
Abstract
High-nickel layered positive electrodes suffer from progressive structural degradation arising from lattice oxygen loss and inherent lattice strain. Although surface coatings are widely used to stabilize lattice-oxygen redox and mitigate electro-chemo-mechanical degradation, achieving coatings with full continuity, robust interfacial bonding, and fast Li+ conductivity remains challenging. Herein, we present a fundamentally different approach to shell formation via a self-derived subtractive strategy, departing from the conventional additive-based coating methods. By accurately applying transient thermal pulses, surface lithium is selectively extracted from layered LiNixCoyMn1-x-yO2 (x = 0.8 ~ 0.9), directly converting the outer region into a coherent spinel-phase shell with tunable thickness. This nanoscale spinel-phase skin forms a robust mortise-and-tenon-like interconnection with the layered bulk, enabling isotropic, high-rate Li+ extraction/insertion while maintaining electronic conductivity throughout cycling. It effectively confines active oxygen intermediates, and suppresses interfacial side reactions and strain evolution under high-potential operation. Therefore, the spinel-phase skin-encapsulated LiNi0.8Co0.1Mn0.1O2 achieves an initial Coulombic efficiency of 95.3% and enables pouch cells with 80.1% capacity retention after 2000 cycles at 180 mA g-1. This strategy is extendable to LiNi0.9Co0.05Mn0.05O2, may open new avenues for advancing nickel-rich positive electrode technologies.
Similar content being viewed by others
Introduction
High-nickel layered oxides, particularly LiNixCoyMn1-x-yO2 (NCM, x ≥ 0.8), are among the most promising positive electrode candidates for next-generation automotive power batteries due to their energy density1,2,3,4. However, rapid capacity degradation remains the most challenging issue in the commercialization of NCM. It is now widely recognized that this degradation originates primarily from lattice oxygen oxidation accompanying the H2-H3 phase transition at elevated potential ( > 4.1 V)5,6. Lattice oxygen oxidation not only leads to gasous oxygen evolution but also generates reactive oxygen intermediates that oxidize the electrolyte, triggering irreversible phase transitions from the layered structure to spinel/rock-salt phases7. These structural changes8, which initiate at the surface and propagate inward, result in a low initial Coulombic efficiency (ICE, 85 ~ 89%). The formation of an electrochemically inactive rock-salt phase at the cathode-electrolyte interface (CEI) significantly impedes Li+ and electron transport, inducing local charge heterogeneity9,10,11,12. Simultaneously, oxygen loss amplifies the intrinsic anisotropic lattice strain of NCM, leading to internal particle cracking6,13,14 and mechanical pulverization15,16. Collectively, these degradation pathways cause the progressive loss of potential plateau and reversible capacity. Thus, suppressing lattice oxygen loss is essential for improving the electrochemical reversibility and longevity of NCM electrodes.
Given that the lattice oxygen oxidation is an intrinsic nature of highly delithiated NCM, an effective strategy must suppress O2 release and mitigate side reactions between active oxygen intermediates and the electrolyte. Surface coating17,18,19,20,21,22,23,24,25 stands out as the most direct and promising solution, but to be effective, the coating must meet various stringent requirements. Ideally, coating layer should provide nearly complete coverage, support high-rate electron and Li+ transport, exhibit extraordinary chemical/ electrochemical stability, and form strong mechanical and chemical bonds with the bulk phase. Traditional coating methods, primarily based on additive processes such as dry and wet chemical deposition of metal oxides17,18,20, fluorides22, phosphides23 and solid Li+ conductor24,25, can largely reduce direct contact between the electrode and electrolyte, but often fail to achieve full coverage and sufficient interfacial bonding. Advanced thin-film deposition techniques such as physical/chemical vapor deposition and atomic layer deposition26,27,28 ensure high coverage and thickness control, yet suffer from low ionic conductivity and lattice mismatch with the underlying NCM.
To overcome these limitations, a fundamentally different strategy is needed. By reverse thinking about the limitations of additive methods, a subtractive strategy29,30,31,32,33,34 that precisely removes certain atoms from the outermost layer of NCM can enable the in situ formation of a self-derived, conformal coating shell. Previous studies have hinted at this potential: Lu30 et al. proposed that Ar/H2 plasma treatment induced oxygen vacancies on the NCM surface, which subsequently transformed into a spinel-phase shell after electrochemical cycles. Similarly, Zhao32 et al. utilized this approach to produce a rock-salt coating on LiCoO2, and Chen34 et al. generated a Li-deficient thin layer on Li-rich layered oxides by adjusting lithium source amount during synthesis. However, these methods still fall short in terms of controllability, scalability and shell integrity, as reflected in only marginal improvements in the ICE.
In this work, we disclose a precisely controlled subtractive strategy using flash Joule heating to quantitatively remove surface lithium/oxygen atoms from NCM. This technique induces an in situ transformation of NCM edges into a coherent and perfectly compatible spinel-phase skin layer with tunable thickness and complete coverage. By precisely controlling the temperature and duration of transient thermal pulses, either spinel-phase or rock-salt-phase coating layers can be selectively formed (Fig. 1). In particular, the nanoscale spinel-phase skin establishes a robust, mortise-and-tenon-like interconnection with the bulk phase, enabling isotropic and high-rate Li+ extraction/insertion and continuous electron transport across the interface during long-term cycling. This conformal shell tightly confines the active oxygen intermediates, eliminates local charge heterogeneity, and significantly suppresses irreversible layered-to-spinel/rock-salt phase transition both at the surface and within the bulk. This greatly contributes to enhancing reaction reversibility, improving the ICE, and mitigating strain evolution and particle cracking. Consequently, the spinel-phase skin-encapsulated LiNi0.8Co0.1Mn0.1O2 achieves an high initial Coulombic efficiency of 95.3% and retains 93.6% capacity after 500 cycles, significantly surpassing the pristine counterpart (89.6% efficiency and 65.2% retention). Moreover, the optimally engineered NCM electrode boosts the retention rate of pouch cells from 52.0% to 80.1% after 2000 cycles. More importantly, the spinel-phase skin is also applicable to higher-nickel NCM electrodes such as LiNi0.9Co0.05Mn0.05O2 and LiNi0.825Co0.115Mn0.06O2. This corrosion-resistant and crack-resistant skin paves the way for enhancing the long-lifespan of high-nickel electrodes.

a Schematic illustration of the customized modulation of the interfacial phase structure of NCM811 via quantitatively stripping surface Li&O atoms using flash Joule heating technology. HRTEM study of temperature-dependent interfacial phase evolution, by varying the temperature with a fixed heat treatment duration (30 s), including: (b) Pristine, (c) 300 °C, (d) 350 °C and (e) 500 °C. The samples treated at different temperatures (X °C) are labeled as FJX. HRTEM study of time-dependent interfacial phase evolution, by extending the pulse width at a fixed temperature (350 °C), including: (f) 15 s, (g) 60 s, (h) 120 s and (i) 300 s. The corresponding Fast Fourier Transform (FFT) and Inverse Fast Fourier Transform (IFFT) images are provided for each condition. Scale bars: 2 nm.
Results
Self-derived growth of phase-customizable skins on NCM811
Figure 1a presents a schematic illustration of the customized modulation of the interfacial phase structure of LiNi0.8Co0.1Mn0.1O2 (NCM811) using flash Joule heating technology. Typically, commercial NCM811 powder was placed into the reaction chamber of an independently developed transient high-temperature equipment (Supplementary Fig S1a) and subjected to flash sintering under an argon atmosphere using thermal pulses with precisely-controlled temperatures and pulse widths. Notably, the NCM811 from a single processing batch can reach 5 ~ 10 g (Supplementary Fig S1b). Under transient thermal pulses, partial lithium and oxygen atoms at the surface/subsurface are sequentially stripped, leading to the formation of Li-deficient or Li&O co-deficient phases30,31,32. Therefore, with increasing transient temperature or prolonged pulse width, the outermost layer of NCM811 particles gradually transforms from a layered phase into spinel phases of varying thickness, and ultimately evolves into a rock-salt phase under severe lithium and oxygen extraction (Fig. 1a).
High-resolution transmission electron microscopy (HRTEM) characterization provides direct evidence for the interfacial evolution of NCM811 under varying thermal pulse treatment. As disclosed in Fig. 1b, the pristine sample shows a consistent R-3m layered phase structure (labeled as L in the box), characterized by a 0.47 nm lattice spacing, from the surface to the interior. As the temperature increases under a fixed pulse width (30 s), surface lithium atoms are gradually thermally stripped. When the temperature reaches 300 °C, the layered structure at the interface begins to transform into the spinel phase (labeled as S in the box, Fig. 1c). Meanwhile, below 400 °C, the thickness of the spinel-phase skin layer increases as the depth of lithium atom extraction grows, expanding from a few nanometers to several tens of nanometers (Fig. 1d and Supplementary Fig. 2). We refer to the samples treated at different temperatures (X °C) as FJX, where FJ stands for Flash Joule heating. Taking the comparison between the pristine sample and FJ350 as an example, scanning electron microscopy (SEM) images show negligible difference in their microscopic morphology (Supplementary Fig. 3). However, the electron paramagnetic resonance33 (EPR) spectra at around 3500 G reveal that the FJ350 sample contains more oxygen vacancies. This indicates that although the layered-to-spinel transition theoretically involves only lithium stripping, it is also accompanied by a certain degree of thermal oxygen extraction during the actual flash sintering process (Supplementary Fig. 4). In addition, the O 1 s X-ray photoelectron spectroscopy (XPS) spectrum of FJ350 also exhibits a more pronounced oxygen defect peak32,33 (Odef), which can likewise be attributed to oxygen vacancies triggered by interfacial oxygen extraction (Supplementary Fig. 5). As the temperature exceeds 500 °C, further delithiation and deoxygenation triggers the transition of the interfacial layer to the rock-salt phase (labeled as R in the box, Fig. 1e), with the thickness increasing as the temperature rises (e.g., 600 °C, Supplementary Fig. 6). Ex situ X-ray diffraction (XRD) patterns demonstrate that under thermal pulses with a 30 s pulse width, a distinct rock-salt phase peak only emerges when the temperature reaches 700 °C (i.e., involving changes in the bulk phase, Supplementary Fig. 7), confirming that Li and O stripping at lower temperatures predominantly occurs in the surface/subsurface regions of the material.
In addition to ex situ HRTEM, we further validated the temperature-dependent interfacial phase correlations via in situ TEM equipped with microchip-based heating devices, confirming that 300 and 450 °C are the critical temperatures for the formation of spinel phase and rock-salt phase, respectively (Supplementary Fig. 8). From another perspective, by extending the pulse width at a fixed temperature, a continuous change in the surface phase structure of NCM811 can also be observed. As displayed in Fig. 1f–i, after 60 s at 350 °C, a transformation from the spinel phase to the rock-salt phase occurs. Note that although interfacial transformations occur, the XRD patterns show negligible changes even when the processing durations exceed 300 s, indicating that transient high-temperature treatments have minimal impact on the bulk crystalline phases of the material, particularly under low-temperature conditions (Supplementary Fig. 9). Interestingly, however, after 30 s of treatment at 350 °C, the calculated I(003)/I(104) intensity ratio reaches its maximum value, corresponding to the lowest degree of Li+/Ni2+ cation mixing (Supplementary Figs. 7 and 9). This demonstrates that the removal of Li and O at the interface still exerts some influence on the bulk crystalline structure of NCM811, particularly affecting the cation mixing, a parameter closely related to the ICE and long-term cycling stability.
It is crucial to emphasize that precisely controlled transient thermal pulse treatment enables the acquisition of numerous kinetically controlled metastable intermediate states, which serve as the key technical foundation for achieving customization and thickness regulation of the NCM811 interfacial phases. In stark contrast, under traditional steady-state thermal sintering conditions (typically lasting several hours at least), these fleeting interfacial phases cannot be captured or preserved, ultimately yielding only thermodynamically controlled rock-salt phase structures (e.g., products at 350 °C for 0.5 h, Supplementary Fig. 10). Undoubtedly, the unique fast heating and rapid cooling mechanisms of the Joule heating technique provide a crucial technical prerequisite for achieving nano-scale precise phase transformation at the electrode material interface.
Spinel-phase skin enables highly reversible NCM811 positive electrodes
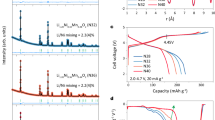
The effect of the phase structure and thickness of the skin shell on the ICE, rate capability, and cycling performance of NCM811 is evaluated in detail through half-cell tests. Figure 2a, 2b present the charge-discharge profiles at 0.1 C (1 C = 180 mA g−1) and corresponding ICE of the NCM811 before and after 30 s of treatment at various temperatures. Apparently, the reversible discharge capacity and ICE of NCM811 both show a trend of first increasing and then decreasing with the rise in pulse temperature, reaching their maximum at 350 °C. Notably, the FJ350 sample discloses an high ICE of 95.3%, surpassing that of the pristine sample (89.6%). This is highly consistent with the lowest degree of Li+/ Ni2+ cation mixing observed in FJ350 (Supplementary Figs. 7 and 9). More effectively, the FJ350 also demonstrates both the highest rate performance and cycling stability at 1 C (Fig. 2c and Supplementary Fig. 11). Moreover, the slopes obtained from cyclic voltammetry profiles at varying scan rates also reveal that FJ350 possesses superior ionic transport properties (Supplementary Fig. 12). This convincingly indicates that a spinel-phase skin with an appropriate thickness can ensure high-rate and isotropic Li+ extraction/insertion across the interface during long-term cycling. In contrast, samples with either too thick or too thin spinel-phase skin coatings (FJ300 and FJ400), although performing better than the pristine sample, are still inferior to FJ350. On one hand, the excessively thin shell lacks sufficient mechanical strength and isolation effect (e.g., incomplete coverage), only partially suppressing lattice oxygen loss and volume effects. On the other hand, the overly thick shell may hinder Li+ transport owing to the raised ionic diffusion barrier, causing adverse ion blockage and polarization. In situ differential electrochemical mass spectrometry (DEMS) was employed to monitor the oxygen evolution of different samples under high potential (e.g., 4.7 V) in order to evaluate the coverage of the spinel-phase shell. As Supplementary Fig. 13 demonstrates, the FJ300 sample with a thin and incomplete spinel-phase shell already exhibits a certain degree of suppression of oxygen evolution during the first charge compared with the pristine sample, but the reduction is limited, decreasing only from 4.6 to 3.1 nmol min-1. In sharp contrast, the oxygen-release signals of the FJ350 and FJ400 electrodes are significantly lower, with FJ350 in particular reduced to as low as 1.2 nmol min−1. This result indicates that the spinel-phase shell can effectively inhibit oxygen evolution even under the high cutoff potential of 4.7 V, directly reflecting both the completeness and the strong spatial confinement effect of the FJ350 coating layer. The suppression of oxygen release is also consistent with the increased Coulombic efficiency and reduced irreversible lithium consumption observed in the electrochemical tests, further demonstrating the critical role of an optimized coating thickness in achieving stable cycling performance. Moreover, samples subjected to higher temperatures, particularly those at 500 °C and above (e.g., FJ500 and FJ600), exhibit a significant reduction in ICE and initial discharge capacity. In particular, the FJ600 sample delivers the lowest discharge capacity of 154.5 mAh g−1, corresponding to a low ICE of 76.0%. According to the semi-quantitative estimation of FJ600 in Supplementary Fig. 14 and Table S1, this deterioration primarily arises from the thick rock-salt reconstruction layer, which blocks Li⁺ transport and exacerbates interfacial deterioration, rather than from the negligible lithium extraction during the flash Joule heating process.

a Initial charge-discharge profiles at 0.1 C (1 C = 180 mA g-1) and (b) corresponding ICEs of NCM811 electrodes before and after 30 s of treatment at various pulse temperatures. c Rate performance of the pristine, FJ300, FJ350, FJ400, FJ500 and FJ600 electrodes. d Initial charge-discharge profiles at 0.1 C and (e) corresponding ICEs of the NCM811 treated at a fixed temperature of 350 °C for various pulse widths. f Cycling performance of the pristine and FJ350 electrodes for 500 cycles at 1 C within 2.7–4.3 V. g Differential capacity (dQ/dV vs. potential) analysis of the pristine and FJ350 electrodes at different cycle numbers. h GITT profile of the charge/discharge processes of the pristine and FJ350 electrodes after 500 cycles and the corresponding DLi+ vs. potential curve. i Digital photograph of a smartphone being charged by a pouch full cell configured with a FJ350 positive electrode and graphite negative electrode. Scale bar: 20 mm. j Long-term cycling performance of the FJ350-based pouch full cell for 2000 cycles at 1 C.
Similarly, samples treated at a fixed temperature of 350 °C for different pulse widths also exhibit electrochemical performance that first increases and then decreases. As manifested in Fig. 2d, 2e, 60 s is both the point at which the interfacial phase transitions from spinel to rock-salt and the turning point for the deterioration of electrochemical performance. The sample treated for 30 s, namely FJ350, possesses the most suitable spinel-phase shell thickness, stands out with the highest ICE and optimal cycling stability among all samples (Supplementary Fig. 15).
Based on the TEM analysis, we performed semi-quantitative measurements and statistical analysis of the average thickness of the spinel-phase shell formed on the surface of NCM811 under different flash-sintering conditions (Supplementary Fig. 16), and correlated these results with the corresponding electrochemical performance. As summarized in Table S2, the thickness of the spinel-phase layer formed under different treatment conditions (pulse temperature and pulse width) varies systematically, generally following the trend of increasing thickness with longer pulse duration or higher treatment temperature. Meanwhile, the thickness of the spinel-phase shell, as well as the associated degree of lithium deficiency, exerts a pronounced influence on the electrochemical performance including ICEs and long-term cycling stability. These findings further demonstrate that an optimally engineered spinel layer, with well-balanced thickness and defect chemistry, enables the FJ350 sample to deliver the most superior electrochemical performance among all treated conditions. As Fig. 2f demonstrates, the FJ350 electrode preserves a discharge capacity of 167.1 mAh g−1 with a retention of 93.6% after 500 cycles at 1 C, far exceeding the 115 mAh g−1 and 65.2% retention observed for the pristine sample. Charge-discharge curves convincingly demonstrate the material advantages of FJ350 in suppressing capacity fading and resisting electrode polarization (Supplementary Fig. 17). The FJ350 electrode exhibits a much slower potential plateau decay, with a drop of only 0.102 mV per cycle, in stark contrast to the pristine electrode, which presents a sharp decay of 0.589 mV per cycle (Supplementary Fig. 18). Apparently, by tightly confining the oxidized lattice oxygen and inhibiting the oxygen loss, the spinel-phase skin-encapsulated FJ350 maintains the average oxidation state of the transition metals in the bulk structure, thereby preserving the operating potential and specific capacity during extended cycling. This can be further confirmed by the differential capacity (dQ/dV vs. potential) analysis. As depicted in Fig. 2g, the H2-H3 phase transition around 4.2 V, associated with c-axis contraction and lattice oxygen oxidation, is significantly diminished in the pristine sample after 500 cycles, indicating severe structural changes and lattice oxygen loss. In sharp contrast, the FJ350 sample maintains a stable H2-H3 redox peak, suggesting reinforced structural integrity and effective stabilization of lattice oxygen, thereby greatly suppressing its release.
The advantage of FJ350 over the pristine sample in reaction kinetics can be elucidated by measuring the Li-ion diffusion coefficient (DLi+). The galvanostatic intermittent titration technique (GITT) profile of the charge/discharge processes of the pristine and FJ350 electrodes and the corresponding DLi+ vs. potential curve indicate that the cycled FJ350 electrode delivers a faster Li+ transport rate (Fig. 2h and Supplementary Fig. 19). Benefiting from the reaction kinetics, the FJ350 electrode retains 81.3% of its capacity after 1000 cycles at a high current density of 2 C. By contrast, the pristine sample suffers rapid capacity degradation, maintaining only 60.3% of its initial capacity after long-term cycling (Supplementary Fig. 20). More encouragingly, the FJ350 electrode demonstrates prominent performance under more harsh operating conditions, such as a potential upper limit of 4.5 V (Supplementary Fig. 21) and a high temperature of 60 °C (Supplementary Fig. 22). As a demonstration of practical application, a pouch full cell with a FJ350 positive electrode and graphite negative electrode was successfully assembled and used to power a smartphone (Fig. 2i). More effectively, within the potential window of 2.7–4.2 V, the FJ350-based pouch cell maintains 80.1% capacity retention after 2000 cycles (Fig. 2j), significantly surpassing that of the pristine NCM811-based pouch cell (52.0%). The charge-discharge curves further demonstrate the advantage of the FJ350 electrode in mitigating capacity fade and resisting electrode polarization, highlighting its long-term cycling stability (Supplementary Fig. 23). Notably, half-cells and pouch cells incorporating the optimized FJ350 exhibit comparable electrochemical performance, especially in cycling stability, compared to counterparts utilizing conventional NCM electrodes reported in the reported literature (Supplementary Fig. 24 and Supplementary Table S3). To further highlight the electrochemical advantages of the self-induced coated FJ350, we conducted comparative studies on the electrochemical performance and DEMS results of FJ350, commercial LiNbO3-coated LiNi0.8Co0.1Mn0.1O2 (LiNi0.8Co0.1Mn0.1O2@LiNbO3), and commercial Li3BO3-coated LiNi0.8Co0.1Mn0.1O2 (LiNi0.8Co0.1Mn0.1O2@B). As shown in Supplementary Figs. 25 and 26, the commercial NCM811 positive electrodes coated with LiNbO3 or Li3BO3 exhibit not only lower initial coulombic efficiencies and poorer cycling stability but also inferior suppression of oxygen release under high potential (4.7 V). In addition, we further compared FJ350 with NCM811 electrodes coated using traditional wet-chemical methods and other representative coating approaches reported in the literature. As presented in Supplementary Table S4, the FJ350 electrode demonstrates comparable improvements in both cycling performance and rate capability. These results collectively highlight that our subtraction-coating strategy not only enhances electrochemical performance but also improves battery stability and extends service life, making it the most effective approach among currently available coating methods.
Samples annealed at 350 °C under an argon atmosphere using a traditional tube furnace (Supplementary Fig. 10) were compared with FJ350. The results show slight improvement compared to the pristine counterpart, but still far inferior to FJ350 (Supplementary Fig. 27). This highlights the inherent limitations of conventional annealing methods, where tube furnace processing fails to capture and preserve the beneficial spinel-phase skin, instead directly leading to rock-salt shell that degrades electrochemical performance. Undoubtedly, transient high-temperature treatment provides a unique and disruptive technological solution for the customized modulation of the interfacial phase structure of NCM811 electrodes. More importantly, this precise interfacial regulation strategy, namely, the transformation from a layered structure to a spinel shell at the interface (Supplementary Fig. 28 and Fig. 29), enhances the reversible capacity and cycling stability of various materials, including polycrystalline LiNi0.825Co0.115Mn0.06O2, polycrystalline LiNi0.9Co0.05Mn0.05O2 (NCM90), and single-crystal LiNi0.8Co0.1Mn0.1O2 (SC-NCM811). These results (Supplementary Fig. 30) highlight the broad applicability of this strategy in tailoring surface atomic composition and stabilizing interfacial phases.
Interfacial and bulk-phase stability during the first cycle
During cycling, delithiation-induced lattice distortion and stress accumulation, along with oxygen loss and cation mixing, can trigger a cascade of phase transitions in the surface layer of the pristine NCM811 electrodes from layered to disordered spinel and rock-salt, leading to continuous deterioration of electrochemical performance. This trend also gradually propagates from the interface into the bulk, resulting in continuous structural degradation of the material. As emphasized in Fig. 3a, the outer layer of the pristine NCM811 begins to exhibit a NiO-like rock-salt phase35 only after the first charge to 4.3 V. Raising the upper limit of charging potential (e.g., 4.7 V) exacerbates this process, as the increased lithium and oxygen vacancy concentrations promote the migration of transition metals, thereby thickening the rock-salt phase transformation (Supplementary Fig. 31). In stark contrast, regardless of the delithiated state at 4.3 V or 4.7 V, the spinel-phase skin on the FJ350 surface stabilizes the three-dimensional framework (Fig. 3b and Supplementary Fig. 32), highlighting its high-potential compatibility and resistance to mechanical deformation. This also indicates that the highly crystalline spinel-phase shell obtained through flash sintering exhibits robust electrochemical stability and does not further transform into the rock-salt phase. This can be further confirmed by measuring the Li distribution in NCM811 after its first charge to 4.3 V. Time-of-flight secondary ion mass spectrometry36 (TOF-SIMS) analysis reveals a heterogeneous distribution of 7Li within the pristine particle (Fig. 3c), which is attributed to the formation of an interfacial inactive rock-salt phase that impedes isotropic Li+ extraction across the surface, leading to local charge heterogeneity9,11, internal structural distortion and localized damage. By contrast, the FJ350 exhibits a uniform 7Li distribution across its entire particle, attributed to low oxygen loss, well-preserved structural integrity and isotropic high-rate Li-ion diffusion channels at the interface (Fig. 3d). Given the above, the coating layer stabilizes the interface by preventing the formation of inactive phases, such as the rock-salt phase. This interfacial stability ensures efficient Li+ transport across the interface and suppresses lattice oxygen loss, thereby enhancing the overall Li⁺ diffusion within the particles. These findings highlight the critical role of the coating in optimizing both interfacial and bulk properties, leading to significantly improved electrochemical performance.

a, b HRTEM images of the pristine and FJ350 electrodes after the first charge to 4.3 V. c, d Spatial distribution maps of 7Li in cross-sections of the pristine and FJ350 electrodes, acquired through TOF-SIMS, after the first charge to 4.3 V. These samples were in the charged state and characterized after collecting the electrode powder from a cell charged to 4.3 V at 0.1 C under 26 °C. e, f In situ XRD spectra of the (003) peak for the pristine and FJ350 electrodes during the initial charge/discharge process in a potential range of 2.7–4.3 V at 0.1 C under 26 °C. g The changes in c-axis length and volume of the pristine and FJ350 electrodes during the first charge process. h, i In situ XRD spectra of the (003) peak and corresponding contour maps of the pristine and FJ350 electrodes during the first charge/discharge process in a potential range of 2.7-4.7 V at 0.1 C under 26 °C, along with (j, k) the fitting results at various states of charge.
In situ XRD spectra coupled with Rietveld refinements further underscore the reinforcing effect of the spinel-phase skin on the structural stability of NCM811. During the first charge process of the pristine electrode, the (003) peak shifts towards lower angles due to the increased repulsion between adjacent oxygen planes37,38, causing the c-axis lattice parameter to expand during charging (Fig. 3e). As the potential approaches 4.3 V, the (003) peak shifts to higher angles, indicating compression of the c parameter at a high delithiation state. During the entire charge process, the (003) peak shifts by 0.42°. During the discharge process, the (003) peak cannot recover to its initial position and ultimately shifts negatively by 0.09°, indicating substantial damage to the crystal structure. In contrast, the FJ350 electrode exhibits a 0.28° shift during the charge process and fully returns to its original peak position after discharging, indicating significantly enhanced structural reversibility (Fig. 3f).
The calculated cell parameters as a function of charge potential are presented in Fig. 3g and Supplementary Fig. 33. The initial expansion of the c-axis is due to increased electrostatic repulsion between oxygen layers as Li⁺ ions are extracted. At higher states of charge, the contraction along the c-axis arises from electron withdrawal by highly oxidized transition-metal ions from the coordinated oxygen atoms, leading to oxygen ion oxidation and a consequent reduction in the repulsive interactions within the delithiated Li layers. Meanwhile, the lattice volume decreases continuously due to dominant in-plane contraction from the shortening of TM-O bonds as the transition metals are oxidized. Furthermore, for the FJ350 sample, the restrained H2-H3 phase mitigates lattice-induced stress accumulation, as the substantial c-axis mismatch between the H2 and H3 phases constitutes a dominant source of mechanical strain during cycling. Quantitatively, the changes in c-axis length, a-axis length and total volume of the FJ350 sample during charging are 2.12%, 1.52%, and 2.85%, significantly lower than that of the pristine electrode (4.31%, 2.15%, and 4.63%). This clearly indicates that the spinel-phase skin indeed helps significantly reduce the lattice anisotropic changes exacerbated by oxygen loss during the H2-H3 phase transition.
To gain deeper insight into how the spinel-phase skin influences the structural evolution and Li-ion transport behavior of NCM811 under more demanding conditions, in situ XRD analysis was additionally carried out at an upper cutoff potential of 4.7 V. As illustrated in Fig. 3h, when the pristine electrode is charged from 4.3 V to 4.7 V, the intensity of the (003) peak significantly weakens, and its shape becomes asymmetrical, indicating the coexistence of H2 and H3 phases and highlighting the dramatic structural changes during the H2-H3 transition39. Even worse, at the fully charged state of 4.7 V, the residue of the H2 phase is still quite evident, revealing that the transition from H2 to H3 has not been fully completed (Fig. 3j). In contrast, only a slight H2 phase is observed at 4.3 V in the FJ350 sample, and as the charging potential increases (e.g., 4.5 V, Fig. 3i and 3k), the phase transition is fully completed21,40. The coexistence of the H2 and H3 phases is likely to lead to lattice mismatch within the particles, causing uneven lithium distribution, which is highly consistent with the TOF-SIMS observations (Fig. 3c). Apparently, the high mechanical strength and rapid reaction kinetics of the spinel-phase skin-encapsulated FJ350 play a key role in promoting the complete transformation of H2-H3. Differential Scanning calorimetry41 (DSC) tests further emphasize the role of the spinel-phase skin in stabilizing the FJ350 structure and suppressing oxygen evolution (Supplementary Fig. 34), primarily reflected in a 32 °C delay in the oxygen release peak compared to the pristine sample, with corresponding heating powers of 705 J g−1 and 1013 J g−1, respectively.
Crack- and corrosion-resistant mechanisms during cycling
The microscopic mechanism by which the spinel-phase skin significantly improves the long-term cycling stability of NCM811 has also been deeply studied. As depicted in Fig. 4a, the cross-sectional SEM image clearly indicates that after 500 cycles, severe cracking and damage appear inside the pristine particle in the discharged state. The enhanced bulk stability of FJ350 can be attributed to interfacial electron transfer between the spinel-phase shell and the bulk Ni-rich lattice, which mitigates c-axis variation and confines lattice oxygen under highly delithiated conditions. Consequently, this process alleviates lattice collapse induced by oxygen release from the surface to the bulk. This undoubtedly accelerates parasitic reactions with the electrolyte, promotes the dissolution and migration of transition metals, and significantly thickens the CEI (Supplementary Fig. 35). In stark contrast, only a small number of stress-induced micropores are observed inside the cycled FJ350 (Fig. 4b), rather than cracks, while a dense and thin CEI film forms on the interface, and the overall structure remains well preserved. The internal cracking and interface deterioration of the cycled pristine and FJ350 electrodes can be semi-quantitatively compared through the electrochemical impedance spectroscopy (EIS) characterization. As manifested in Supplementary Fig. 36 and Table S5, the FJ350 electrode exhibits markedly lower CEI film resistance (Rf) and charge transfer resistance (Rct) at all stages of cycling42. The pronounced reduction in Rct confirm that the spinel-phase skin establishes a percolating electron pathway, synergizing with fast Li+ transport to achieve high rate capability and cycling capacity retention. Further insights into the surface microstructure after cycling were compared by HRTEM analysis and corresponding FFT and IFFT images. As manifested in Supplementary Fig. 37, a 15 nm thick NiO-like rock-salt phase formed on the cycled pristine electrode surface owing to the continuous side reactions with the electrolyte15. In contrast, the FJ350 particle perfectly preserved the spinel-phase skin-encapsulated layered structure even after 500 cycles (Fig. 4c), again confirming the electrochemical stability and mechanical robustness of the flash sintering-induced highly crystalline spinel-phase shell. High angular annular dark field (HAADF) images of the cycled pristine and FJ350 electrodes suggest that the spinel-phase skin adapts well to lattice contraction and expansion and prevents the formation of microcracks (Supplementary Fig. 38), thereby suppressing the interfacial side reactions and the growth of the rock-salt phase and preserving the structural integrity of the NCM811 particles during cycling (Supplementary Fig. 39).

a, b Cross-sectional SEM images of the pristine and FJ350 electrodes after 500 cycles in the discharge state. c HRTEM analysis and corresponding FFT and IFFT images of the cycled FJ350. d, e Depth-profile XPS analysis of the pristine and FJ350 electrodes after 500 cycles. f TOF-SIMS secondary depth profiles (normalized to maximum) of partial species on the cycled pristine and FJ350 electrodes. The insets show the TOF-SIMS 3D reconstruction of C2H3OF−, NiF3−, CoF3−, MnF3− species. g, h The Ni-K edge XANES spectra of the pristine and FJ350 electrodes after 500 cycles. i, j FT-EXAFS spectra of the pristine and FJ350 electrodes after 500 cycles. k–n Wavelet-transformed EXAFS analysis of the pristine and FJ350 before and after 500 cycles. These samples were in the discharged state and characterized after collecting the electrode powder from a cell discharged to 2.7 V after 500 cycles at 1 C under 26 °C.
Depth-profile XPS analysis of the cycled electrodes reveals the CEI components arising from the decomposition of the lithium salts and solvents (Fig. 4d and 4e). The F 1 s XPS peaks18,33 indicate the presence of LiF, C-F and LixPFyOz in the FJ350 sample throughout the cycling process, with a significantly higher LiF content compared to the pristine sample. The higher LiF content in the CEI can largely suppress the decomposition of the electrolyte and LiPF6, contributing to long-term stability. The O 1 s spectra43 consists of four main peaks related to C-O, C = O, P-O and lattice O, respectively. The retention of the lattice O signal at the FJ350 interface validates the effectiveness of the spinel-phase shell in suppressing outer-layer oxygen loss. Moreover, the significantly reduced intensities of the C = O and P-O peaks in the FJ350 sample indicate that the LiF-dominated CEI effectively suppresses the decomposition of solvents (ethylene carbonate and diethyl carbonate) and LiPF6 salt. The LiF-rich CEI not only passivates the interface but also exhibits moderate electronic conductivity, balancing electron blocking for side reaction suppression and sufficient charge transfer for redox reactions. This can be confirmed by the low intensities of CO32− and C = O peaks from ROCO2Li in the C 1 s spectrum16,18 of the FJ350 sample. The C 1 s spectrum further reveals a stronger signal from the organic polymer C-C backbone in FJ350. The polymer components not only enhance the mechanical flexibility and strength of the CEI but also effectively embed the inorganic LiF, contributing to the formation of a thinner and more robust CEI layer. These results strongly indicate that the spinel-phase skin serves as a physical barrier between the highly delithiated cathode and the electrolyte, effectively inhibiting solvent and LiPF6 decomposition as well as active material dissolution, while promoting the formation of an LiF-rich CEI layer, thereby preventing severe structural degradation and interfacial side reactions.
TOF-SIMS analysis further corroborated the above results by revealing the three-dimensional structure and compositional distribution of the CEI layer40,44 (Fig. 4f, Supplementary Figs. 40 and 41). The following lists several major ion fragments, including C2H3OF− (organic species from solvent decomposition), LiF2- and PO2F2- (inorganic species from the LiPF6 decomposition), NiF3−, CoF3− and MnF3− (dissolved products of active materials). The trend of secondary ion distribution with sputtering time can be derived from the normalized intensity plot in Fig. 4f. Apparently, a significantly thinner CEI layer is formed on the surface of the cycled FJ350 electrode. Moreover, the concentrations of both organic and inorganic species (e.g., C2H3OF−, MnF3−, NiF3− and CoF3−) generated from interfacial degradation are markedly reduced on the FJ350 surface, confirming that electrolyte decomposition and metal cation dissolution are greatly suppressed. This interfacial stabilization strategy demonstrates universal effectiveness across different high-Ni positive electrode materials, as evidenced by the TOF-SIMS analyses of LiNi0.9Co0.05Mn0.05O2 (Supplementary Fig. 42) and single-crystal LiNi0.8Co0.1Mn0.1O2 electrodes (Supplementary Fig. 43). Both systems exhibit a thinner CEI layer and markedly reduced fragment intensities, confirming the cross-system applicability of this approach.
Figure 4g, h present the X-ray absorption near-edge structure (XANES) of Ni in the pristine and FJ350 samples after 500 cycles. The Ni-K edge of cycled FJ350 nearly overlaps with that of the pristine sample, demonstrating improved stability in the oxidation state of nickel throughout the cycling process40,45,46,47. By contrast, a pronounced absorption edge shift toward higher energy is observed in the cycled pristine sample, implying incomplete reduction (i.e., irreversible Ni redox behavior) of the NCM811 material. This phenomenon can be attributed to the formation of the rock-salt-type NiO phase at the interface and within the bulk during cycling, which impedes Li+/electron transport and causes localized deactivation. And the underlying cause can be traced back to lattice oxygen loss45,46. Fourier-transformed extended X-ray absorption fine structures (FT-EXAFS) analysis at the Ni-K edge reveals that, after prolonged cycling, the Ni-O and Ni-transition metal (e.g., Co, Mn) coordination distances are noticeably shortened in the pristine sample (Fig. 4i), indicative of lattice contraction, accumulated structural stress, and oxygen vacancy formation. In contrast, the variation in interatomic distances in the cycled FJ350 electrode is nearly negligible, highlighting a highly stable local atomic coordination environment (Fig. 4j). Moreover, the wavelet-transformed EXAFS analysis in Fig. 4k and l reveals a substantial reduction in signal intensity in the high-K region for the pristine sample after 500 cycles, pointing to the degradation of Ni-transition metal coordination and increased structural disorder. In contrast, the cycled FJ350 (Fig. 4m, 4n) retains strong signals in this region, suggesting that the spinel coating layer effectively mitigates coordination degradation and preserves the long-range structural order of the material.
Density functional theory calculations
Based on the structural model presented in Supplementary Fig. 44 (Supplementary Data 1), we performed a comparative analysis of the partial density of states (PDOS) for both pristine LiTMO2 (TM = Ni0.8Co0.1Mn0.1), representing the bulk layered phase, and the spinel-coated LiTMO2@ LiTM2O4 under charged and discharged states48. The PDOS results reveal a notable increase in the electronic density near the Fermi level for the coated material, along with a narrowed band gap and a reduced electronic transition barrier (Fig. 5a). These electronic structure modifications suggest a marked enhancement in electrical conductivity, which is expected to improve the rate performance relative to the uncoated sample. To further understand the redox behavior and structural stability, we analyzed the crystal orbital Hamilton population (COHP) of Ni-O bonds (Supplementary Fig. 45). The integrated COHP (-ICOHP) values indicate that in the fully lithiated state, the Ni-O bonds in LiTMO2@LiTM2O4 possess a stronger bonding strength (-ICOHP = 0.803 eV) than those in pristine LiTMO2 (-ICOHP = 0.761 eV). In the delithiated state, both TMO2 and TMO2@TM2O4 show comparable Ni-O bond strengths. Given that LiTM2O4 generally retains some Li+ during cycling, the LiTMO2@LiTM2O4 heterostructure helps maintain robust bond stability throughout charge and discharge, thereby reinforcing the host framework and suppressing oxygen loss as well as phase degradation. This effect contributes to the inhibition of irreversible oxygen and nickel redox activity. In contrast, the pristine sample exhibits a pronounced decrease in the electronic density of states near the Fermi level under high degrees of delithiation, along with lower -ICOHP values in the lithiated state. These features imply inferior electronic transport and weaker Ni-O bonding during cycling, which can trigger oxygen release and structural collapse, ultimately resulting in rapid capacity fading. These theoretical insights are consistent with experimental observations, i.e., the spinel-coated LiTMO2@LiTM2O4 maintains high structural stability even under high-potential conditions, effectively suppressing oxygen release and irreversible phase transitions.

a PDOS of LiTMO2 with and without spinel coating under charged and discharged states (TM= Ni0.8Co0.1Mn0.1). b Volume expansion rate of the LiTMO2, LiTM2O4 and the LiTMO2@LiTM2O4 composites. c Interlocking ‘mortise-and-tenon’ architecture formed at the LiTMO2/LiTM2O4 interface. Green, light gray, blue, yellow, and red spheres represent Li, Ni, Co, Mn, and O, respectively. The same color code applies to all subsequent panels. d Average Li-ion adsorption energies in LiTM2O4 and LiTMO2@LiTM2O4 under different lithiation states. e Diffusion energy barriers of Li+ in LiTMO2 and LiTMO2@LiTM2O4 under different lithiation states. f, g Energy barriers and diffusion pathways of Li+ at the LiTMO2@LiTM2O4 interface. h Comparison of Fermi levels and work functions (the inset) of TMO2 and TM2O4. i, j Surface oxygen evolution and corresponding atomic strain mapping induced by Li+ intercalation/deintercalation in TMO2 layered structures. k, l Surface oxygen evolution and corresponding atomic strain mapping induced by Li-ion intercalation/deintercalation in TM2O4 spinel structures.
During the lithiation/delithiation process, the system expansion rates of LiTMO2 and LiTM2O4 reach 10.6% and 9.0%, respectively. However, after composite treatment, the volume change of the material significantly reduces to 3.5% (Fig. 5b). This improvement can be attributed to the strong covalent interaction of Ni-O bonds at the spinel/LiTMO2 interface, effectively suppressing the volume effects induced by lithium ions. Notably, after coating formation, the lattice of LiTMO2 expands while that of LiTM2O4 contracts (Fig. 5c). This mortise-and-tenon like locking mechanism significantly reduces lattice distortion during cycling, providing reliable structural support for enhancing the cycling life of the positive material. This is consistent with our experimental observation that FJ350 exhibits significantly lower crystal volume change (Fig. 3g) and higher structural stability (Fig. 4b).
Based on the analysis of average lithium ion adsorption energy in LiTMO2 and LiTM2O4, the spinel phase exhibits a significantly stronger adsorption capability than LiTMO2 throughout the charge/discharge process, especially under lithium-rich conditions (Fig. 5d). This enhanced adsorption behavior is closely related to Li⁺ diffusion kinetics. In line with established studies on layered oxide electrodes49,50, we employed the climbing image nudged elastic band method (CI-NEB) method to evaluate the tetrahedral site hop (TSH) mechanism, which is the dominant Li+ diffusion pathway characterized by a relatively low activation energy. The computed diffusion barrier for TMO2 is approximately 0.3 eV, consistent with reported values. For clarity, schematic illustrations of the TSH pathway from both side and top views are provided. As shown in Fig. 5e, in the high-delithiation state, the LiTMO2 region in the coated sample shows improved lithium adsorption compared to the uncoated material. However, when the spinel coating is in a lithium-rich state, this enhancement diminishes with increasing lithium content. This implies that an excessively thick LiTMO2 outer layer could impede Li+ transport into the bulk and weaken the adsorption contribution from LiTMO2. Therefore, an optimal thickness of the spinel coating is essential to enhance lithium adsorption (Fig. 5d) while establishing an efficient Li+ diffusion channel (Fig. 5e). Moreover, Li+ diffusion across the coating interface exhibits strongly asymmetric energy barriers (Fig. 5f, g), suggesting that extraction of Li⁺ from LiTMO2 is energetically more favorable than insertion. Hence, rational design of the thickness and microstructure of the LiTM2O4 spinel layer is critical to improve structural stability while minimizing adverse effects on lithium-ion transport kinetics. These findings are consistent with the high ICE, rate capability, and cycling performance observed in sample FJ350, which possesses an optimally tuned spinel-phase thickness.
Another significant advantage of spinel LiTM2O4 coating on LiTMO2 lies in its enhancement of surface structural stability. As shown in Fig. 5h, TM2O4 exhibits a lower Fermi level and higher work function compared to TMO2, indicating that electrons reside at lower energy levels and are less likely to be lost, thereby reducing the probability of adverse side reactions. By comparing the behavior of lithium ions at different concentrations entering LiTM2O4 and LiTMO2 (Fig. 5i and 5k, and Supplementary Fig. 46a), the outer and subouter layer oxygen atoms of LiTMO2 lose stability, detach from the lattice, and form clusters with moving lithium ions, which becomes more pronounced with increasing lithium ion concentration. This may increase the risk of forming active oxygen species during charge/discharge processes, leading to crystal structure instability, oxygen evolution, phase transitions, and aggravated side reactions with the electrolyte, ultimately resulting in significant capacity reduction and cycle performance deterioration. In comparison, the surface and subsurface oxygen atoms of spinel LiTM2O4 exhibit structural stability under both low and high lithium concentrations. Particularly at low lithium concentrations, the surface oxygen atoms only display lattice distortion (Fig. 5j, l). Thus, the spinel LiTM2O4 coating (Supplementary Fig. 46b) effectively enhances LiTMO2 stability by inhibiting oxygen release and passivating active sites. Moreover, the stable surface can avoid adverse interactions with the electrolyte, significantly improving the overall stability of the electrode material, further demonstrating its potential as a high-performance positive electrode material.
Discussion
In summary, we have presented a subtractive strategy for engineering a spinel-phase skin on high-nickel NCM electrodes, enabling significant improvements in electrochemical performance and structural stability. With the aid of a self-built flash Joule heating system, surface lithium atoms are precisely and quantitatively stripped under optimized thermal pulse conditions, enabling the in situ formation of a customized spinel-phase shell with tunable thickness and complete coverage. The nanoscale spinel-phase skin forms a robust, mortise-and-tenon-like interconnection with the bulk phase, effectively isolating the NCM electrode from reacting with the electrolyte, suppressing the oxygen loss, and accommodating isotropic and high-rate Li+ extraction/insertion. It also provides a continuous electron-conducting network across the interface during cycling. As a result, the modified LiNi0.8Co0.1Mn0.1O2 electrode exhibits greatly suppressed lattice volume changes (4.63–2.85%), superior resistance to cracking and corrosion, and delivers a high ICE of 95.3% with 93.6% capacity retention after 500 cycles, significantly surpassing the pristine counterpart. Moreover, the optimally engineered positive electrode enables pouch cells to retain 80.1% of capacity after 2000 cycles, compared to 52.0% for the unmodified version. More encouragingly, this spinel-phase skin can also be expanded to NCM with even higher nickel content, e.g., LiNi0.9Co0.05Mn0.05O2 and LiNi0.825Co0.115Mn0.06O2. Our subtractive coating construction concept presents a disruptive and universal strategy that overcomes the limitations of conventional additive methods, offering a potential new pathway to enhance the durability and electrochemical performance of Ni-rich layered positive electrode materials.
Methods
Synthesis: All NCM positive electrode materials were used directly without further pre-treatment. Polycrystalline LiNi0.8Co0.1Mn0.1O2, polycrystalline LiNi0.825Co0.115Mn0.06O2, polycrystalline LiNi0.9Co0.05Mn0.05O2 and single-crystal LiNi0.8Co0.1Mn0.1O2, were purchased directly from Xiamen XTC Intelligent Equipment Technology Inc. ( > 99.5%, battery grade). Flash sintering was conducted using a self-built Joule heating system equipped with a temperature control system and a circulating cooling system. Prior to sintering, 0.5 ~ 2 g of the NCM sample was enclosed in the central cavity formed by two pieces of carbon felt (100 mm × 100 mm), with both ends of the felt connected to the positive and negative poles of the heating clamp. After sealing the reaction chamber, three vacuum cycles were performed to remove residual air and ensure an oxygen-free environment for subsequent treatment. The interfacial structure of the NCM materials was tuned by adjusting the treatment temperature and pulse duration. The reaction chamber was evacuated three times to remove residual air, ensuring an oxygen-free environment for the subsequent treatment. The customized modulation of the interfacial structure of the NCM material was primarily achieved by tuning the temperature and pulse width of the transient thermal pulses. The treatment temperatures ranged from 300 °C to 800 °C, with pulse durations from 15 to 300 s. For comparison, samples were also prepared and tested after conventional heating in a tube furnace at 350 °C for 0.5 to 3 h.
Characterization: The crystal structure of the samples was analyzed by X-ray diffraction (XRD, Thermo Scientific ARL EQUINOX 3500) with Cu-Kα radiation using a scanning rate of 10° min−1 and a step size of 0.02°. For in situ XRD, the slurry was coated onto carbon paper, dried in a vacuum oven, and then assembled into a specially designed Bruker chamber for testing and then the resulting data was fitted by Rietveld refinement (GSAS software). The morphology of the materials before and after cycling was characterized using scanning electron microscopy (SEM, Gemini SEM 500, Zeiss), with the samples directly mounted on conductive adhesive for testing and operated at a beam voltage of 15 kV. Oxygen vacancies were investigated by Electron Paramagnetic Resonance measurement (EPR, Bruker EMXplus-9.5/12), Measurements were performed in the X-band (9.830243 GHz) with a modulation amplitude of 1 G, microwave power of 2 mW and modulation frequency of 100 kHz. The conversion time was set to 60 ms. The interfacial microstructure of pristine, flash-sintered, charged, and cycled NCM samples was characterized using transmission electron microscopy (TEM, FEI Talos F200s) with a beam voltage of 200 kV. Additionally, in situ TEM heating was performed with a Gatan heating holder. The temperature was ramped from room temperature to 450 °C at a rate of 1 °C s−1, with measurements conducted at various intermediate temperatures. The sample was held at each temperature for approximately 10 min. In situ differential electrochemical mass spectrometry (DEMS, Hiden HPR-20) studies were investigated using a specialized cell that replaced the positive and negative button cell casings with an in situ cell. The positive and negative materials, electrolyte, gaskets, and shrapnel selected are identical to those of the button cell. To ensure gas generation during the charging process, each aluminium foil piece with a diameter of 14 mm was loaded with approximately 10 ~ 14 mg of active material. For post-cycling analyses, the cycled cells were disassembled and the electrodes were washed with dimethyl carbonate (DMC) and dried in an Ar-filled glovebox. Cross-sectional samples of the cycled particles were prepared using a triple ion beam cutting machine (Leica EM TIC 3X) and subsequently examined by SEM to examine the internal structure, as well as time-of-flight secondary ion mass spectroscopy (TOF-SIMS, IONTOF M6) to investigate the distribution of 7Li. The thermal stability of the positive electrode material was assessed using differential scanning calorimetry (DSC, STA 409 PC, NETZSCH) with a heating speed of 5 °C min−1 under an Ar atmosphere. The cycled electrodes surface chemical compositions were identified by TOF-SIMS using a Bi3+ primary ion source operated at 30 keV with a beam current of 0.5 pA. X-ray photoelectron spectroscopy with depth profiling (XPS, Thermo Fisher, Escalab Xi + ) was performed using Ar⁺ ion sputtering to etch the electrode surface with a sputtering energy of 2000 eV, a sputtering rate of 0.29 nm s−1 and sputtering depths of 0, 5, 10 and 15 nm. The cycled electrodes were handled in an argon-filled glovebox due to their air sensitivity. After electrochemical cycling, the coin cells were disassembled, and the electrodes were rinsed twice with dimethyl carbonate to remove residual electrolyte. The cleaned electrodes were then dried under an inert atmosphere prior to analysis.
Electrochemical measurements: The NCM positive electrodes were fabricated using a composite material consisting of 90 wt.% active material, 5 wt.% Super P (battery grade, Shenzhen Kejing Co., Ltd.) and 5 wt.% poly(vinylidene fluoride) (PVDF, 99.9%, Sigma-Aldrich) dissolved in N-methyl-2-pyrroidone (99.9%, Canrd Technology Co., Ltd.). The obtained slurry was casted using an automatic coating machine (MSK-AFA-SC200, Shenzhen Kejing Co., Ltd.) onto carbon-coated aluminum foil (thickness of 15 μm, Shenzhen Kejing Co., Ltd.) and dried at 120 °C for 4 h in a vacuum oven. The electrodes were cut using electrode punching machine (MSK-T10, Shenzhen Kejing Co., Ltd.) into discs with a diameter of 11 mm, and the mass loading of the active material was approximately 3.5 mg cm−2. When assembling CR2032-type half cells (316 stainless steel), lithium metal foil (thickness: 1 mm, diameter: 15.8 mm, 99.9%, China Energy Lithium Co., Ltd.) was used as the negative electrode and Celgard 2500 as the separator (thickness: 25 μm, diameter: 19 mm, Canrd Technology Co., Ltd.). The spacer (thickness: 1 mm, diameter: 15.8 mm, Dodochem Technology Co., Ltd.) and spring (thickness: 1.1 mm, diameter: 15.4 mm, Dodochem Technology Co., Ltd.) are made of 316 stainless steel. The electrolyte (Dodochem Technology Co., Ltd.) was composed of 1 M LiPF6 dissolved in ethylene carbonate (EC) and diethyl carbonate (DEC) with a volume ratio of 3:7 and with 2 wt.% vinylene carbonate addition. The electrolyte amount used in the coin cells was 60 µL. Half cells were tested using the Land battery testing system (Wuhan Lanhe Electronics, CT200A) under standard conditions of 2.7–4.3 V at 26 °C, as well as under harsher conditions such as 4.5 V and 60 °C with temperature-controlled incubator. 3-5 batteries were tested in parallel, and their performances are highly consistent. For pouch-cell tests, the negative electrode was composed of 95 wt.% graphite, 1.2 wt.% Super P, 1.8 wt.% sodium carboxymethyl cellulose (99.5%, Shenzhen Kejing Co., Ltd.) and 2 wt.% styrene butadiene rubber (50%, Shenzhen Kejing Co., Ltd.) and then coated onto copper foil (thickness of 4.5 μm, Shenzhen Kejing Co., Ltd.). The negative-to-positive capacity ratio (N/P ratio) for the pouch cell is around 1.12. The electrolyte injection procedure is as follows: The electrolyte is injected with an electrolyte-to-capacity ratio (E/C) of 3.5, which refers to the ratio of the electrolyte weight to the cell capacity (g Ah−1). After injection, the pouch cell was sealed under vacuum and then soaked at 45 °C for 48 h, followed by a formation cycle. After formation, the cell is aged at 45 °C for 24 h. The gas in the pouch is then vented by cutting the gas bag and the cell was resealed under vacuum. Finally, the cell undergoes partial capacity measurement, followed by subsequent electrochemical testing. The pouch cell consisted of 4 layers and full cells ( ~ 200 mAh) were tested between 2.7 and 4.2 V at 1 C (1 C = 180 mA g−1) using the Neware battery testing system (CT-4008T, Neware Technology Co., Ltd.) under an external pressure by gently clamping the cell with a pressure of 37.2 kPa. Pouch-cell cycling tests were performed under ambient conditions without temperature control. Cyclic voltammetry (CV) tests were carried out at scan rates of 0.2, 0.4, 0.6, and 0.8 mV s−1 within a potential window of 2.7–4.5 V using an electrochemical workstation (CHI 660E, Shanghai Chenhua). Galvanostatic intermittent titration technique (GITT) measurements were performed with a constant current for 600 s with data recorded every 10 s followed by 1800 s relaxation with the upper potential of 4.3 V and lower potential of 2.7 V, respectively. Electrochemical impedance spectroscopy (EIS) was conducted using an electrochemical workstation. The measurements were performed after two cycles at 0.1 C and at the 1st, 100th, 200th, 300th, 400th and 500th cycles at 1 C. Prior to EIS analysis, the cells were charged to 4.3 V under constant-potential mode for 3 h, and then impedance spectra were collected over a frequency range of 0.01 Hz to 105 Hz with a perturbation amplitude of 5 mV.
Theoretical methods: All spin-polarized calculations were conducted by using density functional theory (DFT) as implemented in the Vienna Ab initio Simulation Package51,52,53 (VASP), with the projector-augmented wave method54 (PAW) escribing the ion-electron interaction. The valence electrons were taken into account using a plane wave basis set with a kinetic energy cut-off of 520 eV, which guarantees that the total energy was converged when it reached 1 meV per atom. The exchange-correlation interaction of electrons was treated within the generalized gradient approximation (GGA) with Perdew-Burke-Ernzerhof55,56,57 (PBE) functional. Convergence thresholds of 10-6 eV for electronic steps and 10−2 eV Å−1 for ionic relaxation were employed. The van der Waals interaction was considered by the empirical correction of Grimme’s scheme (DFT-D3). The reciprocal space was sampled using a gamma-centered k-grid with a resolution of 0.04 in units of 2π Å−1. For single-point energy calculations, the k-grid resolution was refined to 0.02. Minimum energy pathways were calculated using the CI-NEB58,59. The path threshold for CI-NEB calculations was set to 0.02 eV Å-1, and the insertion points were determined by the displacement of the diffusion atom. The von Mises local shear was calculated to characterize the degree of distortion of surface atoms. Regarding our interface model, the spinel phase formed through in situ transformation maintains crystallographic continuity with the parent layered structure, particularly along the basal plane direction. We focused on the 4-coordinated facets as model surfaces since they represent vulnerable sites where phase transition likely initiates. Stabilizing these critical interfaces through our approach provides substantial improvement in overall performance. The interface model was constructed as a symmetric slab without vacuum layers60,61, with thick atomic regions to bulk-like conditions. Full geometry optimization was performed without constraints to minimize artificial strain. Li-deficient structures were generated using the SQS method62 to approximate random vacancy distributions, with consistent sampling applied for all comparative studies. The adsorption energy of Li+ (Eads) is defined as:
where \({E}_{{{{\rm{total}}}}}\), \({E}_{{{{\rm{TM}}}}{{{{\rm{O}}}}}_{2}/{{{{\rm{TM}}}}}_{2}{{{{\rm{O}}}}}_{4}}\), and \({E}_{\,{{{\rm{Li}}}}}\) denote the total energy of TMO2 or TM2O4 with n Li+, the energy of TMO2 or TM2O4, and the energy of one Li atom, respectively.
Data availability
All data generated or analyzed during this study are included in the published article and its Supplementary Information. Additional data are available from the corresponding authors on request. Source data are provided with this paper.
References
Li, W., Erickson, E. M. & Manthiram, A. High-nickel layered oxide cathodes for lithium-based automotive batteries. Nat. Energy 5, 26–34 (2020).
Liu, J. et al. Pathways for practical high-energy long-cycling lithium metal batteries. Nat. Energy 4, 180–186 (2019).
Xu, C. et al. Bulk fatigue induced by surface reconstruction in layered Ni-rich cathodes for Li-ion batteries. Nat. Mater. 20, 84–92 (2021).
Jiao, S., Wang, J., Hu, Y.-S., Yu, X. & Li, H. High-capacity oxide cathode beyond 300 mAh/g. ACS Energy Lett. 8, 3025–3037 (2023).
Ryu, H.-H., Park, K.-J., Yoon, C. S. & Sun, Y.-K. Capacity fading of Ni-Rich Li[NixCoyMn1–x–y]O2 (0.6≤x≤0.95) cathodes for high-energy-density lithium-ion batteries: bulk or surface degradation? Chem. Mater. 30, 1155–1163 (2018).
Yan, P. et al. Intragranular cracking as a critical barrier for high-voltage usage of layer-structured cathode for lithium-ion batteries. Nat. Commun. 8, 14101 (2017).
Lin, F. et al. Surface reconstruction and chemical evolution of stoichiometric layered cathode materials for lithium-ion batteries. Nat. Commun. 5, 3529 (2014).
Li, W., Asl, H. Y., Xie, Q. & Manthiram, A. Collapse of LiNi1-x-yCoxMnyO2 lattice at deep charge irrespective of nickel content in lithium-ion batteries. J. Am. Chem. Soc. 141, 5097–5101 (2019).
Li, S. et al. Mutual modulation between surface chemistry and bulk microstructure within secondary particles of nickel-rich layered oxides. Nat. Commun. 11, 4433 (2020).
Wang, L. et al. Reaction inhomogeneity coupling with metal rearrangement triggers electrochemical degradation in lithium-rich layered cathode. Nat. Commun. 12, 5370 (2021).
Xu, C. et al. Operando visualization of kinetically induced lithium heterogeneities in single-particle layered Ni-rich cathodes. Joule 6, 2535–2546 (2022).
Yu, R. et al. Layer-by-layer delithiation during lattice collapse as the origin of planar gliding and microcracking in Ni-rich cathodes. Cell Rep. Phys. Sci. 4, 101480 (2023).
Lin, F., Zhao, K. & Liu, Y. Heterogeneous reaction activities and statistical characteristics of particle cracking in battery electrodes. ACS Energy Lett. 6, 4065–4070 (2021).
Wei, Z. et al. In-depth study on diffusion of oxygen vacancies in Li(NixCoyMnz)O2 cathode materials under thermal induction. Energy Storage Mater. 47, 51–60 (2022).
Meng, X.-H. et al. Kinetic origin of planar gliding in single-crystalline Ni-rich cathodes. J. Am. Chem. Soc. 144, 11338–11347 (2022).
Li, J. et al. Dynamics of particle network in composite battery cathodes. Science 376, 517–521 (2022).
Zhao, C. et al. Suppressing strain propagation in ultrahigh-Ni cathodes during fast charging via epitaxial entropy-assisted coating. Nat. Energy 9, 345–356 (2024).
Lu, S.-Q. et al. Surface lattice modulation through chemical delithiation toward a stable nickel-rich layered oxide cathode. J. Am. Chem. Soc. 145, 7397–7407 (2023).
Cheng, J. et al. Improving intrinsic safety of Ni-rich layered oxide cathode by modulating its electronic surface state. Energy Storage Mater. 79, 104332 (2025).
Wang, R. et al. Inhibiting phase conversion and improving cyclic stability of Ni-rich layered oxide by high-valence element concentration gradient doping. Chem. Eng. J. 485, 149827 (2024).
Wang, L., Liu, T., Wu, T. & Lu, J. Strain-retardant coherent perovskite phase stabilized Ni-rich cathode. Nature 611, 61–67 (2022).
Ryu, H.-H., Lim, H.-W., Lee, S. G. & Sun, Y.-K. Near-surface reconstruction in Ni-rich layered cathodes for high-performance lithium-ion batteries. Nat. Energy 9, 47–56 (2024).
Liu, W. et al. Functional passivation interface of LiNi0.8Co0.1Mn0.1O2 toward superior lithium storage. Adv. Funct. Mater. 31, 2008301 (2021).
Yoon, M. et al. Reactive boride infusion stabilizes Ni-rich cathodes for lithium-ion batteries. Nat. Energy 6, 362–371 (2021).
Guo, W. et al. In situ surface engineering enables high interface stability and rapid reaction kinetics for Ni-rich cathodes. eScience 3, 100082 (2023).
Tian, Y. et al. Regulation of interface ion transport by electron ionic conductor construction toward high-voltage and high-rate LiNi0.5Co0.2Mn0.3O2 cathodes in lithium ion battery. Adv. Sci. 11, 2402380 (2024).
Yao, J. et al. Scalable precise nanofilm coating and gradient al doping enable stable battery cycling of LiCoO2 at 4.7 V. Angew. Chem. Int. Ed. Engl. 63, e202407898 (2024).
Guo, H.-J. et al. Surface degradation of single-crystalline Ni-rich cathode and regulation mechanism by atomic layer deposition in solid-state lithium batteries. Angew. Chem. Int. Ed. Engl. 61, e202211626 (2022).
Zhang, C. et al. Quenching-etched surface spinel to passivate layered cathode materials from structural degradation at high potentials. Chem. Mater. 35, 6692–6701 (2023).
Gan, Q. et al. Surface spinel reconstruction to suppress detrimental phase transition for stable LiNi0.8Co0.1Mn0.1O2 cathodes. Nano. Res. 16, 513–520 (2023).
Wang, K. et al. Unraveling the role of surficial oxygen vacancies in stabilizing Li-rich layered oxides. Adv. Energy Mater. 13, 2301216 (2023).
Qi, S. et al. A pre-fatigue training strategy to stabilize LiCoO2 at high voltage. Energy Environ. Sci. 17, 2269–2278 (2024).
Cheng, X. et al. Pre-deoxidation of layered Ni-rich cathodes to construct a stable interface with electrolyte for long cycling life. Adv. Funct. Mater. 33, 2211171 (2023).
Hao, Z. et al. Suppressing bulk strain and surface O2 release in Li-rich cathodes by just tuning the Li content. Adv. Mater. 36, 2307617 (2024).
Liang, C. et al. Insight into the structural evolution and thermal behavior of LiNi0.8Co0.1Mn0.1O2 cathode under deep charge. J. Energy Chem. 65, 424–432 (2022).
Lee, S.-B. et al. Doping strategy in developing Ni-rich cathodes for high-performance lithium-ion batteries. ACS Energy Lett. 9, 740–747 (2024).
Xu, C., Reeves, P. J., Jacquet, Q. & Grey, C. P. Phase behavior during electrochemical cycling of Ni-rich cathode materials for Li-ion batteries. Adv. Energy Mater. 11, 2003404 (2021).
Liu, X. et al. Origin and regulation of oxygen redox instability in high-voltage battery cathodes. Nat. Energy 7, 808–817 (2022).
Wang, W. et al. Optimized in situ doping strategy stabling single-crystal ultrahigh-nickel layered cathode materials. ACS Nano 18, 8002–8016 (2024).
Dai, Z., Li, Z., Chen, R., Wu, F. & Li, L. Defective oxygen inert phase stabilized high-voltage nickel-rich cathode for high-energy lithium-ion batteries. Nat. Commun. 14, 8087 (2023).
Shi, X. et al. Achieving high safety for lithium-ion batteries by optimizing electron and phonon transport. ACS Energy Lett. 8, 4540–4546 (2023).
Zhang, B. et al. Manipulated fluoro-ether derived nucleophilic decomposition products for mitigating polarization-induced capacity loss in Li-rich layered cathode. Angew. Chem. Int. Ed. Engl. 63, e202316790 (2024).
Zhuang, Z. et al. Ultrahigh-Voltage LiCO2 at 4.7 V by interface stabilization and band structure modification. Adv. Mater. 35, 2212059 (2023).
Ryu, H.-H., Lim, H.-W., Kang, G.-C., Park, N.-Y. & Sun, Y.-K. Long-lasting Ni-rich NCMA cathodes via simultaneous microstructural refinement and surface modification. ACS Energy Lett. 8, 1354–1361 (2023).
Zhang, R. et al. Compositionally complex doping for zero-strain zero-cobalt layered cathodes. Nature 610, 67–75 (2022).
Zhang, R. et al. Long-life lithium-ion batteries realized by low-Ni, Co-free cathode chemistry. Nat. Energy 8, 695–702 (2023).
Wang, Z. et al. Contact-electro-catalysis for the degradation of organic pollutants using pristine dielectric powders. Nat. Commun. 13, 130 (2022).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Cryst. 44, 1272–1276 (2011).
Kang, K., Morgan, D. & Ceder, G. First principles study of Li diffusion in I-Li2NiO2 structure. Phys. Rev. B 79, 014305 (2009).
Zhou, Z. et al. First-Principles study on the interplay of strain and state-of-charge with Li-ion diffusion in the battery cathode material LiCoO2. ACS Appl. Mater. Interfaces 15, 53614–53622 (2023).
Kresse, G. Ab initio molecular dynamics for liquid metals. J. Non-Cryst. Solids 192, 222–229 (1995).
Kresse, G. & Hafner, J. Ab initiomolecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Olsson, E., Chai, G., Dove, M. & Cai, Q. Adsorption and migration of alkali metals (Li, Na, and K) on pristine and defective graphene surfaces. Nanoscale 11, 5274–5284 (2019).
Wang, Z. et al. In-situ formed Co nano-clusters as separator modifier and catalyst to regulate the film-like growth of Li and promote the cycling stability of lithium metal batteries. J. Colloid Interface Sci. 660, 226–234 (2024).
Behler, J. Atom-centered symmetry functions for constructing high-dimensional neural network potentials. J. Chem. Phys. 134, 074106 (2011).
Zhou, Z. et al. Tuning the electronic, ion transport, and stability properties of Li-rich manganese-based oxide materials with oxide perovskite coatings: A first-principles computational study. ACS Appl. Mater. Interfaces 14, 37009–37018 (2022).
Zhou, Z. et al. LiNbO3 and LiTaO3 coating effects on the interface of the LiCoO2 cathode: A DFT study of Li-ion transport. ACS Appl. Mater. Interfaces 16, 42093–42099 (2024).
Van de Walle, A. et al. Efficient stochastic generation of special quasirandom structures. Calphad 42, 13–18 (2013).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. 92372101-L.Z., U25A20237-Q.Z., 92472104-Q.Z., 92472203-L.Z.), National Key Research and Development Program of China (grant 2024YFE0209300-Q.Z.), the Fundamental Research Funds for the Central Universities (20720220010-L.Z., 20720230036-J.H.), the National Key Research and Development Program of China (2021YFA1201502-L.Z.), Fujian Provincial Natural Science Foundation of China (2024J01038-J.H.). L. Zhang and Q.B. Zhang acknowledge the support of Nanqiang Young Top-notch Talent Fellowship in Xiamen University. H.P. Yang, Z.F. Sun and Y.H. Zhao contribute equally to this work.
Author information
Authors and Affiliations
Contributions
L. Zhang and Q.B. Zhang conceived the idea and supervised the experiments. H.P. Yang and Y.H. Zhao synthesized the materials and wrote the paper. Z.F. Sun conducted the HRTEM experiments and electrochemical testing. Z.Y. Sun and W.X. Chen conducted the XANES experiments. H.L. Yi, Y. Zhang and J.J. Liu conducted the TOF-SIMS and XPS experiments. H.Q. Wang and Y.X. Mao conducted the in situ XRD and SEM experiments. J.J. Han conducted the theoretical calculations. J.X. Wang, Q.H. Zhao, S.J. Feng and Y. Cao assisted in revising the paper. All the authors discussed and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yi Cheng, Kwangjin Park and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, H., Sun, Z., Zhao, Y. et al. Flash joule heating-induced spinel-phase surface in Ni-rich layered oxide positive electrodes to stabilise lattice oxygen. Nat Commun 17, 4008 (2026). https://doi.org/10.1038/s41467-026-70616-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70616-8
