
Abstract
Chemical innovation is essential for fungi to adapt to ever-changing ecological environments. However, the environmental and evolutionary drivers of fungal metabolic differentiation remain ambiguous. Here, we show the phylogeographic diversity of 1052 Aspergillus flavus strains across four continents, as conducted through phylogenetic and biogeographical analysis, including 544 newly sequenced strains from China. These strains exhibit varying levels of population-specific mycotoxin production, as determined by population metabolomics analysis. We report a toxigenic subpopulation from China, identified through comparative population genomics analysis. Pan-metabolome analysis reveals strong phylogeographic metabolic patterns associated with specific ecological niches. Low-mycotoxin production clades harbor distinct uncharacterized biosynthetic gene clusters and produce different specialized metabolites instead. This discrepancy is only partially explained by variation in biosynthetic pathway genes, and changes in regulation and primary metabolism appear to mainly drive differentiation of specialized metabolite profiles across fungal populations, as indicated by pangenome profiling, metabolites-genome-wide association study, genotype-environment association study, pan-transcriptome analysis, and gene knockout experiments. Altogether, our results reveal how environmental shifts drive the fungal metabolic evolution, and provide insights for predicting the risk of harmful fungal outbreaks and for biogeographically-informed, precise control measures.
Similar content being viewed by others

Introduction
Fungal specialized metabolism has a profound impact on the Earth’s ecosystem1, especially, since the world is currently experiencing rapid anthropomorphic climate change, which is increasing the risk of harmful fungal outbreaks that infect humans, destroy crops or poison foods with their toxic specialized metabolites2. Climate shifts promote fungal evolution, altering host-pathogen interactions and contributing to the emergence of new pathogenic strains3. Fungi are well-known for their capacity to produce a wide range of bioactive compounds. These molecules possess important ecological functions in inter-microbial competition, defense, signaling, nutrient acquisition, and development4. Many fungi secrete mycotoxins to facilitate pathogenesis or to defend a specific ecological niche5. Unfortunately, the mycotoxins, such as aflatoxin B1, mainly produced by the A. flavus, is classified as a Class I carcinogen by the World Health Organization, may lead to poisoning events in humans due to their carcinogenicity. According to FAO data, ~ 25% of the world’s food is contaminated yearly by mycotoxins, causing large socioeconomic losses globally and posing a major challenge to food security and public health6. Moreover, A. flavus is also the second most common opportunistic pathogen causing aspergillosis. More than 2.113 million people suffer from aspergillosis every year worldwide, with a mortality rate of 85.2%7.
An improved understanding of phylogeographic diversity of harmful fungal specialized metabolism would inform the design of precision control strategies to mitigate the growing threat of fungal infection and mycotoxins. The extraordinary diversity of fungal specialized metabolites is maintained by evolutionary adaptations to challenges posed by complex environments8. Specialized metabolite repertoires have been shown to evolve rapidly through the functional divergence, horizontal transfer, and de novo assembly of biosynthetic gene clusters (BGCs) that encode for their production5. Recent computational genome mining efforts, including global analysis of 1000 fungal genomes9, investigation of inter- and intraspecies variation in Aspergillus section Nigri10, and a comparative genomics study of 23 Aspergillus species from section Flavi11 have shown that fungal biosynthetic diversity is vastly untapped. Even within individual fungal species, considerable variation in BGC repertoires exists12, and, sometimes, closely related pathogenic and non-pathogenic strains show distinct differences in their biosynthetic capabilities13. However, little is understood about the processes that shape the biogeographical diversity of fungal specialized metabolism within species under climatic shifts. The hypothesis is that the environment selects for diversification; yet, to what extent the environment really does select for specific specialized metabolic repertoires and their evolutionary direction, and how this is encoded genetically, has remained unclear. While recent key studies showed the existence of considerable pan-genomic diversity of accessory BGCs12,14 and population-specific differences in metabolite production15. Traditionally, BGC variation is considered to be the main driving force; however, other drivers, such as regulatory, primary metabolism and environmental selection of gene variation, have not been systematically studied. Therefore, a transcontinental phylogeographic analysis is required to systematically identify the major genetic and environmental drivers that lead to different metabolic profiles across populations around the globe.
Here, we select the ubiquitous species of broad societal impact, A. flavus, as a model system to analyze the evolutionary drivers of phylogeographic diversity of fungal specialized metabolism. We perform de novo genome sequencing of >550 representative isolates from China and also investigate these strains using untargeted eco-metabolomics; the genomics data were then analyzed together with 508 previously published environmental (n = 412) and clinical (n = 96) A. flavus genomes from nine other countries. We report an aflatoxigenic subpopulations that mainly reside in southern and central China. Strains from different populations exhibit conserved BGC repertoires, yet show phylogeographically distinct specialized metabolite production. Notably, low-aflatoxin clades tend to harbor clade-specific unknown BGCs and were found to produce different metabolites instead, including other mycotoxins, which may prompt a rethinking of food safety management strategies. To our surprise, these differences in metabolic output are only partially explained by polymorphisms of BGC genes, but mainly driven by evolutionary rewiring of key transcriptional regulation and primary metabolism. Genotype-environment association (GEA) analysis further indicates that environmental local adaptation promotes these processes. Altogether, our multi-omics approach reveals evolutionary drivers of the phylogenetic diversity of fungal specialized metabolites in the context of a changing environment.
Results
Multi-omics dataset across 1052 A. flavus strains
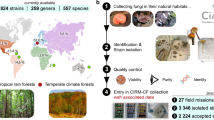
To facilitate dissecting the nature and origins of fungal specialized metabolic differentiation from a comprehensive overview of variation at the species level, 544 representative A. flavus and five Aspergillus section Flavi strains were selected from an in-house strain library from China. The chosen sampling sites represent six climatic ecological zones to maximize their ecological breadth and geographical origins (Fig. 1a and Supplementary Fig. S1a, and see for detailed strain sampling site coordinates in Supplementary Table 1). The analysis workflow of this study is shown in Supplementary Fig. S1b. Subsequently, these isolates were deep-sequenced using Illumina paired-end sequencing with a 97-fold mean sequencing depth and 99% mean mapping rate (Supplementary Fig. S2a). The reads of all isolates were de novo assembled for obtaining the whole genome sequence. The average genome size was 37.9 ± 2.2 Mb, with 47.5 ± 0.3% GC content and 2.6 ± 0.7% repetitive sequence content. In addition, 508 previously sequenced A. flavus and two Aspergillus section Flavi strains genomes were downloaded from NCBI GenBank (Supplementary Fig. S1a and Supplementary Table 1, 2), mainly from three studies: 95 of them originate from a study of USA populations15, 225 isolates from infected plant parts of corn or soil samples16, and 96 representive clinical samples mainly originating from a study by Hatmaker et al.17. The scaffold number of newly sequenced assemblies (median value = 109.5 ± 91.2) is significantly lower than the number (median value = 317 ± 653.9) of draft A. flavus genomes that have been published so far (Supplementary Fig. S2b and Supplementary Table 3), indicating that our genome assemblies are of high quality. We further selected 27 representative strains from a species phylogeny for PacBio long-read sequencing to obtain chromosome-level assemblies for the construction of high-quality pangenomes. The scaffold number (median value = 20.5 ± 4.9) of the 27 PacBio-sequenced strains was significantly lower than the Illumina assembly results (median value = 109.5 ± 91.2), near chromosome-level (Supplementary Fig. S2c, d). The HiC sequencing technology was further used to sequence 8 strains, which assisted in the assembly to the chromosome level. Most single-nucleotide polymorphisms (SNPs) are present at low frequencies, 92% with a minor allele frequency (MAF) < 0.3 (Supplementary Fig. S3a). Extended results on detailed genetic differentiation and diversity characterization, such as InDels (Insertion/Deletion, InDel), structural variation (SVs), and transposon variation in the population, are available in the Supplementary Information. Untargeted metabolomics profiles for >550 Aspergillus sp. isolates were simultaneously acquired by ultra-performance liquid chromatography coupled with Orbitrap Fusion high-resolution mass spectrometry using data-dependent acquisition (DDA) of mass fragmentation spectra (UPLC-HRMS/MS) (see “Methods”). All metabolomes were annotated in detail using a range of computational metabolomics procedures that utilize spectral alignment, spectral similarity networking, and de novo structure prediction(see “Methods”). The raw metabolomic data of 95 strains from the USA generated by Drott et al.14 were downloaded from GNPS. In addition, 28 strains (with three biological replicates each and a total of 84 samples) from different clades with different phenotypes were selected to obtain species-level pan-transcriptome variation profiles. Finally, to conduct genotype-environment association (GEA) analysis, we collect the data of 21 climate variables and two geographical factors from the China weather data website (https://data.cma.cn/) and soil survey data of 24 soil physiological metrics from our lab.

a The population structures and phylogeographic patterns of A. flavus at the transcontinental scale and different environmental sources. The correspondence with previously defined populations is indicated. b Comparison of the latitudinal distribution of different subpopulations. W-clades 4, 5, 6, and 7 consist mainly of aflatoxigenic strains and are distributed in lower latitudes. The latitudinal distribution of the non-aflatoxigenic strain subpopulation was significantly higher than that of the aflatoxigenic strains. W-clades 4, 5 subpopulation (c) Genetic diversity (π value) of clades and genetic differentiation (Fst value) between paired subpopulations. d Distribution of copy number of virulence gene 1 (Afu5g03790) in A. flavus strains from environmental and clinical sample sources.
Discovery of an aflatoxigenic subpopulations from China
To provide a strong foundation for transcontinental-scale phylogeographic analysis, we reconstruct a high-resolution phylogenetic tree from all 1059 genomes (including seven outgroups) using a genome-wide SNP data matrix (including 387,282 SNPs). This facilitates the study of phylogenetic relationships of strains isolated from different geographical origins and niches (Fig. 1a and Supplementary Table 1). The phylogenetic analysis of A. flavus on a transcontinental scale is genetically differentiated into eight subpopulations. W-clade 6 was discovered and contains high-frequency isolates with aflatoxin-producing capacity(APC) sampled mainly from China (Fig. 1a). Previously, Geiser et al.18 constructed a phylogenetic tree of USA A. flavus isolates using three marker genes and divided the population into two groups (I, II). Drott et al.15 recently reported that the USA A. flavus population is subdivided into three genetically differentiated subpopulations (Pops) (Pop A, B, and C) at genome-wide resolution. The study of Hatmaker et al.17, recently inferred that the A. flavus population could be grouped into five populations: Pop A, B, C, D, and S-type, which were also identified in our phylogeny. However, our study at the largest scale to date revealed that of which Pop A was further differentiated into four subpopulations (Pop A1, A2, A3, A4) (Fig. 1a and Supplementary Fig. S4). Specifically, Pop B is synonymous with the W-clade 2 found in this study, and Pop C (containing a mix of clinical and environmental samples) was the same as our W-clade 3. Pop D, equivalent to W-Clade 8, contains the vast majority of clinical samples, as shown by Hatmaker et al. Pop A mainly overlapped with the W-clades 4,5,6,7 of this study17. Notably, 95% of the strains in W-clade 6 were from southern and central China, and only five strains were from the USA, which may result from long-distance migration or geographical spread by atmosphere, with 70% of the strains in W-clade 6 having an aflatoxigenic chemotype. We further construct the neighbor-net network to cross-validate the reliability and accuracy of population structure inference (Supplementary Fig. S4a). The network topology is highly similar to the phylogenetic tree, and the W-clades 4 and 5 strains from China also differentiated into the branches. Principal component analysis (Supplementary Fig. S4b) and population structure inference (Supplementary Fig. S4c) confirm the rationale of dividing the A. flavus population into eight subpopulations. Notably, 94% of the geographical locations of the strains of these clades were from southern and central China in the subtropical or mid-subtropical regions at low latitudes (Fig. 1b). We further characterize the genetic diversity and degree of genetic differentiation among different subpopulations. W-clade 6 has high genetic diversity(π = 0.173, p = 0.01). In contrast, W-clade 2 (also known as Pop B) has the lowest genetic diversity (Fig. 1c). There is a large genetic differentiation (Fst > 0.25) between W-clade 2 and other clades, including W-clade 6 (Fig. 1c). The clades show moderate to large genetic divergence from the other clades (Fig. 1c). In conclusion, this combined transcontinentally sampled whole-genome phylogeny reveals fine-scale population structure and genetic differentiation across the A. flavus species.
Phylogeographic Diversity Patterns of A. flavus
The genetic diversity of A. flavus shows strong associations with the environmental origins of the strains at a continental scale. Phylogenetic analysis reveals that clinical and environmental samples were clearly clustered, showing genetic differentiation of ecological niches. For example, 51% of clinical samples are distributed in W-clade 8, 20% in W-clade 3, 22% in W-clade 5, and the remaining 7% are scattered in other subpopulations. This pattern is consistent with a recent study on the pathogenicity of A. flavus samples from clinical settings17. On the other hand, it is worth noting that 65% of environmental strains from China in W-clade 8 were phylogenetically interspersed with clinical strains, suggesting that environmental non-aflatoxigenic strains of this subpopulation may have the ability to infect humans19. For example, we observe that the genomes of these clinical A. flavus strains (W-clades 3,8) harbored higher copy numbers of homolog virulence gene 1 (Afu5g03790, Iron transport multicopper oxidase fetC) after large-scale homology clustering comparison of 1003 A. flavus genomes and 149 virulence gene protein sequences from A. fumigatus strains that have been reported to infect humans19 (Fig. 1a, d). In addition, the homolog virulence gene 2 (Afu8g00230, Chain A, Verruculogen synthase) is mainly distributed in W-Clade 2 environmental non-aflatoxigenic clade strains. However, overall, the vast majority of the virulence genes were found in both clinical and environmental strains, and the phenomenon of homologs of virulence genes over-represented in clinical strains versus environmental strains was not widespread (Supplementary Fig. S5a, b). In environmental samples, sclerotial size S (W-clade 1) and L morphotypes (W-clades 2-8) with different aflatoxin profiles and niche adaptability characteristics were also clearly classified (Fig. 1a). The phylogenetic relationships show that phylogeographically distinct clades of A. flavus show clear differences in aflatoxin profiles (Fig. 1a). Analyzing from a transcontinental scale, the proportion of non-aflatoxigenic subpopulation strains in W-clades 2 and 8 exceeds 50%. In contrast, W-clades 4, 5, 6, and 7 are dominated by aflatoxigenic strains (Fig. 1a, b). The results show a clear latitudinal gradient pattern, that is, the aflatoxin-producing strains were frequently found in low-latitude areas, whereas the non-aflatoxigenic strains are enriched at higher latitudes both within and between clades (Fig. 1b). Analyzing strains from China alone, 84% aflatoxigenic strains originate from the south subtropical climate zones (Fig. 2a). 62% of the strains isolated from middle subtropical regions had medium or high aflatoxin-producing capacity.

a Phylogenetic tree, field sampling sites and phylogeographic patterns of A. flavus strains from China, and the geographical distribution pattern of aflatoxin-producing capacity. 97% nodes in the phylogenetic tree have a bootstrap value greater than 0.98. b The phylogenetic tree from the population of A. flavus in China. The heatmap contains the five A. flavus metabolic patterns mapping on the phylogenetic tree. Low aflatoxin-producing capacity clades often produce other metabolites instead. c Cyclopiazonic acid (CPA) molecular family mainly found in clades 1 and 3 (see Online Methods for metabolite annotation details). d The highest abundance of the aflatoxin molecular family was observed in Chinese clades 2, 4, 5, and 6, and the lowest abundance was observed in Chinese clades 1 and 3. The geographic information data of China’s map and major climate zones used in the Fig. 2a map comes from the environmental resources and environmental science data platform of the Chinese Academy of Sciences (https://www.resdc.cn). Source data are provided in this paper.
In addition to aflatoxins, pan-metabolome analysis further reveals that phylogeographically distinct clades show extensive metabolic differentiation. Since it is difficult to simultaneously obtain consistent metabolome data from other locations using the same culture conditions and comparable metabolome methods, we used 551 samples from China as a case dataset to discover patterns. A total of 16,432 metabolic features were extracted from 551 metabolome profiles. We find that 36.5% (5996/16,432) core metabolome features were shared between all isolates. We also observe the presence of many clade-specific metabolites. For example, W-clade 4 and W-clade 5 possess 11.3% (1861/16,432) and 4.6% (762/16,432) clade-specific metabolite features, respectively (Supplementary Fig. S6a). Five largely discrete metabolic patterns could be distinguished following differential abundance analysis between different sets of clades based on annotated metabolites (Fig. 2b). We use feature-based molecular networking (FBMN) to organize our LC-HRMS/MS data by grouping mass features with similar mass fragmentation spectra20, accompanied by extensive metabolite annotation efforts using various (GNPS) mass spectral libraries, to boost annotation rates (see Methods). We found that the distribution of a molecular family including cyclopiazonic acid followed a largely opposite trend compared to that of the aflatoxins molecular family (Fig. 2c, d). The highest abundance of the aflatoxin molecular family associated with Chinese clades 2, 4, 5, and 6 (W-clades 4, 5, 6 and 7), while it was found at much lower abundance in Chinese clades 1 and 3 (W-clades 2 and 8) (Fig. 2d). This corroborates recent results by Drott et al.14, who showed for the US population that aflatoxin B1 was produced more in Pop A (W-clades 4, 5, 6 and 7) than in Pop B and C (W-clades 2 and 3). Our data therefore demonstrates that most A. flavus isolates have a wide mycotoxin-producing capacity and that low-aflatoxin producing capacity clades often produce other metabolites, including the mycotoxins of Cyclopiazonic acid (CPA).
Evolution of accessory genes drives metabolic differentiation
Next, we conduct an exhaustive analysis of the possible drivers that affect the formation of phylogeographic diversity patterns of fungal specialized metabolism. A pangenome representation was built based on 977 representative genomes selected from the phylogeny to characterize commonality and uniqueness attributes. These 977 strain sources cover 9 countries, including 75 clinical strains17 and 902 environmental strains isolated from soil, peanuts, corn, cotton, and other sources14,16. Orthology inference revealed that the A. flavus pangenome is composed of 15,628 protein-coding genes, which is 3181 genes more than the reference genome of A. flavus NRRL3357 (GCA_009017415.1)15. These non-redundant orthogroups could be subdivided into core (in all isolates, n = 9584), accessory (in 5–95% of the isolates, n = 5085) and ‘unique’ (in < 5% of the isolates, n = 959) genomes (Fig. 3a) after rigorous removal of bacterial and other prokaryotic sequences by aligning the NR database. Among them, the conserved core genome accounts for 61.3%, the accessory genome accounts for 32.5%, and the unique genes account for the remaining 6.2% of genes. Our results and recently reported 59%16 and 42.5%17 proportion of accessory genomes illustrate that the A. flavus population exhibits significant plasticity. However, the ratio of core metabolome (39%) (Supplementary Fig. S6a) is lower than the core genome (61.3%). The pangenome exhibits a closed trend as the population size increases (Fig. 3b). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis confirms that genes responsible for genetic information processing are dominant in the core genome, alongside genes related to primary metabolism and cellular processes (Fig. 3c). 81% of accessory genes and 72% of unique genes were annotated in biosynthesis of primary and specialized metabolites(Fig. 3d, e). Clustering analysis of the gene gain/loss of accessory genes (Fig. 3f) and pan-transcriptome data indicates corresponding clade-specific expression profiles (Fig. 3g), which may be linked to these accessory gene variations directly or indirectly. In conclusion, pangenome and pan-transcriptome evidence indicate that the evolution of accessory genes could drive metabolic divergence in direct or indirect ways.

a Presence/absence matrix heatmap of 15,628 orthologous genes identified from 977 representative A. flavus isolates genomes. The pangenome contains core (orthogroups present in all isolates), accessory (orthogroups present in 5–95% of the isolates), and unique (orthogroups present in less than 5% of the isolates) genomes. b Rarefaction curve of gene family variation in the pan-genome and core genome as the number of genomes increases. c KEGG enrichment analysis of the core gene set. The false discovery rate (FDR) was calculated based on the nominal P-value from the hypergeometric test. d KEGG enrichment analysis of the accessory gene set. The false discovery rate (FDR) was calculated based on the nominal P-value from the hypergeometric test. e KEGG enrichment analysis of the unique gene set. The false discovery rate (FDR) was calculated based on the nominal P value from the hypergeometric test. f Cluster analysis of the distribution of gain and loss of accessory genes recapitulates clade-specific evolutionary trends. g Clade-specific patterns emerge in the expression profiles of accessory genes in different W-clades.
Variations in BGC genes only partially explain metabolic differentiation
A logical explanation for the differentiation in specialized metabolism could be variation in BGC repertoires between the clades. A total of 52,511 BGCs were identified across all genomes, which were grouped into Gene Cluster Families (1707 GCFs) using BiG-SCAPE (Supplementary Fig. S7a). However, the number of unique BGCs after the first round of automatic deduplication was obviously overestimated, due to e.g., contig breaks inside gene clusters in subsets of the genomes. Therefore, we extracted 2268 core gene protein sequences of BGCs from these GCFs for detailed curated deduplication by pairwise sequence comparison. Finally, 103 unique BGCs were confirmed in the entire A. flavus population by large-scale sequence alignment and manual validation based on BGC class rules, which amounts to 11 uncharacterized BGCs compared with previous A. flavus pan-BGCs studies14. We matched these 103 BGCs with the 92 BGCs reported in previous literature14 and numbered the BGCs’ ID in the same order. The extrapolation results suggest that there is still a relatively large biosynthetic potential within A. flavus, considering the differences in BGC microevolution sites between strains (Supplementary Fig. S7b) and a more stringent sequence similarity threshold to perform similarity clustering and de-duplication on the BGC core gene. The rarefaction curve showed that the entire species could contain ± 120 distinct families of BGCs (Supplementary Fig. S7c).
Previous research by Drott reported that genetic differences in BGCs core gene can result in differences in SMs production14, highlighting how population-specific SNPs and InDels result in clade-specific differences in BGC core gene content and presence/absence. Lind’s analysis of the A. fumigatus population emphasized that the high-frequency variation mainly comes from SNPs and gene gain/loss polymorphisms12. However, partially because most BGCs in A. flavus have not been characterized (16 BGCs products are confirmed by experiments21), previous studies did not statistically or experimentally link core and flanking genes variation to differentiation in the production of known SMs to validate the consequences of genetic variation. To this end, we used a knowledge-based machine learning and mGWAS approach to couple larger paired datasets to comprehensively analyze the variation of BGC core genes and flanking genes and their association with metabolic differentiation patterns. We found that 77% of BGCs core genes showed a conserved pattern, 15% showed presence-absence variation (PAV), and 9% showed a dispersed distribution pattern (Supplementary Fig. S8). For example, there is an overall trend of conservation of aflatoxin BGC in A. flavus (Supplementary Figs. S8, S9) and Aspergillus species (Supplementary Fig. S10) based on large-scale collinearity analysis. This corroborates previous work by Inge et al. in Aspergillus section Flavi, based on comparative genomics11. In addition, by leveraging the power of paired omics integrated analysis using NPLinker22 and based on structural annotations using reference MS/MS spectra of several known metabolites, we were able to link detected metabolites to candidate corresponding BGCs (Table 1). As a positive control, the aflatoxin B1 and B2 MS/MS spectra are associated with this method to the experimentally characterized aflatoxin BGC (MIBiG:BGC0000006) with high metcalf scores of 4.78 and 6.58, respectively (Table 1). Furthermore, vioxanthin and viopurpurin, two dimeric naphthopyrones produced by Aspergillus species23, were annotated in our metabolomics dataset. These specialized metabolites protect filamentous fungi from a wide range of predators23. The biosynthetic pathway of these two structurally similar compounds is still unknown in A. flavus, and the mass spectra of these molecules were putatively linked to the putative naphthopyrone BGC homologous to MIBiG gene cluster BGC0000107 from Aspergillus nidulans (Table 1). Additional plausible BGC-mass spectral links for seven different A. flavus BGCs with metabolites of three different chemical compound classes pave the way to further dissect their detailed biosynthetic pathways and ecological roles using genetic and biochemical studies (Table 1).
To assess the effect of BGC core and flanking gene variation on differences in metabolite production, we compare in pairs 2 experimentally characterized BGCs with corresponding metabolites detected in the metabolome dataset (Supplementary Fig. S11). The results reveal that variations of BGC genes can only partially explain metabolic differentiation. Specifically for the aflatoxin BGC, 34% of W-clade 2 and 14% of W-clade 3,8 strains showed core or multiple peripheral gene loss events (Fig. 4a). However, 44% strains with intact clusters still showed no/low aflatoxin production. This is consistent with Drott et al.’s finding that the loss of aflatoxin BGC core gene occurred only in the PopB population14. For Cyclopiazonic acid (CPA) BGC, 29.8% (34/114) of clade 1 and 14.3% (13/91) of clade 3 strains showed whole BGC loss. These strains, therefore, did not produce CPA. However, 73.3% (11/15), 84.7% (89/105), 70.9% (61/86), and 58.1% (75/129) contain all BGC genes in clades 2, 4, 5, and 6, respectively, while producing little or no CPA. We then analyzed the transcriptional profiles of 58 BGCs (Expressed BGCs genes numbers = 440) of 28 strains in different clades (Supplementary Table 5) and found that 64% (282/440) of BGCs genes were not significantly differentially expressed in different clades, and only 36% (158/440) were significantly differentially expressed (log2|FC | ≥ 1, p.adjust < 0.05) (Supplementary Table 6). The aflatoxigenic Chinese clade 4 (W-clades 5,6) differentially expressed aflatoxin and cyclopiazonic acid pathway genes, while the low-aflatoxins Chinese clade 1 (W-clade 2) highly expressed other BGCs instead, especially kojic acid, aflatrem, and aspirochlorine BGCs (Fig. 4b). KEGG enrichment analysis also demonstrated significant differential expression among different subpopulations in aflatoxin biosynthesis pathways (Fig. 4c–g). In addition, a linear mixed model (mGWAS) method was used to demonstrate the association and contribution rate of genetic variation and metabolic differentiation in BGC. The mGWAS results were intersected with all predicted BGCs genes in A. flavus, and it was further found that SNP variations in 28% (124/440) of BGCs genes were significantly associated with metabolic differentiation (Fig. 5b). Among them, 44 genes showed significant differential expression across clade strains (Fig. 4b). Thus, both analyses can link a subset of variation in BGCs to metabolic differentiation. This suggests that BGC absence/presence variation only partially explains the differential expression at the transcriptional level and the metabolic differentiation outcomes.

a Experimentally characterized BGC core gene presence/absence variation (PAV) only partially explains specialized metabolites’ abundance profiles. For example, in aflatoxin BGC, 34% of W-clade 1 and 14% of W-clade 6 strains showed multiple peripheral gene loss events, 44% strains without core gene loss events still showed no/low aflatoxin production. b The differentially expressed gene ranking dotplot of 58 BGCs of 28 strains in Chinese clade4/clade1. c Results of pathway enrichment analysis of differentially expressed genes in C-clade4/clade1. d Results of pathway enrichment analysis of differentially expressed genes in C-clade4/clade3. e Results of pathway enrichment analysis of differentially expressed genes in C-clade3/clade1. f Results of pathway enrichment analysis of differentially expressed genes in C-clade6/clade3. g Results of pathway enrichment analysis of differentially expressed genes in C-clade6/clade4.

a The differentially expressed gene ranking dotplot of 306 regulatory genes in the Chinese clade 4/clade 1. b Environmental adaptive genes identified by SamBada and Bayenv2 and their intersection. The intersection and union results with mGWAS genes, known regulatory genes, and BGC genes. c Random forest model predicts the variable importance of environmental factors. d Upset graph showing the intersection and union of DEGs of regulators, BGC genes, environmental selection genes, and the mGWAS genes set. e KEGG metabolism level enrichment analysis of 803 environmental adaptability genes. f GO pathway enrichment analysis of 803 environmental adaptability genes.
Environmental change promotes metabolic pattern evolution
The above transcriptome analysis of strains on different clades also revealed that 12 regulators associated with environmental factors such as light (veA and laeA,B), pH (pacC), ion starvation (mscA, mscB, pmr1, hapB, hapE, and hapX), temperature (AFLA_037820), and sensors of carbon/nitrogen (C/N) source (creA and areA) show significant differential expression (Supplementary Table 7 and Fig. 5a). We speculate that geographic environment selects certain functional regulators to accelerate the formation of phylogeographic metabolic diversity. The long-term environmental adaptation of fungi does select for specific specialized metabolic repertoires (Fig. 2b)14,24, for example, the latitudinal gradient pattern of aflatoxins (Fig. 1a, b)24,25. However, which environmental factors drive fungal metabolic differentiation remains poorly understood. Jointly with genetic factors, these environmental factors could shape phylogeographical metabolic differentiation. To explore this possibility, the data of 21 climate variables and two geographical factors were collected from the China weather data website (https://data.cma.cn/) and soil survey data of 24 soil physiological metrics from our lab. These environmental data correspond to the sampling points one by one. Two widely used genotype-environment association (GEA) pipelines in landscape genomics were employed to identify and interpret the signatures of local adaptation across loci. A total of 2651 local adaptive genes were identified (G-Scores > 60, a score threshold) by a logistic regression model using SamBada (Fig. 5b). Bayenv2 identified 3652 SNPs were located in 2336 protein-coding gene regions (Fig. 5b); among these, we identified a total of 803 common adaptively selected genes via two models (Fig. 5b). There are 191 overlaps with 2522 mGWAS genes, 34 intersections with known regulatory genes (n = 306), and 33 overlaps with BGCs genes (n = 441) (Fig. 5b). The important environmental influencers prioritized by a random forest model included soil bulk density, temperature, precipitation, average relative humidity, and soil pH value4 et al. (Fig. 5c), all of which showed a notable correlation with latitude across different years (Supplementary Fig. S12). We further performed differential expression gene (DEGs) analysis from different origins at the transcription level. The genes with the most intersections with DEGs of environmental selection genes were mGWAS genes (n = 67), followed by regulatory genes (n = 10) (Fig. 5d). This demonstrates that environmental changes can promote the evolution of genes associated with different metabolic differentiation patterns (mGWAS) or regulatory genes. It is possible that these environmental factors that vary with the latitudinal gradient jointly shape the phylogeographic metabolic diversity by selecting the above-mentioned environmental selected genetic loci. These environmentally mediated genes have broad functions, mainly related to metabolic regulation (Fig. 5e) and physiological responses (Fig. 5e, f). Among them, 304 environmental selection genes were significantly differentially expressed between strains of different clades. For example, AFLA_057410, a NAD-binding Rossmann fold oxidoreductase, and a NADH-cytochrome b5 reductase of AFLA_029440, and an ATP citrate lyase subunit (Acl) encoded by AFLA_106350 participate in energy metabolism in the tricarboxylic acid cycle, providing energy and precursor materials such as acetyl-CoA for the biosynthesis of specialized metabolites. Fatty acid synthase alpha subunit, encoded by fasA (AFLA_117420) (High Gscore = 90) and fatty acid desaturase of AFLA_089170, known to be linked to the biosynthesis of polyketide specialized metabolites such as aflatoxins, was also detected as a local adaptation locus and in mGWAS. Environmental selection-related signals appear in the tryptophan synthase alpha subunit encoded by AFLA_112810 and a serine/threonine protein phosphatase PPT1 (AFLA_056690); these genes are also significantly differentially expressed between strains from different clades. Previous transcriptome and metabolome enrichment analyses also revealed significant differences in the tryptophan pathway in different clade strains. In addition, a regulatory gene (sfaD, AFLA_093240) for local adaptation (Gscore = 92) encodes a guanine nucleotide-binding protein subunit. Environmental selection signals in their gene intervals and significant differential expression were simultaneously detected. This suggests that environmental changes, such as climate change, can accelerate the evolution of genes involved in primary metabolism and regulators. In terms of genes involved in physiological responses, we detected strong environmental interaction signals between bulk soil density environmental factors and two genes encoding a pH signal transduction protein, palA (AFLA_113560, Gscore = 112), and a pH-response transcription factor, pacC (AFLA_030580, Gscore = 65), using SamBada. In particular, the expression levels of the pacC gene across strains from different clades are significantly different. In addition, four heat shock protein genes, HSPs (AFLA_037820, AFLA_035620, AFLA_084590, AFLA_084460), also showed local adaptation (Gscore = 62 ~ 63). These genes may be related to environmental temperature adaptability, especially the AFLA_037820 gene (heat shock protein Hsp30-like) (Fig. 5a), which has significant differences in expression levels among different geographical origins and clade strains.
Regulatory Variations Drive Metabolic Rearrangement
Deleterious mutations in regulatory genes may trigger larger phenotypic effects through a cascade effect. For example, a partial loss-of-function of the regulatory gene veA has been shown to greatly impact SMs production in fungal co-cultures26. However, some nonsense mutations do not cause metabolic phenotype changes, the mutations in regulatory genes generally cannot be directly associated with metabolic outcomes. We analyzed the differential expression of 306 experimentally validated regulatory genes extracted from peer-reviewed literature. Significant differences were found in the expression of different clades of multiple regulatory genes (Fig. 5a). We demonstrated statistical association of mutation sites by intersecting the 2522 genes previously associated with 60 representative metabolites, for example, aflatoxin B1. We found that mutations in ~ 28% (86/306) of reported regulatory genes were significantly associated with the five metabolic differentiation patterns (Fig. 5d). Among these 86 associated regulatory genes, ~ 30% (26/86) showed significant differential expression (Fig. 5b, d). This illuminated that regulatory variation were significantly associated with metabolic differentiation, significant differences in transcription level was also observed (Supplementary Fig. S13). We further examined the deleterious variation (dividing them into low-, moderate-, and high-impact variants) in 36 major regulatory genes that govern specialized metabolism in filamentous fungi. This analysis revealed that clade-specific deleterious variants appeared in regulatory genes, such as the pathway-specific regulator genes aflS, aflR, and the global regulatory gene veA (Supplementary Fig. S14). Other variations in specific regulatory genes and the distribution patterns of these deleterious variants in the population are described in the supplementary information. Between the clades, 47% (143/306) known regulatory genes were differentially expressed (log2|FC | ≥ 1, p.adjust < 0.05). For example, the expression of aflR aflS, acon, laeB, msnA, nsdD, veA, and creA regulatory genes was significantly different between the aflatoxigenic (Chinese clade4, PopA3) and non-aflatoxigenic (Chinese clade1, PopB) clades (Supplementary Table 7 and Fig. 5a). Excitingly, the functions of a large number of other uncharacterized genes associated with mGWAS have not yet been experimentally verified, providing us valuable avenues for future research (Fig. 5b, d). This also reveals that ~86% (2160/2522) of uncharacterized metabolic regulatory genes or other functional genes associated with metabolic differentiation (Fig. 5b) play an important role in driving metabolic differentiation. To verify the association results at the molecular level, we selected four known (sakA, nsdD, apsA, gprJ) and 12 unknown function putative regulatory genes associated with aflatoxin synthesis by mGWAS and conducted gene knockout experiments in LNZW-1 strains (from Chinese clade 5) (Fig. 6a, b). Nine of these genes had a significant impact on metabolic differentiation. Compared with the wild-type strain, the AF210 (nsdD), AF420 (apsA), and AF890 (uncharacterized gene) gene knockout strains (Fig. 6c) have smaller growth diameters, especially the AF210 and AF420 gene knockout strains (Fig. 6c). Gel electrophoresis images of wild type (top) and transformant (bottom) of the gene of AF210 (nsdD) (Fig. 6d), AF420 (apsA) (Fig. 6e), and AF890 (uncharacterized gene) (Fig. 6f), demonstrating successful gene knockout. The untargeted metabolomic analysis of these mutant strains validated that a single gene mutation can underpin metabolic switches (Fig. 6g). The concentrations of aflatoxin B1, G1, cyclopiazonic acid and kojic acid were significantly downregulated in 9 out of the 16 knockout strains compared to the wild-type strain (Fig. 6h–k). The differential and pathway analysis based on the metabolomes demonstrated that single gene mutations resulted in significant metabolic perturbations (Supplementary Fig. S15a, c, e), mainly enriched in primary metabolism pathways, such as phenylalanine and tryptophan biosynthesis (Supplementary Fig. S15b, d, f). These regulatory mutations changed the chemical profile of deletion strains (Chinese clade 4, PopA) to look more like the chemical profile of strains in Chinese clade 1, PopB.

a Manhattan plot showing SNP sites significantly associated with aflatoxin B1. Association significance was assessed using two-sided Wald tests. Genome-wide significance was defined as p < 5 × 10−8 (Bonferroni correction). b The QQ plot shows that aflatoxin B1 are really affected by these significant sites. The QQ plot compares the observed −log₁₀(p) values to those expected under the null hypothesis of no association. The solid diagonal line represents the expected distribution under the null hypothesis. The shaded area indicates the 95% confidence interval of the expected distribution, calculated based on the order statistics of the uniform distribution. c After knocking out three genes, compared with the wild-type strain, the hyphal growth diameter, and reduced spore production. d Gel electrophoresis images of wild type (top) and transformant (bottom), demonstrating successful AF210 (nsdD) gene knockout. e Gel electrophoresis images of wild type (top) and transformant (bottom), demonstrating successful AF420 (apsA) gene knockout. f Gel electrophoresis images of wild type (top) and transformant (bottom), demonstrating successful AF890 (uncharacterized gene) gene knockout. g The principal component analysis(PCA) classification of the metabolic profiles of the three gene knockout strains. h–k The concentration changes of Aflatoxin B1, G1, Cyclopiazonic acid, and Kojic acid in three knockout strains and wild-type strains. l Genome-wide selective sweep signals screening for Chinese clade 3 versus clade 4 to identify sites subject to natural selection. m Enrichment analysis of natural selection genes identified between Chinese clade 3 and Chinese clade 4 reveals 62% of the genes subject to natural selection are primary-metabolism-related genes.
mGWAS results had linked primary metabolic genes in glycolysis pathways (AFLA_133830 and AFLA_053990) to aflatoxin B1 production (Fig. 6a). The differentially expressed genes of Chinese clades 1 and 4 were significantly enriched in the glycolysis pathway, TCA cycle, and aflatoxin pathway (Fig. 4c), thus supporting this mGWAS association result and the general hypothesis that variations in primary metabolism genes are significantly associated with specialized metabolism differentiation. To further address this hypothesis, we examined evidence from three levels: (1) Genome-wide selective sweep analysis (XP-CLR method27), a well-established method in population genetics natural selection pressure analysis, was used to screen for genes showing signs of recent natural selection in each clade. KEGG enrichment analysis revealed differences in the type or number of metabolic genes selected across subpopulations. 58% to 66% genes with signatures of selection were part of pathways belonging to primary metabolism (Fig. 6l, m and Supplementary Fig. S16a1–14). Among them, amino acid metabolism (such as tryptophan metabolism), lipid metabolism, and carbohydrate metabolism are over-represented (Supplementary Fig. S16b, c1–14). In addition, 17% to 29% genes with signatures of selection were related to specialized metabolism, and again, signatures were often specific to one or a few clades, for example, aflatoxin, aflatrem, and aspirochlorine BGCs. (2) We performed KEGG enrichment analysis on the genes associated with mGWAS of different 60 metabolites in the previous five metabolic patterns and differentially expressed genes from different clades, looking for those primary metabolic genes that are significantly associated with metabolic differentiation. The results show that the top five metabolic pathways with the most genes associated with mGWAS are carbohydrate metabolism, amino acid metabolism, and lipid metabolism. (3) Lastly, we looked at the correspondence (co-clustering) of transcriptome profiles with metabolome profiles of precursor biosynthesis pathways (e.g., amino acids, etc) in primary metabolism across the 28 strains. Differentially expressed genes from different clade strains were also significantly enriched in glycolysis, citrate cycle (TCA cycle), pentose phosphate pathway, tryptophan biosynthesis, fatty acid biosynthesis, glycerolipid metabolism, sphingolipid metabolism, glycosphingolipid biosynthesis, pyruvate metabolism, and aflatoxin biosynthesis(Fig. 4c–g). Correspondingly, the metabolomic data also showed that primary metabolite abundances show consistent clade-specific differences consistent with those of SMs. For example, we found that tryptophan is present at higher levels in Chinese clades 1 and 3 than in clades 2, 4, 5, and 6 (Fig. 7a). The distribution trend of the abundance of cyclopiazonic acid, which incorporates tryptophan as a precursor, is consistent with that of tryptophan (Fig. 7b). Similar patterns exist between metabolic abundances between other primary and specialized metabolites in the corresponding pathway, such as oleic acid ethyl ester, averantin (AVN), versicolorin A and aflatoxin B1. It is also worth noting that many important primary metabolites, including nicotinamide adenine dinucleotide (NAD) and UDP, have higher abundance in Chinese clades 1 and 3 (Fig. 7c, d). The mirror plot demonstrates the high reliability of these four metabolites’ annotations (Fig. 7e–h). Varying abundance of cofactors and associated energy metabolism thus seems to be associated with differences in activity of specialized metabolism28. Based on the above data, we summarized and described a model of the main genetic and environmental drivers of fungal secondary metabolic diversity (Fig. 7i). The evidence emphasizes that the transcription levels of primary metabolic pathways differ between strains. These differences likely ultimately mediate the differentiation of specialized metabolism. Altogether, the above evidence indicates that variation in primary metabolism flux and regulatory gene sequences significant impact on diversity in specialized metabolite production.

The boxplots indicate abundance distributions of primary and specialized metabolites in different clades. a The boxplots showing how, e.g., abundance of precursors like tryptophan is correlated to abundance of associated specialized metabolites, such as (b) alpha Cyclopiazonic acid(CPA). c The boxplots indicate abundance distributions of Nicotinamide adenine dinucleotide(NAD) in different clades. d The boxplots indicate abundance distributions of Uridine-5-diphosphate(UDP) in different clades. e The mirror plot shows the MS/MS spectra of tryptophan in the sample and in the GNPS library, aligned with the cosine score plot. f The mirror plot shows the MS/MS spectra of alpha cyclopiazonic acid(CPA) in the sample and in the GNPS library, aligned with the cosine score plot. g The mirror plot shows the MS/MS spectra of Nicotinamide adenine dinucleotide(NAD) in the sample and in the GNPS library, aligned with the cosine score plot. h The mirror plot shows the MS/MS spectra of Uridine 5 diphosphate (UDP) in the sample and in the GNPS library, aligned with the cosine score plot. i Schematic diagram of regulatory gene variation and metabolic rearrangement. The upper part includes important regulatory genes that interact with different environments. These genes are significantly differentially expressed in different clade strains. The lower part is a diagram of the primary metabolism and specialized metabolism reorganization pathways of strains in different clades, in which differentially expressed primary metabolism regulatory genes are marked in green. Abbreviation: norsolorinic acid (NOR); averantin (AVN); 5′-hydroxyaverantin (HAVN); 5′-oxoaverantin (OAVN); versicolorin B (VERB); versicolorin A (VERA); averufanin (AVF); demethylsterigmatocystin (DHDMST); sterigmatocystin (ST); dihydrosterigmatocystin (DHST); O-methylsterigmatocystin (OMST); dihydro-O-methylsterigmatocystin (DHOMST); aflatoxin B1 (AFB1); aflatoxin B2 (AFB2); aflatoxin G1 (AFG1); aflatoxin G2 (AFG2); Cyclopiazonic acid (CPA).
Discussion
Against the backdrop of global climate change and growing drug resistance, harmful fungi threaten global food supplies by secreting biochemicals such as mycotoxins, and pathogenic fungi are increasingly causing infections of crops and humans29. A warming climate could make fungi more dangerous (such as drug resistance, hypervirulence, infectious, and mycotoxigenic), as increased mutagenesis was observed under the influence of body temperatures that are typically warmer than outside temperatures30 or also drive the emergence of new pathogenic fungi3. It is crucial to understand the environmental adaptability and vulnerability of pathogenic fungi and then deploy effective measures to control threats in advance. Fungal specialized metabolites are an important aspect of environmental adaptation5,21,31. In this study, we carry out a comprehensive biogeographical study and reveal phylogeographic patterns of fungal specialized metabolism at various spatial scales.
The use of non-aflatoxigenic strains as biological control agents to inhibit aflatoxigenic strains has been used with some success32. Our results suggest that, in practice, potential health risks may be overlooked or underappreciated, as most non-aflatoxigenic strains were shown to produce considerable amounts of alternative mycotoxins, and they have the potential to produce many more – yet unknown -specialized metabolites. The presence of a virulence gene homolog specific to a particular population has also been identified, for example, (Afu8g00230, Chain A, Verruculogen synthase), which is predominantly present in W-Clade 2 environmental non-aflatoxigenic clade strains (Fig. 1a). This poses a potential risk to human food safety and public health. As to aflatoxin itself, the different metabolic distribution patterns reveal that aflatoxin production appears to be shaped by long-term geographically bound environmental adaptation. Specifically, high aflatoxigenic strains seem to have a selective advantage in southern regions (low-latitude areas). Strains from the middle subtropical (74% middle- & high-APC) and south subtropical (62% middle- & high-APC) climatic zones have higher APCs. The most notable examples are the strains from W-clade 4, a high-APC clade (Figs. 1, 2), which are mainly sourced from these two climatic zones. Soils are universally recognized to be the natural habitat of A. flavus, but the ecological role of aflatoxin in this environment is complicated. Drott et al. have suggested that a fitness cost when competing with soil microbes33 and a benefit when competing with insects34 may maintain both aflatoxigenic and non-aflatoxigenic chemotypes (i.e., balancing selection). Latitudinal stratification of A. flavus chemical profiles could result from greater density of insects and arthropods at low latitudes selecting for the production of chemical defenses, such as aflatoxin31 and vioxanthin23 to mediate interference competition or mitigate fungivory34. Conversely, as the suppressive effect of soil microbial communities is more pronounced at lower temperatures33, higher latitudes may select for metabolic profiles that mediate microbe-microbe competition. Genotype-environmental association analysis shown that relative humidity is an important environmental factor, together with soil bulk density, temperature, soil pH, and precipitation (Fig. 5c), that may contribute to patterns of phylogeographic metabolic differentiation. The evidence from genome, transcriptome, and metabolome levels supports that environmentally selected metabolic regulation enzymes may be crucial for fungal biosynthesis of specialized metabolites to adapt to complex environmental changes. As global environmental change intensifies, the above-mentioned important environmental factors, such as warmer temperatures, droughts or increased precipitation, and northward shifts of climate zones, could promote pathogenic fungi to evolve to stronger adaptability3 and increased spread and redistribution of fungal spores. Specialized metabolites are important mediators of ecological adaptation31, and we expect that fungal metabolic evolution will be accelerated in the background of global climate change. Furthermore, abiotic stress factors resulting from climate change are expected to weaken the resistance of host crops, rendering them more vulnerable to fungal disease outbreaks and resulting in more frequent mycotoxin contamination29. We report that the identified high-frequency aflatoxigenic clades (W-clade 6) were mainly from the high-temperature and high-humidity areas in central and southern China. As the climate zone moves northward, the abundance of aflatoxigenic strains in high-latitude areas may increase in the future. This study provides insights into local adaptation of harmful fungi and the prediction of fungal evolutionary trends under global warming scenarios; and worryingly, it suggests that aflatoxin (or other potential specialized metabolite) production may become a larger problem in the future due to climate change as predicted by a studies35.
The current study comes with several limitations: while our studied strains cover multiple countries and continents, broader international collaboration would be beneficial so that phylogeographic studies of fungal specialized metabolism could be extended across the globe, particularly in the southern hemisphere. Differences in isolate sampling and demographic histories (e.g., clonal expansions) as nested within geographic samplings could impact the overarching patterns described here. Under the currently used medium culture conditions, metabolite structures of nine of the sixteen experimentally resolved biosynthetic gene clusters (BGCs) could be identified in the metabolome data. We note that a single PDA medium here is unlikely to be sufficient to describe the comprehensive specialized metabolic diversity of fungi, as a significant proportion of BGCs are typically inactive and require specific conditions to be activated, expressed, and biosynthesized4,36. Our pan-transcriptome analysis of 28 strains from different subpopulations under the same culture conditions validated that a large proportion of BGC genes were expressed at low levels or not at all (Supplementary Table 5). Alternatively, limitations in extraction solvents (the protocol used here does not include dedicated lipidome extraction solvents) and separation conditions (e.g., HILIC column for polar metabolites) may have prevented the detection of additional metabolites. In metabolomics data analysis, newly developed AI-driven methods such as the Spectral Denoising Search37 may enhance both the number and accuracy of metabolite annotations, surpassing traditional cosine similarity-based alignment algorithms. Currently, many fungal metabolites that have been isolated and identified, however, still lack high-quality MS/MS spectral information in public databases, which limits the number of specialized metabolites we can identify. Still, our work provides the most comprehensive overview of the diversity of this species thus far and unearthed many clade-specific BGCs and metabolites, empowered by state-of-the-art computational genomics and metabolomics workflows for natural products discovery. Furthermore, integrative omics mining utilizing the paired omics dataset revealed promising leads to connect biosynthetic gene clusters to the products whose production they encode. This will aid in elucidating their biosynthetic pathways and understanding their ecological roles. Nevertheless, we believe our contribution to be a valuable resource for the ecology metabolism-related fungal research community.
In summary, we find that the phylogeographically distinct clades of A. flavus have evolved distinct chemotypes to adapt to spatially heterogeneous ecological niches. We conclude that regulatory and primary metabolic variation underlie the formation process of phylogeographic patterns of fungal specialized metabolism. The phylogenetic patterns and the proposed mechanisms by which they originate enhance our understanding of how fungi adapt to geographic environments with chemical innovation. The genes driving geographic metabolic differentiation identified by mGWAS and GEA analysis in this study will provide candidate gene resources for a subsequent large-scale knockout/gene editing keystone target screen. The discovery also gives insight into the evolutionary trends of toxigenic fungal variation caused by global climate shift and will inform rational design of ‘personalized’ geographical control agents, in order to achieve more accurate and long-term control of harmful fungi, mitigation of the adverse effects of mycotoxins and harmful fungi injection. Our work provides a large-scale annotated genomic and high-resolution metabolomic dataset for the fungal research community and provides insight for eco-metabolomics and biogeography of microbial metabolism research.
Methods
A. flavus strains library construction and mycelium cultivation
We collected soil, peanut, or corn samples across China from 2014 to 2019, and isolated 3567 Aspergillus flavus strains by single spore isolation, morphology combined with ITS sequencing identification. The latitude of sampling points in the China region spans from 24.3 north (Zhanjiang, Guangdong province) to 46.4 (Qiqihar, Heilongjiang province). Longitude from 87.3 E (Changji, Xinjiang Uygur Autonomous Region) to 123.42 (Qiqihar, Heilongjiang Province) in the west. Altitudes range from 5.3 m (Lianyungang, Jiangsu Province) near sea level to 2330 m (Zayü County, Tibet Autonomous Region)24. All strains were isolated from independent samples. Detailed strain sampling site coordinates were described in Supplementary Table 1. We selected ~ 600 representative A. flavus strains from the library as research objects by integrating information such as geographic origin, phylogenetic relationships constructed using ITS sequences, climate zone, and APCs.
A. flavus was incubated on a Potato Dextrose Agar (PDA) medium and then put on Petri dish plates at a constant temperature and humidity incubator in dark conditions at 29 ± 1 °C for 10 days. We used a 0.1% Tween-80 solution to wash the conidia and obtain a suspended spore mixture. Conidia suspension mixture was used, which was quantified using a hemocytometer and a light microscope38. 2.5 × 105 conidia/mL mixture was inoculated into 50 mL of autoclaved liquid medium, containing 0.25% yeast extract, 0.1% K2HPO4, 0.05% MgSO4 · 7H2O, and 10% glucose (pH = 6.0). The liquid culture flasks containing conidia of A. flavus were placed in a shaker (180 rpm) at 29 ± 1 °C in dark conditions. After 5 days, the mycelia were acquired by quickly filtering the cultures from the flasks with multiple layers of cheesecloth and washed with 10 mL of 4 °C saline solution (0.9% NaCl), and the mycelia were squeezed with sterile absorbent paper. Then, A. flavus mycelium was quickly transferred into a 50 mL centrifuge tube and quenched with liquid nitrogen. We used a split strategy to sequence one of the collected mycelium samples for acquiring genome data and the other for metabolome research. The mycelial samples were directly weighed for the DNA extraction experiment. However, for metabolomics experiments, the mycelium samples were freeze-dried and then weighed.
DNA extraction, library preparation, and genome sequencing
We used the following DNA extraction protocol: 200 mg of fungal tissue was weighed in an EP tube with a one-thousandth balance, quenched with liquid nitrogen, and placed in a freezer grinder to homogenize it into powder. 1.2 mL CTAB lysis buffer was added, and the mixture was vortexed for 60 s. The mixture was incubated at 65 °C for 60 minutes. The mixture was cooled and centrifuged at 13,201 × g for 6 min. Then, the supernatant was transferred to a 2.0 mL EP tube, an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1) reagent was added, and the mixture was shaken. The mixtures were centrifuged at 13,201 × g for 15 min, and their supernatant was transferred into a 1.5 mL EP tube. Then, 2/3 volume of − 20 °C pre-chilled isopropanol was added into the tube, shaken up and down to mix evenly, and then put in a − 20 °C refrigerator for more than 2 hours. The mixtures were centrifuged at 13,201 × g for 15 min, and then their supernatant was aspirated into a new 1.5 mL EP tube, supplemented with 750 μL of 75% ethanol, and the pellet was rinsed with a pipette. Finally, the tube was centrifuged at 13,201 × g for 5 min at ambient temperature, and the supernatant was aspirated. After centrifugation, the residual liquid was removed, air-dried for 3-5 min, and solubilized in appropriate amounts of TE solution (EB solution for Pacbio sequencing samples).
For next-generation sequencing (NGS), 1 μg of genomic DNA was taken and disrupted by sonication using a Covaris instrument. Fragment selection was performed on the fragmented samples so that the sample bands were concentrated around 200–400 bp. Then, a reaction system was prepared, the end of the double-stranded DNA was repaired, and an A base was added to the 3’ end. Furthermore, a linker ligation reaction system was constructed to connect the linker to the DNA. A PCR reaction system was then prepared, and a reaction program was set to amplify the ligated products. Amplification products were purified and recovered by magnetic beads. After the PCR product was denatured into single strands, a circularization reaction system was prepared, and the reaction system was fully mixed and incubated to obtain a single-stranded circular product. After digesting the linear DNA molecules that had not been circularized, the final library was obtained. Qualified NGS libraries were performed on the Illumina (HiSeq X-Ten) platform at BGI (Shenzhen, China) to generate the 150 bp paired-end raw reads.
For whole-genome long-read sequencing, 1 μg of genomic DNA was fragmented using a Covaris instrument. The fragments longer than 20 kb were selected for the PacBio library construction according to the manufacturer’s instructions, and qualified libraries were sequenced on the PacBio Sequel II system at BGI (Shenzhen, China).
Quality filtering and genome-wide variation detection
We used SOAPnuke (v1.5.6)39 to filter out adapter contamination reads, PCR duplication reads, and low-quality and ambiguous reads with the parameters “-q 0.2 -l 0.2 -n 0.05 -d” for the NGS data. High-quality sequencing reads from each individual were aligned to the reference genome of A. flavus NRRL 3357 (Accession No. GCA_000006275.2) using Burrows-Wheeler Aligner with the BWA MEM algorithm. Subsequently, duplicate reads and realigned indels were processed using Sentieon (https://www.sentieon.com/), and then the gvcf file of each individual by using the Haplotyper method of Sentieon. GVCFtyper was used to integrate the variations detected from all individuals. Thereafter, SNPs were filtered by GATK(v4.0.2.1), which match the condition “QD < 2.0 | | MQ < 40.0 || MQRankSum < − 12.5 || ReadPosRankSum < − 8.0 | | FS > 60.0 | | SOR > 3.0”. High-quality InDels were filtered under the parameters of “QD < 2.0 || ReadPosRankSum < − 20.0 | | FS > 200.0 | | SOR > 10.0”. Single-nucleotide polymorphism (SNP) variants used for population analysis were further processed by vcftools with parameters “--maf 0.05 --minDP 4 --minGQ 10 --minQ 30 --max-missing 0.99”. The impact of both SNPs and InDels was annotated by SnpEff(v4.3) with default parameters.
De novo assembly, decontamination, and assessment of genome assemblies
For the Illumina sequenced data, to obtain the ideal assembly results, we use SPAdes (v.3.15.1)40 to assemble the high-quality clean reads into genomic sequences with k-mer ranges from 33 to 83 by a step size of 10. Genome assembly for Pacbio RSII sequencing data was performed using wtdbg241 with the parameters “-L 5000 -x preset2”. Clean reads from Illumina sequencing data were used to correct INDEL and SNP errors for the assembly sequences, and Pilon (v 1.24) was used to perform three rounds of polish error correction for possible assembly INDEL and SNP errors with parameters “--diploid --changes –verbose”.
To remove the bacterial contamination sequences, the Non-Redundant Protein Sequence (NR) and Nucleotide Sequence (NT) database (v20180315) were used to align against the assembled genomes using BLAST. The alignment results with E-value < 1e-5, align length > 100 bp, and identity > 70 for NT, > 40 for NR were deemed significant. Sequences not matching to the Aspergillus species were considered as contaminations and then removed to obtain the final assembled genome. The completeness and redundancy of genomes were assessed based on single-copy ortholog analysis with BUSCO (3.0.2) using the Eurotiomycetes_odb database. The Virulence genes were collected from the J. Rhodes et al. research42, and OrthoFinder(v2.4.0)43 with default parameters was used to find homologous genes in China and America A. flavus samples, respectively.
Genome annotation, functional classification, and enrichment analysis
There are two main types of repeats in the genome: one is tandem repeats (Tandem Repeat, TR); the other is interspersed repeats (Transposable element, TE). For repeat region annotation, we used Tandem Repeats Finder (TRF, v4.0) to annotate tandem repeats. TEs’ homolog annotation was carried out by compared with a known repeat sequence library (Repbase, v16.02) by using Repeatmasker and Proteinmask.
Gene structure annotation was performed by two methods: homology annotation (Homolog) and model-based de novo prediction(De novo), respectively, and EVidenceModeler integration software was used to combine different annotation results: (1) homology-based annotation: Five closely related species sequences (GCA_000002655.1, GCA_000184455.3, GCA_009193485.1, GCA_009176385.1, GCA_000006275.2) were selected and downloaded from NCBI, and the genome was annotated using GeneWise; and (2) de novo annotation: gene prediction of assembled genomes using Augustus and GeneMark. The weight of the homologous annotation result was set to 10, and the weight of the two de novo software prediction results was set to 1.
The A. flavus genes were functionally annotated as detailed as possible using the six mainstream functional databases, including NR, Interpro, GO, KOG, KEGG, and SwissProt. We used Blastn (v2.2.31) for NT database (v20180315) annotation with E-value < 1e-5 and identity > 70%, Blastp (v2.2.31) for NR (v20180315), KOG (v20090331), KEGG (v84) and SwissProt (release-2017-09) annotation with E-value < 1e-5 and identity > 40%, and Blast2GO (v2.5.0) combined with nr-based annotation results for GO (v20171220) annotation. The phyper function in R was used for enrichment analysis of GO and KEGG annotations, based on Fisher’s exact test.
Population structure and phylogenetic analysis
By using admixture (v1.3.0)44, we assessed population structure from k = 2 to k = 10 with default parameters. The maximum likelihood estimation was applied without using the prior population information, and the most likely number of ancestors was determined using cross-validation (CV) error. Principal component analysis (PCA) was performed with Plink v1.945, and the first two eigenvectors were plotted. The θπ ratio and population genetic differentiation index (Fst) between each clade were calculated using VCFtools v0.1.13, respectively, with a 5 Kb non-overlapping slide window. By screening the genome in windows, we detected candidate regions with selection signatures with the largest differences in θπ ratio (θπ-clade-i /θπ-clade-j, bottom and top 5%), and the top 5% FST(population differentiation coefficient values) regions between two clades. The intersections of the θπ ratio and FST candidate regions were defined as the regions that were under selective sweep. Then, the genes within or overlapping the regions showing a selection sweep were defined as candidate genes. Linkage disequilibrium (LD) decay of each clade was calculated by using PopLDdecay v3.40 with default parameters.
The genome-wide SNPs were used to construct the phylogenetic tree for 1064 samples using the Neighbor-Joining method using TreeBeST (v1.9.2) with parameters “-b 1000”. For other group phylogenetic analysis, we employed the maximum likelihood method in IQ-TREE2 (v2.0.4)46 with parameters “-alrt 1000 -bb 1000”, then, the tree output was displayed by the iTOL (http://itol.embl.de). A. flavus has three mating types (MAT1-1, MAT1-2, and MAT1-1-MAT1-2), which are encoded MAT alpha 1 (MAT1-1) gene (Genebank Accession Nos: EU357934) and the HMG-box protein MAT1-2 (MAT1-2) gene (Genebank Accession Nos: EU357936)47. We downloaded these two genes from GeneBank and then used Blast to scan the genome for mating typing.
Pangenome construction and analysis
An A. flavus pan-genome included 25 PacBio assembly genomes and 977 NGS assembly genomes was built by EUPAN (v0.44)48. Genomic quality assessment was first performed using QUAST using the longest contiguity genome HNSY-2B (Has the highest N50 value) as a reference. The unaligned contigs from individuals were merged, and then the redundant contigs were removed to generate the non-redundant contigs using CD-HIT (v4.6.3) with sequence identity < 0.9. Subsequently, a BLAST-based method was used to cluster the reference sequences and non-redundant unaligned contigs using the “eupan rmRedundant blastCluster” command with parameter “-c 0.5” to generate the comprehensive A. flavus pan-genome dataset, and the pan-genome sequences were obtained from the previous sequences annotation. We mapped raw reads to the comprehensive sequences to determine gene presence-absence. A “map-to-pan” strategy subsequently was utilized to determine gene presence-absence and gene family presence-absence by using Bowtie2 (v2.2.5). The gene body coverage and CDS coverage of each gene were calculated after mapping the reads to the pan-genome sequence. Finally, we determined gene presence-absence patterns considering gene body coverage value (> 0.8) and CDS coverage values (> 0.95). If one member of a gene family satisfying the above criteria was present in a given A. flavus genome, the gene family was considered as present. The output of the presence/absence variation (PAV) profile file was further used to plot the rarefaction curves and perform statistical analysis.
Biosynthetic gene cluster deduplication and validation
We used antiSMASH49 to predict BGCs across all 651 de novo assembly and annotated A. flavus genomes from China and the USA. BGCs in all genome sequences were clustered to deduplicate similar BGCs sequences into Gene Cluster Families (GCFs) and compared to experimentally characterized reference BGCs from the MIBiG (v2.0) repository using BiG-SCAPE (v1.0.0)50. The biosynthetic gene clusters (BGCs) in all genomes were identified with antiSMASH (v6), using the genome assembly results and gene annotation gff3 files as input files. These samples have corresponding metabolomic data, which facilitates subsequent paired comparisons between BGCs and secondary metabolites. BiG-SCAPE analysis was performed using default settings, separating the analysis according to the BGC product type and creating network directories for each class. In addition, mixing all classes and retaining singletons was also performed to deduplicate similar BGCs sequences into Gene Cluster Families (GCFs) using a threshold of 0.3 to 0.6, with an interval of 0.05, and then identify which BGCs are shared between which strains. This resulted in a total of 52,511 BGCs that were grouped into 1707 GCFs. The core gene protein sequences of these BGCs were further extracted from these 1707 GCFs, and then 2268 core genes were obtained (some BGCs have more than 2 core genes). Pairwise comparisons were performed using diamond (v0.8.23.85) with the following parameters: --evalue 1e-5 --sensitive. Then, according to the comparison results, the core genes were redundancy-filtered according to different similarities (amino acid identity value = 40, 50, 60, 70, 80, 90). After taking their intersection, we further manually checked and removed the duplicates. The network files were visualized using Cytoscape (v3.8.2). The BGC types encoding production of miyakamides and desferri-ferrichrysin were manually annotated, since MIBiG (v2.0) had not yet included these two BGCs. Based on the results, a gene cluster family (GCF) presence/absence matrix was constructed. This was used to screen for conserved BGCs and clade-specific BGCs. To identify clade-specific GCFs, we performed a dimensionality reduction analysis based on the presence/absence matrices of GCFs to obtain the linear combination matrices and compared the combination matrices by principal component analysis (PCA) function in R.4.1.0. Rarefaction curves analysis was constructed by “iNEXT” package of R to extrapolate biosynthetic diversity potential of the whole A. flavus populations. The t-Distributed Stochastic Neighbor Embedding (t-SNE) analyses were conducted with the “Rtsne” package of R.
Biosynthetic gene cluster variation and comparative analysis
Two recently developed pipelines were used for finding clusters of co-located homologous sequences (cblaster v.1.2.951) and visualizing the BGCs comparison (clinker v0.0.2052) for a large-scale study population BGC variation. The specific analysis steps are as follows: 1. Building a cblaster database using the assembled genome and annotation files of each sample with the command “cblaster makedb input.gff myDB”. A total of 570 sample databases were constructed, including an old reference genome version of A. flavus NRLL 3357 and three new versions of A. flavus NRLL 3357 genomes, including GCA_009017415.1, GCA_014117465.1, and GCA_014117485.1. 2. Detecting the presence/absence of the gene cluster in each sample in turn according to the protein sequence of each BGC, to facilitate comparison with BGCs that have been experimentally characterized. BGC protein sequences were predicted by SMURF to determine the BGC start nucleotide of the first biosynthetic gene (5′ end) and the stop nucleotide of the last biosynthetic gene (3′ end) with the AFLA_x gene ID, and these coordinates were manually curated. Then, these BGCs were fed into cblaster as input files to locate and align the BGC in a specific genome with the command “cblaster search -m local -db myDB.dmnd -jdb myDB.json -qf input_pep.fa -p output.html -o output.alignment”. 3. Creating a json file for the next step to extract the best alignment sequence gbk file. 4. Using the cblaster extract_clusters module to extract the gbk files aligned for each BGC in each sample for clinker input. 5. Leveraging cblaster to extract the gbk files of each sample (each BGC) as clinker input; call clinker to get the visualization results of each group of gene clusters. In order to facilitate the subsequent comparative synteny analysis, we selected every 20 genomes as a group for clinker analysis. The BGC core (backbone) gene was identified by antiSMASH 549. The protein sequences of these core genes were then extracted and fed into cblaster to scan all isolate genomes for analyzing the identity values of the BGC core gene. A threshold of 80% was then employed to determine the presence/absence of BGC core genes.
Sample preparation and quality control (QC) for metabolomics analysis
The 566 fungal samples (biological replicate n = 1) were separated to two aliquots, with one for genome sequencing and one for metabolomics data acquisition. In our subsequent subpopulation comparative analysis, we treated different strains within the same subpopulation as biological replicates. The liquid culture flasks containing conidia of A. flavus were placed in a shaker (180 rpm) at 29 ± 1 °C in dark conditions. After 5 days, the mycelia were acquired by quickly filtering the cultures from the flasks with multiple layers of cheesecloth and washed with 10 mL of 4 °C saline solution (0.9% NaCl), and the mycelia were squeezed with sterile absorbent paper. Then, A. flavus mycelium was quickly transferred into a 50 mL centrifuge tube and quenched with liquid nitrogen. The mycelium samples were freeze-dried and then weighed. To obtain metabolite extracts, an optimized two-step extraction method24 is compatible with polar and non-polar metabolites from mycelial samples that maximizes the scope of extraction. The specific steps are as follows: (1) the mycelium samples were freeze-dried, and then 50 mg per lyophilized mycelial sample was extracted by vortexing vigorously for 1 min with 1 mL of extraction solution 1 (methanol: acetonitrile: water 2:2:1 v/v/v), which contained the internal standard of 2-chlorophenylalanine (20.0 µg/mL) and camphanic acid (25.0 µg/mL). (2) We added 6 ~ 8 beads in each tube and homogenized in a ball mill for 4 min at 45 Hz, then it was treated for 5 min with an ultrasound of 700 W (incubated in ice water). Hereafter, all tubes were sonicated for 5 min with 700 W (incubated in ice water). This process was repeated one more time using solution 2 (methanol: dichloromethane: ethyl acetate 1:1:1 v/v/v) to further extract the nonpolar metabolites. (3) The proteins were precipitated by incubating the homogenate for 1 h at − 20 °C followed by centrifugation at 20000 g/min for 10 min at − 20 °C. 1 mL of the resulting supernatant was transferred into a fresh 2 mL LC-MS/MS glass vial for the UPLC-HRMS (Ultra performance liquid chromatography-tandem high-resolution mass spectrometry) analysis. Based on the minimum quality control requirements of the metabolomics community, three quality control methods were used for data quality control throughout the metabolomics analysis process in this study. Quality control measures such as the introduction of quality control samples, the addition of internal standards, and blank samples ensured that we could collect high quality metabolomics raw dataset during this period53. Firstly, two internal standards were added to the extraction solvent to monitor experimental deviations in the sample pre-treatment process to ensure the quality of the data. Meanwhile, a widely used assessment criterion was employed to assess the quality of metabolomic data. Namely, If the data from quality control (QC) samples were all closely clustered to the origin in the PCA scores plot, indicating that the quality of the data was suitable for subsequent analysis. Furthermore, we inserted a sample of blank solvent per 10 samples to monitor the cross-contamination between samples. Quality control (QC) samples were prepared by pipetting 10 µL from each sample extract into a QC injection vial using a 100 µL pipette. We set up a tube containing two extraction solvents as an extraction blank. This tube did not contain any A. flavus mycelium sample, but instead contained 4 ~ 6 pieces of bread and two internal standard solvents. Solvent blank samples were prepared by pipetting 1 mL of pure methanol solvent (99.99%) into the injection vial named “blank”. The 551 samples were divided into 6 batches for analysis using UPLC-HRMS, because the sample tray of the UPLC-HRMS instrument can only accommodate 120 sample vials, and the mobile phase is only sufficient for one batch. A diagram of the sample run sequence shown in our previous research articles of Supplementary Fig. S124. Each batch contains 120 samples, one solvent blank sample, and one extraction blank sample. However, in the LC-MS run sequence, the solvent blank sample and quality control sample will be injected repeatedly every 10 samples to assess cross-contamination between samples and instrument stability. Before collecting mass spectrometry data from A. flavus mycelial samples, we first collected 10 quality control samples as technical replicates to allow the UPLC-HRMS instrument to reach mobile phase equilibrium and a stable state. The mycelial samples and extraction blank samples were collected only once. For the actual mycelial samples, we did not set up technical replicates; we used QC samples to assess the reproducibility of the samples directly. Therefore, each batch generated 12 solvent blank samples and 22 quality control samples, resulting in a total of 72 solvent blank samples and 132 quality control samples across six batches, for which metabolomic data files were obtained. Ultimately, we collected metabolomic data from 673 A. flavus strains. However, due to failures in the genome sequencing process for some strains, we had to discard the metabolomic data for those strains. There were 551 strains that possessed both genomic and metabolomic profiles. The quality control analysis results shown in our previous research articles of Supplementary Fig. S124. The high-quality data were proved by QC replicates, which are tightly clustered in PCA space. We also used the peak alignment results of the metabolite Versicolorin B to assess its distribution in different A. flavus strain samples, quality control samples, and blank samples. We found that cross-contamination was minimal and reproducibility was excellent, indicating that we obtained high-quality metabolomic data24.
Metabolomics data acquisition by UPLC-HRMS
All the isolates’ non-targeted metabolome raw data were collected via our optimized and standardized metabolome platform24,38. A C18 column (Hypersil Gold, 100 mm × 2.1 mm (i.d.)) with 3 µm pore size (Thermo Fisher Scientific, USA) coupled with the Ultimate 3000 system (Dionex, Sunnyvale, CA, USA) was employed for liquid chromatography separation. Mobile phase A is water: methanol (95:5, v/v) containing 0.1% formic acid, and 10 mM ammonium formate. Mobile phase B is composed of methanol: water (95:5, v/v) containing 0.1% formic acid and 10 mM ammonium formate. Both mobile phases were mixed following the program below in the UPLC system at a flow rate of 300 μL/min as a gradient mobile phase for effective separation of different components. The gradient elution program was set to: 0 ~ 1 min: 15% B (mobile phase B), 1 ~ 3 min: 15–50% B, 3 ~ 5 min: 50–70% B, 5 ~ 10 min: 70–100% B, 10 ~ 13 min: 100% B, 15 min: 100 ~ 15%B, and from 15 ~ 20 min: 15% B. Subsequently, 2 μL of the extracted metabolites was injected into Orbitrap Fusion mass spectrometer (Thermo, USA) for mass spectrometry raw data acquisition.
In this study, full scan mass spectrometry (MS1) data were used for relative quantitative analysis, and mass fragmental (MS2) data for metabolites identification and annotation. We acquired the mass spectrometry data containing MS1 and MS2 mass spectra by a full-scan MS coupled with data-dependent fragmentation (DDA) mode under Ultra Performance Liquid Chromatography Tandem Orbitrap Fusion High-resolution Mass Spectrometry (UPLC-HRMS). For parameter optimization, we refer to the optimal method parameter optimization results of François’ lab using LC-Orbitrap fusion mass spectrometry for metabolite annotation54. The positive and negative mode raw data were separately acquired to enhance data quality. The HESI (Heated Electron Spray Ionization) ion source was used to efficiently ionize the UPLC effluent components. The − 1.9 kV capillary voltage and 320 °C ion transfer tube temperature parameter settings were selected. The specific parameters of the ion source are as follows: 320 °C heater temperature for mobile phase volatilization and ionization; sheath gas (Arb): 40; aux gas (Arb): 5; and sweep gas (Arb): 0; spray voltage: static, positive ion: 3.5 kV, negative ion: 3.0 kV. The full-scan MS1 stage was identical to that for the positive and negative models except for polarity. The specific parameters were as follows: Orbitrap detector resolution: 12000, scan range (m/z): 100 ~ 1200, RF lens (%): 60, AGC target: 5.0e5, maximum injection time (ms): 100, and source fragmentation: disabled. The acquisition of mass fragmentation (MS2) spectra requires a smaller number of ions; otherwise, it will affect the number of primary mass spectra (MS1) and finally affect the quantification. Filter parameters included: intensity threshold at 1.0e4, charge state: 1 ~ 2, and dynamic exclusion after 1 time. The exclusion duration was set to 60 seconds. Top speed mode was selected in the data-dependent acquisition mode, and the number of scan event types: 1 s. The DDA section-specific parameter settings were identical as for positive and negative mode, except for the high-energy collision-induced dissociation (HCD) setting. A type of stepped collision energy in HCD activation in positive mode was set at 40 ± 5 eV, and 33% for fragmentation of the isolated precursor ions, and the HCD cell pressure was set to 8 mTor. In negative ionization mode, the value of HCD was set at 30 ± 5 eV in negative mode. Other parameters for both ionization modes included: Orbitrap detector mass resolution of 30,000 FWHM, AGC target: 5.0e4, and the maximum injection time was set to 100. The quadrupole with a narrow isolation window of ± 1 Th to isolate the MS2 ions. Submission of data acquisition sequences, instrument control, and partial data processing task via Xcalibur 4.0 software (Thermo Fisher Scientific).
Metabolome features extraction and data quality control
The metabolome feature extraction, feature alignment, and gap-filling steps were performed in MZmine 4.2755 (Linux version). First, LC-HRMS positive and negative raw mass spectrometry data files were converted to mzXML format by the ProteoWizard using default parameters. In MZmine 4, the batch processing mode (mzwizard tab) was selected to achieve streamlined mass spectrometry data processing. The MS1 and MS2 noise levels were set at 1E5 and 6E3, respectively. The final MS2 noise threshold (6E3) selection was determined by comparing the number of core and unique metabolites identified on the GNPS platform by Feture-Based Molecular Networking (FBMN) analysis across eight preset MS2 thresholds (1E0; 1E2; 1E3; 2E3; 4E3; 6E3; 8E3; 1E4) in the MS2 peak detection step (Supplementary Fig. S25). A minimum group size of scans set 5, a minimum highest intensity set 2E5, and an m/z tolerance set 0.01 Da in the second step of the ADAP chromatogram builder. We selected the baseline cut-off deconvolution algorithm to perform the peak deconvolution with the following settings: min peak height: 1.0E5; peak duration range(min): 0.05-2.0; Baseline level: 1E5; m/z range for MS2 scan pairing (Da): 0.01; RT range for MS2 scan pairing(min): 0.3. We used the isotopic peaks grouper algorithm with an m/z tolerance of 0.01 Da (or 10 ppm) and an RT tolerance of 0.2 min for grouping isotopes. The join aligner module (m/z tolerance = 0.01 Da, weight for m/z = 2, weight for RT = 2, absolute retention time tolerance = 0.2 min) was used to do feature alignment. Feature filtering was performed by feature list rows filter selection, with a minimum peaks in a row = 2, and a minimum peaks in an isotope pattern = 2. The peak gap was filled with the RT = 0.2 min and m/z tolerance of 0.01 Da. Eventually, a.csv quantitative file was exported and then was submitted to GNPS20 and MetDNA56 after modifying the data matrix format for metabolites identification or molecular networking analysis and structural annotation. We followed the metabolomics data quality control methods and data quality assessments as previously reported24. The raw metabolomic data of 95 strains from the United States generated by Drott et al.14 were downloaded from GNPS MassIVE repository (ID no. MSV000087134, https://doi.org/10.25345/C54226). They used a target method of PRM mode to acquire the data on the Thermo Scientific Q Exactive Orbitrap mass spectrometer. For the 94 USA strain samples, they only acquired MS1 information for quantification and then collected 24 qualitative files to acquire the MS/MS spectral information containing target compounds (We finally contacted Drott and Tomás to upload the 24 qualitative files containing the MS/MS mass spectra to the GNPS repository). Drot’s PNAS article only studied 13 experimentally verified pathways for secondary metabolites, but did not study those pathways that have been isolated and identified in A. flavus but whose corresponding unknown BGCs have not yet been resolved. The identification of 13 metabolites was completed by Tomás and Drott, and they provided MS/MS spectra information of 13 secondary metabolites in the PNAS article14 (Supplementary Table S4 and SI Appendix, Supplementary Figs. S5–S29 in this article). Four of the secondary metabolites belong to annotation level 2, and the other nine metabolites belong to annotation level 3. We merged the metabolite quantification table sent to me by Drott with our data matrix based on the MS1 accurate mass ( ± 5 ppm, m/z tolerance window) and MS/MS spectral information from the PNAS literature (annotation level 3). However, this data fusion method does have risks. Therefore, we performed MS1 and MS2 detection using the same Mzmine 4 workflow and FBMN analysis on the updated files containing MS/MS data with the same parameters, aiming to merge the two datasets based on MS1 accurate mass ( ± 5 ppm, m/z tolerance window) and MS/MS spectrum comparison results (annotation level 2). We only retained the MS1 metabolic features that had MS/MS spectra. From the Mzmine analysis results, we can see that Drott et al.’s dataset contains only hundreds of metabolite MS/MS spectra information, which is consistent with their goal of targeting the analysis of a few secondary metabolites (Figure S17). However, compared with the number of MS1 reported in their article14 (Fig. 2A in the PNAS article), the number of metabolic features (MS1) containing MS/MS spectra is small. Therefore, it is difficult to perform FBMN joint analysis with our dataset, rich in MS/MS spectra information. We therefore performed a separate FBMN analysis and only matched 3 metabolites with the GNPS reference library, including aflatoxin B1, cyclopiazonic acid, and aflatrem (GNPS job results link: https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=3532f01f1f8a47c9b2e36eca093b89bc). This is consistent with the results of annotation level 2 shown in Table S4 in their PNAS14 article (annotation level 2), except for the metabolite Ditryptophenaline. We couldn’t find it in the GNPS library. In order to comply with the minimum standards proposed, we therefore selected two A. flavus secondary metabolites, including Aflatoxin B1 and Cyclopiazonic acid ( ± 5ppm, m/z tolerance window, and with MS/MS spectrum alignment mirror plot) for data fusion to verify that the presence/absence variation of the BGC gene in the strains from China and the United States in our manuscript can only partially explain the changes in secondary metabolites.
Metabolite annotation, identification, and molecular networking analysis
Metabolite identification and annotation were performed using a four-step approach. First, we used our in-house mycotoxin library, which contains 262 commercial or isolated chemical standards, and a manually curated compound list based on accurate mass (m/z, ± 5 ppm), MS/MS spectra, retention time, and isotope patterns. If we found a match, we considered this MSI Metabolite Identification Level 1. Second, metabolites were further identified based on accurate mass, isotope pattern, and MS/MS spectra against public databases by spectrum alignment of tandem mass spectrometry (MS/MS), including mzCloud, GNPS20 (all public mass spectral libraries included there), MoNA, MassBank, and NIST. Specifically, tandem mass spectrometry (MS/MS) data of samples were acquired using an Orbitrap Fusion mass spectrometer (Thermo Fisher, USA). To annotate more likely metabolites, we used three widely available commercial or open-source metabolome software, including Compound Discovery 2.0, GNPS, MetDNA3. For Compound Discovery 2.0, the detailed parameter settings of Compound Discovery 2 (Thermo Fisher Scientific, USA) were the same as in our previously reported method24. The detailed steps are as follows: 1) metabolomics raw data were imported into the software of Compound Discovery (CD) 2.1(Thermo Fisher Scientific, USA) to generate a data matrix that consists of the retention time (RT), mass-to-charge ratio (m/z) values, peak intensity, and annotation of metabolites. 2) Metabolite annotation consists of the following steps: ① Unknown peaks were aligned and detected with the parameters of RT tolerance of 0.8 min and 5 ppm mass deviation. Minimum peak intensity set to 10,000 and S/N threshold=3. ② The compounds were annotated with different types of databases. Among them, the mzCloud database was used to identify compounds on the MS/MS level with a mass tolerance of 10 ppm. Compound class set to All, Match ion activation type = False. Chemspider, BioCyc, and KEGG databases were used to annotate features based on exact mass (MS1) with a mass tolerance of 5 ppm. An endogenous metabolites database of 4400 compounds embedded in the CD internal database also was used to annotate metabolites. 3) In addition, an in-house mass spectral library of > 3384 microbial natural products and mycotoxin reference standards was used to search by the search mass lists module in CD. ③mass tolerance set to 5ppm, RT tolerance(min) = 0.05, S/N threshold = 1.5 infill gaps (missing values) module. We used Xcaliber 4.0 (Thermo Fisher Scientific, USA) to manually check and identify the metabolites identified above. The Genesis peak detection method was selected to detect the peak area. Minimum peak height(S/N) set to 3. Metabolic features(m/z) with peak intensity greater than 1E4 in QC raw data files were retained. The metabolites that did not meet the requirements were discarded. Finally, compounds were identified and selected by integrating positive and negative ion modes to build a peak list for subsequent biomarker analysis. For GNPS, feature-based molecular networking (FBMN)20 analysis was performed using MZmine 4 and the GNPS platform to build the molecular network. The .csv quantitative and.mgf mass spectrum profiles file (from MZmine 4 output above) and metadata file were submitted to the GNPS20 platform (http://gnps.ucsd.edu) to generate the molecular network. We used a parent mass tolerance of 0.02 Da and an MS/MS fragment ion tolerance of 0.02 Da to cluster the spectra and to subsequently create consensus spectra from which the network is constructed. All networks where edges were filtered to have a cosine score above 0.6 and more than five matched peaks. The mass spectra in the molecular network were then matched to the mass spectral libraries of GNPS with a threshold score above 0.6 and at least five matched peaks recorded. Other networking parameters were left at default levels. The obtained molecular network was then visualized using Cytoscape (version 3.9.1). For the MetDNA3 software, the metabolites features table (from MZmine 4 output above) and MS/MS data file (.mgf format) were upload and the default parameters were used. Furthermore, those metabolic features not identified by the above three software were searched in the online public databases, METLIN and NIST. Then, the MS/MS spectra of all the above matched metabolite features was exported from Xcalibur, and these matches were manually checked to confirm the annotation, which was considered a level 2 identification. In addition, if metabolite features with MS/MS spectra were not matched in public databases as analyzed by the above tools, we considered them a MSI level 4 unknown identification. Third, to further annotate these unknown metabolic features, we construct a self-built MS1 mass spectra Aspergillus metabolites library from NPAtlas (https://www.npatlas.org/) and Reaxys (https://www.reaxys.com/). We used accurate masses ( ± 5 ppm) to deduplicate the metabolic features using Compound Discovery 2 (Thermo Fisher, USA), and gave an MSI level 3 identification. Fourth, we used the computational metabolomics annotation tools MolDiscovery and Dereplicator+57 to annotate microbial metabolites, mainly peptides natural products. They are both implemented in the GNPS ecosystem, and we used default parameters. The mzCloud, MetDNA2, DEREPLICATOR + , and MOLDISCOVERY annotation results were summarized in Supplementary Fig. S6b. Mirror plots of query and GNPS mass spectral library MS/MS spectra of representative metabolites that show high to decent matching qualities (Supplementary Fig. S6e–j). We uploaded 355 metabolites MS/MS spectral alignment mirror plots to demonstrate the reliability of the metabolites annotation results, which are stored in https://zenodo.org/records/18670081. MetDNA3 only outputs MS/MS similarity score results, and their database is not open source; therefore, we cannot obtain mirror plots. In addition, we provide chromatographic retention times (Supplementary Fig. S27) and MS/MS chromatograms of standards and A. flavus mycelial extracts of six precursor metabolites in the aflatoxin biosynthetic pathway, including 5-methoxysterigmatocystin, Norsolorinic acid, Averantin, Averufin, Versicolorin B, and Versicolorin A (Supplementary Figs. S28–35).
Fungi RNA extraction, sequencing, and transcriptome analysis
We selected 28 representative strains from different clades on the phylogenetic tree for cultivation, with the same culture conditions as our previous research24 and three biological replicates. Transcriptome sample preparation, sequencing, and data analysis were consistent with our previous study methods58. After 3 days of cultivation, we harvested A. flavus hyphae for RNA isolation. The TRIzol method was used to extract the fungi RNA. Firstly, the filamentous fungus was ground to a powder using liquid nitrogen, and the powder was transferred into the 2 mL tube contains 1.5 mL Trizol reagent. The mixture was shaken for 3 min, and placed for 5 min at normal temperature; then it was centrifuged at 10,000 × g for 5 min. 200 μL of chloroform/isoamylalcohol (24:1) was added to the supernatant with 1 mL of lysis reagents. After centrifugation at 10000 × g for 10 min, the supernatant was transferred into another new tube with an equal volume of isopropanol and then refrigerated at − 20 °C for 1 h. After centrifugation at 13600 g for 20 min at 4 degrees celsius, the supernatant was precipitated by 1 mL of 75% ethanol and let dry for 5 min. The RNA pellet was dissolved in 30 ~ 100 microliters of DEPC water or RNase-free water. The concentration of the extracted RNA samples was determined using a Nanodrop system(NanoDrop, Madison, USA), and the integrity of the RNA was examined by the RNA integrity number (RIN) using an Agilent 2100 bioanalyzer (Agilent, Santa Clara, USA). The mRNA was purified with Oligo(dT)-attached magnetic beads and subsequently fragmented into small pieces with a fragment buffer at appropriate temperatures. Then, the first-strand cDNA was generated by PCR, and the second-strand cDNA was synthesized as well. The final product of the reaction was purified through end-repair and subsequent incubation. The cDNA fragments with adapters were amplified by PCR before they were purified with Ampure XP beads. The quality and quantity of the library were assessed using an Agilent 2100 bioanalyzer before it was subjected to DSN treatment. The DSN-treated library’s quality was evaluated using two methods to ensure high-quality sequence data. First, we checked the distribution of the fragment size using the Agilent 2100 bioanalyzer, and secondly, we quantified the library using real-time quantitative PCR (qPCR). The qualified library was amplified on cBot to generate the cluster on the flow cell, and the amplified flow cell was sequenced single-end on the Hiseq 4000. Illumina sequencing was commissioned from the Beijing Genomics Institute(BGI) for RNAseq sequencing.
After obtaining the raw sequencing data, low-quality, adapter-polluted sequences and reads with a high content of unknown base (N) were filtered. The clean reads were mapped to an A. flavus reference genome (GCA_000006275.3) using HISAT and Bowtie 2 with default parameters. The gene expression level for each sample was determined with RSEM59. The FPKM (Fragments Per Kb of exon per Million reads) and TPM method were used to calculate and normalize the expression level of genes. The Dr.Tom (BGI) cloud platform was used to generate a transcriptome expression quantitative data matrix. Gene annotation, GO and KEGG enrichment analysis, GSEA enrichment analysis, and WGCNA analysis were also completed in this platform. Filtering, normalization, and statistical analysis of transcriptome quantification data matrix were performed in Expressanalyst60 with default parameters. The log2-counts per million (logCPM) transformation method was used to normalize. Limma61 R package was selected to do the differential expression analysis against a common control of Clade1, pairwise comparisons, and nested comparisons method in simple metadata penel. To perform multi-factor comparison analysis for complex metadata, we leveraged linear models with covariate adjustments of limma for its high-performance implementation in the Expressanalyst60 cloud platform. We executed the plotting in Windows 10 R.4.1.0 environment with the ggplot2 package (https://r-graph-gallery.com/ggplot2-package.html).
Statistics and reproducibility
To compare the differences between the two groups, we chose the two-sided Wilcoxon test or Student’s t test, with a p-value of P < 0.05. The correction method chosen is Benjamini-Hochberg (BH). The gene pathway enrichment analysis in Fig. 3c–e and Fig. 4c–g was performed using the hypergeometric test. T-tests, principal component analysis (PCA), and Projections to Latent Structures Discriminant Analysis (PLS-DA) model were used to pre-screen differential metabolites between the two clades. Univariate analysis used the Student’s t test, with a p-value of P < 0.05. A variable influence on projection (VIP) value ≥ 1.0 was used as a selection threshold to select differential metabolites in the PLS-DA model. A statistical method specialized for multi-testing, SAM (Significance Analysis of Microarrays) to validate the credibility of differential metabolites between the two clades in metabolome-wide analysis using MetaboAnalystR62 package. The sample-wise permutation (default by the samr package) and distribution-independent ranking tests (based on the Wilcoxon test) were used to ascertain significance (false discovery rate, FDR < 0.05) for the SAM analysis. The default recommended parameters were used in the SAM analysis. The differential metabolites identified and screened by the steps mentioned above with MSI levels 1, 2, and 3 were pooled together. Enrichment analysis and the pathway analysis module in MetaboAnalyst 5.0 were used to perform the metabolite set enrichment analysis (MSEA) as well as metabolic pathway analysis (MetPA), respectively, using all MSI-level-1-and-2-annotated metabolites as input. All the default parameters were used in MetaboAnalyst 5.062. All statistical analyses were performed on a Windows 10 R.4.1.0 environment and using Excel 2019 (Microsoft, USA).
Gene knockout experiments
We referred to the method of Perng-Kuang Chang’s laboratory63 to construct an A. flavus fluorescent engineering strain with the PyrG gene using an aflatoxigenic A. flavus strain LNZW-1A isolated from the peanut field in Zhangwu, Liaoning province. The detailed gene knockout experimental procedure is as follows: (1) Primers were designed based on the target gene sequence using Primer3 (https://www.primer3plus.com/). Primer synthesis was performed by Wuhan Tianyi Huayu Gene Technology Co., Ltd. Primer information used in A. flavus regulatory gene knockout experiments was provided in the supplementary table 19. (2) First, using the A. flavus LNZW-1 genome as a template, the 5′ upstream homologous arm and 3′ downstream homologous arm sequences (each 1000-1200 bp) of the target gene, as well as the full-length pyrG gene, were amplified (reaction system: 1 µL template DNA, 1 µL Primer-F, 1 µL Primer-R, 12.5 µL 2 × Hieff Robust PCR Master Mix, and ddH2O added to a total volume of 25 µL); (3) Amplification procedure: Pre-denaturation at 94 °C for 3 min, denaturation at 94 °C for 10 sec, annealing at 58 °C for 20 sec, extension at 72 °C for 15 sec/kb; denaturation, annealing, and extension were repeated for 35 cycles, followed by a final extension at 72 °C for 5 min; PCR instrument: (Bio-RAD, T100 Thermal Cycler, USA). (4) The upstream homologous arm, the full-length pyrG gene, and the 3’ downstream homologous arm fragment were fused together using the Double-Joint PCR method (reaction system: 1 µL of 5’ upstream homologous arm, 3 µL of full-length pyrG, 1 µL of 3’ downstream homologous arm, 5 µL of 2 × Hieff Robust PCR Master Mix, and ddH2O added to a final volume of 20 µL). (5) Amplification procedure: pre-denaturation at 94 °C for 2 min, denaturation at 94 °C for 30 sec, annealing at 60 °C for 1 min, extension at 72 °C for 3 min, denaturation, annealing and extension repeated for 10 cycles, final extension at 72 °C for 10 min. The genetically transformed fragment was obtained by nested PCR amplification, and the transformed fragment was recovered by gel extraction (E.Z.N.A Gel Extraction Kit, Omega Bio-tek, D2500-02). (6) Finally, the transformed fragments were introduced into A. flavus protoplasts using a polyethylene glycol (PEG)-mediated method to obtain the knockout strain. The mutant strain was then verified for successful gene knockout by amplifying the target gene and its upstream and downstream homologous arm fragments. The preparation methods and composition of the key reagents used in the experiment:
(1) Enzymatic lysis solution: 200 mg Lysing Enzymes (Sigma, USA, 37340-57-1), 50 mg Driselase (Sigma, USA, 85186-71-6), 20 µL β-glucuronidase (85000 µg/mL, Sigma, USA, 9001-45-0), 1.4 g NaCl, 8 µL of 1 mol/L CaCl2, and 10 mmol/L NaH2PO4 (pH 5.8) to a final volume of 20 mL. The lysis solution was filtered through a 0.22 µm filter after preparation. The lysis solution was prepared on ice and used immediately after preparation. (2) 1.2 mol/L STC: 1.2 M Sorbitol, 10 mM Tris-HCl (pH 7.5), 50 mM CaCl2; Weigh 21.86 g of sorbitol, add 1 ml of 1 M Tris-HCl (pH 7.5) solution and 5 ml of 1 M CaCl2 solution, and dilute to 100 ml with deionized water. Sterilize at 121°C for 20 min. (3) 50% PEG: 50 g of polyethylene glycol (PEG4000), 1 mL of 1 M Tris-HCl (pH 7.5) solution, and 1 mL of 1 M CaCl2 solution were heated and stirred until dissolved. Deionized water was then added to a final volume of 100 mL, and the solution was sterilized at 121 °C for 20 minutes. (4) Regeneration medium: 34.2 g sucrose, 0.1 g casein hydrolysate, 0.1 g yeast extract, 1.5 g agar, deionized water to a final volume of 100 mL, sterilized at 121 °C for 20 min. (5) PDA and PDB culture media: Boil 200 g of peeled potatoes, filter the liquid through four layers of gauze, add 20 g of glucose, and adjust the volume to 1 L. Sterilize in aliquots at 121 °C for 20 min. Adding agar (15 g/L) results in PDA medium.
Protoplast preparation: A. flavus spores were inoculated into PDB medium and cultured overnight at 37 °C and 200 r/min in a constant temperature shaker (Shanghai Zhicheng Analytical Instruments Manufacturing Co., Ltd., ZWY-2102C, Shanghai). The mycelia were filtered through four layers of lens paper and washed three times with sterile water. The mycelia were then transferred to lysis buffer and incubated at 30 °C and 100 r/min for 4 h. The number and morphology of protoplasts were checked every half hour using a microscope (Nikon, ECLIPSE E100, Japan) after 2 h. The lysis buffer was filtered through four layers of lens paper into a 50 mL centrifuge tube. The tube was centrifuged at 5000 r/min for 10 minutes at 4 °C in a low-temperature centrifuge (Hunan Xiangyi Laboratory Instruments Development Co., Ltd., H1850R, Hunan). The supernatant was carefully decanted, leaving a white precipitate at the bottom of the tube. 2 mL of STC was added to the opposite side of the precipitate, and the centrifuge tube was gently rotated to wash the precipitate. The washing solution was decanted. An appropriate amount of STC was added, and the precipitate was resuspended by pipetting. The suspension was centrifuged at 5000 r/min at 4 °C for 10 min, and the supernatant was carefully decanted. An appropriate amount of STC was added, and the precipitate was resuspended by pipetting. The protoplast concentration was adjusted to 1 × 10⁶ cells/mL for further use. Add the prepared DNA fragments (total concentration > 5 ng) to a 10 mL centrifuge tube, then add pre-cooled STC buffer to a final volume of 200 µL, and mix thoroughly with a pipette. Add 100 µL of freshly prepared A. flavus protoplasts to the mixture, mix thoroughly with a pipette, and incubate on ice for 30 min. Then, sequentially add 200 µL, 400 µL, and 800 µL of PEG Buffer, gently invert to mix, and incubate on ice for 20 min. Finally, add 1 mL of pre-cooled STC buffer and incubate on ice for 10 min. Add all the transformation mixture to the regeneration medium at approximately 40 °C, mix thoroughly by shaking, and pour onto plates. Invert the plates and incubate in a 30 °C constant temperature incubator (Shanghai Jinghong Experimental Equipment Co., Ltd., DNP-9022, Shanghai) in the dark for 36 hours. Pick the hyphal tips of single colonies and transfer them to PDA medium. After 3 days of culture, extract DNA and amplify the fragments to verify the transformants. Supplementary Figs. 37–39 show the original gel images for the three gene knockout (ΔAF210, ΔAF420, and ΔAF890).
Paired omics data analysis
NPLinker, a metabologenomic analysis software framework22, was employed to link genomic and metabolomic data and connect the identified microbial specialized metabolites to their candidate biosynthetic genomic regions. First, we downloaded the molecular networking results by feature-based molecular networking (FBMN) analysis (metabolomics) of 544 strains (7 strains with low data quality were filtered) from the GNPS platform20. Metabolome data, antiSMASH-predicted BGCs, and BiG-SCAPE-derived GCFs were compiled as input for NPLinker. In the configuration file, we set the Metcalf threshold to 4 (the default value is 2) in order to speed up NPLinker and filter out links with a low Metcalf association score. Other parameters were default. After running NPLinker, the results were examined for promising BGC-mass spectral links in the NPLinker web app running from a Docker instance.
Genotype-environmental analysis
Sambada v0.5.364, based on logistic regression and Bayenv 265 by bayes factor analysis models were recruited to identify putatively adaptive loci associated with environmental factors. A single SNP locus was identified as a candidate locus when the log-likelihood ratios (G scores > 60) were significant. G-Scores were corrected for multiple testing with the Bonferroni method at a 95% confidence level. In addition, Bayenv2, a covariance matrix based on putatively neutral markers, was used to assess the correlations between SNP and environmental variables at the Markov chain Monte Carlo (MCMC) model. Bayesian factor(BFs) was used as an indicator to evaluate the degree of genetic and environmental association. An averaged log10(BF) value > 1.2 is considered a threshold support for the model where parameters significantly affect allele frequencies.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The PacBio sequencing 27 A. flavus strains and the sequencing clean data reads of 566 A. flavus strains reported in this paper have been deposited in the Genome Sequence Archive in the National Genomics Data Center66, China National Center for Bioinformation and Beijing Institute of Genomics, Chinese Academy of Sciences under Bioproject ID: PRJCA012686 that are publicly accessible at https://ngdc.cncb.ac.cn/gsa/browse/CRA008573. The whole genome sequence data reported in this paper have been deposited in the Genome Warehouse in the National Genomics Data Center66, Beijing Institute of Genomics, Chinese Academy of Sciences, and China National Center for Bioinformation, under bioproject PRJCA035534 that is publicly accessible at https://ngdc.cncb.ac.cn/gwh/search/advanced/result?search_category=&search_term=&source=0&query_box=PRJCA035534. Transcriptome data of 84 samples from 28 strains (three biological replicates were set up) have been deposited in the Genome Sequence Archive in National Genomics Data Center66, China National Center for Bioinformation and Beijing Institute of Genomics, Chinese Academy of Sciences under Bioproject ID: PRJCA035512 that are publicly accessible at https://ngdc.cncb.ac.cn/gsa/browse/CRA022539. Metabolome positive and negative mode of raw data of 551 A. flavus strains and 12 other Aspergillus species have been deposited in the MassIVE repository with accession ID no. MSV000096910 (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=7bb1b634f3454639858b80252b1fdab4) and MSV000096982 (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=2524ced05941428bbaa0ddbf0e9a6328), respectively. Spectral data and corresponding metabolic feature abundance tables for 8 different MS2 thresholds and the MS/MS alignment mirror plot of 355 metabolites from GNPS are available at https://zenodo.org/records/1867008167. The previously disclosed genome data of 508 public strains used in this study can be downloaded from the NCBI database based on the metadata information, including bioproject accession, biosample accession, SRA accession numbers, and corresponding references, provided in the supplementary table 1. For the convenience of readers, we have also uploaded these scattered genome files used in this study, along with a supplementary table of this article to https://zenodo.org/records/1867008167. Source data are provided in this paper.
Code availability
This study used open-source scripts or commercial software for data analysis of genome and metabolome data, with the used versions and parameter settings as described in detail in the Methods section. The code to reproduce the partial figures of the manuscript is available at https://github.com/jeep3/AF1000_manuscript_code/tree/master.
References
Case, N. T. et al. Fungal impacts on Earth’s ecosystems. Nature 638, 49–57 (2025).
Iliev, I. D. et al. Focus on fungi. Cell 187, 5121–5127 (2024).
Singh, B. K. et al. Climate change impacts on plant pathogens, food security and paths forward. Nat. Rev. Microbiol. 21, 640–656 (2023).
Keller, N. P. Fungal secondary metabolism: regulation, function and drug discovery. Nat. Rev. Microbiol. 17, 167–180 (2019).
Rokas, A., Mead, M. E., Steenwyk, J. L., Raja, H. A. & Oberlies, N. H. Biosynthetic gene clusters and the evolution of fungal chemodiversity. Nat. Prod. Rep. 37, 868–878 (2020).
Eskola, M. et al. Worldwide contamination of food-crops with mycotoxins: Validity of the widely cited ‘FAO estimate’ of 25. Crit. Rev. Food Sci. Nutr. 60, 2773–2789 (2020).
Denning, D. W. Global incidence and mortality of severe fungal disease. Lancet Infect. Dis. 24, e428–e438 (2024).
Medema, M. H., de Rond, T. & Moore, B. S. Mining genomes to illuminate the specialized chemistry of life. Nat. Rev. Genet. 22, 553–571 (2021).
Robey, M. T., Caesar, L. K., Drott, M. T., Keller, N. P., Kelleher, N. L. An interpreted atlas of biosynthetic gene clusters from 1000 fungal genomes. Proc. Natl. Acad. Sci. USA118, https://doi.org/10.1073/pnas.2020230118 (2021).
Vesth, T. C. et al. Investigation of inter- and intraspecies variation through genome sequencing of Aspergillus section Nigri. Nat. Genet. 50, 1688–1695 (2018).
Kjærbølling, I. et al. A comparative genomics study of 23 Aspergillus species from section Flavi. Nat. Commun.11, 1106 (2020).
Lind, A. L. et al. Drivers of genetic diversity in secondary metabolic gene clusters within a fungal species. PLoS Biol. 15, e2003583 (2017).
Hoogendoorn, K. et al. Evolution and diversity of biosynthetic gene clusters in Fusarium. Front. Microbiol. 9, 1158 (2018).
Drott, M. T. et al. Microevolution in the pansecondary metabolome of Aspergillus flavus and its potential macroevolutionary implications for filamentous fungi. Proc. Natl. Acad. Sci. USA 118, e2021683118 (2021).
Drott, M. T. et al. The frequency of sex: population genomics reveals differences in recombination and population structure of the aflatoxin-producing fungus Aspergillus flavus. mBio11, https://doi.org/10.1128/mbio.00963-20 (2020).
Gangurde, S. S. et al. Aspergillus flavus pangenome (AflaPan) uncovers novel aflatoxin and secondary metabolite associated gene clusters. BMC Plant Biol. 24, 354 (2024).
Hatmaker, E. A. et al. Population structure in a fungal human pathogen is potentially linked to pathogenicity. Nat. Commun.16, 7594 (2025).
Geiser, D. M., Dorner, J. W., Horn, B. W. & Taylor, J. W. The phylogenetics of mycotoxin and sclerotium production in Aspergillus flavus and Aspergillus oryzae. Fungal Genet. Biol. 31, 169–179 (2000).
Barber, A. E. et al. Aspergillus fumigatus pan-genome analysis identifies genetic variants associated with human infection. Nat. Microbiol. 6, 1526–1536 (2021).
Nothias, L. F. et al. Feature-based molecular networking in the GNPS analysis environment. Nat. Methods 17, 905–908 (2020).
Uka, V. et al. Chemical repertoire and biosynthetic machinery of the Aspergillus flavus secondary metabolome: A review. Compr. Rev. Food Sci. Food Saf. 19, 2797–2842 (2020).
Hjorleifsson Eldjarn, G. et al. Ranking microbial metabolomic and genomic links in the NPLinker framework using complementary scoring functions. PLoS Comput. Biol. 17, e1008920 (2021).
Xu, Y. et al. Bis-naphthopyrone pigments protect filamentous ascomycetes from a wide range of predators. Nat. Commun. 10, 3579 (2019).
Xie, H. L. et al. Fungi population metabolomics and molecular network study reveal novel biomarkers for early detection of aflatoxigenic Aspergillus species. J. Hazard. Mater. 424, (2022).
Manabe, M. & Tsuruta, O. Geographical distribution of aflatoxin-producing fungi inhabiting in Southeast Asia. JARQ 12, 224–227 (1978).
Wang, G. et al. Fungal-fungal cocultivation leads to widespread secondary metabolite alteration requiring the partial loss-of-function VeA1 protein. Sci. Adv. 8, eabo6094 (2022).
Chen, H., Patterson, N. & Reich, D. Population differentiation as a test for selective sweeps. Genome Res. 20, 393–402 (2010).
Walsh, C. T., Tu, B. P. & Tang, Y. Eight Kinetically Stable but Thermodynamically Activated Molecules that Power Cell Metabolism. Chem. Rev. 118, 1460–1494 (2018).
Casu, A., Camardo Leggieri, M., Toscano, P. & Battilani, P. Changing climate, shifting mycotoxins: A comprehensive review of climate change impact on mycotoxin contamination. Compr. Rev. Food Sci. Food Saf. 23, e13323 (2024).
Huang, J. et al. Pan-drug resistance and hypervirulence in a human fungal pathogen are enabled by mutagenesis induced by mammalian body temperature. Nat. Microbiol. 9, 1686–1699 (2024).
Kunzler, M. How fungi defend themselves against microbial competitors and animal predators. PLoS Pathog. 14, e1007184 (2018).
Atehnkeng, J., Ojiambo, P. S., Cotty, P. J. & Bandyopadhyay, R. Field efficacy of a mixture of atoxigenic Aspergillus flavus Link:Fr vegetative compatibility groups in preventing aflatoxin contamination in maize (Zea mays L. Biol. Control. 72, 62–70 (2014).
Drott, M. T., Debenport, T., Higgins, S. A., Buckley, D. H. & Milgroom, M. G. Fitness cost of aflatoxin production in aspergillus flavus when competing with soil microbes could maintain balancing selection. mBio10, https://doi.org/10.1128/mbio.02782-18 (2019).
Drott, M. T., Lazzaro, B. P., Brown, D. L., Carbone, I. & Milgroom, M. G. Balancing selection for aflatoxin in Aspergillus flavus is maintained through interference competition with, and fungivory by insects. Proc. R. Soc. B Biol. Sci. 284, 20172408 (2017).
Leggieri, M. C., Toscano, P. & Battilani, P. Predicted aflatoxin B1 increase in Europe due to climate change: actions and reactions at global level. Toxins13, 292 (2021).
Brakhage, A. A. Regulation of fungal secondary metabolism. Nat. Rev. Microbiol. 11, 21–32 (2012).
Kong, F. et al. Denoising search doubles the number of metabolite and exposome annotations in human plasma using an Orbitrap Astral mass spectrometer. Nat. Methods 22, 1008–1016 (2025).
Xie, H. L. et al. Monitoring metabolite production of aflatoxin biosynthesis by Orbitrap Fusion mass spectrometry and a D-optimal mixture design method. Anal. Chem. 90, 14331–14338 (2018).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, 1–6 (2018).
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A. & Korobeynikov, A. Using SPAdes de novo assembler. Curr. Protoc. Bioinform. 70, e102 (2020).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158 (2020).
Rhodes, J. et al. Population genomics confirms acquisition of drug-resistant Aspergillus fumigatus infection by humans from the environment. Nat. Microbiol. 7, 663–674 (2022).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Alexander, D. H. & Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 12, 246 (2011).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic Era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Ramirez-Prado, J. H., Moore, G. G., Horn, B. W. & Carbone, I. Characterization and population analysis of the mating-type genes in Aspergillus flavus and Aspergillus parasiticus. Fungal Genet. Biol. 45, 1292–1299 (2008).
Hu, Z. et al. EUPAN enables pan-genome studies of a large number of eukaryotic genomes. Bioinformatics 33, 2408–2409 (2017).
Blin, K. et al. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87 (2019).
Navarro-Munoz, J. C. et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 16, 60 (2020).
Gilchrist, C.L.M. et al. cblaster: a remote search tool for rapid identification and visualization of homologous gene clusters. Bioinform. Adv. 1, https://doi.org/10.1093/bioadv/vbab016 (2021).
Gilchrist C. L. M., Chooi Y. H. Clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics37, 2473–2475 (2021).
Kirwan, J. A. et al. Quality assurance and quality control reporting in untargeted metabolic phenotyping: mQACC recommendations for analytical quality management. Metabolomics 18, https://doi.org/10.1007/s11306-022-01926-3 (2022).
Barbier Saint Hilaire, P. et al. Evaluation of the high-field orbitrap fusion for compound annotation in metabolomics. Anal. Chem. 90, 3030–3035 (2018).
Schmid, R. et al. Integrative analysis of multimodal mass spectrometry data in MZmine 3. Nat. Biotechnol. 41, 447–449 (2023).
Zhou, Z. et al. Metabolite annotation from knowns to unknowns through knowledge-guided multi-layer metabolic networking. Nat. Commun. 13, 6656 (2022).
Mohimani, H. et al. Dereplication of microbial metabolites through database search of mass spectra. Nat. Commun. 9, 4035 (2018).
Xie, H. et al. Aspergillus flavus’s response to antagonism bacterial stress sheds light on a regulation and metabolic trade-off mechanism for adversity survival. J. Agric. Food Chem. 69, 4840–4848 (2021).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Liu, P. et al. ExpressAnalyst: A unified platform for RNA-sequencing analysis in non-model species. Nat. Commun. 14, 2995 (2023).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47 (2015).
Pang, Z. et al. MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 49, W388–W396 (2021).
Chang, P.-K. et al. Aspergillus flavus GPI-anchored protein-encoding ecm33 has a role in growth, development, aflatoxin biosynthesis, and maize infection. Appl. Microbiol. Biotechnol. 102, 5209–5220 (2018).
Stucki, S. et al. High performance computation of landscape genomic models including local indicators of spatial association. Mol. Ecol. Resour. 17, 1072–1089 (2017).
Günther, T. & Coop, G. Robust identification of local adaptation from allele frequencies. Genetics 195, 205–220 (2013).
Members & Partners, C.-N. Database resources of the national genomics data center, China national center for bioinformation in 2024. Nucleic Acids Res. 52, D18–D32 (2024).
Huali, X. et al. Large-scale multi-omics profiling reveals environmental and evolutionary drivers of fungal phylogeographic and metabolic diversity. Zenodo https://zenodo.org/records/18670081 (2026).
Acknowledgements
This work was supported by the key program of the National Natural Sciences Foundation of China (32030085, U22A20551, 32441047), the major project of Hubei Hongshan Laboratory (2021hszd015), and the major project of Hubei Province Science & Technology (2023BBA002). Z.Q. was supported by grants from the National Science Foundation and the major project of Hubei Hongshan Laboratory. L.P.W. was supported by grants from the National Science Foundation and the major project of Hubei Province Science & Technology. A research visit to X.H.L’s to Wageningen University was supported by the China Scholarship Council. Thanks to Dr. Sandra Smit of the Bioinformatics Group at Wageningen University for helpful discussions in the analysis of genomic data. Thanks to the high-performance computing (HPC) platform of the Oil Crops Research Institute, Chinese Academy of Agricultural Sciences, the high-performance computing (HPC) platform of Hubei University, and the Inner Mongolia High Performance Computing Public Service Platform (IMHPC).
Author information
Authors and Affiliations
Contributions
P.W.L., Q.Z., and H.L.X. conceived of the study and designed the experiments. P.W.L. and Q.Z. provided an experimental platform, grant support, and guidance. M.H.M. and J.J.J.vdH. designed the data analysis plan and provided the guidance for genome and metabolome data analysis. H.L.X. conducted experiments, the data collection, and metabolomic and genomic data in-depth analysis. J.H. contributed the population genetics data analysis. X.L.Z. carried out the gene knockout experiment. J.W.C. contributed to part of the genome data analysis and helpful discussions. X.F.Y. contributed the strains isolation, metadata collection, and provided helpful discussions. C.H.Z. contributed to part of the genome data analysis. F.Z. contributed to genome assembly analysis. J.C.N.M. contributed to part of the BGCs redundancy analysis. J.J. and X.Q.T. contributed to helpful discussions. E.A.H., A.E.B., and A.R. provided the genomic files from previously published articles from their lab, along with helpful discussions and revisions. M.T.D. contributed to Neighbor-Net analysis, helpful discussions and the revisions. H.L.X. wrote the initial draft, and with P.W.L., M.H.M., J.J.J.vdH., N.P.K., and Q.Z. made multiple rounds of revisions to the manuscript together with intensive discussions on its contents. P.W.L., M.H.M., J.J.J.vdH., and Q.Z. revised and proofread the manuscript. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
M.H.M. is a member of the scientific advisory board of Hexagon Bio and Hothouse Therapeutics. J.J.J.vd.H. is a member of the Scientific Advisory Board of NAICONS Srl., Milano, Italy, and consults for Corteva AgriScience, Indianapolis, IN, USA. All other authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Peer review
Peer review information
Nature Communications thanks Christian Martin, Jens Nielsen, and the other anonymous reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xie, H., Hu, J., Zhao, X. et al. Large-scale multi-omics profiling reveals environmental and evolutionary drivers of fungal phylogeographic and metabolic diversity. Nat Commun 17, 4121 (2026). https://doi.org/10.1038/s41467-026-70721-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-70721-8
