
Abstract
Presynaptic accumulation of misfolded α-synuclein (α-syn) and altered synaptic transmission are considered early events in the pathogenesis of Parkinson’s disease (PD), suggesting a potential causal link between these two events. However, the mechanisms by which α-syn aggregation induces synaptic dysfunction and the subsequent progressive neurodegeneration remain elusive. In the present study we leveraged the high temporal resolution of the Light-Inducible Protein Aggregation (LIPA) system in vivo and in human dopaminergic neurons to explore the early sequence of α-syn-induced pathological events leading to synaptopathy. We observed that nigrostriatal axonal transport and presynaptic accumulation of α-syn aggregates altered the activity of different neuronal populations in the mouse striatum. The results of histological and metabolite analyses show that presynaptic accumulation of α-syn induced a shift in the activation pattern of D1- and D2-expressing striatal medium spiny neurons, caused an increase in the size and density of dopaminergic synapses, and disrupted striatal dopamine signaling. Altogether, our findings reveal that the accumulation of α-syn in dopaminergic terminals triggered early presynaptic impairments, which subsequently altered striatal neuronal activity. Our study provides new insights into the molecular mechanisms underlying early synaptopathy in PD.
Similar content being viewed by others


Introduction
α-Synuclein (α-syn) is a ubiquitous protein, particularly enriched at neuronal presynaptic terminals1,2, where it exists in equilibrium between a soluble monomeric form and a membrane-bound, high-order multimeric state3,4. Under physiological conditions, α-syn plays key roles in the synaptic vesicle cycle, including promoting SNARE complex formation, regulating synaptic vesicle trafficking, participating in neurotransmitter release and endocytosis processes, and controlling the clustering of synaptic vesicles within distinct vesicle pools2,5,6,7,8,9,10. Pathologically aggregated α-syn is the primary component of intracellular inclusions known as Lewy bodies (LBs) and Lewy neurites, hallmark features of Parkinson’s disease (PD) and other synucleinopathies, including PD with dementia (PDD), dementia with LBs (DLB), and multiple system atrophy (MSA)11,12,13,14.
Growing evidence suggests that synaptic dysfunction is an early event in the pathogenesis of PD, occurring prior to nigrostriatal axonal and neuronal degeneration15,16, supporting the “dying-back” hypothesis15,17. Additionally, postmortem studies of brains from patients with DLB and other synucleinopathies have revealed that α-syn aggregates appear earlier in presynaptic terminals than in cell bodies18,19, accompanied by a reduction in various presynaptic markers19. Moreover, several imaging studies in PD patients have also confirmed synaptic dysfunction during the prodromal phase, occurring several years before the onset of motor symptoms20,21. Taken together, these observations suggest that aberrant α-syn accumulation at the synaptic terminals might represent an early initiating event in the cascade leading to synaptic dysfunction. Over time, the progressive loss of synaptic integrity directly impacts neuronal activity, disrupting synaptic communication and ultimately resulting in circuit-level defects22. This scenario is strongly supported in the nigrostriatal pathway, which is particularly vulnerable to α-syn aggregation23,24,25. Dopaminergic cells in the substantia nigra pars compacta (SNc) project their long axons to the dorsal striatum where they release dopamine (DA) to modulate the activity of striatal medium spiny neurons (MSNs)25,26,27. The balanced activity between the two main subtypes of MSNs, D1 receptor-expressing MSNs (D1-MSNs), which regulate the basal ganglia direct pathway (dMSNs), and D2 receptor-expressing MSNs (D2-MSNs), which regulate the indirect pathway (iMSNs), is essential for fine control of movement28,29,30,31. However, the abnormal accumulation of α-syn in dopaminergic terminals has been shown to be detrimental to nigrostriatal transmission15,25,32,33. Studies involving the overexpression of α-syn or using α-syn preformed fibrils have shown that there are early changes in the excitability of dopaminergic neurons, leading to alterations in striatal synaptic transmission34,35,36. In addition, other studies have reported that α-syn oligomers prompt the early impairment of long-term potentiation in MSNs, reflecting a decrease in synaptic strength and an alteration of the synaptic plasticity22,36,37.
Although previous research has made significant strides toward understanding the relevance of synaptic impairment in the pathogenesis of PD, the precise sequence of events and underlying mechanisms by which α-syn aggregates induce early synaptic dysfunction in the nigrostriatal pathway remain unresolved. This gap in knowledge is mainly due to the limitations of the experimental models of α-syn aggregation, which often lack temporal resolution and only allow studies of the late stages of aggregation38,39. To overcome this limitation, we developed an optogenetic-based cellular and animal model that allows for the induction and the formation of α-syn aggregates under the control of the blue light, referred to as the light-inducible protein aggregation (LIPA) system40,41. The LIPA system not only enables the formation of pathological LB-like inclusions in neuronal cultures and in the mouse brain, but also allows for the real-time monitoring α-syn aggregation and inclusions formation with unprecedented spatial and temporal resolution40. Interestingly, by combining the LIPA-induced α-syn aggregation system and in vivo Ca2+ imaging in the striatum of mice, we recently reported that the induction of LIPA-α-syn inclusions in the midbrain leads to rapid and sustainable changes in striatal neurons activity, thus highlighting the advantage of the LIPA system for studying the early stages of nigrostriatal transmission alteration40.
In the present work, we leveraged the advantages of the LIPA system to unravel the early sequence of pathological events leading to synaptic impairment and the dysfunction of the nigrostriatal circuit. Using in vivo Ca²⁺ imaging in the striatum of freely behaving mice following light-induced α-syn accumulation, we found that the formation of LIPA-α-syn inclusions in the midbrain differentially affected the activity patterns of striatal MSNs populations, prior to the onset of motor symptoms. Further analyses using immunolabeling revealed that the activities of D1-MSNs and D2-MSNs were unbalanced. To explore the underlying mechanisms, we next investigated synaptic dysfunction and structural remodeling as potential contributors to the altered neuronal activity. Our results showed that the accumulation of LIPA-α-syn in dopaminergic terminals disrupted presynaptic structures, altered synaptic architecture, and impaired dopamine homeostasis. Notably, these pathological changes occurred before the degeneration of dopaminergic axons. Collectively, our results provide new insights into the initial pathophysiological changes within the nigrostriatal circuit triggered by α-syn accumulation, and highlight synaptic impairment as an early event in the cascade that drives the onset and progression of the disease.
Results
The induction of α-syn inclusions in the midbrain results in a rapid spatial reorganization of the striatal neuronal circuitry
To determine how the induction of α-syn accumulation in the SNc dopaminergic neurons affects the functional organization of the nigrostriatal pathway, we analyzed Ca2+ activity in the striatal neurons of freely moving mice following midbrain induction of LIPA-α-syn aggregation40 (Fig. 1A). To do so, we implanted a double optogenetic/mini-endoscopic cannula into C57Bl6 male mice previously injected with AAV2/6-LIPA-α-syn-mCherry into SNc. The optic fiber was positioned just above SNc while a gradient index imaging lens (GRIN) was positioned in the striatum to perform Ca2+ imaging40 (Fig. 1A). We first performed Ca2+ imaging analyses under baseline conditions every other day for 4 days followed by imaging with blue light stimulation every other day for 7 days (Fig. 1B). After the 7-day blue light stimulation period, we assessed Ca2+ activity every other day for 10 more days (Fig. 1B). This experimental design previously revealed that both the synchronization and frequency of striatal neurons are specifically and rapidly impaired following the induction of LIPA-α-syn accumulation in the midbarin40. Based on these observations, we next focused on the baseline day 3 and post-stimulation day 2 and 10 time points to investigate the cellular and molecular consequences of LIPA-α-syn accumulation underlying the striatal responses (Fig. 1B–D).

A Schematic of the double opto-endoscopic system used to assess Ca2+ activity in the striatum of mice and to induce LIPA-α-syn accumulation in the SNc. B Schematic of the experimental protocol used to assess changes in the activity of striatal cells after inducing the formation of LIPA-α-syn inclusions in the SNc of mice. C Maximum projection images of MSNs expressing the Ca2+ indicator GCaMP6s from a 10-min recording session and examples of Ca2+ traces from the three clusters at baseline (scale bar = 20 μm). D Maximum projection images of MSNs expressing the Ca2+ indicator GCaMP6s from a 10-min recording session and examples of Ca2+ traces from the three clusters on post-stimulation (scale bar = 20 μm). E Frequency and amplitude of Ca2+ transients fitted to a Gaussian distribution before and after blue light-induced LIPA-α-syn accumulation. Significant changes in the frequency distribution for post-stimulation days 2 and 10 (two-sided Kolmogorov–Smirnov test with p < 0.0001 for both post-stimulation days) and the distribution of Ca2+ amplitude for post-stimulation day 2 (two-sided Kolmogorov–Smirnov test with p < 0.01) were observed as compared to baseline. F Pie chart representation of the cluster distributions at baseline and on post-stimulation days 2 and 10. Statistical significance was assessed by the Agresti-Caffo independence test. For post-stimulation day 2: Cluster 1: confidence Interval = (0.08, 0.28); p = 0.0004; Cluster 2: confidence Interval = (−0.23,0.02); p = 0.02; Cluster 3: confidence Interval = (−0.17, 0.01); p = 0.09. For post-stimulation day 10: Cluster 1: confidence Interval = (−0.2, 0.0004); p = 0.05; Cluster 2: confidence Interval = (−0.05,0.17); Cluster 3: confidence Interval = (−0.02, 0.13). Note, that while the same trend is observed for both post-stimulation days, the effect is significant for post-stimulation day 2, likely because of higher number of imaged cells (141 cells for post-stimulation day 2 and 110 cells for post-stimulation day 10). G Kernel Density Estimation (KDE) map showing the frequency and amplitude densities of cluster 1 at baseline and clusters 1 and 2 on post-stimulation day 2 (upper panel) and post-stimulation day 10 (lower panel). Note that cells from cluster 1 tended to move toward cluster 2 (the red arrow shows the direction from the center of the KDE map of cluster 1 on post-stimulation day 2 to the center of the KDE map of cluster 2 on post-stimulation day 2). H Examples of traces from a representative analysis of mouse movement showing the speed of displacement and heat maps of ΔF/F Ca2+ signals before and after inducing the accumulation of LIPA-α-syn in the SNc of the mice. I Total mobile and immobile times of mice before and after the formation of LIPA-α-syn inclusions (n = 5). No significant differences were observed in the immobility and mobility times of mice after inducing LIPA-α-syn accumulation. The results are expressed as means ± SEM. Statistical significance was assessed using a one-way ANOVA test followed by Tukey’s multiple comparisons test. Time immobile: F(2, 11) = 0.3, p = 0.8; Time mobile: F(2, 11) = 0.3, p = 0.8. J Representation of the frequency and amplitude of Ca2+ transients of cluster 1 during immobility and mobility before and after inducing LIPA-α-syn accumulation (n = 5 mice). Graphs showing the fold-changes in frequency and amplitude normalized to the immobile periods. The results are expressed as means ± SEM. Statistical significance was assessed using an unpaired two-sided Student’s t test. Baseline: Frequency-Cluster 1: t(154) = 12.6, p < 0.0001; ΔF/F-Cluster 1: t(154) = 0.1, p = 0.9. Post-stimulation Day 2: Frequency-Cluster 1: t(60) = 6.6, p < 0.0001; ΔF/F-Cluster 1: t(60) = 0.6, p = 0.5; Post-stimulation Day 10: Frequency-Cluster 1: t(59) = 5.3, p < 0.0001; ΔF/F-Cluster 1: t(59) = 0.4, p = 0.7. K Representation of the frequency and amplitude of Ca2+ transients of cluster 2 during immobility and mobility before and after inducing LIPA-α-syn accumulation (n = 5 mice). Graphs showing the fold-changes in frequency and amplitude normalized to the immobile periods. The results are expressed as means ± SEM. Statistical significance was assessed using an unpaired two-sided Student’s t-test. Baseline: Frequency-Cluster 2: t(200) = 15.1, p < 0.0001; ΔF/F-Cluster 2: t(200) = 3.5, p < 0.001. Post-stimulation Day 2: Frequency-Cluster 2: t(180) = 9.8, p < 0.0001; ΔF/F-Cluster 2: t(180) = 4.4, p < 0.0001. Post-stimulation Day 10: Frequency-Cluster 2: t(124) = 14.3, p < 0.0001; ΔF/F-Cluster 2: t(124) = 3.9, p < 0.001. L Representation of the frequency and amplitude of Ca2+ transients of cluster 3 during immobility and mobility before and after inducing LIPA-α-syn accumulation (n = 5 mice). Graphs showing the fold-changes in frequency and amplitude normalized to the immobile periods. The results are expressed as means ± SEM. Statistical significance was assessed using an unpaired two-sided Student’s t test. Baseline: Frequency-Cluster 3: t(28) = 7.0, p < 0.0001; ΔF/F-Cluster 3: t(28) = 0.3, p = 0.7. Post-stimulation Day 2: Frequency-Cluster 3: t(36) = 13.6, p < 0.0001; ΔF/F-Cluster 3: t(36) = 1.0, p = 0.3. Post-stimulation Day 10: Frequency-Cluster 3: t(25) = 3.6, p < 0.001; ΔF/F-Cluster 3: t(25) = 1.3, p = 0.2. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
To assess the functional impact of LIPA-α-syn inclusions on the activity of striatal cells, we monitored the frequency and amplitude of Ca2+ events and fitted the results to a Gaussian distribution. In line with our previous data40, our results show that the induction of LIPA-α-syn inclusions prompted a significant shift in the frequency of Ca2+ transients at both post-stimulation days as compared to baseline (Fig. 1E). Interestingly, our results also show that LIPA-α-syn accumulation affected the amplitude of Ca2+ events in a heterogenous manner such that there was a decrease in the lower and medium amplitudes of Ca2+ transients, while higher amplitude Ca2+ events increased even further (Fig. 1E). The same pattern of activity was observed on post-stimulation days 2 and 10 (Fig. 1E), suggesting that LIPA-α-syn-accumulation induced changes appeared as early as post-stimulation day 2 and were largely sustained at post-stimulation day 10. These results also suggest that LIPA-α-syn accumulation might affect different populations of striatal cells in dissimilar ways. We, thus, next assessed the possibility of functionally distinguishing between different populations of striatal neurons by performing a cluster analysis based on the properties of Ca2+ activity, such as amplitude and frequency, and to evaluate the effects of LIPA-α-syn accumulation on these distinct striatal neuronal populations. Under baseline conditions, we distinguished three different clusters. Cluster 1, which consisted of 40.2% of the total number of striatal neurons and exhibited a high amplitude of Ca2+ transients (ΔF/F = 2.49 and frequency = 0.073 Hz); cluster 2, which consisted of 52.1% of the total number of striatal neurons and exhibited a low amplitude of Ca2+ transients (ΔF/F = 1.27 and frequency = 0.069 Hz), and cluster 3, which consisted of the remaining 7.7% of striatal cells and exhibited the highest amplitude of Ca2+ transients (ΔF/F = 4.75 and frequency = 0.067 Hz). Interestingly, the distribution of the clusters differed between baseline and after blue light-induced LIPA-α-syn accumulation at post-stimulation days 2 and 10. After inducing the formation of LIPA-α-syn inclusions, there was a significant increase in the percentage of cells in cluster 2 (64.5%, ΔF/F = 1.15 and frequency = 0.064 Hz) and a significant decrease in cluster 1 (22%, ΔF/F = 2.83 and frequency = 0.062 Hz) at post-stimulation day 2 (Fig. 1F). Only mild, non-significant changes were observed in the percentage of striatal neurons in cluster 3 after the formation of LIPA-α-syn inclusions (13.5%, ΔF/F = 5.61 and frequency = 0.066 Hz) (Fig. 1F). Similarly, at post-stimulation day 10, the percentage of cells in cluster 1 decreased (29.1%, ΔF/F = 3.03 and frequency = 0.063 Hz) and increased, albeit not significantly, in cluster 2 (58.2%, ΔF/F = 1.35 and frequency = 0.063 Hz) (Fig. 1F). To better understand the changes observed in our cluster analysis, we mapped the distribution of Ca2+ dynamics using the Kernel Density Estimation (KDE). Our results show that, after inducing LIPA-α-syn accumulation at both post-stimulation days, cells in cluster 1 tended to move toward cluster 2 (Fig. 1G). These results suggest that the induction of LIPA-α-syn inclusions promoted a shift in the activity of striatal cells in cluster 1, making their activity resemble the Ca2+ dynamics of cells in cluster 2.
Given the role played by striatal neurons, particularly MSNs, in encoding mouse locomotion42,43, we next determined whether the reorganization of striatal neuronal activity was associated with changes in motor behavior. We recorded the behavior of mice placed in an open field arena while Ca2+ activity was being monitored40 and determined whether specific neuronal clusters were more tuned to locomotion and, if so, whether α-syn accumulation disrupted the recruitment of striatal neurons during movement. As reported previously43,44,45, we observed an increase in the activity of striatal cells when the mice were moving (Fig. 1H). No significant changes in the mobile and immobile periods after light-induced accumulation of α-syn at post-stimulation days 2 and 10 were observed (Fig. 1I). This is in line with the timeline of light-induced α-syn accumulation used in our study which was prior to the onset of the characteristic motor symptoms. We next determined whether the activity of striatal cells from a specific cluster was linked to the movement of the mice. As expected, the frequency of Ca2+ transients at baseline was significantly higher during mobile periods in all three clusters (Fig. 1J–L). Similarly, the frequency of Ca2+ transients was significantly higher during mobile periods in all three clusters after light-induced α-syn accumulation at post-stimulation days 2 and 10 (Fig. 1J–L). Only cells in cluster 2 exhibited a significant increase in the amplitude, in addition to changes in the frequency, of Ca²⁺ transients when the mice were moving before and after inducing LIPA-α-syn accumulation (Fig. 1K). However, the increase in the frequency of Ca²⁺ transients during mobile periods on post-stimulation days 2 and 10 was comparable to that at baseline. Cells in cluster 1 and 3 showed no significant differences in the amplitude of Ca2+ transients during immobile and mobile periods at baseline and on post-stimulation days 2 and 10, and the increase in the frequency of Ca2+ transients during mobile periods after inducing LIPA-α-syn accumulation was comparable to that at baseline (Fig. 1J–L). These observations suggest that Ca2+ transients of striatal neurons increased in all clusters during movement and light-induced LIPA-α-syn accumulation induced a functional reorganization of the neuronal network by triggering a shift of striatal cells from cluster 1 to cluster 2, even prior to the development of motor symptoms mimicking prodromal PD.
LIPA-α-syn inclusions induce an imbalance in the direct and indirect striatal pathways
The results of the Ca2+-based neuronal activity analysis show that the early functional disruption induced by LIPA-α-syn inclusions mostly affected two striatal neuronal populations that were defined based on their functional activity. These two populations might be the MSNs that expressed either the D1 DA receptor or the D2 DA receptor (D1-MSNs and D2-MSNs) that govern the basal direct and indirect ganglia pathways, respectively. These neuronal populations are functionally connected to nigrostriatal afferents29. Given that these two neuronal populations represent a large percentage of mouse striatal cells and are highly susceptible to DA modulation30, it is plausible that LIPA-α-syn-induced neuronal alterations primarily affect these two populations. To test this hypothesis, we injected LIPA-α-syn AAV into the SNc of wild-type (WT) mice and, 15 days later, induced LIPA-α-syn inclusions in the SNc by blue light stimulation (Fig. 2A). We assessed the activation of striatal cells by analyzing c-fos expression 10 days post-stimulation (Fig. 2A). We used LIPA-Empty, a LIPA construct lacking the sequence of α-syn, as a control40. The results of c-fos analysis show that there was a significant decrease in the number of activated striatal cells after inducing α-syn accumulation, while no changes were observed in the control groups, LIPA-α-syn non-stimulated and LIPA-Empty stimulated (Fig. 2B, C).

A Schematic of the in vivo experimental approach used to overexpress and induce LIPA-α-syn accumulation in the midbrain of WT mice. The mice were euthanized 10 days after inducing LIPA-α-syn accumulation. B Representative confocal images of c-fos staining in the striatum of mice overexpressing LIPA-α-syn and optogenetically stimulated with blue light, mice overexpressing LIPA-α-syn but not stimulated with blue light, and mice overexpressing LIPA-Empty and stimulated with blue light (scale bar = 10 μm). C Quantification of the number of c-fos+ cells in mice overexpressing LIPA-α-syn and LIPA-Empty constructs stimulated or not with blue light. D Representative confocal images showing activated D1-MSNs in the striatum of mice overexpressing LIPA-α-syn and stimulated with blue light, overexpressing LIPA-α-syn and not stimulated with blue light, and LIPA-Empty stimulated with blue light (scale bar = 10 μm, scale bar magnification = 10 μm). E Quantification of the number of D1 + /c-fos+ cells in the striatum of mice overexpressing the LIPA-α-syn and LIPA-Empty constructs and stimulated or not with blue light. F Representative confocal images showing activated D2-MSNs in the striatum of mice overexpressing LIPA-α-syn stimulated with blue light, overexpressing LIPA-α-syn and not stimulated with blue light, and LIPA-Empty stimulated with blue light (scale bar = 10 μm, scale bar magnification = 10 μm). G Quantification of the number of D2 + /c-fos+ cells in the striatum of mice overexpressing the LIPA-α-syn and LIPA-Empty constructs stimulated or not with blue light. The results are expressed as means ± SEM. LIPA-α-syn stimulated (n = 5-6), LIPA-α-syn not stimulated (n = 5), and LIPA-Empty stimulated (n = 5-6). C, E, G Statistical significance was assessed by one-way ANOVA followed by Tukey’s multiple comparisons test: C F(2, 12) = 8.278, p = 0.0055; E F(2, 13) = 24.41; G F (2, 14) = 9.511; p = 0.0025. *p < 0.05, **p < 0.01, and ****p < 0.0001.
We then immunolabeled D1- and D2-MSNs and assessed the activation of these cells in the striatum. The accumulation of LIPA-α-syn induced a significant decrease in the number of activated D1+ cells (Fig. 2D, E) and a significant increase in the number of activated D2+ cells (Fig. 2F, G), while no changes were observed in the LIPA-α-syn non-stimulated and LIPA-Empty stimulated groups for either D1- or D2-MSNs (Fig. 2D–G). These results are similar to those of our analysis of Ca2+ activity showing that there was a decrease in the number of cells in cluster 1 and an increase in the number of cells in cluster 2. Overall, these results show that the formation of LIPA-α-syn inclusions in the nigrostriatal pathway disrupted the activity of D1- and D2-MSNs, causing an imbalance in the striatal network that mimicked the early stages of PD36. Given that the activity of MSNs in the striatum is tightly controlled by dopaminergic projections from the SNc27,46, our results suggest that the altered activity might be caused by a pre-synaptic dysfunction induced by LIPA-α-syn accumulation.
Accumulation of LIPA-α-syn inclusions at dopaminergic terminals induces synaptic dysfunction in the striatum of mice
Previous studies have reported that synaptic pathology can arise early in the pathogenesis of PD15,16. We thus wondered whether the accumulation of LIPA-α-syn accumulation at DA terminals would induce presynaptic damage, leading to the altered activity observed in striatal cells. We first determined whether the early imbalance observed in the activity of direct- and indirect-striatal neurons was caused by dopaminergic axonal degeneration. To do so, we overexpressed LIPA-α-syn or LIPA-Empty in the SNc of WT mice (Fig. 3A). Fifteen days post-surgery, we induced LIPA-α-syn accumulation by optogenetic stimulation with blue light and assessed tyrosine hydroxylase (TH) and DA transporter (DAT) expression 10 days post-stimulation (Fig. 3A). Since in these experiments we performed an immunohistochemical assessment of the expression pattern of DAT and TH, which is unlikely to be changed at post-stimulation day 2, we analyzed samples only at post-stimulation day 10. We did not detect any differences in the densities of TH (Fig. 3B, C) or DAT (Fig. 3D, E) fibers between the LIPA-α-syn stimulated, LIPA-α-syn non-stimulated, and LIPA-Empty stimulated groups at post-stimulation day 10. These results show that the induction of LIPA-α-syn accumulation in the nigrostriatal pathway did not induce dopaminergic fiber degeneration, suggesting that the abnormal activity observed in MSNs was probably due to a synaptic dysfunction and not to dopaminergic degeneration. We next determined whether the presynaptic accumulation of LIPA-α-syn inclusions could lead to synaptic dysfunction and alterations in the activity of MSNs. To do so, we first confirmed the presence of α-syn in the presynaptic terminals by immunolabeling. At 10 days post-stimulation, we observed a considerable accumulation of α-syn in the axons of the blue light-stimulated LIPA-α-syn group and, to a lesser extent, in the non-stimulated LIPA-α-syn group (Fig. 3F). Interestingly, we did not detect the presence of LIPA-Empty inclusions in the axons of the LIPA-Empty group, suggesting that only LIPA-α-syn was transported to the nigral axonal terminals (Fig. 3F). We then assessed the effect of the presence of LIPA-α-syn inclusions at the synaptic terminals. Interestingly, we observed LIPA-α-syn accumulation colocalizing with the presynaptic marker synaptophysin in the LIPA-α-syn group stimulated with blue light (Fig. 3F arrowhead). However, we observed no such level of colocalization in the LIPA-α-syn non-stimulated group (Fig. 3F arrowhead). An assessment of the number and morphology of the synaptic varicosities using the presynaptic marker synaptophysin showed that the number of synaptophysin varicosities and the surfaces of the overall synapses had significantly increased in the blue light-stimulated LIPA-α-syn group compared to the control groups (Fig. 3G, H). In addition, the accumulation of LIPA-α-syn at the dopaminergic terminals resulted in a significant increase in the expression levels of synaptophysin in the striatum (Fig. 3I). These observations suggest that the accumulation of LIPA-α-syn inclusions at the presynaptic structure might lead to synaptic impairment. Altogether, our results show that synaptic pathology occurred because LIPA-α-syn accumulated in the dopaminergic terminals and highlighted the role of α-syn accumulation in inducing the first event of a cascade leading to neuronal dysfunction prior to axonal neurodegeneration.

A Schematic of the in vivo experimental design of blue light-induced α-syn accumulation in the midbrain of WT mice. The mice were euthanized 10 days after inducing the accumulation of LIPA-α-syn. B Representative epifluorescence microscopic images of striatal fibers stained for TH in mice overexpressing LIPA-α-syn and optogenetically stimulated with blue light (n = 6), mice overexpressing LIPA-α-syn and not stimulated with blue light (n = 6), and mice overexpressing LIPA-Empty and stimulated with blue light (n = 6) (scale bar = 500 μm). C Quantification of TH fiber density in WT mice overexpressing LIPA constructs and stimulated or not stimulated with blue light. The results are expressed as means ± SEM. A one-way ANOVA was used (F(2,15) = 0.6, p = 0.5). D Epifluorescence microscopic images illustrating striatal fibers stained for DAT in mice overexpressing LIPA-α-syn and stimulated with blue light (n = 5), mice overexpressing LIPA-α-syn and not stimulated with blue light (n = 6), and mice overexpressing LIPA-Empty and stimulated with blue light (n = 6) (scale bar = 500 μm). E Quantification of DAT fiber density in WT mice overexpressing LIPA constructs and stimulated or not with blue light. The results are expressed as means ± SEM. A one-way ANOVA was used (F(2,14) = 2.6, p = 0.16). F Representative confocal images of synaptic terminals immunolabeled with the presynaptic marker synaptophysin in the striatum of mice overexpressing LIPA-α-syn and stimulated with blue light, mice overexpressing LIPA-α-syn and not stimulated with blue light, and mice overexpressing LIPA-Empty and stimulated with blue light. Note the extensive accumulation of α-syn labelled using anti-α-syn in the synaptic terminals of mice stimulated with blue light and the colocalization with synaptophysin varicosities (yellow arrowheads) (scale bar = 5 μm). G Quantification of synaptophysin varicosities density, (H) surface of synaptophysin varicosities (I), and synaptophysin fluorescence levels in mice overexpressing LIPA-α-syn and stimulated with blue light (n = 6), mice overexpressing LIPA-α-syn but not stimulated with blue light (n = 6), and mice overexpressing LIPA-Empty and stimulated with blue light (n = 5). The results are expressed as means ± SEM. A one-way ANOVA was used followed by Tukey’s multiple comparisons test: G F(2,14) = 10.4, p = 0.002; H F(2,14) = 8.5, p = 0.004; I F(2,14) = 7.6, p = 0.006. * p < 0.05 and ** p < 0.01. MGV: Mean Grey Value.
Nigral induction of LIPA-α-syn inclusions disrupts dopamine homeostasis in the mouse striatum
Given the early impact of LIPA-α-syn inclusions on striatal neuronal activity, the known susceptibility of these striatal neurons to dopaminergic modulation46,47, and the early synaptic defects observed when LIPA-α-syn accumulation is induced, we determined whether the formation of LIPA-α-syn inclusions would lead to early dysregulation of the neurotransmitter DA in the nigrostriatal pathway21,48. To address this question, we injected an AAV encoding LIPA-α-syn or the control LIPA-Empty in the SNc of WT mice and implanted an optic fiber in the midbrain. Fifteen days later, we induced the formation of LIPA-α-syn inclusions using blue light stimulation for one week and assessed the levels of DA and its metabolites in the striatum of mice by high-performance liquid chromatography (HPLC) (Fig. 4A). Strikingly, we observed that the nigral induction of LIPA-α-syn accumulation resulted in a significant increase in striatal DA content, while no changes were observed in the control groups (LIPA-α-syn –light and LIPA-Empty +light) (Fig. 4B). Unlike the changes in DA content we observed, we did not detect any significant differences in the content of the DA metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) (Fig. 4C) and 3-methoxytyramine (3-MT) across the experimental groups (Fig. 4D). In addition, the striatal DOPAC/DA ratio significantly decreased in mice with LIPA-α-syn inclusions (Fig. 4E), suggesting that while striatal DA levels had increased, the turnover of DA was not affected. The striatal 3-MT/DA ratio was significantly decreased in the blue light-stimulated LIPA-α-syn group compared to mice overexpressing LIPA-Empty and stimulated with blue light, while no differences were observed with the non-stimulated LIPA-α-syn group (Fig. 4F). Altogether these results suggest that the accumulation of LIPA-α-syn induced a deregulation of striatal DA transmission without affecting the catabolism of the neurotransmitter.

A Schematic of the in vivo experimental approach for overexpressing and inducing LIPA-α-syn accumulation in the midbrain of WT mice using blue light stimulation. Mice were euthanized 1 week after starting the protocol to aggregate α-syn. Quantification of striatal DA levels (B) and its metabolites (C) DOPAC, and D 3-MT in the right hemisphere of mice overexpressing LIPA-α-syn and optogenetically stimulated with blue light, mice overexpressing LIPA-α-syn and not stimulated with blue light, and mice overexpressing LIPA-Empty and stimulated with blue light. Quantification of the ratios of (E) DOPAC/dopamine, and F 3-MT/dopamine as an estimate of turnover in mice overexpressing the LIPA-α-syn and LIPA-Empty constructs and stimulated or not with blue light. The results are expressed as means ± SEM. LIPA-α-syn stimulated with blue light (n = 8–9), LIPA-α-syn not stimulated with blue light (n = 7-8), and LIPA-Empty stimulated with blue light (n = 7–8). Statistical significance was assessed by one-way ANOVA followed by Tukey’s multiple comparisons test: B F(2,21) = 5.7, p = 0.01; C F(2,21) = 2.9, p = 0.07; D F(2,21) = 3.6, p = 0.0448; E F(2,20) = 20.2, p < 0.0001; F F(2,20) = 6.6, p = 0.006; p < 0.0001. *p < 0.05, **p < 0.01, and ****p < 0.0001.
LIPA-α-syn inclusions induce early synaptic dysfunction in human dopaminergic neurons
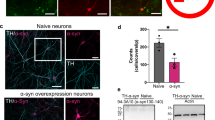
Human-derived induced pluripotent stem cells (hiPSCs) are an excellent model for studying the pathogenesis of PD in vitro49. Combined with our LIPA system, hiPSCs provide an exceptional opportunity to study the early alterations that occur in human cells after the induction of α-syn accumulation. We previously established a rapid, efficient differentiation protocol for turning hiPSCs into mature dopaminergic neurons (iDA) by combining neurogenin-2 programming and midbrain differentiation and maturation media kits50,51,52 (Fig. 5A). To determine whether α-syn inclusions induce an early synaptic dysfunction in human dopaminergic neurons as observed in our in vivo model, at day 28 we overexpressed the LIPA-α-syn construct and the two control constructs, LIPA-Empty and a non-aggregatable form of α-syn LIPA-α-synΔNAC53. in the iDA (Fig. 5A, Supplementary Fig. S1). Two days later, the cells were optogenetically stimulated with blue light to induce α-syn accumulation (Fig. 5A). As previously reported40, only iDA neurons expressing LIPA-α-syn and LIPA-Empty stimulated with blue light form inclusions (Fig. 5B, Supplementary Fig. S1). To evaluate the impact of LIPA-α-syn inclusions on synaptic integrity, we assessed the number and morphology of synaptic varicosities in the neurites of dopaminergic neurons by immunolabelling with the presynaptic marker synaptophysin (Fig. 5B). We observed a colocalization of LIPA-α-syn inclusions with a synaptophysin signal (Fig. 5B yellow arrowhead). In line with the results obtained with our in vivo model, the formation of LIPA-α-syn inclusions induced a significant increase in the number of synaptophysin varicosities as well as in their surface while no changes were observed in the control groups (Fig. 5C, D). Moreover, the accumulation of LIPA-α-syn significantly increased the expression levels of synaptophysin (Fig. 5E). These results suggest that the accumulation of LIPA-α-syn inclusions in the dopaminergic neurites led to early synaptic dysfunction.

A Schematic illustrating the timeline of the differentiation and maturation of hiPSCs to obtain human induced dopaminergic neurons (iDA). At 28 DIV, the iDA were transfected with the LIPA constructs (LIPA-Empty, LIPA-α-syn, and LIPA-α-synΔNAC). Two days later, the iDa were stimulated or not with blue light and the neurons were fixed after 6 h of illumination. B Representative confocal images of human dopaminergic cells transfected with the LIPA-α-syn, LIPA-Empty, and LIPA-α-synΔNAC constructs, stimulated or not with blue light, and immunolabeled with the presynaptic marker synaptophysin (green, white arrowheads). Note the presence of LIPA inclusions only in the neurites of dopaminergic neurons overexpressing LIPA-α-syn and LIPA-Empty (needle-like LIPA-Empty inclusions) stimulated with blue light in addition to the morphological changes observed in the synaptophysin varicosities colocalizing with the LIPA-α-syn inclusions (yellow arrowhead) compared to the synaptophysin varicosities colocalizing with the needle-like LIPA-Empty inclusions (yellow arrowhead) (scale bar = 10 μm; scale bar magnification = 1 μm). Quantification at 30 DIV of (C) the number of synaptophysin varicosities per μm, D the surface of the synaptophysin varicosities, and E the expression levels of synaptophysin in the neurites of dopaminergic neurons overexpressing the LIPA-α-syn, LIPA-Empty, and LIPA-α-synΔNAC constructs stimulated or not with blue light. F Quantification of the number of synaptophysin varicosities (in percentage) colocalizing with mCherry inclusions. G Quantification at 30 DIV of the synaptophysin varicosities surface that colocalizes or not with LIPA mCherry inclusions in the neurites of dopaminergic neurons overexpressing the LIPA-α-syn, LIPA-Empty, and LIPA-α-synΔNAC constructs stimulated or not with blue light. H Quantification at 30 DIV of the expression levels of synaptophysin in the synaptic varicosities that colocalize or not with LIPA mCherry inclusions in the neurites of dopaminergic neurons overexpressing the LIPA-α-syn, LIPA-Empty, and LIPA-α-synΔNAC constructs stimulated or not with blue light. The results are expressed as means ± SEM. LIPA-α-syn stimulated with blue light (n = 102–106), LIPA-α-syn not stimulated with blue light (n = 72–75), LIPA-Empty stimulated with blue light (n = 55–58), LIPA-Empty not stimulated with blue light (n = 77–81), LIPA-α-synΔNAC stimulated with blue light (n = 66–67), and LIPA-α-synΔNAC not stimulated with blue light (n = 71–74). Statistical significance was assessed by one-way ANOVA followed by Tukey’s multiple comparisons test: C F(5,445) = 10.97, p < 0.0001; D F(5,442) = 57.1, p < 0.0001; E F (5,4) = 47.4, p < 0.0001; G F(7,6) = 83.9, p < 0.0001; H F (7,6) = 51.9,p < 0.0001. F Statistical significance was assessed by unpaired two-sided Student’s t test: t(162) = 14.16, p = <0.0001. ****p < 0.0001. Y Yes, N No.
As shown in Fig. 5B, not all the synaptophysin varicosities in the neurites of dopaminergic neurons were positive for LIPA-α-syn or LIPA-Empty aggregates (Fig. 5B yellow and white arrowheads). Indeed, 50% of the total synaptophysin varicosities colocalized with LIPA-α-syn inclusions, while only 11% colocalized with LIPA-Empty inclusions (Fig. 5F). We thus determined whether the increase in the surface of synaptophysin-positive synaptic varicosities was due to the presence of LIPA-α-syn inclusions. Interestingly, the surface of synaptophysin varicosities colocalizing with LIPA-α-syn inclusions was significantly higher than that of non-colocalizing varicosities (Fig. 5G). In addition, the area of synaptophysin varicosities non-colocalizing with LIPA-α-syn inclusions was similar to that of the control groups (Fig. 5G). Moreover, synaptophysin expression levels significantly increased only when synaptophysin varicosities colocalized with LIPA-α-syn inclusions, while no differences of synaptophysin varicosities non-colocalizing with LIPA-α-syn inclusions with the other control groups were observed (Fig. 5H). Of note, the surface of synaptophysin varicosities colocalizing and non-colocalizing with LIPA-Empty inclusions was not significantly different from the other control groups (Fig. 5G). In addition, no differences were observed in the synaptophysin expression of varicosities colocalizing or non-colocalizing with LIPA-Empty inclusions (Fig. 5H). These observations suggest that the presence of LIPA-α-syn inclusions might rapidly trigger synaptic disruption. Overall, these results indicate that, in human dopaminergic neurons, a synaptic dysfunction caused by LIPA-α-syn inclusions occurs early in the progression of the disease and may lead to the progressive neurodegeneration observed in PD. Overall, our results indicated that a dysfunction in nigrostriatal transmission occurs early in the progression of the disease, highlighting the presynaptic structure as one of the first elements affected following α-syn accumulation.
Discussion
Several lines of evidence support the “dying-back” hypothesis in PD, where the pathology starts by affecting presynaptic structures, inducing axonal degeneration, and gradually progressing toward the cell bodies of neurons, eventually culminating in their death15,17. In the present study, we leveraged the temporal resolution of the LIPA system to gain new insights into the early pathological events that occur in the nigrostriatal pathway of mice and in human dopaminergic neurons and that are associated with α-syn aggregation. We provide evidence that LIPA-α-syn inclusions induced in the SNc rapidly accumulated in the dopaminergic terminals in the striatum leading to structural changes in the presynaptic boutons and altering DA levels. We also show that LIPA-α-syn aggregates differentially induced changes in the activity of different striatal populations, suggesting that there were early underlying defects in the nigrostriatal circuit. Additionally, a histological analysis revealed that there was an opposite effect on the activation of D1- and D2-MSNs, as assessed by c-fos labeling, after the induction of the accumulation of LIPA-α-syn. In parallel, our in vitro findings using human dopaminergic neurons also brought to light changes in the architecture of presynaptic structures.
Dopaminergic neurons in the SNc are particularly vulnerable to α-syn accumulation. Research over the last few decades has focused on developing models to understand the effects of α-syn inclusions in this specific population23,54,55,56,57. Previous work by our group has shown that the LIPA system prompts the induction of α-syn inclusions in the dopaminergic neurons of the SNc in mice, reproducing several key features of LBs observed in PD patients40. In addition, an important pathological characteristic of α-syn aggregates is their ability to be transported and spread to different areas of the brain, exacerbating the progression of the disease56,58,59. Our results show that, as early as 10 days following the induction of LIPA-α-syn aggregation in the SNc, there was a substantial accumulation of LIPA-α-syn in the presynaptic terminals in the striatum, suggesting that α-syn aggregates can be transported to the striatum via anterograde transport56,60. Indeed, the accumulation of α-syn aggregates in the presynaptic terminals has also been commonly observed in PD animal models and human tissue19,61,62,63. Given this, the deposition of α-syn in dopaminergic projections might trigger structural and functional synaptic alterations2.
Cumulative evidence suggests that synaptic dysfunction arises early in the progression of PD15,16. However, the mechanisms driving the synaptic pathology remain unclear. In the present study, we uncovered early presynaptic alterations caused by LIPA-α-syn inclusions before the onset of dopaminergic axonal degeneration. Interestingly, our results show that the accumulation of LIPA-α-syn caused a significant increase in the number of synaptic boutons in the striatum. This observation contrasts with previous studies that have reported either no change or a significant decrease in the number of synaptic varicosities associated with α-syn accumulation64,65,66. This discrepancy may stem from the fact that these studies were performed at more advanced stages of the disease where axonal degeneration may have already occurred55,65. Moreover, the increase in presynaptic boutons reported in our study may have uncovered early compensatory synaptic changes aimed at counteracting the malfunction in DA transmission67 or underly early synaptic dysfunction. In addition, previous studies overexpressing α-syn in mice or using brain tissue from patients suffering from DLB have reported an increase in synapse size63,66,68. We also observed an enlargement of the presynaptic structures, which is consistent with the increase in synapse size. In addition to synaptic structural alterations, our findings also show that there was an increase in the expression of synaptophysin resulting from LIPA-α-syn inclusions. Given that synaptophysin is a presynaptic protein localized to synaptic vesicles69,70, it is conceivable that LIPA-α-syn inclusions might cause an abnormal accumulation of synaptic vesicles at the synapses. Although previous ultrastructural analyses have determined that the overall number of synaptic vesicles is not affected and that pathological α-syn induces an accumulation of enlarged synaptic vesicles and changes in the distribution of synaptic vesicles in the synaptic pools, pointing to a malfunction in exocytosis/endocytosis processes or the trafficking of synaptic vesicles7,33,71,72,73,74.
Our results show that LIPA-α-syn accumulation induced early alterations in DA homeostasis. Surprisingly, our findings also show that there was an increase in striatal DA contents without particularly affecting its metabolites. Following its synthesis at synapses, DA is rapidly internalized into synaptic vesicles via VMAT2 to avoid DA oxidation75,76,77. After neurotransmitter release, DA can either be degraded by Monoamine oxidase and Catecholamine-O-methyltransferase enzymes or recycled into synaptic vesicles after reuptake78,79. It is thus conceivable that LIPA-α-syn inclusions induce the trapping of DA in synaptic vesicles, making it inaccessible for enzymatic degradation. Taken together with the alterations observed at the structural level, our results may suggest that LIPA-α-syn inclusions induce an early impairment in DA signaling33. However, the synaptic vesicle cycle is a complex process, and which stages are impacted by LIPA-α-syn aggregates remain to be determined. Given the physiological role of α-syn in synaptic vesicle clustering and neurotransmitter release through the stabilization of the SNARE complex3,4 and observations of changes in synaptic structure and DA homeostasis, LIPA-α-syn inclusions may impact the normal clustering of synaptic vesicles, interfere with their trafficking, or alter DA release by preventing SNARE complex formation7,74,80. Importantly, while our current study provides a steady-state view and primarily focuses on dopamine (DA) content rather than DA release, future experiments combining LIPA with temporally resolved DA release assays, such as genetically encoded DA sensors (e.g., dLight)81,82,83, will be essential to determine how DA release is altered during the early stages of α-syn accumulation at striatal dopaminergic terminals.
As a result of these LIPA-α-syn-induced presynaptic changes, striatal neurons may also undergo early alterations. Indeed, previous work by our team and others has shown that a disruption of the nigrostriatal pathway leads to dysfunctional striatal cell activity36,40,44 and that this can occur early in the progression of the disease36,40. We monitored neuronal activity with single-cell resolution using mini-endoscopes and found that the induction of LIPA-α-syn inclusions in the midbrain not only caused a decrease in the frequency of Ca2+ transients in striatal cells but also distinctly affected the amplitude of their Ca2+ transients. Although Ca2+ transients with low and medium amplitude values decreased, transients with high amplitude values increased. These results suggest that LIPA-α-syn accumulation might affect different populations of striatal cells in distinct ways. The cytoarchitecture of the striatum encompasses various types of neuronal populations, including a large population of GABAergic MSNs (95%) and a small population of interneurons (5%)84,85,86. MSNs, in turn, can be divided into three populations depending on the expression of the DA receptor, i.e., D1-MSNs (or dMSNs), D2-MSNs (or iMSNs), and a smaller proportion of D1/D2-coexpressing MSNs28,87. Our cluster analysis based on the properties of Ca2+ dynamics shows that, under baseline conditions, striatal cells were divided into three different clusters. Cluster 1 and cluster 2 comprised the largest percentage of neurons, making up 40.2% and 52.1% of the total number of striatal neurons recorded, respectively. Given that MSNs constitute the largest population of neurons in the striatum84,85,86, we propose that cells from cluster 1 and cluster 2 could comprise the neuronal populations of D1- and D2-MSNs, respectively. In addition, while some studies that performed Ca2+ imaging in freely behaving mice showed Ca2+ transients with similar frequencies and amplitudes in D1- and D2-MSNs43,44, it is worth noting that the work by Varin et al.88, who recorded the activities of D1- and D2-MSNs using endoscopic imaging, revealed significantly higher activity in D1-MSNs than in D2-MSNs88. Given that our cluster analysis shows that neurons in cluster 1 exhibited a higher frequency of Ca2+ events than neurons in cluster 2, it is conceivable that cluster 1 might consist of D1-MSNs while cluster 2 might consist of D2-MSNs. This hypothesis is supported by the results of our assessment of c-fos+ cells showing that LIPA-α-syn inclusions induced a decrease in the number of activated D1+ cells and an increase in the number of activated D2+ cells, providing support for the changes observed in cluster organization following LIPA-α-syn accumulation. In the classical view, D1 DA receptors are coupled to a Gαs/olf protein, and their activation triggers a downstream signaling cascade that enhancemes the neuronal excitability of D1-MSNs27,89. In contrast, D2 DA receptors are coupled to Gi/o protein, and their activation initiates a downstream signaling cascade that reduces the excitability of D2-MSNs27. The observed reduction in D1 activity concomitant with an increase in D2 activity supports the hypothesis that DA release is diminished, despite elevated tissue DA contents29,90.
Converging lines of evidence have shown that enhanced striatal activity is associated with mouse movement for both D1- and D2-MSNs, suggesting that the coactivation of these two populations of striatal neurons encodes mouse locomotion42,43,44,45. Consistent with this, we found that under both baseline and after light-induced accumulation of α-syn on post-stimulation days 2 and 10 conditions, all three clusters exhibited increased activity during mobile periods. However, the accumulation of LIPA-α-syn did not affect the activity of striatal cells during locomotion at this early stage.
In line with our observations in the mouse brain, we found, in human iDA neurons, that the early accumulation of LIPA-α-syn in dopaminergic axons led to a significant increase in the number of synaptic varicosities, an enlargement of synaptic structures, and an accumulation of the presynaptic marker synaptophysin. Interestingly, changes in synaptic size and synaptophysin expression were specifically associated with the presence of LIPA-α-syn inclusions. This observation contrasts with previous studies that used human dopaminergic neurons derived from patients harboring the A53T mutation to show that there is a decrease in the number of synaptic varicosities65,91. These discrepancies may be attributed to differences in the time points chosen to assess synaptic pathology. Our observations highlight the early synaptic changes caused by LIPA-α-syn accumulation, which may trigger synaptic dysfunction and alter the activity of human dopaminergic neurons, potentially compromising cell viability92.
In conclusion, our study shows that the accumulation of LIPA-α-syn inclusions in the dopaminergic terminals at early stages of the disease triggered synaptic dysfunction and dysregulation in DA homeostasis. In addition, we speculate that the presynaptic impairment observed in mouse brains following α-syn accumulation led to alterations in striatal neuronal activity that occur prior to the onset of dopaminergic fiber neurodegeneration in the striatum and the emergence of the characteristic motor symptoms of PD. Ultimately, these events may progress and result in the development of dopaminergic neurodegeneration.
Methods
Animals
Adult 2- to 3-month-old C57Bl/6 WT male mice (n = 47) (Charles Rivers, Wilmington, MA, USA) were kept on a 12-h light/dark cycle at 22 °C with ad libitum access to food and water. All experiments were approved by the institutional Animal Welfare Committee of Université Laval, protocol 2020–641 to AO, in accordance with the Canadian Council on Animal Care guidelines. All mini-endoscopic imaging coupled with optogenetic stimulation experiments were approved by the Animal Welfare Committee of Université Laval, protocol 2019–020 to AS.
Mice were anesthetized with 2% isoflurane-O2 and were placed in a stereotactic frame (David Kopf Instruments, CA, USA). An incision was made in the skin, and the skull was cleared to visualize the bregma. After drilling a hole above the SNc, the mice received a unilateral 2-μL injection of either AAV2/6-LIPA-α-syn-mCherry or AAV2/6-LIPA-Empty at a rate of 0.2 μL/min. The injections were performed using the following coordinates with respect to the bregma: AP − 3.08 mm, ML − 1.5 mm, and DV − 4.25 mm. Following the viral injection, a wireless optogenetic device (Eicom/Amuza, San Diego, CA, USA) was slowly implanted in the SNc using the following coordinates with respect to the bregma: AP − 3.08 mm, ML − 1.5 mm, and DV − 4.20 mm. The optogenetic device was fixed to the skull using cement.
Fifteen days after the surgery, the mice received 1.76-mW/mm2 10-ms 20-Hz pulses over a 1-h period for 7 days using wireless optogenetic devices (Eicom/Amuza, San Diego, CA, USA). Just before each optogenetic stimulation session, a battery (Eicom/Amuza, San Diego, CA, USA) was directly connected to the implant.
Ca2+ dynamics analysis
All the Ca2+ results described in the present paper have been reanalyzed from previous experiments we conducted40. As previously described40, 2-month-old C57Bl/6 WT male mice (n = 5) (Charles Rivers, Wilmington, MA, USA) received an injection of AAV2/6-LIPA-α-syn-mCherry in the SNc (AP − 3.5, ML + / − 1.2, DV − 4) and AAV2/5 CAG-GCaMP6s in the striatum (AP + 1.5, ML + / − 1.2, DV − 2.5). Two to three weeks later, a double optogenetic/mini-endoscopic cannula was implanted. The GRIN lens was placed above the dorsal striatum and the optic fiber just on top of the SNc. Three weeks after surgery, Ca2+ activity was recorded at a rate of 10 fps in the striatum of freely behaving mice twice for 10 min every other day. Four days after beginning the Ca2+ recordings, the SNc of the mice were subjected to a 1-h stimulation with blue light before the Ca2+ imaging using the following pattern 1.76-mW/mm2 10-ms 20-Hz pulses. After the 7-day stimulation protocol, the mice continued to be imaged every second day for up to 10 days. Ca2+ recorded images were processed as previously described40. Briefly, the raw images were time-averaged in 200-ms bins using ImageJ software. Afterwards, Doric Neuroscience Studio software (Doric Lenses, Quebec City, QC, Canada) was used to correct the images in XY for brain motion, apply ΔF/F0 processing, and subtract the background. ROIs were manually traced around the cell bodies, and the mean fluorescence values were obtained. Ca2+ traces were analyzed using a custom-script created in MATLAB (The MathWorks, Natick, MA, USA) to obtain the mean amplitude and frequency of the Ca2+ transients. Of note, while we cannot formally rule out the possibility that GCaMP6 excitation induced by blue light in the striatum may also contribute to α-syn accumulation at dopaminergic terminals in the striatum, our previous experiments argue against this possibility40. We have previously shown Ca2+ imaging in the striatum without delivering a blue light in the SNc does not induce α-syn accumulation at dopaminergic terminals and changes in Ca2+ activity in the striatum40.
Mouse behavior tests
All the behavior experiments were reported in a previously published paper40 and were reanalyzed for the present study. Briefly, mice (n = 5) implanted with a mini-endoscope were placed in an open-field arena and their behavior was monitored twice for 10 min every other day while Ca2+ activity was being recorded. To record behavior, we used a video camera (30 fps) (Doric Lenses, Quebec City, QC, Canada) positioned above the arena and controlled by Doric Neuroscience Studio software (Doric Lenses, Quebec City, QC, Canada). We used DeepLabCut software93,94 to track the nose; head; left and right ears; upper, center, and lower parts of the body; and base of the tail of the mice. To train the DeepLabCut algorithm, we extracted 20 frames from each of the recorded videos and manually labeled the abovementioned regions of the mice. The network was trained for up to 500,000 iterations and was used to obtain the coordinates of all ROIs in the complete recorded footage. The tracked region of the center part of the body was used to calculate the speed and to measure the time the mice spent immobile and mobile throughout the videos. The percentage of time the mice spent mobile and immobile, along with their speed, was determined using a custom-written script created in python.
Cluster analysis of Ca²⁺ transients
A clustering analysis was performed based on the frequencies and amplitudes of the Ca²⁺ transients. To determine the optimal number of clusters for each condition, we computed three different scores: Calinski-Harabasz, Elbow, and Silhouette. Given the variability in the Calinski-Harabasz and Elbow scores, we used the recommendations resulting from the Silhouette score. The clustering was performed using the K-Means algorithm included in scikit-learn 1.5.0. The Python package seaborn 0.13.2 was used to generate the KDE maps.
To investigate potential trends in cluster activity changes between the mobile and immobile periods, we merged the DeepLabCut results with the raw results, maintaining the labels from the cluster analysis results for each ROI/Animal ID. We first processed the DeepLabCut results to be able to categorize the results between the mobile and immobile periods using a binary scoring system (1 for mobile, 0 for immobile). We then computed the mean values for the Ca²⁺ result columns of interest, frequency and amplitude, before merging the mobility and calcium data. Once combined, we separated the results between mobile and immobile periods for each cluster to obtain movement-specific activity data.
Immunostaining
The mice (n = 17) were deeply anesthetized with an intraperitoneal injection (0.1 mL/30 g) of a mixture of ketamine (100 mg/mL) and xylazine (10 mg/mL). They were then perfused intracardially with PBS followed by 4% paraformaldehyde (PFA). The brains were collected and were post-fixed overnight in 4% PFA at 4 °C. They were then incubated in 20% sucrose-PBS for 24 h at 4 °C. Coronal slices (30-μm thick) of the brains were cut using a microtome (Leica SM2000R), and the slices were stored in cryoprotection solution (0.05 M sodium phosphate (pH 7.3), 30% ethylene glycol, and 20% glycerol) at −20 °C.
Free-floating brain slices were permeabilized with 0.3% Triton X-100 for 30 min at RT and were then blocked with a mixture of 0.3% Triton X-100, 1% bovine serum albumin (BSA), and 5% normal goat serum (NGS) for 2 h at RT. The brain slices were then incubated with the following primary antibodies diluted in a mixture of 0.1% Triton X-100, 1% BSA, and 1% NGS overnight at 4 °C: rat anti-mCherry (16D7) (1:500, Thermo Fisher Scientific, Waltham, MA, USA), mouse anti-human α-syn (LB509) (1:400, Thermo Fisher Scientific, Waltham, MA, USA), guinea pig anti-cfos (1:1000, Synaptic Systems, Göttingen, Germany), mouse anti-TH (1:1000, EMD Millipore, Temecula, CA, USA), rabbit anti-TH (1:1000, Pel-Freez, Rogers, AR, USA), rat anti-DAT (1:500, Millipore Sigma, Burlington, MA, USA), rabbit anti-dopamine receptor 1 (1:250, Novus Biologicals, Centennial, CO, USA), rabbit anti-dopamine receptor 2 (1:250, Proteintech, Rosemont, IL, USA), and rabbit anti-synaptophysin (1:200, Thermo Fisher Scientific, Invitrogen, Waltham, MA, USA) (Table 1). The slices were then incubated with the appropriate secondary antibodies diluted in 0.1% Triton X-100 in PBS for 2 h at RT: Alexa Fluor 488 anti-rabbit (1;1000, Invitrogen, Waltham, MA, USA), Alexa Fluor 488 anti-mouse (1;1000, Invitrogen, Waltham, MA, USA), Alexa Fluor 488 anti-guinea pig (1:250, Jackson ImmunoResearch Laboratories Inc, West Grove, PA, USA), Alexa Fluor 568 anti-rat (1:1000, ThermoFisher, Waltham, MA, USA), Alexa Fluor 633 anti-rabbit (1:250 for DRD1 and DRD2 staining and 1:400 for synaptophysin staining, Invitrogen, Waltham, MA, USA), Mouse IgG (680RD) (1:1000, LI-COR Biosciences, Lincoln, NE, USA), Cyanine5 anti-rat (1:500, Jackson ImmunoResearch Labs, West Grove, PA, USA) (Table 1). Lastly, the brain slices were counterstained with DAPI (1:5000) diluted in PBS and were mounted on Superfrost Plus microscope slides with Fluoromount mounting medium (Thermo Fisher Scientific, Waltham, MA, USA).
We used an antigen retrieval protocol for the immunostaining to assess c-fos activation in D2 positive cells. Briefly, the brain slices were mounted on Superfrost Plus microscope slides and were left to dry overnight. The brain slices were then subjected to an antigen retrieval protocol by incubating them in Tris-EDTA (10 mM Tris base, 1 mM EDTA, 0.05% Tween 20, pH 9.0) for 10 min at 95 °C. The subsequent steps of the protocol were the same as described above.
For the immunohistological analyses of human dopaminergic neurons, the cells were permeabilized with 0.3% Triton X-100 for 10 min at RT and were blocked with a mixture of 0.1% Triton X-100, 1% BSA, and 5% NGS for 1 h at RT. The cells were then incubated with primary antibodies diluted in blocking solution overnight at 4 °C. The following primary antibodies were used: mouse anti-TH (1:1000, EMD Millipore, Temecula, CA, USA) and rabbit anti-synaptophysin (1:1000, Thermo Fisher Scientific, Invitrogen, Waltham, MA, USA). The cells were then incubated with the appropriate secondary antibodies diluted in blocking solution (1:800) for 2 h at RT. Lastly, the cells were counterstained with DAPI (1:5,000), and the coverslips were mounted on Superfrost Plus microscope slides with Fluoromount mounting medium (Thermo Fisher Scientific, Waltham, MA, USA).
Quantification
To evaluate the activation of D1- and D2-MSNs in the striatum of the mice, five random images were acquired from the dorsal striatum of each brain slice using a Zeiss LSM800 confocal microscope (Carl Zeiss, Oberkochen, Germany) equipped with a 40x oil immersion objective (Plan-Apochromat 40x/NA 1.4; Zeiss). Zen software was used to blindly quantify the number of c-fos + /D1-MSNs+ and c-fos + /D2-MSNs + . A cell was considered to be a c-fos + /D1-MSNs+ or a c-fos + /D2-MSNs+ when the c-fos staining was surrounded by D1 or D2 staining, respectively.
To assess the density of TH+ and DAT+ fibers in the striatum, images were acquired using a Zeiss Axio Imager M2 equipped with an ApoTome epifluorescent microscope (Carl Zeiss, Oberkochen, Germany) with a 20x air objective (EC Plan-Neofluar 20x/0.50 M27; Zeiss). Tiling was performed to obtain an image of the entire striatum. ImageJ software was used to manually delimit the area of the dorsal striatum and quantify the mean pixel intensity in the dorsal part of the striatum of the injected hemisphere.
To evaluate synaptic density in the striatum of the mice, six random images were acquired from the dorsal striatum of each brain slice using a Zeiss LSM800 confocal microscope (Carl Zeiss, Oberkochen, Germany) equipped with a 63x oil immersion objective (Plan-Apochromat 63x/NA 1.4; Zeiss). The number of synaptophysin varicosities, surface, and integrated density were blindly analyzed using the 3D Objects Counter plugin from ImageJ software. An intensity threshold of 7842 was determined for all the groups analyzed.
To evaluate the synaptic density in human dopaminergic cells, we acquired images using a Zeiss LSM800 confocal microscope (Carl Zeiss, Oberkochen, Germany) equipped with a 63x oil immersion objective (Plan-Apochromat 63x/NA 1.4; Zeiss) and performed tiling. The synaptophysin varicosities were manually quantified along the neurites of mCherry-expressing neurons using ImageJ software. The number of synaptophysin varicosities was corrected by the length of the neurite, which was also quantified using ImageJ software. We also extracted the values for the mean surface and integrated density of the synaptophysin varicosities.
Striatal biogenic amine assay
Mice (n = 25) were deeply anesthetized with an intraperitoneal injection (0.1 mL/30 g) of ketamine (100 mg/mL), and xylazine (10 mg/mL). They were then intracardially perfused with PBS containing a cocktail of phosphatases (50 mM sodium fluoride and 1 mM sodium pyrophosphate) and protease inhibitors (Sigma-Aldrich, Burlington, MA, USA), and the regions containing the SNc and striatum were separated. The brain area containing the striatum was rapidly frozen in liquid nitrogen and was stored at −80 °C until it was processed for high performance liquid chromatography (HPLC). The brain region including the SNc was immediately fixed in 4% PFA overnight at 4 °C, was incubated in 4% PFA and 20% sucrose for 24 h at 4 °C, and was then processed for immunohistochemistry using the same protocol described in the Immunostaining section.
For HPLC analysis, the right anterior part of the striatum (bregma ~1.18 at ~0.14 mm, Paxinos and Franklin, 200195) was dissected, homogenized in 250 μl of 0.1 N HClO4 (Sigma-Aldrich, Burlington, MA, USA) at 4 °C and then centrifuged at 10,000 × g for 30 min (4 °C) to precipitate proteins. The resulting pellet was homogenized in 250 μl of 0.1 M NaOH, and the protein concentrations were then assayed using a Micro BCA Protein Assay kit (Thermo Scientific, Rockford, IL). The supernatant containing the striatal contents of dopamine and its metabolites DOPAC and 3-MT were measured by HPLC with electrochemical detection. Supernatants of striatal tissue were directly injected into the chromatograph consisting of a Waters 717 plus autosampler automatic injector equipped with a sample heater/cooler set at 4 °C (Waters, Mississauga, ON, Canada), a Waters 515 pump equipped with a C-18 column (Waters Nova-Pak C18, 3 μm, 3.9 mm × 150 cm), and a BAS LC-4C amperometric detector connected to a dual glassy carbon electrode (BASi research Products, West Lafayette, IN, USA). The electrochemical potential was set at 0.8 V with respect to an Ag/AgCl reference electrode. The mobile phase consisted of 0.025 M citric acid, 1.7 mM 1-heptane-sulfonic acid (Sigma-Aldrich, MilliporeSigma Canada Ltd, Oakville, ON, Canada), and 10% methanol (VWR International, Mississauga, ON, Canada), in filtered distilled water, delivered at a flow rate of 0.8 ml/min. The final pH of 3.99 was obtained by addition of NaOH. To prevent bubble formation in the liquid chromatography system, the mobile phase is degassed by bubbling helium through it for a few minutes just before use. Throughout the chromatography process, the amperometric signal from the detector was transferred directly to the Kipp-Zonen BD41 2-channel chart recorder. Peak heights corresponding to the different amines were measured directly on the chromatogram plotted on the chart recorder using a digital scale ruler. Standard curves containing a mixture of different concentrations of dopamine, DOPAC, and 3-MT, dissolved in HClO4, were obtained to measure the concentrations of each amine present in the samples. The amine levels in the samples, expressed in nanogram, were transferred to a Microsoft Excel spreadsheet and converted to nanograms of amine per milligram of protein based on the injection volume in the liquid chromatography system and the protein content for each sample.
Generation of human dopaminergic neurons
We used a previously published protocol51 to generate Ngn2 iNeurons from hiPSCs and to differentiate them into dopaminergic neurons. Prior to the initiation of the differentiation protocol, hiPSCs expressing the doxycycline-inducible Ngn2 cassette were seeded on Matrigel-coated plates (Matrigel Matrix hESC-qualified) (Thermo Fisher Scientific, Waltham, MA, USA) and were kept in mTesr™ Plus medium (Stemcell Technologies, Vancouver, BC, Canada), as previously described50. To differentiate the Ngn2 hiPSC line into iNeurons, one day after seeding the cells were cultured in Day 0/1 medium containing DMEM/F12 (Thermo Fisher Scientific/Gibco, Waltham, MA, USA), N2 (Thermo Fisher Scientific/Gibco, Waltham, MA, USA), B27 (Thermo Fisher Scientific, Waltham, MA, USA), NEAA (Wisent Life Sciences, Saint-Jean-Baptiste, QC, Canada), BDNF (Peprotech, Cranbury, NJ, USA), GDNF (Peprotech, Cranbury, NJ, USA), mouse laminin (Thermo Fisher Scientific, Waltham, MA, USA), and 2 µg/mL doxycycline (Takara Bio Company, Kusatsu, Japan) to induce Ngn2 expression. On day 2 (DIV 2), the cells were dissociated with Accutase (Thermo Fisher Scientific, Waltham, MA, USA), were seeded on coverslips coated with 50 µg/mL of poly-L-ornithine (Sigma-Aldrich, St. Louis, MO, USA) and 10 µg/mL of laminin, and were maintained in Day 2 neurobasal medium (Thermo Fisher Scientific/Life Technologies, Waltham, MA, USA), N2, B27, GlutaMax (Thermo Fisher Scientific, Waltham, MA, USA), NEAA, BDNF, GDNF, mouse laminin, and 2 µg/mL of doxycycline. From DIV 2, iNeurons were induced to differentiate into dopaminergic neurons using a STEMdiff™ Midbrain Neuron Differentiation Kit (Stemcell Technologies, Vancouver, BC, Canada). From DIV 2 to DIV 4, the medium, which was supplemented with 2 µM Ara-C, was replaced every day. From DIV 5 to DIV 8, only half the medium was changed every day. At Day 9 (DIV 9), the medium was completely replaced with midbrain maturation medium from a STEMdiff™ Midbrain Neuron Maturation Kit (Stemcell Technologies, Vancouver, BC, Canada) supplemented with 2 µg/mL of doxycycline. From day 10 (DIV 10) onward, and every two days, half the medium was replaced with fresh maturation medium. At Day 28 (DIV28), the cells were transfected with the LIPA constructs40: LIPA-α-syn (0.5 μg), LIPA-Empty (0.4 μg), and LIPA-α-synΔNAC (0.6 μg) using Lipofectamine® 2000 (Thermo Fisher Scientific, Waltham, MA, USA). The maturation medium was replaced with Opti-MEM medium (Thermo Fisher Scientific, Waltham, MA, USA) 30 min before transfecting the cells. The DNA was combined with the liposome mix at a ratio of 1:3 (DNA:Reagent) and was added to the Opti-MEM medium. Six hours later, the Opti-MEM medium was replaced with maturation medium. Two days post-transfection, the cells were stimulated with blue light (0.3 mW/mm2) for 6 h and were then fixed using 4% PFA and 3% sucrose for 15 min at RT. After three 5-min washes in PBS, the cells were stored in PBS at 4 °C for immunohistological analyses.
Statistical analysis
In vitro assays were performed in three independent experiments for the LIPA-α-syn group, and two independent experiments were carried out for the control groups.
For the in vivo analyses, a one-way ANOVA followed by Tukey’s multiple comparisons test was used to assess differences between the three experimental groups used. In addition, an unpaired two-sided Student’s t test was used to analyze the differences in the frequencies and amplitudes of Ca2+ transients between the mobile and immobile periods. For the in vitro analyses, a one-way ANOVA followed by Tukey’s multiple comparisons test was used to determine the differences between the experimental conditions used. Outliers (values greater than the 1.5 interquartile intervals below the first quartile or 1.5 interquartile intervals above the third quartile) were excluded from the analysis. To test statistical significance for the frequency and amplitude of Ca2+ events, two-sided Kolmogorov–Smirnov test was used. All values are expressed as means ± SEM. The statistical analyses were performed using Prism 9 (GraphPad). p < 0.05 was required for the rejection of the null hypothesis.
Data availability
Data are available from the corresponding author upon reasonable request.
References
Maroteaux, L., Campanelli, J. T. & Scheller, R. H. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8, 2804–2815 (1988).
Sharma, M. & Burre, J. alpha-Synuclein in synaptic function and dysfunction. Trends Neurosci. 46, 153–166 (2023).
Wang, L. et al. alpha-synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr. Biol. 24, 2319–2326 (2014).
Burre, J., Sharma, M. & Sudhof, T. C. alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 111, E4274–E4283 (2014).
Burre, J. et al. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667 (2010).
Somayaji, M. et al. A dual role for alpha-synuclein in facilitation and depression of dopamine release from substantia nigra neurons in vivo. Proc. Natl. Acad. Sci. USA 117, 32701–32710 (2020).
Scott, D. & Roy, S. alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci. 32, 10129–10135 (2012).
Vargas, K. J. et al. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J. Neurosci. 34, 9364–9376 (2014).
Diao, J. et al. Native alpha-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. Elife 2, e00592 (2013).
Calabresi, P., Di Lazzaro, G., Marino, G., Campanelli, F. & Ghiglieri, V. Advances in understanding the function of alpha-synuclein: implications for Parkinson’s disease. Brain 146, 3587–3597 (2023).
Spillantini, M. G. et al. Alpha-synuclein in Lewy bodies. Nature 388, 839–840 (1997).
Goedert, M., Spillantini, M. G., Del Tredici, K. & Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 9, 13–24 (2013).
Lashuel, H. A., Overk, C. R., Oueslati, A. & Masliah, E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14, 38–48 (2013).
Woerman, A. L. et al. alpha-Synuclein: multiple system atrophy prions. Cold Spring Harb. Perspect. Med. 8, https://doi.org/10.1101/cshperspect.a024588 (2018).
Bridi, J. C. & Hirth, F. Mechanisms of alpha-Synuclein Induced Synaptopathy in Parkinson’s Disease. Front. Neurosci. 12, 80 (2018).
Calabresi, P. et al. Alpha-synuclein in Parkinson’s disease and other synucleinopathies: from overt neurodegeneration back to early synaptic dysfunction. Cell Death Dis. 14, 176 (2023).
Dauer, W. & Przedborski, S. Parkinson’s disease: mechanisms and models. Neuron 39, 889–909 (2003).
Marui, W. et al. Progression and staging of Lewy pathology in brains from patients with dementia with Lewy bodies. J. Neurol. Sci. 195, 153–159 (2002).
Kramer, M. L. & Schulz-Schaeffer, W. J. Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J. Neurosci. 27, 1405–1410 (2007).
Gcwensa, N. Z., Russell, D. L., Cowell, R. M. & Volpicelli-Daley, L. A. Molecular mechanisms underlying synaptic and axon degeneration in Parkinson’s disease. Front. Cell Neurosci. 15, 626128 (2021).
Cramb, K. M. L., Beccano-Kelly, D., Cragg, S. J. & Wade-Martins, R. Impaired dopamine release in Parkinson’s disease. Brain 146, 3117–3132 (2023).
Kulkarni, A. S., Burns, M. R., Brundin, P. & Wesson, D. W. Linking alpha-synuclein-induced synaptopathy and neural network dysfunction in early Parkinson’s disease. Brain Commun. 4, fcac165 (2022).
Alegre-Abarrategui, J. et al. Selective vulnerability in alpha-synucleinopathies. Acta Neuropathol. 138, 681–704 (2019).
Minakaki, G., Krainc, D. & Burbulla, L. F. The convergence of Alpha-synuclein, mitochondrial, and lysosomal pathways in vulnerability of midbrain dopaminergic neurons in Parkinson’s disease. Front. Cell Dev. Biol. 8, 580634 (2020).
McGregor, M. M. & Nelson, A. B. Circuit mechanisms of Parkinson’s disease. Neuron 101, 1042–1056 (2019).
Liu, C., Kershberg, L., Wang, J., Schneeberger, S. & Kaeser, P. S. Dopamine secretion is mediated by sparse active zone-like release sites. Cell 172, 706–718.e715 (2018).
Surmeier, D. J., Ding, J., Day, M., Wang, Z. & Shen, W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 30, 228–235 (2007).
Yung, K. K. et al. Immunocytochemical localization of D1 and D2 dopamine receptors in the basal ganglia of the rat: light and electron microscopy. Neuroscience 65, 709–730 (1995).
Gerfen, C. R. & Surmeier, D. J. Modulation of striatal projection systems by dopamine. Annu Rev. Neurosci. 34, 441–466 (2011).
Cui, G. et al. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature 494, 238–242 (2013).
Howe, M. W. & Dombeck, D. A. Rapid signalling in distinct dopaminergic axons during locomotion and reward. Nature 535, 505–510 (2016).
Luk, K. C. et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953 (2012).
Nemani, V. M. et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65, 66–79 (2010).
Dagra, A. et al. alpha-Synuclein-induced dysregulation of neuronal activity contributes to murine dopamine neuron vulnerability. NPJ Parkinsons Dis. 7, 76 (2021).
Hill, E., Gowers, R., Richardson, M. J. E. & Wall, M. J. alpha-Synuclein aggregates increase the conductance of substantia nigra dopamine neurons, an effect partly reversed by the KATP channel inhibitor glibenclamide. eNeuro 8, https://doi.org/10.1523/ENEURO.0330-20.2020 (2021).
Tozzi, A. et al. Dopamine-dependent early synaptic and motor dysfunctions induced by alpha-synuclein in the nigrostriatal circuit. Brain 144, 3477–3491 (2021).
Durante, V. et al. Alpha-synuclein targets GluN2A NMDA receptor subunit causing striatal synaptic dysfunction and visuospatial memory alteration. Brain 142, 1365–1385 (2019).
Fares, M. B., Jagannath, S. & Lashuel, H. A. Reverse engineering Lewy bodies: how far have we come and how far can we go?. Nat. Rev. Neurosci. 22, 111–131 (2021).
Airavaara, M. et al. Back and to the future: from neurotoxin-induced to human Parkinson’s disease models. Curr. Protoc. Neurosci. 91, e88 (2020).
Berard, M. et al. A light-inducible protein clustering system for in vivo analysis of alpha-synuclein aggregation in Parkinson disease. PLoS Biol. 20, e3001578 (2022).
Teixeira, M., Sheta, R., Idi, W. & Oueslati, A. Optogenetic-mediated induction and monitoring of alpha-synuclein aggregation in cellular models of Parkinson’s disease. STAR Protoc. 4, 102738 (2023).
Klaus, A. et al. The Spatiotemporal organization of the striatum encodes action space. Neuron 95, 1171–1180 e1177 (2017).
Barbera, G. et al. Spatially compact neural clusters in the dorsal striatum encode locomotion relevant information. Neuron 92, 202–213 (2016).
Parker, J. G. et al. Diametric neural ensemble dynamics in parkinsonian and dyskinetic states. Nature 557, 177–182 (2018).
Maltese, M., March, J. R., Bashaw, A. G. & Tritsch, N. X. Dopamine differentially modulates the size of projection neuron ensembles in the intact and dopamine-depleted striatum. Elife 10, https://doi.org/10.7554/eLife.68041 (2021).
Surmeier, D. J. et al. The role of dopamine in modulating the structure and function of striatal circuits. Prog. Brain Res. 183, 149–167 (2010).
Calabresi, P., Mercuri, N. B. & Di Filippo, M. Synaptic plasticity, dopamine and Parkinson’s disease: one step ahead. Brain 132, 285–287 (2009).
Calabresi, P., Picconi, B., Tozzi, A. & Di Filippo, M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 30, 211–219 (2007).
Bose, A., Petsko, G. A. & Studer, L. Induced pluripotent stem cells: a tool for modeling Parkinson’s disease. Trends Neurosci. 45, 608–620 (2022).
Zhang, Y. et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798 (2013).
Sheta, R. et al. Combining NGN2 programming and dopaminergic patterning for a rapid and efficient generation of hiPSC-derived midbrain neurons. Sci. Rep. 12, 17176 (2022).
Sheta, R., Teixeira, M., Idi, W. & Oueslati, A. Optimized protocol for the generation of functional human induced-pluripotent-stem-cell-derived dopaminergic neurons. STAR Protoc. 4, 102486 (2023).
Giasson, B. I., Murray, I. V., Trojanowski, J. Q. & Lee, V. M. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 276, 2380–2386 (2001).
Venda, L. L., Cragg, S. J., Buchman, V. L. & Wade-Martins, R. alpha-Synuclein and dopamine at the crossroads of Parkinson’s disease. Trends Neurosci. 33, 559–568 (2010).
Thakur, P. et al. Modeling Parkinson’s disease pathology by combination of fibril seeds and alpha-synuclein overexpression in the rat brain. Proc. Natl. Acad. Sci. USA 114, E8284–E8293 (2017).
Masuda-Suzukake, M. et al. Prion-like spreading of pathological alpha-synuclein in brain. Brain 136, 1128–1138 (2013).
Matsuoka, Y. et al. Lack of nigral pathology in transgenic mice expressing human alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol. Dis. 8, 535–539 (2001).
Braak, H. et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211 (2003).
Li, J. Y. et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 14, 501–503 (2008).
Freundt, E. C. et al. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann. Neurol. 72, 517–524 (2012).
Nakata, Y. et al. Accumulation of alpha-synuclein triggered by presynaptic dysfunction. J. Neurosci. 32, 17186–17196 (2012).
Phan, J. A. et al. Early synaptic dysfunction induced by alpha-synuclein in a rat model of Parkinson’s disease. Sci. Rep. 7, 6363 (2017).
Colom-Cadena, M. et al. Synaptic phosphorylated alpha-synuclein in dementia with Lewy bodies. Brain 140, 3204–3214 (2017).
Perez-Acuna, D., Shin, S. J., Rhee, K. H., Kim, S. J. & Lee, S. J. alpha-Synuclein propagation leads to synaptic abnormalities in the cortex through microglial synapse phagocytosis. Mol. Brain 16, 72 (2023).
Kouroupi, G. et al. Defective synaptic connectivity and axonal neuropathology in a human iPSC-based model of familial Parkinson’s disease. Proc. Natl. Acad. Sci. USA 114, E3679–E3688 (2017).
Gcwensa, N. Z. et al. Excitatory synaptic structural abnormalities produced by templated aggregation of alpha-syn in the basolateral amygdala. Neurobiol. Dis. 199, 106595 (2024).
Blesa, J. et al. Compensatory mechanisms in Parkinson’s disease: Circuits adaptations and role in disease modification. Exp. Neurol. 298, 148–161 (2017).
Boassa, D. et al. Mapping the subcellular distribution of alpha-synuclein in neurons using genetically encoded probes for correlated light and electron microscopy: implications for Parkinson’s disease pathogenesis. J. Neurosci. 33, 2605–2615 (2013).
Wiedenmann, B. & Franke, W. W. Identification and localization of synaptophysin, an integral membrane glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell 41, 1017–1028 (1985).
Jahn, R., Schiebler, W., Ouimet, C. & Greengard, P. A 38,000-dalton membrane protein (p38) present in synaptic vesicles. Proc. Natl. Acad. Sci. USA 82, 4137–4141 (1985).
Scott, D. A. et al. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J. Neurosci. 30, 8083–8095 (2010).
Vargas, K. J. et al. Synucleins Have Multiple Effects on Presynaptic Architecture. Cell Rep. 18, 161–173 (2017).
Froula, J. M. et al. alpha-Synuclein fibril-induced paradoxical structural and functional defects in hippocampal neurons. Acta Neuropathol. Commun. 6, 35 (2018).
Janezic, S. et al. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc. Natl. Acad. Sci. USA 110, E4016–E4025 (2013).
Erickson, J. D., Schafer, M. K., Bonner, T. I., Eiden, L. E. & Weihe, E. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc. Natl. Acad. Sci. USA 93, 5166–5171 (1996).
Umek, N., Gersak, B., Vintar, N., Sostaric, M. & Mavri, J. Dopamine Autoxidation is controlled by acidic pH. Front. Mol. Neurosci. 11, 467 (2018).
Hastings, T. G., Lewis, D. A. & Zigmond, M. J. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc. Natl. Acad. Sci. USA 93, 1956–1961 (1996).
Meiser, J., Weindl, D. & Hiller, K. Complexity of dopamine metabolism. Cell Commun. Signal. 11, 34 (2013).
Xu, H. & Yang, F. The interplay of dopamine metabolism abnormalities and mitochondrial defects in the pathogenesis of schizophrenia. Transl. Psychiatry 12, 464 (2022).
Choi, B. K. et al. Large alpha-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. USA 110, 4087–4092 (2013).
Cosme, C. V., Palissery, G. K. & Lerner, T. N. A dLight-ful New View of Neuromodulation. Trends Neurosci. 41, 566–568 (2018).
Patriarchi, T. et al. An expanded palette of dopamine sensors for multiplex imaging in vivo. Nat. Methods 17, 1147–1155 (2020).
Patriarchi, T. et al. Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science 360, https://doi.org/10.1126/science.aat4422 (2018).
Kreitzer, A. C. Physiology and pharmacology of striatal neurons. Annu. Rev. Neurosci. 32, 127–147 (2009).
Graveland, G. A. & DiFiglia, M. The frequency and distribution of medium-sized neurons with indented nuclei in the primate and rodent neostriatum. Brain Res. 327, 307–311 (1985).
Chuhma, N., Oh, S. J. & Rayport, S. The dopamine neuron synaptic map in the striatum. Cell Rep. 42, 112204 (2023).
Perreault, M. L., Hasbi, A., O’Dowd, B. F. & George, S. R. The dopamine d1-d2 receptor heteromer in striatal medium spiny neurons: evidence for a third distinct neuronal pathway in Basal Ganglia. Front. Neuroanat. 5, 31 (2011).
Varin, C., Cornil, A., Houtteman, D., Bonnavion, P. & de Kerchove d’Exaerde, A. The respective activation and silencing of striatal direct and indirect pathway neurons support behavior encoding. Nat. Commun. 14, 4982 (2023).
Nishi, A., Kuroiwa, M. & Shuto, T. Mechanisms for the modulation of dopamine d(1) receptor signaling in striatal neurons. Front. Neuroanat. 5, 43 (2011).
Klaus, A., Alves da Silva, J. & Costa, R. M. What, if, and when to move: basal ganglia circuits and self-paced action initiation. Annu. Rev. Neurosci. 42, 459–483 (2019).
Stern, S. et al. Reduced synaptic activity and dysregulated extracellular matrix pathways in midbrain neurons from Parkinson’s disease patients. NPJ Parkinsons Dis. 8, 103 (2022).
Virdi, G. S. et al. Protein aggregation and calcium dysregulation are hallmarks of familial Parkinson’s disease in midbrain dopaminergic neurons. NPJ Parkinsons Dis. 8, 162 (2022).
Mathis, A. et al. DeepLabCut: markerless pose estimation of user-defined body parts with deep learning. Nat. Neurosci. 21, 1281–1289 (2018).
Nath, T. et al. Using DeepLabCut for 3D markerless pose estimation across species and behaviors. Nat. Protoc. 14, 2152–2176 (2019).
Paxinos, G., Franklin, K. B. J. & Franklin, K. B. J. The mouse brain in stereotaxic coordinates. 2nd edn, (Academic Press, 2001).
Ikui, A. E., Ueki, N., Pecani, K. & Cross, F. R. Control of pre-replicative complex during the division cycle in Chlamydomonas reinhardtii. PLoS Genet. 17, e1009471 (2021).
Zhou, X. et al. Development of a genetically encoded sensor for probing endogenous nociceptin opioid peptide release. Nat. Commun. 15, 5353 (2024).
Evans, H. T. et al. Light-Activatable, cell-type specific labeling of the nascent proteome. ACS Chem. Neurosci. 15, 3473–3481 (2024).
Jakes, R. et al. Epitope mapping of LB509, a monoclonal antibody directed against human alpha-synuclein. Neurosci. Lett. 269, 13–16 (1999).
Russo, E. E. et al. Evaluation and validation of commercially available dopamine transporter antibodies. eNeuro 10, https://doi.org/10.1523/ENEURO.0341-22.2023 (2023).
Yan, Y. et al. Increased dopamine and its receptor dopamine receptor D1 promote tumor growth in human hepatocellular carcinoma. Cancer Commun. 40, 694–710 (2020).
Bhowmick, S. et al. Intercellular adhesion molecule-1-induced posttraumatic brain injury neuropathology in the prefrontal cortex and hippocampus leads to sensorimotor function deficits and psychological stress. eNeuro 8, https://doi.org/10.1523/ENEURO.0242-21.2021 (2021).
Yan, Y. et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160, 62–73 (2015).
She, X., Gao, Y., Zhao, Y., Yin, Y. & Dong, Z. A high-throughput screen identifies inhibitors of lung cancer stem cells. Biomed. Pharmacother. 140, 111748 (2021).
Illiano, P. et al. Recombinant adeno-associated virus-mediated rescue of function in a mouse model of dopamine transporter deficiency syndrome. Sci. Rep. 7, 46280 (2017).
Acknowledgements
This work was supported by the Canadian Institutes of Health Research (CIHR: PJT-162400) to A.O., (CIHR; PJT-159680) to T.D.P., (CIHR; PJT-186027) to A.S., the Natural Sciences and Engineering Research Council (NSERC; RGPIN-2023-05581) to A.O. A.O. was supported by Junior 2 salary Awards from the Fonds de Recherche du Québec-Santé (FRQS) and la Société Parkinson du Québec. A.S. holds a Canada Research Chair Tier 1 position.
Author information
Authors and Affiliations
Contributions
A.O. and A.S. conceived the study, designed the experiments, and supervised the project. R.R.A. collected and analyzed the in vivo data and data from iPSC-derived neurons. B.R.Q. contributed to data analysis. R.S. conducted the experiments involving iPSC-derived neuron generation. M.M. and T.D.P. collected and analyzed dopamine and dopamine metabolite data. R.R.A., A.O., and A.S. wrote the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rodriguez-Aller, R., Romero-Quineche, B., Morissette, M. et al. Optogenetic-induced α-synuclein accumulation reveals early synaptic dysfunction in experimental models of Parkinson’s disease. npj Parkinsons Dis. 11, 356 (2025). https://doi.org/10.1038/s41531-025-01201-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41531-025-01201-x
