
Abstract
As mRNA technology emerges as a groundbreaking innovation, an increasing number of studies are attempting to increase expression levels by optimizing the structure of mRNA, such as the 5’ or 3’ untranslated regions (UTR). We designed a novel loop structure in the poly(A) tail of an mRNA platform to improve the stability and duration. Protein expression analyses were conducted using bioluminescence, both in vitro and in vivo. Additionally, cellular and humoral responses to different antigens were confirmed using flow cytometry and ELISA of splenocytes and serum isolated from mice immunized with mRNA containing different poly(A) tail structures. The poly(A) tail with a loop structure exhibited higher bioluminescence signals, both in vitro and in vivo, and increased human erythropoietin (hEPO) expression in vivo compared to the other poly(A) tail groups. This indicates that the addition of a loop structure to the poly(A) tail region improves mRNA stability and efficiency. The results of analyzing cellular and humoral immunity by expressing HPV E6 and E7 antigens, and influenza virus HA antigens in mRNA with various poly(A) structures showed no significant difference in T cell immunity and antibody titer. In conclusion, the addition of stable structures, such as a loop to the poly(A) tail, can significantly increase the expression efficiency and stability of mRNA, but the expression difference did not have as much of an impact on the immune response as expected.
Similar content being viewed by others


Introduction
Messenger RNA (mRNA) is a single-stranded molecule transcribed from a strand of deoxyribonucleic acid (DNA)1. Proteins are synthesized through the process of translation, during which ribosomes decode the genetic information carried by messenger RNA (mRNA) into a defined amino acid sequence2. In vitro-transcribed mRNA encoding specific antigens has been used as a prophylactic or therapeutic vaccine against viruses or cancer, especially during the recent COVID-19 pandemic, because it can provoke immune responses by generating proteins3,4,5. Importantly, improving mRNA expression efficiency also offers benefits beyond antigen quantity, including improved durability of expression, reduced dosage requirements, more precise control over protein folding, and broader applications3,6,7,8,9.
mRNA consists of a 5′ cap, 5′ and 3′ untranslated regions (UTRs), and a poly(A) tail; therefore, numerous efforts have been made to optimize these elements for improvement of the translation efficiency10,11,12. The 5′ cap interacts with binding proteins and complexes essential for translation, such as the eukaryotic initiation factor13. Numerous capping reagents have been developed to enhance both translational efficiency and mRNA stability. In addition, the structural features of the 5′ and 3′ UTRs play important roles in mRNA stability and translation11. In particular, while some degree of secondary structure, such as small loops, could be beneficial, excessive or highly complex structures near the 5′ cap may impede translation initiation14. Recently, machine learning approaches have been employed for systematic UTR optimization, which have resulted in significantly improved protein expression15,16. Additionally, incorporating N1-methylpseudouridine has been shown to markedly enhance translation efficiency by evading type I interferon responses17. This modification was a key factor contributing to the development of mRNA vaccines, and was recognized by the awarding of the 2023 Nobel Prize to Katalin Karikó18.
Poly(A) tails are also important components of translation efficiency, stability, and nuclear export19. Poly(A) tails are formed via a two-step process20. The process begins with specific endonucleolytic cleavage at a site determined by the binding of polyadenylation factors, followed by polymerization of the adenosine tail to a species-specific length of approximately 150–250 nucleotides in mammals20,21.
The length and structure of poly(A) tails significantly influence the translation efficiency and stability of mRNA20,21,22. Longer poly(A) tails allow more poly(A)-binding proteins (PABPs) to interact with them, enhancing the translation efficiency of mRNA23,24. Additionally, longer poly(A) tails can extend the lifespan of mRNA by binding more PABPs, which protects it from degradation and slows down the degradation process by deadenylation24. Therefore, most of researches have focused on the optimization of length of poly A tails not the structure22,25.
However, the presence of structural elements, such as poly(U) sequences or stem-loop structures upstream of the poly(A) sequence, can also stabilize the poly(A) tail, thereby extending the half-life of mRNA26. It has been reported that secondary structure like hairpin or double-stranded RNA (dsRNA) exhibits greater stability than linear mRNA or single-stranded RNA (ssRNA)27,28,29. Previous study reported that deadenylation mediated by CCR4–NOT is impeded when poly(U) sequences are incorporated into poly(A) tails or when secondary structures are formed26. Additionally, the incorporation of non-adenosine ribonucleotides into the poly(A) tail also affects the deadenylation process. Certain viral transcripts incorporate non-adenosine ribonucleotides into their poly(A) tails to inhibit deadenylation and enhance their stability24. Therefore, we designed and compared various loop structures containing secondary structure and dsRNA characteristic in the poly(A) tail of mRNA platform.
A loop structure increased translation efficiency both in vitro and in vivo, as evidenced by bioluminescence assays and human erythropoietin (hEPO) expression level analyses. Additionally, we investigated whether the difference in translation efficiency resulting from variations in the poly(A) tail structure could influence immune responses against two types of antigens.
Results
Poly(A) sequence design
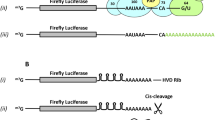
Four types of poly(A) tails were used: A50-Linker-A50 (A50L50LX) and A50-Linker-A50-complementary linker sequence (A50L50LO) were designed for comparation between small loop by pairing with its complementary linker sequence and linear structure in poly A sequence. In addition, A30-Linker-A70 (A30L70) was used in BioNTech30 as a positive control to compare both A50L50LO and A50L50LX (Fig. 1). A120 was also expected to show the translation efficiency of a poly A tail without a linker sequence, consisting solely of adenosine triphosphate (ATP) residues. RNA folding analysis using the RNAfold program predicted that only sequences with complementary sequences would form small loops, while other sequences would form large loops in the poly(A) region. Based on previous studies suggesting that compact RNA structures are more stable and lead to higher protein expression31, we hypothesized that the A50L50LO sequence would yield the best results. To investigate the impact of poly(A) sequences, four mRNA platforms were constructed with varying poly(A) sequences while the antigen remained constant.

Schematic diagram of poly(A) sequence composition and the structure of the poly(A) sequence predicted using RNAfold.
The A50L50LO sequence exhibited the highest luminescence signal in in vitro cell culture systems
To investigate the effects of poly(A) tail sequences on mRNA expression in in vitro cell culture systems, we encoded the firefly luciferase (F/L) gene into the mRNA-poly(A) platform. After in vitro transcription of mRNA with four different poly(A) structures, we purified them and confirmed that there were no significant differences in integrity as assessed by gel electrophoresis (Fig. S1A). These F/L mRNAs were transfected at a concentration of 500 ng/well into four cell lines (Nor10, HeLa, A549, and HepG2) (Fig. 2). Cells were harvested at 6, 24, and 48 h post-transfection, and luminescence signals were measured using a luminescence assay. In HeLa and Nor10 cells, A50L50LO and A30L70 consistently exhibited the highest luminescence signal levels with minimal changes over time. However, in A549 and HepG2 cells, only A50L50LO exhibited a superior luminescence signal. These results indicated that A50L50LO consistently promoted higher expression in all four cell lines.

Nor10, HeLa, HepG2, and A549 cells (n = 3) were transfected with 500 ng of F/L-mRNA-poly(A). Cells were harvested at 6, 24, and 48 h post-transfection for luminescence measurements. Data are presented as mean values ± SD. P values were calculated using one-way analysis of variance (ANOVA) with multiple comparison tests. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
The A50L50LO sequence exhibited the highest protein expression among both F/L and hEPO in an in vivo mouse model
To assess in vivo protein expression via mRNA, mRNA was formulated with lipid nanoparticle (LNP) and all mRNA/LNP formulations were characterized by dynamic light scattering (DLS) after purification (Fig. S1B). We administered 5 μg of F/L-mRNA-poly(A) constructs intramuscularly to C57BL/6 mice and monitored luciferase expression over time using an In Vivo Imaging system (IVIS) (Fig. 3A). Initially, all types of mRNA-poly(A) exhibited similar luminescence signals at 6 h. However, at 24 h, only A50L50LO maintained high expression. This indicated that the A50L50LO sequence consistently promoted the highest luciferase expression in the in vivo mouse model, as shown by both the image and quantitative plots of luminescence (Fig. 3B, C). To assess the impact of different antigens, we encoded hEPO into the mRNA-poly(A) platforms and administered 5 μg intravenously to C57BL/6 mice (Fig. 3D). Serum samples were collected at 2 and 6 h post-injection and hEPO levels were measured using ELISA. Consistent with the previous results, the A50L50LO sequence yielded the highest hEPO expression, followed by a slightly lower expression level in the A30L70 sequence (Fig. 3E). These data demonstrate that the A50L50LO sequence consistently outperformed the other sequences in terms of protein expression across all experimental conditions analyzed.

A Six-week-old C57BL/6 mice (n = 5) were intramuscularly administered 5 μg of F/L-mRNA-poly(A). The nil group was treated with DPBS. At each time point, 3 mg of luciferin was intraperitoneally administered, and bioluminescence was measured using an In Vivo Imaging system (IVIS) after anesthesia with isoflurane. C The luminescence was quantified from the images and B represented graphically. The statistics presented in the graph represent the results of the comparison between A50L50LO and A50L50LX. D Approximately 5 μg hEPO-mRNA-poly(A) was administered intravenously to 6-week-old C57BL/6 mice (n = 5). The nil group was treated with DPBS. E hEPO ELISA was performed on serum obtained 2 and 6 h post-immunization. Data are presented as mean values ± SD. P values were calculated using one-way ANOVA with multiple comparison tests. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Administration of HPV-mRNA poly(A) showed no statistically significant difference in CD8 + T lymphocyte responses across the immunization groups
To compare the immune responses to mRNA-poly(A) and human papilloma virus (HPV) antigens, E6 and E7 were encoded into mRNA-poly(A) platforms, as previously reported5. C57BL/6 mice were intramuscularly administered with HPV-mRNA at 3 μg or 10 μg on two occasions at weekly intervals that could induce sufficient T cell responses as demonstrated in previous research5,32 (Fig. 4A). The mice were sacrificed one week after the last immunization. Splenocytes were harvested and analyzed by flow cytometry, focusing on CD8 + T cells. No significant differences in the levels of HPV antigen-specific CD8 + T cells were observed among the immunized groups, despite all immunization groups exhibiting higher CD8 + T cell activation than the Nil group (Fig. 4B). Furthermore, analysis of pro-inflammatory cytokines (interferon gamma and tumor necrosis factor-alpha) (Fig. 4C) and activation markers (CD69 and CD25) on CD8 + T cells (Fig. 4D) revealed dose-dependent differences but no substantial differences in immune responses among the mRNA-poly(A) platforms.

A HPV-mRNA-poly(A) at doses of 3 μg and 10 μg was administered twice to 6-week-old female C57BL/6 mice (n = 5). The nil group was treated with DPBS. One week after the last administration, splenocytes were harvested for flow cytometry analysis. B Antigen-specific CD8 + T-cells in splenocytes without peptide stimulation. C Cytokine (Interferon (IFN)-gamma, tumor necrosis factor (TNF)-alpha) production and D T cell activation markers in splenocytes stimulated with peptides. Data are presented as mean values ± SD. P values were calculated using a two-way ANOVA. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Administration of HA-mRNA poly(A) with A50L50LO and A30L70 sequences resulted in stronger antibody responses
To investigate the humoral immune response, we replaced the antigen with HA and encoded it on an mRNA-poly(A) platform. Vero and A549 cells were transfected with 5 μg of each construct, and cell lysates were harvested 24 h post-transfection. In both cell lines, the A50L50LO sequence consistently exhibited the highest HA protein expression, as confirmed by western blotting (Figs. 5A and S4). Balb/c mice were intramuscularly administered with HA-mRNA at 5 μg on two occasions at 2 weekly intervals (Fig. 5B). Serum collected 6 hours after the first and second immunizations exhibited comparable IL-6 levels across all poly(A) groups, with no notable differences observed (Fig. 5C). Two weeks after the final immunization, HI assays and ELISA for IgG, IgG1, and IgG2a antibody titers were conducted using serum samples (Fig. 5D, E). The results demonstrated that antibody responses were largely similar among the experimental groups, and the observed differences were not statistically significant.

A Vero and A549 cells were transfected with 5 μg of HA-mRNA- poly(A), followed by protein purification and subsequent western blot analysis 24 h later. B 5 μg HA-mRNA-poly(A) was administered intramuscularly on two occasions to 6-week-old Balb/c mice (n = 5). The nil group was treated with DPBS. C IL-6 ELISA performed using serum collected 6 h after the first and second immunizations. D HI titer in response to HA-mRNA-poly(A). E The IgG, IgG1 and IgG2a antibody titers were examined by ELISA using serum collected two weeks after the last administration.
Discussion
mRNA molecules play critical roles in various applications, including vaccines and therapeutics. However, their inherent instability and short half-life pose challenges in achieving optimal expression levels. Previous reports have revealed several key strategies to enhance mRNA expression, such as optimizing the 5’ or 3’ UTRs to reduce miRNA binding sites or leveraging artificial intelligence technology33,34. Additionally, Liu et al. demonstrated that substituting adenosine residues in the poly(A) tail with non-A nucleotides enhanced translation efficiency19. Accordingly, numerous studies have attempted to enhance mRNA performance through modifications of the UTRs, adjustments to poly(A) tail length, or alterations of nucleotide composition. In this study, we investigated the effect of introducing loop structures into the poly(A) tail region of mRNA. The results revealed that A50L50 with a loop exhibited significantly stronger bioluminescence signals than the other poly(A) groups. Consistently, in vivo experiments using firefly luciferase or hEPO expression confirmed that loop structures demonstrated promising protein expression efficiency compared with other poly(A) tail groups in mice. This suggests that the addition of loop structures to the poly(A) tail region can improve the efficiency of protein expression by enhancing mRNA stability and duration, as observed in the in vivo model.
Recently, mRNA technology has expanded beyond its initial application in infectious disease vaccines and cancer therapeutics3,5,35,36. It is now being explored as an expression platform for antibodies37,38,39 and functional proteins, such as CRISPR-Cas940. Modifying the poly(A) tail region with loop structures could provide significant benefits by improving protein expression, similar to that of the protein described here, compared to other poly(A) platforms. For example, an mRNA platform with this loop structure could be applied to platforms where it would be beneficial to increase expression efficiency, such as the delivery of Cas9 for genome editing, or to systems that express the heavy and light chain genes of antibodies, and would yield better efficiency than conventional expression platforms.
We also examined the immune responses of each mRNA platform to confirm the correlation between expression level and immunogenicity using mRNA containing different poly(A) tails with HPV and influenza virus antigens. The HPV antigen is highly effective in eliciting T-cell responses, making it suitable for evaluating cellular immune responses41. Conversely, the influenza HA antigen predominantly induces antibody responses rather than T-cell responses, rendering it more appropriate for assessing humoral immune responses42. Therefore, in this paper, mRNA with various poly(A) structures were used to express HPV E6 and E7 antigens and influenza HA antigen, respectively, for immune response analysis.
When an mRNA vaccine containing the HPV antigen was administered at doses of 3 μg and 10 μg, no significant differences in cellular immunity were observed between different poly(A) structures at the same dose. However, differences in immune responses were observed depending on the administered dose. This suggests that variations in expression levels resulting from modifications to the poly(A) structure may have a limited impact on immune responses. Importantly, the dose-dependent enhancement of immune responses highlights the intrinsic adjuvanticity of the mRNA-LNP platform, as LNPs not only serve as delivery vehicles but also activate innate immune pathways that enhance adaptive immunity. Therefore, the observed differences in immune responses based on dosage indicate that mRNA-LNP possesses strong adjuvanticity43,44.
In addition, the results of the analysis of humoral immunity using mRNA vaccines expressing HA showed that the HI assay or ELISA antibody titer did not differ significantly depending on the poly(A) structure of the mRNA vaccine. Consistently, IL-6 ELISA conducted as an indirect assessment of innate immune activation showed comparable results across the poly(A) groups. Collectively, these findings suggest that while modifications in the mRNA poly(A) structure influence protein expression levels, they exert minimal impact on the induction of immune responses.
Furthermore, this approach has the potential to be more effective in protein replacement therapy. It may be advantageous as it can induce high levels of protein expression while minimizing unnecessary immune responses, even with a small dose of mRNA-LNP administration. Additionally, this strategy is expected to offer further benefits by more effectively reducing unnecessary immune responses, thereby minimizing adverse effects.
Overall, our findings suggest that modifying the poly(A) tail structure to enhance stability can contribute to improving protein expression and the durability of the mRNA platform. In particular, this modification is expected to be beneficial for studies aimed at in vivo expression of functional proteins, such as antibodies, where expression levels and stability are critical factors.
Methods
Cell
HepG2 (human hepatocellular carcinoma cells) were cultured in RPMI 1640 medium (Cytiva) supplemented with l-glutamine (2 mM). Nor10 (mouse muscle cells), HeLa (human cervical carcinoma cells), and A549 (lung cancer cells) cells were cultured in Dulbecco’s modified Eagle’s medium (Cytiva) supplemented with l-glutamine (4 mM) and glucose (4500 mg/L). All media were supplemented with penicillin (100 IU/mL), streptomycin (100 μg/mL), amphotericin B (0.25 μg/mL), and 10% fetal bovine serum (Gibco). All cells were maintained in a humidified incubator at 37 °C and 5% CO2.
Mice
Six-week-old female C57BL/6 and BALB/c mice were purchased from Daehan Biolink Co. Ltd. (Seoul, South Korea). At the Catholic University of Korea, the animals were housed in specific pathogen-free conditions and exposed to a 12 h light/12 h dark cycle. All experimental animal procedures were approved by the Institutional Animal Care and Use Committee of the Catholic University of Korea (CUK-IACUC-2023-039). The administration route and dosage were determined based on findings from previous studies5,45. Mice used in the experiment were euthanized using CO₂ either at the conclusion of the study or for the purpose of immunological analysis.
mRNA preparation
The DNA template for the mRNA platform contains a 5’UTR, 3’UTR, and poly(A) sequence. Poly(A) sequences (A50L50LO, A50L50LX, A30L70, and A120) were inserted into the mRNA platform using restriction enzymes (EcoR1 and Not1). Detailed information regarding the platform can be found in the patent (KR10–2022-0115033), and the information related to the poly(A) sequence has been provided in the Supplemental Data (Table S1). After the DNA was purified, mRNA was produced using the EZ T7 High Yield In vitro Transcription Kit (EZ027S; Enzynomics, Daejeon, Korea), a cap 1 capping analog (SMARTCAP®, ST PHARM, Seoul, Korea), and N1-methylpseudouridine-5′-triphosphate (m1ΨTP; TriLink, CA, USA) to replace uridine-5′-triphosphate (UTP). After transcription, mRNA was purified using lithium chloride precipitation and dsRNA was eliminated by cellulose purification46. RNA integrity was assessed by gel electrophoresis, and the concentration, pH, and endotoxin levels of the solutions were determined (A39553; Pierce, Thermo Fisher Scientific) (Fig. S1C).
Luciferase assay
HeLa, Nor10, A549, and HepG2 cells seeded into a 48-well plate at 1 × 105 cells per well and cultured at 37 °C for 24 h. Prior to transfection, the medium was replaced with serum-free medium. Following the manufacturer’s instructions, the cells were transfected at 80–90% confluence using Lipofectamine 2000 (11668019; Invitrogen, Thermo Fisher Scientific). For the assay, we mixed 500 ng of RNA (F/L) and 0.5 μl of Lipofectamine per well in 50 μl of Opti-MEM medium (Gibco) and incubated at room temperature for 5 min. Next, the diluted RNA was mixed with diluted Lipofectamine and incubated for 5 min at room temperature. The prepared 48-well plates were transfected. After 24 h of transfection, the cells were harvested and luciferase assays were performed (Promega). All reagents were prepared according to the manufacturer’s instructions. For cell lysis, cells were resuspended in 80 μl of 1× lysis buffer per well. After 15 minutes of lysis, 20 μl of each lysate was transferred to a 96-well white assay plate (Corning Costar Corp., USA), and 100 μl of luciferase assay substrate reagent was added to each test sample. Luminescence was then measured using the Glomax Discover system (Promega).
mRNA-LNP formulation
LNPs were prepared as previously reported. All lipid components, in a molar ratio of 50:10:38.5:1.5 (SM-102: distearoylphosphatidylcholine: cholesterol: 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000), were briefly dissolved in ethanol. The mRNAs were dissolved in sodium citrate buffer (50 mM, pH 4) at a charge ratio of N/P = 6. The formulation of LNPs was conducted using an enCELL-Master V2 (ENPARTICLE, Busan, Korea) by mixing aqueous and organic solutions at a 3:1 ratio with a total flow rate of 10 mL/min. The LNP solution was concentrated via ultrafiltration using an Amicon Ultra centrifugal filter (UFC9030; Merck Millipore, MA, USA) following the manufacturer’s instructions.
mRNA-LNPs were diluted 100-fold in DW for size and PDI measurements. The size and PDI of the LNP were measured using a 12 mm square polystyrene cuvette (DTS0012; suitable for size and PDI) with a PANalytical Zetasizer Ultra (Malvern, USA).
In vivo imaging system
During IVIS scans, mice were anesthetized with a mixture of isoflurane (1.5%) and oxygen. The mRNA platform encoding firefly luciferase (5 μg per mouse) was injected intramuscularly into the mice. d-Luciferin (P1043; Promega, Madison, WI, USA) was administered intraperitoneally (I.P.) at a dose of 150 mg/kg/mice. Bioluminescent images were acquired at 8, 24, 48, and 72 h using an IVIS Spectrum System (LUCI, CELLGENTEK), and the analysis was performed using Living Image software (NEOimage program).
Flow cytometry
Mouse splenocytes (1 × 106 cells/well) were seeded in a 96-well plate and stimulated with 2 µg/well HPV serotype 16 E7 (RAHYNIVTF). Subsequently, the samples were treated with Brefeldin A (Golgi plug, BD Biosciences, Franklin Lakes, NY, USA) for 10 h at 37 °C. The splenocytes were blocked by CD16/CD32 (14-0161-82; Invitrogen) for 15 min at 4 °C and subsequently stained with Live/Dead ™ Fixable Aqua Dead Cell Stain Kit (L34966, Invitrogen, MA, USA), CD8 (100744; Biolegend), CD4 (17-0042-82; invitrogen) for 30 min at 4 °C in the dark. Cells were permeabilized using a Fixation/Permeabilization Solution kit (BD Biosciences) for 1 h at 4 °C in the dark and then stained with IFN-γ (505826; Biolegend), and TNF-alpha (506304; Biolegend) for 30 min at 4 °C in the dark. For each sample, all the antibodies were treated with 0.1 µg. The cells were analyzed using a CytoFlex flow cytometer (Beckman Coulter, Brea, CA, USA), and data analysis was performed using CytExpert (Beckman Coulter).
For other plates, we seeded 1 × 106 cells/well mouse splenocytes in a 96-well plate and stimulated them with 2 µg/well of HPV serotype 16 E7 (RAHYNIVTF). Subsequently, the samples were blocked with CD16/CD32 (14-0161-82; Invitrogen) at 4 °C for 15 min. Thereafter, we stained the cells in the dark at 4 °C for 30 min using Live/Dead™ Fixable Aqua Dead Cell Stain Kit (L34966; Invitrogen, MA, USA), HPV 16 E7 49-57-specific tetramer (RAHYNIVTF), CD8 (100706; Biolegend), CD69 (17-0691-82; Invitrogen), and CD25 (64-0251-82; eBioscience). Each sample was treated with the antibody at a concentration of 0.1 µg.
Western blot analysis
Vero cells (Korea Cell Line Bank, Seoul, Korea) and A549 cells were transfected with 5 μg of HA-mRNA-poly(A) using Lipofectamine 2000® (Invitrogen, Waltham, MA, USA). After 24 h, proteins were extracted from the cell lysates. Cell lysates were prepared with RIPA buffer (89900; Thermo scientific) for protein extraction, and protein concentrations were determined using a BCA assay kit (23227; Thermo scientific). The extracted proteins (10 μg) were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. Membranes were incubated at 25 °C for 1 h in phosphate-buffered saline (PBS) containing 0.1% Tween-20 and 5% skim milk to block nonspecific binding. Thereafter, the membranes were incubated overnight at 4 °C with anti-HA antibody (11684-MM03; sinobiological) and anti-β-actin antibody (3700 S; Cell Signaling Technology). Horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibodies were used as the secondary antibodies. The signal was detected using ECL substrate solution (ECL-PS100; Donginbio, Seoul, Korea).
Enzyme-linked immunosorbent assay
The 96-well plates (32296; SPL, Korea) were coated with 100 ng of HA protein per well and incubated overnight at 4 °C. The wells were washed thrice with 200 µL of PBS containing 0.05% Tween-20 (PBS-T) and then blocked with 100 µL of blocking buffer (1% bovine serum albumin in PBS) at room temperature for 1 h. The diluted serum samples were added to the wells and incubated at room temperature for 2 h. After incubation, the wells were washed thrice with PBS-T. Next, a 1:5000 dilution of horseradish peroxidase (HRP)-conjugated anti-mouse IgG (A90-116P;), IgG1 (A90-105P) and IgG2a (STAR133P; Biorad) antibody was added to the blocking buffer and incubated for 1 h at 20 °C. After five washes with PBS-T, tetramethylbenzidine (TMB) substrate (34021; Thermo Fisher Scientific) was added, and the plate was incubated for 10 min. The reaction was terminated by adding 2 N H2SO4. Optical density (OD) values were measured at 450 nm using the GloMax Explorer microplate reader (Promega, Madison, WI, USA). The hEPO levels were measured using an hEPO DuoSet ELISA kit (DY286; R&D Systems, USA).
HI assay
The mouse serum was treated with a receptor-destroying enzyme (Denka Seiken, Tokyo, Japan) at 37 °C for 16 h. Subsequently, the mixture was inactivated at 56 °C for 30 min to remove nonspecific reactions. All serum samples were serially diluted two-fold with cold PBS in a 96-well V-bottom plate (Corning, NY, USA). A standardized virus suspension (16 HA U/50 μl) was added, and the plate was incubated at 25 °C for 30 min. Chicken red blood cells (3% in 50 μl) were then added, and the plate was incubated at room temperature (25 °C) for approximately 30 min. The geometric mean titer of the 95% confidence interval (CI) of antibodies was expressed as the reciprocal of the highest serum dilution showing complete hemagglutination inhibition.
RNAfold
The RNAfold tool from the ViennaRNA Package 2.0 (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) was used to predict the secondary structure of the mRNA-poly(A). The minimum free energy (MFE) associated with the predicted secondary structure was calculated using the McCaskill method. The structure prediction was performed using hEPO-mRNA-poly(A), and Fig. 1 specifically highlights only the poly(A) region.
Statistical analysis
All data are presented as mean ± standard deviation (SD). Statistical analyses were performed using GraphPad Prism software 9.0. One-way analysis of variance (ANOVA) with Tukey’s multiple comparison test was used to assess statistical significance. Statistically significant differences were defined as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Data availability
The authors confirm that the data supporting the findings of this study are available in the article and as supplementary data. The raw data supporting the findings of this study are available from the corresponding author upon reasonable request.
References
Qin, S. et al. mRNA-based therapeutics: powerful and versatile tools to combat diseases. Signal Transduct. Target. Ther. 7, 166 (2022).
Jiao, L. et al. Ribosome biogenesis in disease: new players and therapeutic targets. Signal Transduct. Target. Ther. 8, 15 (2023).
Pardi, N., Hogan, M. J., Porter, F. W. & Weissman, D. mRNA vaccines—a new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279 (2018).
Fang, E. et al. Advances in COVID-19 mRNA vaccine development. Signal Transduct. Target. Ther. 7, 94 (2022).
Lee, S. et al. mRNA-HPV vaccine encoding E6 and E7 improves therapeutic potential for HPV-mediated cancers via subcutaneous immunization. J. Med. Virol. 95, e29309 (2023).
Jin, L., Zhou, Y., Zhang, S. & Chen, S.-J. mRNA vaccine sequence and structure design and optimization: advances and challenges. J. Biol. Chem. 301, 108015 (2025).
Comes, J. D. G., Pijlman, G. P. & Hick, T. A. H. Rise of the RNA machines—self-amplification in mRNA vaccine design. Trends Biotechnol. 41, 1417–1429 (2023).
Munang’andu, H. M., Fredriksen, B. N., Mutoloki, S., Dalmo, R. A. & Evensen, Ø Antigen dose and humoral immune response correspond with protection for inactivated infectious pancreatic necrosis virus vaccines in Atlantic salmon (Salmo salar L). Vet. Res. 44, 7 (2013).
Johansen, P. et al. Antigen kinetics determines immune reactivity. Proc. Natl. Acad. Sci. USA 105, 5189–5194 (2008).
Schoenmaker, L. et al. mRNA-lipid nanoparticle COVID-19 vaccines: structure and stability. Int. J. Pharm. 601, 120586 (2021).
Asrani, K. H. et al. Optimization of mRNA untranslated regions for improved expression of therapeutic mRNA. RNA Biol. 15, 756–762 (2018).
Leppek, K. et al. Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics. Nat. Commun. 13, 1536 (2022).
Decroly, E., Ferron, F., Lescar, J. & Canard, B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 10, 51–65 (2012).
Lai, W.-J. C. et al. mRNAs and lncRNAs intrinsically form secondary structures with short end-to-end distances. Nat. Commun. 9, 4328 (2018).
Castillo-Hair, S. et al. Optimizing 5’UTRs for mRNA-delivered gene editing using deep learning. Nat. Commun. 15, 5284 (2024).
Zeng, J. et al. Characterization and optimization of 5´ untranslated region containing poly-adenine tracts in Kluyveromyces marxianus using machine-learning model. Microb. Cell Fact. 23, 7 (2024).
Nance, K. D. & Meier, J. L. Modifications in an emergency: the role of N1-methylpseudouridine in COVID-19 vaccines. ACS Cent. Sci. 7, 748–756 (2021).
Pederson, T. Tributaries of the 2023 Nobel Prize in Physiology or Medicine, and lessons learned. RNA. 30, 101–104 (2024).
Liu, Y., Nie, H., Sun, R., Wang, J. & Lu, F. Enhancement of synthetic mRNA translation efficiency through engineered poly(A) tails. bioRxiv. https://doi.org/10.1101/2021.08.30.458298 (2021).
Tian, B., Hu, J., Zhang, H. & Lutz, C. S. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 33, 201–212 (2005).
Brown, C. E. & Sachs, A. B. Poly(A) tail length control in Saccharomyces cerevisiae occurs by message-specific deadenylation. Mol. Cell Biol. 18, 6548–6559 (1998).
Biziaev, N. et al. The impact of mRNA poly(A) tail length on eukaryotic translation stages. Nucleic Acids Res. 52, 7792–7808 (2024).
Xiang, K. & Bartel, D. P. The molecular basis of coupling between poly(A)-tail length and translational efficiency. eLife 10, e66493 (2021).
Passmore, L. A. & Coller, J. Roles of mRNA poly(A) tails in regulation of eukaryotic gene expression. Nat. Rev. Mol. Cell Biol. 23, 93–106 (2022).
Kojima, S., Sher-Chen, E. & Green, C. Circadian control of mRNA polyadenylation dynamics regulates rhythmic protein expression. Genes Dev. 26, 2724–2736 (2012).
Stowell, J. A. W. et al. Reconstitution of targeted deadenylation by the Ccr4-not complex and the YTH domain protein Mmi1. Cell Rep. 17, 1978–1989 (2016).
Bai, M. et al. Influence of diverse storage conditions of double-stranded RNA in vitro on the RNA interference efficiency in vivo insect Tribolium castaneum. Pest Manag. Sci. 79, 45–54 (2023).
Pal, A. & Levy, Y. Structure, stability and specificity of the binding of ssDNA and ssRNA with proteins. PLoS Comput. Biol. 15, e1006768 (2019).
Solodushko, V. & Fouty, B. Terminal hairpins improve protein expression in IRES-initiated mRNA in the absence of a cap and polyadenylated tail. Gene Ther. 30, 620–627 (2023).
Eberle, F., Sahin, U., Kuhn, A., Vallazza, B. & Diken, M. Stabilization of poly(A) sequence encoding DNA sequences. US12054723B2 (2024).
Wen, J. D. et al. Following translation by single ribosomes one codon at a time. Nature 452, 598–603 (2008).
Da Silva, D. M., Martinez, E. A., Bogaert, L. & Kast, W. M. Investigation of the optimal prime boost spacing regimen for a cancer therapeutic vaccine targeting human papillomavirus. Cancers 14, https://doi.org/10.3390/cancers14174339 (2022).
Orlandini von Niessen, A. G. et al. Improving mRNA-based therapeutic gene delivery by expression-augmenting 3’ UTRs identified by cellular library screening. Mol. Ther. 27, 824–836 (2019).
Zhang, H. et al. Algorithm for optimized mRNA design improves stability and immunogenicity. Nature 621, 396–403 (2023).
Lee, Y.-S. et al. Analysis of the immunostimulatory effects of cytokine-expressing internal ribosome entry site–based RNA adjuvants and their applications. J. Infect. Dis. 229, 1408–1418 (2023).
Kim, J.-Y. et al. mRNA vaccine encoding Gn provides protection against severe fever with thrombocytopenia syndrome virus in mice. npj Vaccines 8, 167 (2023).
Deal, C. E., Carfi, A. & Plante, O. J. Advancements in mRNA encoded antibodies for passive immunotherapy. Vaccines 9, https://doi.org/10.3390/vaccines9020108 (2021).
Rybakova, Y. et al. mRNA delivery for therapeutic anti-HER2 antibody expression In Vivo. Mol. Ther. 27, 1415–1423 (2019).
Wu, L. et al. Engineered mRNA-expressed bispecific antibody prevent intestinal cancer via lipid nanoparticle delivery. Bioengineered 12, 12383–12393 (2021).
Stadelmann, C. et al. mRNA-mediated delivery of gene editing tools to human primary muscle stem cells. Mol. Ther. Nucleic Acids 28, 47–57 (2022).
Pinto, L. A. et al. Cellular immune responses to human papillomavirus (HPV)-16 L1 in healthy volunteers immunized with recombinant HPV-16 L1 virus-like particles. J. Infect. Dis. 188, 327–338 (2003).
Deck, R. R., DeWitt, C. M., Donnelly, J. J., Liu, M. A. & Ulmer, J. B. Characterization of humoral immune responses induced by an influenza hemagglutinin DNA vaccine. Vaccine 15, 71–78 (1997).
Alameh, M. G. et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity 54, 2877–2892.e2877 (2021).
Ndeupen, S. et al. The mRNA-LNP platform’s lipid nanoparticle component used in preclinical vaccine studies is highly inflammatory. iScience 24, 103479 (2021).
Park, H. J. et al. Analyzing immune responses to varied mRNA and protein vaccine sequences. NPJ Vaccines 8, 84 (2023).
Baiersdörfer, M. et al. A facile method for the removal of dsRNA contaminant from in vitro-transcribed mRNA. Mol. Ther. Nucleic Acids 15, 26–35 (2019).
Acknowledgements
This study was approved by the Institutional Animal Care and Use Committee of the Catholic University of Korea (CUK-IACUC-2023-039). This research was supported by the Ministry of Food and Drug Safety (22213MFDS421 and RS-2025-02213409, J.H.N.), Korea Disease Control and Prevention Agency (No. 2023-ER1701-00), and partially supported by the Brain Korea 21 Four Program. Schematics were generated with the support of BioRender.
Author information
Authors and Affiliations
Contributions
A.O., H.-J.P., and J.-H.N. conceptualized the study. The methodology was developed by A.O., S.L., J.L., D.H., S.Y., S.L., and G.R. The investigation was conducted by A.O., S.-Y.L., J.L., and E.-J.C. H.-J.P., S.-H.B., S.J., H.C., Y.C., and Y.L. contributed to the provision of materials. S.C., H.-J.C., S.J., J.K., and H.-J.P. curated the data. A.O., S.L., and J.-H.N. were responsible for writing the original draft as well as reviewing and editing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Oh, A., Lee, S., Park, HJ. et al. Loop structure in poly(A) tail of mRNA vaccine enhances antigen translation efficiency and mRNA stability. npj Vaccines 10, 234 (2025). https://doi.org/10.1038/s41541-025-01287-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41541-025-01287-7
