
Abstract
Trastuzumab deruxtecan (T-DXd) is an antibody-drug conjugate successfully used to treat HER2-low and HER2-positive metastatic breast cancer, but resistance consistently develops. Using multivariate Cox proportional hazards in a real-world cohort of 2,799 patients with breast cancer, we aimed to identify clinically relevant T-DXd resistance mechanisms. In patients with samples collected prior to T-DXd treatment, higher expression of ERBB2 (HER2) and lower expression of ABCC1 (an ATP-binding cassette transporter involved in drug efflux) were significantly associated with longer T-DXd-related overall survival (OS); ABCC1 predicted OS independently of HER2. Furthermore, mutations in several genes were enriched in post-T-DXd samples compared to unmatched T-DXd-naïve samples, including ERBB2, NFE2L2 (a transcriptional activator of ABCC1), and KEAP1 (a negative regulator of NFE2L2), indicating plausible resistance mechanisms related to HER2 target levels and ABCC1-mediated drug efflux. Identifying such resistance mechanisms might lead to improved methods of precision oncology and novel therapeutic approaches to overcome resistance.
Similar content being viewed by others
Introduction
Trastuzumab deruxtecan (T-DXd) is an antibody-drug conjugate (ADC) widely used as a treatment for HER2-low and HER2-positive metastatic breast cancer. Preclinical studies have identified multiple potential mechanisms of resistance that can develop at various points in the course of ADC trafficking through cancer cells—from receptor binding, to internalization and processing of the ADC, to efflux of the payload out of the cell, as well as antibody-dependent cellular cytotoxicity (ADCC)1,2,3 (Fig. 1). Despite the knowledge of T-DXd resistance mechanisms generated through preclinical studies, there are few large population-based studies that have explored resistance to this therapy4.

Created in BioRender. Ribeiro, J. (2026) https://BioRender.com/vr77in9.
The availability of real-world data from insurance claims, combined with whole transcriptome sequencing, offers a unique opportunity to evaluate resistance mechanisms across key pathways in an unbiased and comprehensive manner. In this study, we integrated next-generation sequencing (NGS), immunohistochemistry (IHC), and claims data to identify clinically relevant mechanisms of resistance to T-DXd. Our aim was to identify genes involved in key processes of T-DXd’s mechanism of action that are independently predictive of T-DXd benefit, while also considering the impact of the tumor microenvironment5,6. Acknowledging the potential effect the treatment might have on gene expression profiles, we focused our discovery work on specimens collected prior to the start of T-DXd. In addition, we examined mutational events occurring after treatment with T-DXd to evaluate the role of selective evolutionary pressures produced by T-DXd.
Results
Patient characteristics
Among the 2799 tumors investigated, the majority (86.5%, n = 2420) were collected before the initiation of T-DXd treatment. The median length of time between tissue collection and treatment start for these patients was 15.5 months (range: 0.1–192 months). Approximately 14% of patients had received TDM1 prior to T-DXd. Using HER2 IHC/CISH criteria, approximately one-third of patients had HER2-low tumors, while those with HER2-positive and HER2-ultra-low tumors each comprised about 16% of the cohort. Surprisingly, 18% of patients treated with T-DXd had HER2-null tumors, which might be related to tumor heterogeneity, discrepancies in HER2 testing between community centers and centralized laboratories, or off-label use of T-DXd. Out of the total cohort, 13.5% (n = 379) of samples were collected after the initiation of T-DXd. The median length of time between treatment start and tissue collection for these patients was 10.5 months (range: 0.1–44.5 months). There were no matched samples in the analysis (Table 1).
Association of key genes with overall survival following T-DXd
The mechanism of action for T-DXd has been extensively investigated in preclinical and clinical studies, leading to a substantial increase in knowledge about individual pathways in recent years7. We compiled a set of 96 transcriptomic features (RNA signatures and gene expressions) across 11 curated pathways/functions: tumor immune microenvironment (TIME), ATP-binding cassette (ABC) transporter, ADCC pathway, cytoskeleton organization, endocytosis, intracellular trafficking, lysosome pathway, prognostic markers, target dimerization partners, topoisomerases, and tubulins (Supplementary Table S2). We performed multivariate Cox proportional regression analysis to assess the association of these features with T-DXd-specific overall survival (OS) in 1714 pre-T-DXd tumors with RNA expression data available.
Six genes were identified with significant associations between their expression levels and T-DXd OS (q < 0.05, Fig. 2a). Specifically, higher expression levels of ERBB2 (HER2) and FCGR3A (Fc gamma receptor IIIa) were each associated with improved T-DXd-specific OS [hazard ratio, i.e., HR associated with each unit (TPM) change of RNA < 1]. Conversely, increased expression of MKI67 (Ki-67), ABCC1 (ATP Binding Cassette Subfamily C Member 1, also known as multidrug resistance-associated protein 1 or MRP1), ABCA6 (ATP Binding Cassette Subfamily A Member 6), and RAB6A (Ras-related protein Rab-6A) correlated with worse T-DXd-specific OS. Stratifying the entire WTS-tested cohort based on expression quartiles of the six genes revealed an increase in T-DXd-specific OS with the highest ERBB2 expression and a stepwise decrease in T-DXd-specific OS with increased MKI67 and ABCC1 expression (Fig. 2b, c). Kaplan–Meier analysis showed no difference in T-DXd-specific OS according to FCGR3A, ABCA6, and RAB6A expression (Supplementary Fig. S1).

Expression level was treated as a continuous variable in the discovery work. a Hazard ratios are indicated by the circles, with upper and lower 95% confidence intervals represented by the whiskers. Statistical significance (log10 FDR-adjusted p-value) is indicated by the size of the circles, with larger circles indicating greater significance. Kaplan–Meier evaluation of ABCC1 (b), and ERBB2 (c) expression quartiles (Q4: highest expression, Q1: lowest expression) and association with T-DXd-specific overall survival. OS was calculated from start of treatment with T-DXd to last patient contact using insurance claims data. p value was calculated using log-rank test.
Given the generic role of Ki-67 as a prognostic marker of proliferation (supported by our findings that KI67 is prognostic regardless of T-DXd treatment (Supplementary Fig. S2), we focused on the ABC transporter ABCC1 as a potential predictor of resistance to T-DXd. We examined the correlation of ABCC1 with all other 95 features and found that ABCB7, ABCE1, and ABCF2 expression significantly correlated with ABCC1 [Spearman ρ > 0.6 (0.60–0.64)]. Adding interactions of ABCC1 with these three genes showed that ABCC1 remained an independent predictor of T-DXd-specific OS (Supplementary Table S3).
Expression of ERBB2 and ABCC1 across HER2 groups
As the expression of ERBB2 and ABCC1 was top predictors of T-DXd-associated OS, we further investigated their expression patterns in public datasets and in our dataset. Using the cBioPortal8,9, we found that ABCC1 was among the most frequently altered (amplifications and mRNA-high expression relative to diploid) and highly expressed ABC transporter at the RNA level in the Cancer Genome Atlas (TCGA) invasive breast cancer cohort, and its protein level correlated modestly with transcript expression (r ≈ 0.4) (Supplementary Fig. S3a–c). In our cohort, HER2-positive tumors were predominantly enriched among tumors with the top quartile expression of ERBB2. Conversely, HER2-low tumors showed a wide range of ERBB2 expression, with more equal distribution across the ERBB2 expression quartiles (>20% in each category). Similarly, HER2-ultra-low tumors had variable ERBB2 expression, although they comprised a smaller proportion of the highest ERBB2 expression group (Fig. 3a). Expectedly, we saw that ERBB2 TPM was significantly associated with ASCO/CAP HER2 categories10 as described in Supplementary Table S1 (Fig. 3b). Conversely, ABCC1 expression showed no association with HER2 status and remained consistent across all categories (Fig. 3b), which we also observed in TCGA cohort (Supplementary Fig. S3c).

a Distribution of HER2 status (HER2-null, HER2-ultra-low, HER2-low and HER2-positive categories) in ERBB2 quartiles (Q4: highest expression, Q1: lowest expression). Numbers of cases are shown within bars. b HER2 (ERBB2) expression (TPM) was significantly associated with HER2 status (left panel) while ABCC1 expression was independent of HER2 status (right panel).
Association of ERBB2 and ABCC1 with overall survival following T-DXd
Baseline clinical and demographic characteristics of the patient cohort stratified by ABCC1 expression quartiles are shown in Supplementary Table S5. Using Kaplan–Meier survival analysis, we observed that high expression of the drug efflux pump ABCC1 was significantly associated with decreased OS following T-DXd, implicating increased efflux of the cytotoxic deruxtecan payload may reduce T-DXd efficacy. As shown in Fig. 2c, patients in the bottom quartile of ABCC1 expression (Q1) had significantly improved OS compared to the top two quartiles (Q3 and Q4) of expression and numerical increase from Q2 A series of thresholds of ABCC1 expression were tested using 5 percentile increments, which justified using the top quartile as an optimal cutoff to achieve the most significance, meaningful hazard ratios, and optimal patient cohort sizes (Supplementary Table S4). Stratifying patients into ABCC1-High (Q4) and ABCC1-Low (ABCC1 Q1-3) revealed that the ABCC1-Low group had a significantly better OS (Fig. 4a).

Kaplan–Meier curves show median T-DXd-specific overall survival based on a ABCC1 high (Q4) vs. ABCC1 low (Q1-Q3); b HER2 status; c a combination of HER2 status and ABCC1 expression; and d a combination of ERBB2 expression and ABCC1 expression.
As expected, ASCO/CAP-guided HER2 categorization (using IHC and CISH) was strongly associated with T-DXd associated OS (Fig. 4b). Notably, stratifying into ABCC1-High and ABCC1-Low groups further predicted patient outcome within each HER2 category to some degree (Supplementary Fig. S4), particularly for patients with HER2-low and HER2-null tumors (Fig. 4c). Similarly, when patients were stratified using a combination of ERBB2 RNA expression and ABCC1 expression (Fig. 4d), a clear improvement in OS was seen for patients with low ABCC1 expression among both ERBB2-Low and ERBB2-High groups. Together, these findings highlight the importance of ABCC1 status in predicting treatment outcomes, independently of HER2/ERBB2.
To confirm that ABCC1 is a biomarker for T-DXd and not simply prognostic, we leveraged a large cohort of breast tumors with ABCC1 expression and outcome data. All patients were included in this cohort regardless of treatment. When analyzing OS calculated from tissue collection to last contact, there was no difference in OS between ABCC1-high versus ABCC1-low groups (HR = 1.03 (95% CI: 0.99–1.06), p = 0.152). When patients were stratified into quartiles based on ABCC1 expression, OS also did not show any incremental increase with increasing expression (Supplementary Fig. S5).
Post-trastuzumab deruxtecan molecular events
To identify the development of potential T-DXd resistance mechanisms, we compared molecular characteristics of tumors treated with T-DXd prior to tissue collection for NGS testing with tumors treated with T-DXd after tissue collection. As expected, ABCC1 expression was significantly higher in post-treatment samples compared to pre-treatment samples (median: 27 TPM vs. 22 TPM, p < 0.001). In contrast, we did not observe a similar post-treatment increase in a group of patients treated with trastuzumab, suggesting that the upregulation of ABCC1 is T-DXd-specific (Supplementary Fig. S6). In addition, potentially important mutational events occurring with significantly increased frequency in post-T-DXd samples are shown in Fig. 5a. In decreasing order, the most frequent mutational events occurred in ESR1 (q < 0.005), ERBB2 (q < 0.05), SMAD4 (p = 0.01; q = 0.3) NFE2L2 (q < 0.0005), TOP1 (q < 0.05) and KEAP1 (q = 0.074). The mutational landscape of all pre- and post-T-DXd tumors is shown in Supplementary Fig. S7.

a Comparison of pre- and post-treatment molecular profiles. All mutations shown have p < 0.05 (unadjusted p value). *: q < 0.05; **: q < 0.005; ***: q < 0.0005. b Lollipop graphs showing gain-of-function mutations of NFE2L2 and c loss-of-function mutations in KEAP1.
Mutations in ERBB2, such as those that inhibit trastuzumab binding, can induce resistance to HER2-targeted therapies, including T-DXd11,12,13. In our cohort, 78% of ERBB2 mutations occurred in the kinase domain, with similar proportions in pre- and post-treatment samples, implicating ERBB2 alterations as one potential resistance mechanism in addition to HER2 cell surface levels (Supplementary Fig. S8a). Likewise, mutations in TOP1 (topoisomerase 1) have also been reported to promote cross-resistance to ADCs in patients with MBC14. We similarly observed more TOP1 mutations in post-T-DXd-treated samples compared to T-DxD-naïve samples in our cohort (Fig. 5a and Supplementary Fig. S8b).
ESR1 mutations are known to be associated with prolonged aromatase inhibitor therapy in ER-positive breast cancer15, so the increased prevalence of ESR1 mutations here may merely represent a population with longer exposure to endocrine therapy. Consistent with this hypothesis, ESR1 mutations were highly concentrated in ER-positive tumors (18%, n = 260/1449), with far fewer being seen in ER-negative tumors (0.3%, n = 2/662), which are not treated with endocrine therapy. To further investigate this hypothesis, we summarized other pre-collection therapies that the patients received that may have exerted mutational pressures (Supplementary Table S5). Approximately one quarter of patients across all quartiles of ABCC1 expression received hormonal therapy prior to tissue collection, suggesting the plausibility of ESR1 mutations as an effect of this exposure.
NFE2L2 (also known as NRF2) is a transcriptional regulator that upregulates ABCC1, while KEAP1 is a negative regulator of NFE2L216,17. Thus, the gain-of-function mutations in NFE2L2 that we observed (Fig. 5b) are predicted to cause upregulation of ABCC1, while the loss-of-function mutations that we observed in KEAP1 (Fig. 5c) are predicted to promote increased activity of NFE2L2, also causing upregulation of ABCC1. We did not observe a significant difference in ABCC1 expression between tumors with and without NFE2L2 or KEAP1 mutations, but this analysis was not statistically powered (Supplementary Fig. S9).
Validation in ADC-resistant breast cancer cells
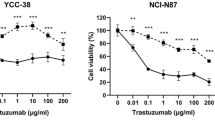
To evaluate the potential of ABCC1 inhibition in reversing ADC resistance, we first assessed the sensitivity of previously characterized HER2+ parental and T-DXd-resistant breast cancer cell lines18 to MK-571. IC50 values for MK-571 ranged from 31.7 to 33.1 μM in parental lines and from 30.2 to 38.5 μM in resistant counterparts (Fig. 6a). Western blot analysis demonstrated increased expression of ABCC1 in resistant line HCC1954-TDXdR compared to the respective parental line (Fig. 6b and Supplementary Fig. S10), supporting a potential role of ABCC1 in mediating resistance to T-DXd; the expression was comparable in the SUM190 resistant and parent lines.

a IC50 values for MK-571 in parental and resistant cell lines. b Western blot of ABCC1 levels in parental and resistant cell lines. β-actin was used as a loading control. Uncropped blots are shown in Supplementary Fig. S10. c MTS cell viability assay. Parental and resistant cell lines were treated with MK-571, T-DXd, or MK-571 + T-DXd for 10 days prior to MTS assay.
Further, combination treatments of MK-571 with T-DXd significantly reduced cell viability in HCC1954-TDXdR cells: co-treatment with MK-571 (20 μM) and T-DXd (133 nM) led to a significant reduction in viability compared to either agent alone (p < 0.0001, Fig. 6c). In SUM190-TDXdR cells, the combination produced a slight viability decrease, with MK-571 alone having little effect. The results in HCC1954 parent/resistant cell lines support the hypothesis that ABCC1-mediated drug efflux contributes to resistance to T-DXd, and its inhibition by MK-571 restores T-DXd sensitivity; the results seen in SUM190 lines suggest potential variations in resistance mechanisms within different contexts and warrant further investigation.
Discussion
T-DXd has significantly improved the outcomes of patients with metastatic breast cancer, both in patients traditionally considered HER2-positive as well as in HER2-low patients19,20,21. Nevertheless, a significant percentage of patients exhibit a priori resistance, and virtually all ultimately develop resistance to this agent. While multiple mechanisms of resistance have been suggested based on preclinical studies and limited clinical studies, there have been few large clinical studies exploring T-DXd resistance4,22.
To address this need, we explored clinical outcomes in a large real-world database of patients treated with T-DXd, whose tumors were profiled using NGS and HER2 IHC/CISH. To the best of our knowledge, this is the largest breast cancer dataset of T-DXd-treated patients to undergo such evaluations. Our basic approach was to follow the passage of T-DXd through breast cancer cells as illustrated in Fig. 1, evaluating the expression of genes associated with cell surface targeting, internalization, intracellular trafficking, lysosomal degradation, topoisomerase inhibition, and drug efflux from the cell. We also evaluated the expression of genes associated with the immune microenvironment. Our findings imply three important mediators of T-DXd resistance: (1) HER2 (ERBB2) target levels or mutations, potentially affecting both T-DXd binding and ADCC; (2) ABCC1-mediated efflux and mutations in genes regulating ABCC1 levels (NEF2L2, KEAP1); and (3) mutations in the gene for the target of the toxic payload of T-DXd, topoisomerase (TOP1) (Fig. 1).
Our work suggests that expression of only a limited number of genes predicts OS in T-DXd-treated patients. Pre-eminent among these is HER2 (ERBB2), whether measured at the protein (IHC) or transcript (NGS) level. At the protein level, median OS progressively improved with increased extracellular target expression, increasing from 10.8 months (HER2-null) to 12.7 months (HER2 ultra-low) to 17.5 months (HER2-low) to 27.3 months (HER2-positive); these results are highly consistent with the results of the phase 2 DAISY trial4. Similarly, we found that ERBB2 gene expression also predicts OS, with the greatest benefit seen in the highest quartile of expression.
When comparing transcript level with protein expression, classic IHC HER2-positive tumors predominantly fall within the top quartile of RNA expression. However, so-called HER2-low tumors show a wide range of ERBB2 expression, comprising more than 20% of each of the four quartiles. Similarly, there is considerable variability in ERBB2 transcript levels among HER2-null, HER2-ultra-low, and HER2-low tumors, suggesting a degree of fluidity and uncertainty of these designations.
In addition to HER2/ERBB2, ABCC1 gene expression appeared to have a moderate but significant effect on outcome in T-DXd-treated patients. ABCC1 (also known as MRP1) is a drug efflux pump that has been suggested to be upregulated in cells that are resistant to ADCs, leading to lower intracellular drug levels for shorter periods of time23. Considering the combination of HER2 and ABCC1, drug resistance might be considered as—in classic pharmacokinetic terms—a function of intracellular concentration (related to HER2) times time (related to ABCC1 drug efflux of T-DXd). Consistent with this concept, we show that the combination of high HER2 and low ABCC1 is associated with the best outcomes, and ABCC1 status stratifies patient outcome within each HER2 category. These findings are also consistent with prior preclinical studies showing that low HER2 levels and high ABCC1 expression mediate resistance to another HER2-targeted ADC, T-DM124,25,26, and may also suggest ABCC1 as a potential therapeutic target. While previous studies have implicated ABC transporters in resistance to various ADCs23,27,28,29, evidence linking a specific pump to T-DXd resistance has remained elusive. Our comprehensive survey of ABC transporter family gene expression identified ABCC1 as an efflux pump that may have particular relevance in mediating T-DXd resistance in breast cancer, offering a strong rationale for its therapeutic targeting. We acknowledge the role other efflux pumps may also play. A recent study in a cohort of 179 patients with breast cancer identified higher expression of both ABCB1 and ABCC1 as associated with shorter duration of T-DXd treatment, and ABCB1 was also associated with shorter T-DXd-specific OS30. However, our extensive survey of all known efflux pumps in the large cohort of T-DXd treated breast tumors showed that ABCB1 expression was not significantly associated with T-DXd OS in either multivariate or univariate analysis, in sharp contrast to ABCC1 expression.
Examining the mutational pressures induced by T-DXd treatment, we conclude that ERBB2 somatic mutations represent a rational potential mechanism of resistance to T-DXd. Previous studies have demonstrated that breast cancers with these mutations are sensitive to (some) kinase inhibitors but not to monoclonal antibody-based therapies, and indeed often lack an external HER2 membrane domain31,32. Similarly, NFE2L2 and KEAP1 mutations represent rational resistance mechanisms for T-DXd through their impact on ABCC1 expression16,17. Interestingly, preclinical studies have shown that camptothecin, a tecan-like topoisomerase I inhibitor, suppresses NFE2L2 expression33,34. Therefore, prolonged exposure to T-DXd may select for mutations in the NFE2L2/KEAP1 pathway—as we observed in our study (Fig. 6B, C)—thereby circumventing this particular mechanism of increased drug sensitivity.
Although these results present plausible resistance mechanisms supported by our clinical outcome findings, there are limitations to this post-T-DXd enrichment analysis. First, patients were exposed to a variety of other therapies at various timepoints that may confound the effects of T-DXd (Supplementary Table S5). Second, we did not have matched patient samples to examine mutational pressures at the individual patient level and therefore cannot establish causality between T-DXd treatment and the emergence of the identified mutations. However, in our large cohort of unpaired samples, we observed expected and previously reported events associated with T-DXd resistance—such as TOP1 mutations14—supporting the validity of our findings. Third, we did not observe differences in ABCC1 levels based on the presence or absence of NFE2L2 or KEAP1 mutations (although this analysis was not well powered). Expression of ABCC1 can be regulated by methylation status that might be relevant in treatment-resistant breast cancers, potentially enabling expression changes without upstream genetic alterations35. In addition, post-transcriptional regulation by microRNAs (e.g., miR-326, miR-133a) may modulate ABCC1 abundance and function, influencing drug efflux independently of genomic events36,37. Therefore, ABCC1 upregulation and T-DXd resistance may occur via multiple converging mechanisms, of which NRF2–KEAP1 dysregulation represents only one pathway. Systematic functional studies will be needed to dissect these diverse regulatory routes and their relative clinical importance. An additional limitation to our study is the limited in vitro mechanistic evidence. While results in one cell line support our conclusions regarding ABCC1, the lack of effect of ABCC1 inhibition on viability of SUM190-TDXdR cells points to context-dependent resistance mechanisms that may not be overcome by blocking ABCC1. Furthermore, we only used HER2+ cell line models and thus cannot infer a similar effect in the context of HER2-low or HER2-ultra-low. Finally, a limitation of real-world data is that we are restricted to information available in insurance claims databases. As claims data lack standard trial endpoints and treatment discontinuation details, treatment-associated OS was used to assess T-DXd benefit as a more objective and clinically meaningful endpoint.
In conclusion, our work suggests that the combination of reduced HER2 cell surface expression and increased ABCC1-mediated efflux is associated with resistance to T-DXd in metastatic breast cancer. It will be of interest to see whether similar resistance patterns occur in other HER2-targeting scenarios, given the recent tissue-agnostic approval of T-DXd for HER2-positive solid tumors by the US Food and Drug Administration38. Future work could involve expanded cohorts and unbiased transcriptomic profiling to place our findings on ABCC1 within the context of broader resistance pathways and identify additional transcriptional programs associated with T-DXd outcomes. The identification of resistance mechanisms might lead to improved precision of administration of this agent and could lead to novel therapeutic approaches to overcome resistance.
Methods
Patient samples
A total of 2799 T-DXd-treated breast cancer samples that underwent comprehensive tumor profiling at Caris Life Sciences (Phoenix, AZ, USA) were included in our analysis. No matched samples were available for analysis. This study was conducted in accordance with the guidelines of the Declaration of Helsinki, the Belmont Report, and the US Common Rule. In accordance with 45 CFR 46.104(d)(4), this study was conducted using retrospective, de-identified clinical data and is considered exempt from Institutional Review Board (IRB) review, with a waiver of patient-informed consent. Waiver of patient consent and exempt status were determined by WCG IRB.
Tumor profiling
Tumor profiling was performed in a CAP/CLIA-certified laboratory, and all results meet standards for clinical reporting. Formalin-fixed paraffin-embedded (FFPE) specimens underwent pathology review to measure percent tumor content and tumor size. Microdissection was performed on all tumor samples to enrich tumor content and ensure comparability across samples. RNA sequencing data were analyzed in the pre-T-DXd samples (n = 2420), and DNA sequencing data were analyzed in all samples (n = 2799).
For whole transcriptome sequencing (WTS), a minimum of 10% of tumor content in the area for microdissection was required. Qiagen RNA FFPE tissue extraction kit was used for extraction, and the RNA quality and quantity were determined using the Agilent TapeStation. Biotinylated RNA baits were hybridized to the synthesized and purified cDNA targets, and the bait-target complexes were amplified in a post-capture PCR reaction. WTS was performed on an Illumina NovaSeq 6000 system (Illumina, San Diego, CA, USA) (RRID:SCR_016387) to an average of 60 M reads. Raw data were demultiplexed by Illumina Dragen BioIT accelerator, trimmed, counted, and sequences aligned to human reference genome hg19 by Spliced Transcripts Alignment to a Reference (STAR) (RRID:SCR_004463)39. Transcripts per million (TPM) values were generated using the Salmon expression pipeline (RRID:SCR_017036)40. Interferon-gamma scores were calculated using an 18-gene signature previously shown to be associated with immune checkpoint inhibitor outcome in various cancer types41. The RNA deconvolution program QuanTISeq (RRID:SCR_022993)42 was used to infer cell populations in the tumor immune microenvironment.
Genomic DNA was sequenced using the NextSeq 500 (RRID:SCR_014983) or NovaSeq 6000 platforms (Illumina, Inc.). For NextSeq sequenced tumors, a custom-designed SureSelect XT assay was used to enrich 592 whole-gene targets (Agilent Technologies, Santa Clara, CA). For NovaSeq sequenced tumors, a panel covering more than 700 clinically relevant genes at high coverage and high read-depth was used, along with another panel designed to enrich for an additional >20,000 genes at lower depth. Variants detected were mapped to reference genome hg19 (592-gene panel) or hg38 (WES) using the Burrows-Wheeler Aligner (BWA 0.7.17) (RRID:SCR_010910) embedded in the analysis tools licensed from Sentieon®. Bioinformatic tools, including SamTools (RRID:SCR_002105), Pindel (RRID:SCR_000560), and snpEff (RRID:SCR_005191) were incorporated to perform variant calling functions, and annotations were standardized to the Human Genome Variation Society format. Filtering was performed to remove benign variants, low-quality and low-depth variants, or variants determined to be unreliable in several analyses, like strand bias and repeat analysis. The 5% variant allele frequency and five alignments supporting a variant were required to call positive variants. Board-certified molecular geneticists interpreted genetic variants identified according to the American College of Medical Genetics and Genomics (ACMG) standards.
HER2 immunohistochemistry (IHC) was performed using the VENTANA® Pathway anti-HER2/neu RxDx (4B5) antibody (Roche (Ventana), Tucson, AZ, USA). HER2 chromogenic in-situ hybridization (CISH) was performed using the INFORM HER-2 Dual ISH DNA Probe Cocktail (Roche (Ventana). IHC was performed on full FFPE sections of glass slides, and results were read by a board-certified pathologist; all scoring followed American Society of Clinical Oncology (ASCO)/College of American Pathologists (CAP) guidelines10 (Supplementary Table S1). HER2 status was determined by a combination of IHC and CISH: HER2-positive: IHC 3+ with >10% of cells positive; HER2-low: IHC 1+ with >10% of cells positive, or IHC 2+ with >10% of cells positive combined with a negative CISH result; HER2-ultra-low: IHC 1+, 2+, or 3+ with ≤10% of cells positive; HER2-null: IHC 0+.
Statistical analysis
Real-world clinical data were obtained from insurance claims, which encompass detailed records of health services that patients received. This includes prescribed medications, performed procedures, and established diagnoses. Overall survival (OS) was calculated from tissue collection to the date of the patient’s last known clinical activity, while treatment-specific OS was defined as the period from the start of treatment with T-Dxd to last contact. Patient death information, when available, was used to calculate patient survival. For patients with no death dates on record, and there were no insurance claims for a period exceeding 100 days, it was inferred that the patient had deceased. Conversely, patients who had a documented clinical activity within 100 days prior to the latest data update were considered censored in the analysis. Kaplan–Meier survival estimates were generated for cohorts defined by molecular characteristics. The Hazard Ratio (HR) was computed utilizing the Cox proportional hazards model, and the significance of differences (p values) in survival times was assessed with the log-rank test. Multivariate analysis was conducted using RNA TPM inputs as continuous variables in a Cox-Proportional Hazard model, with significance determined at p < 0.05. Genes and RNA signatures surveyed for correlation with patient outcomes are shown in Supplementary Table 2. Molecular comparisons were performed using Chi-square tests, and p values were adjusted for multiple comparisons using the Benjamini-Hochberg method (q < 0.05).
Preclinical studies in cell lines
The HER2-positive breast cancer cell lines HCC1954 and SUM190 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and BioIVT (Westbury, NY, USA), respectively. HCC1954 cells were cultured in RPMI-1640 medium (GenDEPOT, Katy, TX, USA) supplemented with 10% fetal bovine serum (FBS; GenDEPOT) and 1% antibiotic-antimycotic solution (GenDEPOT). SUM190 cells were maintained in Ham’s F-12 medium (Thermo Fisher, Waltham, MA, USA) supplemented with 5% FBS, 1% antibiotic-antimycotic, 5 µg/mL insulin (Sigma-Aldrich, St. Louis, MO, USA), and 1 µg/mL hydrocortisone (Sigma-Aldrich). All cell lines were authenticated by short tandem repeat (STR) profiling using primer extension to detect single-base deviations and were confirmed to be free of mycoplasma contamination using the MycoAlert™ Mycoplasma Detection Kit (Lonza, Morristown, NJ, USA).
T-DXd-resistant cell lines (HCC1954-TDXdR and SUM190-TDXdR) were utilized alongside their parental counterparts to evaluate ABCC1 expression and assess the effect of ABCC1 inhibition on reversing T-DXd resistance. As previously characterized, the T-DXd resistant cell lines display reduced ERBB2 gene copy number amplification and reduced HER2 protein and cell surface expression compared to their sensitive counterparts, while retaining dose-dependent growth inhibition to free DXd18. Sulforhodamine B (SRB) was obtained from Sigma–Aldrich, and the ABCC1 inhibitor MK-571 was purchased from MedChemExpress (Monmouth Junction, NJ, USA). For cell viability assays, cells were seeded in 96-well plates and treated with serial dilutions of MK-571, T-DXd, or their combination for 10 days. Cell viability was quantified using the SRB assay, and absorbance was measured using a Tecan Spark microplate reader (Baldwin Park, CA, USA). Viability was expressed as a percentage relative to untreated control cells. Half-maximal inhibitory concentration (IC50) values were calculated using non-linear regression analysis in GraphPad Prism (version 10.0). ABCC1 protein levels were evaluated by Western blotting. Total protein lysates were prepared from both parental and resistant cell lines using RIPA buffer supplemented with protease inhibitors (GenDEPOT). Protein concentration was determined using the BCA assay (Thermo Fisher). Equal amounts of total protein (25 µg) were separated by SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were probed with primary antibodies against ABCC1 (Clone:1G4A2, Proteintech, Rosemont, IL, USA) and β-actin as loading control (Clone: AC-15, Sigma–Aldrich), followed by appropriate HRP-conjugated secondary antibodies (Thermo Fisher). Protein bands were detected using enhanced chemiluminescence and visualized with the Azure 500 imaging system (Azure Biosystems, Dublin, CA, USA).
Data availability
The deidentified sequencing data are owned by Caris Life Sciences and cannot be publicly shared due to the data usage agreement in place. These data will be made available to researchers for replication and verification purposes through our letter of intent process, which is generally fulfilled within 6 months. For more information on how to access this data, please contact Joanne Xiu at [jxiu@carisls.com](mailto:jxiu@carisls.com).
References
Khongorzul, P., Ling, C. J., Khan, F. U., Ihsan, A. U. & Zhang, J. Antibody-drug conjugates: a comprehensive review. Mol. Cancer Res. 18, 3–19 (2020).
Ríos-Luci, C. et al. Resistance to the antibody-drug conjugate T-DM1 is based in a reduction in lysosomal proteolytic activity. Cancer Res. 77, 4639–4651 (2017).
Sung, M. et al. Caveolae-mediated endocytosis as a novel mechanism of resistance to trastuzumab emtansine (T-DM1). Mol. Cancer Ther. 17, 243–253 (2018).
Mosele, F. et al. Trastuzumab deruxtecan in metastatic breast cancer with variable HER2 expression: the phase 2 DAISY trial. Nat. Med. 29, 2110–2120 (2023).
Zhang, L. et al. Antibody-drug conjugates and immune checkpoint inhibitors in cancer treatment: a systematic review and meta-analysis. Sci. Rep. 14, 22357 (2024).
Chang, H. L., Schwettmann, B., McArthur, H. L., & Chan, I. S. Antibody-drug conjugates in breast cancer: overcoming resistance and boosting immune response. J. Clin. Investig. 133, e172156 (2023).
Martín, M. et al. Trastuzumab deruxtecan in breast cancer. Crit. Rev. Oncol. Hematol. 198, 104355 (2024).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013).
Wolff, A. C. et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology–College of American Pathologists guideline update. Arch. Pathol. Lab. Med. 147, 993–1000 (2023).
Chen, W. et al. Abstract LB280: resistance to trastuzumab deruxtecan (T-DXd) in breast cancer via loss of HER2 expression and binding. Cancer Res. 85, LB280–LB (2025).
Liu, D. et al. Diverse ERBB2/ERBB3 activating alterations and coalterations have implications for HER2/3-targeted therapies across solid tumors. Cancer Res. Commun. 5, 680–693 (2025).
Yamaguchi, K. et al. Impact of genomic alterations on efficacy of trastuzumab deruxtecan against human epidermal growth factor receptor-2-positive advanced gastric cancer. JCO Precis. Oncol. 8, e2300681 (2024).
Abelman, R. O. et al. TOP1 mutations and cross-resistance to antibody-drug conjugates in patients with metastatic breast cancer. Clin. Cancer Res. 31, 1966–1974 (2025).
Clatot, F. et al. Kinetics, prognostic and predictive values of ESR1 circulating mutations in metastatic breast cancer patients progressing on aromatase inhibitor. Oncotarget 7, 74448–74459 (2016).
Itoh, K. et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13, 76–86 (1999).
Cao, J. Y. et al. A genome-wide haploid genetic screen identifies regulators of glutathione abundance and ferroptosis sensitivity. Cell Rep. 26, 1544–1556.e8 (2019).
Lee, J. et al. The DNA repair pathway as a therapeutic target to synergize with trastuzumab deruxtecan in HER2-targeted antibody-drug conjugate-resistant HER2-overexpressing breast cancer. J. Exp. Clin. Cancer Res. 43, 236 (2024).
Denkert, C. et al. Clinical and molecular characteristics of HER2-low-positive breast cancer: pooled analysis of individual patient data from four prospective, neoadjuvant clinical trials. Lancet Oncol. 22, 1151–1161 (2021).
Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N. Engl. J. Med. 387, 9–20 (2022).
Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N. Engl. J. Med. 382, 610–621 (2020).
Guidi, L., Pellizzari, G., Tarantino, P., Valenza, C., & Curigliano, G. Resistance to antibody-drug conjugates targeting HER2 in breast cancer: molecular landscape and future challenges. Cancers 15, 1130 (2023).
Gandullo-Sánchez, L., Ocaña, A., & Pandiella, A. Generation of antibody-drug conjugate resistant models. Cancers 13, 4631 (2021).
Loganzo, F. et al. Tumor cells chronically treated with a trastuzumab-maytansinoid antibody-drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol. Cancer Ther. 14, 952–963 (2015).
Li, G. et al. Mechanisms of acquired resistance to trastuzumab emtansine in breast cancer cells. Mol. Cancer Ther. 17, 1441–1453 (2018).
Le Joncour, V. et al. A novel anti-HER2 antibody–drug conjugate XMT-1522 for HER2-positive breast and gastric cancers resistant to trastuzumab emtansine. Mol. Cancer Ther. 18, 1721–1730 (2019).
Yu, S. F. et al. A novel anti-CD22 anthracycline-based antibody-drug conjugate (ADC) that overcomes resistance to auristatin-based ADCs. Clin. Cancer Res. 21, 3298–3306 (2015).
Loganzo, F., Sung, M. & Gerber, H. P. Mechanisms of resistance to antibody-drug conjugates. Mol. Cancer Ther. 15, 2825–2834 (2016).
Chen, R. et al. CD30 downregulation, MMAE resistance, and MDR1 upregulation are all associated with resistance to brentuximab vedotin. Mol. Cancer Ther. 14, 1376–1384 (2015).
Alkassis, S. et al. Transcriptomic biomarkers of therapeutic response to antibody-drug conjugates in metastatic breast cancer: a comprehensive multi-center study. J. Clin. Oncol. 43, 1041 (2025).
Zuo, W. J. et al. Dual characteristics of novel HER2 kinase domain mutations in response to HER2-targeted therapies in human breast cancer. Clin. Cancer Res. 22, 4859–4869 (2016).
Xu, X. et al. HER2 reactivation through acquisition of the HER2 L755S mutation as a mechanism of acquired resistance to HER2-targeted therapy in HER2(+) breast cancer. Clin. Cancer Res. 23, 5123–5134 (2017).
Chen, F. et al. Camptothecin suppresses NRF2-ARE activity and sensitises hepatocellular carcinoma cells to anticancer drugs. Br. J. Cancer 117, 1495–1506 (2017).
Liu, Q. et al. Nrf2 down-regulation by camptothecin favors inhibiting invasion, metastasis and angiogenesis in hepatocellular carcinoma. Front. Oncol. 11, 661157 (2021).
Arrigoni, E., Galimberti, S., Petrini, M., Danesi, R. & Di Paolo, A. ATP-binding cassette transmembrane transporters and their epigenetic control in cancer: an overview. Expert Opin. Drug Metab. Toxicol. 12, 1419–1432 (2016).
Hua, Y. T., Xu, W. X., Li, H. & Xia, M. Emerging roles of MiR-133a in human cancers. J. Cancer 12, 198–206 (2021).
Liang, Z. et al. Involvement of miR-326 in chemotherapy resistance of breast cancer through modulating expression of multidrug resistance-associated protein 1. Biochem. Pharm. 79, 817–824 (2010).
United States Food and Drug Administration. FDA grants accelerated approval to fam-trastuzumab deruxtecan-nxki for unresectable or metastatic HER2-positive solid tumors. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-fam-trastuzumab-deruxtecan-nxki-unresectable-or-metastatic-her2 (2024).
Dobin, A. et al. T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Cristescu, R. et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362, eaar3593 (2018).
Finotello, F. et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 11, 34 (2019).
Acknowledgements
This study was supported by Caris Life Sciences and The University of Hawai’i Cancer Center Support Grant (P30CA071789, Shared Resources: Preclinical Core).
Author information
Authors and Affiliations
Contributions
Conceptualization: G.W.S., J.X., D.B.S., and N.T.U. Methodology: N.T.U., J.L., and D.R.R. Software: N.K. Formal analysis: J.X., J.L., D.R.R., and J.R.R. Investigation: G.W.S., J.X., R.L.M., A.C.S.L., F.M.-B., D.R.R., J.L., and N.T.U. Resources: M.J.O., M.R., and D.B.S. Data Curation: N.K. Writing—original draft: J.X. and J.R.R. Writing—review and editing: J.R.R., J.X., and G.W.S. Visualization: J.X. and J.R.R. Supervision: G.W.S. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
G.W.S., J.X., J.R.R., N.K., M.J.O., M.R., and D.S. are employees of Caris Life Sciences. N.T.U. reports consulting roles (AstraZeneca Pharmaceuticals LP (USA and UK), Bayer AG, Bristol Myers Squibb Company, Carna Biosciences, Inc., CytoDyn Inc., Daiichi Sankyo Co., Ltd., Eisai Co., Ltd., Eli Lilly, Genentech, Inc., Genomic Health, Inc., Gilead Sciences, Inc., Lavender Health, OncoCyte Corporation, Pear Bio, Peptilogics, Inc., Pfizer Inc., Phoenix Molecular Designs, Preferred Medicine, Carisma Therapeutics, Inc., Sysmex Corporation, Takeda Pharmaceutical Company Limited (Japan), and Unitech Medical, Inc.); stock ownership (Pear Bio and Phoenix Molecular Designs); speaker or preceptorship roles (AstraZeneca Pharmaceuticals LP, Chugai Roche, Inc., Daiichi Sankyo Co., Ltd., Kyowa Kirin Co., Ltd., Pfizer Inc., Total Health Conferencing, and Eli Lilly); and research agreements (AnHeart Therapeutics Inc., Eisai Co., Ltd., Gilead Sciences, Inc., Phoenix Molecular Designs, Daiichi Sankyo, Inc., Puma Biotechnology, Inc., Merck & Co., Oncolys BioPharma Inc., OBI Pharma Inc., ChemDiv, Inc., Tolero Pharmaceuticals, Inc., and VITRAC Therapeutics, LLC). J.L. reports scientific advisory role (Astrion Inc.) and sponsor research agreements (Eisai Co., Ltd., OBI Pharma Inc., Duality Biologics, Jazz Pharmaceutical, and CytoDyn Inc.). D.R.R. reports no conflicts of interest. RLM reports consulting/advisory roles (Agendia, Arvinas, AstraZeneca, Daiichi, Genentech, Gilead, Hologic, Lilly, Novartis, Pfizer, Puma, Stemline) and research funding (Gilead, paid to institution). FM reports consulting roles (AstraZeneca Pharmaceuticals, Becton Dickinson, Biocartis NV, Calibr (a division of Scripps Research), Daiichi Sankyo, Dava Oncology, Debiopharm, EcoR1 Capital, eFFECTOR Therapeutics, Elevation Oncology, Exelixis, GT Aperion, Incyte, Jazz Pharmaceuticals, LigaChem Biosciences, Menarini Group, Molecular Templates, Protai Bio, Ribometrix, SystImmune, Tempus, Vir Biotechnology, Zymeworks); advisory committee roles (Cybrexa Therapeutics, go Therapeutics, Guardant Health, Illumen Therapeutics, Kivu Biosciences, LOXO-Oncology, Mersana Therapeutics, Seagen, Theratechnologies, Zentalis Pharmaceuticals); sponsored research (to institution: Jazz Pharmaceuticals, Zymeworks, Aileron Therapeutics, Inc. AstraZeneca, Bayer Healthcare Pharmaceutical, Calithera Biosciences Inc., Curis Inc., CytomX Therapeutics Inc., Daiichi Sankyo Co. Ltd., Debiopharm International, eFFECTOR Therapeutics, Genentech Inc., Guardant Health Inc., Klus Pharma, Takeda Pharmaceutical, Novartis, Puma Biotechnology Inc., Taiho Pharmaceutical Co.); honoraria (Dava Oncology); and other (travel: Cholangiocarcinoma Foundation, Dava Oncology). ACSL reports consulting/advisory roles (AstraZeneca, Stemline, Delphie, Lilly); associated research funding to institution (AstraZeneca, Merck, Pfizer, Genentech); and grants to institution (Gilead).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sledge, G.W., Xiu, J., Mahtani, R.L. et al. Mechanisms of resistance to trastuzumab deruxtecan in breast cancer elucidated by multi-omic molecular profiling. npj Breast Cancer 12, 1 (2026). https://doi.org/10.1038/s41523-025-00868-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41523-025-00868-y
