
Abstract
The Lieb lattice is fundamental in condensed matter physics for hosting exotic electronic and topological states. Through high-throughput computational screening of 1470 binary metal-inorganic frameworks (MIFs), we identified 24 stable Lieb lattice structures, including 22 new materials. These comprise 15 nonmagnetic, 2 ferromagnetic (FM) half-metals, and 7 antiferromagnetic semiconductors, with critical temperatures reaching 877 K. Key electronic features include flat bands, Dirac cones, and van Hove singularities. HfCl₂ and WO₂ are FM half-metals with large spin gaps (5.37 eV and 3.57 eV), enabling full spin polarization. Be₂C and ReF₂ exhibit nodal loops and quasi-flat bands, respectively, hosting nontrivial topology confirmed by edge-state analysis. Nine MIFs are zero-dimensional electrides with work functions as low as 2.64 eV. Thirteen structures are ground-state phases, ensuring stability. These Lieb lattices offer promising platforms for high-temperature electronic, spintronic, and topological applications.
Similar content being viewed by others
Introduction
The Lieb lattice is a two-dimensional structure composed of three atoms arranged in an edge-depleted square pattern within each unit cell1. This arrangement exemplifies geometric frustration and closely resembles the metal-oxygen planes found in cuprate superconductors2,3. In an ideal Lieb lattice, the interference of electron waves creates a unique band structure featuring a Dirac cone intersected by a flat band at the Fermi level4,5,6,7. The Dirac cones are associated with massless charge carriers, while the flat bands result in minimal kinetic energy. Consequently, the Lieb lattice serves as an ideal platform for exploring various physical phenomena, including superconductivity8, itinerant-electron ferromagnetism1,9,10, various topological nontrivial states11,12,13,14,15,16, and polariton condensation17. These distinct electronic structures also hold promise for applications in optics18,19, electronics20, spintronics21, topological Mott insulators22, superconductors23, and energy conversion and storage technologies24.
The unique characteristics of the Lieb lattice have attracted significant research interest, particularly in the fields of strongly correlated systems and the search for real materials that embody this lattice structure. While Lieb lattices have been successfully created in artificial systems such as electronic6, photonic4,5, and cold-atom setups22, realizing them in actual materials remains challenging due to theoretical constraints like the Landau-Peierls-Mermin-Wagner theorem. Theoretically, two covalent organic frameworks (COFs), namely sp2-C-COF and sp2-N-COF, have been recognized as distorted Lieb lattices9,10. Additionally, an experimentally synthesized phthalocyanine-based metal-organic framework (MPc-MOF) has been classified under the Lieb lattice category14. However, these COFs and MOFs are typically trivial insulators because of factors such as large hopping distances, significant wave function overlaps, and interaction beyond nearest neighbors, which hinder the exploration of their exotic quantum states.
Metal-inorganic frameworks (MIFs), with their expansive chemical space and simplified monoatomic layer structures, are promising candidates for hosting Lieb lattices. Recently, a buckled Lieb lattice formed by a tin overlayer on an Al(100) surface was experimentally realized, although it does not exhibit the characteristic electronic features of an ideal Lieb lattice15. Furthermore, the synthesis of planar Cu2N monolayer and substrate-stabilized Pt-P adlayer has demonstrated Lieb lattice morphology with partial flat bands, Dirac cones, and van Hove singularities (VHSs) through molecular beam epitaxy12,13. Despite this progress, Lieb lattices in MIFs are rarely reported, and it is crucial to explore and identify new Lieb lattices within MIFs. Conducting a comprehensive screening of stable Lieb lattice structures in MIFs and understanding of their intrinsic physical properties are essential steps towards advancing scientific knowledge and the practical application of Lieb lattices.
In this study, we present a family of two-dimensional (2D) MIFs featuring stable Lieb lattices with diverse electronic characteristics, identified through high-throughput first-principles calculations and extensive structure search. From 1470 binary inorganic compounds with metal-to-nonmetal ratios of 1:2 and 2:1, we discovered 24 stable MIF-based Lieb lattice structures, 22 of which are newly reported. These include 14 nonmagnetic (NM) metals, one Dirac semimetal (DSM), two ferromagnetic (FM) half-metals, and seven antiferromagnetic (AFM) semiconductors. Notably, the Be2C monolayer is recognized as a Dirac nodal-loop semimetal, and the ReF2 monolayer exhibits flat bands below the Fermi level. Additionally, Au2N, Cu2N, and another Au2N structure display partial flat bands and VHSs in their band structures. The existence of nontrivial topological properties is further confirmed through edge state analysis. Our global structure search also establishes that the Lieb lattices of IrO2, OsO2, RhO2, RuO2, TiF2, WO2, ZrF2, Ag2N, Ag2O, Au2N, Be2C, Cd2B, Mg2B, and Zn2B are global minimum structures. Here, global minimum structure denotes the atomic arrangement possessing the lowest energy across the entire potential energy surface under two-dimensional confinement, while stable structure corresponds to a dynamically stable Lieb lattice. The reproducibility of band structure characteristics and the enhancement of critical temperature have also been demonstrated when Lieb lattices are deposited on a substrate. This study establishes a foundation for further exploration of Lieb lattices in MIFs and their potential applications in various technological fields.
Results
Identification and screening of stable Lieb lattices in MIFs
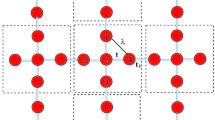
The Lieb lattice is composed of one corner site (A) and two edge-centered sites (B), where site-A coordinates with four atoms and site-B coordinates with two, as illustrated in Fig. 1a. This 2D structure belongs to the P4/mmm (No.123) space group and possesses D4h symmetry. In MIFs featuring a Lieb lattice, both site-A and site-B can be occupied by either metallic or nonmetallic elements. To thoroughly search for stable Lieb lattices in MIFs, we selected 49 elements, including alkali, alkaline earth, transition, and p-block metals, along with 14 nonmetallic elements ranging from hydrogen to iodine (Fig. 1b). This selection resulted in 735 possible combinations where site-A is a metal and site-B is a nonmetal, and another 735 combinations where site-A is a nonmetal and site-B is a metal, totaling 1470 binary MIFs for initial screening (Fig. 1c).

a Crystal structure of the Lieb lattice comprises a single corner site-A, and two edge-centered sites-B, within the unit cell which is marked by yellow dashed lines. b The metal and nonmetal elements adopted in the construction of the Lieb lattice. c Workflow for screening stable Lieb lattices from 1470 metal-inorganic frameworks (MIFs). NM, FM, and AFM represent nonmagnet, ferromagnet, and antiferromagnet, respectively. d The percentage of stable MIFs identified. e The binding energies of screened MIFs. The binding energy of experimental realized Cu2N is indicated by the dashed line.
The screening process involved several steps. First, all 1470 MIFs were optimized to find their most stable structures. Next, the magnetic properties of each optimized structure were assessed to identify their ground states. Third, phonon spectra were calculated to evaluate the dynamic stability of the structure. Finally, ab initio molecular dynamics (AIMD) simulations were performed to test the thermal stability of the candidates. Through this high-throughput screening, only 24 MIFs with stable Lieb lattices were identified (1.63% of the initial structures) (Table 1), underscoring the rarity of Lieb lattices in real materials. Specifically, 10 metal-rich MIFs had site-B occupied by a metal, while 14 nonmetal-rich MIFs had site-A occupied by a metal. Among the 24 stable Lieb lattices, Cu2N and Be2C were previously reported materials11,12. Regarding magnetic properties, 2 materials (HfCl2 and WO2) are ferromagnetic (FM) and 7 materials (MoF2, OsF2, ReF2, RuF2, RuO2, TiF2, and ZrF2) are antiferromagnetic (AFM).
To evaluate the feasibility of synthesizing the screened Lieb lattice, extensive structure searches were conducted. As shown in Supplementary Fig. 1, IrO2, OsO2, RhO2, RuO2, TiF2, WO2, ZrF2, Ag2N, Be2C, Cd2B, Cu2N, Mg2B, and Zn2B were identified as ground-state structures at their stoichiometric ratio. The binding energies of these Lieb lattices range from −1.73 to −6.37 eV per atom. Except for Ag2N, Au2N, Cd2B, Hg2Se, Hg2Te, Mg2B, Zn2B, HgH2, and RhO2, the other 14 MIFs all have more negative binding energies than experimentally realized Cu2N (−3.46 eV per atom). Additionally, Au2N, RuF2, and MoF2 exhibit small relative energies of 0.04 eV, 0.11 eV, and 0.18 eV per atom compared with their global minimum structures, respectively, suggesting that they could also be stabilized through growth on a suitable substrate25. The formation energies of HfCl2, IrN2, OsF2, and ReF2 were calculated to be −0.74 eV, −0.19 eV, −0.16 eV, and -0.39 eV per atom, respectively. These values are comparable to that of the global minimum structure pp-Fe4N2 (−0.24 eV per atom)26, indicating that the formation of the Lieb lattice in these MIFs is thermodynamically favorable. From an energetic perspective, the coplanar Be2B is less stable than its bulk crystal counterpart by 0.86 eV per atom, which is similar to the energy difference between silicene and bulk silicon (0.83 eV per atom)11. We also notice that planar Cu2N, distorted Pt-P, and buckled AlSn alloy Lieb lattices have been experimentally realized on metal substrates through molecular beam epitaxy12,13,15. The results indicate that these MIFs are likely to be fabricated using advanced experimental techniques such as atomic layer deposition and molecular beam epitaxy techniques12,13,15. The subsequent sections provided a detailed analysis of the structural, electronic, magnetic, and topological properties of these stable Lieb lattices.
The lattice constants of the identified Lieb lattices range from 3.28 to 5.83 Å for metal-rich MIFs and from 3.70 to 4.93 Å for nonmetal-rich MIFs (Table 1). Bader charge analysis indicates that electrons are transferred from metal to nonmetal atoms, a trend supported by charge density difference plots (Supplementary Fig. S2). The charge transfer values vary from 0.22 electrons in Hg2Te to 3.11 electrons in Be2B for site-A, and from 0.32 electrons in HgH2 to 2.36 electrons in IrO2 for site-B. These findings demonstrate a strong interaction between metal and nonmetal atoms within the Lieb lattice structures.
Electron localization functions (ELF) maps further highlight the diversity of bonding types within the Lieb lattice (Supplementary Fig. S3). Specifically, HgH2, Hg2Se, and Hg2Te exhibit metallic bonding, characterized by delocalized electrons spread throughout the framework. In contrast, Be2B, Be2C, RhO2, and IrO2 display covalent-like bonding, as evidenced by regions of high ELF value between metal and nonmetal atoms. For other MIFs, the ELF plots show delocalized electrons around metal atoms and localized electrons around nonmetal atoms, with low ELF values in the interstitial regions along metal-nonmetal directions, indicative of typical ionic bonding. Interestingly, excess localized electrons were also observed in several screened MIFs, which is attributed to the diverse bonding characteristics and various potential valence states of transition metal elements. ELF maps (Supplementary Fig. S4) reveal that electrons are spatially localized above and below the transition metal atoms. These excess electrons, isolated from the planar atomic frameworks, indicate that the HfCl2, OsF2, ReF2, RuF2, TiF2, WO2, ZrF2, IrN2, and OsO2 function as zero-dimensional (0D) electrides.
To evaluate lattice stability, phonon spectra were calculated for the 24 stable MIFs (Supplementary Fig. S5). The absence of imaginary frequencies along a high-symmetry path confirms the dynamic stability of these MIFs. Additionally, AIMD simulations were performed to evaluate thermal stability using a 5 × 5 supercell. The simulations revealed that Hg2Se, Hg2Te, and HgH2 could not maintain their lattice structure even at 100 K, making them unsuitable for practical application at room temperature (Supplementary Fig. S6). IrN2 retained its 2D lattice at 100 K after a 5 ps simulation. The lattice of IrO2, RhO2, and ZrF2 remained stable up to 300 K, while HfCl2 and Au2N maintained their structures at 400 K. Except for MoF2 and ReF2, which decomposed above 600 K, the remaining 13 MIFs preserved their 2D networks with only minor energy fluctuations at higher temperatures over the 5 ps simulation period (e.g., TiF2 and RuO2 at 700 K, Be2B and WO2 at 800 K, OsF2 at 900 K, RuF2 at 1000 K, Ag2N at 1200 K, Cd2B and Mg2B at 1300 K, OsO2 at 1400 K, Be2C, Zn2B, and Cu2N at 1500 K). These results indicate that these Lieb lattices are thermally stable and hold promise for a wide range of applications.
Electronic and magnetic properties
To determine the magnetic ground states of the identified MIFs, three magnetic configurations were evaluated, including ferromagnetic (FM), Néel-type antiferromagnetic (AFM-Néel), and stripe-type antiferromagnetic (AFM-stripe) (Fig. 2a). Spin density distributions (Fig. 2b and Supplementary Fig. S7) indicate that spin polarization is primarily attributed to the metal atoms. The local magnetic moments on the metal atoms were calculated to be 1.2 µB in HfCl2, 3.3 µB in MoF2, 2.8 µB in ReF2, 1.8 µB in RuF2, 1.5 µB in TiF2, 1.8 µB in OsF2, 1.6 µB in WO2, 1.2 µB in ZrF2, and 2.5 µB in RuO2. As summarized in Supplementary Table S1, the HfCl2 and WO2 exhibit FM ground states, while OsF2 and ZrF2 adopt the AFM-stripe configuration, with energy difference between FM and AFM states of 109.6, 243.7, −406.5, and −152.3 meV per metal atom, respectively. Additionally, MoF2, ReF2, RuF2, TiF2, and RuO2 were identified as AFM-Néel-type, with exchange interaction energies ranging from −77.7 to −508.0 meV per metal atom. These energy differences are comparable to those observed in the planar pentagon room-temperature ferromagnet Fe4N226, suggesting potentially high critical temperature in these square Lieb lattices.

a Three magnetic configurations were adopted to determine the magnetic ground states of MIFs, with the spin-up and spin-down indicated by red and blue balls, respectively. b Spin density distributions of WO2 (FM), MoF2 (AFM-Néel), and OsF2 (AFM-stripe), respectively, with isosurface values of 0.02 electron/bohr3. c The simulated specific heat capacity and average magnetization per site as a function of temperature for the HfCl2 Lieb lattice based on Monte Carlo simulations. d The estimated critical temperature of magnetic MIFs. The room temperature is marked by a dashed line.
To further investigate the spin dynamics and estimate the critical temperature (Curie temperature TC for ferromagnets and Néel temperature TN for antiferromagnets), Monte Carlo (MC) simulations based on the classical Heisenberg model were performed using the SEU-MTC package27. The spin Hamiltonian used in the simulation is given by:
where E0 represents the energy of the nonmagnetic state and J1 and J2 are the nearest and next-nearest neighbor exchange coupling parameters, respectively, and Si denotes the effective spin at each magnetic site (Fig. 2a). The detailed derivation of J1 and J2 is provided in the Supplementary information, with the calculated values summarized in Supplementary Table S1. TC or TN temperatures were determined from the peaks in the temperature-dependent specific heat CV (T) curve (Fig. 2c and Supplementary Fig. S8). HfCl2 and WO2 exhibit TC values of 360 K and 476 K, respectively, which are significantly higher than that of the Lieb-lattice-like sp2-c-COF (8.1 K)10 and comparable to pp-Fe4N2 (428 K)26 and Janus titanium sulfide (339 to 497 K)28. For the AFM MIFs, except for MoF2, ZrF2, and ReF2 with TN values of 165 K, 256 K, and 295 K, respectively, all other AFM Lieb lattices demonstrate antiferromagnetic ordering above room temperature (Fig. 2d). These high critical temperatures suggest that these MIFs are promising candidates for spintronics applications.
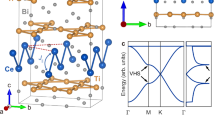
The electronic band structures of the 24 stable MIFs reveal four distinct electronic states, including nonmagnetic metals (NM-M), Dirac semimetals (DSM), FM half-metals (FM-HM), and AFM semiconductors (AFM-SC) (Fig. 3a). Specifically, 14 stable structures are classified as NM-M, 1 MIF as DSM, 2 MIFs as FM-HM, and 7 MIFs as AFM-SC (Fig. 3b). For instance, the band structure of Be2C (Fig. 3c) exhibits two bands crossing at the Fermi level, resulting in two type-I Dirac points along the X-Γ-M path. These Dirac points are not located at high-symmetry points, leading to the formation of a nodal loop with type-I dispersion that encircles the M-point across the entire 2D Brillouin zone, as shown in the three-dimensional (3D) band structure (Fig. 3d), consistent with previous results11. Similarly, Be2B, Cd2B, Mg2B, and Zn2O each possess a pair of Dirac points above the Fermi level (Supplementary Fig. S9), forming type-I nodal loops centered at the M-point (Supplementary Fig. S10). In addition to reproducing the electronic structure characteristics of the synthesized Cu2N12, both Ag2N and Au2N display similar hole-like bands at the M-point and saddle points near the Fermi level at each X-point. These features are indicative of partial flat bands and VHSs in their band structures, which are crucial for various electronic and topological properties.

a, b Schematic diagram of the band structures and the proportion of NM metal (NM-M), Dirac semimetal (DSM), FM half metal (FM-HM), and AFM semiconductor (AFM-SC). c Band structures of Be2C, Ag2N, WO2, RuO2, and TiF2. Dirac points, type-II Dirac points, van Hove singularities, and flat bands are marked by brown, green, red, and cyan dashed lines, respectively. d, e Three-dimensional band structures showing nodal-loop of Be2C and flat bands of TiF2.
Spin-resolved band structures indicate that the spin band gaps of two FM half metals (HfCl2 and WO2) are 5.37 eV and 3.57 eV, respectively. These substantial spin gaps effectively prevent spin flips caused by thermal fluctuation, resulting in a 100% spin-polarization ratio. Notably, HfCl2 and WO2 are FM half-metallic electrides, combining high carrier mobility, 100% spin polarization, and low work function (WF). These properties offer significant advantages for spintronic applications compared to conventional magnets, making these materials highly promising for future technological advancements. Additionally, MoF2, OsF2, RuF2, TiF2, and ZrF2 were identified as AFM semiconductors with band gaps of 3.29 eV, 1.21 eV, 0.77 eV, 2.33 eV, and 0.95 eV, respectively, while ReF2 and RuO2 possess direct band gaps of 2.40 eV and 0.51 eV, respectively. The semiconducting behavior in these seven MIFs is due to the localized electron wave function distribution, i.e., the formation of strong ionic bonds. Noteworthy, OsF2, ReF2, RuF2, TiF2, and ZrF2 are identified as AFM semiconducting electrides, a rare class of materials. Their band gaps are all larger than that of the experimentally synthesized semiconducting electride Sc2C (0.31 eV)29,30. Several Lieb lattices, including HfCl2, MoF2, ReF2, RuF2, and TiF2, exhibit quasi-flat bands near the Fermi level. For instance, the 3D band structure of TiF2 shows a nearly dispersionless quasi-flat band across the entire 2D momentum space (Fig. 3e). This feature provides a promising platform for exploring novel condensed matter phenomena. Furthermore, essential electronic characteristics such as partial flat bands, Dirac cone, and VHS observed in previously reported Lieb lattices are also present in these MIFs (Fig. 3c).
These MIFs also exhibit low WFs, as excess electrons in electrides are typically less tightly bound to the lattice. As summarized in Table 1, most MIFs have WFs comparable to or lower than those of conventional electron emission materials such as Ni (5.35 eV), Pd (5.12 eV), and graphene (4.60 eV)31,32. Notably, MoF2, TiF2, and ZrF2 exhibit exceptionally low WFs of 3.08 eV, 2.93 eV, and 2.64 eV, respectively, even lower than that of the 2D electride Ca2N (~3.4 eV)33. These properties suggest that these MIFs are promising candidates for applications in electron emitters, spintronics, and catalyst promoters34,35,36.
Topological properties
To investigate the topological properties of MIFs with nodal loops, we examined the effects of spin-orbit coupling (SOC), which can induce band openings and inversions. Taking Be2B as an example, the two conduction bands forming a Dirac ring around the M-point are primarily contributed by Be and B atoms, respectively (Fig. 4a). Upon inclusion of SOC, gaps of 1.7 meV and 14.9 meV open at the Dirac point (Fig. 4b), indicating the emergence of a nontrivial topological state. The presence of topological edge states, as highlighted by red contours in Fig. 4c, confirms the topological nontriviality of Be2B. Similarly, nontrivial topological states were identified in Be2C, Mg2B, Cd2B, and Zn2B by analyzing their topological band structures (Supplementary Fig. S11). Although the edge states of Be2B, Mg2B, Cd2B, and Zn2B are above the Fermi level, the energy level engineering of topological states could be realized through a practical electron doping strategy37,38,39.

a, d Projected band structures, (b, e) spin-orbit coupling (SOC) induced gap opening, and (c, f) edge states of Be2B (top panel) and Au2N (bottom panel). The contribution of metal and nonmetal elements is displayed by cyan and red colors, respectively. The edge state crossing of Be2B is highlighted by black dashed circle. The SOC-induced gap opening and the isolated edge state of Au2N are marked by black dash circles and curves, respectively.
In Cu2N, Ag2N, and Au2N, partial flat bands and parabolic bands intersect at the M-points without SOC. The introduction of SOC gaps, these crossing points12, as illustrated for Au2N (Fig. 4d, e). The decomposed band structure of Au2N shows that the crossing bands are predominantly derived from Au atoms, while N atoms contribute saddle points at the X-point (Fig. 4d). The significant gap at the M-point upon SOC inclusion (Fig. 4e) leads to the formation of robust edge states, as demonstrated in Fig. 4f and Supplementary Fig. S11. We notice that the edge states are not attached to bulk bands but exist in isolation, a characteristic that was experimentally observed in a photonic honeycomb lattice40. Remarkably, these isolated edge states remain robust and unaffected by spin-orbit coupling (SOC), maintaining their dispersion and spatial localization regardless of SOC (Fig. S12). Further, the nontrivial topological nature of these states was also confirmed by the Z2 = 1. These edge states verify the nontrivial topological nature of the flat bands in Cu2N, Ag2N, and Au2N Lieb lattices.
Deposition on the substrate
The synthesis of these lattices and their utilization in electronics require substrates. Therefore, we chose representative metallic Au2N, half-metallic WO2, and AFM semiconducting RuO2 to investigate the interaction between the Lieb lattice and the substrate. For Au2N, WO2, and RuO2 Lieb lattices, we selected bulk Au, WO3, and RuO2 as substrates, constructing heterostructures with lattice mismatches of 2.2%, 0.5%, and 4.0%, respectively. After structural optimization, the average distances between the Lieb lattice and the substrate were found to be 2.83 Å, 2.91 Å, and 2.83 Å, respectively, with charge transfers of 0.01, 0.17, and 0.10 electrons per A-site for Au2N, WO2, and RuO2 (Fig. 5a–c). As summarized in Table S2, the Néel-AFM and FM ground states of RuO2 and WO2 are maintained after deposition on a substrate, with spin charge densities predominantly distributed on metal atoms (Fig. 5d, e). The estimated critical temperatures of WO2 and RuO2 further increased to 588 and 1132 K, respectively, indicating a substrate-enhanced magnetic coupling in the Lieb lattice (Fig. 5f). In the absence of substrate effects, the energy differences between distinct magnetic states under applied strains for WO₂ and RuO₂ are summarized in Table S3. Monte Carlo simulations reveal critical transition temperatures of 606 K for WO₂ under 0.5% compressive strain and 697 K for RuO₂ under 4% tensile strain, as shown in Fig. S13. The compressive strain reduces inter-magnetic-site distances, enhancing magnetic coupling, whereas tensile strain increases these distances, weakening the coupling strength. This strain-mediated modulation of exchange interactions directly correlates with the observed elevation of the critical transition temperature in compressed WO₂ and its reduction in tensile-strained RuO₂. The projected band structure of the Lieb lattice on the substrate demonstrates that the relatively flat band and the touching point at the M-point are retained in Au2N while the semiconducting and half-metallic electronic structure of RuO2 and WO2 characters are reproduced on the substrate (Fig. 5g–i). These findings imply that screened Lieb lattices could be realized and applied in devices on a suitable substrate.

Side views of the optimized structural models of (a) Au2N on Au(001), (b) RuO2 on RuO2(001), and (c) WO2 on WO3(001) substrate. Top views of spin charge density distributions of (d) RuO2 and (e) WO2 on substrate with isosurface values of 0.05 and 0.01 electron/bohr3, respectively. f The simulated specific heat capacity as a function of temperature for RuO2 and WO2 based on Monte Carlo simulations. Projected band structures of (g) Au2N, (h) RuO2, and (i) WO2 Lieb lattice on substrates.
Discussion
In summary, our high-throughput calculations screened 1470 MIFs, identifying 24 stable Lieb lattices with diverse electronic and magnetic properties, of which 22 are newly reported. Among these, nine are magnetic MIFs, comprising two FM half-metals and seven AFM semiconductors with band gaps ranging from 0.51 eV to 3.29 eV. Notably, six of these magnetic MIFs exhibit long-range magnetic ordering with a critical temperature above room temperature. Specifically, HfCl₂ and WO₂ show remarkably high critical temperature values of 360 K and 476 K, respectively. Among other compounds, ReF₂ demonstrates a critical temperature of 295 K, while ZrF₂ and MoF₂ exhibit lower critical temperatures of 256 K and 165 K, respectively. Of the fifteen nonmagnetic MIFs, Be2C is a nodal loop semimetal, while Be2B, Cd2B, Mg2B, and Zn2B feature nodal loops centered at the M-point above the Fermi level. These nonmagnetic MIFs also exhibit nontrivial topological edge states when SOC is considered. Additionally, five AFM semiconducting MIFs, two FM half-metallic MIFs, and two NM metallic MIFs demonstrate 0D electride-like behavior, with work functions as low as 2.64 eV. The possibility of synthesizing these Lieb lattices was supported by structure searches and favorable formation energies. Further, the retention of band structure characteristics and the enhancement of critical temperature are observed in Lieb lattices when deposited on a substrate. Our findings significantly expand the library of candidate Lieb lattices and highlight their potential applications in spintronics, topological materials, and electron emission technologies.
Methods
Density-functional theory calculations
The properties of MIFs were calculated using first-principles methods based on density functional theory (DFT), as implemented in the Vienna ab initio simulation package (VASP) code41,42. The projected augmented wave (PAW) method was employed, along with the generalized gradient approximation (GGA) for the exchange-correlation potential, using the Perdew-Burke-Ernzerhof (PBE) parameterization42,43,44. A plane wave cut-off energy of 500 eV was chosen following convergence tests to ensure accuracy. To prevent interactions between periodic images along the z-direction, a vacuum space exceeding 15 Å was added. Geometric optimizations were performed until the total energy and atomic forces converged to within 10−6 eV and 10−2 eV/Å, respectively. Brillouin zone integrations were carried out using Monkhorst-Pack k-point sampling with a spacing of approximately 2π × 0.025 Å−1. Spin-polarized calculations were conducted to determine the magnetic ground state. To accurately evaluate the electronic structure and magnetic properties, the Heyd-Scuseria-Ernzerhof (HSE06) hybrid functional was utilized45. Phonon spectra were calculated using the finite displacement method as implemented in the PHONOPY code46, to ensure the dynamic stability of the structure. Ab initio molecular dynamics (AIMD) simulations were conducted within a NVT canonical ensemble using the Nosé-Hoover thermostat at temperatures of up to 1500 K over 5 ps with a time step of 1 fs to evaluate thermal stability47,48. Previous studies have systematically evaluated the accuracy of the HSE functional for bandgap prediction by benchmarking it against a comprehensive dataset of experimentally characterized materials. These analyses demonstrate that the HSE hybrid functional achieves a mean absolute error (MAE) of less than 0.3 eV for bandgap predictions49. While slight deviations in bandgap estimation may persist, the HSE functional excels at distinguishing between metallic and semiconducting behaviors. Furthermore, the band structure of the Cu2N Lieb lattice calculated using HSE06 functional exhibits good agreement with experimental angle-resolved photoemission spectroscopy (ARPES) measurements12. These validations collectively confirm the reliability of the band structure characteristics derived for the Lieb lattice in this work, though the absolute bandgap values may deviate from experimental values by up to 0.3 eV.
Structure search
Structure searches were executed using the particle swarm optimization algorithm as implemented in the CALYPSO program, which effectively predicts ground-state or metastable structures based solely on chemical compositions26,50,51. A wide range of elements were considered, including 49 metals and 15 nonmetals, to generate MIFs with metal-to-nonmetal atomic ratios of 1:2 and 2:1. Candidate structures were explored using simulation cells containing three or six atoms, corresponding to the unit cell sizes of the targeted Lieb lattices. The structure search was terminated for any generated MIFs exhibiting positive formation enthalpies, as these indicate thermodynamic instability under ambient conditions. This approach ensured that only energetically favorable structures were considered for further analysis.
Topological properties calculations
Maximally localized Wannier functions (MLWFs) were constructed using the WANNIER90 package based on VASP52,53. A 20 × 20 × 1 Gamma-center k-point grid was employed to ensure convergence, and the Wannier basis explicitly included the d-orbitals of transition metal, along with s- and p-orbitals of alkaline earth metal and nonmetal atoms, to accurately reproduce the electronic structure near the Fermi level. The Wannier functions were successfully disentangled within 200 optimization steps, producing a fully converged tight-binding Hamiltonian that shows excellent agreement with the original DFT bands across the entire Brillouin zone. The WANNIERTOOLS package was employed as a post-processing tool to investigate the topological properties of the system by utilizing the tight-binding model derived from the wannier90_hr.dat file generated by WANNIER9054. The edge states of semi-infinite nanoribbons were computed using the recursive Green’s function method, with the number of principal layers set to 3 to ensure proper boundary conditions. The nanoribbon structure was constructed by cleaving the two-dimensional unit cell along the [100]/[010] direction (which are equivalent directions in the Lieb lattice) and subsequently extending it by a factor of 10 to achieve sufficient width.
Monte Carlo simulations
The magnetic critical temperatures were estimated using a Monte Carlo simulation implemented in the SEU-MTC package27. Simulations were performed on a 50 × 50 × 1 supercell with periodic boundary conditions, using a temperature range from 1 to 1000 K with a temperature step of 1 K. The Heisenberg model was employed, and the exchange coupling constants were extracted from DFT calculations. A total of 2 × 106 Monte Carlo steps were adopted to ensure convergence of the simulations. Since no experimental validation currently exists for the magnetic materials reported in this study, direct assessment of the deviation between calculated critical temperatures and experimental values remains unfeasible. Experimentally synthesized monolayer CrI3—a prototypical 2D magnetic semiconductor—was selected as a benchmark system. The Heisenberg model-based simulation yields a value of 42 K27, exhibiting a modest underestimation of 6.67% from experimental observations (45 K)55. Consequently, if CrI3 is taken as a reference standard, the actual critical temperature of the magnetic Lieb lattice reported in this work is likely to slightly exceed our theoretical prediction.
Data availability
The authors declare that the main data supporting the findings of this study are available within the article and its Supplementary Information files. Additional data that support the findings of this study are available from the corresponding author on reasonable requests. The central codes used in this paper are VASP. Detailed information related to the license and user guide is available at [http://www.vasp.at]. The user guide and code for SEU-MTC package is available at [https://physics.seu.edu.cn/jlwang_zh/mtc]. The code of WANNIER90 package is available at [http://wannier.org]. The code of WANNIERTOOLS package is available at [http://wanniertools.org].
References
Lieb, E. H. Two theorems on the Hubbard model. Phys. Rev. Lett. 62, 1201–1204 (1989).
Keimer, B., Kivelson, S. A., Norman, M. R., Uchida, S. & Zaanen, J. From quantum matter to high-temperature superconductivity in copper oxides. Nature 518, 179–186 (2015).
Pan, G. A. et al. Superconductivity in a quintuple-layer square-planar nickelate. Nat. Mater. 21, 160–164 (2022).
Shen, R., Shao, L. B., Wang, B. & Xing, D. Y. Single Dirac cone with a flat band touching on line-centered-square optical lattices. Phys. Rev. B 81, 041410 (2010).
Mukherjee, S. et al. Observation of a localized flat-band state in a photonic Lieb lattice. Phys. Rev. Lett. 114, 245504 (2015).
Slot, M. R. et al. Experimental realization and characterization of an electronic Lieb lattice. Nat. Phys. 13, 672–676 (2017).
Neves, P. M. et al. Crystal net catalog of model flat band materials. npj Comput. Mater. 10, 39 (2024).
Julku, A., Peotta, S., Vanhala, T. I., Kim, D.-H. & Törmä, P. Geometric Origin of Superfluidity in the Lieb-Lattice Flat Band. Phys. Rev. Lett. 117, 045303 (2016).
Cui, B. et al. Realization of Lieb lattice in covalent-organic frameworks with tunable topology and magnetism. Nat. Commun. 11, 66 (2020).
Jiang, W., Huang, H. & Liu, F. A Lieb-like lattice in a covalent-organic framework and its Stoner ferromagnetism. Nat. Commun. 10, 2207 (2019).
Yang, B., Zhang, X. & Zhao, M. Dirac node lines in two-dimensional Lieb lattices. Nanoscale 9, 8740–8746 (2017).
Hu, X. et al. Realization of a two-dimensional checkerboard lattice in monolayer Cu2N. Nano Lett. 23, 5610–5616 (2023).
Wu, W. et al. Realization of a 2D Lieb lattice in a metal–inorganic framework with partial flat bands and topological edge states. Adv. Mater. 36, 2405615 (2024).
Jiang, W., Zhang, S., Wang, Z., Liu, F. & Low, T. Topological band engineering of Lieb lattice in phthalocyanine-based metal–organic frameworks. Nano Lett. 20, 1959–1966 (2020).
Feng, H. et al. Experimental realization of two-dimensional buckled Lieb lattice. Nano Lett. 20, 2537–2543 (2020).
Palumbo, G. & Meichanetzidis, K. Two-dimensional Chern semimetals on the Lieb lattice. Phys. Rev. B 92, 235106 (2015).
Klembt, S. et al. Polariton condensation in S- and P-flatbands in a two-dimensional Lieb lattice. Appl. Phys. Lett. 111, 231102 (2017).
Beugeling, W., Everts, J. C. & Morais Smith, C. Topological phase transitions driven by next-nearest-neighbor hopping in two-dimensional lattices. Phys. Rev. B 86, 195129 (2012).
Danieli, C., Andreanov, A., Leykam, D. & Flach, S. Flat band fine-tuning and its photonic applications. Nanophotonics 13, 3925–3944 (2024).
Niţă, M., Ostahie, B. & Aldea, A. Spectral and transport properties of the two-dimensional Lieb lattice. Phys. Rev. B 87, 125428 (2013).
Ţolea, M. & Niţă, M. Ground state spin and excitation energies in half-filled Lieb lattices. Phys. Rev. B 94, 165103 (2016).
Dauphin, A., Müller, M. & Martin-Delgado, M. A. Quantum simulation of a topological Mott insulator with Rydberg atoms in a Lieb lattice. Phys. Rev. A 93, 043611 (2016).
Wang, H., Yu, S.-L. & Li, J.-X. Spin fluctuations and unconventional pairing on the Lieb lattice. Phys. Lett. A 378, 3360–3365 (2014).
Zhang, T., Jiang, Z. & Rappe, A. M. Hydrogenation of covalent organic framework induces conjugated π bonds and electronic topological transition to enhance hydrogen evolution catalysis. J. Am. Chem. Soc. 146, 15488–15495 (2024).
Liu, L. et al. A metastable pentagonal 2D material synthesized by symmetry-driven epitaxy. Nat. Mater. 23, 1339–1346 (2024).
Zhang, K. et al. Nodal-loop half metallicity in a two-dimensional Fe4N2 pentagon crystal with room-temperature ferromagnetism. Nanoscale 13, 19493–19499 (2021).
Zhang, Y., Wang, B., Guo, Y., Li, Q. & Wang, J. A universal framework for Metropolis Monte Carlo simulation of magnetic Curie temperature. Comput. Mater. Sci. 197, 110638 (2021).
Zhang, K. & Xiaojun, W. Room-temperature ferromagnetism in two-dimensional janus titanium chalcogenides. Acta Chim. Sin. 81, 1142–1147 (2023).
Zhang, X. et al. Magnetic electrides: high-throughput material screening, intriguing properties, and applications. J. Am. Chem. Soc. 145, 5523–5535 (2023).
McRae, L. M. et al. Sc2C, a 2D semiconducting electride. J. Am. Chem. Soc. 144, 10862–10869 (2022).
Takahashi, T., Tokailin, H. & Sagawa, T. Angle-resolved ultraviolet photoelectron spectroscopy of the unoccupied band structure of graphite. Phys. Rev. B 32, 8317–8324 (1985).
Skriver, H. L. & Rosengaard, N. M. Surface energy and work function of elemental metals. Phys. Rev. B 46, 7157–7168 (1992).
Lee, K., Kim, S. W., Toda, Y., Matsuishi, S. & Hosono, H. Dicalcium nitride as a two-dimensional electride with an anionic electron layer. Nature 494, 336–340 (2013).
Kim, S. W., Toda, Y., Hayashi, K., Hirano, M. & Hosono, H. Synthesis of a room temperature stable 12CaO•7Al2O3 electride from the melt and its application as an electron field emitter. Chem. Mater. 18, 1938–1944 (2006).
Lee, S. Y. et al. Ferromagnetic quasi-atomic electrons in two-dimensional electride. Nat. Commun. 11, 1526 (2020).
Kitano, M. et al. Ammonia synthesis using a stable electride as an electron donor and reversible hydrogen store. Nat. Chem. 4, 934–940 (2012).
Zhang, K., Wu, X. & Yang, J. Transition metal dichalcogenide magnetic atomic chains. Nanoscale Adv. 4, 4905–4912 (2022).
Li, Y. et al. Electrically induced topological chirality switching in orbital-engineered covalent organic radical framework monolayers. J. Phys. Chem. Lett. 16, 5188–5194 (2025).
Rosenzweig, P., Karakachian, H., Marchenko, D., Küster, K. & Starke, U. Overdoping graphene beyond the van Hove singularity. Phys. Rev. Lett. 125, 176403 (2020).
Plotnik, Y. et al. Observation of unconventional edge states in ‘photonic graphene. Nat. Mater. 13, 57–62 (2014).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 118, 8207–8215 (2003).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1–5 (2015).
Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 (1984).
Hoover, W. G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 (1985).
Heyd, J., Peralta, J. E., Scuseria, G. E. & Martin, R. L. Energy band gaps and lattice parameters evaluated with the Heyd-Scuseria-Ernzerhof screened hybrid functional. J. Chem. Phys. 123, 174101 (2005).
Wang, Y., Lv, J., Zhu, L. & Ma, Y. CALYPSO: a method for crystal structure prediction. Comput. Phys. Commun. 183, 2063–2070 (2012).
Zhang, K., Lv, H., Wu, X. & Yang, J. Two-dimensional alkali auride bimetallene semiconductors. Sci. China Mater. 67, 1209–1216 (2024).
Mostofi, A. A. et al. An updated version of wannier90: A tool for obtaining maximally-localised Wannier functions. Comput. Phys. Commun. 185, 2309–2310 (2014).
Pizzi, G. et al. Wannier90 as a community code: new features and applications. J. Phys. Conden. Matter 32, 165902 (2020).
Wu, Q., Zhang, S., Song, H.-F., Troyer, M. & Soluyanov, A. A. WannierTools: An open-source software package for novel topological materials. Comput. Phys. Commun. 224, 405–416 (2018).
Huang, B. et al. Layer-dependent ferromagnetism in a van der Waals crystal down to the monolayer limit. Nature 546, 270–273 (2017).
Acknowledgements
This work was supported by the National Natural Science Foundation for Distinguished Young Scholars (22225301, 22503091), the Anhui Provincial Natural Science Foundation (2308085QB51), the CAS Project for Young Scientists in Basic Research (YSBR-004), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0450101), and the Fundamental Research Funds for the Central Universities (WK9990000153). We thank the support from the Super Computer Centre of University of Science and Technology of China and Supercomputing Center of Chinese Academy of Sciences.
Author information
Authors and Affiliations
Contributions
X.W. conceived and designed the study. K.Z. and Y.L. performed the first-principles calculations, data analysis, investigation, and wrote the initial manuscript. All authors participated in discussing and editing the manuscripts. K.Z. and Y.L. contributed equally to this work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, K., Li, Y., Wang, D. et al. Exploring stable Lieb lattices in two-dimensional binary metal-inorganic frameworks: a high-throughput screening approach. npj Comput Mater 12, 10 (2026). https://doi.org/10.1038/s41524-025-01877-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41524-025-01877-y
