
Abstract
α,β-Unsaturated δ-lactones (AUDLs) are key pharmacophores of various polyketides exhibiting potent biological activity. Fostriecin has attracted interest as an anticancer agent, but its structural characteristics have limited its development and motivated investigations into biosynthesis-based production strategies. Here, we elucidate the enzymatic steps responsible for AUDL formation in fostriecin biosynthesis by in vitro reconstitution using complex synthetic substrate surrogates. We demonstrate that the terminal polyketide synthase (PKS) module FosMod8 produces a 3-O-malonyllactone by the unusual bifunctional thioesterase FosTE, which catalyses O-malonylation and subsequent lactonisation. Structural modelling and site-directed mutagenesis reveal two arginine residues in the active site of FosTE that mediate malonyl-CoA binding and transesterification, thereby enabling the domain to mimic PKS acyltransferase chemistry. Additionally, we show that AUDL formation is carried out by the demalonylating enzyme FosM, whose activity strongly depends on prior fostriecin backbone phosphorylation by the broad-specific kinase FosH. This arrangement optimises the biosynthesis of phosphorylated AUDL metabolites by minimising shunt intermediate formation and losses from spontaneous side reactions of sensitive intermediates. This unique enzymatic logic represents a blueprint for other AUDLs and understanding it paves the way for new synthetic strategies to AUDL polyketides using chemoenzymatic synthesis or engineered biosynthesis.
Similar content being viewed by others
Introduction
Polyketides are a diverse class of natural products with significant therapeutic potential, inspiring ongoing drug discovery efforts1. Clinically validated examples such as erythromycin A, amphotericin A and synthetic derivatives such as ixabepilone underscore their medical relevance. However, the structural complexity of many polyketides presents significant challenges for conventional synthetic approaches. This has spurred the development of alternative strategies that leverage the biosynthetic machinery of the producing organisms, including chemoenzymatic synthesis and mutasynthesis2,3. A detailed mechanistic understanding of polyketide biosynthesis at both the genetic and enzymatic levels is essential to enable and refine these approaches.
In bacteria, non-aromatic polyketides are mainly assembled by type I polyketide synthases (PKS) consisting of multiple modules with reducing activity, known as assembly line proteins. These make use of similar domains as fatty acid synthases (FAS)4,5. Ketosynthase (KS), acyltransferase (AT), and acyl carrier protein (ACP) domains cooperatively elongate polyketide chains using malonyl-CoA-type building blocks, followed by reductive loop processing by ketoreductase (KR), dehydratase (DH) and enoylreductase (ER) domains5,6. Thioesterase (TE) domains release PKS products as carboxylic acids or macrocycles. The enormous structural diversity of polyketides made by these systems results from the variety of different malonyl-CoA-derived building blocks and reductive loop architectures. In contrast to FAS, not each PKS module contains KR, DH and ER domains leading to the presence of ketones, hydroxyl groups and olefins in the products.
α,β-Unsaturated δ-lactone (AUDL)-containing polyketides are a pharmacologically highly interesting subgroup7,8. AUDLs serve as covalent warheads for nucleophilic attack by cysteine / lysine residues. The broad activity profile of AUDL polyketides results from their interaction with different targets, modulated by the C-5 substituent of the lactone. Prominent members are the phosphorylated protein phosphatase 1/2 inhibitors fostriecin (1), the phoslactomycins (2a) and cytostatins (3)9, which show effective antitumor activity in vivo. Non-phosphorylated AUDLs like the lactomycins (2b), the leptomycins (4a), the anguinomycins (4c) and others10 have also shown anti-tumour, antifungal and antiviral activity due to selective interaction with targets such as the β-tubulin subunit7, cathepsin B10 or exportin 111,12,13. The pharmacological potential of these compounds prompted the development of various total syntheses, which enabled SAR studies and subsequent clinical trials in the case of fostriecin14. Though the latter were abandoned due to storage instability and unpredictable purity of fostriecin preparations, interest in the development of improved analogues persisted15,16,17. While the AUDL of fostriecin was found to be essential as a Michael acceptor for attack by Cys269 of type 2 A protein phosphatase (PP2A), the phosphate ester serves as a mimic of the natural phosphothreonine substrate, highlighting them as moieties of particular importance. 8-OH, 11-OH and the triene provide hydrogen bonding/hydrophobic interactions, the disruption of which reduced the inhibitory activity on PP2A.
The gene clusters of fostriecin (fos in Streptomyces pulveraceus), the phoslactomycins (plm in Streptomyces sp. HK-803, pn in Streptomyces platensis SAM-0654 and Streptomyces auratus AGR0001), the lactomycins (lac in Streptomyces sp. ACT232) and the leptomycins (lep in Streptomyces sp. ATS1287) have already been sequenced10,18,19,20,21,22,23. Information on the annotation of individual genes and the biosynthetic tailoring processes were gained by knockout experiments in the fos, pn and plm clusters18,19,24,25. Individual knockouts of the genes coding for the kinase FosH and the oxidising enzymes FosJ, FosK and FosG led to the isolation of diverse, mostly 9-O phosphorylated AUDL and malonyllactone intermediates of different oxidation states. FosJ and FosH apparently act first, whereby 9-O phosphorylation is essential for the activity of FosK. FosG is only poorly active on 1 to yield a C-5-hydroxylated PD 113,271. In vitro studies on FosH and FosJ revealed relaxed substrate specificity of both enzymes, which was used for the development of a chemoenzymatic synthesis comprising both enzymes18,26. Knockout of the homologous fosM and plmT2 in the fostriecin and phoslactomycin producers gave compounds 11 and 13, respectively, showing their involvement in late-stage demalonylation (Fig. 1b, c)18,24,25. Introduction of fosM into the ΔplmT2 mutant restored demalonylation, underlining the high similarity of the two biosynthetic processes. Direct confirmation of the demalonylation activity of FosM or one of its homologues by in vitro experiments is, however, still lacking25.

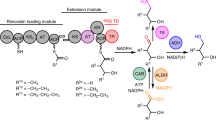
a Structures of AUDL-containing polyketides and key structural motifs of fostriecin (1). b The polyketide scaffold is assembled by a modular Type IPKS and subsequently modified by various tailoring enzymes to yield fostriecin (fos, 1). fosA-F = genes encoding for the Fos-PKS, FosH = homoserine kinase, FosK = cytochrome P450 monooxygenase, FosJ = cytochrome P450 monooxygenase, FosM = NAD-dependent epimerase/dehydratase family protein18. c 13 and 14 were isolated from a ΔplmT2 knockout mutant of the phoslactomycin producer Streptomyces sp. HK-803. d possible AT or TE-catalysed scenarios for O-malonylation by Fos-PKS domains.
The biosynthetic introduction of the malonyl group is not clarified. The Fos-PKS contains a KR and a TE (FosTE) in module 8, suggesting the nonaketide intermediate 7 as the primary PKS product18. Cyclisation and malonylation would then lead to the O-malonyllactone 8, whereby inline or post-PKS O-malonylation is conceivable. No isolated transferases or ligases are encoded in the fos-cluster, indicating the involvement of a PKS domain or an enzyme coded outside of the fos biosynthetic gene cluster in O-malonylation. For phoslactomycin biosynthesis, it was proposed that O-malonylation is catalysed by an AT domain of the Plm-PKS25.
Malonyl transfer from malonyl-CoA has been reported for various PKS domains and homologous enzymes. Besides canonical PKS-AT domains, trans-acting ATs are known to act as malonyl introducers into assembly line proteins, for example malonyl-CoA:ACP ATs of FAS, which prime ACPs of modular27 or iterative28,29 PKSs within the same organism. Self-malonylation is as a common activity of ACPs in type II PKS30. Biosynthetic O-malonylation of small molecule substrates has been reported for ATs, such as the steroid/phenol/N-acylethanolamine glycoside-malonylating PMAT1 from Arabidopsis thaliana31,32,33 and KSs, such as CerJ from the biosynthesis of the glycosylated polyketide cervimycin34. Esterification of polyketide and peptide backbone hydroxyls was described for ATs35,36, KSs37 and assembly line-integrated TEs, the latter participating in O-acylations in different scenarios38,39,40,41,42,43,44. In the biosynthetic pathways of FR900359, necroxime A and salinamide, a side chain is attached to a mature macrocycle by a TE domain (TrsA-TE / NecA-TE / Sln9-TE) of a separate non-ribosomal peptide synthetase (NRPS) module38,39,40. The inline O-acylating Cong-TE, Ela-TE and ThiA-TE catalyse intermolecular transacylation of one polyketide monomer onto a hydroxyl of a second, ACP-bound monomer, leading to macrodiolide or methylorsellinic acid trimer formation, respectively42,43,44. OocS TEB from the penultimate module of the oocydin trans-AT PKS O-acetylates a 3-hydroxythioester intermediate, but lacks thioester hydrolysis activity41. OocS TEB showed broad tolerance for different acyl-CoAs in vitro, but did not accept malonyl-CoA. Dedicated malonyl-transferring TE domains are not yet known in biosynthetic pathways.
Given this precedent, various types of PKS domains could in principle be involved in the malonyl transesterification required for AUDL formation. However, the biosynthesis framework of fostriecin points to a scenario inconsistent with the properties of the domains described to date. While KSs and ATs can be catalytically competent for both esterification and malonyl transfer, this has only been demonstrated for tailoring enzymes that process free molecules. TE domains have so far neither shown malonyl transfer activity nor bifunctionality associated with tolerance for ACP- and CoA-bound substrates45.
Here, we show a complete reconstruction of the AUDL assembly process and refine the proposal for the timing of the final stages of fostriecin biosynthesis. By enzymatic in vitro assays using synthetic substrate surrogates, we show that FosTE exhibits bifunctionality, catalysing O-malonylation followed by δ-lactonisation. Demalonylation by FosM occurs only after previous polyketide chain phosphorylation by the broad-specific kinase FosH. Investigations on the substrate specificity suggest that this tightly controlled series of events likely improves the production of bioactive AUDL-containing metabolites, minimising the impact of shunt product formation by spontaneous decomposition of sensitive O-malonylated intermediates.
Results
Assignment of O-malonylation activity to the PKS Module 8
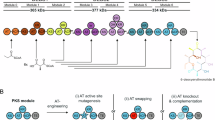
To confirm the PKS involvement in O-malonylation, we started with a reconstitution of the FosMod7 and FosMod8 activity. The holo-forms of FosMod7, FosMod8 and the fusion protein FosMod7-FosTE were heterologously produced in E. coli BAP1 and purified. Compounds 16, 17, 19, 21, 25, 28 and 31, representing surrogates of anticipated key intermediates and useful reference compounds, were chemically synthesised (Supplementary information)46. Assay analysis was performed by LC-MS analysis, accompanied by detailed LC-MS/MS and LC-HRMS analyses in cases when no standards were available. Surrogate 16 resembles the most characteristic structural features of the FosMod7 precursor 5, with the N-acetyl cysteamine (SNAC) thioester mimicking the ACP attachment via the 4’-phosphopantetheinyl arm and the alkyl chain at C7 representing the hydrophobic polyene tail (Figs. 1a,2a, b). 16 was incubated for 3 h with FosMod7 and FosMod8 with an excess of malonyl-CoA and NADPH. Small quantities of malonyllactone 27 (1%) and AUDL 28 (5%), alongside O-malonylated 22, 23a, and 23b were detected (Fig. 2a). Stepwise reconstitution of the two-module reaction was undertaken by incubating 16 with FosMod7-FosTE and incubating 17 with FosMod8. Precursor 16 was converted by 26% into carboxylic acid 3-l-19 by FosMod7-FosTE, with no other elongated or O-malonylated products being detectable (Fig. 2b). Incubation of 17 with FosMod8 gave distinct hydrolysis to 19, but also small amounts of the elongated 27, 28, 23, 20 and 24, of which 27 and 23 represent O-malonylated species (Fig. 2c).

a Reaction scheme for activity assays with surrogate 16, FosMod7, FosMod8 and FosTEII (top). LC-MS analysis of assay with FosMod7, FosMod8 and FosTEII (3 h), product references 27 and 28, control reaction without enzymes (3 h) (left). Incubation of 16 with FosMod7, FosMod8 and increasing FosTEII equivalents (right). b Reaction scheme for activity assays with surrogate 16, FosMod7-FosTE and FosTEII (top). LC-MS analysis of assay with FosMod7-FosTE and FosTEII (16 h and 3 h), product reference (3-l-19) and the control reaction without enzymes (16 h) (left). Incubation of 16 with FosMod7-FosTE and increasing FosTEII equivalents (right). c Reaction scheme for activity assays with surrogate 17, FosMod8 and FosTEII (top). LC-MS analysis of assay with FosMod8 and FosTEII (3 h), product references 27 and 28, control reaction without enzymes (3 h). Incubation of 17 with FosMod8 and increasing FosTEII equivalents (right). Extracted ion chromatograms (EICs) are shown and the mass/charge ratios (m/z) used for each compound are listed in Supplementary Table 4. HRMS/MS data is shown in Supplementary Figs. 33–61. Conversion rates were approximated from EIC data and are shown as mean ± standard deviation (s.d) from three independent, separately prepared reactions (n = 3).
To obtain stronger evidence for O-malonylation by FosMod8, the reaction conditions were optimised towards higher conversion into 27 and 28. We turned our interest towards the oligomerisation state of the PKS modules as the activity of type I PKS modules is strongly linked to their homodimeric state in solution47,48,49. SEC analysis showed that higher-order oligomers were present alongside the homodimeric forms for all constructs immediately after purification and that, particularly in the case of FosMod8, their proportion increased significantly during incubation (Supplementary Figs. 2, 5, 8). Upon refinement of the purification protocol, the homodimeric forms of the modules exhibited enhanced structural integrity and stability, leading to significantly higher product formation (Supplementary Figs. 4, 7, 10). 25% conversion of 16 by FosMod7+FosMod8 and 39% conversion of 17 by FosMod8 into the O-malonylated 27, along with ~ 5–8% of the AUDL 28 were observed (Fig. 2b, c).
To further improve conversion efficiency by rapid clearing of the assembly line, FosTEII was introduced50. FosTEII is a close homologue of the proof-reading TE PnG from the pn cluster. Erb et al. have previously shown by in vitro reconstitution that PnG can release intermediate and terminal polyketides from the Pn-PKS assembly line, thus improving in vitro production by more than one order of magnitude. Adding FosTEII to the assays resulted in a further concentration-dependent boost in conversion of 4.8–8.5% across all three reactions. Together with the improvement achieved by the refined protein purification protocol, this enabled a detailed analysis of the complex product profile (Fig. 2). In addition to 27 and 28, O-malonylated dihydroxyacid 23, as well as β-keto-γ-hydroxyacid 20 and β-ketolactone 24 were found as relevant elongation products in the improved FosMod8 reaction with 17, showing that any β-hydroxythioester intermediate formed underwent efficient O-malonylation. These compounds also emerged in the assay of 16 with FosMod7 and FosMod8, where they were accompanied by numerous minor components, including the SNAC adduct 26 as well as 19, 21, 22 and 25, owing to the more complex nature of this conversion. The latter are presumably formed by inaccurate substrate recognition and conversion by the modules, such as the processing of 16 by FosMod8 that was confirmed to give 22 in a separate incubation (Supplementary Fig. 16b). Notably, these shunt intermediates account for approximately 50% of the crude product with the presence of 22 and 23 providing further evidence for an O-malonylation activity of FosMod8. Gratifyingly, the improved PKS module reaction conditions also allowed a reaction upscaling that gave access to sufficient amounts of the synthetically inaccessible key intermediate 27 for a full characterisation by NMR and LC-HRMS(/MS) after semipreparative HPLC purification (Supplementary Figs. 33, 34, 85, 86). These results demonstrate that O-malonylation is an intrinsic activity of the fostriecin PKS. The presence of 27 and 23 in all assays containing FosMod8 strongly indicates an O-malonylation activity of this module. We then focused on identifying the specific domain harbouring this non-canonical PKS activity.
FosTE in module 8 is responsible for O-malonylation
We created the FosMod8 mutant FosMod8(S159A) in which FosTE was inactivated by replacing its catalytic serine (S159) with alanine via site-directed mutagenesis. Incubation of FosMod8(S159A) with substrate 17 showed a complete abolishment of the formation of 27 and 28. Instead, the β-keto carboxylic acid 20 and β-keto lactone 24 emerged, resulting from spontaneous hydrolysis and lactonisation immediately after elongation (Fig. 3a, c). Previous studies have shown that changes in the interactions between PKS domains can impair ketoreduction51. In this case, we hypothesise that ketoreduction occurs in concert with self-malonylation of the TE catalytic serine, such that abolishment of the latter may disrupt KR–TE communication and consequently slow ketoreduction. The formation of the β-hydroxythioester intermediate was thus forced by in trans-supplementation of FosKR8. This indeed accumulated dihydroxycarboxylic acid 21 alongside the mixture of ketones 20, 24 and 29 (Fig. 3d). The absence of O-malonylated intermediates 27, 23a and 23b and the AUDL 28, which were found in the reaction with wild-type FosMod8 (Fig. 2c), suggests that the TE domain plays a pivotal role in the O-malonylation. Consistent with the expected disruption of specific lactonisation activity by the S159A mutation, β-hydroxy lactone 25 was also not detected. Adding isolated FosTE to the reaction of 17 with FosMod8(S159A) and FosKR8 restored the formation of malonyl lactone 27 and AUDL 28, along with significant hydrolysis to 19 (Fig. 3e). Although the amounts of 27 and 28 formed were small, this clearly indicates the involvement of FosTE in both processes. It is a known feature of cyclising TE domains that they revert to their hydrolytic activity in vitro in response to significant deviations from the natural environment52,53, as given here due to the simplified substrate surrogate and the absence of the PKS module environment.

a Reaction scheme for in vitro malonylation assays with FosMod8 and mutant variants, using surrogate 17. b EIC of activity assay with wild-type FosMod8. c Activity assay with FosMod8(S159A). d Activity assay with FosMod8(S159A), in trans-supplemented by FosKR8. e Activity assay with FosMod8(S159A), in trans-supplemented by FosKR8 and FosTE. f Isolated reference 27. g Synthetic reference 28. h Activity assay with FosMod8(JerTE). i Activity assay with FosMod8(R198L/R227L). j Synthetic reference 25. See supplementary Figs. 33–61 for HRMS(/MS) data.
FosTE was also swapped by the monofunctional, lactonising JerTE domain, which forms a β-keto-δ-lactone that is stabilised by subsequent O-methylation in jerangolid biosynthesis54,55. Incubation of FosMod8(JerTE) with surrogate 17 yielded dicarboxylic acid 21 and hydroxylactone 25 as the sole elongation products. The absence of 27 or other malonylated by-products (Fig. 3h) supports that neither FosAT8 nor another FosMod8 domain is responsible for O-malonylation and that FosTE possesses dual O-malonylation–lactonisation activity. The self-malonylation activity of FosTE was demonstrated by incubating the domain with malonyl-CoA, resulting in a mass shift of 86 Da, which is consistent with malonyl-FosTE half-ester formation (Supplementary Fig. 67).
Two non-canonical arginine residues are essential for transmalonylation by FosTE
To elucidate structural features underlying this bifunctionality, we generated AlphaFold 3 models of the dimeric FosTE domain56. From five predicted structures, the two models with the highest confidence scores were selected for further analysis. In line with structurally resolved TE domains57,58,59,60, the binding pockets in both models form a channel connecting the phosphopantetheinyl-binding site (entrance) and the polyketide-binding site (exit), with the catalytic triad positioned centrally within this tunnel (Fig. 4b, c). This architecture allows entry of the acyl-ACP from one side and lactone formation in the acyl cavity at the opposite end. Strikingly, two arginine residues (R198 and R227) were identified, protruding into the acyl cavity at the exit side (Fig. 4b–e).

a Structure of phosphopantetheine thioester 30 used for molecular docking simulations. b AlphaFold 3 model of the FosTE active site showing an extended substrate tunnel into which ligand 30 was docked. c AlphaFold 3 model of the FosTE active site showing a more constricted substrate tunnel due to the positioning of R198 and R227, into which malonyl-CoA was docked. d Docking simulation of thioester 30 in the extended tunnel (b), showing interactions with the catalytic triad (S159, D186, H281; magenta) and characteristic arginine residues (R198, R227; blue). e Docking simulation of malonyl-CoA in the constricted cavity (c), showing interactions with the catalytic triad (S159, D186, H281; magenta) and characteristic arginine residues (R198, R227; blue). Hydrophobicity profiles of the substrate tunnels (b, c) were predicted using CavitOmix/PyMOL, with red areas representing hydrophobic regions and blue areas representing hydrophilic regions.
The first model (Fig. 4b) features a larger (~ 900 Å3), more extended cavity compared to the second model (Fig. 4c), which exhibits a narrower binding pocket and a more constricted tunnel (~ 770 Å3). This observation is consistent with the proposed requirement for FosTE to accommodate two structurally distinct substrates at different steps of the biosynthetic pathway. While the proposed FosTE precursor 7 contains an extended polyketide moiety, the malonyl group of malonyl-CoA is substantially smaller. For this reason, surrogate 30 was first docked into the binding site of the first model (Fig. 4b, d). In this configuration, R198 and R227 are positioned in proximity to the β-O-malonyl group, forming a stabilising hydrogen-bonding network that could support substrate binding and orientation. The thioester carbonyl carbon is located near the catalytic S159, consistent with a transacylation step, and the δ-hydroxyl group is positioned close to the same carbonyl carbon, facilitating δ-lactone formation.
When malonyl-CoA was docked into the same model, the malonyl moiety localised near R198 and R227, but the thioester carbonyl carbon remained too distant from S159 to permit nucleophilic attack (Supplementary Fig. 28). Consequently, malonyl-CoA was instead docked into the narrower pocket of the second model (Fig. 4c, e). In this model, the substrate channel is more spatially restricted, and the exit region is sterically constricted, leaving insufficient volume to accommodate a bulky polyketide moiety such as that of thioester 30 (Fig. 4c). We therefore propose that this second model captures a more closed state with an active-site geometry compatible with malonyl-CoA binding. Docking of malonyl-CoA into this model positions the thioester carbonyl carbon in close proximity to S159, consistent with nucleophilic attack during transmalonylation, while the terminal carboxylate forms electrostatic interactions with the side chains of R198 and R227 (Fig. 4e). These interactions likely ensure proper anchoring of the malonyl moiety and enhance the electrophilicity of the carbonyl carbon, promoting efficient transmalonylation. This finding aligns with the established function of conserved arginine residues in AT domains, which mediate extender unit recognition and substrate binding61,62,63,64.
To substantiate the molecular docking results, a FosMod8(R198L/R227L) double mutant and the single mutants FosMod8(R198L) and FosMod8(R227L) were created and reacted with surrogate 17, malonyl-CoA and NADPH (Fig. 3i and Supplementary Fig. 18). Compared to wild-type FosMod8, mutant FosMod8(R227L) produced significantly lower amounts of malonyl-lactone 27, along with higher levels of β-ketolactone 24, β-hydroxylactone 25 and hydrolysis product 19 (Supplementary Fig. 18d). In contrast, the R198L single mutation completely disrupted the module’s elongating activity, leaving only residual thioester hydrolysis (Supplementary Fig. 18c). The mutation appears to impair the module’s catalytic function, possibly by disrupting domain interactions in a manner comparable to the S159A mutant (Fig. 3c–e). In the reaction of the FosMod8(R198L/R227L) double mutant, β-hydroxylactone 25 was observed along with dihydroxycarboxylic acid 21 and carboxylic acid 19 without any traces of 27 and 28 (Fig. 3i), an outcome that is most similar to the reaction of 17 with FosMod8(JerTE). The replacement of both polar and positively charged arginine residues by leucines thus appeared to partially restore lactonisation/hydrolysis but not O-malonylation activity. Probably, hydrophobic interactions between leucines stabilise the active site, but do not provide the electrostatic interactions crucial for binding and activation of the malonyl group.
Notably, the homologues PlmTE (phoslactomycin PKS) and LepTE (leptomycin PKS) conserve arginine residues at equivalent positions (Supplementary Figs. 29, 31)19,20,21,22,65. This observation aligns with multiple sequence alignment, showing that these TEs possess a GxSxS consensus motif, contrasting with the canonical GxSxG motif found in most TEs (Supplementary Fig. 30). This divergence in active-site architecture and sequence motifs, together with the mutation experiments on FosTE, strongly supports their classification as an evolutionarily distinct subclass of TEs with dual activity.
Timing of O-malonylation and lactonisation
Backbone hydroxyl acylation by C-terminal TE domains has been described to take place on assembly line-bound42,43,44 and free macrolactone38,39,40 precursors. To narrow down the timing of O-malonylation by FosTE, in vitro assays with surrogates mimicking putative biosynthetic precursors before and after PKS release were performed (Fig. 5a, b). The SNAC thioester analogue of ACP thioester 7 is prone to undergo spontaneous lactonisation, so that the non-hydrolysable analogue 31 was synthesised, which also avoids undesired self-acylation of FosTE due to the known specificity of TEs for thioesters. Upon incubation of 31 with malonyl-CoA and FosMod8 or FosTE, we indeed observed the formation of the O-malonylated species 32a and 32b (Fig. 5c left, Supplementary Fig. 19). The relatively low conversion by both enzymes can be attributed to the limited ability of the oxoester to mimic all structural features of the biosynthetic precursor and an additional loss of interdomain interactions in the case of isolated FosTE. The formation of regioisomers is consistent with the emergence of 23a and 23b in the incubations of 17 with FosMod8 and can be attributed to spontaneous acyl migration, a well-documented phenomenon in mono-acylated polyols (Fig. 2a and Supplementary Figs. 40–45)66,67. The minor formation of dihydroxycarboxylic acid 21 and hydroxylactone 25 also occurred in an enzyme-free reference experiment pointing towards spontaneous hydrolysis/lactonisation occurring under the assay conditions (Supplementary Fig. 17). Most importantly, the incubation of 31 with FosMod8(JerTE), FosMod8(R198L/R227L), FosMod8(S159A) and multiple other point mutants did not give any malonylation to 32, demonstrating reliance on S159, R198 and R227 (Fig. 5c left and Supplementary Fig. 19). By contrast, O-malonylation was observed with an FosMod7-FosTE hybrid module, demonstrating that FosTE can retain its dual activity in a different protein environment if confronted with a substrate of suitable length. Incubation of 31 with FosAT8 and the tridomain FosKS8-AT8-KR8 did not show formation of 32, thus ruling out a contribution of FosAT8 to backbone O-malonylation.

a Proposed mechanism for β-O-malonylation prior to δ-lactonisation. Self-malonylation of catalytic S159 with malonyl-CoA is followed by malonyl transfer onto the C-3 hydroxyl group of the polyketide intermediate. The ACP-bound polyketide is transacylated onto the catalytic S159 of the TE for subsequent cyclisation and off-loading. b Proposed mechanism for β-O-malonylation of released δ-lactone. ACP-bound polyketide is transacylated onto catalytic S159 of the TE for subsequent δ-lactonisation. The malonyl moiety is then transacylated from malonyl-CoA onto S159 and transferred onto the C-3 hydroxyl group of the δ-lactone. c Reaction scheme and EICs for the in vitro reactions of oxoester 31 with FosMod8, FosTE, FosMod8(S159A), FosMod8(R198L/R227L), FosAT8, FosKS8-AT8-KR8, the no-enzyme control reaction and the substrate (31) (left). Reaction scheme and EICs for the in vitro reactions of lactone 25 with FosMod8, FosTE, FosMod8(S159A), FosMod8(R198L/R227L), a negative control, and the product reference 27 (right). Supplementary Figs. 44, 45, 52, 53 for HRMS(/MS) data.
The continued presence of lactone 25 in the crude reaction mixture is a strong indicator that an open-chain intermediate like 7 is the authentic O-malonylation precursor in biosynthesis, as high affinity and efficient conversion of this close surrogate of 15 would be expected. Accordingly, an incubation of lactone 25 with FosMod8 or FosTE in the presence of malonyl-CoA gave no formation of O-malonylated lactone 27 (Fig. 5c right). In conclusion, these results clearly demonstrate that FosTE-catalysed O-malonylation occurs on the ACP-bound intermediate.
Reconstitution of malonate elimination and 9-phosphorylation
Incubating malonyllactone 27 with purified FosM resulted in increased AUDL 28 formation by ~10% compared to the enzyme-free negative control (Fig. 6a). Demalonylation of advanced fostriecin and phoslactomycin biosynthesis intermediates is known from previous studies and was also detected in all our in vitro experiments (Fig. 6a–c, e)18,24,25. Incubation of 27 with FosH and FosM gave much more pronounced malonate elimination into 34 (Fig. 6b), while a single incubation of 27 with FosH showed full conversion of the starting material into phosphorylated malonyllactone 33 along with 34 from spontaneous elimination. FosM virtually fully converted 33 into 34 (Fig. 6c). Notably, FosM showed significantly higher activity towards phosphoryl lactone 33 compared to lactone 27 (Fig. 6a, c), proving that 9-phosphorylation is important, if not essential for efficient FosM-mediated processing. These results confirm previous indications18,24 that FosM-catalysed demalonylation strictly succeeds 9-OH phosphorylation in fostriecin biosynthesis.

a EIC of FosM reaction with malonyllactone 27, and the no-enzyme control reaction. b Simultaneous incubation of FosH and FosM with 27, and the negative control without FosM. c Reaction of FosM with 33 and the negative control without enzyme. d FosH reaction with AUDL 28 and negative control without enzyme. e FosMod8 and FosTEII were incubated with substrate 17, malonyl-CoA, and NADPH for 3 h. Subsequently, FosH, ATP, and MgCl2 were added to the reaction, and the mixture was incubated for an additional hour (top). Full reconstitution: FosMod8 and FosTEII were incubated with substrate 17, malonyl-CoA, and NADPH for 3 h. Subsequently, FosH, FosM, ATP, and MgCl2 were added to the reaction, and the mixture was incubated for an additional hour (bottom). Dashed arrows illustrate the emergence of side products 37 and 24 during the reactions. Supplementary Figs. 36–39 for HRMS(/MS) data.
FosH also converted AUDL 28 into 34 to a similar degree as 27 and 28, which aligns with the functional relevance of the 9-phosphate group for the bioactivity of fostriecin (Fig. 6d)17,68. The tolerance of FosH likely provides biosynthetic robustness, ensuring consistent phosphorylation. Individual incubations of 16, 17, 25 and 31 with FosH revealed monophosphorylation of all assay compounds, underpinning the relaxed substrate specificity of FosH (Supplementary Fig. 20). The hypothesis that phosphorylation might additionally occur during the final stages of PKS processing was effectively ruled out during a full reconstitution of the AUDL formation process (Fig. 6e). When 17 was reacted with FosMod8 and FosTEII followed by addition of FosH and FosM after 3 h, the 9-phosphorylated AUDL 34 was observed as the main product with 37 as the major side product. Excluding FosM shifted the main product to 33, while still accumulating similar amounts of 37. This compound was exclusively observed when 17 was preincubated with FosH for 16 h, followed by the addition of FosMod8 and FosTEII, demonstrating that 9-phosphorylation prohibits acceptance by FosMod8 (Supplementary Fig. 21).
Overall, the AUDL is biosynthesised by a highly ordered sequence of ketide elongation, O-malonylation, lactonisation, 9-phosphorylation and demalonylation, which is controlled by the substrate specificity of the participating enzymes. This unique biosynthetic arrangement optimises the yield of fostriecin by coupling the formation of the two relevant structural elements of the pharmacophore. At the same time, shunt products resulting from spontaneous demalonylation of 8, 9, dephospho-10 or dephospho-11 can be ‘rescued’ for effective processing owing to the broad substrate tolerance of FosH. The hydroxylations at C-4, C-8 and C-18 appear to be dispensable for the catalytic activity of FosH and FosM, suggesting that FosG, FosJ and FosK can interfere at multiple stages of the tailoring process to yield fostriecin and its analogues PD113,270 and PD113,271.
Discussion
The AUDL is a pharmacophoric moiety found in various polyketides with exceptional bioactivity14. In this study, we have elucidated the biosynthesis of the AUDL in fostriecin using in vitro experiments with purified enzymes and complex synthetic substrate surrogates. Reconstituting the reaction of the PKS modules FosMod7 and FosMod8 led to the identification of a β-O-malonylation activity embedded within the C-terminal FosTE domain. Evidence for this activity was on the one hand, provided by an in-trans complementation experiment of a hydrolysis / malonylation-incompetent FosMod8 mutant with isolated FosTE, which restored the formation of malonyllactone 27 and AUDL 28 from 17. Furthermore, FosMod8, a lone-standing FosTE and the hybrid module FosMod7-FosTE O-malonylated 31, a non-hydrolysable analogue of the FosMod8 elongation product, showing that this activity is directly linked to the presence of the FosTE domain. Replacing FosTE with the canonical JerTE domain from jerangolid biosynthesis accordingly yielded a hybrid module (FosMod8(JerTE)), which was able to extend 17 and hydrolyse or lactonise the resulting intermediate, but incompetent to O-malonylate 21, 25 or 31. Incubation of 31 with FosAT8, FosKS8-AT8-KR8, and FosTE-mutated FosMod8 constructs containing an intact AT demonstrated that, despite the AT domain’s natural function as a canonical malonate introducer in PKS, it does not play a specific role in O-malonylation. No O-malonylation of the hydroxylactone 25 by FosTE could be reconstituted, strongly pointing towards malonyllactone 8 and not 15 being the first post-PKS intermediate. The strict order of O-malonylation before lactonisation is also supported by the exhaustive O-malonylation of all hydroxyls formed by FosKR8 in the conversion assays of 17 with wild-type FosMod8 (Fig. 3b).
Structural modelling of FosTE revealed two distinct active-site conformations with arginine residues R198 and R227 positioned to enable the transmalonylation activity (Fig. 4b–e). These residues appear to cooperatively orchestrate malonyl-CoA binding and electrophilic activation, mimicking AT domain chemistry within the FosTE scaffold. Point mutations of R198 and/or R227 in FosMod8 led to a sharp drop or complete abolishment of malonyllactone 27 formation starting from 17. The R198L/R227L double mutant instead accumulated the dihydroxycarboxylic acid 21 and β-hydroxy lactone 25 alongside hydrolysis product 19, giving a product profile most similar to that of FosMod8(JerTE) bearing the canonically-acting TE (Fig. 2f). Along these lines, all R→L point mutants of FosTE and FosMod8 lost their ability to O-malonylate 31. This emphasises the dominant role of R198 and R227 for transmalonylation, which is accompanied by further, more subtle differences, like the presence of a distinctive GxSxS motif in FosTE that diverges from the highly conserved GxSxG motif commonly found in canonical TE domains69. Intriguingly, these mutations are conserved in homologous TEs from the phoslactomycin and leptomycin biosynthetic pathways (Supplementary Figs. 24 and 25), indicating a shared evolutionary trajectory among TE domains specialised for installing the AUDL warhead (Supplementary Fig. 32). FosTE exhibits catalytic abilities that exceed those of all hydroxyl-esterifying TE domains reported to date. In contrast to TrsA-TE, NecA-TE and Sln9-TE, FosTE is bifunctional and acts on a PKS intermediate rather than a free molecule38,39,40. Cong-TE, Ela-TE and ThiA-TE are also inline-acylating, but they only use related ACP-bound acyl building blocks provided by their own assembly line42,43,44. FosTE additionally processes a CoA-bound malonate, introducing a previously unseen pathway for malonyl incorporation, formerly confined to AT and ACP domains5,30,70. TEB domains are the only other known type of assembly line integrated TEs that accept CoA thioesters41. They do, however, not process malonates or ACP-bound acyls and are also monofunctional. Collectively, these features suggest the existence of a new evolutionarily refined subclass of bifunctional TE domains with evolved β-O-malonylation activity, which expands the known catalytic repertoire of type I PKS-TEs45.
Our findings also refine previous assumptions about the sequence and specificity of post-PKS tailoring events in fostriecin biosynthesis. The maturation of the O-malonylated PKS intermediate to fostriecin (1) involves the consecutive action of FosTE, the kinase FosH and the demalonylase FosM, precisely guided by their substrate specificities. The malonate elimination activity of FosM was virtually inexistent with the malonyllactone 27 and requires prior C-9 phosphorylation by the broad-specific kinase FosH to become markedly enhanced (Fig. 6b, c). Although FosH phosphorylated various surrogates of PKS and post-PKS intermediates, its role during biosynthesis could clearly be restricted to a post-PKS enzyme as the phosphate ester in 37 prohibited processing by FosMod8. This biosynthetic arrangement could ultimately optimise the formation of the 9-phosphorylated AUDL pharmacophore, given the inevitable spontaneous elimination of malonyllactones to AUDLs. In addition to the FosM-catalysed demalonylation of phosphorylated malonyllactones, the relaxed substrate specificity of FosH enables a second pathway via recovery of non-phosphorylated AUDL shunt products. All processes involved in AUDL formation and phosphorylation appear to be independent of the oxidation state of the biosynthesis intermediates, suggesting flexible timing of their action within the tailoring process.
Future research will focus on more refined mechanistic investigations to unravel the complex processes underlying FosMod8 catalysis. Elucidating the interplay of FosTE self-malonylation and the assembly of the polyketide chain could, for example, explain the observed coupling between KR and TE activities and help to improve the in vitro performance of FosMod8. High-resolution crystal structures of relevant enzymes will be essential to capture individual states and guide in vitro validation. Furthermore, a more precise mechanistic assignment of individual amino acid residues within FosTE will advance understanding of the catalytic cycle.
Investigations into the specificity of the pharmacophore-forming enzymes will provide critical insights for optimised application in chemoenzymatic synthesis. Building on the scalability of transformations with FosH, FosJ, FosM and FosMod8 demonstrated by us and others, these findings will enable the development of multienzyme systems for late-stage enzymatic assembly of the fostriecin pharmacophore. This knowledge will also be crucial for biosynthetic engineering to obtain novel AUDL polyketides. Investigating FosTE/FosMod8 homologues from the Pn, Plm and Lep PKSs will broaden the knowledge base and the scope of application.
In summary, this study reveals the full reconstitution of AUDL biosynthesis, a structural motif that is part of the pharmacophore of a subgroup of polyketides with exceptional bioactivity and high relevance for drug development. New biosynthetic enzymology was uncovered by in vitro characterisation of the bifunctional lactonisation–O-malonylating PKS domain FosTE and the demalonylating tailoring enzyme FosM, which together set up the AUDL fragment from a 3-hydroxythioester PKS intermediate. The demalonylation is reliant on previous kinase-catalysed 9-phosphorylation, thus linking and optimising the formation of both essential entities of the pharmacophore. These findings lay the groundwork for new strategies of metagenomic discovery, engineered biosynthesis and chemoenzymatic synthesis to access phosphorylated AUDL natural products and analogues26.
Methods
Additional methods, materials and spectra are available in the Supplementary Information.
Cloning procedure
All synthetic gene sequences used in this study were codon-optimised for expression in E. coli and purchased from Genscript, Genewiz or Merck. Side-directed mutagenesis (SDM) was performed according to the literature using primers listed in Supplementary Table 271. To produce His-tagged fusion proteins, the corresponding gene sequences were amplified by PCR (Peqlab Peqstar thermocycler) using the primers listed in Supplementary Table 2. All PCR products were purified using Monarch Spin PCR & DNA Cleanup Kit or Monarch DNA Gel Extraction Kit (New England Biolabs) according to the manufacturer’s instructions. Insert fragments were cloned into the target vector pET-28a(+) (linearised with NdeI and XhoI for N-terminal tags, NcoI and XhoI for C-terminal tags) using Gibson Assembly (Invitrogen). Gibson Assembly reaction mixtures or PCR products for SDM were subsequently introduced into chemically competent E. coli TOP10 cells for plasmid propagation. Plasmid DNA was isolated from transformed E. coli TOP10 cells using a Monarch Plasmid Miniprep Kit (New England Biolabs) according to the manufacturer’s instructions. All DNA sequences were verified via Sanger sequencing (Microsynth). For heterologous expression, the verified plasmid DNA was introduced into chemically competent E. coli BL21(DE3) or BAP1 cells. Information on the oligonucleotide and plasmids used in this study are also available in the Supplementary Data 2 and 3 files.
Heterologous protein production and purification
Unless otherwise specified, transformed E. coli BL21(DE3) and BAP1 cells were cultivated in terrific broth medium supplemented with 0.1%(v/v) kanamycin (60 mg/mL stock solution) in 0.5 L flasks at 37 °C and 180 rpm until the optical density at 600 nm (OD600) reached 0.7 – 0.9. For expressions at 15 – 20 °C, the cultures were cooled at 4 °C for 20 min prior to induction, which was a critical step for the soluble production of FosMod8 variants. For expression at 30 °C, the inducer was added directly upon reaching the desired OD600. The cells were generally harvested and washed by centrifugation at 5000 × g and 4 °C for 20 min.
For protein purification all steps were performed at 4 °C or on ice, and buffers A (30 mM Tris-HCl, 500 mM NaCl, 10 mM imidazole, 10%(w/v) glycerol, pH 7.5), B (30 mM Tris-HCl, 500 mM NaCl, 500 mM imidazole, 10%(w/v) glycerol, pH 7.5), C (50 mM HEPES, 100 mM NaCl, 2.5 mM EDTA, 20%(w/v) glycerol, pH 7.8), D (200 mM sodium phosphate, pH 7.5), E (200 mM sodium phosphate, pH 6.8), F (50 mM sodium phosphate, 50 mM NaCl, 10%(w/v) glycerol, pH 7.5), G (50 mM sodium phosphate, 50 mM NaCl, 500 mM imidazole, 10%(w/v) glycerol, pH 7.5), H (50 mM sodium phosphate, 50 mM NaCl, pH 7.5), I (50 mM sodium phosphate, 1 M NaCl, pH 7.5), J (100 mM sodium phosphate, pH 7.5), K (50 mM sodium phosphate, 50 mM NaCl, 10 mM imidazole, 10%(w/v) glycerol, pH 7.5) and L (100 mM sodium phosphate, pH 8) were used. When applicable, reducing agents were freshly prepared and added to the buffers just before starting the purification process. Protein samples were always concentrated using 3, 10 or 30 kDa molecular weight cut-off (MWCO) ultra centrifugal filters (Merck) by centrifugation at 5000 × g and 4 °C.
To produce FosH and FosKR8, gene expression in E. coli BL21(DE3) was induced with a final concentration of 0.5 mM (FosH) or 0.3 mM (FosKR8) isopropyl-β-d-thiogalactopyranoside (IPTG) and the cultures were shaken at 20 °C (FosH) or 30 °C (FosKR8) with 180 rpm for 20 h. The collected cells were resuspended in buffer A with 0.1 g cell pellet/mL and disrupted by sonication on ice (45% amplitude, 10 s sonication, 10 s hold, 10 min per 20 mL cell suspension). Cell debris was removed by centrifugation (16000 × g, 4 °C, 30 min) and the crude lysate was subsequently filtered (0.45 μm, polytetrafluorethylen (PTFE) syringe filter from Labsolute) for immobilised metal chelate affinity chromatography (IMAC) purification of target proteins by fast protein liquid chromatography (FPLC), using a 5 mL HisTrap FF Ni-NTA column (Cytiva). The column was washed with 20 column volumes (CV) of 10% buffer B in buffer A. The target protein was eluted using a linear gradient of 10 → 100% buffer B in buffer A over 5 CV. The pooled fractions were concentrated to a maximum volume of 6 mL and subjected to a pre-equilibrated (2 CV buffer E) 25 mL desalting column containing Sephadex G-25 resin (Cytiva) for buffer exchange with an isocratic gradient (1 CV buffer E). The combined, protein containing fractions were concentrated using a 10 kDa MWCO ultra-centrifugal filter and immediately used for activity assays or aliquoted and flash frozen in liquid N2 before storing at − 80 °C.
FosM, FosAT8 and MatB were produced in E. coli BL21(DE3) by inducing expression with 1 mM (FosM), 0.1 mM (FosAT8) or 0.5 mM (MatB) IPTG and the cultures were incubated at 15 °C with 180 rpm for 22 h. The harvested cells were resuspended in buffer A with 0.1 g cell pellet/mL and disrupted by sonication on ice (45% amplitude, 30 s sonication, 30 s hold, 10 min per 20 mL cell suspension). Cell debris was removed by centrifugation (16000 × g, 4 °C, 30 min), and the crude lysate was subsequently filtered (0.45 μm, PTFE syringe filter from Labsolute) for IMAC purification by FPLC (5 mL HisTrap FF Ni-NTA column from Cytiva). The column was washed with 10 CV of buffer A. The target protein was eluted using a linear gradient of 0 → 100% buffer B in buffer A over 5 CV. The pooled fractions were concentrated to a maximum volume of 6 mL and subjected to a pre-equilibrated (2 CV buffer E for FosM and FosAT8, buffer C for MatB) 25 mL desalting column containing Sephadex G-25 resin (Cytiva) for buffer exchange (1 CV buffer E for FosM and FosAT8, buffer C for MatB). The fractions were concentrated using a 10 kDa MWCO ultra-centrifugal filter and immediately used for activity assays or aliquoted and flash frozen in liquid N2 before storing at − 80 °C.
FosTEII production in E. coli BL21(DE3) was performed with 0.1 mM IPTG and 180 rpm at 15 °C for 22 h. The harvested cells were resuspended in buffer K with 0.1 g cell pellet/mL and disrupted by sonication on ice (45% amplitude, 10 s sonication, 10 s hold, 10 min per 20 mL). Cell debris was removed by centrifugation (16000 × g, 4 °C, 30 min), and the crude lysate was filtered (0.45 μm, PTFE syringe filter from Labsolute) for IMAC purification of target proteins by FPLC (5 mL HisTrap FF Ni-NTA column from Cytiva). The column was washed with 10 CV of buffer K. The target protein was eluted using a linear gradient of 0 → 100% buffer G in buffer K over 5 CV. Protein containing fractions were concentrated to a maximum volume of 500 µL (10 kDa MWCO ultra-centrifugal filter) and subjected to a pre-equilibrated (2 CV buffer D) Superdex 75 10/300 GL column (Cytiva) for further purification (1 CV buffer D). Fractions containing the monomeric protein were concentrated using a 10 kDa MWCO ultra-centrifugal filter and immediately used for activity assays or aliquoted and flash frozen in liquid N2 before storing at − 80 °C.
For FosMod7 and FosMod7-FosTE, expression in E. coli BAP1 was induced using 0.1 mM IPTG, and the cultures were incubated at 15 °C and 180 rpm for 19 h. Production of FosMod8 and its mutant variants (FosMod8(S159A), FosMod8(R198L), FosMod8(R227L), and FosMod8(R198L/R227L)) was carried out by induction with 0.7 mM IPTG and 180 rpm at 15 °C for 19 h. For FosKS8-AT8-KR8, expression in E. coli BL21(DE3) was carried out with 0.1 mM IPTG and 180 rpm at 15 °C for 22 h.
In the initial purification protocol for FosMod7, FosMod7-FosTE and FosMod8, harvested cells were resuspended in buffer A (0.1 g cell pellet/mL buffer) and disrupted by sonication on ice (45% amplitude, 30 s sonication, 30 s hold, 10 min per 1 g cell pellet). Cell debris was removed by centrifugation at 13000 × g for 30 min at 4 °C. The resulting crude lysate was filtered through a 0.45 μm PTFE syringe filter (Labsolute) and subjected to FPLC IMAC purification (5 mL HisTrap FF Ni-NTA column from Cytiva). The column was washed with 10 CV of buffer A. The target protein was eluted using a linear gradient of 0 → 100% buffer B in buffer A over 5 CV, and the corresponding fractions were subjected to a PD-10 desalting column (Cytiva) for buffer exchange in buffer J and concentrated with a 10 kDa MWCO ultra-centrifugal filter and immediately used for activity assays.
Using the optimised protocol for isolating FosMod7, FosMod8, and their mutant variants (FosMod7-FosTE, FosMod8(S159A), FosMod8(R198L), FosMod8(R227L), and FosMod8(R198L/R227L)), harvested cells were resuspended at a concentration of 0.1 g cell pellet per mL of buffer F supplemented with 1 mM Tris-(2-carboxyethyl)-phosphine (TCEP). The cells were disrupted by sonication on ice (45% amplitude, 10 s sonication, 10 s hold for 10 minutes per 15 mL cell suspension). Cell debris was removed by centrifugation (16000 × g, 4 °C, 30 min), and the crude lysate was filtered (0.45 μm, PTFE syringe filter from Labsolute) for IMAC purification by FPLC (5 mL HisTrap FF Ni-NTA column from Cytiva). The column was washed with 20 CV of 8.5% buffer G + 1 mM TCEP in buffer F + 1 mM TCEP. The target protein was eluted using a linear gradient of 8.5 → 100% buffer G + 1 mM TCEP in buffer F + 1 mM TCEP over 5 CV. The protein containing fractions were pooled, diluted with an equal volume of buffer H and centrifuged at 17000 × g and 4 °C for 10 min before loading onto a pre-equilibrated (5 CV buffer H + 0.2 mM TCEP) 5 mL HiTrap Q FF anion exchange column (Cytiva). After loading the protein sample, the column was washed with 5 CV of buffer H + 0.2 mM TCEP. Homodimeric species of the modules were separated from higher order oligomers using a step gradient (45% (6 CV) → 60% (3 CV) → 100% (3 CV) buffer I + 0.2 mM TCEP in buffer H + 0.2 mM TCEP). Fractions containing the dimeric proteins (eluting at 45% buffer I + 0.2 mM TCEP) were pooled and concentrated with a 30 kDa MWCO ultra-centrifugal filter and immediately used for activity assays or aliquoted and flash frozen in liquid N2 before storage at − 80 °C.
For FosMod8(JerTE), the expression in E. coli BAP1 was induced with 0.3 mM IPTG and 180 rpm at 15 °C for 18 h. The harvested cells were resuspended in buffer F supplemented with 1 mM TCEP (0.1 g cell pellet/mL buffer) and disrupted by sonication on ice (45% amplitude, 10 s sonication, 10 s hold, 10 min per 20 mL). Cell debris was removed by centrifugation (17000 × g, 4 °C, 30 min), and the crude lysate was filtered (0.45 μm, PTFE syringe filter from Labsolute) for IMAC purification by FPLC (5 mL HisTrap FF Ni-NTA column from Cytiva). The column was washed with 20 CV of 7% buffer G + 1 mM TCEP in buffer F + 1 mM TCEP. Protein elution was performed using a linear gradient a 70% step gradient of buffer G + 1 mM TCEP in buffer F + 1 mM TCEP over 3 CV. Target protein containing fractions were pooled and concentrated to a volume of 1 mL using a 30 kDa MWCO ulta-centrifugal filter and subsequently subjected to a pre-equilibrated (2 CV buffer E + 0.2 mM TCEP) HiLoad 16/600 Superose 6 pg preparative column (Cytiva) for size exclusion chromatography (SEC) purification (1 CV buffer E + 0.2 mM TCEP). Fractions containing the homodimeric protein were concentrated using a 30 kDa MWCO ultra-centrifugal filter and immediately used for activity assays or aliquoted and flash frozen in liquid N2 before storing at − 80 °C.
For FosTE and its mutant variants (R198L, R227L and R198L/R227L), the expression in E. coli BL21(DE3) was induced with 1 mM IPTG and 180 rpm at 15 °C for 19 h. The harvested cells were resuspended in buffer F with 1 mM TCEP (0.2 g cell pellet/mL buffer) and disrupted by sonication on ice (50% amplitude, 10 s sonication, 10 s hold, 10 min per 15 mL). Cell debris was removed by centrifugation (17000 × g, 4 °C, 30 min), and the crude lysate was filtered (0.45 μm, PTFE syringe filter from Labsolute) for IMAC purification by FPLC (5 mL HisTrap FF Ni-NTA column from Cytiva). The column was washed with 20 CV of 8% buffer G + 1 mM TCEP in buffer F + 1 mM TCEP, and the proteins were eluted with a linear gradient of 8 → 100% buffer G + 1 mM TCEP in buffer F + 1 mM TCEP over 5 CV. Target protein containing fractions were concentrated to a volume of 1 mL, using a 10 kDa MWCO ultra-centrifugal filter. Subsequently, the sample was subjected to a pre-equilibrated (2 CV buffer D + 0.2 mM TCEP) HiLoad 16/600 Superose 6 pg preparative column (Cytiva) for SEC purification (1 CV buffer D + 0.2 mM TCEP). Fractions containing the monomeric protein were concentrated using a 10 kDa MWCO ultra-centrifugal filter (Merck) and immediately used for activity assays or aliquoted and flash frozen in liquid N2 before storing at − 80 °C.
Holo-FosACP7 and holo-FosACP8 production in E. coli BAP1 was performed with 0.1 mM IPTG and 180 rpm at 15 °C for 19 h. The harvested cells were resuspended in buffer F with 0.1 g cell pellet/mL and disrupted by sonication on ice (45% amplitude, 10 s sonication, 10 s hold, 10 min per 20 mL). Cell debris was removed by centrifugation (16000 × g, 4 °C, 30 min), and the crude lysate was filtered (0.45 μm, PTFE syringe filter from Labsolute) for IMAC purification by FPLC (5 mL HisTrap FF Ni-NTA column from Cytiva). The column was washed with 20 CV of 7% buffer G in buffer F. The target proteins were isocratically eluted with 60% buffer G in buffer F (3 CV), and the corresponding fractions were concentrated to a volume of 500 µL, using a 3 kDa MWCO ultra-centrifugal filter. The protein sample was subsequently loaded onto a pre-equilibrated (2 CV buffer L) Superdex 75 10/300 GL column (Cytiva) for SEC (1 CV buffer L). Fractions containing the target protein were concentrated using a 3 kDa MWCO ultra-centrifugal filter and flash frozen in liquid N2 before storing at − 80 °C
For PikTE production, the transformed E. coli BL21(DE3) cells were cultivated in 2 × yeast extract tryptone (dYT) medium supplemented with 0.1%(v/v) kanamycin (60 mg/mL stock solution) in 0.5 L flasks at 37 °C and 180 rpm until the OD600 reached 0.4 – 0.6. Gene expression was induced with 0.1 mM IPTG, followed by incubation at 15 °C and 180 rpm for 22 h. The harvested cells were then resuspended in buffer F at a concentration of 0.1 g cell pellet per mL and lysed by sonication on ice (50% amplitude, 10 s sonication, 10 s hold, 10 min per 15 mL). Cell debris was removed by centrifugation (16000 × g, 4 °C, 30 min), and the crude lysate was filtered (0.45 μm, PTFE syringe filter from Labsolute) for IMAC purification by FPLC (5 mL HisTrap FF Ni-NTA column from Cytiva). The column was washed with 20 CV of 8% buffer G in buffer F. The target protein was eluted using a linear gradient of 8 → 100% buffer G in buffer F over 5 CV, and the pooled fractions were concentrated to a maximum volume of 6 mL prior to subjection to a pre-equilibrated (1 CV buffer E) 25 mL desalting column containing Sephadex G-25 resin (Cytiva) for buffer exchange (2 CV buffer E). Target protein containing fractions were concentrated using a 10 kDa MWCO ultra-centrifugal filter and immediately used for activity assays or aliquoted and flash frozen in liquid N2 before storing at − 80 °C.
Analytical-scale enzymatic in vitro assays
In vitro assays with thioester 16 were set up with a total volume of 50 μL in a 200 mM sodium phosphate buffer, pH 6.8, containing 2%(v/v) DMSO, 1 mM 16, 5 mM NADPH and 5 mM malonyl-CoA. Reactions either contained 9 µM of FosMod7-FosTE or a combination of FosMod7 and FosMod8, each with a concentration of 9 µM. In cases where FosTEII was included in the reaction, it was added at a concentration of 0.9 µM, 2.25 µM, 4.5 µM, 9 µM or 18 µM. All reactions were incubated at 25 °C and 300 rpm for 3 h or 16 h.
Assays with thioester 17 were set up with a total volume of 50 μL in a 200 mM sodium phosphate buffer, pH 6.8, containing 2%(v/v) DMSO, 1 mM 17, 9 µM module enzyme (FosMod8, FosMod8(S159A), FosMod8(R198L/R227L), FosMod8(R198L), FosMod8(R227L) or FosMod8(JerTE)), 5 mM NADPH and 5 mM malonyl-CoA. In cases where FosTEII was added to the reaction, it was added at a concentration of 0.9 µM, 2.25 µM, 4.5 µM, 9 µM or 18 µM. In FosMod8(S159A) reactions with FosKR8 and FosTE, one or both domains were included at concentrations of 18 µM each. All reactions were incubated at 25 °C and 300 rpm for 3 h.
Assays using δ-lactone 25 or oxoester 31 were prepared in a total volume of 50 μL with 200 mM sodium phosphate buffer at pH 6.8, 2%(v/v) DMSO, 1 mM substrate (25 or 31), 5 mM malonyl-CoA and 18 µM module enzyme (FosMod8, FosMod8(S159A), FosMod8(R198L/R227L), FosMod8(R198L), FosMod8(R227L), FosMod8(JerTE) FosMod7-FosTE or FosKS8-AT8-KR8). For assays involving stand-alone domains (FosTE, FosAT8, FosTE(R198L), FosTE(R227L), or FosTE(R198L/R227L)), the proteins were included at 60 µM each. All reactions were incubated at 25 °C and 300 rpm for 16 h.
FosH reactions were generally prepared in a volume of 50 μL with 0.5 mM substrate, 1 mM ATP, 5 mM MgCl2 and 60 μM FosH in 200 mM sodium phosphate buffer, pH 6.8 and incubated at 25 °C and 300 rpm for 1 h. FosM reactions were generally prepared in a volume of 50 μL with 0.5 mM substrate and 50 μM FosM in 200 mM sodium phosphate buffer, pH 6.8 and incubated at 25 °C and 300 rpm for 1 h. In experiments where FosH and FosM were applied simultaneously, each mixture contained 0.5 mM substrate, 1 mM ATP, 5 mM MgCl2, 60 μM FosH, and 50 μM FosM, all in 200 mM sodium phosphate buffer (pH 6.8). The reactions were set up with a total reaction volume of 50 µL and incubated for 1 h at 25 °C and 300 rpm.
In FosH reactions with substrate 17, where FosMod8 and FosTEII were subsequently added, the following protocol was employed: Initially, FosH (50 µM), ATP (1 mM), MgCl2 (1 mM), and substrate 17 (1 mM) were incubated in 200 mM sodium phosphate buffer (pH 6.8) containing 2% DMSO for 16 h. Thereafter, FosH was removed from the reaction mixture by centrifugation using a 10 kDa molecular weight cut-off filter (14000 × g). To the resulting filtrate, FosMod8 (9 µM), FosTEII (2.25 µM), malonyl-CoA (5 mM), and NADPH (5 mM) were added, adjusting the final volume to 80 µL. The mixture was incubated for an additional 5 h at 25 °C and 300 rpm.
For reactions of FosMod8 with substrate 17, in which FosH and/or FosM were subsequently added, the following protocol was used: FosMod8 (9 µM), FosTEII (2.25 µM), substrate 17 (1 mM), malonyl-CoA (5 mM), and NADPH (5 mM) were incubated in 200 mM sodium phosphate buffer, pH 6.8 (supplemented with 2% DMSO) for 3 h at 25 °C and 300 rpm in a 50 µL reaction volume. Subsequently, FosH (35 µM), along with ATP (1 mM) and MgCl2 (1 mM), was added, resulting in a final volume of 60 µL, and incubation was continued for an additional hour under the same conditions (25 °C, 300 rpm). In reactions where FosM was included in addition to FosH, FosM was added at a final concentration of 20 µM (in 60 µL total volume) simultaneously with FosH, followed by incubation for 1 h at 25 °C and 300 rpm.
In general, after completion of the reaction, enzymes were removed prior to analysis by U(H)PLC-(HR)MS using 10 or 30 kDa centrifugal filters at 14000 × g (MERCK) for at least 30 min. The filter membrane was washed with an equal volume of water, and the filtrates were combined and centrifuged again for 5 minutes at 17000 × g before analysis.
All analytical-scale in vitro experiments were performed at least two times with similar results, and representative data are shown.
Self-malonylation of FosTE with Malonyl-CoA
Reactions containing 5 µM FosTE were prepared in 200 mM NaH2PO4 buffer (pH 6.8), supplemented with 5 mM malonyl-CoA and 1 mM TCEP, in a total reaction volume of 100 µL. Samples were incubated for 4 h at 25 °C with continuous agitation at 300 rpm. Following incubation, each sample was buffer-exchanged three times into 50 mM ammonium formate (pH 4.0), using 10 kDa MWCO spin filters. Intact protein analysis was conducted by U(H)PLC-HRMS, injecting 1 µg of protein per run. Chromatographic separation comprised a 3 min pre-equilibration under initial gradient conditions, followed by a linear gradient from 30% → 80% MeCN + 0.1% DFA in H2O + 0.1% DFA between 1.5-15 min. The column was then washed from 15-16 min with 80% → 90% MeCN + 0.1% DFA in H2O + 0.1% DFA and held at 90% MeCN + 0.1% DFA in H2O + 0.1% DFA from 16–18 min. A YMC-Triart Bio C4 column (1.9 μm particle size, 2.1 × 50 mm, 300 Å pore size) was employed for chromatographic separation. The column oven temperature was maintained at 40 °C throughout the analysis. Data analyses were perfromed using UniDec72.
Chemical synthesis and semi-preparative chemoenzymatic reactions
Details of all chemical syntheses (compounds 16, 25, 28, and 31), semi-preparative chemoenzymatic reactions (compounds 19, 27, 21, and malonyl-CoA), and small-scale chemoenzymatic reactions (compound 33), as well as their corresponding spectral data, are provided in the Supplementary Information.
Molecular modelling and docking
Protein structures were predicted using AlphaFold 3with 200 seeds per protein. All ligand structures were initially built in Chem3D Professional 17.1 (PerkinElmer). Docking calculations were performed using AutoDock Vina 1.1.2, and results were analysed in PyMOL 3.4.1 (Schrödinger). Prior to docking, ligands and receptors were prepared by adding polar hydrogens and defining selected rotatable and non-rotatable bonds. For ligand docking, grid boxes were placed in the substrate binding sites as follows: phosphopantethein thioester 30, grid centre x = − 5.58, y = − 15.66, z = 10.13; grid size 22.8 Å × 13.68 Å × 10.13 Å; binding affinity − 8.0 kcal/mol. For malonyl-CoA, grid center x = − 0.13, y = 22.82, z = − 13.45; grid size 24.4 Å × 24.4 Å × 17.4 Å; binding affinity − 10.4 kcal/mol. The exhaustiveness parameter was set to 40. Protein cavities were predicted and visualised using the PyMOL plug-in CavitOmiX.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are provided in the Supplementary Information. The fostriecin and jerangolid gene sequences used in this study were acquired from the NCBI GenBank under accession numbers HQ434551.1 and DQ897668, respectively. Amino acid sequences for multiple sequence alignments were acquired from the MIBiG and the NCBI database. All data are available from the corresponding author upon request. Source data are provided inh this paper.
References
Davison, E. K. & Brimble, M. A. Natural product derived privileged scaffolds in drug discovery. Curr. Opin. Chem. Biol. 52, 1–8 (2019).
Kirschning, A. & Hahn, F. Merging chemical synthesis and biosynthesis: a new chapter in the total synthesis of natural products and natural product libraries. Angew. Chem. Int. Ed. 51, 4012–4022 (2012).
Friedrich, S. & Hahn, F. Opportunities for enzyme catalysis in natural product chemistry. Tetrahedron 71, 1473–1508 (2015).
Grininger, M. Enzymology of assembly line synthesis by modular polyketide synthases. Nat. Chem. Biol. 19, 401–415 (2023).
Hertweck, C. The biosynthetic logic of polyketide diversity. Angew. Chem. Int. Ed. 48, 4688–4716 (2009).
Weissman, K. J. Polyketide stereocontrol: a study in chemical biology. Beilstein J. Org. Chem. 13, 348–371 (2017).
Yang, J. et al. Pironetin reacts covalently with cysteine-316 of α-tubulin to destabilize microtubule. Nat. Commun. 7, 12103 (2016).
Lawhorn, B. G. et al. Total synthesis and evaluation of cytostatin, its C10−C11 diastereomers, and additional key analogues: impact on PP2A inhibition. J. Am. Chem. Soc. 128, 16720–16732 (2006).
Tohyama, S. et al. The potent protein phosphatase 2A inhibitors aminocytostatins: new derivatives of cytostatin. J. Antibiot. 74, 743–751 (2021).
Sun, Y. et al. Lactomycins A–C, dephosphorylated phoslactomycin derivatives that inhibit cathepsin B, from the marine-derived streptomyces sp. ACT232. Mar. Drugs 16, 70 (2018).
Kudo, N. et al. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. USA 96, 9112–9117 (1999).
Fleta-Soriano, E. et al. The myxobacterial metabolite ratjadone A inhibits HIV infection by blocking the Rev/CRM1-mediated nuclear export pathway. Microb. Cell Factories 13, 17 (2014).
Gerth, K., Schummer, D., Höfle, G., Irschik, H. & Reichenbach, H. Ratjadon: a new antifungal compound from Sorangium cellulosum (myxobacteria) production, physio-chemical and biological properties. J. Antibiot. 48, 973–976 (1995).
Trost, B. M., Knopf, J. D. & Brindle, C. S. Synthetic strategies employed for the construction of fostriecin and related natural products. Chem. Rev. 116, 15035–15088 (2016).
Jong, R. S. et al. Phase I and pharmacokinetic study of the topoisomerase II catalytic inhibitor fostriecin. Br. J. Cancer 79, 882–887 (1999).
Lê, L. H. et al. Phase I and pharmacokinetic study of fostriecin given as an intravenous bolus daily for five consecutive days. Invest. New Drugs 22, 159–167 (2004).
Swingle, M. R. et al. Structure-activity relationship studies of fostriecin, cytostatin, and key analogs, with PP1, PP2A, PP5, and (β12–β13)-Chimeras (PP1/PP2A and PP5/PP2A), provide further insight into the inhibitory actions of fostriecin family inhibitors. J. Pharmacol. Exp. Ther. 331, 45–53 (2009).
Kong, R. et al. Elucidation of the biosynthetic gene cluster and the post-PKS modification mechanism for fostriecin in streptomyces pulveraceus. Chem. Biol. 20, 45–54 (2013).
Chen, Y.-L. et al. Identification of phoslactomycin biosynthetic gene clusters from Streptomyces platensis SAM-0654 and characterization of PnR1 and PnR2 as positive transcriptional regulators. Gene 509, 195–200 (2012).
Palaniappan, N., Kim, B. S., Sekiyama, Y., Osada, H. & Reynolds, K. A. Enhancement and selective production of phoslactomycin B, a protein phosphatase IIa inhibitor, through identification and engineering of the corresponding biosynthetic gene cluster. J. Biol. Chem. 278, 35552–35557 (2003).
Hu, Z., Reid, R. & Gramajo, H. The leptomycin gene cluster and its heterologous expression in Streptomyces lividans. J. Antibiot. 58, 625–633 (2005).
Hu, Z. & Reid, R. Biosynthetic Gene Cluster for Leptomycins. (2005).
Han, X. et al. Genome sequence of streptomyces auratus strain AGR0001, a phoslactomycin-producing actinomycete. J. Bacteriol. 194, 5472–5473 (2012).
Liu, X., Kong, R., Niu, M., Qiu, R. & Tang, L. Identification of the post-polyketide synthase modification enzymes for fostriecin biosynthesis in streptomyces pulveraceus. J. Nat. Prod. 76, 524–529 (2013).
Palaniappan, N., Alhamadsheh, M. M. & Reynolds, K. A. cis-Δ2,3-Double bond of phoslactomycins is generated by a Post-PKS tailoring enzyme. J. Am. Chem. Soc. 130, 12236–12237 (2008).
Jiang, Y., Yu, X. & Renata, H. Concise synthesis of fostriecin and analogs through late-stage chemoenzymatic installation of their key pharmacophores. J. Am. Chem. Soc. 147, 25454–25461 (2025).
Wesener, S. R., Potharla, V. Y. & Cheng, Y.-Q. Reconstitution of the FK228 biosynthetic pathway reveals cross talk between modular polyketide synthases and fatty acid synthase. Appl. Environ. Microbiol. 77, 1501–1507 (2011).
Ishikawa, F., Sugimoto, H. & Kakeya, H. In vitro investigation of crosstalk between fatty acid and polyketide synthases in the andrimid biosynthetic assembly line. ChemBioChem 17, 2137–2142 (2016).
Bisang, C. et al. A chain initiation factor common to both modular and aromatic polyketide synthases. Nature 401, 502–505 (1999).
Arthur, C. J. et al. Self-malonylation is an intrinsic property of a chemically synthesized type II polyketide synthase acyl carrier protein. Biochemistry 44, 15414–15421 (2005).
Gan, S. et al. The acyltransferase PMAT1 malonylates brassinolide glucoside. J. Biol. Chem. 296, https://doi.org/10.1016/j.jbc.2021.100424 (2021).
Taguchi, G. et al. Malonylation is a key reaction in the metabolism of xenobiotic phenolic glucosides in Arabidopsis and tobacco. Plant J. 63, 1031–1041 (2010).
Khan, B. R. et al. Malonylation of Glucosylated N-Lauroylethanolamine A NEW PATHWAY THAT DETERMINES N-ACYLETHANOLAMINE METABOLIC FATE IN PLANTS*. J. Biol. Chem. 291, 27112–27121 (2016).
Bretschneider, T. et al. A ketosynthase homolog uses malonyl units to form esters in cervimycin biosynthesis. Nat. Chem. Biol. 8, 154–161 (2012).
Xie, X., Watanabe, K., Wojcicki, W. A., Wang, C. C. C. & Tang, Y. Biosynthesis of lovastatin analogs with a broadly specific acyltransferase. Chem. Biol. 13, 1161–1169 (2006).
Moss, S. J. et al. Identification of Asm19 as an acyltransferase attaching the biologically essential ester side chain of ansamitocins using N-desmethyl-4,5-desepoxymaytansinol, not maytansinol, as its substrate. J. Am. Chem. Soc. 124, 6544–6545 (2002).
Kwon, H.-J. et al. C-O Bond formation by polyketide synthases. Science 297, 1327–1330 (2002).
Niehs, S. P. et al. Mining symbionts of a spider-transmitted fungus illuminates uncharted biosynthetic pathways to cytotoxic benzolactones. Angew. Chem. Int. Ed. 59, 7766–7771 (2020).
Hermes, C. et al. Thioesterase-mediated side chain transesterification generates potent Gq signaling inhibitor FR900359. Nat. Commun. 12, 144 (2021).
Ray, L., Yamanaka, K. & Moore, B. S. A peptidyl-transesterifying type I thioesterase in salinamide biosynthesis. Angew. Chem. Int. Ed. 55, 364–367 (2016).
Fraley, A. E. et al. Structure of a promiscuous thioesterase domain responsible for branching acylation in polyketide biosynthesis. Angew. Chem. Int. Ed. 61, e202206385 (2022).
Zhou, Y. et al. Iterative mechanism of macrodiolide formation in the anticancer compound conglobatin. Chem. Biol. 22, 745–754 (2015).
Zhou, Y., Prediger, P., Dias, L. C., Murphy, A. C. & Leadlay, P. F. Macrodiolide formation by the thioesterase of a modular polyketide synthase. Angew. Chem. Int. Ed. 54, 5232–5235 (2015).
Ji, Q., Xiang, H., Wang, W.-G. & Matsuda, Y. Mechanism behind the programmed biosynthesis of heterotrimeric fungal depside thielavin A. Angew. Chem. Int. Ed. 63, e202402663 (2024).
Cantu, D. C., Chen, Y. & Reilly, P. J. Thioesterases: A new perspective based on their primary and tertiary structures: Thioesterases: A New Perspective. Protein Sci 19, 1281–1295 (2010).
Derra, S., Hoffmann, J. & Hahn, F. Synthesis of biomimetic thioesters for studies of ketoreductase domains from the biosynthesis of cytotoxic polyketides. Synlett https://doi.org/10.1055/a-2216-4521 (2024).
Dutta, S. et al. Structure of a modular polyketide synthase. Nature 510, 512–517 (2014).
Bagde, S. R., Mathews, I. I., Fromme, J. C. & Kim, C.-Y. Modular polyketide synthase contains two reaction chambers that operate asynchronously. Science 374, 723–729 (2021).
Smith, J. L., Skiniotis, G. & Sherman, D. H. Architecture of the polyketide synthase module: surprises from electron cryo-microscopy. Curr. Opin. Struct. Biol. 31, 9–19 (2015).
Geyer, K., Hartmann, S., Singh, R. R. & Erb, T. J. Multiple functions of the type II thioesterase associated with the phoslactomycin polyketide synthase. Biochemistry 61, 2662–2671 (2022).
Mortison, J. D., Kittendorf, J. D. & Sherman, D. H. Synthesis and biochemical analysis of complex chain-elongation intermediates for interrogation of molecular specificity in the erythromycin and pikromycin polyketide synthases. J. Am. Chem. Soc. 131, 15784–15793 (2009).
Pinto, A., Wang, M., Horsman, M. & Boddy, C. N. 6-Deoxyerythronolide B synthase thioesterase-catalyzed macrocyclization is highly stereoselective. Org. Lett. 14, 2278–2281 (2012).
Roß-Taschner, T. et al. Highly stereoselective biocatalytic one-pot synthesis of chiral saturated oxygen heterocycles by integration of a biosynthetic heterocyclase into multiple-enzyme cascades. ACS Catal. 13420–13428 https://doi.org/10.1021/acscatal.4c03692 (2024).
Hahn, F. & Guth, F. M. The ambruticins and jerangolids – chemistry, biology and chemoenzymatic synthesis of potent antifungal drug candidates. Nat. Prod. Rep. 37, 1300–1315 (2020).
Rydzek, S. et al. Chemoenzymatic synthesis of plant-derived kavalactone natural products by dynamic resolution using a biosynthetic O-methyltransferase tailoring enzyme. ChemCatChem e202400883 https://doi.org/10.1002/cctc.202400883 (2024).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Tsai, S.-C. et al. Crystal structure of the macrocycle-forming thioesterase domain of the erythromycin polyketide synthase: Versatility from a unique substrate channel. Proc. Natl. Acad. Sci. USA 98, 14808–14813 (2001).
Zhou, Y. et al. Structural and mechanistic insights into chain release of the polyene PKS thioesterase domain. ACS Catal. 12, 762–776 (2022).
McCullough, T. M. et al. Substrate trapping in polyketide synthase thioesterase domains: structural basis for macrolactone formation. ACS Catal. 14, 12551–12563 (2024).
Scaglione, J. B. et al. Biochemical and structural characterization of the tautomycetin thioesterase: analysis of a stereoselective polyketide hydrolase. Angew. Chem. Int. Ed. 49, 5726–5730 (2010).
Reeves, C. D. et al. Alteration of the substrate specificity of a modular polyketide synthase acyltransferase domain through site-specific mutations. Biochemistry 40, 15464–15470 (2001).
Kalkreuter, E. et al. Computationally-guided exchange of substrate selectivity motifs in a modular polyketide synthase acyltransferase. Nat. Commun. 12, 2193 (2021).
Zhang, F. et al. Structural insights into the substrate specificity of acyltransferases from salinomycin polyketide synthase. Biochemistry 58, 2978–2986 (2019).
Liew, C. W. et al. Crystal structure of the acyltransferase domain of the iterative polyketide synthase in enediyne biosynthesis *. J. Biol. Chem. 287, 23203–23215 (2012).
Geyer, K., Sundaram, S., Sušnik, P., Koert, U. & Erb, T. J. Understanding substrate selectivity of phoslactomycin polyketide synthase by using reconstituted in vitro systems. ChemBioChem 21, 2080–2085 (2020).
Lassfolk, R. et al. Acyl group migration in pyranosides as studied by experimental and computational methods. Chem. Eur. J. 28, e202200499 (2022).
Gorantla, J. N., Santhi, M., Hua, Y. & Cairns, J. R. K. Total synthesis of ceramides and β-O-glucosylceramides via intramolecular fatty acyl group migration. New J. Chem. 46, 3270–3276 (2022).
Della-Felice, F., Bartolomeu, A., de, A. & Pilli, R. A. The phosphate ester group in secondary metabolites. Nat. Prod. Rep. 39, 1066–1107 (2022).
Keatinge-Clay, A. T. The structures of type I polyketide synthases. Nat. Prod. Rep. 29, 1050–1073 (2012).
West, A.-K. R. & Bailey, C. B. Crosstalk between primary and secondary metabolism: Interconnected fatty acid and polyketide biosynthesis in prokaryotes. Bioorg. Med. Chem. Lett. 91, 129377 (2023).
Liu, H. & Naismith, J. H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol 8, 91 (2008).
Marty, M. T. et al. Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal. Chem. 87, 4370–4376 (2015).
Acknowledgements
Funding for this work was provided by the Boehringer Ingelheim Foundation (Exploration Grant to F.H.) and the VolkswagenStiftung (Momentum Grant to F.H.). We thank Central Analytics of the Department of Chemistry as well as the North Bavarian NMR Centre (NBNC) at the University of Bayreuth. We thank the chair of Biochemistry I at the University of Bayreuth for protein identity analysis by MS/MS.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
L.N. and F.H. conceptualised and designed the project. L.N. and M.S. carried out gene cloning, gene expression and protein production. L.N. performed bioinformatic analysis, structural modelling, enzymatic assays and their analysis by LC-(HR)MS and NMR spectroscopy. L.S., J.H. and D.B. carried out chemical synthesis and product analysis. L.N. and F.H. collaboratively wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks anonymous reviewers for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nguyen, L.N.K.T., Schlotte, L., Hoffmann, J. et al. Covalent warhead assembly in fostriecin biosynthesis involves malonylation-lactonisation by a bifunctional thioesterase and enzymatic demalonylation. Nat Commun 17, 2365 (2026). https://doi.org/10.1038/s41467-026-70144-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70144-5
