
Abstract
Clinical observations of patients with congenital heart disease carrying SMAD2 genetic variants revealed correlations with multi-organ impairments at the developmental and functional levels. Many patients with congenital heart disease present with glomerulosclerosis, periglomerular fibrosis and albuminuria. It remains largely unknown whether SMAD2 variants associated with congenital heart disease can directly alter kidney cell fate, tissue patterning and organ-level function. Here we investigate the role of pathogenic SMAD2 variants in podocytogenesis, nephrogenic cell lineage specification and glomerular filtration barrier function using a combination of CRISPR-based disease modelling, stem cell and microfluidic organ-on-a-chip technologies. We show that the abrogation of SMAD2 results in altered patterning of the mesoderm and intermediate mesoderm cell lineages, which give rise to nearly all kidney cell types. Following further differentiation of intermediate mesoderm cells, the mutant podocytes failed to develop arborizations and interdigitations. A reconstituted glomerulus-on-a-chip system showed substantial albumin leakage, as observed in glomerulopathies. This study implicates chronic heart disease-associated SMAD2 mutations in kidney tissue malformation that might inform targeted regenerative therapies.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
The authors declare that all data supporting the findings of this study are included in the article and Supplementary Information. Raw patient data are available from the corresponding author, subject to approval from the Institutional Review Board of Harvard Medical School. The patient data have been previously published in refs. 12,29,30. Data on the clinical studies listed in the tables were obtained from refs. 3,4,11,16,34,37. Source data for clinicopathological information on patients summarized in Tables 1 and 2 are cited in the tables. The scRNA sequencing data analysis source data for ref. 106 are available at https://cellxgene.cziscience.com/collections/f7cecffa-00b4-4560-a29a-8ad626b8ee08; those for ref. 107 at Gene Expression Omnibus (GEO) accession numbers GSE151302, GSE195460 and GSE131882 (https://cellxgene.cziscience.com/collections/b3e2c6e3-9b05-4da9-8f42-da38a664b45b); those for ref. 108 at https://cellxgene.cziscience.com/collections/120e86b4-1195-48c5-845b-b98054105eec; those for ref. 109 at GEO accession number GSE183279 (https://cellxgene.cziscience.com/collections/bcb61471-2a44-4d00-a0af-ff085512674c) and those for ref. 110 at GEO accession number GSE151302 (https://cellxgene.cziscience.com/collections/9b02383a-9358-4f0f-9795-a891ec523bcc). The bubble plot was generated using the CellxGene online data viewer. Western blot stain-free blots, stain-free gels and chemiluminescent images are presented in Supplementary Figs. 4–8. Source data are provided with this paper.
Change history
20 November 2025
In the version of this article initially published, in the third paragraph of Results, the current callout to “Extended Data Fig. 2a; Fig. 1d,e” was originally incorrect, while in the seventh paragraph of Results, the current callout to “Extended Data Fig. 3b; Fig. 2c–f” was originally incorrect. The text is updated in the HTML and PDF versions of the paper.
References
Loh, K. M. et al. Mapping the pairwise choices leading from pluripotency to human bone, heart, and other mesoderm cell types. Cell 166, 451–467 (2016).
Musah, S., Bhattacharya, R. & Himmelfarb, J. Kidney disease modeling with organoids and organs-on-chips. Annu. Rev. Biomed. Eng. 26, 383–414 (2024).
Micha, D. et al. SMAD2 mutations are associated with arterial aneurysms and dissections. Hum. Mutat. 36, 1145–1149 (2015).
Granadillo, J. L. et al. Variable cardiovascular phenotypes associated with SMAD2 pathogenic variants. Hum. Mutat. 39, 1875–1884 (2018).
Loeys, B. L. et al. Aneurysm syndromes caused by mutations in the TGF-β receptor. N. Engl. J. Med. 355, 788–798 (2006).
Cannaerts, E. et al. Novel pathogenic SMAD2 variants in five families with arterial aneurysm and dissection: further delineation of the phenotype. J. Med. Genet. 56, 220–227 (2019).
Bikbov, B. et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 395, 709–733 (2020).
Morton, S. U., Quiat, D., Seidman, J. G. & Seidman, C. E. Genomic frontiers in congenital heart disease. Nat. Rev. Cardiol. 19, 26–42 (2022).
Dovjak, G. O. et al. Abnormal extracardiac development in fetuses with congenital heart disease. J. Am. Coll. Cardiol. 78, 2312–2322 (2021).
Gabriel, G. C., Pazour, G. J. & Lo, C. W. Congenital heart defects and ciliopathies associated with renal phenotypes. Front. Pediatr. 6, 175 (2018).
Rajpal, S. et al. Association of albuminuria with major adverse outcomes in adults with congenital heart disease: results from the Boston Adult Congenital Heart Biobank. JAMA Cardiol. 3, 308–316 (2018).
Zaidi, S. et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 498, 220–223 (2013).
Homsy, J. et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350, 1262–1266 (2015).
Scholes, G. B., Zannino, D., Kausman, J. Y. & Cheung, M. M. H. Altered in utero kidney development in newborns with congenital heart disease. Pediatr. Res. 85, 644–649 (2019).
Gupte, P. A., Vaideeswar, P. & Kandalkar, B. M. Cyanotic nephropathy—a morphometric analysis. Congenit. Heart Dis. 9, 280–285 (2014).
Dimopoulos, K. et al. Prevalence, predictors, and prognostic value of renal dysfunction in adults with congenital heart disease. Circulation 117, 2320–2328 (2008).
Greenberg, J. H. et al. Kidney outcomes 5 years after pediatric cardiac surgery: the TRIBE-AKI Study. JAMA Pediatr. 170, 1071–1078 (2016).
Nugent, J. et al. Assessment of acute kidney injury and longitudinal kidney function after hospital discharge among patients with and without COVID-19. JAMA Netw. Open 4, e211095 (2021).
Li, A. H. et al. Genetic architecture of laterality defects revealed by whole exome sequencing. Eur. J. Hum. Genet. 27, 563–573 (2019).
Hobbs, C. A. et al. Genetic epidemiology and nonsyndromic structural birth defects: from candidate genes to epigenetics. JAMA Pediatr. 168, 371–377 (2014).
Rees, L., Schaefer, F., Schmitt, C. P., Shroff, R. & Warady, B. A. Chronic dialysis in children and adolescents: challenges and outcomes. Lancet Child Adolesc. Health 1, 68–77 (2017).
Woolf, A. S. A molecular and genetic view of human renal and urinary tract malformations. Kidney Int. 58, 500–512 (2000).
Strong, A. et al. Expanding the phenotypic spectrum of Mendelian connective tissue disorders to include prominent kidney phenotypes. Am. J. Med. Genet. A 185, 3762–3769 (2021).
Billett, J., Majeed, A., Gatzoulis, M. & Cowie, M. Trends in hospital admissions, in-hospital case fatality and population mortality from congenital heart disease in England, 1994 to 2004. Heart 94, 342–348 (2008).
Keller, G., Zimmer, G., Mall, G., Ritz, E. & Amann, K. Nephron number in patients with primary hypertension. N. Engl. J. Med. 348, 101–108 (2003).
Boo, H. J. et al. The presence of simple renal cysts is associated with an increased risk of albuminuria in young adults. Korean J. Intern. Med. 37, 425–433 (2022).
Sevillano, A. M. et al. Multiple kidney cysts in thin basement membrane disease with proteinuria and kidney function impairment. Clin. Kidney J. 7, 251–256 (2014).
Chen, J., Ma, X., Xu, D., Cao, W. & Kong, X. Association between simple renal cyst and kidney damage in a Chinese cohort study. Ren. Fail. 41, 600–606 (2019).
Jin, S. C. et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 49, 1593–1601 (2017).
Xiao, F. et al. Functional dissection of human cardiac enhancers and noncoding de novo variants in congenital heart disease. Nat. Genet. 56, 420–430 (2024).
Passwell, J. et al. Abnormal renal functions in cyanotic congential heart disease. Arch. Dis. Child. 51, 803–805 (1976).
Burlet, A., Drukker, A. & Guignard, J.-P. Renal function in cyanotic congenital heart disease. Nephron 81, 296–300 (1999).
Esch, J. J., Salvin, J. M., Thiagarajan, R. R., Del Nido, P. J. & Rajagopal, S. K. Acute kidney injury after Fontan completion: risk factors and outcomes. J. Thorac. Cardiovasc. Surg. 150, 190–197 (2015).
Gillesén, M. et al. Chronic kidney disease in patients with congenital heart disease: a nationwide, register-based cohort study. Eur. Heart J. Open 2, oeac055 (2022).
Anne, P., Du, W., Mattoo, T. K. & Zilberman, M. V. Nephropathy in patients after Fontan palliation. Int. J. Cardiol. 132, 244–247 (2009).
Sharma, S. et al. Assessment of kidney function in survivors following Fontan palliation: kidney function following Fontan palliation. Congenit. Heart Dis. 11, 630–636 (2016).
Opotowsky, A. R. et al. Estimated glomerular filtration rate and urine biomarkers in patients with single-ventricle Fontan circulation. Heart 103, 434–442 (2017).
Broda, C. R. et al. Renal dysfunction is associated with higher central venous pressures in patients with Fontan circulation. Congenit. Heart Dis. 13, 602–607 (2018).
Wilson, T. G. et al. Hepatic and renal end-organ damage in the Fontan circulation: a report from the Australian and New Zealand Fontan Registry. Int. J. Cardiol. 273, 100–107 (2018).
Özsu, E., Yeşiltepe Mutlu, G., Yüksel, A. B. & Hatun, Ş Features of two cases with 18q deletion syndrome. J. Clin. Res. Pediatr. Endocrinol. 6, 51–54 (2014).
Hochberg, E. P. et al. Case 11-2017—a 61-year-old woman with leg swelling, back pain, and hydronephrosis. N. Engl. J. Med. 376, 1461–1471 (2017).
Turleau, C., Chavin-Colin, F., Narbouton, R., Asensi, D. & Grouchy, J. D. Trisomy 18q-. Trisomy mapping of chromosome 18 revisited. Clin. Genet. 18, 20–26 (1980).
Nomura, M. & Li, E. Smad2 role in mesoderm formation, left–right patterning and craniofacial development. Nature 393, 786–790 (1998).
Weinstein, M. et al. Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor smad2. Proc. Natl Acad. Sci. USA 95, 9378–9383 (1998).
Musah, S. Decoding cell fate: human models reveal how SMAD2 variants shape development. Nat. Rev. Genet. 26, 586 (2025).
Ward, T. et al. Modeling SMAD2 mutations in induced pluripotent stem cells provides insights into cardiovascular disease pathogenesis. J. Am. Heart Assoc. 14, e036860 (2025).
Faial, T. et al. Brachyury and SMAD signalling collaboratively orchestrate distinct mesoderm and endoderm gene regulatory networks in differentiating human embryonic stem cells. Development 142, 2121–2135 (2015).
Kispert, A. & Herrmann, B. G. The Brachyury gene encodes a novel DNA binding protein. EMBO J. 12, 3211–3220 (1993).
Kispert, A., Koschorz, B. & Herrmann, B. G. The T protein encoded by Brachyury is a tissue-specific transcription factor. EMBO J. 14, 4763–4772 (1995).
Mendjan, S. et al. NANOG and CDX2 pattern distinct subtypes of human mesoderm during exit from pluripotency. Cell Stem Cell 15, 310–325 (2014).
Ten Berge, D. et al. Wnt signaling mediates self-organization and axis formation in embryoid bodies. Cell Stem Cell 3, 508–518 (2008).
Henderson, N. C., Rieder, F. & Wynn, T. A. Fibrosis: from mechanisms to medicines. Nature 587, 555–566 (2020).
Musah, S. et al. Mature induced-pluripotent-stem-cell-derived human podocytes reconstitute kidney glomerular-capillary-wall function on a chip. Nat. Biomed. Eng. 1, 0069 (2017).
Bhattacharya, R., Bonner, M. G. & Musah, S. Harnessing developmental plasticity to pattern kidney organoids. Cell Stem Cell 28, 587–589 (2021).
Ranghini, E. J. & Dressler, G. R. Evidence for intermediate mesoderm and kidney progenitor cell specification by Pax2 and PTIP dependent mechanisms. Dev. Biol. 399, 296–305 (2015).
Qiu, C. et al. A single-cell time-lapse of mouse prenatal development from gastrula to birth. Nature 626, 1084–1093 (2024).
Lam, A. Q. et al. Rapid and efficient differentiation of human pluripotent stem cells into intermediate mesoderm that forms tubules expressing kidney proximal tubular markers. J. Am. Soc. Nephrol. 25, 1211–1225 (2014).
Meng, X. M. et al. Smad2 protects against TGF-β/Smad3-mediated renal fibrosis. J. Am. Soc. Nephrol. 21, 1477–1487 (2010).
Samson, A. L. et al. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat. Commun. 11, 3151 (2020).
Edeling, M., Ragi, G., Huang, S., Pavenstädt, H. & Susztak, K. Developmental signalling pathways in renal fibrosis: the roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 12, 426–439 (2016).
Zeisberg, M. et al. BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 9, 964–968 (2003).
Schnell, J., Achieng, M. & Lindström, N. O. Principles of human and mouse nephron development. Nat. Rev. Nephrol. 18, 628–642 (2022).
Perico, L., Conti, S., Benigni, A. & Remuzzi, G. Podocyte–actin dynamics in health and disease. Nat. Rev. Nephrol. 12, 692–710 (2016).
Wharram, B. L. et al. Altered podocyte structure in GLEPP1 (Ptpro)-deficient mice associated with hypertension and low glomerular filtration rate. J. Clin. Invest. 106, 1281–1290 (2000).
Kim, Y. H. et al. GLEPP1 receptor tyrosine phosphatase (Ptpro) in rat PAN nephrosis. Nephron 90, 471–476 (2002).
Tossidou, I. et al. Tyrosine phosphorylation of CD2AP affects stability of the slit diaphragm complex. J. Am. Soc. Nephrol. 30, 1220–1237 (2019).
Bhattacharya, R. et al. A novel TRPC6 mutation causes autosomal dominant FSGS. Preprint at bioRxiv https://doi.org/10.1101/2025.02.11.637765 (2025).
Guo, J.-K. WT1 is a key regulator of podocyte function: reduced expression levels cause crescentic glomerulonephritis and mesangial sclerosis. Hum. Mol. Genet. 11, 651–659 (2002).
Kavsak, P. et al. Smad7 Binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGFβ receptor for degradation. Mol. Cell 6, 1365–1375 (2002).
Fukasawa, H. et al. Down-regulation of Smad7 expression by ubiquitin-dependent degradation contributes to renal fibrosis in obstructive nephropathy in mice. Proc. Natl Acad. Sci. USA 101, 8687–8692 (2004).
Chung, A. C. K. et al. Disruption of the Smad7 gene promotes renal fibrosis and inflammation in unilateral ureteral obstruction (UUO) in mice. Nephrol. Dial. Transplant. 24, 1443–1454 (2009).
Wang, W., Koka, V. & Lan, H. Y. Transforming growth factor-β and Smad signalling in kidney diseases. Nephrology 10, 48–56 (2005).
Li, Z. et al. 3D culture supports long-term expansion of mouse and human nephrogenic progenitors. Cell Stem Cell 19, 516–529 (2016).
Bonse, J. et al. Nuclear YAP localization as a key regulator of podocyte function. Cell Death Dis. 9, 850 (2018).
Burt, M. A., Kalejaiye, T. D., Bhattacharya, R., Dimitrakakis, N. & Musah, S. Adriamycin-induced podocyte injury disrupts the YAP-TEAD1 axis and downregulates Cyr61 and CTGF expression. ACS Chem. Biol. 17, 3341–3351 (2022).
Haley, K. E. et al. YAP translocation precedes cytoskeletal rearrangement in podocyte stress response: a podometric investigation of diabetic nephropathy. Front. Physiol. 12, 625762 (2021).
Schwartzman, M. et al. Podocyte-specific deletion of yes-associated protein causes FSGS and progressive renal failure. J. Am. Soc. Nephrol. 27, 216–226 (2016).
Holbourn, K. P., Acharya, K. R. & Perbal, B. The CCN family of proteins: structure–function relationships. Trends Biochem. Sci. 33, 461–473 (2008).
Musah, S. et al. Glycosaminoglycan-binding hydrogels enable mechanical control of human pluripotent stem cell self-renewal. ACS Nano 6, 10168–10177 (2012).
Furukawa, K. T., Yamashita, K., Sakurai, N. & Ohno, S. The epithelial circumferential actin belt regulates YAP/TAZ through nucleocytoplasmic shuttling of merlin. Cell Rep. 20, 1435–1447 (2017).
Aragona, M. et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 154, 1047–1059 (2013).
Campbell, K. N. et al. Yes-associated protein (YAP) promotes cell survival by inhibiting proapoptotic dendrin signaling. J. Biol. Chem. 288, 17057–17062 (2013).
Meliambro, K. et al. The Hippo pathway regulator KIBRA promotes podocyte injury by inhibiting YAP signaling and disrupting actin cytoskeletal dynamics. J. Biol. Chem. 292, 21137–21148 (2017).
Beck, B. et al. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell 16, 67–79 (2015).
Kuppe, C. et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 589, 281–286 (2021).
Lasagni, L., Lazzeri, E. & Shankland, S. J. Podocyte mitosis—a catastrophe. Curr. Mol. Med. 13, 13–23 (2013).
Atchison, L. et al. iPSC-derived endothelial cells affect vascular function in a tissue-engineered blood vessel model of Hutchinson–Gilford progeria syndrome. Stem Cell Rep. 14, 325–337 (2020).
Roye, Y. et al. A personalized glomerulus chip engineered from stem cell-derived epithelium and vascular endothelium. Micromachines 12, 967 (2021).
Okafor, A. E., Bhattacharya, R. & Musah, S. in iPSCs in Tissue Engineering, Vol 11 (ed. Birbrair, A.) 329–370 (Elsevier, 2021).
Robert, B., Zhao, X. & Abrahamson, D. R. Coexpression of neuropilin-1, Flk1, and VEGF164 in developing and mature mouse kidney glomeruli. Am. J. Physiol. Renal Physiol. 279, F275–F282 (2000).
Eremina, V. et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Invest. 111, 707–716 (2003).
Kalejaiye, T. D., Holmes, J. A., Bhattacharya, R. & Musah, S. in Regenerative Nephrology 2nd edn (ed. Goligorsky, M. S.) 331–351 (Elsevier, 2022).
Dittrich, S. et al. Renal impairment in patients with long-standing cyanotic congenital heart disease. Acta Paediatr. 87, 949–954 (1998).
Krull, F. et al. Renal involvement in patients with congenital cyanotic heart disease. Acta Paediatr. Scand. 80, 1214–1219 (1991).
Zheng, J., Yao, Y., Han, L. & Xiao, Y. Renal function and injury in infants and young children with congenital heart disease. Pediatr. Nephrol. 28, 99–104 (2013).
van der Linde, D. et al. Birth prevalence of congenital heart disease worldwide. J. Am. Coll. Cardiol. 58, 2241–2247 (2011).
Henry, G. L. & Melton, D. A. Mixer, a homeobox gene required for endoderm development. Science 281, 91–96 (1998).
Nishita, M. et al. Interaction between Wnt and TGF-LN signalling pathways during formation of Spemann’s organizer. Nature 403, 781–785 (2000).
Hofbauer, P. et al. Cardioids reveal self-organizing principles of human cardiogenesis. Cell 184, 3299–3317.e22 (2021).
Wagner, K.-D. et al. An inducible mouse model for PAX2-dependent glomerular disease: insights into a complex pathogenesis. Curr. Biol. 16, 793–800 (2006).
Dressler, G. R. et al. Deregulation of Pax-2 expression in transgenic mice generates severe kidney abnormalities. Nature 362, 65–67 (1993).
Murer, L. et al. Expression of nuclear transcription factor PAX2 in renal biopsies of juvenile nephronophthisis. Nephron 91, 588–593 (2002).
Fang, N.-W. et al. Incidence and risk factors for chronic kidney disease in patients with congenital heart disease. Pediatr. Nephrol. 36, 3749–3756 (2021).
Le Jemtel, T. H. et al. Direct evidence of podocyte damage in cardiorenal syndrome type 2: preliminary evidence. Cardiorenal Med. 5, 125–134 (2015).
Heyer, J. et al. Postgastrulation Smad2-deficient embryos show defects in embryo turning and anterior morphogenesis. Proc. Natl Acad. Sci. USA 96, 12595–12600 (1999).
Duan, W.-J., Yu, X., Huang, X.-R., Yu, J. & Lan, H. Y. Opposing roles for Smad2 and Smad3 in peritoneal fibrosis in vivo and in vitro. Am. J. Pathol. 184, 2275–2284 (2014).
Sharma, A. et al. CRISPR/Cas9-mediated fluorescent tagging of endogenous proteins in human pluripotent stem cells. Curr. Protoc. Hum. Genet. 96, 21.11.1–21.11.20 (2018).
Musah, S., Dimitrakakis, N., Camacho, D. M., Church, G. M. & Ingber, D. E. Directed differentiation of human induced pluripotent stem cells into mature kidney podocytes and establishment of a glomerulus chip. Nat. Protoc. 13, 1662–1685 (2018).
Kulkarni, R., Teves, M. E., Han, A. X., McAllister, J. M. & Strauss, J. F. Colocalization of polycystic ovary syndrome candidate gene products in theca cells suggests novel signaling pathways. J. Endocr. Soc. 3, 2204–2223 (2019).
Palikaras, K., SenGupta, T., Nilsen, H. & Tavernarakis, N. Assessment of dopaminergic neuron degeneration in a C. elegans model of Parkinson’s disease. STAR Protoc. 3, 101264 (2022).
Li, R. et al. Mapping single-cell transcriptomes in the intra-tumoral and associated territories of kidney cancer. Cancer Cell 40, 1583–1599.e10 (2022).
Wilson, P. C. et al. Multimodal single cell sequencing implicates chromatin accessibility and genetic background in diabetic kidney disease progression. Nat. Commun. 13, 5253 (2022).
Stewart, B. J. et al. Spatiotemporal immune zonation of the human kidney. Science 365, 1461–1466 (2019).
Lake, B. B. et al. An atlas of healthy and injured cell states and niches in the human kidney. Nature 619, 585–594 (2023).
Muto, Y. et al. Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nat. Commun. 12, 2190 (2021).
Karki, S. et al. Incidental finding of dextrocardia with situs inversus and absent left kidney: a case report. J. Nepal Med. Assoc. 60, 196–199 (2022).
Oka, H., Nakau, K., Kajihama, A. & Azuma, H. Assessment of potential renal dysfunction in patients with congenital heart disease after biventricular repair. Korean Circ. J. 48, 418–426 (2018).
Murakami, T. et al. Glomerular function depends on the period of cyanosis in adult patients with tetralogy of Fallot. J. Am. Coll. Cardiol. 67, 982 (2016).
Ekulu, P. M., Kazadi-wa-Kazadi, O., Lumbala, P. K. & Aloni, M. N. Nephrotic syndrome in a child suffering from tetralogy of Fallot: a rare association. Case Rep. Pediatr. 2015, 1–3 (2015).
Hehir, D. A. et al. Acute kidney injury following surgery for congenital heart disease: role of urine biomarkers, renal perfusion pressure, and somatic oxygen saturation. J. Am. Coll. Cardiol. 63, A589 (2014).
Mel, E., Davidovits, M. & Dagan, O. Long-term follow-up evaluation of renal function in patients treated with peritoneal dialysis after cardiac surgery for correction of congenital anomalies. J. Thorac. Cardiovasc. Surg. 147, 451–455 (2014).
Inatomi, J., Matsuoka, K., Fujimaru, R., Nakagawa, A. & Iijima, K. Mechanisms of development and progression of cyanotic nephropathy. Pediatr. Nephrol. 21, 1440–1445 (2006).
Adedoyin, O. T. & Afolabi, J. K. Sudden deterioration in the renal function of an African child with cyanotic congenital heart disease. J. Natl Med. Assoc. 98, 287–289 (2006).
Flanagan, M. F., Hourihan, M. & Keane, J. F. Incidence of renal dysfunction in adults with cyanotic congenital heart disease. Am. J. Cardiol. 68, 403–406 (1991).
Shaw, N. J., Brocklebank, J. T., Dickinson, D. F., Wilson, N. & Walker, D. R. Long-term outcome for children with acute renal failure following cardiac surgery. Int. J. Cardiol. 31, 161–165 (1991).
Ingelfinger, J. R., Kissane, J. M. & Robson, A. M. Glomerulomegaly in a patient with cyanotic congenital heart disease. Am. J. Dis. Child 120, 69–71 (1970).
Acknowledgements
S.M. is a recipient of the Whitehead Scholarship in Biomedical Research, a Chair’s Research Award from the Department of Medicine at Duke University, a MEDx Pilot Grant on Biomechanics in Injury or Injury Repair, a Burroughs Wellcome Fund PDEP Career Transition Ad Hoc Award, a Duke Incubation Fund from the Duke Innovation & Entrepreneurship Initiative, a Genentech Research Award and a George M. O’Brien Kidney Center Pilot Grant (P30 DK081943), and NIH Director’s New Innovator Award (Award Number DP2DK139544), which supported the study. R.B. is a recipient of the Lew’s Predoctoral Fellowship in the Center for Biomolecular and Tissue Engineering (CBTE) at Duke University (T32 Support NIH Grant T32GM800555). We acknowledge the National Heart, Lung, and Blood Institute Pediatrics Cardiac Genomics Consortium (PCGC) investigators (R01 HL151257 (C.E.S.), 1UM1HL098166 (J.G.S.) and others) for their support and expertise. T.W. is a recipient of the Ruth L. Kirschstein National Research Service Award (NRSA) T32 Fellowship (2T32 HL 7208-46 A1). J.G.S. is supported by Foundation Leducq 16 CVD 03, and C.E.S. is supported by the Howard Hughes Medical Institute. We thank D. W. Cain from the Duke Human Vaccine Institute (DHVI) Flow Cytometry Facility for assistance with the flow cytometry experiments and data analyses; J. Faust from Evident and L. Cameron from the Department of Biology, Duke University, for helping with the glomerulus-on-a-chip microscopy; Y. Gao for assistance with the live microscopy setup; X. Mou for plasma treating the microfluidic organ chips; and Y. Roye for technical assistance with endothelial cell differentiation. We also thank all members of the Musah Lab and M. Pachino from the Graduate Communication Center (GCC) at Duke University for helpful comments on the paper.
Author information
Authors and Affiliations
Contributions
S.M. and T.W. conceived the idea for this work. S.M., T.W., J.G.S. and R.B. discussed the research approach and strategy. T.W. performed CRISPR–Cas9 experiments, generated the SMAD2 mutant human iPS cell lines used in this study, analysed relevant data and interpreted the results with input from J.G.S. and C.E.S. R.B. performed stem cell differentiation experiments, analysed the data and interpreted all relevant results with input from T.D.K. and S.M. R.B. and S.M.L. performed qPCR analyses. R.B., T.D.K. and H.A. performed western blot analyses. R.B. performed microscopy and microfluidic organ chip experiments and analysed the data. T.D.K. performed protein quantification, endothelial cell differentiation and EdU analysis. T.D.K. and R.B. performed CCK-8 cell viability assay. A.M. performed cell migration analyses from data generated by R.B. and also provided new strategies for visualizing the data. R.B., T.W., T.D.K. and S.M. interpreted the study results. R.B. wrote the initial draft of the paper with input from the other authors. S.M., T.W. and S.M.L. edited the paper. T.W. was supervised by J.G.S. and C.E.S. R.B., T.D.K., A.M., S.M.L. and H.A. were supervised by S.M.
Corresponding author
Ethics declarations
Competing interests
S.M. is an inventor on a patent regarding podocyte differentiation held by Harvard University (US20210338736A1; US Patent App 17/366,827, 2021). S.M. is also an inventor on a pending patent application regarding engineered microphysiological systems with in vivo-like tissue structure and function held by Duke University. The other authors declare no competing interests.
Peer review
Peer review information
Nature Biomedical Engineering thanks Azhar Mohamad, Ryuji Morizane and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Derivation and characterization of SMAD2 mutant cell lines.
a, Schematic representation of SMAD2 mutation installation in human iPS cells. b, Introduction of SMAD2 variants into human iPS cells was confirmed by Sanger Sequencing. c, Representative phase contrast images of WT, SMAD2−/−, and SMAD2mh1/mh1 iPS cells. Scale bar, 650 µm. d, Representative immunoblots showing loss of SMAD2 protein in the mutant cells. e, Karyotypic analysis of WT and mutant cell lines after 4 passages post-gene edits. n = 20 cells from each cell line. f, Representative phase contrast and immunofluorescent images from 3 independent experiments showing the expression of the pluripotency marker OCT4 (red). DAPI (blue) labels the nuclei, and the scale bar represents 150 µm.
Extended Data Fig. 2 Mesoderm derivation.
a, Immunoblots showing expression of SMAD2, SMAD3, pSMAD3, and SMAD4 in the mesoderm cells. Quantifications represent mean ± SEM; pSMAD3 was increased in SMAD2−/− (P = 0.0396) and SMAD2mh1/mh1 (P = 0.0076) mesoderm cells. SMAD3 was increased in SMAD2−/− (P = 0.0874) and SMAD2mh1/mh1 (P = 0.0164) mesoderm cells. n = 3 independent experiments. b, Schematic representation of the derivation of posterior mesoderm cells. c, Representative phase contrast images of WT, SMAD2−/−, and SMAD2mh1/mh1 iPS cell-derived posterior mesoderm. Scale bar, 275 µm. d, Immunoblots showing expression of mesoderm lineage-specific markers in the differentiated mesoderm derived with or without Activin A pulses. n = 2 independent experiments. e, RNA expression of posterior mesoderm-specific genes. Data are mean values ± SEM; n = 3 independent experiments. f, Quantification of cell-secreted Activin A on Days 1 and 2 during posterior mesoderm induction. Data are mean values ± SEM; n = 2 independent experiments. We performed a P < 0.05 one-way ANOVA with Dunnett’s multiple comparison test (a, e). Color symbols shown in the histograms of Figs. a and e represent a biological replicate. Same colors in Fig. e indicate 3 independent wells of mesoderm cells from each biological replicate. Representative images from 3 independent experiments are shown in Fig. c. Images of all the stain-free gels and stain-free blots can be found in Supplementary Fig. 6a. Source data are available for this figure.
Extended Data Fig. 3
IM derivation. a, Immunostaining for Actin and DAPI. b, Representative whole-well scan of differentiated IM cells after 14 d of induction. c, Snapshots of the velocity field obtained from Particle Image Velocimetry analysis of divergence of WT and mutant IM cells. The arrows indicate local divergence to the mean direction of migration. d, Immunoblots showing expression of EMT markers SNAI1 and TWIST in IM cells. n = 2 independent experiments. e, Representative immunoblots showing expression of pSMAD1, SMAD2, SMAD3, and SMAD4 in the IM cells. n = 3 independent experiments. pSMAD1 was increased in SMAD2−/− (P = 0.0019) and SMAD2mh1/mh1 (P = 0.0046) IM cells. SMAD3 was increased in SMAD2−/− (P = 0.0368) and SMAD2mh1/mh1 (P = 0.0659) IM cells. f, Flow cytometry analysis and g, quantitation of PE-SNAI1 and BV421-WT1. n = 3 independent experiments. h, Flow cytometry plot and i, quantitation showing co-expression of necroptosis marker AF488-MLKL and BV421-WT1. n = 3 independent experiments. j, Immunoblots showing expression of MLKL oligomers, and k, αSMA in differentiating IM cells. n = 2 independent experiments. n = 2 independent experiments. l, RNA expression analyses for canonical and noncanonical WNT target genes and m, mesenchymal genes in IM cells. n = 3 independent experiments. Data are presented as mean ± SEM (Figs. e, g, i, l, m). We performed a P < 0.05 one-way ANOVA with Dunnett’s multiple comparison test (e, g, i, l) and a paired two-tailed t-test (m). Color symbols shown in the histograms of Figs. e, g, i, l, and m represent a biological replicate. Same colors in Figs. l and m indicate 3 independent wells of IM cells from each biological replicate. Representative images from 3 independent experiments are shown in Figs. (a, b) Scale bar: 100 µm (a). For immunoblots, see supplementary Figs. 6b, and 7. Source data is available for this figure.
Extended Data Fig. 4 Podocyte derivation.
a, Phase contrast images showing podocyte foot processes (WT) and effacement-like phenotype (mutants). b, Fluorescence intensity quantification of podocyte lineage-specific markers, n = 3 independent experiments. c, Immunostaining for Nephrin (green), CD2AP (red), and WT1 (yellow) in differentiated podocytes. DAPI, shown in grey, labels the nuclei. The nuclear intensity of WT1 was significantly downregulated in the mutant podocytes. n = 3 independent experiments. d, Representative immunoblots and e, quantitation of pSMAD1, SMAD2, pSMAD3, SMAD3, SMAD6, and SMAD7 in podocytes across 3 biologically independent experiments. f, Immunostaining for YAP (magenta) and PAX2 (green). The nuclear intensity of PAX2 was significantly increased in the mutant podocytes. n = 3 independent experiments. g, Immunostaining for Actin. Top right: zoomed-in regions indicate actin dissolution and subsequent podocyte effacement. h, CCK-8 cell viability assay. n = 3 independent experiments. i, Bubble plot comparing the expression of markers in healthy and diseased human kidney podocytes. j, SMAD3 knockdown podocytes underwent foot process effacement (indicated with orange arrows) and cell death. k, Cell size was significantly reduced after SMAD3 knockdown. n = 3 independent experiments. l, Cell viability was significantly reduced after SMAD3 knockdown. n = 3 independent experiments. m, Immunoblots showing expression of SMAD3 and Synaptopodin after SMAD3 knockdown. n = 2 biologically independent experiments. Data are presented as mean ± SEM (Figs. b, c, e, f, h, I, k, l). We performed a P < 0.05 one-way ANOVA with Dunnett’s (b, c, e, g, h) and Sidak’s (k, l) multiple comparison tests. Color symbols shown in the histograms of Figs. b, c, e, f, h, k, and l represent a biological replicate. Representative images from 3 independent experiments are shown in Figs. (a, c, f, j) and 2 independent experiments are shown in Fig. g. Scale bar: 150 µm (a), 100 µm (c, f, j). For immunoblots, images of all the stain-free gels and stain-free blots are shown in Supplementary Fig. 8a. Source data are available for this figure.
Extended Data Fig. 5 Endothelial cell derivation.
a, RNA expression of mesenchymal markers ICAM1, Smoothelin, and Vimentin in WT and mutant endothelial cells. Data are mean values ± SEM; n = 3 independent experiments. ICAM1 was increased in SMAD2−/− (P = 0.0009) and SMAD2mh1/mh1 (P = 0.0078) endothelial cells. Smoothelin was increased in SMAD2−/− (P < 0.0001) and SMAD2mh1/mh1 (P = 0.0020) endothelial cells. Vimentin was increased in SMAD2−/− (P < 0.0001) and SMAD2mh1/mh1 (P < 0.0001) endothelial cells. We performed a P < 0.05 one-way ANOVA with Dunnett’s multiple comparison test. Same colors indicate 3-4 independent wells of endothelial cells from each biological replicate. b, Representative immunofluorescent images showing the expression of mesenchymal markers Transgelin (cyan) and TWIST (red) in endothelial cells after 2 weeks of maintenance culture. n = 2 independent experiments. Scale bar, 100 µm.
Extended Data Fig. 6 Recapitulating the glomerular capillary wall using a glomerulus-on-a-chip device.
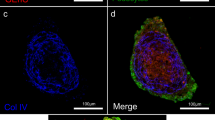
a, Schematic representation of the development of glomerulus-on-a-chip cultures. b, Example photograph of the experimental setup for glomerulus-on-a-chip cultures connected to two reservoirs for the cell culture media for the urinary (podocyte induction media) and capillary (CultureBoost-R) channels. These microfluidic glomerulus-on-a-chip were connected to media reservoirs via low-retention inlet and outlet tubing, using a peristaltic pump. Media was recirculated and replenished every alternate day. c, Immunostaining for Nephrin (magenta)- and PECAM1 (cyan)-positive podocytes and endothelial cells, respectively. d, Single-channel images of Nephrin+ podocytes (magenta, urinary channel) and PECAM1+ endothelial cells (cyan, capillary channel) along the z-axis. e, ‘Y’ junction of the chips to visualize the incoming apical and basal channels before they overlay at the interface. f, Immunostaining for Nephrin (red) in WT, SMAD2−/−, and SMAD2mh1/mh1 podocytes on the glomerulus-on-a-chip platform cultured in the absence of the endothelial cells. DAPI, shown in grey, labels the nuclei. Representative images from 3 independent experiments are shown in Figs. (c, d, f). Scale bars: 50 µm (c), 30 µm (d), 100 µm (e, f).
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–8, Tables 1 and 2, and Notes 1–3.
Supplementary Video 1 (download AVI )
Time-lapse video of differentiating WT intermediate mesoderm cells.
Supplementary Video 2 (download AVI )
Time-lapse video of differentiating SMAD2−/− intermediate mesoderm cells.
Supplementary Video 3 (download AVI )
Time-lapse video of differentiating SMAD2mh1/mh1 intermediate mesoderm cells.
Source data
Source Data Figs. 1–5 and Extended Data Figs. 2–5 (download XLSX )
Statistical source data.
Source Data Figs. 1–4 and Extended Data Figs. 1–4 (download PDF )
Unprocessed western blots.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bhattacharya, R., Ward, T., Kalejaiye, T.D. et al. Engineered human induced pluripotent stem cell models reveal altered podocytogenesis in congenital heart disease-associated SMAD2 mutations. Nat. Biomed. Eng (2025). https://doi.org/10.1038/s41551-025-01543-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41551-025-01543-0
