
Abstract
Accurate tracking of bacterial strains that stably engraft in faecal microbiota transplant (FMT) recipients is critical for understanding the determinants of strain engraftment, evaluating correlations with clinical outcomes and guiding the development of therapeutic consortia. While short-read sequencing has advanced FMT research, it faces challenges in strain-level de novo metagenomic assembly. Here we describe LongTrack, a method that uses long-read metagenomic assemblies for FMT strain tracking. LongTrack shows higher precision and specificity than short-read approaches, especially when multiple strains co-exist in the same sample. We uncovered 648 engrafted strains across six FMT cases involving patients with recurrent Clostridioides difficile infection and inflammatory bowel disease. Furthermore, long reads enabled assessment of the genomic and epigenomic stability of engrafted strains at the 5-year follow-up timepoint, revealing structural variations that may be associated with strain adaptation in a new host environment. Our findings support the use of long-read metagenomics to track microbial strains and their adaptations.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
All sequencing data generated for this study are available via the Sequence Read Archive under BioProject (PRJNA1101882). Source data are provided with this paper.
Code Availability
Customized scripts of LongTrack and a tutorial with supporting data are available via GitHub at https://github.com/fanglab/LongTrack. Source data are provided with this paper.
References
Feuerstadt, P. et al. SER-109, an oral microbiome therapy for recurrent Clostridioides difficile infection. N. Engl. J. Med. 386, 220–229 (2022).
Khanna, S. et al. Efficacy and safety of RBX2660 in PUNCH CD3, a phase III, randomized, double-blind, placebo-controlled trial with a Bayesian primary analysis for the prevention of recurrent Clostridioides difficile infection. Drugs 82, 1527–1538 (2022).
Ooijevaar, R. E., Terveer, E. M., Verspaget, H. W., Kuijper, E. J. & Keller, J. J. Clinical application and potential of fecal microbiota transplantation. Annu. Rev. Med. 70, 335–351 (2019).
Li, S. S. et al. Durable coexistence of donor and recipient strains after fecal microbiota transplantation. Science 352, 586–589 (2016).
Pamer, E. G. Fecal microbiota transplantation: effectiveness, complexities, and lingering concerns. Mucosal Immunol. 7, 210–214 (2014).
Bethlehem, L. et al. Microbiota therapeutics for inflammatory bowel disease: the way forward. Lancet Gastroenterol. Hepatol. 9, 476–486 (2024).
Paramsothy, S. et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet 389, 1218–1228 (2017).
Rossen, N. G. et al. Findings from a randomized controlled trial of fecal transplantation for patients with ulcerative colitis. Gastroenterology 149, 110–118.e4 (2015).
Sevcikova, A. et al. The impact of the microbiome on resistance to cancer treatment with chemotherapeutic agents and immunotherapy. Int. J. Mol. Sci. 23, 488 (2022).
Davar, D. et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 371, 595–602 (2021).
Zhao, S. et al. Adaptive evolution within the gut microbiomes of healthy people. Cell Host Microbe 25, 656–667.e8 (2019).
Lloyd-Price, J. et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 550, 61–66 (2017).
Porcari, S. et al. Key determinants of success in fecal microbiota transplantation: from microbiome to clinic. Cell Host Microbe 31, 712–733 (2023).
Ianiro, G. et al. Variability of strain engraftment and predictability of microbiome composition after fecal microbiota transplantation across different diseases. Nat. Med. 28, 1913–1923 (2022).
Aggarwala, V. et al. Precise quantification of bacterial strains after fecal microbiota transplantation delineates long-term engraftment and explains outcomes. Nat. Microbiol. 6, 1309–1318 (2021).
Duvallet, C. et al. Framework for rational donor selection in fecal microbiota transplant clinical trials. PLoS ONE 14, e0222881 (2019).
Podlesny, D. et al. Identification of clinical and ecological determinants of strain engraftment after fecal microbiota transplantation using metagenomics. CR Med. 3, 100711 (2022).
Luo, C. et al. ConStrains identifies microbial strains in metagenomic datasets. Nat. Biotechnol. 33, 1045–1052 (2015).
Smillie, C. S. et al. Strain-tracking reveals the determinants of bacterial engraftment in the human gut following fecal microbiota transplantation. Cell Host Microbe 23, 229–240 (2018).
Schmidt, T. S. B. et al. Drivers and determinants of strain dynamics following fecal microbiota transplantation. Nat. Med. 28, 1902–1912 (2022).
Olm, M. R. et al. InStrain enables population genomic analysis from metagenomic data and sensitive detection of shared microbial strains. Nat. Biotechnol. 39, 727–736 (2021).
Blanco-Míguez, A. et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 41, 1633–1644 (2023).
Van Rossum, T., Ferretti, P., Maistrenko, O. M. & Bork, P. Diversity within species: interpreting strains in microbiomes. Nat. Rev. Microbiol. 18, 491–506 (2020).
Gounot, J.-S. et al. Genome-centric analysis of short and long read metagenomes reveals uncharacterized microbiome diversity in Southeast Asians. Nat. Commun. 13, 6044 (2022).
Sczyrba, A. et al. Critical Assessment of Metagenome Interpretation—a benchmark of computational metagenomics software. Nat. Methods 14, 1063–1071 (2017).
Tidjani Alou, M. et al. State of the art in the culture of the human microbiota: new interests and strategies. Clin. Microbiol. Rev. https://doi.org/10.1128/cmr.00129-19 (2020).
Moss, E. L., Maghini, D. G. & Bhatt, A. S. Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nat. Biotechnol. 38, 701–707 (2020).
Gehrig, J. L. et al. Finding the right fit: evaluation of short-read and long-read sequencing approaches to maximize the utility of clinical microbiome data. Microb. Genomics 8, 000794 (2022).
Kim, C. Y., Ma, J. & Lee, I. HiFi metagenomic sequencing enables assembly of accurate and complete genomes from human gut microbiota. Nat. Commun. 13, 6367 (2022).
Vicedomini, R., Quince, C., Darling, A. E. & Chikhi, R. Strainberry: automated strain separation in low-complexity metagenomes using long reads. Nat. Commun. 12, 4485 (2021).
Agustinho, D. P. et al. Unveiling microbial diversity: harnessing long-read sequencing technology. Nat. Methods 21, 954–966 (2024).
Chen, L. et al. Short- and long-read metagenomics expand individualized structural variations in gut microbiomes. Nat. Commun. 13, 3175 (2022).
Tourancheau, A., Mead, E. A., Zhang, X.-S. & Fang, G. Discovering multiple types of DNA methylation from bacteria and microbiome using nanopore sequencing. Nat. Methods 18, 491–498 (2021).
Beaulaurier, J. et al. Metagenomic binning and association of plasmids with bacterial host genomes using DNA methylation. Nat. Biotechnol. 36, 61–69 (2018).
Beaulaurier, J., Schadt, E. E. & Fang, G. Deciphering bacterial epigenomes using modern sequencing technologies. Nat. Rev. Genet. 20, 157–172 (2019).
Kong, Y. et al. Critical assessment of DNA adenine methylation in eukaryotes using quantitative deconvolution. Science 375, 515–522 (2022).
Fang, G. et al. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol. 30, 1232–1239 (2012).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Li, D., Liu, C.-M., Luo, R., Sadakane, K. & Lam, T.-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Kent, A. G., Vill, A. C., Shi, Q., Satlin, M. J. & Brito, I. L. Widespread transfer of mobile antibiotic resistance genes within individual gut microbiomes revealed through bacterial Hi-C. Nat. Commun. 11, 4379 (2020).
Bae, M. et al. Akkermansia muciniphila phospholipid induces homeostatic immune responses. Nature 608, 168–173 (2022).
Wu, Z. et al. Akkermansia muciniphila ameliorates Clostridioides difficile infection in mice by modulating the intestinal microbiome and metabolites. Front. Microbiol. 13, 841920 (2022).
Leylabadlo, H. E. et al. The critical role of Faecalibacterium prausnitzii in human health: an overview. Microb. Pathogen. 149, 104344 (2020).
Quévrain, E. et al. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s disease. Gut 65, 415–425 (2016).
Chen-Liaw, A. et al. Gut microbiota strain richness is species specific and affects engraftment. Nature 637, 422–429 (2025).
Durrant, M. G. & Bhatt, A. S. Microbiome genome structure drives function. Nat. Microbiol. 4, 912–913 (2019).
Kong, Y., Mead, E. A. & Fang, G. Navigating the pitfalls of mapping DNA and RNA modifications. Nat. Rev. Genet. 24, 363–381 (2023).
Noinaj, N., Guillier, M., Barnard, T. ravisJ. & Buchanan, S. K. TonB-dependent transporters: regulation, structure, and function. Annu. Rev. Microbiol. 64, 43–60 (2010).
Dingemans, J. et al. The deletion of TonB-dependent receptor genes is part of the genome reduction process that occurs during adaptation of Pseudomonas aeruginosa to the cystic fibrosis lung. Pathog. Dis. 71, 26–38 (2014).
Partridge, S. R., Kwong, S. M., Firth, N. & Jensen, S. O. Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. https://doi.org/10.1128/cmr.00088-17 (2018).
Maqbool, A. et al. The substrate-binding protein in bacterial ABC transporters: dissecting roles in the evolution of substrate specificity. Biochem. Soc. Trans. 43, 1011–1017 (2015).
Sánchez-Romero, M. A. & Casadesús, J. The bacterial epigenome. Nat. Rev. Microbiol. 18, 7–20 (2020).
Beaulaurier, J. et al. Single molecule-level detection and long read-based phasing of epigenetic variations in bacterial methylomes. Nat. Commun. 6, 7438 (2015).
Oliveira, P. H. & Fang, G. Conserved DNA methyltransferases: a window into fundamental mechanisms of epigenetic regulation in bacteria. Trends Microbiol. 29, 28–40 (2021).
Seib, K. L., Srikhanta, Y. N., Atack, J. M. & Jennings, M. P. Epigenetic regulation of virulence and immunoevasion by phase-variable restriction-modification systems in bacterial pathogens. Annu. Rev. Microbiol. 74, 655–671 (2020).
Oliveira, P. H. et al. Epigenomic characterization of Clostridioides difficile finds a conserved DNA methyltransferase that mediates sporulation and pathogenesis. Nat. Microbiol. 5, 166–180 (2020).
Lopez-Siles, M., Duncan, S. H., Garcia-Gil, L. J. & Martinez-Medina, M. Faecalibacterium prausnitzii: from microbiology to diagnostics and prognostics. ISME J. 11, 841–852 (2017).
Wu, H. et al. Strategies for high cell density cultivation of Akkermansia muciniphila and its potential metabolism. Microbiol. Spectr. 12, e0238623 (2024).
Zhernakova, D. V. et al. Host genetic regulation of human gut microbial structural variation. Nature 625, 813–821 (2024).
West, P. T., Chanin, R. B. & Bhatt, A. S. From genome structure to function: insights into structural variation in microbiology. Curr. Opin. Microbiol. 69, 102192 (2022).
Furuta, Y. et al. Methylome Diversification through changes in DNA methyltransferase sequence specificity. PLoS Genet. 10, e1004272 (2014).
Srikhanta, Y. N., Fox, K. L. & Jennings, M. P. The phasevarion: phase variation of type III DNA methyltransferases controls coordinated switching in multiple genes. Nat. Rev. Microbiol. 8, 196–206 (2010).
Ji, P., Zhang, Y., Wang, J. & Zhao, F. MetaSort untangles metagenome assembly by reducing microbial community complexity. Nat. Commun. 8, 14306 (2017).
Cao, L. et al. mEnrich-seq: methylation-guided enrichment sequencing of bacterial taxa of interest from microbiome. Nat. Methods 21, 236–246 (2024).
Sarkar, A. et al. Microbial transmission in the social microbiome and host health and disease. Cell 187, 17–43 (2024).
Kolmogorov, M. et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat. Methods 17, 1103–1110 (2020).
Wu, Y.-W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. G. T. D. B. - Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2020).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 158 (2018).
Antipov, D., Raiko, M., Lapidus, A. & Pevzner, P. A. Metaviral SPAdes: assembly of viruses from metagenomic data. Bioinformatics 36, 4126–4129 (2020).
Tang, X., Shang, J., Ji, Y. & Sun, Y. PLASMe: a tool to identify PLASMid contigs from short-read assemblies using transformer. Nucleic Acids Res. 51, e83 (2023).
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114 (2018).
Rodriguez-R, L. M. et al. An ANI gap within bacterial species that advances the definitions of intra-species units. mBio 15, e0269623 (2023).
Marçais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944 (2018).
Kokot, M., Dlugosz, M. & Deorowicz, S. KMC 3: counting and manipulating k-mer statistics. Bioinformatics 33, 2759–2761 (2017).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Yang, C., Chu, J., Warren, R. L. & Birol, I. NanoSim: nanopore sequence read simulator based on statistical characterization. GigaScience 6, gix010 (2017).
Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner (Lawrence Berkeley National Laboratory, 2014); https://sourceforge.net/projects/bbmap/
Sedlazeck, F. J. et al. Accurate detection of complex structural variations using single molecule sequencing. Nat. Methods 15, 461–468 (2018).
Smolka, M. et al. Detection of mosaic and population-level structural variants with Sniffles2. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-02024-y (2024).
Acknowledgements
This work was supported in part by the staff and resources of the Microbiome Translational Center, Center for AI-Driven Genomic and Microbiome Medicine and Department of Scientific Computing at the Icahn School of Medicine at Mount Sinai. This work was supported by grant no. R35 GM139655 (G.F.) from the National Institutes of Health. G.F. is a Hirschl Research Scholar by Irma T. Hirschl/Monique Weill-Caulier Trust and a Nash Family Research Scholar.
Author information
Authors and Affiliations
Contributions
Initiated study: G.F. and J.J.F. Developed methodology: Y.F. and G.F. Data analysis: Y.F., J.J.F. and G.F. Data interpretation: Y.F., M.N., V.A., E.A.M., M.K., L.C., J.J.F. and G.F. Collection and interpretation of clinical data: M.A.K., T.J.B., S.P., N.O.K. and A.G. Supervised research: G.F. Wrote first draft of paper: Y.F. and G.F. Approved paper: all authors.
Corresponding author
Ethics declarations
Competing interests
S.P. has served as a consultant/steering committee member for Vedanta Biosciences and has received speaker/advisory board fees from AbbVie, Dr Falk Pharma, Ferring, Janssen and Takeda. J.J.F. is a consultant for Vedanta Biosciences and Genfit. The other authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
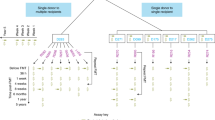
Extended Data Fig. 1 Description of sample collection and FMT study design for CDI patients.
Stool samples were collected from three donors and four recipients at multiple time points. For the illustration of LongTrack, strain tracking was performed for long-read MAGs assembled from donors and recipients using short-read metagenomic data generated across all the samples shown in the figure (Methods). The Venn diagram displays the number of strains that were recovered by long-read (LR) MAGs (yellow color) or isolate genomes from bacterial culture (red color) from 3 donors and 2 recipients.
Extended Data Fig. 2 Illustration of LongTrack workflow’s implementation in this study for generating strain-level long-read MAGs.
Initially, the raw long reads from donor and pre-FMT recipient underwent metagenomic assembly using the metaFlye (v2.9). The resulting Metagenome-assembled genomes (MAGs) were then partitioned into individual bins corresponding to the species level using MaxBin (v2.0). Strainberry (v1.1) was used to separate the contigs into strain level based on haplotype phasing for each species-level bin. Subsequently, metaWRAP (v1.3.2) was employed to bin the separated contigs into strain level. Finally, the strain-level long-read MAGs with completeness of more than 10% were chosen for FMT strain tracking in post-FMT recipients (Methods).
Extended Data Fig. 3 Comparative evaluation of long-read MAGs and short-read MAGs from Donor 1.
A. Completeness levels of 124 long-read MAGs (LR MAGs) and 135 short-read MAGs (SR MAGs) from Donor 1 reveal no significant differences. B. Contamination levels of 124 LR MAGs and 135 SR MAGs from Donor 1 assessed using CheckM, showing significantly lower contamination in LR MAGs (p = 0.043; two-sided Wilcoxon rank sum test; p < 0.05, *). C. Contig N50 (kb) comparison for 124 LR MAGs and 135 SR MAGs Donor 1, showing significantly higher N50 in LR MAGs (p = 2.2e-16; two-sided Wilcoxon rank sum test; p < 0.01, **).
Extended Data Fig. 4 Comparative evaluation of long-read MAGs and short-read MAGs from Donor 1.
A. Schematic of contamination level by mapping long-read and short-read MAG to its corresponding isolate genome, calculated as the percentage of unaligned bases contamination; B. Comparative analysis of contamination levels determined by comparing it to the isolate genome and by using the CheckM tool. A total of 60 long-read (LR) MAGs from Donors 1–3 and Recipients 1–4, and 23 short-read (SR) MAGs from Donor 1 (D1) were analyzed. (C–D) Comparison of contamination levels in corresponding strains between LR MAGs and SR MAGs from D1. (C) Bar plots illustrate contamination levels in corresponding strains between LR MAGs and SR MAGs for species with multiple co-existing strains and species with a single strain, emphasizing the consistently lower contamination observed in LR MAGs. D. Contamination levels for 31 LR MAGs and 23 SR MAGs from D1, categorized by species with either a single strain or multiple co-existing strains. LR MAGs consistently demonstrated lower contamination levels (p-value = 0.0048 for single strain; p-value = 0.0086 for multiple strain; two-sided permutation test on the mean difference, n = 10,000 permutations; p-value < 0.01, **) E. Genomic similarity of shared strains between the long-read (LR) MAGs/short-read (SR) MAGs and their corresponding isolate genomes determined by k-mer similarity (Methods).
Extended Data Fig. 5 Evaluation of FMT strain tracking between LongTrack and StrainFinder as a representative of SNP-based inference approach.
A. For the donor sample D1, in silico spike-in dataset was designed with 3% Clostridium tyrobutyricum and 2% Turicibacter sanguinis added to the microbiome dataset. For the post-FMT recipient R1, the spike-in data set A (low abundance) had a composition of 0.25% Clostridium tyrobutyricum and 0.1% Turicibacter sanguinis, while the spike-in data set B (high abundance) had a composition of 2.5% Clostridium tyrobutyricum and 1% Turicibacter sanguinis (Methods). B. The strain tracking results indicate that the SNP-based approach, StrainFinder, works well for strains with relatively modest-to-high abundance (data set B), but tends to miss strains with modest-to-low abundance (data set A). The chart represents the presence (green) or absence (gray) of strains in post-FMT recipients based on spike-in design (considering ground truth), LongTrack with long-read MAGs, or StrainFinder. Discrepancies with the ground truth are indicated by an “X” marker.
Extended Data Fig. 6 Sensitivity evaluation of LongTrack and culture-based approach (Strainer).
The sensitivity evaluation of LongTrack and the culture-based approach (Strainer) was performed using the union of all strains detected either through isolate genomes or MAGs as the reference set. To evaluate sensitivity across different microbial abundance levels, the reference set was stratified by relative abundance thresholds, which provides a comprehensive benchmark that encompasses the diversity of detectable strains, including those uniquely identified by each method. Numbers in brackets on the x-axis indicate the number of reference strains (n) in each bin.
Extended Data Fig. 7 LongTrack uncovered engraftment of cultured strains across FMT cases in rCDI patients.
A, Proportional engraftment of donor strains (PEDS) for shared cultured strains is shown across individual post-FMT recipients at various time points of the 5-year follow up. B, Proportional persistence of recipient strains (PPRS) for shared cultured strains is shown across individual post-FMT recipients at various time points of the 5-year follow up.
Extended Data Fig. 8 LongTrack uncovered uncultured of engrafted strains of rCDI.
The long-read MAGs uncultured by the previous study were utilized for strain tracking to determine if they were present in the post-FMT recipients for FMT cases of CDI: The results of tracking across three FMT cases D1- > R1 (A), D2- > R3 (B), and D3- > R4 (C), which describe the presence (green) or absence (gray) of strains based on LongTrack.
Extended Data Fig. 9 Reverse strain tracking of earlier samples based on long-read MAGs assembled from post-FMT recipients using LongTrack.
The long-read MAGs assembled from post-FMT recipient samples were utilized for strain tracking to determine if they were also present in the donor and pre-FMT recipient metagenomic data using LongTrack (Reverse tracking). The results of reverse tracking across three FMT cases: D1 - > R1, D2 - > R3 and D3 - > R4 are shown in A-C, which describe the presence (green) or absence (gray) of strains based on LongTrack. (D-F) Illustration of bacterial strains that may have been acquired from environmental sources, as determined by long-read MAGs assembled from the five-year post-FMT recipients’ samples but absent in all the earlier samples. The results across three FMT cases: D1 - > R1, D2 - > R3 and D3 - > R4 are shown in D-F, which describe the presence (green) or absence (gray) of strains based on LongTrack.
Extended Data Fig. 10 Description of samples collection and FMT study design for two IBD patients.
Stool samples were collected from six donors and two recipients at multiple time points. The two recipients (IBD R5 and IBD R6; orange) had received FMT from six donors (IBD D4 - D9; purple and gray). For the illustration of LongTrack, strain tracking was performed for long-read MAGs assembled from two donors (IBD D4 and D5; purple) using short-read metagenomic data generated across all the samples shown in the figure (Methods).
Supplementary information
Source data
Source Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 4 (download XLSX )
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Fan, Y., Ni, M., Aggarwala, V. et al. Long-read metagenomics for strain tracking after faecal microbiota transplant. Nat Microbiol 10, 3258–3271 (2025). https://doi.org/10.1038/s41564-025-02164-8
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41564-025-02164-8
