
Abstract
Ulcerative colitis (UC) is a chronic inflammatory bowel disease characterized by mucosal inflammation, immune dysregulation, and gut microbiota dysbiosis. Using a dextran sulfate sodium (DSS) mouse model, we applied an integrated multi-omics approach comprising untargeted metabolomics, colonic transcriptomics, and 16S rRNA sequencing to evaluate the effects of the prebiotic stachyose (STA). STA supplementation alleviated colitis and coincided with marked increases in several medium-chain fatty acids (MCFAs), including azelaic acid and 3-hydroxyoctanoic acid. Transcriptomic profiling revealed modulation of fatty acid metabolism genes (e.g., Mfsd2a, Anxa1) and enrichment of pathways such as PPAR signaling and fatty acid degradation. Microbiota analyses showed enrichment of Ruminiclostridium_9, Roseburia, and Christensenellaceae, with partial restoration of microbial diversity and functions. Integrated correlations linked specific taxa, MCFAs, and host lipid-related genes. While associative and requiring mechanistic validation, these findings suggest colonic MCFAs as candidate mediators of the protective effects of STA in experimental colitis.
Similar content being viewed by others


Introduction
Ulcerative colitis (UC), a subtype of inflammatory bowel disease (IBD), is a chronic condition characterized by diffuse mucosal inflammation of the colon, leading to symptoms such as recurrent diarrhea, abdominal pain, rectal bleeding, and weight loss1,2, which collectively impair patients’ quality of life3. The etiology and pathogenesis of UC are complex and multifactorial, involving immune dysregulation4, genetic predisposition5, environmental factors6, dietary influences7, and significant dysbiosis of gut microbiota8. The incidence of UC is rising globally, particularly among young individuals, posing a significant public health challenge5.
Mounting evidence indicates that gut microbiota dysbiosis contributes to UC development and progression9. For instance, Lactobacillus acidophilus and Clostridium butyricum mitigate colitis in mouse models by enhancing gut barrier function and reducing inflammation10, while the probiotic mixture VSL#3 similarly reduces colonic inflammation and strengthens epithelial barrier integrity in experimental models11. Additionally, recent research highlights the therapeutic potential of Akkermansia muciniphila in improving UC outcomes12. In contrast, a reduction in key gut bacteria, such as Roseburia hominis and Faecalibacterium prausnitzii, is associated with UC development, underscoring their role in maintaining gut health13. Collectively, these findings highlight the critical importance of restoring gut microbiota balance for effective UC management, laying a solid foundation for novel therapeutic strategies like STA.
Stachyose (STA), a naturally occurring oligosaccharide abundant in legumes, has emerged as a candidate prebiotic14. STA resists digestion in the upper gastrointestinal tract and undergoes fermentation in the colon15,16,17, enriching beneficial taxa such as Bifidobacterium18 and Lactobacillus19. Our previous work demonstrated that STA supplementation alleviates DSS-induced colitis by modulating gut microbiota and enhancing butyrate–PPARγ signaling20. However, whether STA exerts broader metabolic effects beyond short-chain fatty acids (SCFAs) remains unclear.
Recent evidence suggests that medium-chain fatty acids (MCFAs; typically C6–C12) play underappreciated roles in gut homeostasis. Unlike SCFAs, MCFAs are rapidly oxidized by colonocytes and also act as signaling molecules involved in lipid metabolism and immune regulation21,22. For instance, azelaic acid and 3-hydroxyoctanoic acid have been implicated in anti-inflammatory pathways via PPAR activation or G-protein–coupled receptors23,24. These observations raise the possibility that diet–microbiota interactions could influence colonic MCFA pools and thereby modulate host responses during colitis.
In this study, we employed an integrated multi-omics strategy, including untargeted metabolomics, transcriptomics, and 16S rRNA sequencing, to investigate how STA modulates the microbiota metabolite host axis in a DSS-induced UC mouse model. We report that STA alleviates colitis symptoms, enriches SCFAs-producing microbes, and markedly elevates colonic levels of several MCFAs. These metabolic shifts are accompanied by transcriptional activation of lipid metabolism and PPAR signaling pathways. Together, our findings extend the understanding of STA anti-colitic mechanisms beyond SCFAs-mediated immune regulation and highlight MCFAs as potential mediators linking prebiotic fermentation to host metabolic protection.
Results
STA is associated with improvements in DSS-induced UC symptoms
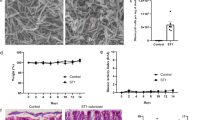
In our previous studies, we demonstrated that STA supplementation alleviated colitis features in mice20. In this study, we investigated the preventive and therapeutic effects of STA at a dose of 400 mg/kg/day in a DSS-induced colitis model (Fig. 1A). Mice were orally administered STA or vehicle for three weeks, with DSS exposure during the final week to induce colitis. As anticipated, the DSS challenge resulted in significant body weight loss (Fig. 1B), increased DAI (Fig. 1C), and pronounced colon shortening (Fig. 1D and Fig. S1A) compared to the negative control (NC) group, confirming the successful establishment of the UC model. STA supplementation coincided with noticeable mitigation of these adverse effects, indicating a potential protective role (Fig. 1B−D and Fig. S1A).

Schematic of experimental design. Male C57BL/6J mice (6 weeks old, n = 24) were acclimated for 1 week and then randomly assigned to three groups (n = 8 per group): NC (no treatment), DSS (2.5% DSS in drinking water during week 3), and STA (STA at 400 mg/kg/day orally for 3 weeks with DSS challenge during week 3) (A). Changes in body weight (B). DAI is evaluated daily based on weight loss, stool consistency, and fecal blood (C). Colon length at sacrifice (D). Representative colon sections and histological score of H&E staining (E, F). Concentrations of pro- and anti-inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-10) were measured in serum (G–J). In bar graphs, bar height represents the mean with error bars ± SD. In line graphs, each data point represents the mean with error bars ± SD. Statistical analysis was performed using one-way ANOVA with Tukey post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Histopathological analysis reinforced these observations. H&E staining revealed reduced inflammatory cell infiltration, partial restoration of crypt architecture, and improved mucosal integrity in STA-treated mice (Fig. 1E, F). Similarly, Alcian blue staining f demonstrated better preservation of goblet cell populations and more continuous mucin layer coverage compared to the pronounced mucosal atrophy and goblet cell loss in DSS-only mice (Fig. S1B, C). Cytokine profiling revealed that DSS exposure triggered robust elevations in pro-inflammatory cytokines (TNF-α, IL-1β, IL-6), while IL-10 levels showed no significant difference between the NC and DSS groups (Fig. 1G–J). Interestingly, STA supplementation markedly upregulated IL-10 compared with both NC and DSS groups, suggesting a shift toward a more anti-inflammatory cytokine environment. Together, these data indicate that STA administration coincides with attenuation of inflammatory damage and partial restoration of mucosal immune balance in DSS-induced UC.
STA is associated with broad metabolic shifts, including increased MCFAs
Given the close interplay between gut microbiota and metabolites in maintaining colonic homeostasis, we hypothesized that STA supplementation may reshape metabolic pathways disrupted by DSS-induced colitis. To test this, we performed untargeted fecal metabolomics using LC-MS/MS. Supervised PLS-DA revealed clear separation between DSS and STA groups in both positive (Fig. 2A) and negative (Fig. 2B) ion modes, indicating substantial remodeling of the fecal metabolome with STA treatment. Differential analysis identified 591 upregulated and 230 downregulated metabolites in positive mode, and 240 upregulated and 79 downregulated metabolites in negative mode (|log2FC | > 0.5, p < 0.05; Fig. 2C, D). Pathway enrichment analysis of upregulated metabolites (VIP > 1, |log2FC | > 0.5, p < 0.05) revealed involvement of multiple metabolic pathways, including fatty acid biosynthesis, vitamin B6 metabolism, tryptophan metabolism, bile acid biosynthesis, and purine metabolism (Fig. 2E and Fig. S2A). Notably, fatty acid biosynthesis ranked among the most enriched pathways, prompting further investigation of fatty acid dynamics. Heatmap visualization of differentially expressed organic acids (Fig. 2F) highlighted clear clustering and notable remodeling of organic acid profiles following STA treatment, with key metabolites validated by VIP rankings (Fig. S2B–E).

PLS-DA score plots in positive (A) and negative (B) ion modes showing clear separation between DSS and STA groups. Volcano plots of differentially abundant metabolites in positive (C) and negative (D) ion modes. Red dots represent significantly upregulated metabolites, blue dots represent significantly downregulated metabolites (|log₂FC | > 0.5, p < 0.05, Wilcoxon rank-sum test). RAMDB enrichment analysis of significantly altered metabolites (VIP > 1, |log₂FC | > 0.5, p < 0.05, Wilcoxon rank-sum test), highlighting fatty acid biosynthesis and other enriched pathways (E). Heatmap of differentially expressed organic acids, including fatty acids, between DSS and STA groups showing distinct clustering patterns (F).
To better understand the nature of these changes, differentially expressed fatty acids were classified by chain length (Fig. 3A). MCFAs emerged as the most consistently upregulated group in STA-treated mice. Several of these MCFAs have been reported to exert bioactive effects. Azelaic acid is clinically used for inflammatory skin conditions17; 3-hydroxyoctanoic acid functions as an HCA3 receptor agonist with immunomodulatory properties25; traumatic acid exhibits antioxidant activity and promotes collagen synthesis26; and suberic acid has been linked to enhanced collagen production27. These functional attributes suggest potential roles in mucosal repair and immune modulation. To explore relationships between these metabolites and UC phenotypes, Spearman correlation analyses (|r| > 0.6, p < 0.05) were performed (Fig. 3B). Most upregulated MCFAs showed positive correlations with colon length, body weight, Alcian blue staining intensity, and serum IL-10 levels in STA-treated mice. Conversely, these metabolites were negatively correlated with pro-inflammatory cytokines (IL-1β, IL-6, TNF-α), and some, such as traumatic acid, suberic acid, and dodecanedioic acid, were inversely associated with DAI scores. Taken together, these findings demonstrate that STA supplementation is associated with increased levels of MCFAs that track closely with improved histological, physiological, and inflammatory readouts. While causality cannot be inferred, these metabolites emerge as strong candidates for further mechanistic exploration.

STA supplementation coincides with increased MCFAs that correlate with improved UC phenotypes. Heatmap showing differential fatty acids classified by chain length between DSS and STA groups, with marked upregulation of MCFAs following STA treatment (Wilcoxon rank-sum test). SCFA, short-chain fatty acid; MCFA, medium-chain fatty acid; LCFA, long-chain fatty acid; VLCFA, very long-chain fatty acid (A). Spearman correlation heatmap between MCFAs and colitis-related phenotypic indicators, including colon length, body weight, histological scores, and serum cytokines. Red and blue colors indicate positive and negative correlations, respectively (B). *p < 0.05, **p < 0.01, and ***p < 0.001.
STA impacts the transcriptome of mice with DSS-induced UC
To further investigate the molecular changes associated with STA treatment in UC, we conducted RNA sequencing on the intestinal epithelium of mice (|log2FC | > 1, p-value < 0.05), identifying 832 upregulated and 319 downregulated genes (Fig. 4A). Hierarchical clustering of these DEGs demonstrated clear separation between STA and DSS groups, reflecting distinct gene expression patterns induced by STA (Fig. 4B). Subsequently, GOBP enrichment analysis (Fig. 4C) revealed significant enrichment of pathways related to fatty acid metabolism, such as fatty acid biosynthetic processes, unsaturated fatty acid biosynthetic processes, long-chain fatty acid transport, and fatty acid transport. These findings suggest that STA treatment is associated with alterations in fatty acid-related metabolic processes in the colonic epithelium of DSS-induced mice. Additionally, Fig. 4D illustrates the expression patterns of fatty acid metabolism-related DEGs, showing upregulation of genes such as Mfsd2a and Anxa1, alongside downregulation of Fabp7 and Cyp2e1. KEGG enrichment analysis (Fig. 4E) further highlighted pathways such as the PPAR signaling pathway, fatty acid degradation, and fatty acid metabolism. To explore potential links between MCFAs and transcriptomic changes, we performed a Spearman correlation analysis between MCFAs levels and fatty acid metabolism-related DEGs (Fig. 4F). The hierarchical clustering heatmap revealed significant positive correlations between Mfsd2a and Anxa1 with azelaic acid, 3-hydroxyoctanoic acid, and 11-aminoundecanoic acid. In contrast, genes such as Cyp2e1, Adipoq, Hadh, Aldh1b1, and Fmo1 exhibited negative correlations with MCFAs. These transcriptomic and correlation patterns highlight a close association between STA treatment, MCFAs levels, and lipid metabolism-related pathways, suggesting that lipid metabolic reprogramming may be a key feature of the host response to STA in DSS-induced colitis.

Volcano plot of the DEGs DSS and STA groups (|log2FC | > 1, p-value < 0.05, unpaired Student’s t-test). In total, 832 genes were upregulated and 319 downregulated (A). Heatmap of DEGs showing distinct clustering of STA and DSS groups, indicating clear transcriptomic separation (B). GOBP pathway enrichment analysis of upregulated DEGs highlighting fatty acid biosynthetic and transport processes (C). Expression patterns of representative fatty acid-related DEGs between groups (unpaired Student’s t-test) (D). KEGG pathway enrichment analysis of fatty acid-related DEGs, indicating enrichment in PPAR signaling, fatty acid degradation, and fatty acid metabolism (E). Spearman correlation heatmap of fatty acid-related genes and MCFAs (F), with positive correlations in red and negative in blue; color intensity corresponds to the Spearman correlation coefficient (r). *p < 0.05, **p < 0.01, and ***p < 0.001.
STA modulated the gut microbiota composition of mice with DSS-induced UC
Given that STA is a prebiotic and the development of UC is closely associated with gut microbiota dysbiosis20, we investigated the impact of STA on the gut microbiota by performing 16S rRNA gene sequencing on fecal samples. Principal coordinate analysis (PCoA) based on distance metrics revealed clear separation of microbial communities among the NC, DSS, and STA groups (Fig. 5A), indicating that DSS-induced colitis and STA treatment are associated with substantial alterations in the overall microbial composition. To further evaluate these changes, α-diversity was assessed using ACE, Chao1, Simpson, and Shannon indices (Fig. 5B, C and Fig. S3A, B). Consistent with previous findings, DSS exposure significantly reduced microbial richness and evenness, reflecting a state of dysbiosis. Interestingly, STA treatment partially restored these indices, suggesting a partial rebalancing of microbial diversity, even though diversity levels did not completely return to those of the NC group. At the phylum level (Fig. 5D), DSS markedly increased the abundance of Bacteroidetes (Fig. 5E) while reducing Firmicutes (Fig. 5F) and Saccharibacteria (Fig. 5G); STA treatment appeared to counteract these shifts to some extent. Similarly, the Firmicutes/Bacteroidetes (F/B) ratio, a recognized marker of gut health28, was reduced in the DSS group and showed a non-significant upward trend following STA administration (Fig. 5H). Analyses at the class, order, and family levels (Fig. S4A–C) revealed consistent patterns, further illustrating the broad regulatory effects of STA on the microbial community. Differential abundance analysis using LEfSe (Fig. 5I, J) and a taxonomic heatmap (Fig. S4D) identified 56 bacterial taxa with significant shifts across the groups. Among these, several taxa associated with fiber degradation and beneficial metabolic functions, such as Ruminiclostridium_9 and Acetatifactor, were enriched in the STA group. Conversely, inflammation-associated taxa, including Alloprevotella, Parasutterella, and Ruminococcus_1, were enriched in the DSS group. These compositional patterns suggest that STA administration is associated with a shift toward a microbial community that more closely resembles a healthy gut profile.

PCoA analysis of β-diversity using robust Aitchison distance showing distinct microbial community separation (A). α-diversity indices (ACE and Chao1), reflecting microbial richness across NC, DSS, and STA groups (B, C). Relative abundances of phylum-level gut microbiota (D). Relative abundance of Bacteroidetes (E) Firmicutes (F), and Saccharibacteria (G). Firmicutes/Bacteroidetes ratio (H). I Differentially enriched gut microbiota identified by LEfSe (LDA > 2.0). J Cladogram from LEfSe analysis illustrating taxonomic differences among groups. Data are presented as mean ± SD. Statistical analysis was performed using one-way ANOVA with Tukey post hoc test. *p < 0.05, **p < 0.01, and ***p < 0.001.
Because our metabolomics and transcriptomics analyses highlighted substantial changes in fatty acid metabolism, we next investigated whether microbial functional capacities might also shift in this direction. Functional predictions using PICRUSt2 revealed alterations in microbial pathways related to fatty acid metabolism (Fig. 6A–D). Specifically, enzymes such as K13767 were downregulated, while others, including K00079, K01054, and K00801, each associated with lipid metabolism, were upregulated in the STA group. This suggests that STA supplementation may be linked to changes in microbial metabolic potential relevant to fatty acid handling. Finally, we performed Spearman correlation analysis (|r | > 0.6, p < 0.05) between the levels of differentially abundant MCFAs and microbial taxa (Fig. 6E). Positive correlations were observed between Ruminiclostridium_9, Roseburia, Christensenellaceae, and Acetatifactor with MCFAs such as 3-hydroxyoctanoic acid and 3-oxolauric acid, whereas Coriobacteriaceae_UCG-002, Alloprevotella, and Ruminococcus_1 showed significant negative correlations. These associations highlight a potential linkage between STA-mediated microbiota shifts and the altered MCFAs profiles, providing clues for future functional studies aimed at dissecting microbial contributions to host metabolic outcomes.

Functional prediction of fatty acid metabolism-related enzymes using PICRUSt2 showing altered abundances of K13767, K00079, K01054, and K00801 (A–D). Spearman correlation heatmap of differentially abundant bacterial taxa and MCFAs (E), with positive correlations in red and negative in blue; values represent Spearman correlation coefficients (r). Data are presented as mean ± SD. Statistical analysis was performed using one-way ANOVA with Tukey post hoc test. *p < 0.05, **p < 0.01, and ***p < 0.001.
Systematic molecular correlations across multi-omics integrating STA-mediated changes in microbiome, metabolome, and host transcriptome
To gain a deeper systems-level perspective, we integrated the microbiome, metabolome, and transcriptome datasets to identify core interactions associated with STA treatment. In the microbiota–metabolite correlation network (Fig. 7A), 13 MCFAs and 18 bacterial taxa formed interconnected nodes, with taxa such as Ruminiclostridium_9, Roseburia, and Ruminococcus_1 showing strong positive correlations with several MCFAs. These co-occurrence patterns suggest that certain bacterial taxa may play a central role in shaping the observed metabolite landscape. In the metabolite–gene correlation network (Fig. 7B), 12 MCFAs were associated with 13 host genes involved in fatty acid metabolism. Notable examples include positive correlations between Anxa1 and Mfsd2a with azelaic acid, 3-hydroxyoctanoic acid, and 11-aminoundecanoic acid, and negative correlations between Fabp7 and traumatic acid. These relationships suggest that specific lipid metabolism genes may respond in parallel with shifts in colonic MCFAs, though causality cannot be inferred from these data. To visualize these integrated relationships, we constructed a Sankey plot (Fig. 7C) incorporating 17 bacterial taxa, 11 MCFAs, and 13 fatty acid metabolism-related DEGs. This comprehensive view underscored the co-variation between microbial taxa such as Ruminiclostridium_9, Roseburia, Alloprevotella, and Ruminococcus_1 with key MCFAs and lipid-related host genes, including Fmo1, Fabp7, Anxa1, Ehhadh, Mfsd2a, and Cyp1b1.

Correlation network of core gut microbiota (hexagons) and MCFAs (circles) (A). Correlation network of core host genes (diamonds) and MCFAs (circles) (B). Positive and negative associations are represented by red and blue lines, respectively. Sankey plot integrating gut microbiota, MCFAs, and host genes, showing directional interactions within the STA-mediated regulatory axis (C). *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Taken together, these multi-omics correlations outline a framework in which STA treatment is consistently associated with coordinated shifts in microbiota composition, MCFAs profiles, and host lipid metabolism pathways. While these findings do not establish causation, they generate a testable hypothesis that microbiota–metabolite–host interactions could be a critical component of the physiological response to STA (Fig. 8).

STA supplementation remodels gut microbiota, reprograms microbial fatty acid metabolism, increases MCFAs, and activates fatty acid-related host genes such as Mfsd2a and Anxa1. These interactions restore mucosal homeostasis, enhance anti-inflammatory IL-10, and suppress pro-inflammatory cytokines, collectively alleviating DSS-induced colitis.
Discussion
Ulcerative colitis (UC) is a chronic inflammatory bowel disease characterized by mucosal inflammation, immune dysregulation, and gut microbiota imbalance, affecting millions of people worldwide5,29. Patients frequently experience relapsing symptoms such as diarrhea, abdominal pain, and rectal bleeding, which severely impair quality of life. Current therapies, including immunosuppressants, remain limited in efficacy and are associated with significant adverse effects30. Recent studies have highlighted the complex interplay between the gut microbiota, microbial metabolites, and intestinal immune homeostasis, suggesting that prebiotics may offer a dietary means to modulate these interactions and relieve symptoms31,32,33. Notably, levels of certain metabolites, including medium-chain fatty acids (MCFAs), are significantly reduced in UC patients and are associated with more severe inflammation34.
Building on this context, our integrated multi-omics analysis reveals a novel protective mechanism of stachyose (STA) in DSS-induced colitis. Unlike our previous work focusing on a butyrate–PPARγ axis20, the present study demonstrates that STA treatment markedly elevated colonic levels of several MCFAs (such as azelaic acid and 3-hydroxyoctanoic acid) and upregulated host genes related to lipid metabolism and PPAR signaling. MCFAs can enter colonocyte mitochondria to serve as efficient energy substrates and signaling molecules21,24,34,35,36, a role that complements the established immunomodulatory actions of short-chain fatty acids (SCFAs). Transcriptomic enrichment analyses further indicated activation of PPAR-regulated lipid oxidation pathways in STA-treated mice. Thus, beyond the known SCFAs-driven immune pathways, our findings suggest a parallel microbiota–MCFAs–host lipid metabolism axis through which STA may reprogram gut fermentation to favor MCFAs accumulation while enhancing host lipid-handling capacity.
The multi-omics data also hint that gut microbes might contribute to the MCFAs increase, although causality is unproven. STA treatment promoted the enrichment of several fiber-degrading, SCFAs-producing bacteria (such as Roseburia37 and Ruminiclostridium38,39), while simultaneously suppressing inflammation-associated taxa, including Parasutterella40, Alloprevotella41, and Coriobacteriaceae_UCG-00242. These compositional shifts positively correlated with colonic MCFAs levels, a co-variation reminiscent of prior observations linking gut microbiota profiles with SCFAs/MCFAs pools43,44. Our correlation network analysis highlighted core taxa (such as Roseburia and Ruminiclostridium_9) tightly associated with STA-induced MCFAs. However, there is currently no direct evidence that typical gut bacteria produce these specific MCFAs, so the microbial contribution remains speculative.
Nevertheless, the literature supports a potentially protective role for MCFAs in the colon. Notably, caprylate (C8:0) and related MCFAs are rapidly oxidized by colonocytes45 and induce energy-metabolism genes (such as Fabp2 and Pdk4) and PPARγ expression20,24,34,46,47,48. In agreement, we observed that STA-induced MCFAs (azelaic acid, 3-hydroxyoctanoic acid) correlated with increased expression of epithelial-protective genes (such as Mfsd2a and Anxa1) and with activation of the PPAR pathway. Moreover, 3-hydroxyoctanoic acid is known to be an endogenous ligand for the human HCA3 receptor49, which mediates anti-inflammatory signaling in innate immune cells. Collectively, these reports and our associations suggest that raising colonic MCFAs may help alleviate inflammation by providing additional fuel to epithelial cells and engaging PPARγ-dependent anti-inflammatory mechanisms.
Taken together with previous findings, our results suggest that STA-driven prebiotic fermentation does not simply increase the overall output of SCFAs but also alters the metabolite profile toward higher levels of MCFAs. In other words, STA may lead the microbiota to generate a different mix of SCFAs and MCFAs rather than an exclusive increase in SCFAs. Emerging evidence indicates that combinations of SCFAs and MCFAs can act synergistically to support gut health, for instance, by improving resistance to pathogens44, and that longer-chain fatty acids often exert stronger immunopathological effects compared with shorter-chain fatty acids50. By promoting the production of both SCFAs and MCFAs, STA could provide the intestinal epithelium with additional energy substrates and signaling lipids (such as azelaic acid and 3-hydroxy fatty acids)23,24 that facilitate mucosal repair. From this perspective, evaluating the balance between SCFAs and MCFAs, rather than SCFAs alone, may uncover new aspects of how gut metabolic homeostasis relates to inflammation resolution.
Importantly, we have not directly traced the source of the elevated MCFAs. Dietary fats, host lipid metabolism, and microbial fermentation all can contribute to MCFA pools21,51,52,53, and these sources cannot currently be distinguished in vivo. Recent studies even indicate that the capacity of gut bacteria to produce MCFAs in situ is unclear45. Thus, the STA-induced increases in azelaic acid and 3-hydroxyoctanoic acid could originate from microbial fermentation, host β-oxidation, or both. Still, several considerations hint that microbes might play a role: for example, MCFAs are rapidly absorbed in the gut, so dietary caprylate is unlikely to reach the cecum in appreciable amounts21,22,24,36,43. In addition, the main component of bedding, cellulose54, does not stimulate the production of either SCFAs or MCFAs45. Some yeast strains can produce caprylate, and certain anaerobic bacteria (such as Eubacterium limosum and related Clostridia)22 are known to ferment short substrates into MCFAs under strict anaerobic conditions. Thus, it is plausible that STA shifts gut fermentation such that more carbon is channeled into MCFAs production, but this hypothesis awaits direct testing. Moreover, a key limitation of this study is that we did not experimentally validate the functional role of these elevated MCFAs in ameliorating colitis. Although their correlations with epithelial-protective genes and PPAR signaling are intriguing, causality has not been established. Future targeted studies, such as isotope tracing, germ-free or defined-consortium studies, and direct MCFA supplementation or inhibition, will be necessary to determine the microbial versus host origins of these metabolites and to test their causal roles.
In summary, our multi-omics analysis suggests that STA supplementation alleviates DSS-induced colitis in part by elevating intestinal MCFAs and activating host lipid metabolism pathways. This effect complements the previously identified butyrate–Treg/PPARγ axis20 and broadens our understanding of how prebiotics can shape host–microbe metabolic interactions. While causality remains to be established, the concurrent enrichment of SCFAs-producing taxa, the rise in MCFAs, and the upregulation of PPAR-related genes point to a cooperative network of microbial and host processes. Overall, these findings underscore the complex interplay between diet, microbiota, and host metabolism in UC, and highlight MCFAs as candidate mediators of the protective effects of stachyose.
Methods
Compliance with ethics requirements
All experimental procedures involving animals were approved by the Ethics Committee of Qingdao Medical College at Qingdao University (QDU-AEC-2024461), in accordance with the institutional guidelines for the ethical use of animals.
Materials
DSS (molecular weight 36–50 kDa, catalog no. 160110) was purchased from MP Biomedicals (Santa Ana, CA, USA). STA (purity >80% by HPLC) was obtained from Solarbio Life Sciences (Beijing, China). ELISA kits for TNF-α, IL-1β, IL-6, and IL-10 were acquired from Thermo Fisher Scientific (Waltham, MA, USA).
Animal study
Male C57BL/6J mice (6 weeks old, n = 24) were obtained from Jinan Pengyue Laboratory Animal Breeding Co., Ltd. (Jinan, China) and acclimated for 7 days in a specific pathogen-free facility under standard conditions (temperature: 22 ± 2 °C; humidity: 50–60%; 12-h light/dark cycle; ad libitum access to standard chow and water). Following acclimation, the mice were randomly allocated into three groups (n = 8 per group): (1) NC group, receiving no additional treatment for 3 weeks; (2) DSS group, exposed to DSS-induced colitis during the third week; and (3) STA group, administered STA at 400 mg/kg/day orally for 3 weeks, with DSS-induced colitis initiated in the third week. Colitis was induced by providing 2.5% (w/v) DSS in the drinking water for 7 days during the third week. The Disease Activity Index (DAI) was assessed daily by evaluating body weight changes, stool consistency, and fecal blood presence to monitor the severity of colitis. At the end of the experiment, mice were anesthetized with inhaled isoflurane (#R510-22-10, Shenzhen Ruiwode Life Technology, Shenzhen, China). Blood samples were then collected via retro-orbital puncture, followed by euthanasia via cervical dislocation. The colon was subsequently obtained for length measurement and histological examination.
Histological assessment
Colon tissue segments were fixed in 10% formalin, then embedded in paraffin and sectioned into 5 μm slices. For morphological assessment, sections were stained with hematoxylin and eosin (H&E). Additional paraffin sections were dewaxed and stained with Alcian blue. Both H&E- and Alcian blue-stained sections were observed using a light microscope (OLYMPUS, Tokyo, Japan) at magnifications of 40× and 200×. Inflammation severity in H&E-stained sections was scored in a blinded manner using a validated histological scoring system. Alcian blue staining was quantitatively analyzed using ImageJ software.
Measurement of inflammatory cytokines in serum
Blood samples were processed by centrifugation at 3000 rpm for 15 min at 4 °C to isolate serum. Concentrations of inflammatory cytokines (TNF-α, IL-1β, IL-6, and IL-10) were determined using ELISA kits in accordance with the protocol provided by the manufacturer.
Untargeted metabolomics analysis
Fecal samples (100 mg) were mixed with 1 mL of a pre-chilled extraction solvent consisting of methanol, acetonitrile, and water (2:2:1, v/v), then processed through freeze-thaw cycles, homogenization, vortex mixing, and ultrasonication for 10 min at 4 °C. To precipitate proteins, the mixture was incubated at −20 °C for 1 h and subsequently centrifuged at 13,000 rpm for 15 min at 4 °C. The supernatant (1 mL) was dried under vacuum, reconstituted in 150 µL of acetonitrile/water (1:1), sonicated at 4 °C for 10 min, and centrifuged again under the same conditions.
Metabolomic profiling was conducted using a Thermo Scientific UHPLC system equipped with a T3 column coupled to a Thermo QExactive mass spectrometer. The mobile phases were configured as follows. In positive ion mode, acetonitrile (A) and 0.1% formic acid in water (B) were used, while in negative ion mode, acetonitrile (A) and 5 mM ammonium acetate in water (B) were employed. The gradient elution profile was set as follows: 0–1 min, 1% B; 1–13 min, linear increase to 99% B; 13–16 min, maintained at 99% B; 16–16.5 min, decreased to 1% B; and 16.5–20 min, equilibrated at 1% B. The flow rate was 0.25 mL/min, with injection volumes of 3 µL in positive mode and 5 µL in negative mode.
H-ESI source was operated with the following parameters: spray voltage at 3.5 kV and −2.8 kV, capillary temperature at 320 °C, probe heater temperature at 300 °C, sheath gas flow at 40 units, auxiliary gas flow at 10 units, and S-lens RF level at 50. Mass spectrometry data were collected in full scan mode over a mass range of m/z 70–1000 at a resolution of 140,000 fwhm, with data-dependent MS/MS scans at 17,500 fwhm resolution for up to eight precursor ions, using collision energies of 20, 35, and 50 eV. Both positive and negative ionization modes were utilized, employing Full MS and data-dependent MS (ddMS) acquisition modes.
Transcriptome analysis
Colon tissue (150 mg) was pulverized, and total RNA was isolated using an RNA extraction kit (#AC0202, Sparkjade). RNA integrity and quality were evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The RNA library was prepared, and its purity and concentration were assessed using an Agilent 2100 Bioanalyzer and a Quant-iT PicoGreen dsDNA Assay Kit. Single-end sequencing of the quantified library was performed on an Illumina Genome Analyzer platform (BerryGenomics Co., Ltd., Beijing, China). Differentially expressed genes (DEGs) were identified using R software. A volcano plot was generated to visualize gene expression differences, with DEGs defined by the criteria of |log2FC | > 1 and p-value < 0.05. For functional annotation, DEGs were mapped to the KEGG database (https://www.genome.jp/kegg/) to identify enriched pathways. GO analysis was conducted using the R package “org.Hs.eg.db” as the reference annotation set. Enrichment analysis of the DEGs was performed with the R package “clusterProfiler”, with statistical significance set at p < 0.05 and a false discovery rate (FDR) < 0.25.
16S rRNA sequencing and PICRUSt2 functional prediction
Each mouse was housed individually in separate cages and allowed to defecate naturally. Fresh fecal pellets were collected directly from individual mice immediately after defecation to avoid contamination. During collection, mice were placed in clean, sterile cages without bedding, and fecal pellets were picked up using sterile forceps. Each sample was transferred into a pre-labeled sterile cryogenic tube, snap-frozen in liquid nitrogen (or placed on dry ice), and then stored at −80 °C until further analysis. Total genomic DNA was isolated from the fecal samples using the MagPure Stool DNA KF Kit B (product number MD5115) from Magen Company, Guangzhou, with DNA quality and integrity verified by agarose gel electrophoresis. The V3-V4 region of the bacterial 16S rRNA gene was amplified using PCR, followed by the preparation of a sequencing library.
The valid sequences obtained were processed using QIIME2 software to merge overlapping sequences. Amplicon sequence variants (ASVs) were identified using a denoising approach, and representative sequences were selected for further analysis. The taxonomic classification of ASVs was performed against the SILVA 16S rRNA reference database (version 123) using the NovoMagic cloud platform (Shenzhen, Guangdong, China). Functional profiles of the microbial communities were inferred using PICRUSt2, with ASVs mapped to the Greengenes database (version 13.5) to predict KEGG orthology-based metabolic pathways.
Statistical analysis
Data analysis was performed using GraphPad Prism and R software. Results are presented as mean ± standard deviation (SD). Normality of data distribution was assessed using the Shapiro–Wilk test. Comparisons between two groups were performed using an unpaired Student’s t-test, while one-way analysis of variance (ANOVA) followed by Tukey’s Honestly Significant Difference (HSD) test was used for comparisons among three or more groups. Relationships between variables were assessed using Spearman's rank correlation analysis. Statistical significance was defined as p-value < 0.05 across all analyses.
Data availability
The authors confirm that the datasets supporting the findings of this study are publicly accessible at the GitHub repository: https://github.com/KIMGENSOO/UC-STA/tree/master. The bioinformatics analysis scripts used in this study are available from the lead author upon reasonable request. Additional information necessary for reanalyzing the data presented in this manuscript can be obtained from the corresponding author upon reasonable request.
References
Kobayashi, T. et al. Ulcerative colitis. Nat. Rev. Dis. Prim. 6, 74 (2020).
Ungaro, R., Mehandru, S., Allen, P. B., Peyrin-Biroulet, L. & Colombel, J. F. Ulcerative colitis. Lancet 389, 1756–1770 (2017).
Voelker, R. What is ulcerative colitis?. JAMA 331, 716 (2024).
Porter, R. J., Kalla, R. & Ho, G. T. Ulcerative colitis: recent advances in the understanding of disease pathogenesis. F1000Research https://doi.org/10.12688/f1000research.20805.1 (2020).
Le Berre, C., Honap, S. & Peyrin-Biroulet, L. Ulcerative colitis. Lancet 402, 571–584 (2023).
de Souza, H. S. & Fiocchi, C. Immunopathogenesis of IBD: current state of the art. Nat. Rev. Gastroenterol. Hepatol. 13, 13–27 (2016).
Keshteli, A. H., Madsen, K. L. & Dieleman, L. A. Diet in the pathogenesis and management of ulcerative colitis; a review of randomized controlled dietary interventions. Nutrients https://doi.org/10.3390/nu11071498 (2019).
Wu, X., Zhang, T., Zhang, T. & Park, S. The impact of gut microbiome enterotypes on ulcerative colitis: identifying key bacterial species and revealing species co-occurrence networks using machine learning. Gut Microbes 16, 2292254 (2024).
Khan, I. et al. Alteration of gut microbiota in inflammatory bowel disease (IBD): cause or consequence? IBD treatment targeting the gut microbiome. Pathogens https://doi.org/10.3390/pathogens8030126 (2019).
Wang, Y. et al. Lactobacillus acidophilus and Clostridium butyricum ameliorate colitis in murine by strengthening the gut barrier function and decreasing inflammatory factors. Benef. Microbes 9, 775–787 (2018).
Kumar, M., Kissoon-Singh, V., Coria, A. L., Moreau, F. & Chadee, K. Probiotic mixture VSL#3 reduces colonic inflammation and improves intestinal barrier function in Muc2 mucin-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 312, G34–G45 (2017).
Xue, L. et al. The effects of live and pasteurized Akkermansia muciniphila on DSS-induced ulcerative colitis, gut microbiota, and metabolomics in mice. Food Funct. 14, 4632–4646 (2023).
Machiels, K. et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 63, 1275–1283 (2014).
Yadav, M. K., Kumari, I., Singh, B., Sharma, K. K. & Tiwari, S. K. Probiotics, prebiotics and synbiotics: Safe options for next-generation therapeutics. Appl. Microbiol. Biotechnol. 106, 505–521 (2022).
Zaman, S. A. & Sarbini, S. R. The potential of resistant starch as a prebiotic. Crit. Rev. Biotechnol. 36, 578–584 (2016).
Ashaolu, T. J., Ashaolu, J. O. & Adeyeye, S. A. O. Fermentation of prebiotics by human colonic microbiota in vitro and short-chain fatty acids production: a critical review. J. Appl. Microbiol. 130, 677–687 (2021).
Liao, Y. et al. Azelaic acid regulates the renin-angiotensin system and improves colitis based on network pharmacology and experimentation. ACS Omega 8, 15217–15228 (2023).
Mu, W. et al. Taeniasis impacts human gut microbiome composition and function. ISME J. https://doi.org/10.1093/ismejo/wrae213 (2024).
He, L. et al. Stachyose modulates gut microbiota and alleviates dextran sulfate sodium-induced acute colitis in mice. Saudi J. Gastroenterol. 26, 153–159 (2020).
He, N. et al. Stachyose exerts anticolitis efficacy by re-balancing Treg/Th17 and activating the butyrate-derived PPARγ signaling pathway. J. Agric. Food Chem. 72, 12171–12183 (2024).
Zhang, R. et al. Inducible adhesion and biofilm formation in Salmonella linked to adaptive fatty acid metabolism. Food Res. Int. 206, 116006 (2025).
Fan, L. et al. Ca:Mg ratio, medium-chain fatty acids, and the gut microbiome. Clin. Nutr. 41, 2490–2499 (2022).
Eschrig, S. et al. LORE receptor homomerization is required for 3-hydroxydecanoic acid-induced immune signaling and determines the natural variation of immunosensitivity within the Arabidopsis genus. N. Phytol. 242, 2163–2179 (2024).
Huang, L., Gao, L. & Chen, C. Role of medium-chain fatty acids in healthy metabolism: a clinical perspective. Trends Endocrinol. Metab. 32, 351–366 (2021).
Peters, A. et al. Hydroxycarboxylic acid receptor 3 and GPR84 - Two metabolite-sensing G protein-coupled receptors with opposing functions in innate immune cells. Pharmacol. Res. 176, 106047 (2022).
Gao, J. et al. Traumatic acid inhibits ACSL4 associated lipid accumulation in adipocytes to attenuate high-fat diet-induced obesity. FASEB J. 37, e23278 (2023).
Kang, W., Choi, D., Son, B., Park, S. & Park, T. Activation of OR10A3 by suberic acid promotes collagen synthesis in UVB-irradiated dermal fibroblasts via the cAMP-Akt pathway. Cells https://doi.org/10.3390/cells11243961 (2022).
Stojanov, S., Berlec, A. & Štrukelj, B. The influence of probiotics on the firmicutes/bacteroidetes ratio in the treatment of obesity and inflammatory bowel disease. Microorganisms https://doi.org/10.3390/microorganisms8111715 (2020).
Lewis, J. D. et al. Incidence, prevalence, and racial and ethnic distribution of inflammatory bowel disease in the United States. Gastroenterology 165, 1197–1205.e1192 (2023).
Gros, B. & Kaplan, G. G. Ulcerative colitis in adults: a review. JAMA 330, 951–965 (2023).
He, N. et al. Gut commensal antibiotic-resistant Parabacteroides goldsteinii ameliorates mouse colitis through valine-isobutyrate metabolism. Research https://doi.org/10.34133/research.0867.
Li, X. et al. Therapeutic effect of Faecalibacterium longum CM04-06 on DSS-induced ulcerative colitis in mice. J. Appl. Microbiol. https://doi.org/10.1093/jambio/lxaf119 (2025).
Mu, M. et al. Multiomics analysis of bacteroides cellulosilyticus anticolitis via gut microbiota metabolite-mediated PI3K-Akt signaling pathway. J. Agric. Food Chem. 73, 16333–16347 (2025).
De Preter, V. et al. Faecal metabolite profiling identifies medium-chain fatty acids as discriminating compounds in IBD. Gut 64, 447–458 (2015).
Chen, Q. et al. Zinc laurate protects against intestinal barrier dysfunction and inflammation induced by ETEC in a mice model. Nutrients https://doi.org/10.3390/nu15010054 (2022).
Schönfeld, P. & Wojtczak, L. Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J. Lipid Res. 57, 943–954 (2016).
Scarborough, M. J. et al. Increasing the economic value of lignocellulosic stillage through medium-chain fatty acid production. Biotechnol. Biofuels 11, 200 (2018).
Dai, M. et al. Caproicibacter sp. BJN0012, a potential new species isolated from cellar mud for caproic acid production from glucose. J. Biotechnol. 388, 11–23 (2024).
Yue, F. et al. Constant light exposure alters gut microbiota and short-/medium-chain fatty acids and aggravates PCOS-like traits in HFD-fed rats. Obesity 30, 694–706 (2022).
Sun, J. et al. Ribonuclease 4 functions as an intestinal antimicrobial protein to maintain gut microbiota and metabolite homeostasis. Nat. Commun. 15, 5778 (2024).
Huang, L. et al. 5-Aminosalicylic acid ameliorates dextran sulfate sodium-induced colitis in mice by modulating gut microbiota and bile acid metabolism. Cell. Mol. Life Sci. 79, 460 (2022).
Tang, Q. et al. Endogenous Coriobacteriaceae enriched by a high-fat diet promotes colorectal tumorigenesis through the CPT1A-ERK axis. NPJ Biofilms Microbiomes 10, 5 (2024).
Fan, Z. et al. Dietary fatty acids promote gut health in weaned piglets by regulating gut microbiota and immune function. Front. Microbiol. 16, 1558588 (2025).
Kumar, A. et al. Organic acid blends improve intestinal integrity, modulate short-chain fatty acids profiles and alter microbiota of broilers under necrotic enteritis challenge. Anim. Nutr. 8, 82–90 (2022).
Gregor, A., Auernigg-Haselmaier, S., Trajanoski, S., König, J. & Duszka, K. Colonic medium-chain fatty acids act as a source of energy and for colon maintenance but are not utilized to acylate ghrelin. Nutrients https://doi.org/10.3390/nu13113807 (2021).
Wang, S. Z. et al. A medium-chain fatty acid analogue prevents endotoxin liver injury in a murine model. Sci. Rep. 15, 13645 (2025).
Gaete, P. V. et al. Medium-chain fatty acids modify macrophage expression of metabolic and inflammatory genes in a PPAR β/δ-dependent manner. Sci. Rep. 13, 11573 (2023).
Li, S. et al. Alleviation of Limosilactobacillus reuteri in polycystic ovary syndrome protects against circadian dysrhythmia-induced dyslipidemia via capric acid and GALR1 signaling. NPJ Biofilms Microbiomes 9, 47 (2023).
Ahmed, K. Biological roles and therapeutic potential of hydroxy-carboxylic Acid receptors. Front. Endocrinol. 2, 51 (2011).
Kunkemoeller, B. et al. The source of dietary fat influences anti-tumour immunity in obese mice. Nat. Metab. 7, 1630–1645 (2025).
Roopashree, P. G., Shetty, S. S. & Suchetha Kumari, N. Effect of medium chain fatty acid in human health and disease. J. Funct. Foods 87, 104724 (2021).
Sam, Q. H. et al. The divergent immunomodulatory effects of short chain fatty acids and medium chain fatty acids. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22126453 (2021).
Peters, A. et al. Natural biased signaling of hydroxycarboxylic acid receptor 3 and G protein-coupled receptor 84. Cell Commun. Signal. 18, 31 (2020).
Gregor, A. et al. Cage bedding modifies metabolic and gut microbiota profiles in mouse studies applying dietary restriction. Sci. Rep. 10, 20835 (2020).
Acknowledgements
This study was supported by the Shandong Provincial Youth Entrepreneurship Program for Colleges and Universities (2024KJJ042, 2021KJ075) and Shandong Provincial Natural Science Foundation (ZR2022MH217).
Author information
Authors and Affiliations
Contributions
N. H., S. L., and F. Z. designed the experiments. N. H. and S. L. provided funding support. G. K. and K. C. carried out experiments and analyzed the data. K. C. and Z. X. performed bioinformatics analysis. X. W., H. Z., Y. F., and Z. Z. performed animal experiments and sample collection. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, G., Chen, K., Xing, Z. et al. Stachyose attenuates DSS-induced colitis and is associated with elevated colonic medium-chain fatty acids: an integrated multi-omics analysis. npj Sci Food 9, 237 (2025). https://doi.org/10.1038/s41538-025-00604-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41538-025-00604-7
