Although [NCA]CF is inherently achiral, its suspension in chloroform displayed a clear positive Cotton effect at 680 nm (Fig. 3a), corresponding to the Q-band adsorption of NCA. Scanning electron microscopy (SEM; Fig. 3b) revealed the formation of entangled helical fibers with a statistical predominance of P-type over M-type (Supplementary Movie 4), indicating the occurrence of mirror symmetry breaking. This unusual phenomenon was reproducibly observed across different reaction temperatures and time (Supplementary Fig. 14a, b). Note that the molar ratio of NCA to DCTH significantly influenced the measured circular dichroism (CD) intensity (Fig. 3c, Supplementary Fig. 14c). Furthermore, the helical sense of the as-formed fibers obtained by CDTA became less distinct with the addition of excess DCTH (Supplementary Figs. 15 and 16). This weakening, coupled with the observed shortening of nucleation period as the molar fraction of DCTH increased (Fig. 3c and Supplementary Fig. 17), demonstrates the nucleation as a critical stage to drive mirror symmetry breaking in an achiral CDTA system.

a Circular dichroism (CD) spectra of the suspensions in chloroform of the isolated [NCA]CF (green) and [NCrac]CF (purple) obtained by CDTA, together with NCA crystallites (orange) obtained by solution crystallization (SC). b False color SEM images of the washed [NCA]CF. P- and M-helical fibers are colored with red and blue, respectively. Scale bar, 5 μm. c Plots of CD intensity at 680 nm and nucleation time against the molar ratio of NNA to DCTH ([NNA]/[DCTH]). d CD spectra of the suspensions in chloroform of the isolated [NCA]CF obtained by CDTA with (R)-terpinen-4-ol (R-dopant, red) or (S)-terpinen-4-ol (S-dopant, blue). e False color SEM images of the washed [NCA]CF obtained by CDTA with R-dopant (left) or S-dopant (right). P- and M-helical fibers are colored with red and blue, respectively. Scale bars, 10 μm. f Percentages of P-(red) and M-(blue) helical fibers obtained by CDTA of NNA with R-dopant (left) or S-dopant (right). The error bars represent the mean ± s.d. of three replicates. p values were calculated by the two-tailed t-test.
In fact, this unexpected symmetry-breaking effect is unique to CDTA. Under thermodynamic control, the NCA crystallites, which prepared by conventional solution crystallization (SC) in N, N-dimethylformamide, adopted an identical C2/m columnar lattice (Supplementary Fig. 18a and Supplementary Table 4) but showed totally silent CD signal (Fig. 3a). Their non-helical rod-like morphology (Supplementary Fig. 18b) further verified that the mirror symmetry breaking in helices can only be induced by CDTA under non-equilibrium conditions. This phenomenon extended to a racemic CDTA system. We heated a mixture of equimolar NNR and NNS to yield the [NCrac]CF (Supplementary Fig. 19), whose CD spectrum similarly showed a positive Cotton effect at 680 nm (Fig. 3a), with P-helical excess as quantified by SEM (Supplementary Movie 5). While the observed CD activity is largely intrinsic, it should be noted that linear dichroism potentially contributes to introduce superimposed artifacts (Supplementary Fig. 20), which distort the measured CD profiles away from a common multimodal shape49,50.
We pointed out that the mirror symmetry breaking in achiral CDTA can be modulated by external asymmetric sources, such as chiral dopants and circularly polarized light (CPL). Preliminary studies revealed that CDTA reactions using an NNA precursor (75 mol%) in the presence of DCTH (12.5 mol%) and (R/S)-terpinen-4-ol (12.5 mol%) produced twisted yet thinner [NCA]CF (Supplementary Fig. 21). The introduction of (R)-terpinen-4-ol dopant into an achiral CDTA system gave rise to an enhanced positive Cotton effect at 680 nm (Fig. 3d), whereas a (S)-terpinen-4-ol dopant instead inverted the CD signal, yielding a negative chiroptical response (Fig. 3d). SEM images (Fig. 3e) showed that CDTA carried out in the asymmetric matrices selectively produces helical fibers with P- or M-helicity (Fig. 3f). Similarly, the right-handed CPL, when applied during CDTA in the nucleation phase, significantly enhanced the CD intensity of the resulting [NCA]CF compared with that formed in non-irradiated regions, while the left-handed CPL produced the opposite CD signal (Supplementary Fig. 22). We confirmed that the screw direction of these helices is determined by the handedness of these external asymmetric sources mediated by enantiophobic interactions51,52.
CDTA-induced mirror amplification
Given the stereogenic centers in their side chains, we also examined the supramolecular chirality of enantiopure NC products obtained by CDTA. As expected, the [NCR]CF and [NCS]CF suspensions displayed perfect mirror-image CD spectra, showing positive and negative Cotton effects, respectively (Fig. 4a). SEM analysis confirmed the formation of single-handed helices, wherein NCR assembled into exclusively right-handed P-type helical columns while NCS formed left-handed M-type ones (Fig. 4b). It is worth mentioning that the CDTA-derived NCs show more intense chiroptical response and helical sense than their SC counterparts (Fig. 4a, Supplementary Fig. 23), indicating that CDTA enhances supramolecular chirality in enantiopure assemblies.

a CD spectra of the suspensions in chloroform of the isolated [NCR]CF (dark red), [NCS]CF (dark blue) obtained via CDTA, together with the NCR (light red) and NCS (light blue) crystallites obtained via SC. b False color SEM images of an isolated fiber of [NCR]CF (left) and [NCS]CF (right). P- and M-helical fibers are colored with red and blue, respectively. Scale bars, 5 μm. c Plot of net helicity against the ee of NNR and NNS mixtures with linear fit (R > 0.99). d Time-dependent CD spectral change profiles at 680 nm of the reaction mixtures obtained by pure NNR (dark red), pure NNS (dark blue), and NNA premixed with [NCR]CF (light red) and [NCS]CF (light blue) as chiral seeds, sandwiched with glass plates upon heating at 185 °C. e False color SEM images of heterofibers obtained by seed-induced CDTA of NNA using [NCR]CF (left) and [NCS]CF (right) as chiral seeds. P- and M-helical fibers are colored with red and blue, respectively. Scale bars, 5 μm. f Plot of net helicity against the %-molar fraction of [NCR]CF (red) and [NCS]CF (blue) chiral seeds in a hot melt of NNA with the corresponding fits (R > 0.99).
To unravel the mirror-amplification mechanism in CDTA, we performed the majority-rule experiments upon heating the mixtures of NNR and NNS precursors with varying enantiomeric excess (ee) at 185 °C for 12 h. Mirror-image CD spectra (Supplementary Fig. 24) of the resulting [NC]CF revealed a linear correlation between the ee and net helicity, as quantified by normalized CD intensity at 680 nm (Fig. 4c). The P/M-helical fibers coexisted across ee values (Supplementary Fig. 25) due to chiral self-sorting or supramolecular deracemization. The absence of nonlinear amplification conclusively rules out a majority-rule effect in CDTA-driven chiral amplification.
To gain insights into the induction of single-handed helicity in CDTA, we in situ monitored solid-state CD spectra by heating enantiopure NN precursors at 185 °C on glass plates (Supplementary Fig. 26). As shown in Fig. 4d, the CD intensity of [NCR]CF slowly rose in the nucleation stage, followed by rapid amplification with fiber elongation, while unexpectedly, [NCS]CF exhibited a delay in this critical transition. This temporal correlation indicated that crystalline nuclei may serve as chiral templates to propagate supramolecular helicity53,54. As one supporting observation for the above hypothesis, when the isolated [NCR]CF or [NCS]CF seeds were added to molten NNA, newly formed NCA fibers grew epitaxially from both termini of these chiral seeds without a nucleation lag (Supplementary Fig. 27). Likewise, CD kinetics showed immediate signal amplification due to the lack of incubation (Fig. 4d). SEM confirmed the heterofibers inherited the helicity from seeds (Fig. 4e). These results revealed that CDTA can transfer and amplify supramolecular chirality from the enantiopure seeds to an achiral system.
With seed-induced chirality transfer in mind, we performed S&S experiments via seeding-induced CDTA (see method), where [NCR]CF or [NCS]CF seeds (sergeants) with varying molar fractions were heated in the molten NNA (soldier) at 185 °C. We found that a small sergeant fraction (<10 mol%) yielded strong Cotton effects in the CD spectra of the soldier (Supplementary Fig. 28a, b). By contrast, no amplification was achieved in control experiments without CDTA feature (Supplementary Fig. 28c, d). In fact, the net helicity exceeded 0.92 upon merely adding ~6 mol% sergeant, showing exceptional S&S amplification (Fig. 4f). Fitting these data to a cooperative chain model55,56 (Supplementary Note 1) derived two key thermodynamic parameters, the helix reversal penalty (HRP) and the mismatch penalty (MMP). We observed asymmetry in HRP values (15.9 kJ mol–1 vs. 13.3 kJ mol–1) for these two sergeant enantiomers (Fig. 4f). Nevertheless, their MMPs were comparable though subject to higher fitting uncertainty due to the extensive variation observed in the graph for this mismatch penalty (Supplementary Fig. 29).
Theoretical calculations
Computational studies based on the semiempirical GFN2-xTB method57 were carried out on decameric columnar models of NC derivatives to elucidate their helix origins. Geometry optimization of these supramolecular columns revealed distinct molecular packing parameters (Fig. 5). In the homochiral columns, NCR exhibits intermolecular distances of ~3.3 Å with a tilt angle of ~22° (Fig. 5a), whereas NCS shows closer π–π stacking (~3.1 Å) due to a greater tilting angle of ~35° (Fig. 5b). Similarly, their amide groups twist ~30° from the NC plane to form H-bonds with ranging from 1.9 to 2.2 Å (Supplementary Fig. 30a, b). Notably, NCR and NCS preferentially adopt CCW and CW stacking orientations, with average rotation angles of +5.5° and –5.3°, respectively (Fig. 5a, b and Supplementary Fig. 31), resulting in well-defined helical structures. This intrinsic helicity originates from J-type π-stacking, which is reinforced by H-bond-assisted molecular staggering together with steric repulsion from chiral side chains. Despite the lack of chiral centers in the side chains, achiral decameric columns still adopt helical configurations via J-type π–π stacking and H-bonding interactions (Fig. 5c, d and Supplementary Fig. 30c, d). Yet the optimized helical sense in achiral NCA is relatively weak, exhibiting smaller rotation angles of either +2.4° or –1.2° (Fig. 5c, d and Supplementary Fig. 31). To validate the driving role of H-bonding interactions in helical structures, we further extended the computational modeling to supramolecular columns composed of twenty monomers, including NC cores (Supplementary Fig. 32a, b) and amide-functionalized NC derivatives without side chains (Supplementary Fig. 32c, d). These simulations revealed that J-aggregation into helical architectures surely requires persistent long-range H-bonding networks formed by amide groups. Furthermore, a theoretical possibility of P–M helical inversion is identified (Supplementary Fig. 32c, d), providing a mechanistic basis for the mirror-symmetry perturbation during CDTA.

Top (upper) and side (bottom) views on the optimized geometries of supramolecular helical columns with decamer stacks of NCR (a), NCS (b) and NCA (c, d) molecules based on GFN2-xTB method. The branched side chains of all NC molecules are omitted for clarity.
Discussion
We experimentally demonstrate that CDTA induces mirror symmetry breaking and chiral amplification in columnar helices. Unlike supramolecular chiral systems under thermodynamic equilibrium24,25, this work describes an autocatalytic reaction in which achiral or racemic precursors transform into helically organized products under non-equilibrium conditions. Here, we are curious about two fundamental issues: (1) how the mirror symmetry breaking originates in racemic or even achiral CDTA systems, (2) how the supramolecular chirality is transferred and propagated within the resulting highly ordered helical architecture.
Regarding to the second issue, we propose a plausible hypothesis based on a template-assisted replication mechanism. As illustrated in Fig. 1b, the closely packed helical columns comprise a J-type π-stack of NC molecules. At their cross-sectional edges, NN molecules may be preorganized in either a CCW or CW orientation via π–π stacking and H-bonding interactions. This preorganization enables efficient autocatalytic cyclotetramerization into NC products. Subsequently, new NN molecules are preorganized on the newly formed edges, facilitating directional chirality transfer. Repetition of these steps elongates [NC]CF with specific handedness, ultimately yielding supramolecular chirality. Given that the non-fibrous regions of the reaction mixture remain colorless (Fig. 2b–e), newly formed NC likely remains attached to the fiber ends. We hypothesize that any initial helical bias introduced during nucleation is efficiently propagated and amplified into a dominant handedness during fiber elongation. The as-formed helical nuclei are stabilized by being trapped in a crystalline state under non-equilibrium conditions, where molecular diffusion is restricted. Consequently, the columnar lattices can act as templates for chirality propagation.
Returning to the first question, our findings unveil the mechanistic role of kinetics during the nucleation stage preceding helical fiber growth. In achiral CDTA systems with tunable amounts of DCTH, CD signals remain barely detectable when a large number of non-helical primary nuclei emerge abruptly (Supplementary Fig. 33 and Supplementary Table 5). This outcome clearly indicates that mirror symmetry breaking is primarily governed by kinetic control over the nucleation process. We envisage that a helical imbalance develops only after primary nuclei form. Mirror symmetry is therefore broken once CDTA subsequently amplifies this mirror bias that emerges during secondary nucleation.
A final question is why the observed symmetry breaking prefers P-helical nucleation. In principle, P- and M-helices should be thermodynamically and kinetically equivalent24,58,59. Unexpectedly, the observed asynchrony in the kinetic profiles of enantiopure NC products (Figs. 2f and 4d) defies existing theoretical frameworks. Given the essential role of DCTH in CDTA, especially during nucleation, we speculate, based on the following evidence, that a potential asymmetric source is present in the DCTH additive. First, CDTA using NNA precursor in the absence of DCTH yielded only non-helical, straight fibers with no chiroptical response (Supplementary Fig. 34), analog to the NCA crystallites obtained via the SC protocol (Fig. 3a). Second, when the SC protocol was carried out with DCTH, the resulting NCA crystallites exhibited distinct helical sense, yet still lacking a chiroptical signature (Supplementary Fig. 35). Third, kinetic analysis using an autocatalytic model (Supplementary Note 2) reveals that under homochiral conditions for producing enantiopure NCR and NCS, P-helical nuclei form earlier and faster than their M-type counterparts (Supplementary Fig. 36a, b). This kinetic difference may be attributed to the favorable influence of a potential R-chiral substance on P-helix formation (Supplementary Fig. 36c, d). We therefore reason that the commercial DCTH should contain a trace amount of certain R-chiral “impurity” (Supplementary Fig. 37), which possibly serves as the origin of the chiral asymmetry60.
Asymmetric autocatalysis has long been theorized as a driver for molecular homochirality. In this work, we establish an experimental paradigm in which CDTA achieves mirror symmetry breaking and chiral amplification at the supramolecular level. By means of nucleation–elongation self-assembly under non-equilibrium conditions, CDTA kinetically controls a right-handed bias in helical fibers during secondary nucleation in an achiral or racemic system, which rapidly develops into dominant P-helicity via template-assisted replication. Through transfer of chiral seed handedness, CDTA significantly amplifies supramolecular chirality in achiral products based on the sergeants-and-soldiers principle. Furthermore, CDTA-induced mirror symmetry breaking and amplification can be modulated by external asymmetric perturbation. Considering its applicability to other π-conjugated macrocyclic systems, CDTA not only offers a pathway for imparting chirality to crystalline materials from achiral components but also provides unique insights into the origin of universal homochirality.
Methods
Chemicals and materials
Unless otherwise noted, reagents were used as received from Alfa Aesar [potassium thioacetate, N-hydroxysuccinimide (NHS) and 1,8-diazabicyclo (5.4.0) undec-7-ene (DBU)], Bide Pharmatech Ltd. [1-bromobutane, (R)-1-bromo-2-methylbutane and (S)-1-iodo-2-methylbutane], Meryer [(R)-terpinen-4-ol and (S)-terpinen-4-ol], Tokyo Chemical Industry [2,3-dibromo-6,7-dicyanonephthalene] and Adamas [N, N-dicyclohexylcarbodiimide (DCC), trifluoroacetic acid (TFA), triethylamine (Et3N), 1-dodecanethiol (DCTH), anhydrous potassium carbonate (K2CO3), methyl 3,4,5-trihydroxybenzoate and 2-bromoethylamine hydrobromide]. Other anhydrous solvents for synthesis were used as received from Macklin, such as tetrahydrofuran (THF), N, N-dimethylformamide (DMF), methanol (MeOH), methylecyclohexane (MCH), dichloromethane (CH2Cl2) and chloroform (CHCl3). Potassium bromide (KBr) plates (5.0 × 5.0 mm2) were purchased from JACSO. The details of synthetic procedures and characterization data for NN precursors are provided in Supplementary Methods.
Characterization
1H and 13C{1H} NMR spectra were recorded on a Bruker AVANCE NMR spectrometer, operating at 600 MHz and 151 MHz, respectively, where chemical shifts (in ppm) were determined with respect to tetramethylsilane as an internal reference. High-resolution electrospray ionization (HR-ESI) mass spectra were recorded by a Thermo Scientific exactive benchtop LC/MS Orbitrap mass spectrometer. Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry was performed with an Applied Biosystems MDS SCIEX 4800 Plus MALDI-TOF/TOF analyzer using trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene] malono-nitrile (DCTB) as the matrix. FT-IR spectra were recorded at 25 °C by a JASCO FT/IR-4100 FT-IR spectrometer equipped with attenuated total reflection (PRO450-S). Polarized FT-IR spectra were recorded at 25 °C using a JASCO FT/IR-4100 FT-IR spectrometer connected to an Irtron IRT-5000 microscope unit. All samples were cast on a KBr plate for measurement within the tested window of 50 × 50 μm2. Electronic absorption spectra and time-dependent UV–vis spectra obtained by detection at a fixed wavelength were recorded with a JASCO V-760 UV–vis spectrophotometer with an INSTEC HCS402 hot stage, on which a hot melt of NN precursors was sandwiched between two identical glass plates (18 × 18 mm2). CD was performed with a Chirascan spectrometer (Applied Photophysics, UK). The solution-state spectra were recorded in the continuous mode between 850 and 250 nm, with a wavelength increment of 1 nm, a response time of 0.5 s, and a bandwidth of 1 nm. The quartz cuvettes (10 mm) with screw caps were used. Samples (0.25 g L−1) were suspended in chloroform. LD spectra were measured with a JASCO J-1500 spectropolarimeter with a LD attachment. DSC was performed with a Mettler–Toledo DSC star system, where temperature and enthalpy were calibrated with In (430 K, 3.3 J mol−1) and Zn (692.7 K, 12 J mol−1) standard samples using sealed Al pans. The heating and cooling scan rate was 10 °C min−1. DSC profiles were analyzed using the Mettler–Toledo star software system. OM and POM were performed with an OLYMPUS BX53M polarizing optical microscope equipped with a high-definition camera. PXRD measurements were performed with a Rigaku SmartLab powder X-ray diffractometer equipped with a 3 kW Cu anode (Cu-Kα radiation, λ = 1.541 Å). SEM was performed using a Hitachi SU8010 field emission scanning electron microscope operated with an accelerating electron beam voltage of 5 kV. Laser with a wavelength of 680 nm was generated from a supercontinuum laser source (Wuhan Yangtze Soton Laser Co., Ltd, 430‒2400 nm) filtered by an acousto-optic tunable filter (Wuhan Yangtze Soton Laser Co., Ltd, 400‒2000 nm). The linear polarizer and the quarter-wave plate were used to convert the incident beam to CPL with an intensity of ~0.06 mW cm−2.
CDTA synthesis of naphthalocyanines
A powdery sample of NN precursors (0.5 mg) premixed with DCTH additive was sandwiched between two identical glass plates (18 × 18 mm2). Unless otherwise noted, the mole ratio of NN precursors to DCTH is 3:1. Upon heating to its isotropic state, a hot melt of NN precursors fully wetted between glass plates and kept at 185 °C for 24 h. After being cooled to 25 °C, the reaction mixtures were washed with MeOH (5 mL) to remove unreacted monomer and side products. Accordingly, the crystalline fibers of NC products were obtained.
NCA: 1H NMR (600 MHz, d5-pyridine with zinc acetate) δ (ppm) = 10.11 (s, 8H, NC-H), 9.37 (s, 8H, NHCO), 8.88 (s, 8H, NC-H), 7.84 (s, 16H, Ph-H), 4.59 (d, J = 4.8 Hz, 16H, CH2NH), 4.28–3.88 (m, 64H, PhSCH2 and Ph-OCH2), 1.66 (dp, J = 35.8, 6.8 Hz, 48H, CH2), 1.37 (hept, J = 6.7, 6.3 Hz, 48H, CH2), 0.82 (dt, J = 26.3, 7.4 Hz, 72H, CH3). 13C NMR (151 MHz, d5-pyridine with zinc acetate) δ (ppm) = 177.75 (NC), 168.08 (NHCO), 153.26 (PhO), 152.89 (PhO), 141.02 (PhS), 132.28 (PhCO), 129.89 (NC), 120.60 (Ph), 106.40 (Ph), 72.76 (PhOCH2), 68.55 (PhOCH2), 40.43 (CH2NH), 33.43 (PhSCH2) 32.41 (CH2), 31.43 (CH2), 29.76 (CH2), 22.57 (CH2), 19.25 (CH2), 19.20 (CH2), 13.73 (CH3), 13.66 (CH3). MALDI-TOF MS (m/z): [M + H]+ calcd. for C216H291N16O32S8, 3880.25; found: 3880.94. [M + Na]+ calcd. for C216H290N16O32S8Na, 3902.25; found: 3902.01.
NCR: 1H NMR (600 MHz, d5-pyridine with zinc acetate) δ (ppm) = 10.15 (s, 8H, NC-H), 9.36 (s, 8H, NHCO), 8.86 (d, J = 17.8 Hz, 8H, NC-H), 7.85 (d, J = 8.6 Hz, 16H, Ph-H), 4.60 (d, J = 31.5 Hz, 16H, CH2NH), 4.29–3.58 (m, 64H, PhSCH2 and Ph-OCH2), 1.90–1.58 (m, 40H, CH2), 1.45–1.13 (m, 48H, CH2), 1.08–0.64 (m, 96H, CH3). 13C NMR (151 MHz, d5-pyridine with zinc (II) acetate) δ (ppm) = 178.22 (NC), 168.06 (NHCO), 153.36 (PhO), 153.28 (PhO), 141.01 (PhS), 132.21 (PhCO), 129.85 (NC),106.48 (Ph), 106.32 (Ph), 73.53 (PhOCH2), 72.82 (PhOCH2), 68.56 (PhOCH2), 40.35 (CH2NH), 34.87 (CH2CHCH3), 33.40 (PhSCH2), 32.45 (CH2), 31.44 (CH2), 26.04 (CH2), 22.71 (CH2), 19.26 (CH2), 19.24 (CH2), 16.42 (CH3), 13.76 (CH3), 13.67 (CH3), 11.20 (CH3). MALDI-TOF MS (m/z): [M + H]+ calcd. for C224H307N16O32S8, 3992.08; found: 3992.47. [M + Na]+ calcd. for C224H306N16O32S8Na, 4014.47; found: 4014.03.
NCS: 1H NMR (600 MHz, d5-pyridine with zinc acetate) δ (ppm) = 10.14 (s, 8H, NC-H), 9.36 (s, 8H, NHCO), 8.85 (s, 8H, NC-H), 7.85 (d, J = 10.3 Hz, 16H, Ph-H), 4.60 (d, J = 32.7 Hz, 16H, CH2NH), 4.30–3.69 (m, 64H, PhSCH2 and Ph-OCH2), 1.92–1.58 (m, 40H, CH2), 1.44–1.08 (m, 48H, CH2), 1.03–0.70 (m, 96H, CH3). 13C NMR (151 MHz, d5-pyridine with zinc (II) acetate) δ (ppm) = 177.95 (NC), 168.06 (NHCO), 153.36 (PhO), 153.28 (PhO), 141.01 (PhS), 132.19 (PhCO), 129.85 (NC), 106.48 (Ph), 106.32 (Ph), 73.53 (PhOCH2), 72.82 (PhOCH2), 68.56 (PhOCH2), 40.43 (CH2NH), 34.87 (CH2CHCH3), 33.44 (PhSCH2), 32.45 (CH2), 31.44 (CH2), 26.04 (CH2), 22.64 (CH2), 19.26 (CH2), 19.24 (CH2), 16.42 (CH3), 13.76 (CH3), 13.67 (CH3), 11.20 (CH3). MALDI-TOF MS (m/z): [M + H]+ calcd. for C224H307N16O32S8, 3992.08; found: 3992.47. [M + Na]+ calcd. for C224H306N16O32S8Na, 4014.47; found: 4014.03.
Solution crystallization of naphthalocyanines
A powdery sample of NC (5 mg) products was added into DMF (30 mL) or the mixed solvent of DMF (30 mL) and DCTH (1 μL). After completely dissolving NCs upon heating in a sealed tube, the resulting solution was slowly cooled to room temperature. The yellow-green colored crystallites of NC were obtained for overnight.
Time-dependent spectroscopic measurements
A powdery sample (0.5 mg) of NN precursors premixed with DCTH (1/3 equiv.) was prepared. For seed-induced chirality transfer experiments, a small amount (4 mol%) of chiral [NC]CF seeds was added into the above mixtures prior to heating. Then, upon heating to the isotropic state, a hot melt of the reaction mixture was fully wetted between two glass plates and was kept at 185 °C, which were placed perpendicular to the light path of the spectrometers. The glass plates were rotated within the plane to eliminate the birefringence and the possible angle dependence of the CD signal. The solid-state CD and UV–vis spectra were recorded at different time.
CPL modulation on CDTA reaction
A powdery sample (0.5 mg) of NNA precursors premixed with DCTH (1/3 equiv.) was sandwiched between two glass plates (18 × 18 mm2), and the resulting reaction mixture was heated at 185 °C for 1 h. The glass substrate was then placed perpendicular to the light path and irradiated with left-handed or right-handed CPL at 680 nm for 2 h. The distance from the lamp to the glass plates was set at 5 cm. The setup was maintained in a darkened environment to prevent interference from external light. After cooling to 25 °C, the CPL-irradiated reaction mixtures sandwiched with glass plates were directly subjected for solid-state CD measurements and optical microscopy.
Sergeant-and-soldiers experiments
Typically, active crystalline seeds of chiral NCR or NCS as sergeant were prepared by chopping the as-formed [NCR]CF or [NCS]CF in methanol. To the resulting solution was added NNA precursor as soldier in different molar ratios and DCTH (1/3 equiv.), and then was cast onto a glass plate and air-dried at 25 °C. Another glass plate was covered and then heated at 185 °C for 12 h. After cooling to 25 °C, the reaction mixtures sandwiched with glass plates were directly subjected for solid-state CD measurements.
Theoretical calculations
To study atomic-level details, all supramolecular columns were optimized using the semiempirical GFN2-Xtb method57 using the xtb software61 (version 6.7.1) and visualized using Visual Molecule Dynamics software62. GFN2-Xtb incorporates anisotropic electrostatic interactions through cumulative atomic multipole moments and a self-consistent D4 dispersion model63,64. This method provides an excellent balance between computational cost and accuracy and is particularly suitable for investigating large molecular systems such as decameric columnar models of NC derivatives to elucidate their helical origins. As for the initial models of homochiral supramolecular columns, neither molecular tilting nor rotation was preset for restriction. As for the initial models of achiral supramolecular columns, however, the molecular rotation in ±2° is preset for restriction. The rotation angle is defined as a dihedral angle between two planes of NC cores rotating about a central axis along out-of-the-plane direction, wherein the rotation angle in a CCW direction is positive (+) while that in a CW direction should be negative (−). As for the 20-mer-consisted supramolecular columns, having an interlayer distance of 3.5 Å, the intermolecular distance is preset for 4.5 Å without predefined rotational restriction. The specific configuration of hydrogen bonds is a key factor that determines the helical structures. Therefore, we defined and calculated the bond lengths and bond angles of the hydrogen bonds, where the bond angle is defined as the angle between the N‒H and C=O connections, and the bond length is defined as the distance between H and O. The tilt angle is defined as a dihedral angle between the plane of NC cores and the xy-plane. The interplanar distance is defined as the distance between the parallel planes of adjacent NC cores.
Data availability
The experimental data supporting the findings of this study are available within the Article and its Supplementary information files. The computational data generated in this study have been deposited in the GitHub database under accession code [https://git@github.com/ChenQxuan/2026-2.git]. All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Blackmond, D. G. The origin of biological homochirality. Cold Spring Harb. Perspect. Biol. 11, a032540 (2019).
Sallembien, Q., Bouteiller, L., Crassous, J. & Raynal, M. Possible chemical and physical scenarios towards biological homochirality. Chem. Soc. Rev. 51, 3436–3476 (2022).
Frank, F. C. On spontaneous asymmetric synthesis. Biochim. Biophys. Acta 11, 459–463 (1953).
Kiliszek, A. & Rypniewski, W. The emergence of biological homochirality. Acta Biochim. Pol. 70, 481–485 (2023).
Blackmond, D. G. Asymmetric autocatalysis and its implications for the origin of homochirality. Proc. Natl. Acad. Sci. USA 101, 5732–5736 (2004).
Soai, K., Shibata, T., Morioka, H. & Choji, K. Asymmetric autocatalysis and amplification of enantiomeric excess of a chiral molecule. Nature 378, 767–768 (1995).
Kawasaki, T., Suzuki, K., Hakoda, Y. & Soai, K. Achiral nucleobase cytosine acts as an origin of homochirality of biomolecules in conjunction with asymmetric autocatalysis. Angew. Chem. Int. Ed. 47, 496–499 (2008).
Athavale, S. V., Simon, A., Houk, K. N. & Denmark, S. E. Demystifying the asymmetry-amplifying, autocatalytic behaviour of the Soai reaction through structural, mechanistic and computational studies. Nat. Chem. 12, 412–423 (2020).
Soai, K. The Soai reaction and its implications with the life’s characteristic features of self-replication and homochirality. Tetrahedron 124, 133017 (2022).
Geiger, Y. One Soai reaction, two mechanisms? Chem. Soc. Rev. 51, 1206–1211 (2022).
Kawasaki, T., Takamatsu, N., Aiba, S. & Tokunaga, Y. Spontaneous formation and amplification of an enantioenriched nitrile: a chiral precursor for Strecker amino acid synthesis. Chem. Commun. 51, 14377–14380 (2015).
Feng, Y. & Philp, D. A molecular replication process drives supramolecular polymerization. J. Am. Chem. Soc. 143, 17029–17039 (2021).
Bare, G. A. K. & Joyce, G. F. Cross-chiral, RNA-catalyzed exponential amplification of RNA. J. Am. Chem. Soc. 143, 19160–19166 (2021).
Deng, M., Yu, J. & Blackmond, D. G. Symmetry breaking and chiral amplification in prebiotic ligation reactions. Nature 626, 1019–1024 (2024).
Kurata, M. & Yoshizawa, A. The formation of a chiral supramolecular structure acting as a template for chirality transfer. Chem. Commun. 56, 8289–8292 (2020).
Das, A., Ghosh, S. & George, S. J. Chiroptical amplification of induced circularly polarized luminescence in nucleotide-templated supramolecular polymer. Angew. Chem. Int. Ed. 62, e202308281 (2023).
Kondepudi, D. K., Kaufman, R. J. & Singh, N. Chiral symmetry breaking in sodium chlorate crystallization. Science 250, 975–976 (1990).
Buhse, T. et. al. Spontaneous deracemization. Chem. Rev. 121, 2147–2229 (2021).
Sanada, K. et al. Chiral symmetry breaking of monoacylated anhydroerythritols and meso-1,2-diols through crystallization-induced deracemization. Angew. Chem. Int. Ed. 61, e202201268 (2022).
Romeo, A. et al. Kinetic control of chirality in porphyrin J-aggregates. J. Am. Chem. Soc. 136, 40–43 (2013).
Romeo, A. et al. Effect of zinc cations on the kinetics of supramolecular assembly and the chirality of porphyrin J-aggregates. Chem. Sci. 8, 961–967 (2017).
Heinemann, U. & Roske, Y. Symmetry in nucleic-acid double helices. Symmetry 12, 737 (2020).
Liu, M., Zhang, L. & Wang, T. Supramolecular chirality in self-assembled systems. Chem. Rev. 115, 7304–7397 (2015).
García, F., Gómez, R. & Sánchez, L. Chiral supramolecular polymers. Chem. Soc. Rev. 52, 7524–7548 (2023).
Huang, S., Chen, Y., Ma, S. & Yu, H. Hierarchical self-assembly in liquid-crystalline block copolymers enabled by chirality transfer. Angew. Chem. Int. Ed. 57, 12524–12528 (2018).
Reppe, T. et al. Spontaneous mirror symmetry breaking in benzil-based soft crystalline, cubic liquid crystalline and isotropic liquid phases. Chem. Sci. 11, 5902–5908 (2020).
Zhang, Q., Wang, W., Zhou, S., Zhang, R. & Bischofberger, I. Flow-induced periodic chiral structures in an achiral nematic liquid crystal. Nat. Commun. 15, 7 (2024).
Yoshida, M. et al. Vortex-flow-directed chiral macroscopic ordering of platelet nanostructures formed via the supramolecular assembly of platinum complexes with bis(phenylisoxazolyl)benzene. J. Am. Chem. Soc. 147, 30674–30683 (2025).
Micali, N. et al. Selection of supramolecular chirality by application of rotational and magnetic forces. Nat. Chem. 4, 201–207 (2012).
Sang, Y. et al. Ultrasound-directed symmetry breaking and spin filtering of supramolecular assemblies from only achiral building blocks. Angew. Chem. Int. Ed. 62, e202215867 (2023).
Xu, Y. et al. Enantioselective synthesis of helical polydiacetylene by application of linearly polarized light and magnetic field. Nat. Commun. 5, 5055 (2014).
Kim, J. et al. Induction and control of supramolecular chirality by light in self-assembled helical nanostructures. Nat. Commun. 6, 6959 (2015).
Sun, J. et al. Control over the emerging chirality in supramolecular gels and solutions by chiral microvortices in milliseconds. Nat. Commun. 9, 2599 (2018).
Čopar, S., Kos, Ž, Emeršič, T. & Tkalec, U. Microfluidic control over topological states in channel-confined nematic flows. Nat. Commun. 11, 59 (2020).
Wen, X., Du, S., Zhang, L. & Liu, M. Chiral deep eutectic solvents enable full-color and white circularly polarized luminescence from achiral luminophores. Angew. Chem. Int. Ed. 62, e202311816 (2023).
Karunakaran, S. C., Cafferty, B. J., Weigert-Muñoz, A., Schuster, G. B. & Hud, N. V. Spontaneous symmetry breaking in the formation of supramolecular polymers: implications for the origin of biological homochirality. Angew. Chem. Int. Ed. 58, 1453–1457 (2019).
Jiang, H., Jiang, Y., Zhang, L., Guo, Z. & Liu, M. Symmetry breaking and amplification in a self-assembled helix from achiral trans-3-nitrocinnamic acid. J. Phys. Chem. C 122, 12559–12565 (2018).
Wang, F., Gan, F., Shen, C. & Qiu, H. Amplifiable symmetry breaking in aggregates of vibrating helical molecules. J. Am. Chem. Soc. 142, 16167–16172 (2020).
Cao, H., Zhu, X. & Liu, M. Self-assembly of racemic alanine derivatives: unexpected chiral twist and enhanced capacity for the discrimination of chiral species. Angew. Chem. Int. Ed. 52, 4122–4126 (2013).
Zhu, X. et al. Homochiral nanotubes from heterochiral lipid mixtures: a shorter alkyl chain dominated chiral self-assembly. Chem. Sci. 10, 3873–3880 (2019).
Wen, X. et al. Achiral solvent inversed helical pathway and cosolvent controlled excited-state “majority rule” in enantiomeric dansulfonamide assemblies. Small 20, e2401954 (2024).
Preuss, M. D. Functionalization of supramolecular polymers by dynamic covalent boroxine chemistry. Angew. Chem. Int. Ed. 63, e202402644 (2024).
Van Zee, N. J., Mabesoone, M. F. J., Adelizzi, B., Palmans, A. R. A. & Meijer, E. W. Biasing the screw-sense of supramolecular coassemblies featuring multiple helical states. J. Am. Chem. Soc. 142, 20191–20200 (2020).
Kosikova, T. & Philp, D. Exploring the emergence of complexity using synthetic replicators. Chem. Soc. Rev. 46, 7274–7305 (2017).
Yang, S. et al. Enantioselective self-replicators. J. Am. Chem. Soc. 145, 16889–16898 (2023).
Chen, Z. et al. Solvent-free autocatalytic supramolecular polymerization. Nat. Mater. 21, 253–261 (2022).
Wu, H., Chen, Q.-X., Su, Y. & Chen, Z. The role of hydrogen bonds in thermally responsive crystallization-driven template autocatalysis. Angew. Chem. Int. Ed. 63, e202404838 (2024).
Fang, T. et al. Direct observation of spherulitic covalent triazine frameworks grown in hot superacid matrices. Small 21, e09494 (2025).
Hirschmann, M., Merten, C. & Thiele, C. M. Treating anisotropic artefacts in circular dichroism spectroscopy enables investigation of lyotropic liquid crystalline polyaspartate solutions. Soft Matter 17, 2849–2856 (2021).
Tsuda, A. et al. Spectroscopic visualization of vortex flows using dye-containing nanofibers. Angew. Chem. Int. Ed. 46, 8198–8202 (2007).
Shen, Z., Jiang, Y., Wang, T. & Liu, M. Symmetry breaking in the supramolecular gels of an achiral gelator exclusively driven by π‒π stacking. J. Am. Chem. Soc. 137, 16109–16115 (2015).
Kang, J. S. et al. Circularly polarized light can override and amplify asymmetry in supramolecular helices. J. Am. Chem. Soc. 144, 2657–2666 (2022).
Weissbuch, I. & Lahav, M. Crystalline architectures as templates of relevance to the origins of homochirality. Chem. Rev. 111, 3236–3267 (2011).
Deck, L.-T., Hosseinalipour, M. S. & Mazzotti, M. Spontaneous emergence of homochiral suspensions from racemic solutions via stochastic nucleation. J. Am. Chem. Soc. 147, 18826–18839 (2025).
Gestel, J. V., Schoot, P. V. D. & Michels, M. A. J. Amplification of chirality in helical supramolecular polymers. Macromolecules 36, 6668–6673 (2003).
Gestel, J. V. Amplification of chirality in helical supramolecular polymers: The majority-rules principle. Macromolecules 37, 3894–3898 (2004).
Bannwarth, C., Ehlert, S. & Grimme, S. GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput. 15, 1652–1671 (2019).
Huang, S., Yu, H. & Li, Q. Supramolecular chirality transfer toward chiral aggregation: asymmetric hierarchical self-assembly. Adv. Sci. 8, 2002132 (2021).
Castro, V. I. B., Reis, R. L., Pires, R. A. & Pashkuleva, I. Bioinspired helical systems with defined chirality assembled from discrete peptide and glycan amphiphiles. Chem. Soc. Rev. 54, 8325–8344 (2025).
Lahav, M. et al. Parity violating energetic difference and enantiomorphous crystalsp-caveats; reinvestigation of tyrosine crystallization. Orig. Life Evol. Biosph. 36, 151–170 (2006).
Bannwarth, C. et al. Extended tight-binding quantum chemistry methods. WIREs Comput. Mol. Sci. 11, e1493 (2021).
Humphrey, W. & Schulten, K. V. M. D. Visual molecular dynamics. J. Mol. Graphics 14, 33–38 (1996).
Caldeweyher, E., Bannwarth, C. & Grimme, S. Extension of the D3 dispersion coefficient model. J. Chem. Phys. 147, 034112 (2017).
Caldeweyher, E. et al. Generally applicable atomic-charge dependent London dispersion correction. J. Chem. Phys. 150, 154122 (2019).
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 22271173 to Z.C.), Guangdong Basic and Applied Basic Research Foundation (No. 2023A1515011902 to Z.C.), Guangdong Provincial Department of Science and Technology (Nos. 2023ZT10L039 and 2021QN02C070 to Z.C.), the scientific research startup fund (No. QD2021029C to Z.C.) and cross-disciplinary research and innovation fund (No. JC2022006 to Z.C.) from Tsinghua Shenzhen International Graduate School (SIGS). We appreciate Ms. Xinyue Gao and Prof. Qinghua Song (Tsinghua SIGS) for providing the technical assistance on the CPL experimental setup. The Testing Technology Center of Materials and Devices of Tsinghua SIGS and the Public Testing Center of Southern University of Science and Technology are thanked for providing the technical assistance on characterization.
Author information
Authors and Affiliations
Contributions
H.W. and Z.C. conceived the idea and designed the experiments. H.W. conducted the synthesis, characterization, and measurements. Q.C. performed the theoretical calculation. D.G. conducted the partial synthesis. All authors participated in the data analysis and discussion. Z.C. led the project. H.W., Q.C., and Z.C. drafted the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Haifeng Yu and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, H., Chen, Q., Gao, D. et al. Crystallization-driven template autocatalysis induces mirror symmetry breaking and amplification. Nat Commun 17, 3277 (2026). https://doi.org/10.1038/s41467-026-70105-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70105-y
