
Abstract
The 3D genome organization plays a key role in regulating interactions among chromosomal loci. While Chromosome Conformation Capture (3C)-based methods have provided static snapshots of chromatin architecture, the kinetics of chromosomal encounters in live cells remain poorly characterized. In this study, we employ Chemically Induced Chromosomal Interaction (CICI) to measure encounter times between multiple loci pairs in G1-arrested budding yeast. Our results show that chromosome motion closely follows the Rouse polymer model, with similar diffusion parameters at all tested loci. Surprisingly, we find that long-range intra-chromosomal encounters occur significantly faster than inter-chromosomal encounters at similar 3D distances. Using targeted depletion experiments, we identify condensin, but not cohesin, as the complex mostly responsible for these rapid intra-chromosomal interactions. This is further supported by Hi-C analysis, which reveals that condensin promotes long-distance intra-chromosomal interactions in G1 yeast. Through polymer simulations, we estimate that condensin extrudes chromatin at ~2 kb/s with a density of one complex per 1-2 Mb and a processivity of 120-220 kb. These findings uncover a novel role for condensin in shaping the interphase genome organization and provide new insights into chromosomal search dynamics in vivo.
Similar content being viewed by others


Introduction
Precise spatial and temporal regulation of nuclear processes, such as DNA replication, transcription, and repair, is essential for the survival of all organisms1. Increasing evidence highlights the critical role of 3D genome organization in orchestrating these processes2. Recent advances in Chromosomal Conformation Captures (3C)-based techniques and DNA Fluorescence In Situ Hybridization (FISH) assays have revealed intricate genome structures with unprecedented scale and resolution3,4,5. While highly insightful, these assays are typically conducted on fixed cells, providing only static snapshots of chromatin contacts rather than capturing the dynamics of chromosomal interactions in live cells.
The “encounter time” or “first passage time” between two loci, which reflects the speed at which different chromosomal regions interact with each other, is a key kinetic parameter for many genomic processes. For instance, the speed at which an enhancer locates its target promoter likely affects the rate of gene activation, while the time required for homologous search dictates the efficiency of double-strand break (DSB) repair. Given its functional significance, cells have evolved mechanisms to regulate chromosomal encounters. Chromosomes exhibit sub-diffusive motion in the nucleus6, and properties such as diffusion coefficient, confinement radius, and average 3D distance between the loci pair can have a major impact on their encounter rate. For example, DSBs increase the genome-wide chromosome mobility, which is correlated with more efficient homologous repair7. Another factor that can influence encounter rate is loop extrusion, a process driven by Structural Maintenance of Chromosomes (SMC) complexes, such as cohesin and condensin8. By extruding DNA until stalling at CTCF binding sites, cohesin is thought to promote chromosomal contacts within the topologically associating domains (TADs) while reducing contacts across TAD boundaries in higher eukaryotes9. Although SMC-mediated loop extrusions have been studied in vitro, mainly on naked DNA10,11,12, the speed, frequency, and processivity of loop extrusion in the chromatin context in live cells remain elusive. It is therefore unclear how loop extrusion quantitatively affects chromosomal interaction dynamics in vivo.
Accurate measurement of encounter times between chromosomal loci pairs is critical for understanding how cells regulate the interaction dynamics. In principle, this information can be obtained by fluorescently tagging two loci, tracking their motion through time-lapse imaging, and evaluating the time needed for the pair to approach within a certain proximity threshold. However, due to limited time and spatial resolution, this method may fail to capture every encounter, and the loci within the threshold may not form genuine interactions. A method we previously developed, Chemically Induced Chromosomal Interaction (CICI)13, allows us to overcome these limitations. By inserting LacO and TetO arrays into target genomic loci and expressing LacI-FKBP12 and TetR-FRB fusion proteins, stable interactions can be induced between the two loci upon rapamycin addition through FKBP12-FRB dimerization. The prolonged co-localization of the loci pair over time facilitates robust detection of encounter events and enables accurate quantification of encounter times.
In addition to experimental approaches, polymer physics provides valuable tools for studying chromosomal motion. In particular, the Rouse model, in which chromatin is approximated as a homogeneous series of beads connected by springs, has been shown to capture the main features of yeast chromosome dynamics14,15,16. Fitting Rouse model predictions to experimental data reveals chromatin properties like stiffness and persistence length. Incorporating loop extrusion into this framework and comparing it with experimentally measured encounter times allows us to infer key parameters of loop extrusion, including extrusion rate, frequency, and processivity.
In this study, we tracked the motion of 13 pairs of chromosomal loci in G1-arrested budding yeast through live-cell imaging and measured their encounter times using CICI, which we refer to as the CICI formation time (CFT). We found that the loci motion can be well-described by the Rouse polymer model, with diffusion parameters being largely homogeneous across all tested loci. Interestingly, CFTs of intra-chromosomal pairs are significantly shorter than those of inter-chromosomal pairs when separated by the same 3D distances, and only the inter-chromosomal encounter rates are consistent with the Rouse model prediction. Through targeted depletion experiments, we identified condensin, but not cohesin, as the key determinant of accelerated intra-chromosomal encounters in G1 yeast. Hi-C analysis further reveals that condensin depletion reduces intra-chromosomal contacts for regions separated by over ~100 kb, supporting its role in promoting long-range intra-chromosomal interactions. Finally, by integrating the imaging and Hi-C data with loop extrusion simulations, we estimated that condensin extrudes chromatin with rates ~2 kb/s, processivity between 120–220 kb, and a very low density of 1 condensin per 1–2 Mb. Overall, our findings identify a novel role for condensin in shaping interphase yeast genome organization and provide quantitative insights into how chromatin encounters are modulated inside cells.
Results
Measuring chromosome dynamics and the encounter time of chromosomal loci pairs using CICI
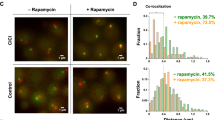
We previously developed CICI to induce interactions between two selected chromosomal loci13. In a rapamycin-resistant yeast strain, we express four fusion proteins, LacI-FKBP12, TetR-FRB, LacI-GFP, and TetR-mCherry, which bind to LacO and TetO arrays inserted into the target loci (Fig. 1A). FKBP12 and FRB dimerize in the presence of rapamycin, leading to strong multivalent interactions between the two arrays. The GFP and mCherry fusion proteins enable visualization of the two arrays as green and red “chromatin dots”. By conducting time-lapse imaging of these dots in live cells, we can track the movement of the two loci and measure the CICI formation time (CFT). As chromatin dynamics and CICI formation are cell-cycle dependent13,17, all measurements below are carried out in G1-arrested cells (“Methods”). This also ensures that both LacO and TetO arrays are present as a single copy in each nucleus, simplifying subsequent data analysis.

A, B CICI scheme. The CICI strain contains LacO and TetO arrays (green and red dots in B) inserted into ChrXV at indicated locations (pair 1). Black dot represents the ChrXV centromere. C Typical time-lapse images taken under three conditions: −rapamycin (−rapa) with 10 s interval (top row), -rapa with 2 min interval (middle), and +rapa with 4 min interval (bottom). Cell contours and detected dots are marked by white circles. D Quantification of the dot distances in (C). CICI formation time (CFT) of the +rapa trace is marked by the gray arrow. After CFT, the two dots show continuous co-localization (distance below 0.38 µm marked by the dotted line). E Population-averaged dot distances ±rapa. Red and purple: −rapa with two acquisition rates. Blue: +rapa. F Relative mean square distance (RMSD) of the two dots ±rapa. Color scheme is the same as (E) except the +rapa data are split at CFTs (before: light blue, and after: dark blue). Power-law fit of the −rapa data is shown in the plot. G Diffusion rate before and after CFT. The bar shows the mean RMSD at Δt = 18 s. Note a 2-fold decrease of the RMSD after CICI formation. H Cumulative probability of CFT over time. The dots are measurements, and the curve is the exponential fit.
We inserted the LacO and TetO into the right arm of ChrXV (separated by 533 kb) (pair 1) and performed time-lapse imaging with and without rapamycin at intervals of 4 and 2 min per frame, respectively (Fig. 1B, C). To capture chromosome dynamics with higher temporal resolution, images were also acquired at 10 s intervals in the −rapamycin condition (Fig. 1C, D). Distances between the LacO and TetO dots were extracted from these images (“Methods”). Without rapamycin, the two dots diffuse in the nucleus with fluctuating distances. Upon rapamycin addition, the dots initially display the same random motion before showing continuous co-localization, indicating CICI formation (Fig. 1D). CICI formation is also supported by the population-averaged dot distance (Fig. 1E). In the absence of rapamycin, the average distance remains ~0.77 μm over time; following rapamycin addition, this distance progressively decreases as the two arrays become co-localized in a larger fraction of cells.
To quantify the diffusion properties of the arrays, we computed the relative mean squared displacement (RMSD) of the two dots as a function of time (“Methods”). The RMSD from the -rapamycin condition shows consistent time-dependence for images acquired at different frame rates (Fig. 1F). These data at short time points are best fitted by a power law function, \({RMSD}(t)=0.036{t}^{0.50}\), with the exponent 0.5 perfectly matching the prediction of the Rouse polymer model15. In the long-time limit, RMSD is slightly lower than the \({t}^{1/2}\) curve (Fig. 1F), which is most likely due to the finite tethering by the DNA between the two loci and the confinement by the nuclear envelope. These results agree with previous MSD measurements of chromosome dynamics in yeast, showing that its chromosomes behave like a Rouse polymer14,15,16,18.
For the +rapamycin traces, we extracted the CFTs based on continuous co-localization and analyzed the traces before and after CFTs separately (“Methods”). The average RMSD before CFT is the same as that in the -rapamycin condition (Fig. 1F), indicating that rapamycin does not affect the diffusion properties before the two arrays encounter. After CFT, RMSD becomes constant over time as the relative distance between the two dots is constrained (Fig. 1F). Taking advantage of the short lag time between the acquisition of red and green images, we can use the RMSD value to estimate the diffusion constant after CICI formation (“Methods”). Remarkably, the diffusion rate of the interacting arrays is ~50% lower than the unlinked ones (Fig. 1G), which is exactly what we expect based on the Rouse model and Langevin equation (“Methods”).
The time-lapse imaging cannot capture CICI formations that occur too fast (<10 min) or too slow (>95 min). For example, out of the 1026 cells that reliably yield CFTs, 257 have already formed CICI in the first frame (7–10 min post rapamycin addition), and 50 show no CICI formation till the end of the movie. To quantify CFT more accurately, we plotted the cumulative CFT probability of the cell population (Fig. 1H). This histogram can be well-fitted by a single exponential function \(y=0.97(1-{e}^{-(t-2.2)/21.6})\), indicating that the average CFT for this loci pair is 21.6 ± 0.4 min. Overall, these data demonstrate that the CICI method can be used to measure chromosome dynamics and encounter times between selected loci.
CFTs between intra- and inter-chromosomal pairs have distinct distance-dependence
To understand how CFTs change with variables such as loci distance and diffusion constant, we extended our analysis in Fig. 1 to 11 other loci pairs (pairs 2–12) on ChrIV or ChrXV (Fig. 2A). These loci pairs were selected to cover different configurations (intra-chromosomal on the same or different arms, or inter-chromosomal), linear distances, and nucleus localizations (based on the Rabl configuration). We imaged these loci in G1 cells ± rapamycin to measure their diffusion and CICI formation kinetics. The RMSDs of all these pairs exhibit a time-dependence similar to the pair measured in Fig. 1, with exponents around 0.5 and scaling factors between 0.039–0.053 μm2/s (Fig. 2B, C and Supplementary Fig. 1A). Some fitted parameters show statistically significant differences, as indicated by non-overlapping error bars. This may reflect each locus’s position relative to structural anchors such as centromeres or telomeres. For inter-chromosomal pairs, RMSD fitting may also be influenced by large-scale relative chromosome motion that is not fully canceled out between the two dots. Nevertheless, these differences are small, and we conclude that diffusion dynamics are relatively homogeneous across all tested loci.

A Configurations of CICI pairs 2-12, which are divided into intra-arm, inter-arm, and inter-chromosomal groups. B RMSDs as a function of time for pair 2, 8, and 9. Other pairs are shown in Fig. S1A. The color scheme is the same as in Fig. 1F. C The best-fit scaling factor and exponent of the RMSDs for all pairs. Error bars represent 95% confidence interval. N = 4628, 1297, 1419, 1907, 1396, 1489, 734, 1118, 2073, 1280, 1421, and 1576 cells are used for the fitting parameters of pairs 1–12. D The ratio of the RMSDs before and after CICI formation (after/before) for all pairs. Orange line marks the 0.5 ratio. Error bars represent 95% confidence interval. E Cumulative CFT probabilities for pair 2, 8, and 9. The single exponential fit is shown in each panel. Other pairs are shown in Supplementary Fig. 1B. F Mean CFTs as a function of mean dot distances. The gray curve is the fit of the inter-chromosomal data with the power law: \({CFT}\propto {\left({dis}-0.1{\mu m}\right)}^{3.1}\). Y error bars represent 95% confidence interval of the CFT fit, and the X error bars are the standard error of the dot distance. N = 4628, 1297, 1419, 1907, 1396, 1489, 734, 1118, 2073, 1280, 1421, and 1576 cells are used for the dot distance measurement of pairs 1–12. N = 1026, 609, 692, 701, 801, 700, 360, 569, 1292, 914, 780, and 699 cells are used for the CFT measurement of pairs 1–12.
After rapamycin addition, CICI is successfully induced in a large fraction of cells within 90 min across all tested loci pairs. The diffusion rates of the co-localized LacO/TetO dots drop to ~50% in comparison to the unpaired ones, further supporting the formation of CICI (Fig. 2D). The cumulative CFT curves for all pairs follow single exponential functions but with distinct time constants (Fig. 2E and Supplementary Fig. 1B). Since diffusion properties of all tested loci are largely uniform, the variable CICI formation times should primarily reflect variations in the 3D distances between loci pairs. To illustrate this, we plotted the CFTs against the average distance between each pair (Fig. 2F). Interestingly, intra-arm and inter-chromosomal CFTs display markedly different distance dependencies: loci within the same chromosome arm encounter each other significantly faster than loci on different chromosomes, even when their average physical separations are comparable. The inter-arm pairs (pairs 7 and 8) exhibit intermediate CFTs between those of intra-arm and inter-chromosomal pairs (Fig. 2F).
For two freely diffusing dots with \({MSD}\propto {t}^{1/2}\), the average encounter time is expected to scale with the fourth power of the mean distance between the two dots, i.e., \({CFT}\propto {{dis}}^{4}\) (“Methods”). Due to confinement, the encounter time should be faster than in a free solution as the region to explore is more restricted. CFTs of the inter-chromosomal pairs can be best fitted by \({CFT}\propto {\left({dis}-0.1{\mu m}\right)}^{3.1}\) (gray curve in Fig. 2F), which agrees well with the theoretical prediction (0.1 μm is likely due to the finite size of the two arrays). CFTs between intra-chromosomal pairs, however, are much less sensitive to distance (Fig. 2F). When separated by the same 3D distance over 0.65 µm, intra-chromosomal encounters occur significantly faster than the inter-chromosomal ones. Note that all the intra-chromosomal pairs tested here are over 150 kb and 0.5 μm apart, well above the previously estimated persistence length of the interphase yeast chromatin (10–20 kb and 0.05–0.2 μm)19. Therefore, despite the physical linkage, the two loci on the same chromosome should move independently. If encounters were purely diffusion-mediated, we would expect intra- and inter-chromosomal pairs to behave similarly at comparable distances. The observed difference suggests that intra-chromosomal encounters are selectively facilitated by an additional mechanism.
Condensin, but not cohesin, is primarily responsible for accelerated intra-chromosomal interactions in G1 cells
Loop extrusion by SMC complexes may account for the observation in Fig. 2F. By translocating along DNA and forming loops within individual chromosomes, this activity could selectively promote the encounter of intra-chromosomal loci. It may also explain the reduced effect on inter-arm pairs, as centromeres are known to act as barriers to loop extrusion20. However, two major loop extruding complexes, cohesin and condensin, are both thought to have low activities in G1 yeast. Cohesin subunits are transcriptionally repressed in G1, and they fail to load efficiently onto chromosomes due to continuous cleavage by the Esp1 separin21. Major condensin subunits are transcriptionally stable and exhibit largely unchanged binding patterns across the cell cycle22. However, the abundance of functional condensin complexes is also reduced in G1 due to the limiting Cap-G subunit, YCG123. Moreover, condensin binding is concentrated at specialized genomic regions, such as rDNA, centromere, and recombination enhancer (RE) locus, and its established loop-extrusion activity is primarily associated with these regions, e.g., in forming centromere-rDNA loops during anaphase20,24,25,26. Therefore, whether either complex contributes to CICI formation kinetics along regular chromosome arms during G1 is not clear.
To test the function of SMC complexes in CICI formation kinetics, we rapidly depleted Smc1 or Smc4, the ATPase subunits of cohesin and condensin, respectively, and measured the resulting CFTs in G1 cells (“Methods”). Consistent with previous reports27,28, Smc1 tagged with auxin-inducible degron (AID) shows partial degradation even in the absence of auxin (IAA), with protein level reduced to ~71% of WT (Fig. 3A). After 1 h of IAA treatment, Smc1 level further drops to ~26% of WT. We compared CFTs across all three conditions: WT, AID − IAA, and AID + IAA. Smc1 depletion does not significantly affect the motion of pairs 1 and 4 in the absence of rapamycin (Supplementary Fig. 2); the CFTs and average distances of these pairs also remain similar across all conditions (Fig. 3B). Notably, pair 1, which forms CICI much faster than inter-chromosomal pairs with similar 3D distances (Fig. 2F), retains the same rate in the absence of cohesin. These data argue that cohesin does not play a major role in the rapid CICI formation observed between intra-chromosomal loci.

A Western blot of V5-tagged Smc1 and Smc4 in a -TIR1 (E3 ligase) strain, or a +TIR1 strain with or without 1 h IAA treatment. The undigested percentage relative to the –TIR1 control is shown (average of two biological replicates). B CFTs (left) and mean dot distances (right) for pairs 1 and 4 in WT and Smc1-AID strain ±IAA. Error bars represent the standard error of the dot distance and 95% confidence interval of the CFT fit. N = 4628 and 1907 cells are used for the dot distance measurement of WT pairs 1 and 4. N = 851 and 1262 cells are used for the dot distance measurement of Smc1-AID strain −IAA pairs 1 and 4. N = 1136 and 1349 cells are used for the dot distance measurement of Smc1-AID strain +IAA pairs 1 and 4. N = 1026 and 701 cells are used for the CFT measurement of WT pairs 1 and 4. N = 439 and 347 cells are used for the CFT measurement of Smc1-AID strain −IAA pairs 1 and 4. N = 457 and 533 cells are used for CFT measurement of Smc1-AID strain +IAA pairs 1 and 4. C, D Mean dot distances (top) and CFTs (bottom) for the labeled pairs in WT and Smc4-AID strain ±IAA. Error bars represent the standard error of the dot distance and 95% confidence interval of the CFT fit. N = 4628, 1907, 1396, 734, 1118, 2073, 1280, and 1576 are used for the dot distance measurement of WT pairs 1, 4, 5, 7, 8, 9, 10, and 12. N = 1450, 847, 1146, 1293, 1111, 1252, 1429, and 1043 are used for the dot distance measurement of Smc4-AID strain −IAA pair 1, 4, 5, 7, 8, 9, 10, and 12. N = 1901, 832, 1204, 1481, 972, 1453, 1640, and 1033 are used for the dot distance measurement of Smc4-AID strain +IAA pairs 1, 4, 5, 7, 8, 9, 10, and 12. N = 1026, 701, 801, 360, 569, 1292, 914, and 699 are used for the CFT measurement of WT pairs 1, 4, 5, 7, 8, 9, 10, and 12. N = 1527, 423, 609, 623, 469, 714, 615, and 551 are used for the CFT measurement of Smc4-AID strain −IAA pairs 1, 4, 5, 7, 8, 9, 10, and 12. N = 2469, 426, 627, 816, 423, 628, 549, and 588 are used for the CFT measurement of Smc4-AID strain +IAA pairs 1, 4, 5, 7, 8, 9, 10, and 12. E Mean CFTs as a function of mean dot distance for labeled pairs in WT (left), Smc4-AID strain −IAA (mid), and Smc4-AID strain +IAA (right). Y error bars represent 95% confidence interval of the CFT fit, and the X error bars are the standard error of the dot distance. The gray curve is the fit of all data shown here with the power law: \({CFT}\propto {\left({dis}-0.1{\mu m}\right)}^{3.3}\). For CFT in (B, D), statistical significance was determined using bootstrap resampling with replacement for 100,000 iterations. For dot distance in (B, C), statistical analysis is based on two-tailed students’ t test.
We next carried out similar measurements in the absence of condensin subunit Smc4. Smc4-AID is depleted to ~93% of the WT level in −IAA and reaches ~18% in +IAA (Fig. 3A). We first evaluated the effect on pair 1, 4, and 5 with the two loci located on the same chromosome arm (Fig. 2A). Depletion of condensin does not significantly affect the dynamics of these loci in the absence of rapamycin (Supplementary Fig. 3). The effect on the mean distances between these pairs is also small (Fig. 3C). Strikingly, we observed large increases in CFTs after IAA treatment in comparison to the WT and −IAA conditions for pair 1 (from ~22 to ~35 min, P-value = 8.0e-5) and pair 4 (from ~18 to ~26 min, P-value = 2.7e-3) (Fig. 3D and Supplementary Fig. 4). For an unknown reason, CFT of the pair 5 is shorter in the Smc4-AID strain in the absence of IAA than in WT cells. Nevertheless, treatment with IAA in the AID strain still significantly increased CFT from ~16 to ~21 min (P-value = 9.8e-3). We also performed the same measurements on the inter-arm pairs 7 and 8. Pair 8, which has a longer 3D distance, shows a moderate and statistically insignificant increase upon condensin depletion (Fig. 3D and Supplementary Fig. 4). These data indicate that condensin accelerates intra-chromosomal encounters, especially for loci on the same chromosome arm.
To determine whether the condensin-dependent acceleration of contact formation is specific to intra-chromosomal interactions, we extended our analysis to inter-chromosomal pairs 9, 10, and 12. For all three pairs, we observed a mild, statistically insignificant decrease in CFTs in the +IAA condition compared to −IAA (Fig. 3D and Supplementary Fig. 4). In other words, the effect of condensin on inter-chromosomal encounters is small, and if anything, the trend is in the opposite direction of what we observed for intra-chromosomal pairs.
As a summary, we plotted CFT as a function of mean dot distance for WT and Smc4-AID strains with or without IAA for all the pairs tested (Fig. 3E). The difference between intra- and inter-pairs becomes smaller in the Smc4-AID strains, even in the absence of IAA, likely due to the background degradation of Smc4. Strikingly, this difference is largely abolished in the +IAA condition with an overall fit of \({CFT}\propto {\left({dis}-0.1 \, {\mu m}\right)}^{3.3}\) (Fig. 3E). Together, these results support a model where all loci can rely on diffusion to establish interactions, but intra-chromosomal, especially intra-arm, communications can be further accelerated by condensin-mediated loop extrusion.
Condensin depletion reduces genome-wide long-distance intra-chromosomal contacts in G1 yeast
Most studies of condensin focus on M phase and/or at specific genomic loci. Even in mitotic yeast cells, depletion of condensin only has a mild effect on genome-wide contact frequencies29. Since our data support condensin-mediated loop extrusion during G1, condensin may also affect yeast interphase genome organization. To test this idea, we performed Hi-C experiments on WT and Smc4-AID strains in G1-arrested cells (Supplementary Fig. 5A–D). Hi-C signals show minor changes in the Smc4-AID strain compared to WT in the absence of IAA, likely reflecting the basal degradation of Smc4. More pronounced changes in Hi-C profiles are observed after IAA treatment (Supplementary Fig. 5C–E). We therefore focus on the comparison between the high degradation condition (Smc4-AID, +IAA) and WT in the following analyses, and the comparisons between Smc4-AID + IAA and −IAA are also shown.
When averaged across all chromosomes, condensin depletion results in decreased intra-chromosomal interactions (Supplementary Fig. 5E). For example, the average Hi-C signals for loci pairs separated by 200 kb (both intra-arm and inter-arm) are reduced by ~15% in the absence of condensin (Supplementary Fig. 5E). This is consistent with our observation that condensin accelerates interactions between intra-chromosomal loci pairs. Notably, the impact of condensin depletion varies among individual chromosome arms. For the right arms of ChrIV and XV, where most CFT measurements were performed, condensin depletion leads to a greater reduction in Hi-C signals on ChrXV (16% over 200 kb) than ChrIV (12% over 200 kb) (Fig. 4A, B). This difference may be due to variations in condensin density, as Smc4 and Brn1 (another condensin subunit) ChIP-exo data show higher enrichment of condensin binding over ChrXV than ChrIV (Fig. 4C)30. This may also explain the smaller change of CFT for pair 5 after condensin depletion (Fig. 3E).

A Hi-C contact matrices over chrIV and chrXV in G1-arrested WT and Smc4-AID strain ±IAA. The log2 fold change between Smc4-AID +IAA and WT is shown on the right. B Pair-wise contact frequencies as a function of genomic distances in WT or Smc4-depleted conditions. Region pairs within the right arms of chrIV and ChrXV, or across between the left and right arms, are used for this calculation. The lower panels show the Smc4 ±IAA / WT and Smc4+IAA/−IAA ratio. C ChIP-exo data of Smc4 and Brn1 (another condensin subunit) on ChrIV and ChrXV. D Inter-chromosomal contact frequencies between ChrIV and ChrXV in G1-arrested WT strain and Smc4-AID strain ±IAA. N = 10051, 9523, and 9732 contacts for WT, Smc4 −IAA, and Smc4 +IAA. Box plot notation: center line, median; box limits, 25th and 75th percentiles; boundaries of the whiskers, minimum and maximum values; points, outliers. Statistical analysis is based on two-tailed students’ t test.
In contrast to intra-chromosomal interactions, inter-chromosomal interactions remain largely unaffected by condensin depletion except for a few specialized regions (see below). There is no significant difference in inter-chromosomal contact frequencies when pooled over all chromosomes or between ChrXV and ChrIV (Fig. 4D and Supplementary Fig. 5F). This is again consistent with the small and non-significant effect of condensin depletion on inter-chromosomal CFTs observed above (Fig. 3D).
Consistent with previous findings that condensin is enriched over centromeres, RE, and rDNA, we found that condensin depletion has particularly pronounced effects on the conformation of pericentric regions, ChrIII, and ChrXII, respectively (Supplementary Fig. 5C, D). Within ~200 kb of centromeres, condensin depletion reduces the interactions between centromeres and the flanking regions in cis but enhances the interactions between different centromeres in trans (Supplementary Fig. 6A). This pattern supports a model where condensin compacts and individualizes pericentromeric regions. Similar effects have been reported in mitotically arrested cells29, and our results suggest this centromeric organization is maintained during G1. On ChrIII, which contains all loci involved in the mating-type switch, condensin depletion led to a broad reduction in interactions between the left and right arms, including the key contacts between RE, HML, and the MAT locus (Fig. 5A). This observation aligns with condensin’s function in promoting loop extrusion and chromosomal interactions near these loci20, as well as mating-type switch between MAT and HML25,31,32.

Hi-C contact matrices for ChrIII (A) and ChrXII (B) in G1-arrested WT strain and Smc4-AID strain ±IAA. Key loci on these chromosomes, including centromeres (block dot), HML, RE, MAT, HMR, and rDNA, are labeled. Log2 fold change between Smc4-AID +IAA and WT is shown on the right. The intersection of the two yellow dashed lines in A marks the interaction between HML/RE and MAT, which is decreased in Smc4 +IAA. The one in (B) corresponds to the interaction between CICI pair 13 (C), which is increased in Smc4 +IAA. C Schematic configuration of intra-chromosomal CICI pair 13, with one array on each side of rDNA. D Mean dot distance and CFT for pair 13 in WT and Brn1-AID strain ±IAA. Brn1 (another condensin subunit) is depleted instead of Smc4 because the SMC4 gene is located very close to the LacO array of pair 13, making it difficult to genetically manipulate without interfering with the locus. Same statistical test is used as in Fig. 3. Error bars represent the standard error of the dot distance and 95% confidence interval of the CFT fit. N = 955, 997, 1131 cells for the dot distance measurement of WT, Brn1-AID strain -IAA, and Brnd1-AID strain +IAA. N = 848, 488, 491 cells for the CFT measurement of WT, Brn1-AID strain -IAA, and Brnd1-AID strain +IAA. Error bars represent the standard error of the dot distance and 95% confidence interval of the CFT fit. For CFT, statistical significance was determined using bootstrap resampling with replacement for 100,000 iterations. For dot distance, statistical analysis is based on two-tailed students’ t test. E Mean dot distance and CFT vs Hi-C contact frequency for all CICI pairs tested. The data taken in WT and Smc4+IAA condition are shown in black and red. The power-law fits of these data are shown in gray curves: \({dis}=0.068{({cf}+0.0008)}^{-0.33}+0.1\), where 0.1 μm represents the finite size of the arrays (same as in Fig. 2F), and \({{{\rm{CFT}}}}=\,0.075{({cf}+0.0008)}^{-0.9}\). Error bars represent the standard error of the dot distance and 95% confidence interval of the CFT fit.
On ChrXII, which harbors the rDNA repeats, condensin depletion significantly reduces the self-association of a region between the centromere and rDNA repeats, allowing it to interact more with the rest of ChrXII in cis and other chromosomes in trans (Fig. 5B and Supplementary Fig. 5D). This effect is already evident in the −IAA condition and is only moderately enhanced with IAA treatment (Fig. 5B and Supplementary Fig. 5D). These results suggest that the region between the centromere and rDNA is normally condensed and insulated from the rest of the genome. This insulated state seems to be very sensitive to condensin concentration and is disrupted even with mild condensin depletion in −IAA. Supporting this model, an intra-chromosomal CICI pair spanning the rDNA locus (pair 13) exhibits an accelerated encounter rate in both −IAA and +IAA conditions (Fig. 5C, D), and the majority of this acceleration already occurs in −IAA. Notably, this effect is in stark contrast to the trend observed for other intra-chromosomal pairs, where condensin depletion typically slows encounter kinetics (Fig. 3E).
To further connect our imaging and Hi-C measurements, we plotted the CFTs and mean dot distances (\({dis}\)) against the corresponding Hi-C contact frequencies (\({cf}\)) for all CICI pairs tested. As expected, higher contact frequencies are associated with shorter dot distances and faster encounters (Fig. 5E). These trends hold true in both WT and condensin-depleted cells. Quantitatively, CFT and 3D distance should follow power-law relationships with contact frequencies: \({CFT}\propto {{cf}}^{\alpha }\) and \({dis}\propto {{cf}}^{\beta }\), where the \({CFT}\) exponent \(\alpha\) should be around −1, and the \({dis}\) exponent \(\beta\) should be between −0.25 and −0.333. After adjusting \({cf}\) by adding a constant to correct for the baseline level, we found that our data yield best-fit parameters of \(\alpha \approx -0.9\) and \(\beta \approx -0.33\) (Fig. 5E), in excellent agreement with theoretical predictions.
Polymer simulations with sparse extruders recapitulate enhanced search time and moderate compaction
Our experiments show that condensin depletion significantly prolongs search times between distal intra-chromosomal loci, while having only a modest effect on Hi-C contact frequencies and 3D distances. To understand how condensin can accelerate search, we developed a polymer model of yeast chromosomes with active loop extrusion with the following goals: (1) to determine whether loop extrusion by condensin can accelerate search, while having little effect on average separations and Hi-C, (2) to estimate the parameters of condensin-mediated loop extrusion in yeast, and (3) to gain insights into the role of loop extrusion in facilitating long-range genomic interactions with implications for other organisms, e.g., enhancer-promoter communication in vertebrates.
Largely unconstrained polymers were shown to be adequate models for yeast chromosome organization15,34 and dynamics14,16. We confirmed this in our system by showing that experimentally measured MSD values for pairs of loci separated by different genomic distances are perfectly fit by the theoretical two-point MSD for the Rouse chain (Supplementary Fig. 7A). Thus, we modeled chromosomes as identical, flexible bead-and-spring polymers diffusing within a spherical volume with 1.5 µm radius at a physiological volume density of 15%. For WT simulation, we also incorporated condensin-mediated loop extrusion (see below).
We first calibrated our model without loop extrusion to reproduce experimentally measured quantities: diffusion dynamics, average distances, and the search times in condensin-depleted cells (+IAA experiments) (see “Methods” and Supplementary Fig. 7B, C). Once calibrated, we introduced loop extrusion characterized by the velocity of extrusion (\(v\)), the average loop size (\(\lambda\)), and the average separation of extruders (\(d\)). We then assessed how extrusion affected search times, mean distances, and contact probabilities between loci pairs, comparing the results to experiments with condensin (−IAA).
The modest effect that we observe of condensin depletion on Hi-C contact frequencies led us to explore the sparse loop extrusion regime, where the separation between extruders is much larger than the average loop size (\(d\)»\(\lambda\), Fig. 6A, B)35. In this regime, only a small fraction of the chain is extruded into loops, leaving large gaps of free polymer in between. We explored extrusion velocities in the range consistent with in vitro10 and in vivo estimates36, 1–4 kb/s, and performed polymer simulations with variable loop sizes and separation at low loop density (\(\lambda /d\) between 0.05 and 0.2) (Fig. 6C–E and Supplementary Fig. 8). As expected, we found that increasing the coverage by extruded loop (\(\lambda /d\)) leads to smaller distances and faster search (different curves on Fig. 6C, D). Importantly, the effect on the search time is much more pronounced (15-40% reduction) than the effect on pairwise distances (3–10% reduction) (Fig. 6E). Consistent with experimental data, we also found that loop extrusion does not accelerate trans-chromosomal interactions (Fig. 6F, G). We conclude that sparse loop extrusion could indeed underlie the observed effects.

A Procedure of simulation. B Schematic representation of chromatin chain looped by extruders, and the corresponding 1D arch diagram for a dense loop conformation (top) and sparse conformation (bottom). The average separation between loops d and loop size λ are depicted. Average search time (C) and distance (D) between two monomers separated by 360 kb (corresponding to the genomic separation of pair 4) with loop extrusion at v = 2 kb s−1, shown for different values of loop size λ and loop separation d. Dashed lines indicate the experimentally measured search times in cells with condensin (−IAA, green) and without condensin (+IAA, gray), with shaded regions representing the corresponding 1.5 times the experimental error bars reported in Fig. 3. E The same simulation results as in (C, D), but with dot distance plotted against search time. Simulations where both the dot distance and search time fall within 1.5 times the experimental error are annotated. CFT (F) and average distance (G) for simulated trans pairs with best matching extrusion parameters. H Simulated contact frequency as a function of genomic distance with extrusion that matched experimental data (orange, green, purple) and without extrusion (red). I Ratio of simulated contact frequency (−extrusion divided by +extrusion) using the same parameters as in (H).
Next, we sought to achieve quantitative agreement between the model and experimental data for pair 4, where condensin-mediated acceleration is most pronounced, and the arrays are positioned far from the centromere. We found that reproducing experimental CFT and distance data together put considerable constraints on the loop extrusion parameters. The best agreement with experimental data was obtained with an average condensin loop size of \(\lambda \approx\) 120–220 kb, and the average density of extruders of 1 per \(d\approx \,\)1-2 Mb, yet always with the loop coverage of \(\lambda /d\approx\) 1/10 (Fig. 6E, green shading). This loop size is highly consistent with the one estimated based on Hi-C contact frequency, ∼170 kb20. Interestingly, we also found that the velocity of extrusion \(v\) should be relatively high, \(\ge \,\,\)2 kb/s, as simulations with slower velocities consistently failed to reproduce the experimental data (Supplementary Fig. 8A–C).
The simulations described above are modeled based on symmetric, two-sided loop extrusion. To account for the possibility that yeast condensin may extrude unidirectionally10,37, we also implemented a one-sided extrusion model (Supplementary Fig. 9A). Across six representative parameter sets, search statistics and average distances between arrays remained unchanged compared to the two-sided case (Supplementary Fig. 9B), indicating that our parameter estimates are not sensitive to the extrusion mode. Importantly, this equivalence arises because first-passage times are primarily governed by the distribution of loop lengths, which is independent of whether DNA is reeled in from one side or two sides. However, this equivalence holds only in the absence of strong extrusion barriers. In their presence, extrusion dynamics become critical: a two-sided extruder stalls on one side but continues extrusion from the other, while a one-sided extruder stalls completely, which may perturb the loop-length distribution. This distinction is not a concern in our genomic context, where barriers are weak (Fig. S10). In contrast, in mammalian genomes where strong barriers such as CTCF play crucial functional roles, the specific extrusion dynamics could influence encounter times between regulatory elements whose interactions are constrained by these barriers.
We also found that the best-fit parameters (\(v=\,\,\) 2 kb/s and \(\lambda /d\) = 0.1) can reproduce changes in Hi-C contact probability upon condensin loss (for the arm containing pair 4), with a somewhat larger effect in simulations than in Hi-C (the fold change peaking at ~35% vs. ~20% in Hi-C) (Fig. 6H, I). Thus, we found that loops of yeast condensin are comparable to those of vertebrate cohesin, yet they are much sparse in yeast G1, as was suggested earlier by models that fit Hi-C data29. The processivity (\(\lambda\)) is also comparable to that of vertebrate condensin I, and speeds are similar to vertebrate condensins36,38.
Finally, our simulations provide mechanistic insight into how condensin-mediated loop extrusion accelerates long-distance contacts between chromosomal elements. We identify two classes of processes that enhance search efficiency (Fig. 7A). The first involves a single extruder landing between the two elements and extruding a substantial portion of the intervening genome, thereby bringing the elements into close proximity. Such a large extrusion event allows the two elements to find each other rapidly (~1–3 min) (Fig. 7B and Supplementary Fig. 11). The second mechanism is due to multiple extrusion events that all moderately shorten the linker, collectively compacting the fiber everywhere and thus accelerating the search (Fig. 7C and Supplementary Fig. 11). In that case, extrusion events are not immediately followed by contact formation (Fig. 7C). While the first mechanism leads to rapid search, such large extrusion events are rare; small extrusion events are more frequent, but their individual contributions are small (Fig. 7D). To dissect the effect of different extrusion size, we performed simulations where we selectively removed extrusion events of specific sizes (“Methods” and Supplementary Fig. 11B). These analyses reveal that both small and large events contribute to faster search, with large events playing a disproportionately important role (Fig. 7D). Ultimately, it is the combined effect of both mechanisms that drives the observed acceleration in chromosomal encounter.

A Schematic representation of both extrusion-mediated acceleration processes (green: large events, blue: small events). Top: Time series of the genomic region around the arrays. Each pair of lines represents the positions of the anchors of a single extruding condensin. Middle: Corresponding effective 1D distance between the arrays (defined schematically), showing shortening due to condensins landing in between. Bottom: Corresponding 3D distance between arrays with 3D contact highlighted. B Top: Average effective 1D distance between the arrays aligned to the onset of large extrusion events (defined as remaining tether <180 kb). Faded lines show individual traces. Bottom: Average contact probability per track aligned to large extrusion event onset (green) compared to random times in simulations without extrusion (gray). High contact probability is present at 3 min (time to extrude to the arrays) following condensin binding. C Same as (B), but for small extrusion events (remaining tether > 180 kb). D CFT simulation with specific extrusion events removed to quantify the contributions of large and small loops. For example, “0–90 kb remaining tether” represents removing all extrusion events with remaining tether > 90 kb.
Discussion
Functional long-distance chromosomal interactions in the native genome, such as looping between distal enhancers and their target promoters, tend to be rare and dynamic39,40,41. This dynamic nature, coupled with the limited time and spatial resolution of imaging technologies, makes it very challenging to accurately detect interaction events. Consistent with this, polymer simulations have demonstrated that simple thresholding of the enhancer-promoter distance or Gaussian mixture modeling can lead to a substantial overestimation of the looped fraction18. In contrast, the interactions generated by CICI are mediated by dimerization between long arrays associated with hundreds of molecules. We expect such multivalent interactions to be stably maintained as long as the arrays are partially occupied. Indeed, while CICI junctions can be disrupted during anaphase due to chromosome segregation13, they remain remarkably stable in G1 cells, with virtually no disruption during our imaging period. This stability enables us to score interaction events and quantify first passage times with high confidence.
Our CFT measurements reveal that intra-chromosomal encounters in G1 yeast occur more rapidly than inter-chromosomal ones, and this difference relies on the presence of condensin, but not cohesin, consistent with their roles through the cell cycle of in S.cerevisiae. Depletion of condensin also selectively reduces long-distance contact frequencies within individual chromosomes. While we cannot directly observe condensin activity inside cells, we propose that loop extrusion is a highly plausible mechanism underlying the accelerated intra-chromosomal encounters due to the following reasons. First, condensin-mediated loop extrusion has been directly visualized in vitro using single-molecule assays10, indicating that condensin has loop-extrusion activities. Second, loop extrusion has the unique capability to differentially impact intra- vs. inter-chromosomal interactions. Third, changes in the contact probability curves upon condensin depletion were consistent with the loss of extruded loops as seen in simulations. The selective effect on intra-chromosomal encounters would be difficult to explain through alternative mechanisms such as chromosome tethering through protein-protein interactions. Finally, incorporating the measured extrusion rates into our polymer model yields simulations that closely match both our CFT and Hi-C data, and provide estimates of condensin extrusion characteristics (e.g., velocity ~2 kb/s), consistent with independent measurements of 1–3 kb/s10,38.
While previous studies have shown that condensin in budding yeast plays specialized roles at particular genomic regions, such as rDNA, the mating-type locus, and centromeres20,42, our analyses extend these observations by revealing condensin’s broader influence on 3D genome organization along regular chromosome arms. Our best-fit parameters of \(d\) and \(\lambda\) indicate that loops are relatively sparse in individual cells, with only ~10 actively extruding loops covering ~10% of the genome. The scarcity of chromosome loops is consistent with small changes in Hi-C signals. The condensin binding rate per unit genomic length can be estimated as \(v/\lambda d\), corresponding to one condensin loading event every 1 to 10 min per Mb. These parameters further imply that only a small fraction of condensin molecules (estimated at 1000–2000 per cell) are chromatin-bound and/or engaged in loop extrusion on chromosome arms at any given moment. This aligns with previous measurements showing rapid condensin turnover in G1 (~2 s residence time)43, indicating that most condensin molecules do not remain bound long enough to extrude large loops. Interestingly, even such a low density of loop extrusion events, when combined with long processivity and high extrusion speed, is sufficient to promote long-range genomic communications. We find that this acceleration is primarily driven by rare, large extrusion events that leave the remaining separation between two loci below ~100 kb. This mechanism likely applies to how cohesin-mediated loop extrusion facilitates enhancer-promoter communication in metazoans.
We found that condensin, rather than cohesin, mediates loop extrusion in G1-phase yeast. Conversely, previous studies have reported that cohesin, not condensin, drives the genome-wide compaction of mitotic chromosomes in yeast, although condensin still contributes to the compaction44,45 and plays critical roles in regions like the nucleolus26,29. These findings are in striking contrast to the well-established roles of these complexes in metazoans and fission yeast, where cohesin is the primary loop extruder during interphase, and condensin is mainly responsible for mitotic chromosome compaction46,47. The reason behind this functional divergence is unknown.
Nevertheless, cohesin loop extrusion in higher eukaryotes may also lead to selective acceleration of intra-chromosomal encounters. Many well-characterized distal enhancers and their target genes—such as the locus control region and the β-globin genes—reside on the same chromosome48. Similarly, long-range promoter-promoter interactions in Drosophila tend to occur within individual chromosomes49. Our findings suggest that loop extrusion may regulate gene expression by modulating the frequency of encounters between genes and their distal regulatory elements. Supporting this idea, cohesin activity has been shown to vary in neurons to control the expression of cell surface proteins by altering their enhancer-promoter interactions50. Moreover, our results in Fig. 2F indicate that the enhancement of encounter rates by loop extrusion is distance-dependent. Intra- and inter-chromosomal encounter rates converge for pairs separated by small 3D distances, suggesting that interactions between proximal loci occur predominantly through diffusion. An active, ATP-driven process like loop extrusion is only required to promote encounter between distal loci. Consistently, cohesin was found to be dispensable for genes activated by nearby enhancers, but is essential for the full expression of genes regulated by distal enhancers51,52.
Unlike higher eukaryotes, budding yeast genes generally lack distal enhancers, yet a few hundred genes exhibit mild expression changes upon condensin depletion in G153. These genes tend to be located near condensin-binding sites53, suggesting that condensin depletion may disrupt the 3D organization of these genes, e.g., by altering their contacts with other loci. Given that gene clustering in yeast has a modest effect on gene expression54,55, it is plausible that some of the gene expression changes result from altered 3D genome conformation. The detailed mechanism underlying these effects requires further investigation.
Loop extrusion by condensin may have functions beyond gene regulation, and the best example is its well-documented function in regulating the mating type switch in G1 yeast. This is further supported by our Hi-C data showing significantly reduced contact frequencies along ChrIII in the absence of condensin. Together with impaired mating type switch efficiency in condensin-depleted strains25,31,32, this supports a model where condensin accelerates and stabilizes the interactions between the HML and the MAT locus and therefore promotes their recombination. A similar principle applies to V(D)J recombination during mouse B cell development, where cohesin-based loop extrusion is modulated to generate different contacts between the VH genes and the recombined DJH segments to diversify the antibody repertoire56.
Method
Plasmid and strain construction
We used the same background strain and the plasmids as described in our previous works13. Specifically, in this study, we started with the background strain containing tor1 and fpr mutations, REV1pr-LacI-GFP and REV1pr-TetR-mCherry were inserted into the HIS3 locus, and REV1pr-LacI-FKBP12 and REV1pr-TetR-FRB were inserted into the ADE2 locus. For the two arrays, we started with the plasmids containing 192X TetO repeats (pSR11) and 256X LacO repeats (pSR13) as the template. We assembled homologous sequences from various loci on ChrIV and ChrXV with the two template plasmids. The reconstructed plasmids were cloned into stable E. coli (Invitrogen MAX Efficiency Stbl2 Competent Cells, 10268019) and were checked for length to be the same as original template. The plasmids were then transformed into a yeast background strain. After every step of transformation, the insertion loci were confirmed by PCR. Each strain was checked under fluorescence microscope to confirm that it contains only one pair of red and green dot in G1. For strains with auxin-induced degradation, we tagged the C-terminus of endogenous Smc1 or Smc4 with AID and a V5 tag, and introduced OsTIR1 to the cells57. Depletion of the tagged protein was achieved by adding IAA (Sigma, I2886) to a final concentration of 500 μM. Strains were incubated with IAA for 1 h prior to microscopy measurement. Depletion was confirmed by Western blot. See Supplementary Table 1 for detailed plasmid and strain information.
Fluorescence microscopy
CICI strains were cultured in synthetic media SCD to log phase, lightly sonicated to disperse cell clusters, and then incubated in SCD + 5 μM alpha factor (Zymo, Y1001) for ~2.5 h to arrest the cells in G1 phase. For experiments involving auxin-induced degradation, cells were first treated with α-factor for 1.5 h, followed by the addition of IAA for another 1 h. G1 arrest was confirmed microscopically by the presence of shmoo formation in a large fraction of cells. G1-arrested cells were then placed onto 1.2% agarose pads prepared with or without 1 ng/μl rapamycin (Sigma, 53123-88-9). We imaged these samples using our published time-lapse microscopy protocol58. For the +rapamycin samples, we typically started imaging within 5–10 min of rapamycin application. For each frame, three images were acquired sequentially in the order of phase, mCherry, and GFP fluorescence.
For the –rapamycin condition, two imaging acquisition settings were used. In the faster acquisition mode, two z-stacks were captured with 0.7 μm spacing and 0.1 s fluorescence exposure. Each fluorescence channel required ~5 s to finish, and the time interval between frames was 10 s. In the slower acquisition mode, the same spacing and exposure were used, but with three z-stacks, and the interval between frames was extended to 120 s. For the +rapamycin condition, the time of initial rapamycin exposure was defined as t = 0 min. Imaging was performed using seven z-stacks with 0.6 μm spacing and an exposure time of 0.12–0.15 s. Each fluorescence channel took approximately 18 s, and the interval between frames was set to 240 s.
Image analysis
We used the same MATLAB software developed in our previous works to annotate cells, detect dot positions and dot distances54,58. Briefly, cell contours were identified based on phase contrast images. For each pixel within a cell boundary, the program scanned across the z-stacks and recorded the maximum fluorescence intensity for both mCherry and GFP channels. These maximum intensity values were compiled into a 2D projection image for each channel. Fluorescent dots were then identified as pixel clusters with intensities significantly above background, and their coordinates were recorded. All detected dots were manually reviewed, and visually incorrect assignments were removed, especially when more than one red or green dot per cell was detected. As expected, the 2D projection yields smaller RMSD values compared to measurements in 3D, a trend that is confirmed by our polymer simulations. Although this approach may introduce some false-positive co-localization events, where foci are aligned in x–y but spatially separated along the z-axis, this limitation is mitigated by defining CICI formation based on continuous co-localization over consecutive time points (see below).
To identify CFT, we applied the following procedure. First, we excluded traces that met any of these criteria: interrupted in more than 60% of frames, lacked a detectable signal for over 1 h, or contained fewer than six valid data points. Second, we scanned the remaining traces for continuous colocalization events, defined as dot distances below 0.38 μm for at least four consecutive frames. If such a pattern was detected, the first frame in which it occurred was designated as the CFT. We applied this method to −rapamycin condition and detected CFT in 0.9% of traces, which was the false discovery rate. Third, we assessed whether disassociation occurred after CFT by identifying the first frame in which the dot distance exceeded 0.5 μm following initial colocalization. In the majority of cases, colocalization persisted after CFT. In traces showing multiple rounds of association and disassociation, only the initial association event was counted to avoid double-counting.
RMSD before and after CICI formation
To quantify the diffusion properties of the chromosome arrays, we used relative mean squared displacement (RMSD), which is defined as
\(\vec{r}_{r}(t)\) and \(\vec{r}_{g}(t)\) are the position of red and green dot respectively. \(\vec{R}(t)=\vec{r}_{g}(t)-\vec{r}_{r}(t)\) is the relative displacement between the two dots. If we ignore the correlations between \(\vec{r}_{r}\) and \(\vec{r}_{g}\),
The RMSDs were derived from time-lapse imaging with three different frame rates. We averaged over all location datapoints separated by the same \(t\). The fitting of RMSD before CICI formation agrees well with the Rouse model, where \({MSD}\left(t\right)={6k}_{B}T{\left(\pi \kappa \gamma \right)}^{-1/2}{t}^{1/2}\).
After CICI formation, the two arrays ideally should be at the same position: \(\vec{r}_{r}(t)=\vec{r}_{g}(t)\) and thus \({\vec{R}} \left(t\right)=0\). However, in our time-lapse imaging, the mCherry and GFP fluorescence channels were not imaged simultaneously, and therefore \(\vec{R}(t)=\vec{r}_{g}(t+\Delta t)-\vec{r}_{r}(t)\). This time lag \(\Delta t\) depends on the number of z stack in our time-lapse imaging, and for +rapamycin condition, it is about 18 s. Substituting \(\vec{R}\left(t\right)\) into the RMSD equation, we get:
In other words, RMSD after CICI formation is a constant determined by the total diffusions of two arrays at time \(\Delta t\), which can be compared with RMSD before CICI formation at \(t=\Delta t\) (Fig. 1G). In Rouse model, when two beads come together, the friction coefficient of the dimer \(\gamma\) is twice the monomer. The spring constant \(\kappa\) is also doubled. Therefore, we expect the coefficient of MSD in Rouse model, \({6k}_{B}T{\left(\pi \kappa \gamma \right)}^{-1/2}\), to decrease by two folds. That is indeed what we observed experimentally.
Prediction of the first passage time
MSD of Rouse motion is \(D{t}^{1/2}\), where \(D\) is the diffusion constant. This means that the array explores roughly a spherical region with radius of \({D}^{1/2}{t}^{1/4}\) within time \(t\), which consists of lattice sites \(\propto {D}^{3/2}{t}^{3/4}\). Meanwhile, the lattice sites visited by the dot should be \(\propto t\). In a long time limit, the array visits each site infinitely often, which is known as “compact exploration”59. Therefore, the array should interact with its target once its explored radius is comparable to the distance between them: \({d=D}^{1/2}{t}^{1/4}\). In other words, CFT should have roughly a quartic dependence on the distance.
Hi-C experiments
Hi-C was adapted from a previously published protocol32. Yeast were incubated in 200 mL SCD medium until OD660 reached 0.3 and arrested in G1 with 5 μM alpha factor for 2.5 h, with a final OD of ~0.4. Cells were fixed with 3% formaldehyde (Fisher, RSOF0010-250A) for 20 min at 25 °C and then quenched with 0.2 M glycine (Fisher, 194825) for 20 min at room temperature. Cells were collected by centrifugation and washed with SCD. Cell pellets were resuspended in 1 mL TBS (Tris-buffered saline), 1% Triton X-100 (Sigma, 9036-19-5) and 1X protease inhibitor cocktail (Thermo Fisher, 87786). Cell lysate was generated by adding 500 μL acid-washed glass beads (Sigma, G8772) and vortexing for 25 min at 4 °C. Chromatin was recovered through centrifugation, washed with 1 mL TBS, resuspended in 500 μL 10 mM Tris-HCl buffer and digested with DpnII (NEB, R0543L) overnight at 37 °C. Digested DNA fragments were filled in with biotin-labeled dATP by incubating with Klenow enzyme (NEB, M0212), biotin-14-dATP (Thermo Fisher, 19524016), dCTP, dTTP, and dGTP (Thermo Fisher, R0181) for 4 h at room temperature. The biotin-filled DNA fragments were ligated by T4 DNA ligase (NEB, M0202L) for 4 h at 16 °C. Crosslink was reversed by incubation with proteinase K (Thermo Fisher, EO0492) at 65 °C overnight. DNA was purified by phenol-chloroform extraction. Biotin-labeled, un-ligated fragment ends were removed by incubating with T4 DNA Polymerase (NEB, M0203), dATP and dGTP for 4 h at 20 °C. DNA was cleaned by DNA clean and concentrator-5 kit (Zymo, D4014) and sheared by Diagenode Biorupter Pico (EZ mode, 30 s on, 30 s off, 15 cycles). Biotin-labeled DNA was enriched by MyOne™ streptavidin C1 beads (Thermo Fisher, 65001). Hi-C libraries were prepared with NEBNext Ultra II DNA Library Prep Kit. Next generation sequencing was performed on NextSeq 2000, and 100 million of 50 bp paired-end reads were generated for each replicate. Hi-C data analysis was performed with HiC-pro (version 3.1.0)60, cooler (version 0.10.0)61, cooltools (version 0.7.0)62 and HiCExplorer (version 3.7.2)63.
FACS analysis
Cycling or arrested cells were harvested at an OD of 0.4, and 1 ml culture from each condition was used for FACS analysis64. Cells were fixed with ethanol overnight and stained with 2.5 µM Sytox Green in 50 mM sodium citrate buffer containing 20 µg/mL RNase A. After proteinase K treatment and overnight incubation, the stained cells were pulse sonicated to reduce cell aggregates and transferred to 5 ml tubes for FACS analysis using the Cytek Aurora (Cytek Biosciences) analyzer (Huck Flow Cytometry Facility). The instrument was configured using conventional mode, and gain settings were optimized for FSC, SSC and Sytox Green signals. Single cells were gated using FSC-A/H and Sytox Green-A/H bivariant plots. The DNA intensity (Sytox Green) was displayed in a histogram using a linear scale with the G1 peak near the first quartile and the G2 peak near the second quartile of the histogram axis. For each sample, 50,000 cells were recorded, and the data files were analyzed using Kaluza software (Beckman Coulter). A cell cycle analysis tool using the Michael H. Fox algorithm was used for quantitative analysis of G1, S, and G2 populations in single cells.
Loop extrusion simulation
Polymer simulation
Polymer simulations were conducted using OpenMM and the polychrom library (http://github.com/mirnylab/polychrom). Each simulation comprised ten freely-jointed polymers, each consisting of 1800 beads, where each bead represents 1 kb of chromatin. The polymers were confined within a spherical potential with a radius 50 times the bond length. Monomeric subunits were connected into a linear polymer through pairwise harmonic bond interactions. Condensin positions, simulated independently as described below, introduce additional harmonic bonds bridging specific monomers. Monomeric subunits repel each other through weak excluded-volume-like interactions. The viscosity of the implicit solvent was set to ensure Brownian dynamics across time scales longer than the shortest polymer relaxation time.
Condensin dynamics simulation
Simulations of condensin were performed using the lefs-cython library (https://github.com/mirnylab/lefs-cython/). Chromatin is modeled as a 1D lattice, with each site representing a monomer from the 3D simulation. Each chromatin-bound condensin occupies two lattice sites, corresponding to its anchor points on the DNA. The total number of condensins, \(N\), present in each simulation is constant and determines the typical distance between two condensins (i.e., centers of the loops) \(d=\frac{L}{N}\), and the linear density of condensin is given by \({d}^{-1}=\frac{N}{L}\). Bound condensins extrude symmetrically, at a constant velocity \(v\). Once extrusion has started, condensins unbind DNA at a rate \({k}_{u}\). When the heads of two condensins collide, they block each other at the site of collision while the other head continues extruding. If a condensin anchor reaches one end of a polymer, it is unloaded. Each time a condensin unbinds, another one binds at a random location. Consequently, the binding rate per lattice site of condensin, \({\kappa }_{b}\), is related to the condensin density and unbinding rate through the relation \({\kappa }_{b}=\frac{N{k}_{u}}{L}\). The processivity, i.e., the average loop length of unobstructed condensin, is given by \(\lambda=\frac{v}{{k}_{u}}\).
Recent studies have shown that budding yeast condensin extrudes DNA asymmetrically and unidirectionally. In our simulations, however, condensin is implemented as symmetric extruders. This simplification is justified because, in the absence of barriers, symmetric and one-sided extrusion generate identical loop distributions for a given processivity and density and therefore yield equivalent contact dynamics and first-passage time statistics. To confirm this, we simulated one-sided extrusion across six representative parameter sets and found no difference in probe distances or search time statistics (Fig. S9).
Simulations with size-selective extruder removal
To isolate the effects of different extrusion event sizes, we performed simulations where extruders were selectively removed based on the remaining length of DNA tether between the arrays during the extrusion process. We defined an extrusion event as a contiguous moment in time where the effective 1D distance between the arrays drops below its equilibrium value due to one or several loop extruders. Using extrusion tracks from simulations that best fit the experimental data, we identified all such events and categorized them by the minimal remaining 1D distance between the arrays reached during the event (Fig. 7A). We then removed all extruders involved in extrusion events outside the desired range and re-ran the polymer simulation with the filtered extrusion track.
Calibration of time and length scales
Simulations were conducted without extruders, initially using arbitrary time and length scales. These were later rescaled against the extrusion-free (condensin-depleted) experiments. The rescaling was performed by matching the two-point MSD of two beads in the simulation to the experimentally measured two-locus MSD, with bead separation in the simulation chosen to correspond to the genomic separation of the pair in the experiment. Because experimental trajectories were measured in two dimensions, simulated 3D trajectories were projected onto 2D before MSD and dot distance analysis. Note, however, that the CICI method measures the encounter time of the beads in 3D space. Consequently, 3D distance was used when computing encounter times in simulations, as described below.
Simulation CFT and array reaction radius calibration
To reproduce CICI formation time quantitatively, we compute the average time for the 3D distance between bead pairs to drop below a threshold value, representing the interaction radius of the arrays, starting from a randomly chosen time point that represents the time of rapamycin introduction. The reaction time depends on the reaction radius—a larger reaction radius makes it more likely for arrays to encounter and react, thus reducing the reaction time. Since the interaction radius of the arrays is unknown, we determine its value by computing the simulation CFT across a range of interaction radii and selecting the value that best matches the experimental CFT. We averaged the CFT across multiple pairs of monomers with the same bead separation, taking advantage of the statistical equivalence of simulated polymers and their translational symmetry along the polymer backbone. Again, by symmetry, since our simulations neglect peripheral tether, all pairs of monomers located on different polymers are statistically equivalent (modulo boundary effects). The CFT for trans pairs was computed from randomly selected pairs of monomers located on different polymers.
Contact probability
Contact frequency curves were computed from the simulated conformations using the polykit library (https://github.com/open2c/polykit).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The Hi-C sequencing data generated in this study have been deposited into the Gene Expression Omnibus (GEO) database under accession code GSE296183: (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE296183). Source data are provided with this paper.
Code availability
Custom scripts for CICI-related imaging analysis, including dot identification, distance, RMSD, and CFT quantification, are available at https://github.com/yzl452/CFT_imaging. Simulation code used in this study is publicly available at GitHub: https://github.com/timothyfoldes/yeast_chromosomal_search_time/ and archived at Zenodo: https://doi.org/10.5281/zenodo.1880210165.
References
Bentovim, L., Harden, T. T. & DePace, A. H. Transcriptional precision and accuracy in development: from measurements to models and mechanisms. Development 144, 3855–3866 (2017).
Galouzis, C. C. & Furlong, E. E. Regulating specificity in enhancer–promoter communication. Curr. Opin. Cell Biol. 75, 102065 (2022).
Fortin, J.-P. & Hansen, K. D. Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data. Genome Biol. 16, 1–23 (2015).
Nora, E. P. et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012).
Denker, A. & De Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes Dev. 30, 1357–1382 (2016).
Marshall, W. et al. Interphase chromosomes undergo constrained diffusional motion in living cells. Curr. Biol. 7, 930–939 (1997).
Miné-Hattab, J. & Rothstein, R. Increased chromosome mobility facilitates homology search during recombination. Nat. Cell Biol. 14, 510–517 (2012).
Hoencamp, C. & Rowland, B. D. Genome control by SMC complexes. Nat. Rev. Mol. Cell Biol. 24, 633–650 (2023).
Wutz, G. et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 36, 3573–3599 (2017).
Ganji, M. et al. Real-time imaging of DNA loop extrusion by condensin. Science 360, 102–105 (2018).
Davidson, I. F. et al. DNA loop extrusion by human cohesin. Science 366, 1338–1345 (2019).
Pradhan, B. et al. The Smc5/6 complex is a DNA loop-extruding motor. Nature 616, 843–848 (2023).
Du, M., Zou, F., Li, Y., Yan, Y. & Bai, L. Chemically induced chromosomal interaction (CICI) method to study chromosome dynamics and its biological roles. Nat. Commun. 13, 757 (2022).
Hajjoul, H. et al. High-throughput chromatin motion tracking in living yeast reveals the flexibility of the fiber throughout the genome. Genome Res. 23, 1829–1838 (2013).
Arbona, J.-M., Herbert, S., Fabre, E. & Zimmer, C. Inferring the physical properties of yeast chromatin through Bayesian analysis of whole nucleus simulations. Genome Biol. 18, 1–15 (2017).
Socol, M. et al. Rouse model with transient intramolecular contacts on a timescale of seconds recapitulates folding and fluctuation of yeast chromosomes. Nucleic Acids Res. 47, 6195–6207 (2019).
Heun, P., Laroche, T., Shimada, K., Furrer, P. & Gasser, S. M. Chromosome dynamics in the yeast interphase nucleus. Science 294, 2181–2186 (2001).
Gabriele, M. et al. Dynamics of CTCF-and cohesin-mediated chromatin looping revealed by live-cell imaging. Science 376, 496–501 (2022).
Bystricky, K., Heun, P., Gehlen, L., Langowski, J. & Gasser, S. M. Long-range compaction and flexibility of interphase chromatin in budding yeast analyzed by high-resolution imaging techniques. Proc. Natl. Acad. Sci. USA 101, 16495–16500 (2004).
Piveteau, V. et al. Condensin loop extrusion properties, roadblocks, and role in homology search in S. cerevisiae. Preprint at bioRxiv https://doi.org/10.1101/2024.09.12.612585 (2024).
Uhlmann, F., Lottspeich, F. & Nasmyth, K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 400, 37–42 (1999).
D’Ambrosio, C. et al. Identification of cis-acting sites for condensin loading onto budding yeast chromosomes. Genes Dev. 22, 2215–2227 (2008).
Doughty, T. W., Arsenault, H. E. & Benanti, J. A. Levels of Ycg1 limit condensin function during the cell cycle. PLoS Genet. 12, e1006216 (2016).
Johzuka, K. & Horiuchi, T. The cis element and factors required for condensin recruitment to chromosomes. Mol. Cell 34, 26–35 (2009).
Dinda, M. et al. Fob1-dependent condensin recruitment and loop extrusion on yeast chromosome III. PLoS Genet. 19, e1010705 (2023).
Lazar-Stefanita, L. et al. Cohesins and condensins orchestrate the 4D dynamics of yeast chromosomes during the cell cycle. EMBO J. 36, 2684–2697 (2017).
Natsume, T., Kiyomitsu, T., Saga, Y. & Kanemaki, M. T. Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep. 15, 210–218 (2016).
Yesbolatova, A. et al. The auxin-inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nat. Commun. 11, 5701 (2020).
Schalbetter, S. A. et al. SMC complexes differentially compact mitotic chromosomes according to genomic context. Nat. Cell Biol. 19, 1071–1080 (2017).
Rossi, M. J. et al. A high-resolution protein architecture of the budding yeast genome. Nature 592, 309–314 (2021).
Li, M., Fine, R. D., Dinda, M., Bekiranov, S. & Smith, J. S. A Sir2-regulated locus control region in the recombination enhancer of Saccharomyces cerevisiae specifies chromosome III structure. PLoS Genet. 15, e1008339 (2019).
Li, Y., Lee, J. & Bai, L. DNA methylation-based high-resolution mapping of long-distance chromosomal interactions in nucleosome-depleted regions. Nat. Commun. 15, 4358 (2024).
Wang, S. et al. Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, 598–602 (2016).
Tjong, H., Gong, K., Chen, L. & Alber, F. Physical tethering and volume exclusion determine higher-order genome organization in budding yeast. Genome Res. 22, 1295–1305 (2012).
Goloborodko, A., Marko, J. F. & Mirny, L. A. Chromosome compaction by active loop extrusion. Biophys. J. 110, 2162–2168 (2016).
Samejima, K. et al. Functional analysis after rapid degradation of condensins and 3D-EM reveals chromatin volume is uncoupled from chromosome architecture in mitosis. J. Cell Sci. 131, jcs210187 (2018).
Shaltiel, I. A. et al. A hold-and-feed mechanism drives directional DNA loop extrusion by condensin. Science 376, 1087–1094 (2022).
Samejima, K. et al. Rules of engagement for condensins and cohesins guide mitotic chromosome formation. Science 388, eadq1709 (2025).
Cattoni, D. I. et al. Single-cell absolute contact probability detection reveals chromosomes are organized by multiple low-frequency yet specific interactions. Nat. Commun. 8, 1753 (2017).
Jusuf, J. M. et al. Genome-wide absolute quantification of chromatin looping. Preprint at bioRxiv, https://doi.org/10.1101/2025.01.13.632736 (2025).
Bintu, B. et al. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362, eaau1783 (2018).
Wang, B.-D., Eyre, D., Basrai, M., Lichten, M. & Strunnikov, A. Condensin binding at distinct and specific chromosomal sites in the Saccharomyces cerevisiae genome. Mol. Cell. Biol. 25, 7216–7225 (2005).
Thadani, R., Kamenz, J., Heeger, S., Munoz, S. & Uhlmann, F. Cell-cycle regulation of dynamic chromosome association of the condensin complex. Cell Rep. 23, 2308–2317 (2018).
Analikwu, B. T. et al. Telomeres stall DNA loop extrusion by condensin. Cell Rep. 44, 115900 (2025).
Guerin, T. M. et al. Condensin-mediated chromosome folding and internal telomeres drive dicentric severing by cytokinesis. Mol. Cell 75, 131–144 e3 (2019).
Kakui, Y., Rabinowitz, A., Barry, D. J. & Uhlmann, F. Condensin-mediated remodeling of the mitotic chromatin landscape in fission yeast. Nat. Genet. 49, 1553–1557 (2017).
Hirano, T. Condensins: universal organizers of chromosomes with diverse functions. Genes Dev. 26, 1659–1678 (2012).
Levings, P. P. & Bungert, J. The human β-globin locus control region: a center of attraction. Eur. J. Biochem. 269, 1589–1599 (2002).
Levo, M. et al. Transcriptional coupling of distant regulatory genes in living embryos. Nature 605, 754–760 (2022).
Kiefer, L. et al. WAPL functions as a rheostat of Protocadherin isoform diversity that controls neural wiring. Science 380, eadf8440 (2023).
Calderon, L. et al. Cohesin-dependence of neuronal gene expression relates to chromatin loop length. Elife 11, e76539 (2022).
Kane, L. et al. Cohesin is required for long-range enhancer action at the Shh locus. Nat. Struct. Mol. Biol. 29, 891–897 (2022).
Lancaster, L., Patel, H., Kelly, G. & Uhlmann, F. A role for condensin in mediating transcriptional adaptation to environmental stimuli. Life Sci. Alliance 4, e202000961 (2021).
Du, M., Zhang, Q. & Bai, L. Three distinct mechanisms of long-distance modulation of gene expression in yeast. PLoS Genet. 13, e1006736 (2017).
Lee, J. et al. Transcription factor condensates, 3D clustering, and gene expression enhancement of the MET regulon. eLife 13, RP96028 (2024).
Dai, H.-Q. et al. Loop extrusion mediates physiological Igh locus contraction for RAG scanning. Nature 590, 338–343 (2021).
Shetty, A., Reim, N. I. & Winston, F. Auxin-inducible degron system for depletion of proteins in Saccharomyces cerevisiae. Curr. Protoc. Mol. Biol. 128, e104 (2019).
Zou, F. & Bai, L. Using time-lapse fluorescence microscopy to study gene regulation. Methods 159, 138–145 (2019).
Redner, S. A guide to first-passage processes (Cambridge University Press, 2001).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 1–11 (2015).
Abdennur, N. & Mirny, L. A. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 36, 311–316 (2020).
Open2C et al. Cooltools: enabling high-resolution Hi-C analysis in Python. PLoS Comput. Biol. 20, e1012067 (2024).
Ramírez, F. et al. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat. Commun. 9, 189 (2018).
Rosebrock, A. P. Analysis of the budding yeast cell cycle by flow cytometry. Cold Spring Harb. Protoc. 2017, https://doi.org/10.1101/pdb.prot088740 (2017).
Foldes, T. Simulation and analysis code for: condensin accelerates long-range intra-chromosomal interactions. Zenodo https://doi.org/10.5281/zenodo.18802101 (2026).
Acknowledgements
We acknowledge all members in Bai lab and Mirny Lab for insightful comments on the manuscript. We also want to thank the members of the Center of Eukaryotic Gene Regulation at PSU for discussions and technical support. This work is supported by the National Institutes of Health (T32 GM125592 to Y.L., R35 GM139654 to L.B., NIH R01GM114190 to L.M.) and the National Science Foundation (MCB- 2016266 to L.B., NSF 2210558 to L.M., NSF 2044895 to L.M.). LM is also a Simons Investigator.
Author information
Authors and Affiliations
Contributions
F.Z. and Y.L. designed and performed most of the experiments; C.S. contributed to the experiments; F.Z., Y.L., and L.B. performed data analysis and wrote the manuscript. T.F., H.D.P., and L.M. performed and analyzed the polymer simulations.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kerry Bloom and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zou, F., Li, Y., Földes, T. et al. Condensin accelerates long-range intra-chromosomal interactions. Nat Commun 17, 4020 (2026). https://doi.org/10.1038/s41467-026-70538-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70538-5
