
Abstract
Machine learning and computational methods can accelerate materials discovery by accurately predicting material properties at low cost. Nevertheless, input data to algorithms and structure model parameters remains a key obstacle. The limitations of conventional battery materials could be overcome by high-entropy materials, a unique class of special valuable materials. The knowledge of designing the crystal structure of high-entropy materials is advancing the design and fabrication of new materials for batteries and supercapacitors, even before chemical synthesis, through the use of learning algorithms and quantum computing. In this review, we first focus on quantum computing and the structure of high-entropy materials, especially high-entropy MXenes. We then discuss how to encode and decode the crystal structure of materials, which is a key factor in creating a database for high-entropy materials. We also discuss how to utilize deep learning algorithms for material discovery prior to synthesis, as well as how to employ these algorithms to identify high-entropy materials suitable for batteries and supercapacitors. Finally, we discuss the potential of new quantum computing and artificial intelligence approaches for determining the structure of high-entropy materials in the energy fields.

Similar content being viewed by others
Introduction
Applications and discoveries of new materials have a major role in advancing human civilization. Discovering novel material components is an important area of study since traditional methods and resources cannot keep up with the rapid advancements in science and technology1. Its composition matters most in determining a material’s properties and applications2. The prediction of material formation is generally an empirical approach, relying on laboratory results and trial-and-error methods that are time-consuming and costly. On the other hand, the chemical and physical interactions involved in the formation of new materials, along with some related events, are often too complex for existing theories to fully explain. One potential avenue to investigate some of these issues is the potential for teaching concepts to computers through learning algorithms and big data3. Nowadays, the “4th industrial revolution” and the “4th paradigm of science” will be lighted by the development of machine learning (ML) techniques, the next generation of computers, and big data4. The evolution of science with different approaches, from experimental, which is called the first paradigm, and the second paradigm, which is the generalization of physics and chemistry theories, to the third and fourth paradigms, which emerged with the advent of computers and artificial intelligence (AI) algorithms, is depicted in Fig. 1.

Science has evolved, with different approaches ranging from experimental examination (paradigm one), generalization of theories of physics and chemistry (paradigm two), simulation of complex phenomena by computers and computational tools (paradigm three), and artificial intelligence (paradigm four). The quantum computer was taken from the NICS Ltd.
Technologies are undergoing new developments due to the emergence of high-entropy materials (HEMs). High-entropy design in electrochemical energy storage has demonstrated beneficial effects on battery materials, including reduced volumetric expansion and decreased dependence on critical metals5. Battery research and development currently costs billions of dollars, leading to significant improvements in energy density and reliability. As a result of improved batteries and electrical and electronic equipment, laptops, mobile phones, and electric and hybrid vehicles (EVs) have made significant progress. The development of new materials for batteries must be accelerated. Battery materials must improve in terms of energy density, capacity, cycle life, charging speed, safety, and reliability6. Since the first HEM was suggested in 2004, emerging single-phase crystalline solid solutions with several primary elements in equimolar or nearly equimolar ratios have been designated as HEMs7. Metals8, HE-MXene9, oxides10, and sulfides11 are among the numerous classes of HEMs that have been found. An example is the HE-alloy (HEA) structural design approach, which creates HEAs with Mg alloys to show exceptional physical properties while balancing strength and ductility12. HEAs are extremely strong, frequently exceeding the properties of typical alloys due to severe lattice distortion that limits dislocation movement and increases strength2. The primary objective of HEA research to date has been to enhance mechanical properties like hardness, elastic modulus, shear modulus, tensile strength, and coefficient of thermal expansion13,14,15.
HEMs featuring a range of compositions have surfaced as new functional materials recently16; these materials find uses in HEM as biomedical materials17, catalysts18, supercapacitors19, batteries20, and magnetics21. One of the challenges in designing materials with a wide range of compositions and uses is figuring out which ratios and components work best to create the desired phase structure and functional qualities. The most effective method for creating novel material structures up until this point has been through experimental work22. Nevertheless, due to its lengthy processing time, the traditional screening approach faces an enormous challenge in the creation of materials for complicated material structures like high-entropy compounds (HEC). For example, an effective alloy screening process is needed to address problems like the synthesis of distinct phases in HEAs due to the extremely high number of potential compositions23.
Three main challenges explain why predicting new HEMs from fundamental principles is challenging. Initially, even with sophisticated sampling methods, entropy is an extremely challenging parameter to parameterize and estimate from first principles24. In the next step, a material has more competing phases the more of its constituent elements there are. Even without considering entropy, computing the competing phases and evaluating their relative stability in about the HEM is quite computationally complex25. In the third step, supercells are frequently required for modeling random disorder phases, which are often wide enough to enable a high-throughput first-principles screening of functional features across the vast field of possibilities26. Nowadays, comprehensive research efforts into the effects of HEM on batteries have led to the creation of HE metal oxides as Li-ion battery anodes10,27. HE-Li-ion cathodes have now been added to this; they performed better than commercial materials in terms of energy density and rate capability28. Solid-state ionic conductors are comparable to HEMs in terms of ionic conductivity, which has led to the exploration of Li-ion and Na-ion solid electrolyte applications29,30,31. In batteries and supercapacitors (B/S), computational techniques and data science have made material discovery extremely effective. Essentially, using traditional methods to assess the synthesis potential of HEMs based on the periodic table necessitates plenty of computations32. HEMs can have billions of states and molar ratios. Thus, for HEMs used in energy applications such as B/S, an array of states and screening techniques can be considered. Before synthesis, it is crucial to examine various states to determine the enthalpy of mixing for various mixed phases. ML algorithms are beneficial tools for material discovery nowadays but require reliable data. HEMs remain challenging to study utilizing data, and existing datasets such as the Genome databases33 only provide a limited amount of data in this field. Consequently, the necessity of building trustworthy databases in HEMs is emphasized. Material discovery has historically relied on empirical data. Significant improvements were made as data from experiments gradually extended in the form of mathematical and conceptual theories. The development of computational tools, such as supercomputers, has enabled the accurate prediction of material properties based on scientific principles and theories34.
Therefore, it is essential to investigate the complex atomic arrangements of special materials for energy applications, such as B/S and to seek solutions to the grand challenges in their design and synthesis, thereby discovering new structures of these materials6. Before synthesizing HEMs, it is essential to examine their electronic structure and the relationships between their structures. It can be challenging to describe the structure of reactive surfaces with flaws. Current knowledge on how the composition of elements and synthesis methods affect the structure and properties of HEMs is minimal. Given the need to develop and investigate the grand challenges in this field, we aim to showcase the most recent developments in materials discovery, utilizing computational techniques and AI to synthesize and predict the structure and properties of HEMs for energy applications. We also explore the potential of computational methods combined with data science to identify new structures of HEMs, along with the challenges of encoding material structures as input for AI algorithms in this field. Our aim is to introduce innovative methods for representing the structure of crystalline materials and to develop new learning algorithms, enabling AI to predict the structures of HEMs. Our review emphasizes computational approaches and highlights recent advancements in material discovery, using artificial intelligence to predict HEM structures in the energy sector before synthesis. Despite the increasing computational studies and consequently more papers on HEMs, relatively few studies have been conducted with a focus on machine learning in this area. The search results for the ML discussion were largely limited to HEAs, and very few papers were found in the energy and battery fields. Recognizing this research gap, and to open the door for further research, we directly addressed the key role of defining the crystal structure of materials, including HEMs, and designing the crystal structure to take into account the inputs of the ML algorithm. Despite recent progress in data science and ML algorithms, we believe these developments will significantly impact the discovery of new materials, especially HEMs for energy applications.
Computational modeling of HEM structure
Quantum computational methods are a tool for understanding complex phenomena, and previously unknown properties of synthesized HEMs can be understood using quantum computing techniques. Material databases and experimental datasets document a wide range of properties of known materials. These resources are then utilized to sort through the data to uncover intriguing and previously undiscovered qualities in known material compositions. In this section, we provide an overview of the quantum computational and molecular methodologies used to investigate HEMs in cathodes, anodes, electrolytes, and electrodes for rechargeable B/S. Computational methods are necessary to fully understand the complex behavior of HEMs, particularly in electrochemical systems where experimental methods are hindered by the complexity of the materials’ reaction and the conditions under which they operate35. The computational design and analysis of HEMs have proven to be a valuable approach for understanding and predicting the behavior of these materials across various applications36. In electrochemical contexts, modeling and simulation techniques for HEMs can be categorized into two primary areas: HEM design and electrochemical performance. These approaches facilitate a detailed examination of critical properties, including chemical composition, formation energies, defect concentrations, and transport mechanisms—factors that directly influence the stability, conductivity, and electrochemical performance of HEM-based energy storage systems. Table 1 presents the theoretical models and simulation techniques employed in HEM studies, with a focus on properties relevant to B/S applications. For example, methods that address chemical composition and defect dynamics are vital for predicting ionic conductivity and charge retention, while approaches that model thermomechanical behavior offer insights into structural integrity under cycling conditions. Computational frameworks also encompass the interaction between chemical reactivity and electronic structure, which is essential for optimizing charge transfer and minimizing energy loss. By integrating these diverse methodologies, it becomes feasible to evaluate the relationship between HEM design and electrochemical efficiency, providing a comprehensive understanding of material performance in advanced energy storage technologies. Therefore, the key objective of this part is to discuss the computational methods for the atomic and molecular simulation of HEMs and high-entropy 2D MXene on B/S. The following sections will discuss the strategies of Density Functional Theory (DFT), including ion adsorption using molecular simulation, adsorption energy, and atomic structural stability of HEMs on B/S materials.
Density functional theory
Over the past 50 years, DFT has emerged as the most dominant and successful method for the ab initio computational study of materials’ ground-state properties. DFT is particularly useful for calculations that aid in designing and evaluating HEM electrochemical performance37. The cornerstone of DFT was established by the work of Hohenberg and Kohn38, who demonstrated that the electronic ground-state total energy can be expressed as a functional of the ground-state charge density, and by Kohn and Sham39, who showed that the ground-state charge density can be determined through a set of self-consistent one-body equations. Within the framework of Kohn-Sham DFT, the complex problem of many interacting electrons is reduced to a one-electron Schrödinger equation for a fictitious system of non-interacting particles with the same density37. DFT provides a precise framework for determining the fundamental characteristics of a system in its lowest energy state, including properties such as the arrangement of molecules and crystals, the ability to withstand deformation, the behavior of vibrations, and the comparative stability of various structural phases40. The accuracy of DFT calculations is critically dependent on the selection of density functionals, which must be rigorously validated rather than applied indiscriminately. Among the commonly employed functionals are semi-local DFT methods (including LDA, GGA, and meta-GGA), GGA + U, and the hybrid HSE06 functional41. Semi-local functionals frequently encounter challenges in accurately representing electron correlation effects. LDA and GGA often significantly underestimate ion insertion voltages due to their inadequate treatment of electronic correlation, which stems from the inability of LDA/GGA to accurately address the self-interaction of electrons, particularly when transitioning from delocalized to localized states42. Consequently, semi-local functionals may yield lower predicted reaction energies and voltages. The DFT + U method43 mitigates this issue by adjusting the fractional occupations of Kohn-Sham orbitals, thereby providing a more accurate representation of electronic correlation effects.
There are many challenges for DFT simulation of HEM structures; the first path is the selection of computational methods. Different computational methods exist for accurate prediction of materials such as structural, electronic, and thermodynamic properties, and their selection can be important while addressing the computational challenges arising from the complex interactions between several transition metals in the crystal structure of HEMs. In these cases, the exchange correlation function (XC) plays a pivotal role due to the nature of the bonding and electronic interactions as well as the quantum parameters of the system44. Pseudopotentials, by replacing the core-electron interactions with effective potentials, increase computational efficiency and preserve the accuracy of the valence electrons45. In addition, dispersion corrections take into account weak van der Waals interactions, which are crucial for modeling intermolecular forces in HEM crystal structures46. Table 2 describes the available computational methods and the rationale for their selection, highlighting their effectiveness in reducing issues such as excessive d-electron delocalization and underestimation of weak interactions, making them suitable for high-throughput screening of HEM compounds.
Formation and electronic structure
The structural stability of B/S materials plays a major role in determining their cycling lifespan. The stability of B/S materials can be evaluated by determining their formation energy47, Gibbs free energy48, and phonon dispersion spectrum49. Formation energy is a quantitative measure of the energy change when a crystal or combination of its constituent elements is formed in their standard states50. The formation energy of a chemical can be calculated using the following Eq. (1)47:
Here \({E}_{{HEM}}\) (eV) denotes the total energy of the HEM, and the stoichiometric coefficients, \({n}_{i}\), indicate the number of atoms of each constituent element i present in the HEM. \({E}_{i}\)(eV) is the energy of each constituent element i in its standard state. A low formation energy suggests that a compound can be synthesized more easily and cost-effectively. Lin et al.51 illustrated this idea by effectively synthesizing a high-entropy O3-type layered compound, NaCu0.1Ni0.3Fe0.2Mn0.2Ti0.2O2 (NCNFMT), employing the Pechini technique52 for utilization as a cathode in sodium-ion batteries (NaB). They used DFT simulations to determine the compound formation energy before synthesis. Figure 2a shows the ensuing negative formation energy of −0.82 eV/f.u. Validated the viability of synthesizing NCNFMT. Binding energy indicates the strength of interaction between atoms or molecules within a compound53. The high-entropy metal oxide HEMO-1, which is composed of Ni, Mg, Cu, Zn, and Co, was created by Zheng et al. with the express purpose of serving as a chemical anchor to immobilize lithium polysulfides in lithium- sulfur (Li-S) batteries54. The (100) plane of HEMO-1, as shown in Fig. 2b, has a significantly higher binding energy (−6.916 eV) to Li2S6 than Ketjen Black (KB) carbon (−0.742 eV), according to DFT calculations. This finding highlights the stronger polar-polar interactions between polysulfides and HEMO-1. Among the critical characteristics that contribute to the formation of HEMs are thermodynamic parameters55.

a Depiction of the O3-type layered structure of Na [Cu0.1Ni0.3Fe0.2Mn0.2Ti0.2] O2, including the formation energy per formula unit (f.u.). The quinary-color site represents the disordered site randomly occupied by Cu, Ni, Fe, Mn, and Ti cations with occupation probabilities of 10%, 30%, 20%, 20%, and 20%, respectively. Reproduced with permission51 Copyright 2022, Elsevier. b Geometry configuration of Li2S6 binding to HEMO-1, with O, Mg, Zn, Cu, Co, Li, and S atoms marked in red, white, orange, green, blue, purple, luminous yellow, and light green, respectively. Simulated bond distances of S-Ni and Li-O are 2.276 Å and 1.842 Å, respectively. Additionally, the geometry configuration of Li2S6 binding to bare KB carbon is illustrated, with carbon, lithium, and sulfur atoms marked in brown, luminous yellow, and light green, respectively. A comparison of the calculated binding energies of Li2S6 with HEMO-1 and KB carbon is presented. Ultraviolet/visible absorption spectra of a Li2S6 solution before and after adding HEMO-1 and KB carbon are shown, with an inset photograph displaying the visual adsorption result using HEMO-1 and bare KB carbon. Reproduced with permission54 Copyright 2019, Elsevier. c Calculated free energy plots of the ORR under alkaline conditions for Fe12Ni23Cr10Co25Mn30, Fe12Ni23Cr10Co27Mn28, Fe12Ni23Cr10Co30Mn25, and Fe12Ni23Cr10Co35Mn20. Reproduced with permission59 Copyright 2023, American Chemical Society.
The Gibbs free energy is used to determine the phase stability and electrochemical performance of high-entropy materials. In fact, the Gibbs free energy of mixing, expressed in Eq. 2, can be used as a thermodynamic index for the formation of the multicomponent crystal structure of HEMs56. In this equation, ΔGmix represents the difference in Gibbs free energy between the multicomponent material and its constituent elements, and the parameter ΔHmix represents the enthalpy of mixing at the absolute temperature (T) and R represents of the gas constant. The parameter xi is the mole fraction of the ith component and ΔSconf is the structural entropy difference ΔSvib and ΔSelec are the vibrational and electronic entropy differences. If the value of ΔGmix is negative, it represents a spontaneous mixing process. This value is often driven by ΔSconf, which is a value greater than the enthalpy of mixing (ΔHmix > 0). This entropy-based stabilization allows HEMs to form crystalline structures with chemical bonds that are usually undesirable in conventional intermetallic alloys or regular crystals. By considering the contributions of ΔSvib and ΔSelec, this model reveals the complex interactions within these complex multicomponent systems56.
In this regard, to evaluate the energy by minimizing the non-equilibrium Gibbs free energy (G*), Eq. 3 can be used at constant temperature (T) and pressure (P) and a given volume (V)57. In this equation, Uel represents the total electronic energy and Fvib is the vibrational free energy using the DFT computational method. The value of Fvib can be calculated using the quasi-harmonic approximation or non-harmonic approaches based on the density of phonon states58. This equation can quantify the energy difference at each stage of the electrochemical processes and interpret the stability and electrochemical performance of HEM-based energy applications at each stage. Equations (2) and (3) can be used to interpret the stability of HEM structures and their performance for energy storage.
In zinc-air battery production, the G calculation is essential for optimizing the critical yet complex oxygen conversion reactions at the air electrode. Cao et al. analyzed the oxygen reduction reaction (ORR) free energy of Fe12Ni23Cr10Co55−xMny/CNT catalysts at 1.23 V in an alkaline environment using DFT calculations. As seen in Fig. 2c, the catalyst (x:30, y:25) had a Gibbs free energy of 0.32 eV in the OOH* step, which was much lower than that of the other variations (x:27, y:28), (x:25, y:30), and (x:35, y:20)59.
The thorough examination of electronic structure using Electron Localization Function (ELF)60, Density of States (DOS)61, Projected Density of States (PDOS), and band structure62 computation is essential for comprehending diverse material features, such as electrical conductivity, optical response, and overall electronic behavior63. The ELF, denoted as η(r)64, gives values ranging from 0 to 1. Higher values of η(r) suggest a stronger electron localization, whereas η(r) = 0.5 represents a condition resembling a uniform electron gas with Pauli repulsion. Strong covalent bonding is associated with higher ELF values, whereas metallic bonding is often found when η(r) is close to 0.5. In cases when η(r) is less than 0.5, weaker bonding is recommended. Concerns about irreversible phase transitions and structural deterioration in O3-type cathode materials should be addressed65. When redox-inactive materials are used as pinning agents, HEMs exhibit extremely reversible phase shifts66.
An Na-B cathode with this combination, NaNi0.2Fe0.2Mn0.35Cu0.05Zn0.1Sn0.1O2, has been suggested by Wang et al.67 It has six metallic elements that occupy the TMO2 layer (TM: transition metals). The oxygen atoms are anchored to the TMO2 layer by their confined environment, which is coordinated with the redox-inactive elements Sn, Mn, and Zn, as seen in Fig. 3a. The anchoring effect inhibits the gliding of the TMO2 layer during the intercalation and deintercalation processes of Na+ ions, thereby enhancing the structural stability of the cathode material. The electronic band structure provides information about the energy levels an electron within a crystal structure can occupy41. The band gap is closely related to electronic conductivity, determining whether a material is a conductor, semiconductor, or insulator. The DOS and PDOS represent the number of different states at a particular energy level that electrons can occupy, measured as the number of electron states per unit volume per unit energy41. Advantageous of the distinct cocktail effect68, the interplay between various metal atoms in high-entropy oxides (HEOs) leads to a markedly expanded d-band and reduced degeneracy in contrast to monometallic oxides (see Fig. 3b(I)). A composite with Co0.6Ni0.6Fe0.6Mn0.6Cu0.6O4 (Co-HEO) was proposed by Du et al.69 as a cathode material for aqueous zinc-ion batteries (AZIBs). The cocktail effect in HEOs arises from the synergistic interaction of multiple metal cations, which creates chemical and structural disorder that significantly alters the electronic DOS. This disorder affects the interactions of the Zn2+ ion, such that the cocktail effect enhances the redox processes and hysteresis compared to single-metal oxides and makes the structural degradation more pronounced (see Fig. 3b(I)). The complex composition of Co-HEO, with five metal cations (Co, Ni, Fe, Mn and Cu), creates significant lattice disorder, leading to the smoother DOS profile observed in Fig. (3b(II)) compared to the peak DOS of single metal oxides. This reduction in degeneracy increases the availability of electronic states near the Fermi level and facilitates electron transfer through enhanced mutation or scattering processes. Figure (3b(III)) further illustrates this effect and shows the energy band structures of Co3O4 and Fe2O3 compared to HEO. The HEO structure has a denser and more continuous band structure near the Fermi level (around 0 eV), indicating a higher DOS. This broadened DOS can reduce the effective band gap or create defect-like states, increase carrier mobility in semiconductors or stabilize transitions in conducting materials. This improved transfer supports efficient charge transfer, which likely contributes to the performance of Co-HEO as a cathode in AZIBs, allowing for faster zinc ion transport/separation and improved electrochemical stability. Due to numerous atomic configurations in Co-HEO, the DOS profile, as seen in Fig. 3b(II), is inherently smoother and more featureless than monometallic oxides, suggesting lesser degeneracy. Furthermore, compared to monometallic oxides, Co-HEO exhibited more effective electron transport because of the increased density of energy states, as shown by the energy band calculations in Fig. 3b(III).

a Schema view of the optimized crystal structure of HE cathode material, ELF of valence electrons viewed along (001) lattice planes, and Schematic diagram of the O3-P3-O3 reversible phase transition of NaNi0.2Fe0.2Mn0.35Cu0.05Zn0.1Sn0.1O2 cathode material. Reproduced with permission67 Copyright 2024, Wiley-VCH GmbH. b(I) Schematic representation of electrode materials: traditional metal-oxide structure electrode materials and HEO electrode materials with multiple electron paths in AZIBs, illustrating the cocktail effect, b(II) DOS profiles for Co-HEO and M₃O₄ (M = Fe, Co, Mn); b(III) energy band diagrams for Co-HEO and mono-metal oxides. Reproduced with permission69 Copyright 2023, Wiley-VCH GmbH.
Adsorption and diffusion
Ion adsorption during supercapacitors’ charging and discharging process (between electrodes) and ion batteries (between cathodes and anodes) could be analyzed using DFT to get important insights into binding energies and the structural and electrical performance of B/S systems. The DFT-calculated adsorption energies for single atoms (A) relative to clean surfaces and isolated atoms are calculated according to Eq. (4)70.
In Eq. (4), * denotes an active site on the surface of high-entropy materials, and *A denotes the adsorbed atom A. The existence of oxygen vacancies71, which provide more active sites for electrochemical processes and improve performance, is one of the factors influencing adsorption energies. Liu et al.72 investigated the electronic characteristics of synthesized (Mg Co Ni Cu Zn) O and the impact of oxygen vacancies on Li+ adsorption energy. According to Fig. 4a(I), oxygen vacancies greatly increased Li+ adsorption because of stronger electronic coupling and increased charge density around Li+ at the vacancy sites. As a result, the adsorption energies of −2.648 eV and −2.5 eV were obtained, as opposed to −1.919 eV for samples without defects. The charge redistribution behavior was revealed by charge density changes, as shown in Fig. 4a(II). Built-in electric fields at vacancy sites provide a negatively charged zone that increases Coulomb attraction for Li ions. The type of the adsorbate or ion, the electrode material’s surface shape73, and the interaction between the adsorbate or ion and the electrode surface are additional factors that affect adsorption energy74. High-entropy spinel (Fe, Ni, Mn, Mg, Zn)3O4 has been employed as a cathode material in rechargeable aluminum batteries by Kang et al.75. The adsorption energies of Fe₂O₃ (−3.33 eV), MnO₂ (−3.76 eV), and NiO (−3.46 eV) reflect their individual interactions with AlCl4⁻, while the high-entropy spinel (Fe, Mn, Ni, Zn, Mg)3O4, with an adsorption energy of −3.54 eV, demonstrates a stronger overall interaction, indicating enhanced reactivity compared to these single metal oxides (see Fig. 4b). Additionally, the high-entropy spinel with oxygen vacancies, exhibiting an adsorption energy of −3.54 eV, shows a less negative value than the perfect spinel (−3.95 eV). Owing to the adjustment of oxygen vacancies, the high-entropy spinel with vacancies (−3.54 eV) has a lower formation energy than the perfect spinel (−3.95 eV), which contributes to a more balanced adsorption energy. This adjustment facilitates efficient AlCl4⁻ extraction from the host and promotes its smooth transport. The nudged elastic band (NEB) method76 and the modified climbing image nudged elastic band (CINEB) method77 are frequently utilized to simulate diffusion kinetics. The NEB method is used to find the minimum energy path between two states in a system, providing insights into transition states and reaction mechanisms. The CINEB method improves upon NEB by refining the highest energy state to determine the transition state accurately. The migration barriers of Na ions are a critical parameter in the performance of cathodes for sodium-ion batteries. Using CINEB calculations, Yang et al.78 investigated the edge-face-edge migration barriers in a Na0.6(Ti0.2Mn0.2Co0.2Ni0.2Ru0.2) O2 cathode. As shown in Fig. 4c, the Na migration barriers vary among different Na(f)TM-TM sites. Specifically, Na(f)Ni-Ni sites have the lowest migration barriers at 89 meV, while Na(f) Ni-Co and Na(f)Co-Co sites exhibit slightly higher barriers of 103–105 meV. Sites containing Ti and Mn showed intermediate diffusion barriers. However, the presence of Ru significantly increased the migration barriers. Ionic diffusivity79 can be determined using the energy barriers computed from the CINEB or NEB methods, as shown in Eq. (5)80.

a(I) Atomic structure models of the (LiMgCoNiCuZn)O and the metal oxide with vacancies ((LiMgCoNiCuZn)O-Vo); a(II) Adsorption energies of Li on the various metal oxides; charge density difference for Li+ in MO and charge density difference for Li+ in MO-Vo, including the impact of metal ion substitution (LiMgCoNiCuZn)O. Reproduced with permission ref. 72 Wiley-VCH GmbH.; b structural diagrams depicting the adsorption energies of AlCl4⁻ on various samples, including Fe2O3, MgO, MnO2, NiO, ZnO, a perfect spinel, and (Fe, Mn, Ni, Zn, Mg)3O4 with an oxygen vacancy. Reproduced with permission ref. 75 Wiley-VCH GmbH.; c Single Na hopping migration barriers (in meV) for Na(e)–Na(f)–Na(e) at various Na(f) site compositions at the fully charged limit and iso surfaces illustrating the probability density distribution of Na ions (yellow). Reproduced with permission ref. 78 Copyright 2020, American Chemical Society; d structural representations of Li2Ti3(B0.25C0.25N0.25O0.25)2O2 and Li4Ti3(B0.25C0.25N0.25O0.25)2O2, including top and side views, and ELF of the (110) plane for the compounds and schematic diagrams showing the OCVs. Reproduced with permission ref. 82 Copyright 2023, American Chemical Society.
Here T is the temperature (K), Eb is the energy barrier, kB is the Boltzmann constant, and D0 is a pre-exponential component unique to the material. Shi et al. have studied niobium doping in the P2-type Na0.78Ni0.31Mn0.67Nb0.02O2 NaB cathode active material to improve Na-ion mobility81. The CINEB calculations reported Na⁺ diffusion coefficients (\({D}_{{{Na}}^{+}}\)) for P2-NaMNNb of 1.08 × 10⁻⁶ cm² s⁻¹ at room temperature (RT, 25°C) and 9.74 × 10⁻⁸ cm² s⁻¹ at low temperature (LT, −40 °C). These values are approximately one order of magnitude higher than those for P2-NaMN, which are 1.60 × 10⁻⁷ cm² s⁻¹ at RT and 8.59 × 10⁻⁹ cm² s⁻¹ at LT. Thus, niobium doping significantly enhances reaction kinetics and Na-ion mobility at both temperatures.
The calculation of open circuit voltage (OCV) is essential, as it represents a battery’s highest possible output voltage, providing critical insights into battery efficiency and performance. OCV at finite temperatures can be obtained by calculating the average voltage within the range of n1 < n < n2 using Eq. (6)42.
Here, EHEM(n1) is the total energy of HEM with n1 ions (eV), EHEM(n2) is the total energy of HEM with n2 ions (eV), n1 is the initial number of ions (unitless), n2 is the final number of ions (unitless), \({E}_{{ion}}\) is the energy of one ion (eV), and e is the elementary charge (Coulombs). Zhang et al. have investigated the OCVs of Ti3C2 and Ti0.75V0.75Cr0.75Mo0.75C2 nanosheets as anode materials for zinc-ion batteries82. As can be shown in Fig. 4d, Ti0.75V0.75Cr0.75Mo0.75C2 exhibits greater OCVs—up to 1.18 V with 4 Zn atoms—than Ti3C2, which shows 0.93 V under comparable circumstances. Due to its higher electric potential and strong Zn contacts, Ti0.75V0.75Cr0.75Mo0.75C2 typically achieves an OCV of 0.63 V, compared to 0.54 V for Ti3C2. This suggests that Ti0.75V0.75Cr0.75Mo0.75C2 has promise as a zinc-ion battery anode material.
Molecular dynamics
HEMs, characterized by their multi-element compositions, exhibit Ionic diffusivity79 and kinetic properties83, among other phenomena in HEMs, directly correlate with atomic motions, particularly in electrochemistry. In typical molecular dynamics (MD) simulations, the nuclei are modeled as classical particles, and material properties are derived from statistical averages of atomic trajectories influenced by interatomic forces84. MD simulations provide a powerful tool to study these properties by modeling atomic motions and interactions. Classical MD, which uses empirical force fields85, ab initio-derived potentials, or machine-learning force fields (MLFF)86 to compute interatomic forces based on predefined potentials, treats nuclei as classical particles and derives material properties from statistical averages of atomic trajectories. It is widely applied to high-entropy electrolytes to investigate ion diffusion83,87,88,89, solvation structures, and interfacial stability, as well as the structural stability of crystalline HEOs and HEAs90,91,92,93. In contrast, ab initio MD (AIMD), which calculates forces by solving coupled equations of motion for classical nuclear and quantum electronic degrees of freedom using DFT with Kohn-Sham orbitals94, is better suited for cathode and anode materials, offering insights into electronic structures, diffusion barriers, and redox processes in HEMs. Table 3 details the specific parameters required for these simulations to accurately capture the complex atomic and electronic interactions in HEMs.
Conductivity in HEMs is critical for their performance in applications like solid-state electrolytes and battery cathodes/anodes, as it governs the mobility of charge carriers (ions or electrons) influenced by the material’s complex, multi-component structure. It is quantified experimentally via techniques like impedance spectroscopy95 or computationally using the Nernst-Einstein equation (NE)96, which connects conductivity to diffusion coefficients.
Accurately simulating HEMs using MD necessitates a realistic representation of potential energy as a function of atomic coordinates. In classical MD, atomic interactions are generally derived by fitting potentials or forcefields to experimental data or reference ab initio calculations. Common approaches include central-force many-body potentials, such as the embedded-atom method (EAM), and modified EAM potentials97. However, these methods often neglect the electronic degrees of freedom, which are crucial for accurately modeling processes such as chemical reactions. To address this limitation, AIMD methods were introduced, which calculate forces by solving coupled equations of motion for both classical nuclear degrees of freedom and ground-state electronic degrees of freedom, as described by Kohn-Sham orbitals98.
To investigate diffusion parameters in HEMs, DFT-based computational methods such as NEB and CINEB provide accurate energy barriers and diffusion pathways in small systems but are computationally intensive. In contrast, MD simulation methods are preferred for large-scale systems and analyze the ion motion through the mean square displacement (MSD)94. This displacement of ions over time is shown in Eq. (7)99, where |Rion(t)-Rion(0)|2 represents the displacement of ions over time. Using the Einstein relation, diffusion coefficients are extracted from the MSD and can be used to study ion dynamics and structural interactions in HEMs to optimize battery performance.
Li-ion distribution and diffusion pathways in high-entropy solid electrolytes (LPSBr, HE-LPSBr, and HE-LPSBrC) were investigated using AIMD simulations by Li et al.100. The enhancement in Li+ mobility is evident from the diffusion coefficients derived from MSD plots, which are 0.74, 0.87, and 1.64 Å2/ps for LPSBr, HE-LPSBr, and HE-LPSBrCl, respectively. As shown in Fig. 5a, the Li⁺ diffusion coefficient increases with higher configurational entropy (ΔSconf), indicating that greater disorder between Br- and S2- in LPSBr facilitates more frequent Li⁺ interchange jumps, thereby enhancing Li+ conductivity. This improvement in ionic conductivity in HEMs can be attributed to cation disorder, which creates site-energy overlap and promotes ion percolation31. Additionally, HEMs exhibit greater tolerance for lattice distortions compared to their low-entropy counterparts. These distortions allow significant perturbations in the energy landscape for ion diffusion, enabling the formation of percolating diffusion pathways. Such carefully engineered lattice distortions can lead to orders-of-magnitude increases in ionic conductivity, as initially demonstrated in HEO-based materials5.

a Schematic illustration of the structure and calculated entropy change (ΔS conf) for LPSBr, HE-LPSBr, and HE-LPSBrCl. Arrhenius plot of ionic conductivities and diffusivity coefficients for LPSBr, HE-LPSBr, and HE-LPSBrCl. Reproduced with permission ref. 100 Copyright 2024, Wiley-VCH GmbH. b Charge density difference for lithium on the HEO surface, with projections for Li-(MgCoNiCuZn) O (001). RDFs for the pristine structure and the lithiated structures. Reproduced with permission ref. 102 Copyright 2023, Elsevier B.V.
Radial distribution functions (RDFs) derived from MD simulations provide information on how particle density changes with distance from a reference particle101. Analyzing RDFs allows for a comprehensive understanding of ions’ spatial distribution and correlation relative to other ions or electrode surfaces. Liu et al. examined the use of (MgCoNiCuZn)O as an anode material for Li-ion batteries102. RDF plots (see Fig. 5b) show that in the pristine structure, the coordination environments of the five cations are similar, with no significant density variation between the first and second coordination shells, indicating stability without bond disruption or formation. In contrast, the lithiated structure exhibits increased Mg-O bond strength, suggesting that Mg mainly provides structural support and contributes minimally to electron transfer during lithium incorporation. In fact, in MD and AIMD simulation methods, the distribution and diffusion paths of ions in solid electrolytes can be examined. The conductivity of ions can be effectively studied through their diffusion coefficients in HEM configurations, providing a comprehensive understanding of the spatial distribution of ions and their correlation with other ions or electrode surfaces.
Since graphene’s experimental isolation in 2004, numerous other two-dimensional (2D) materials, including TM oxides, dichalcogenides, and TM carbides/nitrides, have garnered significant research attention103. MXene 2D materials104 are highly sought after in electrochemical applications due to their excellent electric conductivity, redox potential, high packing density, and advantageous surface chemistry, which includes electrocatalytic activity, chemical inertness, and polarity105,106. These characteristics have been extensively studied for their potential in electrochemical energy storage.
In 2021, a high-entropy approach combined with MXene led to the synthesis of a new HEM, high-entropy MXene (HE-MXene), to enhance electrochemical performance107. The development of HE-MXene materials can be traced through several key milestones. First, the MAX phases were invented in 1970 by Nowotny et al. as H-phases or M2BX108. This was followed by the rebirth of these phases in 1993 when Barsoum et al. introduced the formula Mn+1AXn (n = 1-4), which evolved into the term MAX phase, where M represents an early TM, A is a group 13 or 14 element, and X is carbon and/or nitrogen109. The next significant development occurred in 2011 when Gogotsi et al. successfully prepared the MXene material Ti3C2 by immersing and exfoliating Ti3AlC2 (a MAX phase) in hydrofluoric acid, which led to the formation of a 2D lamellar crystal similar to graphene and introduced the term MXene104. Although HE-MXenes with four or more TM elements were reported more recently, they derived from high-entropy MAX phases, which were not independently explored before their 2D MXene counterparts107. The utilization of the high-entropy strategy, which encompasses the combined impacts of cocktail effects, lattice distortions, and structure stabilization, can improve the electrochemical performance of MXene materials110. In a study conducted by Liang et al., the adsorption and catalytic properties of HE-MXene, TiVNbMoC3 (doped graphene composites coated on a commercial Celgard separator), were compared to those of Ti4C3 in Li-S batteries as separators using DFT calculations9. As shown in Fig. 6a–c, HE-MXene was found to have a lower energy barrier of 0.027 eV for Li+ diffusion compared to 0.030 eV for Ti4C3 and also showed a significantly lower energy barrier of 0.017 eV for Li2S decomposition, whereas Ti4C3 had an energy barrier of 0.191 eV. Gibbs free energy calculations (see Fig. 6d) indicates that the reduction of Li2S8 to Li2S6 on HE-MXene is spontaneous and more favorable than on Ti4C3, where this reduction is non-spontaneous and requires higher energy. Despite the reduction process typically being endothermic, HE-MXene demonstrated a decrease in Gibbs free energy from 21.08 eV to 19.06 eV compared to Ti4C3, indicating enhanced thermodynamic favorability for the sulfur reduction step. Since the discovery of HE-MXenes, there have been limited studies on their performance in energy applications such as B/S. Table 4 presents an informative summary of the latest atomistic simulations on HE-MXenes, emphasizing the significant discoveries regarding their effectiveness in battery applications. HE-MXenes have demonstrated potential applications in zinc-air, lithium-ion, lithium-sulfur batteries, and supercapacitors, as shown in Table 4. Simulations utilizing DFT and AIMD suggest that their low diffusion barriers, moderate adsorption energies, and thermodynamic stability may enhance ion transport and redox reactions. In lithium-sulfur and lithium-ion batteries, HE-MXenes could enable faster ion movement and greater storage capacities, although their long-term performance remains uncertain. Regarding supercapacitors, they seem to show structural stability and acceptable electrochemical properties. While these findings highlight promising characteristics, further experimental validation is essential to confirm their practical benefits and long-term reliability.

a, b Energy pathways for Li+ ion diffusion on HE-MXene and Ti4C3, highlighting the diffusion barriers. c Diffusion barriers for the decomposition of Li2S on HE-MXene and Ti4C3. d DFT-calculated Gibbs free energy diagram for sulfur reduction on Ti4C3 and HE-MXene. Insets show the optimized structure configurations. Reprinted with permission. Reference9 Copyright 2024, American Chemical Society.
DFT methods can capture the thermodynamic and electronic properties of HEMs, especially in 2D systems. For accurate structure prediction, combining random structure generation with DFT validation may enhance pre-synthesis modeling. Plane-wave PBE functionals111, with vdW corrections112 and spin polarization113, are better suited for 2D HE systems due to weak interlayer forces and unpaired electron states. However, standard DFT struggles with long-range dispersion and achieving accurate band gaps. Using vdW-corrected XC functionals for structural relaxation114, followed by hybrid functionals or GW methods115, offers improved precision. Nevertheless, GW calculations in 2D systems face challenges from dielectric function convergence and vacuum layer effects, which significantly alter optical properties116. MD simulations further reveal mechanical stability in HEMs but require accurate interatomic potentials. Reactive force field (ReaxFF) is better for describing chemical reactions and structural evolution117, while EAM potentials capture mechanical properties more effectively118. Selecting the appropriate potential depends on the specific behavior being modeled. Combining DFT with MD can address structural and dynamic complexities in HE-2D materials, supporting more reliable predictions for next-generation battery applications. The data obtained from the atomic structure simulation can be used as input for the ML algorithm to predict the lowest ground state energy level of the proposed structure before synthesis. This is the first step towards forecasting the optimal crystal structure of the HEMs prior to synthesis. This area is discussed in detail in the section.
Machine learning on HEM for energy applications
Engineering research on material structure design has moved towards the computational path in the current two decades. Currently, we can focus on the structure of the materials, and we can move away from the numerous and time-consuming traditional tests, thereby reducing errors to the unique design of the material structure119. Therefore, with a scientific approach based on modern laws of physics, chemistry, and advanced mathematics, we can develop more sophisticated models of material structure120. With the development of new computing tools and ML algorithms that operate based on big data, it is possible to set goals to understand and design materials aligned with the desired objectives121. ML algorithms are trained on data and utilize underlying patterns and relationships to make new predictions122. ML methods aim to extract the mapping between the relationships among forces, physical and chemical structures of materials, and their performance, thereby establishing a new pattern among all parameters123.
As mentioned in earlier sections, we first presented computational methods and atomic and molecular simulation of materials, focusing on HEMs for energy applications such as B/S. In this section, we will discuss how to connect atomic data obtained from computational methods to learning algorithms. This section is developed with the perspective of providing solutions for the design of HEMs in the energy domain and introduces advanced methods for discovering the structure of materials before synthesis. Material structure encoding techniques, together with the advances in ML algorithms discussed in this section, offer the potential to link learning algorithms with quantum computing to discover new material structures, especially in HEMs. This section aims to discuss the current advances in the field of HEM structure discovery and to present techniques for predicting the crystal structure of materials, as relatively few studies have been conducted in this area. Furthermore, the key role of defining the crystal structure of materials, including HEMs, and designing the crystal structure of materials to take into account the inputs to the learning algorithm is reviewed and discussed in this section.
The primary learning techniques for HEM phase determination and synthesis involve feature engineering, data collection, model tuning, model launch, training and selection, experimental validation, and data set completion. These data points are derived from the DFT computation or MD simulation results and fed into ML algorithms124. Vast data is analyzed using several ML algorithms, such as random forests (RF)125, decision trees (DT), support vector machines (SVM), and neural networks (NN)126, which also uncover intricate multi-component correlations127,128. This approach could be used to predict variables associated with HEO/HEA/HEM, such as oxygen vacancy content, phase stability, ionic conductivity, CALPHAD phase diagram computation, and chemical binding energy129,130,131. The electrochemical performance of materials with varying compositions can be predicted and modeled using ML algorithms. Simulating and forecasting performance under identical operating circumstances is possible by training the algorithm model on current experimental data. This enables the quick screening and optimization of various material compositions and architectures, significantly accelerating the experimental exploration process132.
The goal of discovering and designing HEMs for batteries is to achieve the highest efficiency, thereby enhancing battery performance. The solution of computational methods before synthesis is the fastest and best way to create the best material structure of B/S. Due to the complexity of battery materials, inverse design is computationally very complex. Various learning algorithm models have been used to generate stable and synthesizable materials. In a very practical study, Zeni et al.133 introduce the MatterGen ML model, a diffusion-based manufacturing model that allows for the design of stable and diverse minerals across the periodic table while satisfying a wide range of property constraints. Unlike previous manufacturing approaches, MatterGen significantly improves performance—producing materials that are more than twice as likely to be novel and stable and ten times closer to their minimum ground state energy. The model works by repeatedly modifying the types of atoms, atomic coordinates, and lattice parameters and can be fine-tuned using adapter modules to target specific chemical compositions, symmetries, and physical properties such as magnetic density. In fact, MatterGen’s versatility allows it to outperform traditional methods such as substitution and random structure search in generating stable, unique, and novel materials. The model can design materials that simultaneously satisfy multiple constraints, such as combining high magnetic density with low supply chain risk. As an experimental validation, they successfully synthesized one of the MatterGen-produced structures whose measured specificity was within 20% of the predicted target, demonstrating the model’s potential as a fundamental generative framework for inverse design. These models can work very well for complex structures of high-entropy materials and are a way to reduce synthesis time. In another study, an advanced ML algorithm for generating realistic crystal structures that preserve symmetry and accurately approximate the initial ground state configurations via the diffusion crystal probabilistic autoencoder (DP-CDVAE) was introduced by Pakornchote et al.134. Building on the original CDVAE architecture that uses score matching, the DP-CDVAE algorithm applies diffusion probabilistic (DP) to more effectively denoise atomic coordinates. The model combines two components: a variational autoencoder (VAE) that predicts lattice parameters and atomic number, and a diffusion module that corrects fractional atomic coordinates using a denoising diffusion probabilistic model (DDPM). While maintaining comparable reconstruction and production quality to CDVAE, DP-CDVAE achieves a significant improvement in energy accuracy, producing structures with an average energy difference of 68.1 meV/atom lower than those produced by CDVAE, compared to the structures relaxed by DFT. With this improved capability, DP-CDVAE can be used to produce crystal structures, such as those of HEMs, that more accurately reflect their true ground-state properties. In recent research work, Batatia et al. presented MACE-MP-0, a general-purpose atomistic ML model that enables stable MD simulations on a wide range of molecules and materials with near-ab initio accuracy135. Traditional ML-based force fields have significantly improved atomistic simulations but are still limited by the need for extensive system-specific training and poor transferability between chemical systems. To overcome this challenge, this model serves as a baseline model for various atomistic applications, providing reliable and transferable FF predictions that apply to almost all structures of materials, including crystalline structures such as metal organic framework (MOF), zeolite, CeO2, and others.
The structural information of the materials includes a description of their properties, and our goal is to optimize the conductivity, work cycling stability, and real synthesis ratio stability of these materials. Therefore, to relate the structure of the materials to goals such as material properties, it is necessary to identify the chemical components and the structure of the materials that we expect to be synthesized. The first step in discovering and designing materials is to develop descriptors or key features that match the materials by building an accurate model between the descriptors and the characteristics of our ultimate goals, such as material properties. Thus, our objective in this part is to provide the process of coding and encoding material structure, which is a beneficial first step in searching HEM’s battery. We could expect increased precision for material design and reverse engineering by developing precise descriptors of the material’s structure.
Structure-property relationship discovery
In this section, we focus on methods that design material structures based on target properties, including generative models and optimization algorithms, and present unique ML frameworks that have been used to generate and predict stable crystal structures. These results demonstrate the potential of ML and interatomic learning to identify novel structures in HEMs. This recommendation also makes large-scale learning a practical step for exploring chemical spaces with more than four distinct elements and predicting their properties.
The emergence of contemporary AI applications has the potential to fundamentally change and increase the role of computational methods in materials engineering sciences. As mentioned earlier, regarding the fourth paradigm of science and the fourth industrial revolution, researchers in the field of materials science are now entering this field by utilizing ML algorithms to understand nonlinear processes through the combined methods of computing and data science. Material design by ML algorithms relies on descriptors that are understandable to computers by encoding the structure of materials136. Material structure encoding has made significant advances, but we remain far from achieving perfect methods. We first introduce how to encode material structures as input to ML algorithms, where we consider the existing methods and potential to accelerate research into discovering new material structures, especially HEMs for energy storage applications.
Since 1952, when Miller and Fletcher of American Cyanamid fitted special keys to a standard typewriter, the chemical structure encoding has been accomplished (see Fig. 7a–d). The Army’s Chemical Typewriter inspired the first automated hardware initiatives intended for encoding molecules. The y and x coordinates of the molecule were typed on this typewriter each time a key was touched, and when the proper circuit was used, all of this data was punched onto the paper tape. The machine was just meant to create visually appealing structures for its card files. They asserted that typing a structure takes, on average, 2 min137.

a View of mechanical typewriter used for encoding chemical structures (Army Chemical Typewriter); b Typed symbols from the Army Chemical Typewriter; c Walter Reed format; d Encoded structure for codeine. Reproduced with permission from ref. 137 Copyright 1963, American Chemical Society; e A workflow progress cycle for computational material structure. Here, the algorithm’s input (solution of the Schrödinger equation combined with the local optimization of the atomic forces) is the computation of a material structure’s physical characteristics, represented by the first input box, and the material’s property would be the output of this algorithm. The chemical composition as input is transferred to the output (structure-property), which includes structural predictions and the composition of potential components, in the second input box using the global optimization technique. While enough data is available and a suitable model has evolved, ML algorithms could be used to predict composition, structure, and properties, as demonstrated by the third input box. Stages one through four are a list of the four primary steps in training a ML model, along with their kinds and selection criteria.
Mapping structure and composition into descriptors that can be quickly supplied as input data to a ML algorithm is crucial for encoding material structure. As a result, considering the simplicity and precision of a material’s structure is crucial while creating its structural characteristics. The computational expenses can be decreased using a ML strategy based on data from the material structure. Greater flexibility in the representation of the links between structure and material characteristics requires structural features of materials with reduced dimensions138. Determining the physical attributes required for the representations, such as atomic bonding, periodicity, symmetry, and interatomic distances, is the first step toward achieving this goal. Calculating the physical characteristics of an input structure is a basic paradigm in the first-generation method. This is typically done by combining local optimization of the atomic forces with an approximation of the Schrödinger equation. In the second-generation method, a chemical composition input is translated to an output that includes predictions of the structure or ensemble of structures, and the combination of components is done using an optimization algorithm. The new third-generation method of ML algorithms that can forecast characteristics, composition, and structure as long as enough data is available and a suitable model has been trained. Figure 7e illustrates the fundamental phases of creating a model for material structure138. The mentioned workflow architecture can be used to successfully apply ML to material structure discovery.
Nevertheless, efficient feature engineering is a key component of ML algorithms for HEM compositions. This dependence on feature production, transformation, or structure selection captures the fundamental physics and chemical characteristics of these intricate systems. To enhance the quality and interpretability of the findings, the data can be preprocessed to eliminate unnecessary data before being filtered to rank features and reduce dimensions. There are actually two main and general features139. The main atomic structure features describe the atomic structure of individual atoms in a material and provide detailed information about nuclear arrangement, bonding, and coordination. Examples of these include orbitals and atomic structural properties, which capture the distribution of neighboring atoms around a central atom. In contrast, the general features represent the overall composition and structure of the material and reflect the collective atomic properties, providing a holistic view of the system. Common general features include the Coulomb matrix and dispersion transform140. All of these are described in detail in section “Inverse materials design”.
Inverse materials design
Despite extensive literature searches, there remains a notable lack of reported HEMs created by ML for energy applications. Therefore, in this section, we outline crystal structure generation and reverse engineering strategies, proposing their use in the inverse design of HEMs for energy-related applications such as B/S. To overcome the limitations of computational cost, nowadays, ML methods have been developed for the inverse design of crystalline solids. Inverse design in materials refers to the user’s ability to define the properties of the target material and infer the material that matches those properties. There are two major approaches to the inverse design of crystalline solids by ML algorithms: (i) global optimization and (ii) generative models141. In the global optimization algorithm, known or randomly counted combinations are modified using a set of rules to design new combinations. The initial selection of material structures and elements limits the heuristic capacity of the global optimization algorithm. In generative models’ algorithms, which learn a given data distribution, all the materials of the training set are directly modeled into a probabilistic representation from which new materials can be sampled142. In this regard, Ren et al. developed a Fourier Transformed Crystal Properties (FTCP) framework for the general inverse design of mineral crystals. They were inspired by the 2D diffraction fingerprint representation of Ziletti et al.143. In this algorithm, the encoder encodes the crystals in the training set in a continuous probabilistic hidden space, and the decoder decodes each vector in the hidden space into its corresponding crystals. During training, the target learning branch jointly organizes the hidden space to reflect a continuous change in material properties. They proposed representing an infinite 3D crystal with a reversible structure, utilizing a crystallographic representation of crystallographic information file (CIF) features that encompass both real-space and Fourier-transformed features in reciprocal space (see Fig. 8a).

a A view of the FTCP matrix model that has both real and inverse space features. Real space includes properties such as CIF properties (element matrix - description of constituent elements, network matrix, description of network parameters conditions, site coordinate matrix - description of fractional coordinates of sites, site occupancy matrix - description of element occupancy at each site, and elemental attribute matrix - descriptors elemental). The cross-space features include the elemental descriptor Zi (i for each site) for all N locations in the unit cell along different spatial frequencies, hkl (Miller indices) via spatial discrete Fourier transform. The distance of each hkl (point k) from (000) to “dhkl” is also recorded in the cross-space features. In the variational autoencoder (VAE) architecture, using inverse FTCP representation to inverse design the encoder plus decoder architecture of a typical VAE, the latent space is also connected to a target learning branch for feature mapping, which reflects the feature gradients and the latent space structured with the corresponding feature. Reproduced with permission ref. 143 Copyright 2021, Elsevier. b Map of a MEGNet module graph for the set of atomic attributes (V), bond attributes E, and global state attributes u. In the first update step, the bond attributes are updated. Information flows from atoms that form the bond, the state attributes, and the previous bond attribute to the new bond attributes. Similarly, the second and third steps update the atomic and global state attributes, respectively, by information flow among all three attributes. The final result is a new graph representation. Reproduced with permission ref. 144 Copyright 2019, American Chemical Society. c Construction of the crystal graph, in this method crystals are converted to graphs with nodes representing atoms in the unit cell and edges representing atom connections. Nodes and edges are characterized by vectors corresponding to the atoms and bonds in the crystal, respectively. Structure of the CNN on top of the crystal graph (R convolutional layers, L1, and L2 hidden layers are built on top of each node, resulting in a new graph with each node representing the local environment of each atom), Reproduced with permission ref. 145 Copyright 2018, American Physical Society.
Another method for creating an innovative crystal structure is to build MEGNet models144 that consistently perform well across a wide range of objective qualities for both molecules and crystalline forms (see Fig. 8b). Atoms and the bonds that connect them are naturally represented as graphs, and the sequential update scheme of graph networks naturally facilitates information flow between atoms, bonds, and global states. This technique further extends these models to state-dependent and data-limited features by incorporating global state inputs and transfer learning of elemental embeddings. The above generalizations provide a strong foundation for creating universal property models that will accelerate the identification of new materials and overcome some significant limitations in the application of ML in chemistry and materials research. When graph network ML techniques are applied to expedite the design of crystalline materials, they typically require the creation of manually crafted feature vectors or intricate conversions of atomic coordinates for the input of crystal structures. These requirements either restrict the model to particular crystal types or impede the provision of chemical insight.
Figure 8c illustrates how Tian et al.145 built a global and interpretable display of crystalline materials using a crystal graph convolutional neural network (CNN) architecture for directly learning material attributes from the bonding of atoms in a crystal. Their approach yielded exact DFT predictions for eight distinct crystal characteristics with varying compositions and structures. This approach can be recommended for HE-2D MXene’s material crystal structures in battery or supercapacitor applications, as it can accurately predict many crystal structure properties.
It is necessary to use atomistic modeling to model molecular systems properly. To find out about the energy, shape, and curvature of the system’s potential energy surface, we need to solve the Schrödinger equation for the molecular electronic Hamiltonian146. Using this approach, the molecule is represented as a collection of nuclear charges and Cartesian coordinates corresponding to the locations of the atoms in 3-D147. Hamiltonians use only a molecule’s physics and atomic constants, but Coulomb matrix representations use Coulombic forces between atoms’ charges. Utilizing simple nuclear charges and atomic locations, Rupp et al.148 developed an ML model to forecast the atomization energies of a wide range of organic compounds. Driven by the nuclear repulsion factor in the molecular Hamiltonian and free atom energies, they created a model that depicts molecules as Coulomb matrices. They made a method for resolving the molecular Schrödinger equation, which translated into a more straightforward nonlinear statistical regression issue. Various methods have been proposed for prediction in the category of displaying molecular structure: fingerprint142, simplified molecular-input line-entry system (SMILES)149, bag of bond150, and symmetry functions151. Since each atom is viewed as a node and bonds as edges, molecules are naturally represented as graphs according to the empirical bonding rules. SMILES strings149, a 1D text encoded with a predetermined grammar, are one of the standards for molecular diagrams. More complex representations employ a weighted graph instead of text encoding and include a range of vector properties on nodes and edges, including charge, distance, and link type. Alberts et al.152 employed the SMILES code to directly predict molecular structure using a transformer model trained on chemical formulas and infrared (IR) spectra. Before fine-tuning the model using real spectra from the National Institute of Standards and Technology153 IR database, they trained the model on simulated infrared spectra using molecular dynamics and the polymer consistent force field154. Although SMILES representations are often employed for discrete molecular systems, by concentrating on representative molecule fragments or cluster models, they may be modified to mimic the local bonding environments and surface reactivity of HEMs. Metal-oxygen or metal-nitrogen units are examples of surface-active motifs or tiny coordination complexes that may be isolated from HEMs and encoded via SMILES for use in ML models. With this method, reactive sites and ligand interactions on HEM surfaces may be efficiently screened, providing information on their interfacial characteristics and catalytic behavior without the need for complete crystalline representations. The challenge of evaluating surface molecular adsorption interactions is one of the recent obstacles in finding an experimentally measurable and theoretically calculable descriptor for assessing adsorption surface interactions. Wang et al.155 showed that the electric dipole moment can be used to establish structure-property relationships for molecular adsorption on metal catalyst surfaces. In addition to providing a method for designing efficient catalysts, they applied an ML (NN) to a big dataset of quantum computations (DFT dataset). In this way, quantum computation and learning algorithms can design HEM structures and effectively identify energy band factors. An advanced approach to material discovery leveraging AI involves generative models. Ruiming et al.156 introduced WyCryst, an innovative AI-driven model that emulates feature-based VAE models while adhering to symmetry constraints and incorporating an automated DFT-based framework. This model facilitates both large-scale exploration of mineral crystal structures and the development of materials that maintain structural symmetry.
One crucial problem for complicated datasets, such as crystalline compounds, is creating low-dimensional representations of input crystal structures using chemical insights. Jiang et al. introduced atom-specific stable homology (ASPH), an individual representation of crystal structures that can represent pairwise and many-body interactions and indicate the topology-property interactions of a group of atoms at different scales157. Using an algebraic topology-based method developed by ML algorithms. ASPH utilizes this framework for periodic systems and provides a range of topological fingerprints particular to individual atoms in the crystal cell. The mentioned ML algorithm, utilizing gradient-boosted regression trees, was trained using the Open Quantum Materials Database (OQMD)158 and the Inorganic Crystal Structure Database (ICSD)159. The OQMD databank has over 200K158 and 300K160 DFT-calculated crystal structures and has a wide range of potential B/S applications and energy material types to model and predict new stable ternary compounds. Another data bank is the Materials Project161. The MP has become a cornerstone of data-driven materials science, providing a sustainable, open-source software ecosystem that accelerates materials discovery and design. MP has also supported the development of ML models for predicting electronic densities of states and X-ray spectra and released datasets such as Matbench162, a benchmark suite for evaluating ML algorithms. The mentioned bank dataset can serve as a new route to create new crystal structures of HEMs for energy applications such as B/S.
The DiffCSP (Diffusion Crystal Structure Prediction) model is one of the most advanced new generations in the field of predicting and generating the crystal structure of materials, which is developed based on diffusion models163. By learning from large databases such as MP161, this model can generate a stable crystal structure from the input chemical composition. Unlike traditional methods that were limited to only direct prediction or reconstruction of structures similar to training data, DiffCSP starts the structure construction process from a random state and gradually reaches the final crystal structure by removing noise. The result of this process is structures that, in addition to high energy stability, have much greater diversity and accuracy than previous methods. As mentioned, CDVAE, which is based on a variational autoencoder with crystallographic constraints, is capable of reconstructing structures but offers less accuracy in unknown compounds. Among these methods, DiffCSP163 and DiffCSP++164, using the inverse diffusion process, are not only capable of generating stable and physically stable structures but also cover a wide range of energies and chemical compositions. This feature makes them a powerful tool for discovering new HEMs and optimizing crystal structures, which can be a key step before more accurate calculations such as DFT.
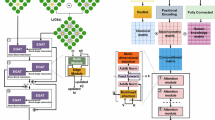
Another strategy for inverse materials design and material discovery, which can be recommended especially for HEMs, is the use of a deep-learning (DL) algorithm. In the following, we present unique DL frameworks that have been used to create and predict stable crystal structures. These results suggest the potential of ML and inter-atomic learning for identifying new structures in HEMs. This recommendation would also make large-scale learning a feasible step for exploring chemical spaces with more than four distinct elements and predicting their characteristics. To address the challenge of forecasting material properties without crystal structures, Goodall and Lee165 developed a physically motivated DL framework using the Deep Ensemble (DE) approach. This approach uses only stoichiometry as input and automatically learns suitable and systematically improvable descriptors from data. Using robust graphs to depict material compositions was their main methodological breakthrough. They demonstrated how creating material compositions greatly enhances the model’s functionality compared to alternative structural methods. They emphasized the significance of uncertainty estimation in material science applications and demonstrated how their model can generate practical uncertainty estimates using a DE. Through extensive active learning, Amil et al. scaled up the DL for materials research, which can direct the discovery of new materials123. Two key elements supported their method: (i) They developed techniques for producing a variety of candidate structures, such as random structure search166 and novel symmetry-aware partial substitutions (SAPS). (ii) They also employed cutting-edge graph neural networks (GNNs) to enhance the modeling of material characteristics. The graph networks for materials exploration (GNoME) were utilized to filter potential structures after being trained on the supplied data, and the mentioned network is illustrated in Fig. 9a. To validate model predictions and provide a data flywheel for training more resilient models on larger datasets in the subsequent active learning cycle, the energy of the filtered candidates was calculated using DFT results. After grouping structures, they prioritized polymorphs for DFT assessment. They create 100 random structures for the assessment using ab initio random structure searching (AIRSS)166 and then use GNoME to filter compositions under relaxed restrictions. Models forecast energy in both frames, and a threshold is selected according to the relative stability (decomposition energy) of competing phases. DFT calculations were employed for the assessment, which quantified the number of stable materials identified and the accuracy of the predicted stable materials in comparison to the Materials Project database33. Based on the string crystal representation, Hang et al.167 have created a simplified linear input crystal encoding system that can reconstruct over 40 K structurally and chemically diverse crystal structures. This system has high potential and will be a useful tool for finding HEMs in energy applications such as B/S.

a A schematic representation of the GNoME-based discovery demonstrates how DFT and model-based filters work together as a data flywheel to enhance forecasts, Reproduced with permission ref. 123 Copyright 2023, Springer Nature Limited; b A schema view of the SCANN technique to how a local structure is represented recursively. To learn the representations of different local structures in a material, the SCANN is created by stacking local attention layers and embedding layers. These local structures’ attention scores are evaluated at the readout stage using a global attention layer. The attention score shows how much focus should be placed on a local structure to effectively depict the material and forecast its physical characteristics. Based on the representations of its local structures and the accompanying attention ratings, the material representation is blended linearly. The material representation is subjected to fully connected (FC) layers to estimate the material’s properties, Reproduced with permission ref. 168 Copyright 2023, Springer Nature Limited.
The self-consistent attention neural network (SCANN), using the DL algorithm, was first presented by Vu et al.168. To facilitate the anticipation and comprehension of material characteristics, the SCANN focuses on expressing material structures through the local structures of atoms, utilizing learned weights. The primary objective of SCANN was to obtain consistent representations of these regional structures in the material through a recursive process. These models were correctly merged to comprehensively summarize the material structure. To capture the intricate and nonlinear interactions between the models and attributes, the SCANN architecture was specifically designed with a fully linked layer. Key elements that aid in comprehending the structure-property links of the material are identified by the global attention (GA) scores of the local structures, which are derived from the global attention layer. Figure 9b thoroughly illustrates the suggested SCANN architecture. SCANN utilizes attention methodologies to learn from material datasets, forecast material attributes, and decipher the fundamental features of material structures. SCANN learns representations of atomic local structures in a self-consistent way and makes accurate property estimates by recursively applying attention to nearby local structures. Results from experiments using five datasets of molecular and crystalline material structures showed how well SCANN predicted various material characteristics. Their structures are shown in Fig. 9b. At first, the accessible crystals were modified to create structural candidates. By modifying ionic replacement possibilities to prioritize discovery and utilizing the developed SAPS to permit the effective use of incomplete substitutes, they significantly expanded the collection of substitutions. During active learning, this expansion produced over 109 possibilities; the resultant structures were filtered using GNoME with uncertainty quantification by deep ensembles and volume-based test-time augmentation169.
The ability of the GNN model to efficiently encode and use the geometric information in the connections of atoms with bonds as edges gives it a clear advantage in material property prediction145. Inter-bond interactions are represented as line graph edges in the Atomistic Line Graph Neural Network (ALIGNN) model, which goes beyond the original abstraction level170. This novel method has allowed ALIGNN to outperform several other modern models, proving its superiority in predicting material properties. The ability of the GNN model to encode complex HEM structures can be highly effective in predicting the connections between atoms through bonds as edges.
In the computational modeling section, we noted that classical MD force fields often assume atomic charges to be fixed, limiting their ability to investigate complex electronic interactions in crystal structures. In this regard, we noted that AIMD achieves quantum-level accuracy by explicitly solving electronic structures, but it is computationally challenging for large-scale simulations in terms of cost and computational time. To overcome this problem, machine learning interatomic potentials (MLIPs) such as ænet171 has emerged as efficient methods today, providing accuracy close to that of the DFT method at a much lower computational cost. More advanced GNN-based MLIPs such as MACE-MP135, and M3GNet172 further improve the accuracy by incorporating physical symmetries and long-range interactions. However, most existing MLIPs—including ænet —has difficulty accounting for the effects of ionic valence and charge states that critically affect bonding, coordination, and electronic structure, especially in transition metal systems. Since charge and spin states strongly influence atomic interactions, the Crystal Hamiltonian Graph Neural Network (CHGNet) introduces site-specific magnetic moments as charge state constraints, enhancing the regularization of the latent space, allowing for accurate modeling of electronic interactions and charge dynamics in complex materials173.
In the approach to studying the structure of materials from an energetic perspective, the importance of the valence of an ion arises from the fact that it can form very different bonds with its environment, depending on the number of electrons. In traditional MLIP algorithms, the elemental label is considered as the basic chemical identity, and the different valence states of transition metal ions, as different elements, behave differently with respect to each other. The charge state is also a degree of freedom that can generate configurational entropy, and its optimization can lead to charge and ion motion that must be considered in the data labeling.
The challenge of considering electrostatic charge in the MLIP algorithm is essential for calculating energy. To overcome this challenge, the charge is defined as an atomic property, namely the atomic charge, which can be inferred from the inclusion of magnetic moments (magmoms—initial magnetic moment). Deng et al.173 showed that by explicitly including location-specific magmas as charge state constraints in the CHGNet, both latent space regularization can be enhanced and electronic interactions can be accurately captured.
In general, the potential of the MLIP algorithm can be divided into descriptor-based FFs and GNN-based models. In a broader definition, MLFFs include descriptor-based interatomic models and DL algorithms86. In fact, these models are supervised ML regression models in the learning algorithm field that are used for interatomic potentials. Furthermore, these models can accelerate the MD simulation process. In the following, we present a subset of these models, suitable for analyzing the crystal structure of HEMs within the B/S applications. More details and descriptions of all these models are provided by Ryosuke and Saori. Descriptor-based models such as Gaussian Approximation Potential (GAP)174, Moment Tensor Potential (MTP)175, and Spectral Neighbor Analysis Potential (SNAP)176 rely on hand-crafted descriptors (such as Smooth Overlap of Atomic Positions (SOAP)177 or moment tensors) that encode atomic and interatomic environments. The high accuracy but comes with a high computational cost. The MTP model uses a linear regression model to extend local environments to moment tensors that can be systematically improved, offering good accuracy and efficiency. The SNAP model uses a linear regression model to use spectral coefficients with linear regression, providing fast and robust simulations suitable for large-scale MD simulations. The Deep-MD model uses a deep NN regression method and deep potential software to learn potential energy levels from ab initio and DFT data178. The total energy of the system is calculated as the sum of local atomic energies, each predicted by an NN algorithm. Compared to models such as GAP or MTP, the Deep-MD model requires more extensive datasets but offers higher throughput in terms of scalability. All of the techniques discussed in this section have a high potential for use in the discovery of high-performance HEM’s structures prior to synthesis for B/S materials, as there has been some recent research on the structure discovery of HEM’s battery.
Challenges and outlook
The costs of chemical reaction modeling grow exponentially with increasing system scale. One technology solution to get around this barrier has been suggested: quantum computers. Many issues involving quantum physics or chemistry cannot be solved by classical computer simulations; therefore, utilizing quantum technology to address these difficulties may be a viable approach. Accurate predictions from classical computers may become possible with the development of efficient classical theory in quantum chemistry and mechanical processes. For instance, when we undertake real-world experiments and obtain correct approximations for exchange-correlation operations, DFT enables precise predictions of molecular characteristics38,179. Since quantum error-correction costs for fault-tolerant quantum computing systems are so low, fault-tolerant quantum computers now have the potential to affect society in the long run180.
Even though noisy intermediate-scale quantum (NISQ) systems180 have been suggested as a potential means of achieving a quantum advantage soon, quantum machine learning (QML) future applicability remains uncertain. The ability to precisely produce quantum data and correctly store it in quantum memory will be provided by quantum algorithms, such as quantum simulation algorithms181,182. In this case, the quantum computer learns directly from the data, making QML the ideal model for learning, inferring, and generating predictions from quantum data183. Combining quantum computing and AI may revolutionize future technologies and create a new school of thought about AI based on generative models. In this context, quantum algorithms will have exponential progress over classical algorithms in the field of generative models184.
One of the challenges ahead is evaluating and estimating the quantum, logical, and physical computing resources required to perform quantum chemistry simulations. Modules that can accurately estimate the qubit coupling, error correction, and error rate in terms of physical computation are needed. Matthew et al. evaluated how many gates, physical qubits, and runtime (in qubits per second) are required for ternary compounds and small molecules under different hypothetical architectures185. The results show that even for small molecules, the number of resources is very large, with a large number of gates T on the order of 107 to 1015. They emphasized that although fault-tolerant quantum algorithms (such as quantum phase estimation) require huge resources and are therefore not yet available for NISQ, NISQ methods (such as the variational quantum eigensolver (VQE)186 and quantum kernel approaches) remain vital in the short term. Hence, although they did not provide a direct demonstration of a realistic NISQ prototype on materials for battery discovery or a comparison with classical baselines on the same data, it does provide an important measure of the resource gap between current quantum approaches and the requirements of materials-scale problems. Thus, researchers who intend to apply NISQ, VQE or quantum core workflows to HEMs for the purpose of discovering novel materials for batteries can use QREChem to estimate the time to implementation of such prototypes, thus building a bridge towards hybrid workflows that compare quantum outputs with classical DFT/ML baselines.
Our perspective is that HEMs could be immediately converted from their fundamental analog form to a quantum digital form in the future, allowing for the direct acquisition of quantum data. Next, utilizing fault-tolerant quantum computing and error-corrected quantum communications, this data may be sent to quantum networks for distributed and/or centralized processing with QML models. QML will soon get to a point where it is comparable to current ML, where data is gathered from several databases, moved to central cloud computing, and used to train ML models. Similar to how huge classical ML emerged in the present era of plentiful data, one might anticipate that QML will become more widely used in the fault-tolerant period because of the easy access to quantum data.
The synthesis of HEMs has made significant progress, but translating this into advanced materials for B/S remains a challenge. As a result of quantum theories, learning algorithms, and powerful supercomputers, we are making great strides in identifying novel material structures before lab synthesis. To further advance the development of the battery industry, efforts to identify new structures of HEMs require data science based on quantum theories to understand the structure of materials fundamentally. Given the immiscibility caused by elemental differences and compositional complexity in HEMs, predicting their structure and performance before synthesis using non-equilibrium approaches under different process conditions is a complex task. A fundamental understanding of surface energies, defects, elemental distribution, electronic structures, and chemical reaction pathways is only possible by combining quantum theories and learning algorithms. Material structure encoding can be a beneficial step in this direction, helping to grasp atomic structure better and turn it into input data for material discovery using AI. Despite recent research in this area, there are still issues that hinder broader research goals from synergizing the knowledge of material discovery in the energy domain, especially for HEMs. In this regard, we aim to identify future research horizons where future studies could be fruitful, enabling researchers and materials research institutions to provide valuable contributions.
-
The structure and ability to capture spatial and angular information of adsorption configurations can play a key role in designing descriptors for encoding. In addition to accurately depicting interatomic interactions, graph theory and algebraic topology are also useful for studying chemical reactions. Using Coulomb matrices and IR descriptors together can enable a powerful representation of crystals.
-
Integration of crystal structures would be very useful as a means of forming and updating crystal structure databases for HEMs. For example, the ICSD contains all mineral crystal structure records published since 1913, including their atomic coordinates. The integration of various HEM structures into a single collection would be highly beneficial, and this could be accomplished by extending an invitation to all researchers.
-
Exploring chemical spaces with more than four unique elements and forecasting their properties is made feasible by the large-scale generation and prediction of novel stable HEMS structures using DL algorithms. A significant methodological advancement in creating new HEMs for new B/S compositions is the use of reaction stoichiometry as input, which automatically learns suitable and systematically improved descriptors from the database.
-
Future materials research to determine the optimal structure for the energy field may heavily rely on building a database and sharing simulated data, as the integration of atomic simulation DFT and AIMD data of HEMs as input to learning algorithms is highly productive.
From the perspective of data science, the research on HEMs is challenging due to the scarcity of reliable datasets for prediction or the development of valuable algorithms. Major material Genome databases5 don’t have much data on B/S and have few details on HEMs. This is due to the complexity of multicomponent HEM systems, which have a large compositional space, resulting in a significant dispersion of the data. Indeed, the lack of standard representations for chemical disorder, the diversity of synthesis and characterization techniques, and the limited availability of benchmark datasets make model reproducibility and validation difficult. Furthermore, the detailed structural descriptors required to account for all the complex thermodynamic and structural interactions simultaneously present in the complex structures of HEMs are currently not available. Although existing databases, such as MP145 and OQMD158,160, provide useful information on the fundamental structures of crystalline materials, they cover only a small fraction of multicomponent elemental systems and are primarily intended for ordered crystalline compounds. Current databases do not have the breadth of the typical HEM compositional structure, which often consists of five or more components. Additionally, the relationship between the complex configuration phases of materials and the composite structure of HEM materials is not adequately addressed by these databases, and most available data focus on equilibrium structures or ideal calculations. Predictive models require new datasets that clearly represent the complex configuration structures of HEMs and ideally contain a bank of computational and experimental data that can be combined. The aforementioned limitations collectively limit the capacity of ML algorithms to synthesize and accurately predict the properties or stability of HEMs for innovative materials applications in the energy field. To support HEM research, it is necessary to develop their structural material datasets. To improve our understanding of HEMs and their characteristics, these aspects necessitate further development in dataset generation and data science techniques.
Data availability
No datasets were generated or analyzed during the current study.
References
Wan, X. et al. Machine learning paves the way for high entropy compounds exploration: challenges, progress, and outlook. Adv. Mater. 37, 2305192 (2025).
Sharma, P., Dwivedi, V. K. & Dwivedi, S. P. Development of high entropy alloys: a review. Mater. Today Proc. 43, 502–509 (2021).
Hutson, M. Bringing machine learning to the masses. Science 365, 416–417 (2019).
Jablonka, K. M., Ongari, D., Moosavi, S. M. & Smit, B. Big-data science in porous materials: materials genomics and machine learning. Chem. Rev. 120, 8066–8129 (2020).
Ouyang, B. & Zeng, Y. The rise of high-entropy battery materials. Nat. Commun. 15, 973 (2024).
Aykol, M., Herring, P. & Anapolsky, A. Machine learning for continuous innovation in battery technologies. Nat. Rev. Mater. 5, 725–727 (2020).
Yeh, J. et al. Nanostructured high-entropy alloys with multiple principal elements: novel alloy design concepts and outcomes. Adv. Eng. Mater. 6, 299–303 (2004).
Sohn, S. et al. Noble metal high entropy alloys. Scr. Mater. 126, 29–32 (2017).
Liang, Q. et al. High-entropy MXene as bifunctional mediator toward advanced Li–S full batteries. ACS Nano 18, 2395–2408 (2024).
Rost, C. M. et al. Entropy-stabilized oxides. Nat. Commun. 6, 8485 (2015).
Zhang, R.-Z., Gucci, F., Zhu, H., Chen, K. & Reece, M. J. Data-driven design of ecofriendly thermoelectric high-entropy sulfides. Inorg. Chem. 57, 13027–13033 (2018).
Yang, Y. et al. Achieving exceptional strength and ductility combination in a heterostructured Mg-Y alloy with densely refined twins. J. Mater. Sci. Technol. 189, 132–145 (2024).
Rogachev, A. S. et al. Structure and properties of equiatomic CoCrFeNiMn alloy fabricated by high-energy ball milling and spark plasma sintering. J. Alloy. Compd. 805, 1237–1245 (2019).
Rao, Z. et al. Unveiling the mechanism of abnormal magnetic behavior of FeNiCoMnCu high-entropy alloys through a joint experimental-theoretical study. Phys. Rev. Mater. 4, 14402 (2020).
Cai, Y. P., Wang, G. J., Ma, Y. J., Cao, Z. H. & Meng, X. K. High hardness dual-phase high entropy alloy thin films produced by interface alloying. Scr. Mater. 162, 281–285 (2019).
Liu, X., Zhang, J. & Pei, Z. Machine learning for high-entropy alloys: progress, challenges and opportunities. Prog. Mater. Sci. 131, 101018 (2023).
Castro, D., Jaeger, P., Baptista, A. C. & Oliveira, J. P. An overview of high-entropy alloys as biomaterials. Metals 11, 648 (2021).
Xin, Y. et al. High-entropy alloys as a platform for catalysis: progress, challenges, and opportunities. ACS Catal. 10, 11280–11306 (2020).
Hussain, I. et al. High entropy alloys as electrode material for supercapacitors: a review. J. Energy Storage 44, 103405 (2021).
Xu, H. et al. Nano high-entropy alloy with strong affinity driving fast polysulfide conversion towards stable lithium sulfur batteries. Energy Storage Mater. 43, 212–220 (2021).
Chaudhary, V., Chaudhary, R., Banerjee, R. & Ramanujan, R. V. Accelerated and conventional development of magnetic high entropy alloys. Mater. Today 49, 231–252 (2021).
Wang, Q., Velasco, L., Breitung, B. & Presser, V. High-entropy energy materials in the age of big data: a critical guide to next-generation synthesis and applications. Adv. Energy Mater. 11, 2102355 (2021).
Ye, Y. F., Wang, Q., Lu, J., Liu, C. T. & Yang, Y. High-entropy alloy: challenges and prospects. Mater. Today 19, 349–362 (2016).
Baldock, R. J. N., Pártay, L. B., Bartók, A. P., Payne, M. C. & Csányi, G. Determining pressure-temperature phase diagrams of materials. Phys. Rev. B 93, 174108 (2016).
Cordell, J. J., Pan, J., Tamboli, A. C., Tucker, G. J. & Lany, S. Probing configurational disorder in ZnGeN 2 using cluster-based Monte Carlo. Phys. Rev. Mater. 5, 24604 (2021).
Dey, D., Liang, L. & Yu, L. Mixed enthalpy–entropy descriptor for the rational design of synthesizable high-entropy materials over vast chemical spaces. J. Am. Chem. Soc. 146, 5142–5151 (2024).
Wang, Q. et al. Multi-anionic and-cationic compounds: new high entropy materials for advanced Li-ion batteries. Energy Environ. Sci. 12, 2433–2442 (2019).
Lun, Z. et al. Cation-disordered rocksalt-type high-entropy cathodes for Li-ion batteries. Nat. Mater. 20, 214–221 (2021).
Li, Y. et al. A lithium superionic conductor for millimeter-thick battery electrode. Science 381, 50–53 (2023).
Jung, S.-K. et al. Unlocking the hidden chemical space in cubic-phase garnet solid electrolyte for efficient quasi-all-solid-state lithium batteries. Nat. Commun. 13, 7638 (2022).
Zeng, Y. et al. High-entropy mechanism to boost ionic conductivity. Science 378, 1320–1324 (2022).
Hautier, G., Fischer, C. C., Jain, A., Mueller, T. & Ceder, G. Finding nature’s missing ternary oxide compounds using machine learning and density functional theory. Chem. Mater. 22, 3762–3767 (2010).
Jain, A. et al. Commentary: The Materials Project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Pyzer-Knapp, E. O. et al. Accelerating materials discovery using artificial intelligence, high performance computing and robotics. npj Comput. Mater. 8, 84 (2022).
Ma, Y. et al. High-entropy energy materials: challenges and new opportunities. Energy Environ. Sci. 14, 2883–2905 (2021).
Jiang, D., Xie, L. & Wang, L. Current application status of multi-scale simulation and machine learning in research on high-entropy alloys. J. Mater. Res. Technol. 26, 1341–1374 (2023).
Louie, S. G., Chan, Y.-H., da Jornada, F. H., Li, Z. & Qiu, D. Y. Discovering and understanding materials through computation. Nat. Mater. 20, 728–735 (2021).
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev. 136, B864 (1964).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133 (1965).
Marzari, N., Ferretti, A. & Wolverton, C. Electronic-structure methods for materials design. Nat. Mater. 20, 736–749 (2021).
He, Q., Yu, B., Li, Z. & Zhao, Y. Density functional theory for battery materials. Energy Environ. Mater. 2, 264–279 (2019).
Zhou, F., Cococcioni, M., Marianetti, C. A., Morgan, D. & Ceder, G. First-principles prediction of redox potentials in transition-metal compounds with LDA+ U. Phys. Rev. B—Condens. Matter Mater. Phys. 70, 235121 (2004).
Meredig, B., Thompson, A., Hansen, H. A., Wolverton, C. & Van de Walle, A. Method for locating low-energy solutions within DFT+ U. Phys. Rev. B—Condens. Matter Mater. Phys. 82, 195128 (2010).
Devi, R., Singh, B., Canepa, P. & Sai Gautam, G. Effect of exchange-correlation functionals on the estimation of migration barriers in battery materials. npj Comput. Mater. 8, 160 (2022).
Borlido, P., Doumont, J., Tran, F., Marques, M. A. L. & Botti, S. Validation of pseudopotential calculations for the electronic band gap of solids. J. Chem. Theory Comput. 16, 3620–3627 (2020).
Stöhr, M., Van Voorhis, T. & Tkatchenko, A. Theory and practice of modeling van der Waals interactions in electronic-structure calculations. Chem. Soc. Rev. 48, 4118–4154 (2019).
Jain, A. et al. Formation enthalpies by mixing GGA and GGA+ U calculations. Phys. Rev. B—Condens. Matter Mater. Phys. 84, 45115 (2011).
Hatzenbichler, L., Zeisl, S., Clemens, H. & Holec, D. Phase stability of TiAl-based BCC high entropy alloys. Intermetallics 158, 107893 (2023).
Thomas, J. A., Turney, J. E., Iutzi, R. M., Amon, C. H. & McGaughey, A. J. H. Predicting phonon dispersion relations and lifetimes from the spectral energy density. Phys. Rev. B—Condens. Matter Mater. Phys. 81, 81411 (2010).
Nepal, N. K., Adhikari, S., Neupane, B. & Ruzsinszky, A. Formation energy puzzle in intermetallic alloys: random phase approximation fails to predict accurate formation energies. Phys. Rev. B 102, 205121 (2020).
Lin, C.-C. et al. In-situ X-ray studies of high-entropy layered oxide cathode for sodium-ion batteries. Energy Storage Mater. 51, 159–171 (2022).
Athayde, D. D. et al. Review of perovskite ceramic synthesis and membrane preparation methods. Ceram. Int. 42, 6555–6571 (2016).
Rohlfing, M. & Bredow, T. Binding energy of adsorbates on a noble-metal surface: exchange and correlation effects. Phys. Rev. Lett. 101, 266106 (2008).
Zheng, Y. et al. A high-entropy metal oxide as chemical anchor of polysulfide for lithium-sulfur batteries. Energy Storage Mater. 23, 678–683 (2019).
Meng, Z., Xu, Z., Tian, H. & Zheng, W. Insights into high-entropy material synthesis dynamics criteria based on a thermodynamic framework. Mater. Horiz. 10, 3293–3303 (2023).
Manzoor, A., Pandey, S., Chakraborty, D., Phillpot, S. R. & Aidhy, D. S. Entropy contributions to phase stability in binary random solid solutions. npj Comput. Mater. 4, 47 (2018).
Pham, T.-L. et al. Structural-stability study of antiperovskite Na 3 O Cl for Na-rich solid electrolyte. Phys. Rev. Appl. 19, 34004 (2023).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1–5 (2015).
Cao, X. et al. FeNiCrCoMn high-entropy alloy nanoparticles loaded on carbon nanotubes as bifunctional oxygen catalysts for rechargeable zinc-air batteries. ACS Appl. Mater. Interfaces 15, 32365–32375 (2023).
Savin, A., Nesper, R., Wengert, S. & Fässler, T. F. ELF: the electron localization function. Angew. Chem. Int. Ed. Engl. 36, 1808–1832 (1997).
Schwarz, K. Band structure and chemical bonding in transition metal carbides and nitrides. Crit. Rev. Solid State Mater. Sci. 13, 211–257 (1987).
Narang, P., Garcia, C. A. C. & Felser, C. The topology of electronic band structures. Nat. Mater. 20, 293–300 (2021).
Jain, A., Shin, Y. & Persson, K. A. Computational predictions of energy materials using density functional theory. Nat. Rev. Mater. 1, 1–13 (2016).
Levämäki, H. & Vitos, L. Electron localization function implementation in the exact muffin-tin orbitals method. Phys. Rev. B 103, 35118 (2021).
Guo, H. et al. Predominant P3-type solid–solution phase transition enables high-stability O3-type Na-Ion cathodes. ACS Appl. Mater. Interfaces 16, 27352–27359 (2024).
Sen, S., Palabathuni, M., Ryan, K. M. & Singh, S. High entropy oxides: mapping the landscape from fundamentals to future vistas: focus review. ACS Energy Lett. 9, 3694–3718 (2024).
Wang, B. et al. High-entropy phase stabilization engineering enables high-performance layered cathode for sodium-ion batteries. Adv. Energy Mater. 14, 2401090 (2024).
Wang, K. et al. Synergy of cations in high entropy oxide lithium ion battery anode. Nat. Commun. 14, 1487 (2023).
Du, K. et al. High entropy oxides modulate atomic-level interactions for high-performance aqueous zinc-ion batteries. Adv. Mater. 35, 2301538 (2023).
Calle-Vallejo, F., Martínez, J. I., García-Lastra, J. M., Rossmeisl, J. & Koper, M. T. M. Physical and chemical nature of the scaling relations between adsorption energies<? format?> of atoms on metal surfaces. Phys. Rev. Lett. 108, 116103 (2012).
Cool, T., Bartol, A., Kasenga, M., Modi, K. & García, R. E. Gibbs: phase equilibria and symbolic computation of thermodynamic properties. Calphad 34, 393–404 (2010).
Liu, X. et al. Kinetically accelerated lithium storage in high-entropy (LiMgCoNiCuZn) O enabled by oxygen vacancies. Small 18, 2200524 (2022).
Onawole, A. T., Hussein, I. A., Carchini, G., Sakhaee-Pour, A. & Berdiyorov, G. R. Effect of surface morphology on methane interaction with calcite: a DFT study. RSC Adv. 10, 16669–16674 (2020).
Mohammadi, M. D., Abdullah, H. Y., Kalamse, V. & Chaudhari, A. Adsorption of alkali and alkaline earth ions on nanocages using density functional theory. Comput. Theor. Chem. 1204, 113391 (2021).
Kang, R. et al. Configurational entropy strategy enhanced structure stability achieves robust cathode for aluminum batteries. Small 20, 2305998 (2024).
Sheppard, D. & Henkelman, G. Paths to which the nudged elastic band converges. J. Comput. Chem. 32, 1769–1771 (2011).
Sheppard, D., Xiao, P., Chemelewski, W., Johnson, D. D. & Henkelman, G. A generalized solid-state nudged elastic band method. J. Chem. Phys. 136, 074103 (2012).
Yang, L. et al. Multiprincipal component P2-Na0. 6 (Ti0. 2Mn0. 2Co0. 2Ni0. 2Ru0. 2) O2 as a high-rate cathode for sodium-ion batteries. JACS Au 1, 98–107 (2020).
Jiang, J., Cao, D., Jiang, D. & Wu, J. Time-dependent density functional theory for ion diffusion in electrochemical systems. J. Phys. Condens. Matter 26, 284102 (2014).
Trachta, M., Volný, T., Rubes, M., Bulanek, R. & Bludský, O. QM-MM approach to the accurate description of slow diffusion in porous materials. J. Phys. Chem. C. 127, 8856–8863 (2023).
Shi, Q. et al. Niobium-doped layered cathode material for high-power and low-temperature sodium-ion batteries. Nat. Commun. 13, 3205 (2022).
Zhang, L. et al. First-principles studies on high-entropy Ti0. 75V0. 75Cr0. 75Mo0. 75C2 MXene nanosheets as anode materials in Zinc-ion batteries. ACS Appl. Nano Mater. 6, 20812–20822 (2023).
Wang, Q. et al. High entropy liquid electrolytes for lithium batteries. Nat. Commun. 14, 440 (2023).
Tuckerman, M. E. & Martyna, G. J. Understanding modern molecular dynamics: techniques and applications. J. Phys. Chem. B 104, 159–178 (2000).
Harrison, J. A. et al. Review of force fields and intermolecular potentials used in atomistic computational materials research. Appl. Phys. Rev. 5, 031104 (2018).
Unke, O. T. et al. Machine learning force fields. Chem. Rev. 121, 10142–10186 (2021).
Wang, Q., Wang, J., Heringa, J. R., Bai, X. & Wagemaker, M. High-entropy electrolytes for lithium-ion batteries. ACS Energy Lett. 9, 3796–3806 (2024).
Wang, X. et al. Size-induced high entropy effect for optimized electrolyte design of lithium-ion batteries. Adv. Mater. 37, e14068 (2025).
Kim, S. C. et al. High-entropy electrolytes for practical lithium metal batteries. Nat. Energy 8, 814–826 (2023).
Feng, R., Liaw, P. K., Gao, M. C. & Widom, M. First-principles prediction of high-entropy-alloy stability. npj Comput. Mater. 3, 50 (2017).
Anand, G., Wynn, A. P., Handley, C. M. & Freeman, C. L. Phase stability and distortion in high-entropy oxides. Acta Mater. 146, 119–125 (2018).
Xie, L., Brault, P., Thomann, A.-L. & Bauchire, J.-M. AlCoCrCuFeNi high entropy alloy cluster growth and annealing on silicon: a classical molecular dynamics simulation study. Appl. Surf. Sci. 285, 810–816 (2013).
Krouna, S. et al. Atomic-scale insights into the thermal stability of high-entropy nanoalloys. Adv. Mater. 37, 2414510 (2025).
He, X., Zhu, Y., Epstein, A. & Mo, Y. Statistical variances of diffusional properties from ab initio molecular dynamics simulations. npj Comput. Mater. 4, 18 (2018).
Sidebottom, D. L. Colloquium: understanding ion motion in disordered solids from impedance spectroscopy scaling. Rev. Mod. Phys. 81, 999–1014 (2009).
France-Lanord, A. & Grossman, J. C. Correlations from ion pairing and the Nernst-Einstein equation. Phys. Rev. Lett. 122, 136001 (2019).
Li, J., Fang, Q. & Liaw, P. K. Microstructures and properties of high-entropy materials: modeling, simulation, and experiments. Adv. Eng. Mater. 23, 2001044 (2021).
Yao, N., Chen, X., Fu, Z.-H. & Zhang, Q. Applying classical, ab initio, and machine-learning molecular dynamics simulations to the liquid electrolyte for rechargeable batteries. Chem. Rev. 122, 10970–11021 (2022).
Sun, Y. et al. Boosting the optimization of lithium metal batteries by molecular dynamics simulations: a perspective. Adv. Energy Mater. 10, 2002373 (2020).
Li, W. et al. High-entropy argyrodite-type sulfide electrolyte with high conductivity and electro-chemo-mechanical stability for fast-charging all-solid-state batteries. Adv. Funct. Mater. 34, 2312832 (2024).
Da Silva, D. A. C. et al. Effect of conductivity, viscosity, and density of water-in-salt electrolytes on the electrochemical behavior of supercapacitors: molecular dynamics simulations and in situ characterization studies. Mater. Adv. 3, 611–623 (2022).
Liu, X. et al. Molten salt synthesis, morphology modulation, and lithiation mechanism of high entropy oxide for robust lithium storage. J. Energy Chem. 86, 536–545 (2023).
Wang, J., Malgras, V., Sugahara, Y. & Yamauchi, Y. Electrochemical energy storage performance of 2D nanoarchitectured hybrid materials. Nat. Commun. 12, 3563 (2021).
Naguib, M. et al. Two-dimensional nanocrystals produced by exfoliation of Ti3AlC2. in MXenes 15–29 (Jenny Stanford Publishing, 2023).
Bonaccorso, F. et al. Graphene, related two-dimensional crystals, and hybrid systems for energy conversion and storage. Science 347, 1246501 (2015).
Xu, X. et al. Progress and perspective: MXene and MXene-based nanomaterials for high-performance energy storage devices. Adv. Electron. Mater. 7, 2000967 (2021).
Nemani, S. K. et al. High-entropy 2D carbide mxenes: TiVNbMoC3 and TiVCrMoC3. ACS Nano 15, 12815–12825 (2021).
Nowotny, H. Structural chemistry of refractory metallic compounds. Angew. Chem. Int. Ed. Engl. 9, 173–174 (1970).
Barsoum, M. W. & El-Raghy, T. Synthesis and characterization of a remarkable ceramic: Ti3SiC2. J. Am. Ceram. Soc. 79, 1953–1956 (1996).
Schweidler, S. et al. High-entropy materials for energy and electronic applications. Nat. Rev. Mater. 9, 266–281 (2024).
Csonka, G. I. et al. Assessing the performance of recent density functionals for bulk solids. Phys. Rev. B—Condens. Matter Mater. Phys. 79, 155107 (2009).
Bucko, T., Hafner, J., Lebègue, S. & Angyan, J. G. Improved description of the structure of molecular and layered crystals: ab initio DFT calculations with van der Waals corrections. J. Phys. Chem. A 114, 11814–11824 (2010).
Kvashnin, Y. O. et al. Exchange parameters of strongly correlated materials: extraction from spin-polarized density functional theory plus dynamical mean-field theory. Phys. Rev. B 91, 125133 (2015).
Terentjev, A. V., Constantin, L. A., Artacho, E. & Pitarke, J. M. Comparison of dispersion-corrected exchange-correlation functionals using atomic orbitals. Phys. Rev. B 100, 235439 (2019).
van Setten, M. J., Weigend, F. & Evers, F. The GW-method for quantum chemistry applications: theory and implementation. J. Chem. Theory Comput. 9, 232–246 (2013).
Yadav, A., Acosta, C. M., Dalpian, G. M. & Malyi, O. I. First-principles investigations of 2D materials: challenges and best practices. Matter 6, 2711–2734 (2023).
Senftle, T. P. et al. The ReaxFF reactive force-field: development, applications and future directions. npj Comput. Mater. 2, 1–14 (2016).
Daw, M. S., Foiles, S. M. & Baskes, M. I. The embedded-atom method: a review of theory and applications. Mater. Sci. Rep. 9, 251–310 (1993).
Rao, Z. et al. Machine learning–enabled high-entropy alloy discovery. Science 378, 78–85 (2022).
Zhang, Z. et al. A multimodal robotic platform for multi-element electrocatalyst discovery. Nature 647, 390–396 (2025).
Mashhadimoslem, H. et al. Computational and machine learning methods for CO2 capture using metal–organic frameworks. ACS Nano 18, 23842–23875 (2024).
Mashhadimoslem, H. et al. Development of predictive models for activated carbon synthesis from different biomass for CO2 adsorption using artificial neural networks. Ind. Eng. Chem. Res. 60, 13950–13966 (2021).
Merchant, A. et al. Scaling deep learning for materials discovery. Nature 624, 80–85 (2023).
Zhang, Y. et al. Unsupervised discovery of solid-state lithium ion conductors. Nat. Commun. 10, 5260 (2019).
Sturman, J., Yim, C.-H., Baranova, E. A. & Abu-Lebdeh, Y. Communication—design of LiNi0. 2Mn0. 2Co0. 2Fe0. 2Ti0. 2O2 as a high-entropy cathode for lithium-ion batteries guided by machine learning. J. Electrochem. Soc. 168, 50541 (2021).
Çolak, A. B. An innovative study on high entropy energy storage mg-Y-Ni-cu systems: machine learning-driven optimization of electrical cycling in Ni-MH battery alloys. J. Energy Storage 107, 114958 (2025).
Zhang, J. et al. Rational design of high-entropy ceramics based on machine learning–a critical review. Curr. Opin. Solid State Mater. Sci. 27, 101057 (2023).
Huang, E.-W. et al. Machine-learning and high-throughput studies for high-entropy materials. Mater. Sci. Eng. R. Rep. 147, 100645 (2022).
Baldassarri, B. et al. Oxygen vacancy formation energy in metal oxides: high-throughput computational studies and machine-learning predictions. Chem. Mater. 35, 10619–10634 (2023).
Mao, H., Chen, H.-L. & Chen, Q. TCHEA1: A thermodynamic database not limited for “high entropy” alloys. J. Phase Equilib. Diffus. 38, 353–368 (2017).
Machaka, R. Machine learning-based prediction of phases in high-entropy alloys. Comput. Mater. Sci. 188, 110244 (2021).
Lv, C. et al. Machine learning: an advanced platform for materials development and state prediction in lithium-ion batteries. Adv. Mater. 34, 2101474 (2022).
Zeni, C. et al. A generative model for inorganic materials design. Nature 639, 624–632 (2025).
Pakornchote, T. et al. Diffusion probabilistic models enhance variational autoencoder for crystal structure generative modeling. Sci. Rep. 14, 1275 (2024).
Batatia, I. et al. A foundation model for atomistic materials chemistry. J. Chem. Phys. 163, 184110 (2025).
Li, S. et al. Encoding the atomic structure for machine learning in materials science. Wiley Interdiscip. Rev. Comput. Mol. Sci. 12, e1558 (2022).
Feldman, A., Holland, D. B. & Jacobus, D. P. The automatic encoding of chemical structures. J. Chem. Doc. 3, 187–189 (1963).
Butler, K. T., Davies, D. W., Cartwright, H., Isayev, O. & Walsh, A. Machine learning for molecular and materials science. Nature 559, 547–555 (2018).
Chen, A., Zhang, X. & Zhou, Z. Machine learning: accelerating materials development for energy storage and conversion. InfoMat 2, 553–576 (2020).
Faber, F., Lindmaa, A., Von Lilienfeld, O. A. & Armiento, R. Crystal structure representations for machine learning models of formation energies. Int. J. Quantum Chem. 115, 1094–1101 (2015).
Zunger, A. Inverse design in search of materials with target functionalities. Nat. Rev. Chem. 2, 121 (2018).
Ziletti, A., Kumar, D., Scheffler, M. & Ghiringhelli, L. M. Insightful classification of crystal structures using deep learning. Nat. Commun. 9, 2775 (2018).
Ren, Z. et al. An invertible crystallographic representation for general inverse design of inorganic crystals with targeted properties. Matter 5, 314–335 (2022).
Chen, C., Ye, W., Zuo, Y., Zheng, C. & Ong, S. P. Graph networks as a universal machine learning framework for molecules and crystals. Chem. Mater. 31, 3564–3572 (2019).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Shirley, J. H. Solution of the Schrödinger equation with a Hamiltonian periodic in time. Phys. Rev. 138, B979 (1965).
Bowler, D. R. & Miyazaki, T. Methods in electronic structure calculations. Rep. Prog. Phys. 75, 36503 (2012).
Rupp, M., Tkatchenko, A., Müller, K.-R. & Von Lilienfeld, O. A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 108, 58301 (2012).
Weininger, D., Weininger, A. & Weininger, J. L. SMILES. 2. Algorithm for generation of unique SMILES notation. J. Chem. Inf. Comput. Sci. 29, 97–101 (1989).
Hansen, K. et al. Machine learning predictions of molecular properties: accurate many-body potentials and nonlocality in chemical space. J. Phys. Chem. Lett. 6, 2326–2331 (2015).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Alberts, M., Laino, T. & Vaucher, A. C. Leveraging infrared spectroscopy for automated structure elucidation. Commun. Chem. 7, 268 (2024).
Lemmon, E. W., Huber, M. L. & McLinden, M. O. NIST standard reference database 23. Ref. fluid Thermodyn. Transp. Prop. (REFPROP), version 9, (2010).
Sun, H., Mumby, S. J., Maple, J. R. & Hagler, A. T. An ab initio CFF93 all-atom force field for polycarbonates. J. Am. Chem. Soc. 116, 2978–2987 (1994).
Wang, X. et al. Electric dipole descriptor for machine learning prediction of catalyst surface–molecular adsorbate interactions. J. Am. Chem. Soc. 142, 7737–7743 (2020).
Zhu, R., Nong, W., Yamazaki, S. & Hippalgaonkar, K. WyCryst: Wyckoff inorganic crystal generator framework. Matter 7, 3469–3488 (2024).
Jiang, Y. et al. Topological representations of crystalline compounds for the machine-learning prediction of materials properties. npj Comput. Mater. 7, 28 (2021).
Saal, J. E., Kirklin, S., Aykol, M., Meredig, B. & Wolverton, C. Materials design and discovery with high-throughput density functional theory: the open quantum materials database (OQMD). Jom 65, 1501–1509 (2013).
Belsky, A., Hellenbrandt, M., Karen, V. L. & Luksch, P. New developments in the Inorganic Crystal Structure Database (ICSD): accessibility in support of materials research and design. Struct. Sci. 58, 364–369 (2002).
Kirklin, S. et al. The Open Quantum Materials Database (OQMD): assessing the accuracy of DFT formation energies. npj Comput. Mater. 1, 1–15 (2015).
Horton, M. K. et al. Accelerated data-driven materials science with the Materials Project. Nat. Mater. 1, 11 (2025).
Cheng, G., Gong, X.-G. & Yin, W.-J. Crystal structure prediction by combining graph network and optimization algorithm. Nat. Commun. 13, 1492 (2022).
Jiao, R. et al. Crystal structure prediction by joint equivariant diffusion. Adv. Neural Inf. Process. Syst. 36, 17464–17497 (2023).
Jiao, R., Huang, W., Liu, Y., Zhao, D. & Liu, Y. Space group constrained crystal generation. arXiv Prepr https://doi.org/10.48550/arXiv.2402.03992 (2024).
Goodall, R. E. A. & Lee, A. A. Predicting materials properties without crystal structure: deep representation learning from stoichiometry. Nat. Commun. 11, 6280 (2020).
Pickard, C. J. & Needs, R. J. Ab initio random structure searching. J. Phys. Condens. Matter 23, 53201 (2011).
Xiao, H. et al. An invertible, invariant crystal representation for inverse design of solid-state materials using generative deep learning. Nat. Commun. 14, 7027 (2023).
Vu, T.-S. et al. Towards understanding structure–property relations in materials with interpretable deep learning. npj Comput. Mater. 9, 215 (2023).
Behler, J. Constructing high-dimensional neural network potentials: a tutorial review. Int. J. Quantum Chem. 115, 1032–1050 (2015).
Choudhary, K. & DeCost, B. Atomistic line graph neural network for improved materials property predictions. npj Comput. Mater. 7, 185 (2021).
López-Zorrilla, J. et al. ænet-PyTorch: a GPU-supported implementation for machine learning atomic potentials training. J. Chem. Phys. 158, 164105 (2023).
Chen, C. & Ong, S. P. A universal graph deep learning interatomic potential for the periodic table. Nat. Comput. Sci. 2, 718–728 (2022).
Deng, B. et al. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nat. Mach. Intell. 5, 1031–1041 (2023).
Klawohn, S. et al. Gaussian approximation potentials: theory, software implementation and application examples. J. Chem. Phys. 159, 174108 (2023).
Podryabinkin, E., Garifullin, K., Shapeev, A. & Novikov, I. MLIP-3: active learning on atomic environments with moment tensor potentials. J. Chem. Phys. 159, 084112 (2023).
Thompson, A. P., Swiler, L. P., Trott, C. R., Foiles, S. M. & Tucker, G. J. Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials. J. Comput. Phys. 285, 316–330 (2015).
Bartók, A. P., Kondor, R. & Csányi, G. On representing chemical environments. Phys. Rev. B—Condens. Matter Mater. Phys. 87, 184115 (2013).
Zhang, L., Han, J., Wang, H. & Car, R. E. W. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics. Phys. Rev. Lett. 120, 143001 (2018).
Kohn, W. Nobel Lecture: electronic structure of matter—wave functions and density functionals. Rev. Mod. Phys. 71, 1253 (1999).
Babbush, R. et al. Focus beyond quadratic speedups for error-corrected quantum advantage. PRX quantum 2, 10103 (2021).
Berry, D. W., Childs, A. M., Cleve, R., Kothari, R. & Somma, R. D. Simulating Hamiltonian dynamics with a truncated Taylor series. Phys. Rev. Lett. 114, 90502 (2015).
Georgescu, I. M., Ashhab, S. & Nori, F. Quantum simulation. Rev. Mod. Phys. 86, 153–185 (2014).
Lvovsky, A. I., Sanders, B. C. & Tittel, W. Optical quantum memory. Nat. Photonics 3, 706–714 (2009).
Gao, X., Zhang, Z.-Y. & Duan, L.-M. A quantum machine learning algorithm based on generative models. Sci. Adv. 4, eaat9004 (2018).
Otten, M. et al. QREChem: quantum resource estimation software for chemistry applications. Front. Quantum Sci. Technol. 2, 1232624 (2023).
Peruzzo, A. et al. A variational eigenvalue solver on a photonic quantum processor. Nat. Commun. 5, 4213 (2014).
Schoen, J. M. Augmented-plane-wave virtual-crystal approximation. Phys. Rev. 184, 858 (1969).
Velický, B. Theory of electronic transport in disordered binary alloys: coherent-potential approximation. Phys. Rev. 184, 614 (1969).
Kim, J. Applicability of special quasi-random structure models in thermodynamic calculations using semi-empirical Debye–Grüneisen theory. J. Alloy. Compd. 650, 564–571 (2015).
Sorkin, V., Tan, T. L., Yu, Z. G. & Zhang, Y. W. Generalized small set of ordered structures method for the solid-solution phase of high-entropy alloys. Phys. Rev. B 102, 174209 (2020).
Tian, F., Lin, D.-Y., Gao, X., Zhao, Y.-F. & Song, H.-F. A structural modeling approach to solid solutions based on the similar atomic environment. J. Chem. Phys. 153, 034101 (2020).
Latimer, K., Dwaraknath, S., Mathew, K., Winston, D. & Persson, K. A. Evaluation of thermodynamic equations of state across chemistry and structure in the materials project. npj Comput. Mater. 4, 40 (2018).
Anelli, A., Engel, E. A., Pickard, C. J. & Ceriotti, M. Generalized convex hull construction for materials discovery. Phys. Rev. Mater. 2, 103804 (2018).
Sivak, J. T. et al. Discovering high-entropy oxides with a machine-learning interatomic potential. Phys. Rev. Lett. 134, 216101 (2025).
Braams, B. J. & Bowman, J. M. Permutationally invariant potential energy surfaces in high dimensionality. Int. Rev. Phys. Chem. 28, 577–606 (2009).
Truhlar, D. G., Garrett, B. C. & Klippenstein, S. J. Current status of transition-state theory. J. Phys. Chem. 100, 12771–12800 (1996).
Messina, L., Castin, N., Domain, C. & Olsson, P. Introducing ab initio based neural networks for transition-rate prediction in kinetic Monte Carlo simulations. Phys. Rev. B 95, 64112 (2017).
Wu, Q. & Van Voorhis, T. Direct calculation of electron transfer parameters through constrained density functional theory. J. Phys. Chem. A 110, 9212–9218 (2006).
Rybkin, V. V. Franck–Condon theory of quantum mechanochemistry. J. Phys. Chem. A 121, 5758–5762 (2017).
Stott, M. J. & Zaremba, E. Linear-response theory within the density-functional formalism: application to atomic polarizabilities. Phys. Rev. A 21, 12 (1980).
Cervinka, C., Fulem, M., Stoffel, R. P. & Dronskowski, R. Thermodynamic properties of molecular crystals calculated within the quasi-harmonic approximation. J. Phys. Chem. A 120, 2022–2034 (2016).
Cuffari, D. & Bongiorno, A. Calculation of mode grüneisen parameters made simple. Phys. Rev. Lett. 124, 215501 (2020).
McGaughey, A. J. H., Jain, A., Kim, H.-Y. & Fu, B. Phonon properties and thermal conductivity from first principles, lattice dynamics, and the Boltzmann transport equation. J. Appl. Phys. 125, 011101 (2019).
Wagner, M. Expansions of nonequilibrium Green’s functions. Phys. Rev. B 44, 6104 (1991).
Pellegrini, C. & Sanna, A. Ab initio methods for superconductivity. Nat. Rev. Phys. 6, 509–523 (2024).
Grimaldi, C., Pietronero, L. & Strässler, S. Nonadiabatic superconductivity. II. Generalized Eliashberg equations beyond Migdal’s theorem. Phys. Rev. B 52, 10530 (1995).
Gibson, D. A., Ionova, I. V. & Carter, E. A. A comparison of Car—Parrinello and Born—Oppenheimer generalized valence bond molecular dynamics. Chem. Phys. Lett. 240, 261–267 (1995).
de Oliveira, C. A. F., Hamelberg, D. & McCammon, J. A. Coupling accelerated molecular dynamics methods with thermodynamic integration simulations. J. Chem. Theory Comput. 4, 1516–1525 (2008).
Quhe, R., Nava, M., Tiwary, P. & Parrinello, M. Path integral metadynamics. J. Chem. Theory Comput. 11, 1383–1388 (2015).
Kang, J. & Wang, L.-W. First-principles Green-Kubo method for thermal conductivity calculations. Phys. Rev. B 96, 20302 (2017).
Baroni, S., De Gironcoli, S., Dal Corso, A. & Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515 (2001).
Sheng, T., Nie, H., Xie, Y., Wang, L. & Li, J. Strain and kinetics synergy in octahedral high-entropy cathodes enables ultra-durable sodium-ion batteries. Small 21, e07895 (2025).
Wen, Z. et al. Boosting Li− CO2 battery performance via high-entropy alloy catalysts: insights into configurational entropy effect. Angew. Chem. 137, e202424121 (2025).
Han, F. et al. High rate and long-cycle life of lithium–sulfur battery enabled by high D-band center of high-entropy alloys. ACS Nano 19, 9182–9195 (2025).
Cho, J.-H. & Scheffler, M. Ab initio pseudopotential study of Fe, Co, and Ni employing the spin-polarized LAPW approach. Phys. Rev. B 53, 10685 (1996).
Rezaei, N., Alaei, M. & Oganov, A. R. Evaluating SCAN and r 2 SCAN meta-GGA functionals for predicting transition temperatures in antiferromagnetic materials. Phys. Rev. B 111, 144406 (2025).
Chevrier, V. L., Ong, S. P., Armiento, R., Chan, M. K. Y. & Ceder, G. Hybrid density functional calculations of redox potentials and formation energies of transition metal compounds. Phys. Rev. B—Condens. Matter Mater. Phys. 82, 75122 (2010).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+ U study. Phys. Rev. B 57, 1505 (1998).
Liechtenstein, A. I., Anisimov, V. I. & Zaanen, J. Density-functional theory and strong interactions: orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 52, R5467 (1995).
Audouze, C., Jollet, F., Torrent, M. & Gonze, X. Comparison between projector augmented-wave and ultrasoft pseudopotential formalisms at the density-functional perturbation theory level. Phys. Rev. B—Condens. Matter Mater. Phys. 78, 35105 (2008).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Tkatchenko, A. & Scheffler, M. Accurate molecular van der Waals interactions from ground-state electron density<? format?> and free-atom reference data. Phys. Rev. Lett. 102, 73005 (2009).
Angioletti-Uberti, S., Ceriotti, M., Lee, P. D. & Finnis, M. W. Solid-liquid interface free energy through metadynamics simulations. Phys. Rev. B—Condens. Matter Mater. Phys. 81, 125416 (2010).
Jinnouchi, R. & Minami, S. Machine learning force fields in electrochemistry: from fundamentals to applications. ACS Nano 19, 22600–22644 (2025).
Sun, H., Ren, P. & Fried, J. R. The COMPASS force field: parameterization and validation for phosphazenes. Comput. Theor. Polym. Sci. 8, 229–246 (1998).
Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996).
Krief, M. & Ashkenazy, Y. Calculation of elastic constants of embedded-atom-model potentials in the NVT ensemble. Phys. Rev. E 103, 63307 (2021).
Thompson, A. P. et al. LAMMPS-a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 271, 108171 (2022).
Van Der Spoel, D. et al. GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 (2005).
Mattsson, A. E., Armiento, R., Schultz, P. A. & Mattsson, T. R. Nonequivalence of the generalized gradient approximations PBE and PW91. Phys. Rev. B—Condens. Matter Mater. Phys. 73, 195123 (2006).
Deák, P., Aradi, B., Frauenheim, T., Janzén, E. & Gali, A. Accurate defect levels obtained from the HSE06 range-separated hybrid functional. Phys. Rev. B—Condens. Matter Mater. Phys. 81, 153203 (2010).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. b 59, 1758 (1999).
Hafner, J. Ab-initio simulations of materials using VASP: density-functional theory and beyond. J. Comput. Chem. 29, 2044–2078 (2008).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantumsimulations of materials. J. Phys. Condens. matter 21, 395502 (2009).
Wang, H. et al. High-entropy Na-deficient layered oxides for sodium-ion batteries. ACS Nano 17, 12530–12543 (2023).
Seong, H. W., Lee, M. S. & Ryu, H. J. First-principles study for discovery of novel synthesizable 2D high-entropy transition metal carbides (MXenes). J. Mater. Chem. A 11, 5681–5695 (2023).
Jin, Z. et al. Rugged high-entropy alloy nanowires with in situ formed surface spinel oxide as highly stable electrocatalyst in Zn–air batteries. ACS Mater. Lett. 2, 1698–1706 (2020).
Cai, T. et al. High-entropy layered oxide cathode enabling high-rate for solid-state sodium-ion batteries. Nano Micro Lett. 16, 10 (2024).
Walczak, K. et al. NaMn0. 2Fe0. 2Co0. 2Ni0. 2Ti0. 2O2 high-entropy layered oxide–experimental and theoretical evidence of high electrochemical performance in sodium batteries. Energy Storage Mater. 47, 500–514 (2022).
Zhou, P. et al. High-entropy P2/O3 biphasic cathode materials for wide-temperature rechargeable sodium-ion batteries. Energy Storage Mater. 57, 618–627 (2023).
Meng, Z. et al. A general strategy for preparing hollow spherical multilayer structures of Oxygen-Rich vacancy transition metal Oxides, especially high entropy perovskite oxides. Chem. Eng. J. 457, 141242 (2023).
Wang, X. L., Kim, E. M., Senthamaraikannan, T. G., Lim, D.-H. & Jeong, S. M. Porous hollow high entropy metal oxides (NiCoCuFeMg) 3O4 nanofiber anode for high-performance lithium-ion batteries. Chem. Eng. J. 484, 149509 (2024).
Sari, F. N. I. et al. Electronic structure modification induced electrochemical performance enhancement of bi-functional multi-metal hydroxide. Electrochim. Acta 439, 141616 (2023).
Wei, Y. et al. Lithium storage characteristic of nanoporous high-entropy alloy@ high-entropy oxide with spin-dependent synergism of cations. Chem. Eng. J. 476, 146881 (2023).
He, R. et al. A CrMnFeCoNi high entropy alloy boosting oxygen evolution/reduction reactions and zinc-air battery performance. Energy Storage Mater. 58, 287–298 (2023).
Li, K. et al. High-entropy MXene Ti3 (B0. 25C0. 25N0. 25O0. 25) 2O2 as anode materials for lithium-ion batteriess: insight from first principles. Mater. Today Commun. 38, 108255 (2024).
Li, K. et al. First-principles calculation on the lithium storage properties of high-entropy MXene Ti 3 C 2 (N 0.25 O 0.25 F 0.25 S 0.25) 2. Dalt. Trans. 52, 18323–18331 (2023).
Lei, N. et al. Anion-intercalated supercapacitor electrode based on perovskite-type SrB0. 875Nb0. 125O3 (B= Mn, Co). Chem. Eng. J. 421, 127790 (2021).
Huang, S. et al. High-entropy transition metal phosphorus trichalcogenides for rapid sodium ion diffusion. Adv. Mater. 36, 2405170 (2024).
Gu, Z. et al. An advanced high-entropy fluorophosphate cathode for sodium-ion batteries with increased working voltage and energy density. Adv. Mater. 34, 2110108 (2022).
Liu, J. et al. Entropy tuning stabilizing P2-type layered cathodes for sodium-ion batteries. Adv. Funct. Mater. 34, 2315437 (2024).
Xiao, B. et al. High-entropy oxides as advanced anode materials for long-life lithium-ion Batteries. Nano Energy 95, 106962 (2022).
Zhou, S. et al. Enhanced Li+ diffusion and lattice oxygen stability by the high entropy effect in disordered-rocksalt cathodes. Angew. Chem. 135, e202311930 (2023).
Liu, Z. et al. Achieving a deeply desodiated stabilized cathode material by the high entropy strategy for sodium-ion batteries. Angew. Chem. Int. Ed. 63, e202405620 (2024).
Ma, W. et al. A new Ti2V0. 9Cr0. 1C2Tx MXene with ultrahigh gravimetric capacitance. Nano Energy 96, 107129 (2022).
Lei, X. et al. Si-based high-entropy anode for lithium-ion batteries. Small Methods 8, 2300754 (2024).
Ge, X. et al. High-entropy doping boosts ion/electronic transport of Na4Fe3 (PO4) 2 (P2O7)/C cathode for superior performance sodium-ion batteries. Small 19, 2302609 (2023).
Zhou, Y. et al. Reversible oxygen redox chemistry in high-entropy P2-type manganese-based cathodes via self-regulating mechanism. ACS Appl. Mater. Interfaces 16, 33539–33547 (2024).
Li, S., Tong, L., Zhang, B. & Fu, X. First-principles study of high-entropy sulfides and their alkali metal-doped modification as cathode material for sodium-ion batteries. ChemPhysChem 25, e202300999 (2024).
Acknowledgements
This work was supported by Khalifa University and a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery grant. This work is supported by NSERC Discovery grant.
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mashhadimoslem, H., Karimi, P., Elkamel, A. et al. Toward high entropy material discovery for energy applications using computational and machine learning methods. npj Comput Mater 12, 50 (2026). https://doi.org/10.1038/s41524-025-01918-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41524-025-01918-6
