
Abstract
The innate immune response involves interferons (IFNs), antiviral cytokines that upregulate numerous IFN-stimulated genes, many of which have uncharacterized functions and mechanisms. Here we performed transcriptomic profiling of lung tissues from wild-type and IFNAR−/− mice infected with SARS-CoV-2 and single-cell RNA sequencing of bronchoalveolar lavage fluid and peripheral blood mononuclear cells from patients with COVID-19. We identified O-GalNAc transferase 2 (GALNT2), an N-acetylgalactosaminyltransferase, as an antiviral IFN-stimulated gene restricting the replication of multiple coronaviruses and influenza A viruses in vitro and in vivo, contributing to viral clearance and reducing disease severity. Mechanistically, GALNT2-dependent O-linked glycosylation may regulate viral glycoprotein proteolytic processing and impair viral growth by blocking virus–cell fusion. In addition, we found that serine residues at 810/813 in the viral spike protein undergo O-glycosylation and function as the primary genetic determinants of sensitivity or evasion towards GALNT2. Human genetic data analysis revealed that individuals with GALNT2 variants that lost antiviral function had elevated risk of hospitalization following SARS-CoV-2 infection. This study establishes GALNT2 as an antiviral factor against some respiratory virus infections.
Similar content being viewed by others

Main
Respiratory viruses, especially coronaviruses (CoVs) and influenza A viruses (IAVs), have been the major pathogens causing global pandemics1 and zoonotic spillover posing persistent threats to human public health. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus responsible for the coronavirus disease 2019 (COVID-19) pandemic, has deeply impacted global health, culture and prosperity, causing over seven million deaths2. Similarly, the first influenza pandemic of the twenty-first century emerged in 2009, rapidly disseminating worldwide and resulting in a minimum of 200,000 fatalities3. The innate immune response, interferons (IFNs) and the IFN-stimulated genes (ISGs), represents the first line of defence against viral assault4. Strong evidence showed that delayed or impaired type I interferon (IFN-I) response is a hallmark of both severe COVID-19 and influenza infections5,6,7: (1) 3.5% of patients with life-threatening COVID-19 expressed deleterious allelic variants of genes within the IFN system8, (2) anti-IFN-neutralizing autoantibodies were detected in nearly 15% of patients with critically severe COVID-19 (ref. 9), (3) recombinant IFNs possess therapeutic potential with proper timing and administration10 and (4) the markedly lower risk of children developing severe COVID-19 correlates with a stronger IFN immune response compared with adults11. The individual ISGs induced by IFNs were associated with protection from severe COVID-19, showing that this antiviral defence is a major component of protective antiviral responses5,12. Therefore, there is great interest in understanding how individual ISGs play a critical role in restricting viral infections.
Herein, we identified O-linked N-acetylgalactosamine (O-GalNAc) transferase 2 (GALNT2) as an ISG that promotes viral clearance and reduces the severity of and susceptibility to SARS-CoV-2 as well as IAV infections, both in vitro and in vivo. GALNT2 provides broad-spectrum and significant protection of the infected hosts via a mechanism by modulating O-linked glycosylation of coronavirus spikes and IAV haemagglutinin, and therefore interfering with viral proteolytic processing and impairing viral growth by blocking membrane fusion. These results provide experimental evidence for a critical role of the GALNT2-based antiviral defence in controlling a variety of respiratory virus infections.
Results
GALNT2 deficiency is associated with increased and prolonged SARS-CoV-2 replication in mice and patients
Previously, we successfully generated a mouse infection model of SARS-CoV-2 by exogenous delivery of human angiotensin-converting enzyme 2 (hACE2) with a replication-deficient adenovirus (Ad5-hACE2)13. Compared with the wild-type (WT) mice, the IFN-I receptor deficiency (IFNAR−/−) mice showed about tenfold higher viral loads, greater weight loss, enhanced inflammatory cell infiltration into the lungs and delayed virus clearance. Canonical IFN-I signalling activates the signal transducer and activator of transcription (STAT) pathway and produces ISGs. To identify the ISGs that restrict SARS-CoV-2 infection, we then performed transcriptional profiling of homogenized lungs of mice infected with SARS-CoV-2 and observed 28 potential ISGs that were fivefold lower expressed in IFNAR−/− mice than those in WT mice, including several well-known antiviral genes4,12,14 (Extended Data Fig. 1a,b). To assess the antiviral activity of these genes, we overexpressed each of the ISGs in Huh7 cells, including a positive control LY6E (ref. 15). Intriguingly, we found that O-GalNAc transferase 2 (GALNT2), an enzyme involved in the transfer of N-acetyl-D-galactosamine onto proteins, was the most pronounced gene that inhibits SARS-CoV-2 infection (Extended Data Fig. 1c,d). Indeed, after SARS-CoV-2 infection, WT mice showed rapidly increased GALNT2 protein expression in the lungs starting from 2 days post infection (d.p.i.), reaching the peak at 4 d.p.i., which correlates with virus clearance and rapid recovery. Conversely, the observed correlation between decreased GALNT2 protein level and elevated viral loads in IFNAR−/− mice persisted until 6 d.p.i., suggesting that dampened GALNT2 expression may lead to prolonged SARS-CoV-2 replication (Extended Data Fig. 1e–g).
To further elucidate the potential relationship between GALNT2 expression and severity of COVID-19, we conducted a comprehensive analysis of single-cell RNA sequencing (scRNA-seq) data obtained from bronchoalveolar lavage fluid (BALF) and peripheral blood mononuclear cell (PBMC) samples combining our data and published datasets16,17,18,19,20,21,22,23,24,25,26. Our study encompassed three distinct groups: healthy controls (HC) and patients with mild or severe COVID-19 (Fig. 1a,b and Extended Data Fig. 2a–c). We observed an increase in GALNT2 expression among SARS-CoV-2-infected patients when compared with the HC group in BALF cells. In particular, GALNT2 expression levels in ciliated cells, which serve as the primary targets for SARS-CoV-2 infection, were clearly higher in patients with mild COVID-19 than in severe cases (Fig. 1c and Extended Data Fig. 2d). Furthermore, the dynamic expression of GALNT2 throughout the course of infection was achieved by analysing PBMCs from SARS-CoV-2-infected patients at various stages of infection, including the initial infection period (T1), conversion phase (T2) and recovery (T3). Our single-cell RNA-seq analysis revealed an increase in GALNT2 expression during T2 of patients with mild COVID-19, compared with levels observed in patients with severe COVID-19 (Fig. 1d). Similarly, we detected higher GALNT2 protein expressions in the PBMC samples at T2 (Extended Data Fig. 2e). This finding suggested a role of GALNT2 in facilitating viral clearance, thus curbing disease severity and hastening recovery. Unlike well-known ISGs, GALNT2 is widely expressed in the lung and other tissues both at the RNA and protein levels. In SARS-CoV-2-infected patients, GALNT2 is markedly upregulated in multiple cell types and tissues, and the upregulation pattern of GALNT2 differs from that of classical ISGs (Fig. 1c and Extended Data Figs. 2e and 3a–e), suggesting a distinct regulatory response during SARS-CoV-2 infection.

a, A schematic overview of the scRNA-seq design. The composition and characteristics of PBMC and BALF patient samples included in the analysis are shown in Extended Data Fig. 2a–c. b, The uniform manifold approximation and projection (UMAP) of the cells from the BALF of HCs (n = 28), patients with moderate COVID-19 (n = 5) and patients with severe COVID-19 (n = 59). Each dot represents a cell, and different cell types are shown in different colours. Canonical markers for different cell types, including T, NK, B and ciliated cells in BALF are shown in dot plots. c, Comparison of GALNT2 expression in pseudo-bulk (left panel) and ciliated cells (right panel) of BALF samples using scRNA-seq. The box plots show the median (centre line) and the 25th and 75th percentiles (bounds of the box), and the whiskers extend to the smallest and largest values within 1.5 times the interquartile range from the box. Each dot represents an individual, and the y-axis represents the average expression level of GALNT2 across all cells or specifically in ciliated cells from that individual. Pearson correlation coefficients and P values by two-tailed test are indicated. The P values were calculated by two-tailed Wilcoxon signed-rank test. The pseudo-bulk analysis provides an overall expression trend across all cells, while the cell-type-specific analysis highlights distinct regulation in ciliated cells. d, Comparison of GALNT2 dynamic expression in PBMC samples using scRNA-seq. Each open dot represents an individual, and the y-axis represents the average expression level of GALNT2 in a specific cell type from that individual. A solid dot represents the average expression. The sampling time points are denoted as T1 (initial infection period), T2 (conversion phase) and T3 (recovery, after negative testing for SARS-CoV-2 virus). Samples from the same individual are connected by dashed lines. The P values were calculated by two-tailed Student’s t-test. e,f, Analysis of genetic variation at the GALNT2 gene locus between SARS-CoV-2-infected patients and control populations using data from the RGC COVID-19 results (e). The forest plot shows the risk of COVID-19 associated with GALNT2 SNP rs76000797 (f). P values were calculated by two-tailed Firth logistic regression test, with appropriate covariates and genomic control. Error bars indicate the 95% confidence interval. See Methods for details.
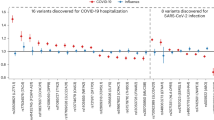
We next analysed COVID-19-related genome-wide association studies (GWAS) and found that the GALNT2 locus harbours single-nucleotide polymorphisms (SNPs), suggestive of an association of virus susceptibility with severity (Fig. 1e,f), as well as IAV infections, including H1N1 (ref. 27) and H7N9 (ref. 28) (Extended Data Fig. 4a,b). In the COVID-19 Host Genetics Initiative GWAS meta-analysis, carriers of V554M showed an elevated risk of hospitalization, suggesting that impaired GALTN2 function may decrease antiviral capacity (Extended Data Fig. 4c).
Identification of GALNT2 as an ISG with broad antiviral activity
To evaluate the antiviral activity of GALNT2 in vitro, we overexpressed GALNT2 in Huh7 cells and found that GALNT2 overexpressed does not impair cell viability and cell growth compared with controls (Extended Data Fig. 1h–k). Interestingly, GALNT2 markedly restricted SARS-CoV-2 (WT and Omicron) replication at various time points (Fig. 2a and Extended Data Fig. 5a–d). Conversely, endogenous GALNT2 knockout by CRISPR–Cas9 in Huh7 (GALNT2−/−) cells (Extended Data Fig. 4f) prominently facilitated SARS-CoV-2 replication (Fig. 2c). To further determine whether GALNT2 has broad-spectrum antiviral activity, cells were infected with various CoVs in Huh7 cells (Fig. 2a) and IAVs in A549 cells (Fig. 2b and Extended Data Fig. 4g). We found that overexpression of GALNT2 significantly inhibited viral replication of both CoVs and IAVs. By contrast, CoVs and IAVs replicated more efficiently and produced more infectious particles in the GALNT2−/− cells (Fig. 2c and Extended Data Fig. 4i–k). Interestingly, GALNT2−/− cells showed similar susceptibility to viral replication as STAT1−/− cells, indicating that GALNT2 is a crucial antiviral effector. Furthermore, reconstitution of GALNT2 expression in knockout cells restored its antiviral activity (Fig. 2c), showing that both endogenous and exogenous GALNT2 were able to broadly limit CoV and IAV replication. Interestingly, the four GALTN2 variants (V554M, V552M, D314A, Q216H) in the GnomAD database showed abrogated ability to inhibit SARS-CoV-2 or IAV infections (Extended Data Fig. 4d,e).

a, Huh7 cells were transfected with plasmids encoding GALNT2 (EV and eGFP as the controls). At 24 h post-transfection, cells were infected with CoVs including SARS-CoV-2 WT and Omicron BA.5 at an MOI of 0.01, HCoV-OC43 at an MOI of 0.05 and HCoV-229E at an MOI of 0.001. The viral titres were detected at 24, 48 and 72 hours post infection (h.p.i.) by FFA. Data are means ± s.d. from n = 3 independent experiments. P values are from two-tailed Student’s t-test. b, A549 cells were transfected with plasmids encoding GALNT2 (EV and eGFP as the controls). At 24 h post-transfection, cells were infected with IAVs at an MOI of 0.01, including H1N1 (PR8 and CA04), H3N2 (HK68) and H9N2 (G1_PB2E627K). Viral titres were determined in MDCK cells at indicated time points by FFA. Data are means ± s.d. from n = 3 independent biological repeats. P values are from two-tailed Student’s t-test. c, Huh7 (Ctrl), Huh7 GALNT2−/− or Huh7 STAT1−/− cells were infected with SARS-CoV-2 (MOI = 0.05, 24 h), HCoV-OC43 (MOI = 0.05, 48 h) and HCoV-229E (MOI = 0.001, 24 h). Viral infection efficiency was detected by IFA. A549 (Ctrl) and A549 GALNT2−/− cells were infected with H1N1 pdmCA04 (MOI = 0.03, 24 h), H3N2 HK68 (MOI = 0.01, 24 h) and H9N2 G1_PB2E627K (MOI = 0.01, 24 h). Viral infection efficiency was detected by IFA. Reconstitution of GALNT2 in the knockout cells by transient transfection and infection efficiency was determined by IFA. Data are means ± s.d. from n = 3 or 6 independent biological repeats. P values were from two-tailed Student’s t-test. d, Huh7 cells infected with WT SARS-CoV-2 (MOI = 0.1) for 0 h (mock), 24 h and 48 h. GALNT2 expression levels were determined by western blot analysis. e, THP-1 cells were treated with 1 μg ml−1 IFNα2, IFNβ or IFNλ. At 24 h post-treatment, GALNT2 expressions in cell lysates were analysed by western blot. f, Western blot analysis of the mouse GALNT2 and STAT1 expressions in SARS-CoV-2 WT and IFNAR−/− mouse lungs at 4 d.p.i. Western blots (d–f) were representative of three independent biological repeats. NS, not significant. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
As SARS-CoV-2 infection can induce robust IFN-I and upregulate GALNT2 expressions (Fig. 2d and Extended Data Fig. 5e), we hypothesized that GALNT2 might function as an ISG. Indeed, we found that GALNT2 mRNA and protein levels were markedly increased in Huh7 cells after HCoV-OC43 and IAV infections (Extended Data Fig. 5f,h), indicating that viral infections augment GALNT2 expression. Furthermore, we also observed upregulated expression of GALNT2 in THP-1 cells after the treatment with IFNα2, IFNβ or IFNλ, respectively (Fig. 2e). On the contrary, the induction of GALNT2 was almost abolished in the Huh7 STAT1−/− cells after IFNα2 treatment (Extended Data Fig. 5i), consistent with impaired GALNT2 protein expression observed in the IFNAR−/− mice after SARS-CoV-2 infection (Fig. 2f). Lastly, after treatment with IFNβ in Huh7, Huh7 GALNT2−/− and Huh7 STAT1−/− cells followed by PR8 infection, GALNT2 clearly functioned as a crucial effector and contributed to the restriction of viral infection by IFNs (Extended Data Fig. 5j). The identification of IFN-stimulated response elements and gamma-interferon activation site elements within the promoter of GALNT2 further supports this conclusion (Extended Data Fig. 5k). Together, these findings reveal that GALNT2 is an ISG and induced by viral infection or IFN stimulation.
GALNT2 protects mice from SARS-CoV-2 and IAV infections in vivo
We next aimed to evaluate GALNT2-mediated IAV and SARS-CoV-2 restriction in vivo. Firstly, to determine whether GALNT2 plays a prominent role in controlling IAV infection in vivo, GALNT2−/− mice were generated (Extended Data Fig. 6a–c). We infected mice with a sublethal dose of PR8 and found that susceptibility of GALNT2−/− mice was reflected by increased weight loss, decreased survival rate, increased viral loads and more severe histological changes in the lungs compared with WT mice (Fig. 3a and Extended Data Fig. 6d–f), showing the protective effect of GALNT2 upon IAV infection. Furthermore, SARS-CoV-2-infected GALNT2−/− mice showed higher viral loads in the lungs compared with WT mice although no significant weight difference was observed (Fig. 3b). These results showed that GALNT2 had potent antiviral activity in vivo.

a, WT (n = 12) and GALNT2−/− (n = 10) C57BL/6 mice were infected with 350 FFU of PR8 H1N1 and monitored for 14 days for weight changes and survival. Two independent experiments were performed. Viral titres (n = 5) in the lungs collected at 3 d.p.i. were determined in MDCK cells by FFA. b, WT (n = 6) and GALNT2−/− (n = 5) C57BL/6 mice were infected with 5 × 103 FFU of SARS-CoV-2. Viral titres in the lungs collected at 2 d.p.i. were determined in Vero E6 cells by FFA. c, Groups of C57BL/6 mice were transduced with either 3.3 × 1011 genome copies (GC) of AAV9-EV (n = 11) or AAV9-GALNT2 (n = 10) intranasally. The mice were infected with 1,000 FFU of H1N1 PR8 14 days post-transduction and were monitored for 14 days for weight changes and survival. Two independent experiments were performed. Viral titres in the lungs collected at 3 d.p.i. (n = 6) and 5 d.p.i. (n = 5 or 6) were determined in MDCK cells by FFA. d, Groups of C57BL/6 mice were intranasally transduced with 3.3 × 1011 GC of AAV9-EV or AAV9-GALNT2 (n = 7). The mice were infected with 1 × 105 FFU of SARS-CoV-2 (WT) 14 days post-transduction. Weight changes were monitored for 14 days. Viral titres (n = 6) in the lungs collected at 2 d.p.i. and 4 d.p.i. were determined in Vero E6 cells by FFA. e, Groups of IFNAR−/− (n = 5) or STAT1−/− (n = 6) mice were transduced intranasally with 3.3 × 1011 GC of AAV9-EV or AAV9-GALNT2. The mice were infected with 1 × 105 FFU of SARS-CoV-2 (WT) 14 days post-transduction. Viral titres in the lungs collected at 2 d.p.i. were determined in Vero E6 cells by FFA. Data are mean ± s.d. P values were from one-tailed Student’s t-test in a, b and d and two-tailed Student’s t-test in c and e. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
As overexpression of human GALNT2 induced strong and broad-spectrum protection against respiratory virus infections in vitro, we then determined the potential of GALNT2 expression as an antiviral strategy in vivo. Using serotype 9 adeno-associated viral (AAV9) gene transfer vectors29, we delivered AAV9-GALNT2 to WT mice intranasally and observed a specifically increase in GALNT2 RNA and protein compared with the AA9-empty vector (EV) transduced mice (Extended Data Fig. 7a,b,g). We subsequently infected the WT mice with PR8 or SARS-CoV-2. Compared with EV-transduced mice, 50% of GALNT2-transduced mice survived the viral challenge lethal dose and showed only minimal body weight loss, lower viral loads in the lungs, less lung tissue damage and lower levels of inflammatory cytokine production, alveolar wall thickening and pulmonary oedema (Fig. 3c,d and Extended Data Fig. 7c–f,h–k). Lastly, we examined whether GALNT2 could protect immunodeficient mice against SARS-CoV-2 infection. For this purpose, we infected GALNT2-transduced IFNAR−/− and STAT1−/− mice with SARS-CoV-2 and found ~10-fold lower titres in the lungs (Fig. 3e), supporting the antiviral potential of GALNT2 in immunodeficient individuals.
GALNT2 specifically O-glycosylates residues upstream of the spike S2′ cleavage site
GALNT2 is one of the 20 N-acetylgalactosaminyltransferases (GalNAc-Ts) that initiate the O-glycosylation modification of specific protein substrates30. Although GALNT2 was reported to have important roles in metabolic regulation and cancer biology31,32, the antiviral function of O-linked glycosylation by GALNT2 has never been reported. We investigated whether the GALNT2-dependent antiviral effect is mediated by regulating host genes through comprehensive RNA-seq and proteomics and glycoproteomics analyses. As depicted in Extended Data Fig. 8a–c, our findings showed that GALNT2 overexpression exerts minimal influence on the expression of a limited subset of host genes at both the RNA and protein levels. The overexpression of GALNT2, an important member of the O-GalNAc transferase family, did increase the O-glycosylation levels of a selected group of substrate proteins, including a few proteins in the endoplasmic reticulum; this might be a consequence of cellular stress caused by overexpression, but has not been reported with viral infections. The robust antiviral activity observed suggests that GALNT2-dependent O-glycosylation might directly target viral proteins and determine the antiviral phenotype.
The SARS-CoV-2 spike is firstly processed at the S1/S2 cleavage site by proprotein convertase furin during viral assembly, while TMPRSS2 makes additional cleavages at the spike S2′ site during ACE2-dependent viral entry and cell–cell fusion, also known as syncytium formation. Interestingly, we observed that the progeny viral particles produced in GALNT2-overexpressed cells notably impaired the plaque-forming size of SARS-CoV-2 (Extended Data Fig. 5c,d). Therefore, we speculated that GALNT2 may exert its function by blocking spike-mediated virus–cell fusion. We therefore hypothesized that GALNT2 could glycosylate one of those protease sites, which might affect spike proteolytic processing and regulate virus entry.
Using an in vitro glycosylation assay, we tested the enzymatic activity of GALNT2 towards synthetic peptides encompassing those protease sites. As shown in Fig. 4a, GALNT2 glycosylated the peptide sequence specifically near the TMPRSS2 (S2′) cleavage sites, but not the furin (S1/S2) cleavage sites. Relevant to phylogenetic analysis, we had meticulously chosen nine representative GALNT genes and examined the in vitro enzymatic activity of other GALNT members and found that GALNT2 was the only GalNAc-T capable of adding O-glycosylation upstream of the S2′ cleavage site (Extended Data Fig. 9a,b). Using liquid chromatography coupled with tandem mass spectrometry (LC–MS/MS), we further determined that GALNT2 specifically modifies residues corresponding to serine 810 and 813 (S810/813) of the WT SARS-CoV-2 spike (Fig. 4b,c). Next, we performed LC–MS-based glycoprofiling of the overexpressed extracellular domain (ECD) of the spike protein and confirmed that at least one of the in vitro glycosylated residues, S813, exists on recombinant spike proteins (Fig. 4d). Subsequently, we checked whether the inability of the previously identified missense mutations of GALNT2 (V554M, V552M, D314A, Q216H) to inhibit SARS-CoV-2 infection was due to impaired enzymatic activity of GALNT2 towards S810/813 of the spike protein using the in vitro glycosylation assay. Interestingly, we did observe a noticeable decrease of glycosylated peptide with all four variants of GALNT2 when the reaction time was reduced to 3 h (Extended Data Fig. 9c,d). However, due to the limited quantitation capability of matrix assisted laser desorption ionization-time of flight (MALDI-TOF)-based mass spectrometry, we could not rule out the possibility of those missense mutations affecting GALNT2 through other mechanisms.

a, MALDI-TOF spectrum analysis of GalNAcylation reactions catalysed by purified GALNT2 (GalNAc-T2) on the synthetic multibasic peptide of the SARS-CoV-2 spike protein (S2′ (TMPRSS2): 800–819, S1/S2 (furin): 674–693). An increase of 203 Da corresponding to the addition of one GalNAc residue was indicated in the site of O-glycosylation by GALNT2. b,c, ETD-MS2 spectrum analysis of the O-GalNAcylated peptide from the GALNT2 reaction on the S810/S813-O-GalNAcylated peptide. The mass of c- and z- fragment ions unambiguously assigned the GalNAc modification to S810 (b) and S813 (c) (labelled with a yellow square above the peptide sequence). The GalNAc residues are denoted as yellow squares according to the Consortium for Functional Glycomics standard. d, Full-length SARS-CoV-2 spike was co-transfected in HEK293T cells with GALNT2 and analysed with LC–MS/MS as described above. e, A schematic showing the GALNT2-dependent O-glycosylated sites on the WT SARS-CoV-2 spike. NTD, N-terminal domain; RBD, receptor binding domain; HR1, heptad repeat 1; CH, central helices; CD, connector domain; TM, transmembrane. f, A schematic showing the biotin-based pulldown of an O-glycosylated spike from cell lysates using helix pomatia agglutinin (HPA). g, Immunoblots of spike obtained from HPA pulldown of the HEK293T cell lysates. Western blots (WB) were representative of three independent biological repeats. Quantitative data from the representative western blot are shown below. h, Immunoblots of spike obtained from HPA pulldown of the Calu3 cell lysates overexpressing 100 ng or 400 ng EV or GALNT2, infected with authentic WT SARS-CoV-2 (MOI = 0.1) for 24 h. Western blots were representative of three independent biological repeats. Quantitative data from representative western blot are shown below. i, MALDI-TOF analysis of GalNAcylation reactions catalysed by purified GALNT2 on the synthetic multibasic peptide of H1N1 HA proteins. j, Immunoblots of HA protein, obtained from HPA pulldown of the HEK293T cell lysates co-expressing HA and EV or GALNT2. Western blots were representative of three independent biological repeats. Quantitative data from a representative western blot are shown below. k, Immunoblots of HA protein, obtained from HPA pulldown of the Calu3 cell lysates overexpressing 100 ng EV or GALNT2, infected with PR8 H1N1 (MOI = 0.1) for 24 h. Western blots were representative of three independent biological repeats. Quantitative data from representative western blot are shown below.
GALNT2 inhibits proteolytic processing of viral glycoproteins
As GALNT2-mediated glycosylation occurs adjacent to the S2′ cleavage site on the SARS-CoV-2 spike protein (Fig. 4e), we first validated its occupancy on the full-length spike using the biotin-conjugated helix pomatia agglutinin (biotin–HPA), a lectin specific for O-GalNAc, in conjunction with streptavidin magnetic beads (Fig. 4f). Indeed, biotin–HPA readily pulled down the full-length spike and S2 from HEK293T cell lysates co-transfected with the SARS-CoV-2 WT spike and GALNT2, with or without the lysosomal inhibitor bafilomycin A1 (Fig. 4g), or GALNT2-expressing Calu3 cells infected with authentic WT SARS-CoV-2 (Fig. 4h). As GALNT2 overexpression was found to inhibit IAV infection and potential O-glycosylation sites are present in both CoVs and IAV haemagglutinin (HA)33, we tested whether membrane-bound HA could be O-glycosylated by GALNT2. Firstly, we confirmed GALNT2-mediated O-glycosylation of IAV HAs (PR8 and WSN) GALNT2 using MALDI-TOF analysis (Fig. 4i). Biotin–HPA pulldown assay showed strong HA detection from cells co-expressing PR8 HA and GALNT2, as well as in Calu3 cells infected with PR8 (Fig. 4j,k).
GALNT2 impairs viral growth by blocking virus–cell fusion
We next investigated whether GALNT2-mediated modification influences S2′ cleavage. In cells expressing both spike and ACE2, the S2′ cleavage fragment was clearly detected. However, co-expression of GALNT2 particularly reduced S2′ cleavage compared with controls in both HEK293T (Fig. 5a) and HEK293T-TMPRSS2 cells (Fig. 5b). To exclude potential indirect effects of GALNT2 on the upstream S1/S2 furin cleavage site, we tested a spike mutant with a deletion (ΔRRAR) at the S1/S2 junction. GALNT2 still effectively reduced S2′ cleavage in this mutant (Fig. 5c), confirming that GALNT2 mediated O-glycosylation of spike protein at the S810/S813 sites and inhibits its proteolytic activation.

a–c, Immunoblots of the SARS-CoV-2 full-length S, cleaved S2′, derived from HEK293T (a) and HEK293T-TMPRSS2 (b) cell lysates, co-transfected with plasmids encoding a spike (S) of SARS-CoV-2 or ΔRRAR spike mutant (c), ACE2, empty vector (EV) or GALNT2 for 24 h. Western blots were representative of three independent biological repeats. Quantitative data from a representative western blot are shown below. d, A schematic showing the Cre-loxP-based fusion assay. Fluorescent images of the mCherry reporter expression and cell nuclei (Hoechst33342), captured from spike and Cre co-expressing HEK293T cells in the presence of EV or GALNT2, co-cultured with control or ACE2 cells with stop-mCherry for 24 h. Images were representative of 3 individual repeats. Scale bars, 50 μm. Cell–cell fusion was quantified as RLUs, derived from co-cultured HEK293T cell lysates using a stop-luciferase reporter. Data are means ± s.d. from n = 14 repeats. P values were from two-tailed Student’s t-test. e, Immunoblots of the SARS-CoV-2 full-length S and S2 collected from the supernatant and lysates of HEK293T cells SARS-CoV-2 pseudovirus preparation. The spike was co-expressed with EV or GALNT2 for 24 h, followed by infection of VSV pseudovirus for 24 h. Supernatant and cell lysates were collected and detected by western blots. Western blots were representative of three independent biological repeats. f, Luciferase activity detected from HEK293T-AEC2 cells infected with SARS-CoV-2 pseudovirus, prepared from EV or GALNT2-expressing cells. Data are means ± s.d. from n = 4 independent biological repeats. P values from two-tailed Student’s t-test. g, Immunoblots of S and S2 on SARS-CoV-2 VLP supernatants, prepared from HEK293T cells co-expressing spike, envelope (E), membrane (M), nucleocapsid (N) and combined with EV, GALNT2 for 72 h. Western blots were representative of three independent biological repeats. h, A schematic showing the quantification of SARS-CoV-2 VLP entry into HEK293T-ACE2 cells overexpressing the LgBiT NanoLuc reporter. RLUs were derived from HEK293T-ACE2 LgBiT cells infected with SARS-CoV-2 VLPs for 8 h. Data are means ± s.d. from n = 3 individual repeats. P values were from two-tailed Student’s t-test. i, Immunoblots showing full-length PR8 HA and cleaved HA1 and HA2 expression in HEK293T cells co-transfected without or with TMPRSS2 and GALNT2 for 24 h. Blots are representative of three individual repeats. The red arrow indicates the cleaved HA1 and HA2. j,k, GALNT2 inhibits the fusogenic activity of the HA protein from the avian influenza virus strain H5N6 (j) and H7N9 (k) with polybasic cleavage sites. Fluorescent images of the GFP reporter expression captured from HA and GFP co-expressing HEK293T cells in the presence of EV or GALNT2 for 24 h. Images were representative of 3 individual repeats. Scale bars, 50 μm. Individual syncytium derived from HA-mediated cell–cell fusion was quantified as relative fluorescent dots. Data are means ± s.d. from n = 20 repeats. P values from two-tailed Student’s t-test. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
Because GALNT2 inhibits proteolytic cleavage of viral glycoproteins, we hypothesized that it may impair cleavage-dependent membrane fusion. To assess the fusogenic capacity of the spike protein in the presence of GALNT2, we performed qualitative and quantitative cell–cell fusion assays using a Cre-loxP reporter system34. While EV-transfected spike cells readily formed syncytia with ACE2 cells showing strong mCherry and luciferase signals, co-expression of spike and GALNT2 reduced both signals by approximately 50% (Fig. 5d), indicating a significant inhibition of spike-mediated membrane fusion. To determine whether GALNT2 also impairs the entry of progeny virions, we first used a vesicular stomatitis virus (VSV)-based pseudovirus system and confirmed that GALNT2-expressing cells produced pseudoviruses with reduced infectivity (Fig. 5e,f). We further used a SARS-CoV-2 virus-like particle (VLP)-HiBiT system, in which VLPs bearing HiBiT-tagged nucleocapsid (N) were produced with or without GALNT2. After normalizing VLPs by N protein, we found that VLPs from GALNT2-expressing cells showed considerably reduced entry, as measured by the NanoLuc activity (Fig. 5g,h). This supports the observed reduced viral entry after multiple replication cycles in GALNT2-expressing cells (Fig. 2a).
Similarly, when co-expressing HA, TMPRSS2 and GALNT2, the levels of cleaved HA1 and HA2 were noticeably reduced compared with the control, suggesting that GALNT2 also modulates HA proteolysis (Fig. 5i). Furthermore, cell–cell fusion assays showed that GALNT2 overexpression notably reduced fusion activity mediated by HAs from highly pathogenic H5N6 and H7N9 strains (Fig. 5j,k). Overall, these results showed that GALNT2-mediated O-glycosylation of viral surface glycoproteins restricts membrane fusion and inhibits viral entry.
The primary targets of GALNT2 against SARS-CoV-2 infection
We next investigated whether the SARS-CoV-2 spike serine 810/813 residues are the primary targets of GALNT2. We observed a transient occurrence of I813 natural substitution in the SARS-CoV-2 B.1.36.25 towards the end of 2020, then this variant quickly disappeared (Fig. 6a). To eliminate the influence of S810-mediated glycosylation, we introduced double S810/813I or S810/813A substitutions into the spike protein and rescued recombinant viruses using an authentic viral system35 (Fig. 6b). Unexpectedly, we observed a noticeable attenuation in viral titres and plaque size for both recombinant viruses when compared with the WT SARS-CoV-2 (Fig. 6c–e) in either Vero E6 or Huh7 cells. Meanwhile, both recombinant viruses showed strong resistance to the restrictions of GALNT2 in vitro and in vivo (Fig. 6f,g). These results revealed that S810/813 of the spike has a key role in determining the sensitivity or resistance to GALNT2. Previously, mutations at residue 813 were found to reduce TMPRSS2-mediated entry and viral fitness owing to decreased usage of TMPRSS2 for viral entry36. Substitutions at S810/S813 may prevent O-glycosylation, allowing the virus to evade GALNT2-dependent antiviral activity, but also evidently lower viral fitness by impairing TMPRSS2-dependent entry and membrane fusion.

a, The temporal dynamics of SARS-CoV-2 S813 site mutation frequency (https://outbreak.info/situation-reports?muts=S:S813I&pango). b, A schematic showing the generation of SARS-CoV-2 recombinant viruses and confirmed by next generation sequencing (NGS). c, The recombinant viruses were amplified in Vero E6 cells, and titres were determined by PFA. Images were representative of viral plaque forming for recombinant viruses. Data are means ± s.d. from (n = 50 for each virus) the diameter of plaques. P values are from two-tailed Student’s t-test. d,e, Vero E6 cells (d) and Huh7 cells (e) were infected with SARS-CoV-2 WT and recombinant viruses at an MOI of 0.01. Viral titres were detected at indicated time points by PFA. Data are means ± s.d. from n = 3 or 4 independent experiments. P values were from two-tailed Student’s t-test. f, Huh7 cells were transfected with plasmids encoding GALNT2 (EV as the control). At 24 h post-transfection, cells were infected with SARS-CoV-2 WT and the recombinant viruses. Viral titres were detected at indicated time points by PFA. Data are means ± s.d. from n = 3 independent experiments. P values were from two-tailed Student’s t-test. g, Groups of C57BL/6 mice (n = 5) were intranasally transduced with 3.3 × 1011 GC of AAV9-EV or AAV9-GALNT2. Mice were infected with 5 × 104 plaque-forming unit (PFU) of SARS-CoV-2 (WT, S810/813A, S810/813I) 14 days post-transduction. Viral titres in the lungs collected at 2 d.p.i. and 4 d.p.i. were determined in Vero E6 cells by PFA. Data are presented as means ± s.d. h, MALDI-TOF analysis of GalNAcylation reactions catalysed by purified GALNT2 on the synthetic multibasic peptide of SARS-CoV-2 spike proteins (WT, S810/813A and S810/813I). Reactions were performed and analysed as described in Methods. An increase of 203 Da corresponds to an addition of one GalNAc residue observed with GALNT2 in WT spike protein. i, Streptavidin pulldown of HPA–biotin mixed with SARS-CoV-2 pseudoviral particles incorporated with SARS-CoV-2 WT or S810/813A spikes. Supernatants containing the SARS-CoV-2 pseudoviral particles derived from EV or GALNT2-expressing HEK293T cells were collected and verified for SARS-CoV-2 spike incorporation and assembly of VSV-M. Normalized SARS-CoV-2 pseudovirus supernatants were gently mixed with 5 μg ml−1 biotin–HPA overnight at 4 °C. Pulldown samples attached on the streptavidin magnetic beads were washed three times in NP-40 lysis buffer before boiling in the 2× Laemmli loading buffer. Protein samples were then separated by SDS-PAGE and detected by immunoblotting of the full-length spike and S2. Western blots were representative of three independent biological repeats. Quantitative data from representative western blot are shown below. j, Fluorescent images of HEK293T control or HEK293T-ACE2 cells carrying the stop-mCherry reporter, co-cultured with cells expressing spike (SARS-CoV-2 WT or S810/813I mutant) co-expressed with EV or GALNT2. Images are representative of three individual repeats. Scale bars, 20 μm. All data are mean ± s.d. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. Panel b created with BioRender.com.
We subsequently investigated whether the mutated 810/813 of the spike undergoes O-glycosylation. As anticipated, the in vitro glycosylation assay showed a significant reduction in the ability of the spike S810/813A and S810/813I mutations to undergo O-glycosylation modulation by GALNT2 (Fig. 6h). Consistently, HPA pulldown of S810/813A spike pseudoviruses derived from GALNT2-expressing cells showed reduced S2 binding to the streptavidin magnetic beads (Fig. 6i). It is intriguing to observe that the S810/813I spike showed a pronounced reduction in cell–cell fusion, irrespective of GALNT2 expression (Fig. 6j). Finally, we introduced S810/813I mutations into the visual viral replicon37 and observed strict resistance of the SARS-CoV-2_Ndel_S810/813I virus to GALNT2 antiviral activity. Furthermore, overexpression of GALNT2 notably reduced syncytium formation for WT SARS-CoV-2, but not the S810/813I mutated replicon (Extended Data Fig. 10a–d). Therefore, our findings indicated that GALNT2-mediated innate immunity shows a highly conserved antiviral effect, rendering the virus incapable of evading it through mere mutations (Extended Data Fig. 10e).
In summary, our data elucidate GALNT2 as an antiviral ISG. Our study fills a crucial gap by showing that an innate immune factor can directly modulate viral glycoprotein O-glycosylation, a post-translational modification that affects viral growth.
Discussion
Since the beginning of the COVID-19 pandemic, key questions remain about what factors contribute to the variability in disease severity, ranging from mild cases to severe manifestations and even fatality. Despite the identification of numerous genes associated with the clinical severity of COVID-19 through GWAS and scRNA-seq analyses in patients or clinical samples38,39, there remains a paucity of experimental validation. In our study, we observed a significant correlation between GALNT2 expression levels and the timing of disease progression and determined disease severity in SARS-CoV-2-infected patients. Our data have further identified GALNT2 as an ISG that exerts regulatory control over the replication of CoVs and IAVs. GALNT2-dependent O-glycosylation has a crucial role in regulating the proteolytic activation of CoV spikes and IAV HA. The O-glycosylation process mediated by GALNT2 primarily occurs in close proximity to the cleavage site of viral glycoproteins, thereby hindering the proteolytic activation and subsequent fusion with the cell membrane. In general, viruses could evade antiviral ISGs through rapid mutations, such as Mx1 (ref. 28) and BTN3A3 (ref. 40). However, the introduction of the 810/813 mutations yielded a substantial decrease in viral replication, which has consistently highlighted the paramount importance of serine 813 in facilitating TMPRSS2 recognition and viral fusion. We and others previously have discovered that O-glycosylations near the furin site of the spike by GalNAc-T1, GalNAc-T3 and GalNAc-T7 affected the furin cleavage process through O-glycosylation41,42. GALNT2 specifically modifies S810/813 residue near the S2′ cleavage site, thereby inhibiting both S2′ cleavage and subsequent membrane fusion. Together, our findings provide evidence that innate immunity can modulate O-linked glycosylation and inhibit proteolytic activation of viral glycoproteins, resulting in broad-spectrum antiviral effects that restrict respiratory viruses.
Methods
Ethics and biosafety
All procedures and methods were conducted in compliance with regulations at Guangzhou Medical University. The infectious animal study was reviewed and approved by institutional animal care and use committees of the First Affiliated Hospital of Guangzhou Medical University (2022114). The donors of the blood samples used in this study provided written informed consent. Routine surveillance samples were detected in the biosafety level 2 (BSL-2) laboratory. The animal experiments were conducted in the BSL-2 (IAV) and BSL-3 (SARS-CoV-2) laboratories. All human study participants were approved by the First Affiliated Hospital of Guangzhou Medical University (ES-2020-65, ES-2023-015-01). Written informed consent was obtained from all recovered SARS-CoV-2-infected patients, and approval was obtained from the institutional review boards of the First Affiliated Hospital of Guangzhou Medical University. We also obtained institutional review board approval from the Health Commission of Guangdong Province to use the anonymized healthy blood donor samples described in this study. The SARS-CoV-2 recombinant viruses, including the S810/813A and S810/813I viruses, were generated and validated in the BSL-3 laboratory of the University of Iowa. All experiments strictly followed standard operating procedures, and all waste materials were autoclaved and verified before disposal.
Cell lines
THP-1 (TIB-202), A549 (CRM-CCL-185), HRT-18 (CCL-244), MDCK (CCL-34), Vero E6 (CRL-1586), Calu3 (HTB-55), Caco2 (HTB-37) and HEK293T (CRL-3216) cells were purchased from the American Type Culture Collection (ATCC). Huh7 (JCRB0403) cells was obtained from the Japanese Collection of Research Bioresources (JCRB) Cell Bank. A549, MDCK, Vero E6, Huh7 and Caco2_N cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS) and 100 U ml−1 penicillin–streptomycin (P–S; Gibco). Calu3 cells were maintained in minimum essential medium (MEM) (Gibco) supplemented with 20% FBS, 0.1 mM MEM non-essential amino acid solution (MEAA, Gibco) and 100 U ml−1 P–S. HRT-18 and THP-1 cells were maintained in Roswell Park Memorial Institute 1640 (Gibco) supplemented with 10% FBS and 100 U ml−1 P–S.
Viruses
The SARS-CoV-2 variants, including the WT and Omicron BA.2, were isolated from SARS-CoV-2-infected patients and preserved in the Guangzhou Customs District Technology Center BSL-3 Laboratory. The SARS-CoV-2 Omicron BA.5 strains were preserved by Guangdong Provincial Center for Disease Control and Prevention. All SARS-CoV-2 viral stocks were generated in Vero E6 cells and titrated by focus-forming assay (FFA). The viral stocks of HCoV-OC43 (VR-1558) and HCoV-229E (VR-740) were generated in HRT-18 and Huh7 cells, respectively, and titrated by FFA. All work with SARS-CoV-2 was conducted in the BSL-3 laboratories of the Guangzhou Customs District Technology Center.
All of the viral stocks of IAV, including A/Puerto Rico/8/1934(H1N1) (PR8), A/WSN/1933(H1N1) (WSN), A/California/04/2009(H1N1) (CA04), A/Hong Kong/1968(H3N2) (HK68), A/Aichi/2/1968(H3N2) (Aichi) and A/Quail/Hong Kong/G1/97 (H9N2) (G1 and G1_PB2E627K), were generated and titrated in MDCK cells. All viruses were purified by plaque assay and confirmed by next-generation sequencing.
Mice
STAT1−/− and IFNAR−/− C57BL/6 mice were purchased from Jackson Laboratories. IFNAR−/− BALB/c mice were generated by back-crossing IFNAR−/− C57BL/6 with WT BALB/c mice for at least ten generations. GALNT2−/− C57BL/6 mice were generated and purchased from GemPharmatech and maintained in the animal care facilities at Guangzhou Medical University. All protocols were approved by the institutional animal care and use committees of Guangzhou Medical University. All mice were housed in a specific pathogen-free facility under a 12-h light–12-h dark cycle, at a constant temperature of 22 ± 2 °C and relative humidity of 50 ± 10%. Mice aged 6–8 weeks old were used for viral infections.
scRNA-seq data analysis
Clinical data and samples from SARS-CoV-2-infected patients were obtained from the First Affiliated Hospital of Guangzhou Medical University, collected under the ethical framework of informed consent. Patient and sample information is provided in Supplementary Table 1. Patient classification was based on infection severity: those requiring intensive care unit treatment were categorized as severe cases, while others were classified as mild cases. The phases of infection (T1, T2 and T3) are defined as follows: T1 corresponds to the sample collected when patients arrived at the hospital, typically right after symptom onset, representing the acute infection period. T2 denotes the period 7–11 days after symptom onset for patients with mild COVID-19 and 15–30 days after symptom onset for patients with severe COVID-19. T3 signifies the period after negative testing for the SARS-CoV-2 virus, usually occurring 20–40 days after symptom onset for patients with mild COVID-19 and 30–60 days after symptom onset for patients with severe COVID-19. Peripheral blood samples underwent single-cell capture and downstream library construction using the 5′ single-cell kit from 10xGenomics. To process the scRNA-seq data, we used the scTE pipeline43 for 10× data, using the default scTE parameters to generate the molecule count matrix. To ensure data quality, the count matrix underwent a light filtration step, excluding cell barcodes with low counts and potential doublets. Specifically, cells with fewer than 200 detected genes, more than 5,000 detected genes or over 20% mitochondrial counts were removed from the analysis. Subsequently, the expression matrix underwent normalization, dimensionality reduction and subpopulation analysis, all performed using the Seurat (v4) toolkit44. For correction of potential batch effects, we applied the Harmony algorithm. Cellular subpopulations were defined using the SingleR package45, which leverages reference transcriptomic datasets of pure cell types to infer the origin of individual single cells independently. Small subgroups accounting for less than 0.5% of the cell number were discarded. In addition, when comparing the mean gene expression level of each cellular subgroup, samples with fewer than ten cells in that particular subgroup were excluded from the analysis.
Publicly available single-cell transcriptome sequencing data of bronchoalveolar lavage (BALF) samples from both HCs and SARS-CoV-2-infected patients were obtained from the studies referenced16,17,18,19,20,21,22,23,24,25,26. To ensure uniform data quality across all studies, we applied consistent quality control criteria: (1) cells with a detected gene count ranging from 300 to 6,000, (2) unique molecular identifier (UMI) count between 300 and 200,000, and (3) a percentage of mitochondrial genes less than 10%. Following these quality control measures, a total of 336,329 cells originating from 92 patients were retained. This encompassed 196,957 cells from 59 severe SARS-CoV-2-infected patients, 9,424 cells from 5 mild SARS-CoV-2-infected patients, and 129,948 cells from 28 healthy controls. For data processing, the expression matrix underwent normalization, reduction and subpopulation analysis using the Seurat (v4) toolkit44. We used the harmony algorithm for batch effect correction. The cellular subgroups were defined based on marker expressions: macrophage cell (CD68), T cell (CD3D), B cell (CD79A), secreted epithelial cell (SCGB1A1) and ciliated cell (FOXJ1).
When comparing the mean gene expression level of each cellular subgroup, samples with fewer than ten cells in that particular subgroup were excluded from the analysis. Pseudo-bulk expression was calculated by summing the raw counts of each gene across all cells from each individual, followed by library size normalization.
GWAS data analysis
The GWAS results of SNPs within the GALNT2 locus were acquired from the Regeneron Genetic Center (RGC; https://rgc-covid19.regeneron.com) for Fig. 1e,f, showing the result of UKB study, the largest one of the RGC. In the context of the comparison between SARS-CoV-2-infected patients who were hospitalized and those who were not (Extended Data Fig. 4c), GWAS results were sourced from the Host Genetics Initiative (https://www.covid19hg.org/) (Release5, COVID19_HGI_B1_ALL_eur_leave_23andme_20210107.txt.gz). For the GWAS analysis, H7N9 and H1N1 viruses were depicted in Extended Data Fig. 4a,b and the P values of SNPs were obtained from the respective H7N9 (ref. 28) and H1N1 (ref. 27) study in Supplementary Table 2.
RNA extraction, reverse-transcription PCR and real-time PCR
Total RNA from cells or tissues was extracted using the Universal RNA Purification Kit (EZBioscience) following the manufacturer’s instructions. For RT-PCR assays, PrimeScript RT Master Mix (Takara, RR036A) was used to generate the cDNA template. Real-time PCR was performed using 2 × Colour SYBR Green qPCR Master Mix (ROX2 plus, EZBioscience). Primers used in this study are listed in Supplementary Table 3. All qPCRs were performed using a QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific).
Cell–cell fusion assay
SARS-CoV-2 spike-mediated fusion assays were performed using HEK293T cells seeded in a 24-well plate format. Plasmids encoding spike WT and S810A/S813A mutants were co-transfected with empty vector (EV) or GALNT2 using Lipofectamine 8000 (Beyotime). For the Cre/loxP bioluminescence and mCherry reporter assays, plasmids encoding the WT or mutant spike, Cre and EV or GALNT2 were co-transfected into donor cells; plasmids encoding stop-luciferase or stop-mCherry were co-transfected with EV or human ACE2 into the recipient cells. At 24 h after transfection, donor and recipient cells were resuspended into DMEM with 2% FBS and 100 U ml−1 P–S and then mixed and co-cultured at 1:1 ratio in 96-well plates for 8 h, before being lysed in ice-cold NP-40 buffer. Firefly luciferase activity was measured in relative luminescence units (RLU) using a commercial substrate (YEASEN, 11404ES80).
For influenza HA-mediated fusion, HEK293T cells were co-transfected with H5N6 (A/Sichuan/06689/2021) or H7N9 (A/Guangdong/Th008/2017) HA, green fluorescent protein (GFP) and EV or GALNT2. After 24 h, cells were stimulated with HEPES buffers (20 mM HEPES, 150 mM NaCl, 2 mM CaCl2 and 1 mM MgCl2) at pH 7.0 (normal) or pH 5.0 (low pH) for 10 min at 37 °C, before being reperfused with complete culture media for 2 h. Syncytium formation was imaged live, and fusion was quantified by measuring the GFP-positive syncytium area (μm²) using ImageJ.
Biotin–HPA pulldown assay
HEK293T cells co-expressing spike and EV or GALNT2 were lysed on ice for at least 20 min in a NP-40 lysis buffer containing 0.5% (v/v) NP-40, 25 mM Tris pH 7.3, 150 mM NaCl, 5% glycerol and 1× EDTA-free protease inhibitor cocktail (Targetmol). Crude lysates were clarified by centrifugation at 500 g before being pre-cleared in the presence of BeyoMag streptavidin magnetic beads (Beyotime, P2151) for 1 h at 4 °C. Input samples were obtained from pre-cleared lysate mixes, and resultant lysates were then incubated with 5 μg ml−1 biotin–HPA or biotin-conjugated bovine serum albumin control overnight at 4 °C. Pulldown samples on the streptavidin magnetic beads were thoroughly washed before boiling in 2× Laemmli loading buffer. Proteins were then separated by reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and detected by immunoblotting of spike S2.
FFA
Viral titres were determined by FFU assay as described previously13. Briefly, cells were seeded in 96-well plates 1 day before infection. Then, viruses were serially diluted and used to inoculate cells at 37 °C for 1 h. Inocula were then removed before adding 100 μl 1.6% (for coronavirus) or 0.9% (for IAV) carboxymethylcellulose warmed to 37 °C per well. After 24 h (SARS-CoV-2/HCoV-229E/IAVs) or 48 h (HCoV-OC43), cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. Cells were then incubated with rabbit anti-SARS-CoV-2 nucleocapsid protein polyclonal antibody (Sino Biological, 40143-T62), rabbit anti-HCoV-OC43 nucleocapsid protein polyclonal antibody (Sino Biological, 40643-T62), rabbit anti-HCoV-229E nucleocapsid protein polyclonal antibody (Sino Biological, 40640-T62) and mouse anti-IAV (N protein) mouse serum, respectively. This was followed by HRP-labelled goat anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories, 111-035-144) or HRP-labelled goat anti-mouse secondary antibody (Jackson ImmunoResearch Laboratories, 115-035-146). The foci were visualized by TrueBlue Peroxidase Substrate (KPL) and counted with an ELISPOT reader (Cellular Technology). All primary antibodies were used at a dilution of 1:2,000 for FFA. All secondary antibodies were used at a dilution of 1:5,000 for FFA.
Viral plaque-forming assay
Viruses from cell or lung homogenates were serially diluted in DMEM. Subsequently, 200 μl of diluted viruses was inoculated onto Vero E6 cells in 12-well plates for 1 h. Plates were gently rocked every 15 min. After the inoculum was discarded, the infected cells were overlaid with 0.6% agarose containing 2% FBS. After 3 days, the cells were fixed with 10% formaldehyde, and the plaques were counted after staining with 0.1% crystal violet.
Packaging of SARS-CoV-2 VLPs
SARS-CoV-2 M, E, N or N-HiBiT were cloned into pcDNA3.1 plasmids. HEK293T cells were co-transfected with a plasmid cocktail (S:M:E:N:EV/GALNT2 = 2:4:1:5:2). After 6 h, the medium was replaced with fresh 2% FBS-containing medium, and cells were incubated for 48 h. Both VLP-containing supernatants and cell lysates were collected to assess spike and N incorporation into virions, followed by LgBiT-based viral entry quantification.
HPA pulldown of SARS-CoV-2 pseudoviral particles
Supernatants containing the SARS-CoV-2 pseudoviral particles derived from EV or GALNT2-expressing HEK293T cells were collected and verified for SARS-CoV-2 spike incorporation and assembly of VSV-M. Normalized SARS-CoV-2 pseudovirus supernatants were gently mixed with 5 μg ml−1 biotin–HPA overnight at 4 °C. Pulldown samples attached to the streptavidin magnetic beads were washed three times in the NP-40 lysis buffer before boiling in the 2× Laemmli loading buffer. Protein samples were then separated by SDS-PAGE and detected by immunoblotting of the full-length spike and S2.
Pseudotyped particle preparation and infection
HEK293T cells were co-transfected with plasmids encoding SARS-CoV-2 spike protein along with either EV or GALNT2. At 24 h post-transfection, the cells were infected with a VSV-based pseudovirus bearing a firefly luciferase reporter at an multiplicity of infection (MOI) of 3–5 for 1 h. After inoculation, the cells were washed four times with growth medium and incubated for an additional 24 h. Supernatants containing the pseudovirus were collected to confirm spike and VSV-M protein assembly before being used for viral entry quantification.
IFNα2, IFNβ and IFNλ treatment
Huh7 and Huh7-STAT1−/− cells were seeded in 12-well plates. THP-1 cells were treated with phorbol-12-myristate-13-acetate (PMA, 100 ng ml−1) and then seeded in 12-well plates. Huh7, THP-1 and Huh7 STAT1−/− cells were stimulated with 1 μg ml−1 IFN-α2, IFN-β, or IFN-λ (Sino Biological). At 6 h or 24 h post-stimulation, the whole-cell lysates were collected for GALNT2 expression detection using western blot or RT-qPCR.
The in vitro GalNAc transferase activity assay
The peptides covering the S2′ (800–819) and S1/S2 (674–693) cleavage sites of the SARS-CoV-2 spike protein were custom synthesized by GenScript and used as substrates for the in vitro GalNAc-T activity assay under the standard condition46,47,48,49. Briefly, in vitro glycosylation assays were performed at 37 °C in a final volume of 25 μl containing 25 mM sodium cacodylic acid (pH 7.4), 10 mM MnCl2, 0.25% Triton X-100, 2 mM UDP-GalNAc and 10 μg of substrate peptide and initiated by adding 0.1 μg of individually purified GalNAc-Ts. The reactions were quenched at 3 h or 12 h by adding 100 μl of 1% TFA and analysed by MALDI-TOF MS. A Muc1-derived peptide (HGVTSAPDTRPAPGSTAPPA) was used as the positive control, and the in vitro activity of representative GlaNac-Ts from different subfamilies was examined, including GalNAc-T1, T2, T3, T4, T5, T7, T11 and T18 in our previous study42. Similar experiments were done for IAV PR8 (HA, 328–350) and WSN (HA, 327–349) peptides. The potential O-glycosylation sites for GALNT2 are present in both CoVs and IAVs in Supplementary Tables 4 and 5.
For glycosylation site identification, the glycopeptide products were purified and desalted by Stage-Tip, and analysed using an Easy-nLC 1200 system coupled to an Orbitrap Fusion Tribrid Mass Spectrometer equipped with a Nanospray Flex ion source (Thermo Fisher Scientific). The samples were dissolved in 0.1% FA and separated on a single analytical column (ReproSil-Pur C18-AQ, 1.9 μm, 75 μm inner diameter, 20 cm length) at a flow rate of 300 nl min−1 over a 120-min gradient from 3% to 32% mobile phase B (mobile phase A: water with 0.1% FA; mobile phase B: 80% acetonitrile, 0.1% FA and 19.9% water). The mass spectrometer was set to acquire full-scan MS spectra (355–1,700 m/z) for a maximum injection time of 100 ms at a mass resolution of 50,000 and an automated gain control target value of 5 × 105. The MS2 analyses were performed in the positive ion mode using data-dependent acquisition with dynamic exclusion set to 60 s at an exclusion window of 10 ppm. The top 10 most abundant multiply charged precursors from the full scan were selected for fragmentation via electron-transfer–collision-induced dissociation in the orbitrap at 50,000 resolutions. The electron-transfer–collision-induced dissociation experiment was carried out with calibrated charge-dependent electron-transfer dissociation (ETD) parameters and supplemental activation (SA) collision energy of 30%. The automated gain control target was set to 5.0 × 104, and the maximum injection time was 75 ms.
For the site-specific glycopeptide identification, the MS/MS was analysed by Proteome Discoverer 2.5 software using Sequest HT as a searching engine. HexNAc at serine/threonine was used for variable modifications. Precursor mass tolerance was set to 10 ppm, and fragment ion mass tolerance was set to 0.02 Da. All spectra of interest were manually inspected for correct peptide identification and glycosite localization.
Generation of the S810/813I and S810/813A SARS-CoV-2 mutated viruses
Lambda red recombination with I-Scel homing endonuclease was used in this study to introduce mutations into pBAC-SARS-CoV-2, as previously described35. Briefly, forward and reverse primers, designed with 60 bp homology to virus sequence and incorporating mutations of S810/813A or S810/813I, were synthesized. Thermo Fisher Scientific primer oligos are shown in Supplementary Table 6. PCR products were amplified from the pEP-Kan-I-SceI plasmid and electroporated into GS1783 Escherichia coli cells containing pBAC-SARS-CoV-2. Bacterial clones were treated with arabinose to induce the expression of the I-SceI restriction enzyme. Verified clones were confirmed through sequencing. Vero E6 cells were transfected with recombinant pBAC (1 μg) using Lipofectamine 3000. When cytopathic effects reached 70%, recombinant viruses were collected and titrated on Vero E6 cells by plaque-forming assay (PFA).
Generation of recombinant SARS-CoV-2 replicon
Yeast VL6-48B was used for transformation-associated recombination assembly. E. coli EPI300 was selected for yeast or bacterial artificial chromosome (YAC or BAC) amplification. The cDNAs (Wuhan-Hu-1, MN908947) of SARS-CoV-2 GFP/ΔN were synthesized from GenScript (the N gene is replaced with a gene of GFP). The cDNA of SARS-CoV-2 GFP/ΔN was divided into six fragments (designated F1 to F6). The S810/813I mutations were introduced into fragment 5 by overlap PCR. The transformation-associated recombination vector pGFCS was linearized by two PCRs using primer pairs THU7285/THU7090 and THU7068/THU7089 (designated to F7 and F8). Yeast colonies from pGFCS transformants were picked and determined by PCR. The trVLP was rescued following the precious study37. Subsequently, 20 µg of viral genomic RNA and 20 µg N mRNA were mixed and electroporated into Caco2-N cells (8 × 106) by a single electrical pulse with settings of 270 V at 950 μF using a GenePulser apparatus (Bio-Rad). P0 virus was collected at 24 h post-electroporation and infected Caco2-N cells for virus amplification. All primers are shown in Supplementary Table 7. The SARS-CoV-2 replicons were determined by immunofluorescence assay (IFA).
Statistical analyses
Biochemical experiments in vitro were routinely repeated at least three times. All western blot experiments with quantification were performed by a minimum of three times with independent biological samples and analysed by ImageJ. Student’s t-tests were used to analyse differences in mean values between groups using GraphPad Prism 9.0.1. Wilcoxon rank sum tests were performed in the scRNA-seq data. Specific statistical tests, numbers of animals and/or replicates, and further definitions of precision measures can be found in the respective figure legends or method details. P values <0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry proteomics data (Fig. 4d and Extended Data Fig. 8c) have been deposited to the ProteomeXchange Consortium via the PRlDE partner repository with the dataset identifier PXD051101. The next-generation RNA sequence data reported in this paper are deposited at Gene Expression Omnibus database with accession number GSE301783. The scRNA-seq data first reported in our previously published data are deposited and accessible via the NCBI (https://www.ncbi.nlm.nih.gov) under accession number PRJNA1099166. All data supporting the findings of this study are available in the Article, Supplementary Information or from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Terrier, O. et al. Influenza viruses and coronaviruses: knowns, unknowns, and common research challenges. PLoS Pathog. 17, e1010106 (2021).
Lamers, M. M. & Haagmans, B. L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 20, 270–284 (2022).
Pandemic H1N1 2009 (WHO Regional Office for South-East Asia, 2009).
Schoggins, J. W. et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 (2011).
Savan, R. & Gale, M. Innate immunity and interferon in SARS-CoV-2 infection outcome. Immunity 56, 1443–1450 (2023).
Ciancanelli, M. J. et al. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 348, 448–453 (2015).
Hadjadj, J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724 (2020).
Zhang, Q. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370, eabd4570 (2020).
Bastard, P. et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370, eabd4585 (2020).
Yang, L. et al. Potential role of IFN-α in COVID-19 patients and its underlying treatment options. Appl. Microbiol. Biotechnol. 105, 4005–4015 (2021).
Loske, J. et al. Pre-activated antiviral innate immunity in the upper airways controls early SARS-CoV-2 infection in children. Nat. Biotechnol. 40, 319–324 (2022).
Wickenhagen, A. et al. A prenylated dsRNA sensor protects against severe COVID-19. Science 374, eabj3624 (2021).
Sun, J. et al. Generation of a broadly useful model for COVID-19 pathogenesis, vaccination, and treatment. Cell 182, 734–743.e5 (2020).
Liu, G. et al. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nat. Microbiol. 6, 467–478 (2021).
Pfaender, S. et al. LY6E impairs coronavirus fusion and confers immune control of viral disease. Nat. Microbiol. 5, 1330–1339 (2020).
Pisu, D. et al. Single cell analysis of M. tuberculosis phenotype and macrophage lineages in the infected lung. J. Exp. Med. 218, e20210615 (2021).
Mould, K. J. et al. Airspace macrophages and monocytes exist in transcriptionally distinct subsets in healthy adults. Am. J. Respir. Crit. Care Med 203, 946–956 (2021).
Morse, C. et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 54, 1802441 (2019).
Liegeois, M. et al. Airway macrophages encompass transcriptionally and functionally distinct subsets altered by smoking. Am. J. Respir. Cell Mol. Biol. 67, 241–252 (2022).
Liao, M. et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 26, 842–844 (2020).
Grant, R. A. et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature 590, 635–641 (2021).
Chua, R. L. et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 38, 970–979 (2020).
Bost, P. et al. Deciphering the state of immune silence in fatal COVID-19 patients. Nat. Commun. 12, 1428 (2021).
Wauters, E. et al. Discriminating mild from critical COVID-19 by innate and adaptive immune single-cell profiling of bronchoalveolar lavages. Cell Res. 31, 272–290 (2021).
Zhu, A. et al. Robust mucosal SARS-CoV-2-specific T cells effectively combat COVID-19 and establish polyfunctional resident memory in patient lungs. Nat. Immunol. 26, 459–472 (2025).
Reyfman, P. A. et al. A novel MIP-1-expressing macrophage subtype in BAL fluid from healthy volunteers. Am. J. Respir. Cell Mol. Biol. 68, 176–185 (2023).
Garcia-Etxebarria, K. et al. No major host genetic risk factor contributed to A (H1N1) 2009 influenza severity. PLoS ONE 10, e0135983 (2015).
Chen, Y. et al. Rare variant MX1 alleles increase human susceptibility to zoonotic H7N9 influenza virus. Science 373, 918–922 (2021).
Samaranch, L., Pérez-Caamás, A., Soto-Huelin, B., Sudhakar, V. & Ledesma, D. Adeno-associated viral vector serotype 9-based gene therapy for Niemann–Pick disease type A. Sci. Transl. Med. 11, 1–15 (2019).
Bennett, E. P. et al. Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology 22, 736–756 (2012).
Zilmer, M. et al. Novel congenital disorder of O-linked glycosylation caused by GALNT2 loss of function. Brain 143, 1114–1126 (2020).
Khetarpal, S. A. et al. Loss of function of GALNT2 lowers high-density lipoproteins in humans, nonhuman primates, and rodents. Cell Metab. 24, 234–245 (2016).
Limburg, H. et al. TMPRSS2 is the major activating protease of influenza A virus in primary human airway cells and influenza B virus in human type II pneumocytes. J. Virol. 93, e00649–00619 (2019).
Yu, S. et al. SARS-CoV-2 spike engagement of ACE2 primes S2′ site cleavage and fusion initiation. Proc. Natl Acad. Sci. USA 119, e2111199119 (2022).
Li, P. et al. Nsp3-N interactions are critical for SARS-CoV-2 fitness and virulence. Proc. Natl Acad. Sci. USA 120, e2305674120 (2023).
Lei, R. et al. Functional and antigenic characterization of SARS-CoV-2 spike fusion peptide by deep mutational scanning. Nat. Commun. 15, 4056 (2024).
Ju, X. et al. A novel cell culture system modeling the SARS-CoV-2 life cycle. PLoS Pathog. 17, e1009439 (2021).
Niemi, M. E. et al. Mapping the human genetic architecture of COVID-19. Nature 600, 2021–477 (2021).
Pairo-Castineira, E. et al. Genetic mechanisms of critical illness in COVID-19. Nature 591, 92–98 (2021).
Pinto, R. M. et al. BTN3A3 evasion promotes the zoonotic potential of influenza A viruses. Nature 619, 338–347 (2023).
Zhang, L. et al. Furin cleavage of the SARS-CoV-2 spike is modulated by O-glycosylation. Proc. Natl Acad. Sci. USA 118, e2109905118 (2021).
Wang, S. et al. Sequential glycosylations at the multibasic cleavage site of SARS-CoV-2 spike protein regulate viral activity. Nat. Commun. 15, 4162 (2024).
He, J. et al. Identifying transposable element expression dynamics and heterogeneity during development at the single-cell level with a processing pipeline scTE. Nat. Commun. 12, 1456 (2021).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019).
Hagen, K. G. T. et al. Characterization of a UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase that displays glycopeptide N-acetylgalactosaminyltransferase activity. J. Biol. Chem. 274, 27867–27874 (1999).
De Las Rivas, M. et al. The interdomain flexible linker of the polypeptide GalNAc transferases dictates their long-range glycosylation preferences. Nat. Commun. 8, 1959 (2017).
Kong, Y. et al. Probing polypeptide GalNAc-transferase isoform substrate specificities by in vitro analysis. Glycobiology 25, 55–65 (2015).
Ye, Z., Mao, Y., Clausen, H. & Vakhrushev, S. Y. Glyco-DIA: a method for quantitative O-glycoproteomics with in silico-boosted glycopeptide libraries. Nat. Methods 16, 902–910 (2019).
Acknowledgements
We thank all study participants who donated blood and BALF samples to our research. We thank C. Ka Pun Mok (assistant professor, The Chinese University of Hong Kong) who provided IAVs, including A/Puerto Rico/8/1934 (H1N1) (PR8), A/WSN/1933 (H1N1) (WSN), A/California/04/2009 (H1N1) (CA04), A/Hong Kong/1968 (H3N2) (HK68), A/Aichi/2/1968 (H3N2) (Aichi) and A/Quail/Hong Kong/G1/97 (H9N2) (G1 and G1_PB2E627K). This study was supported by grants from the National Key Research and Development Program of China (2022YFC2604100 to J.S.), National Natural Science Foundation of China (82495200, 82495203 and 82025001 to Jincun Zhao; 82402594 to W.R.; T2450074 to Y.M.; 81971500 to Jingxian Zhao; 31971213 to Shengjun Wang; 82472247 to Shaobo Wang; 82402593 to S.Y.; 92478126 to Y. Yuan and W.R.), State Key Laboratory of Respiratory Disease (SKLRD-Z-202304 to Q.H., SKLRD-Z-202508 to S.Y.), Major Project of Guangzhou National Laboratory (SRPG22-002 to Shaobo Wang and Jincun Zhao, QNPG23-03 to S.Y.), The Guangzhou Science and Technology Plan Project (202103000029 to Y.M., 2023A1515010152 to S.Y., The Science and Technology Project of General Administration of Customs, P.R. China (2021HK141to L.Z.), Technology innovation group project of Foshan 2019 (FS0AA-KJ919-4402-0027 to T.W.), Korea Institute of Planning and Evaluation for Technology in Food, Agriculture and Forestry (IPET) through Animal Disease Management Technology Advancement Support Program, funded by the Ministry of Agriculture, Food and Rural Affairs (MAFRA) (122012-2, 122060-2 to J.-E.P.), NIH, R01. (AI129269 to S.P.).
Author information
Authors and Affiliations
Contributions
W.R., Jingxian Zhao, Y.M., Shaobo Wang and Jincun Zhao led and conceived of this study. W.R., J.Y., S.Y., C.W., J.S., F.L., W.T. and H.H. led the molecular virology work. Q.H., Q.Y., K.Z. and F.C. guided the GWAS and scRNA-seq analyses. A.Z. and Z.C. directed the SARS-CoV-2 clinical data collection and analysis. Y.M., Y.H., Shengjun Wang, Y. Yuan, H.T. and L.S. headed the O-linked glycosylation work. S.P., P.L. and R.P. designed the SARS-CoV-2 recombinant virus experiments. Q.D. and Y. Yu designed the SARS-CoV-2 replicon experiments. W.R., J.S., F.L., Yanqun Wang, Zhaoyong Zhang, J. Zhuo, Z.Z., R.B., H.Z. and T.W. guided the animal experiments. W.R., L.Z., D.L., D.W., K.Y., Yiliang Wang, J.D. and Y.Z. directed the SARS-CoV-2 experiments in BSL-3. W.R., J.Y., S.Y., Shaobo Wang, Shengjun Wang, S.P. and Jincun Zhao conducted formal analysis. W.R., J.S., Jingxian Zhao, Y.M., Shaobo Wang, Jincun Zhao, Q.H., Yanqun Wang, S.Y., L.Z., S.P., T.W., Y. Yuan and J.-E.P. acquired funding. Q.D., S.P., Jingxian Zhao, Y.M., Shaobo Wang and Jincun Zhao supervised the study. W.R., Q.H., Shaobo Wang and Jincun Zhao wrote the original draft.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks Frank Kirchhoff, Andreas Pichlmair and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Impaired type I IFN response lead to increased and prolonged SARS-CoV-2 infection in vivo.
a, A schematic showing the mouse infection model of SARS-CoV-2 by exogenous delivery of human ACE2 with a replication-deficient adenovirus (Ad5-hACE2). b, IFNAR-/- (n = 3) and WT (n = 3) were intranasally transduced with 2.5×108 FFU of Ad5-hACE2. Five days post transduction of Ad5-hACE2, mice were infected with 1×105 PFU of SARS-CoV-2 (WT). The lungs were harvested at 2 days post infection and performed mRNA-seq. The transcriptional profiling of homogenized lungs are expressed as normalized read counts to WT mice. c, The genes showed in (b) were homo sapiens codon-optimized and synthetized de novo into pcDNA3.1 vector. Huh7 cells were transfected with plasmids encoding these genes (EV and eGFP as the controls). At 24 h post transfection, cells were infected with SARS-CoV-2 (WT, MOI = 0.05) for 48 h. Viral infection efficiency was determined by IFA. Data were normalized to EV. Data are means ± s.d. from n = 3 independent experiments. d, Huh7 cells were transfected with plasmid encoding GALNT2 (EV as the control), and GALNT2 protein expression level was detected at 24 h post transfection by western blots. Representative IFA images that GALNT2 inhibits SARS-CoV-2 infection in Huh7 cells. SARS-CoV-2 N was stained using indirect fluorescent antibody (green), nuclei are stained with DAPI (blue). Western blots and IFA images were representative of three independent biological repeats. e, GALNT2 and STAT1 protein levels of WT and IFNAR-/- mice (n = 3) at 2, 4, and 6 days post infection were determined by western blots. f, Related to (e), comparison viral titres and GALNT2 protein levels of WT mice at 2, 4, and 6 d.p.i. n = 3 mice per group. GALNT2 protein levels were normalized to that in WT mice (4 d.p.i.). g, Related to (e), comparison viral titres and GALNT2 protein levels of IFNAR-/- mice at 2, 4 and 6 d.p.i. n = 3 mice per group. GALNT2 protein levels were normalized to that in WT mice (4 d.p.i.). h, Huh7 cells were transfected with plasmids encoding GALNT2-HA (EV as the control) for 36 h. Cells were then fixed with 4% paraformaldehyde and stained using Anti-HA-tag antibodies conjugated with Allophycocyanin (APC). Cells were analyzed by IFA. Scale bars are indicative of 20 μm. i, Huh7 and HEK293T cells were transfected with plasmids encoding EV, eGFP and GALNT2-HA for 24 h. GALNT2 expression levels were detected by flow cytometry and data analysis was performed using the FlowJo 10.4 (LCC). Data are means ± s.d. from n = 3 independent experiments. j, Huh7 cells were transfected with plasmids encoding GALNT2 (EV and eGFP as the controls. At 24 h post transfection, the cell viability was measured by CCK-8 and normalized to Huh7 WT cells. Data are means ± s.d. from n = 12 independent experiments. k, Huh7 cells were transfected with plasmids encoding GALNT2 (EV and eGFP as the controls. At 24 h post transfection, cells were seeded in 24-well plates at 1×104 cells/well. Cell growth is counted at indicated time points. Data are means ± s.d. from n = 3 independent experiments. P values from two-tailed Student’s t-test.
Extended Data Fig. 2 Single-cell transcriptional profiling of BALF and PBMCs from HDs and SARS-CoV-2 infected patients.
a, Canonical markers for different cell types in BALF were shown in UMAP and dot plots. b, The UMAP plots were split by different studies. c, Comparison of GALNT2 expression in BALF samples using scRNA-seq. Each dot represents an individual, and the y-axis represents the average expression level of GALNT2 in a specific cell type from that individual. d, Comparison of GALNT2 expression in Myeloid, T and B, ciliated and secretory cells of BALF samples using scRNA-seq. Each dot represents an individual, and the y-axis represents the average expression level of GALNT2 across all cells or specifically in ciliated cells from that individual. The box plots show the median (center line), the 25th and 75th percentiles (bounds of the box), and the whiskers extend to the smallest and largest values within 1.5 times the interquartile range from the box. The p values were calculated by two-tailed Wilcoxon signed-rank test. ((ns): no significant, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001). e, Protein expression of GALNT2 in the PBMC samples at conversion phase (T2) from mild and severe patients were detected by western blots. Western blots were representative of three independent biological repeats. Quantitative data from representative western blot is shown. Data are means ± s.d. from n = 3 independent experiments. P values from two-tailed Student’s t-test. ((ns): no significant, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001).
Extended Data Fig. 3 Expression and regulation of GALNT2 and classical ISGs across tissues and COVID-19 severity groups.
a, RNA expression patterns of OAS1, MX1, and GALNT2 across human tissues (data from the Human Protein Atlas; https://www.proteinatlas.org), visualized as z-score normalized heatmaps (red: higher expression, blue: lower expression). Scatter plots below show pairwise expression correlations between genes, with locally estimated scatterplot smoothing (LOESS) fits (blue line) and 95% confidence intervals (gray shading). Pearson correlation coefficients and p-values by two-tailed test are indicated. b, Protein-level expression of OAS1, MX1, and GALNT2 across human tissues, from the Human Protein Atlas. Expression levels are categorized as not detected(grey), low (light blue), medium (blue), or high (dark blue) by HPA database. c, Left: Comparison of OAS1 expression in ciliated epithelial cells from bronchoalveolar lavage fluid (BALF) across COVID-19 severity groups based on scRNA-seq data. The box plots show the median (center line), the 25th and 75th percentiles (bounds of the box), and the whiskers extend to the smallest and largest values within 1.5 times the interquartile range from the box. Each dot represents an individual; the y-axis shows average OAS1 expression per individual. Right: Longitudinal OAS1 expression in peripheral blood mononuclear cell (PBMC) subsets (B cells, T cells, and monocytes) across healthy controls (HC) and patients with mild or severe COVID-19. Open dots represent individual-level averages; solid dots represent group means. Time points: T1 (acute phase), T2 (conversion), T3 (recovery; see Methods for definitions). Samples from the same individual are connected by dashed lines. P values were calculated using two-tailed Wilcoxon signed-rank test (left) or two-tailed Student’s t-test (right). d, Comparison of MX1, SOCS1, STAT1, and USP18 expression in ciliated BALF cells across clinical severity groups. Each dot represents one individual; the y-axis shows average gene expression. The box plots show the median (center line), the 25th and 75th percentiles (bounds of the box), and the whiskers extend to the smallest and largest values within 1.5 times the interquartile range from the box. P values by two-tailed Wilcoxon signed-rank test. e, Summary of differential protein expression for the indicated genes across tissues in SARS-CoV-2 infected patients versus controls, based on published proteomics data65. Red indicates significant upregulation (FDR < 0.05); gray, not significant. ((ns): no significant, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001).
Extended Data Fig. 4 GALNT2 associated with the severity or susceptibility of SARS-CoV-2 and IAV infections.
a, GALNT2 association results in influenza A virus H7N9 genome-wide association studies (GWAS). Mx1 gene (left panel) as the control. Red plots represent the SNPs associated with virus infection. b, GALNT2 association results in influenza A virus H1N1 genome-wide association studies (GWAS). Mx1 gene (left panel) as the control. Red plots represent the SNPs associated with virus infection. P values from (a) and (b) were calculated by two-tailed Firth logistic regression test, with appropriate covariates and genomic control. c, Analysis of genetic variation at the GALNT2 gene locus between SARS-CoV-2 infected patients and control populations using data from Host Genetics Initiative (https://www.covid19hg.org/). Population frequency information for the SNP was sourced from https://www.ncbi.nlm.nih.gov/snp/rs2273970. P values were calculated by two-tailed Firth logistic regression test, with appropriate covariates and genomic control. d, Huh7 cells were transfected with plasmids encoding GALNT2 (WT or four mutants), and cells then infected with SARS-CoV-2 (MOI = 0.5) and viral infection efficiency after 24 h.p.i. by IFA. Data are means ± s.d. from n = 3 or 5 independent experiments. P values from two-tailed Student’s t-test. ((ns): no significant, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001). e, Huh7 cells were transfected with plasmids encoding GALNT2 (WT or V554M), and cells then infected with H1N1 PR8 (MOI = 0.01) and viral infection efficiency after 24 h.p.i. by IFA. Data are means ± s.d. from n = 3 independent experiments. P values from two-tailed Student’s t-test. ((ns): no significant, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001). f, GALNT2 protein expression levels in Huh7 OE-GALNT2 (GALNT2-stable cells) and Huh7 GALNT2-/- cells were detected by western blot. Western blots were representative of three independent biological repeats. g, GALNT2 protein expression levels in A549 OE-GALNT2 (GALNT2-stable cells) and A549 GALNT2-/- cells were detected by western blot. Western blots were representative of three independent biological repeats. h, STAT1 protein expression levels in Huh7 STAT1-/- and A549 STAT1-/- cells were detected by western blot. Western blots were representative of three independent biological repeats. i, Huh7 WT, GALNT2-/- and reconstitution of GALNT2 in the knockout cells by transient transfection (eGFP as the control) were infected with HCoV-OC43 at MOI 0.05. Supernatants were harvested at 24, 48 and 72 h.p.i. and viral titres were determined in HRT-18 cells by FFA. Data are means ± s.d. from n = 3 independent experiments. j, Huh7 WT, GALNT2-/- and reconstitution of GALNT2 in the knockout cells by transient transfection (eGFP as the control) were infected with HCoV-229E at MOI 0.001. Supernatants were harvested at 24, 48 and 72 h.p.i. and viral titres were determined in Huh7 cells by FFA. Data are means ± s.d. from n = 3 independent experiments. k, A549 WT (Ctrl) and A549 GALNT2-/- cells were infected with IAVs at MOI of 0.01, including H1N1 (PR8 and CA04) and H3N2 (HK68). Supernatants were harvested at indicated time points and viral titres were determined on MDCK cells by FFA. Data are means ± s.d. from n = 3 independent experiments. Data are means ± s.d. from n = 3 independent experiments. P values from two-tailed Student’s t-test. (ns): no significant, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001).
Extended Data Fig. 5 GALNT2 is an antiviral ISG.
a-b, AAV9-EV and AAV9-GALNT2 transduced Calu3 cells. At 48 h post transduction, cells were infected with SARS-CoV-2 WT at MOI 0.01, supernatants were harvested at 48 h.p.i. and viral titres were determined in Vero E6 cells by FFA. Images of viral foci were captured by ELISPOT. P values from two-tailed Student’s t-test. c-d, Foci forming sizes (n = 20) were measured and analyzed. e, Quantative PCR analysis of GALNT2 mRNA in Huh7 cells after SARS-CoV-2 infection at MOI of 0.5. Data are means ± s.d. from n = 3 independent experiments. f, THP-1 cells were infected HCoV-OC43 at MOI of 1. The mRNA expression levels of GALNT2 was analyzed by qPCR at indicated time points. Data are means ± s.d. from n = 3 independent experiments. g, Huh7 cells were infected with HCoV-OC43 for 6 (n = 3) or 24 h (n = 3), respectively. GALNT2 and phosphorylated STAT1 (pSTAT1) protein expression levels were detected by western blot. h, A549 cells were infected with PR8 H1N1 at MOI of 0.01. Cell lysates were harvested at 6 and 24 h.p.i., respectively. GALNT2 and pSTAT1 protein expression levels were detected by western blot. Western blots were representative of three independent biological repeats. i. Huh7 STAT1-/- and Huh7 cells (Ctrl) were treated with 5 μg/mL of IFN-α2 for 24 h. The mRNA levels of GALNT2, STAT1 and OAS1 were detected by qPCR. Data are means ± s.d. from n = 3 independent experiments. j, Huh7, Huh7 GALNT2-/- and Huh7 STAT1-/- cells were treated with 100 ng/mL of IFN-β for 24 h. Then cells were infected with PR8 H1N1 at MOI of 0.01. Viral infection efficiency was detected the by IFA. Data are means ± s.d. from n = 6 independent experiments. k, Schema depicting the presence and position of GAS and ISRE elements in the promoter region (2 kb upstream from the transcription initiation site) of human GALNT2 gene. All Western blots were representative of three independent biological repeats. Data are mean ± s.d. P values from two-tailed Student’s t-test. ((ns): no significant, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001). Figure created with BioRender.com.
Extended Data Fig. 6 Increased lethality and pathogenicity of H1N1 PR8 in GALNT2-/- mice.
a, Using CRISPR/Cas9-mediated gene editing technology, two single guide RNAs (sgRNAs) were designed targeting the second exon and the region between the second and third exons of the GALNT2 gene. b, Sequencing analysis revealed the complete knockout of the second exon of GALNT2. c, WT (n = 5) and GALNT2-/- (n = 4) C57BL/6 mice were intranasally treated by 2 μg IFN-α2. Lungs were harvested at 3 d.p.i. GALNT2 protein expression levels were determined by western blot. Western blots were representative of three independent biological repeats. d, WT (n = 5) and GALNT2-/- C57BL/6 (n = 5) mice were intranasally infected with 350 FFU of PR8 H1N1. Lungs were harvested at 3 d.p.i. e, Quantitative PCR analysis of Il-1β mRNA in the lungs from (d). f, HE staining of the infected lungs of WT (n = 5) or GALNT2-/- (n = 5) C57BL/6 mice infected with PR8 H1N1 at 3 d.p.i. The scale bars are indicative of 100μm and 1000μm respectively. Data are mean ± s.d. P values from two-tailed Student’s t-test in (b), (d). ((ns): no significant, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001).
Extended Data Fig. 7 Overexpression of GALNT2 alleviates pneumonia induced by IAV and SARS-CoV-2 infection.
a, Groups of C57BL/6 mice (n = 2) were either transduced with 3.3×1011 GC of AAV9-EV or AAV9-hGALNT2 intranasally. Lungs were harvested at 14 d.p.i. GALNT2 protein expression levels were monitored by western blot. Western blots were representative of three independent biological repeats. b-c, AAV9-EV or AAV9-GALNT2 transduced C57BL/6 mice (n = 5 or 6) infected with 1000 FFU PR8 H1N1. Three days post infection, GALNT2 expression levels (b) and PR8 M (c) gene mRNA in the lungs were analyzed by qPCR. Two of five mice in AAV9-GALNT2 group could not be detected by RT-qPCR (RT-qPCR Cq values above 38 were considered as negative). d, AAV9-EV or AAV9-GALNT2 transduced C57BL/6 mice infected with 1000 FFU PR8 H1N1. Three days post infection, lungs were harvested. Representative images of infected lungs were shown. e, HE staining of the lungs from (d). The scale bars are indicative of 1000μm and 500μm respectively. f, Quantitative PCR analysis of Il-1β mRNA in the lungs from (b). g-h, Groups of C57BL/6 mice were either transduced with 3.3×1011 GC of AAV9-EV or AAV9-GALNT2 (n = 5 or 6) intranasally and subsequently transduced with 2.5×108 FFU of Ad5-hACE2 after 9 days post AAV transduction. Five days after secondary transduction, mice were infected with 1×105 FFU of SARS-CoV-2 (WT). GALNT2 and SARS-CoV-2 N genes mRNA expression levels in the lungs were determined by qPCR. RT-qPCR Cq values above 38 were considered as negative. i, Representative images of infected lungs from (g-h). j, HE staining of the lungs of from (g-h). The scale bars are indicative of 100μm and 1000μm respectively. k, Quantitative PCR analysis of Il-1β mRNA in the lungs from (g-h). Data are mean ± s.d. P values from two-tailed Student’s t-test. ((ns): no significant, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001).
Extended Data Fig. 8 Overexpression of GALNT2 in A549 has a minimal impact on host genes.
a, Scatter plot showing relative up- and down-regulated genes, identified by RNA-seq analysis of GALNT2-overexpressing A549 cells. GALNT2 expression was validated by western blots (Extended Data Fig. 4g). b, Proteomics: Volcano plot illustrates GALNT2-regulated proteins from the Stable Isotope Labeling By Amino Acids In Cell Culture (SILAC) experiment. Greater than 2-fold change in abundance upon GALNT2 overexpression and lower than 0.05 adjusted p value were set as the threshold criteria for GALNT2-regulated proteins (see method). c, Glycoproteomics: Volcano plot illustrates GALNT2-regulated O-glycosylation sites from the SILAC experiment. Greater than 2-fold increase in abundance upon GALNT2 overexpression and lower than 0.05 adjusted p value were set as the threshold criteria for GALNT2-regulated sites (see method). Pearson correlation coefficients and p-values by two-tailed test are indicated.
Extended Data Fig. 9 GALNT2-dependent O-linked glycosylation at S810/813 of spike.
a, Soluble GalNAc-T1, T2, T3, T4, T5, T7, T11 and T18 were overexpressed in FreeStyleTM HEK293T cells and purified by Ni-NTA chromatography. All proteins were analyzed by Western blots. Western blots were representative of three independent biological repeats. b, MALDI-TOF analysis of GalNAcylation reactions catalyzed by purified GalNAc-Ts on the synthetic spike S2’ (TMPRSS2) peptides. Reactions were performed and analyzed as described in the Methods. An increase of 203 Da corresponds to addition of one GalNAc residue was only detected O-glycosylation by GALNT2. c, Soluble GALNT2 (WT and four mutations) were overexpressed in FreeStyleTM HEK293T cells and purified by Ni-NTA chromatography. All proteins were analyzed by Western blots. Western blots were representative of three independent biological repeats. d, MALDI-TOF analysis of GalNAcylation reactions catalyzed by purified GALNT2 on the synthetic spike S2’ (TMPRSS2) peptides. Reactions were performed (3 h) and analyzed as described in the Methods. An increase of 203 Da corresponds to addition of one GalNAc residue was obvious detected O-glycosylation by WT GALNT2, but an apparent decline in four mutants.
Extended Data Fig. 10 The S810/813I of spike evades GALNT2 antiviral function in a SARS-CoV-2 replicon system.
a, Constructing a reverse genetic complementary SARS-CoV-2 replicon system. The N gene is deleted from the viral genome and replaced with a GFP reporter gene. Through in vitro ligation and transcription, a SARS-CoV-2 RNA genome lacking the N gene but carrying the GFP reporter gene is obtained. Then RNA genome is electroporated into Caco2 cells stably expressing the viral N protein (Caco2-N, packaging cell line), allowing the production of SARS-CoV-2-NdeI replicon. Here we generated 2 replicons, comprising SARS-CoV-2 WT and S810/813I spike mutations. b, Fluorescent and bright field images of SARS-CoV-2 replicons, including WT or S810/813I. Images were representative of three independent biological repeats. Scale bars are indicative of 200 μm. c, Caco2-N cells were infected by WT SARS-CoV-2 or S810/813I replicon at MOI of 0.01 for 24 or 48 h in presence of EV or GALNT2. Viral titres were determined by TCID50. Data are means ± s.d. from n = 3 independent experiments. P values from two-tailed Student’s t-test. d, Caco2-N cells were infected with SARS-CoV-2_Ndel-WT and SARS-CoV-2_Ndel-S810/813I viruses at MOI of 0.01 for 48 h in presence of EV or GALNT2. Caco2-N cells were infected with SARS-CoV-2_Ndel-WT and SARS-CoV-2_Ndel-S810/813I viruses at MOI of 0.01 for 48 h in presence of EV or GALNT2. The GFP-positive cells were quantified of syncytia among total cells in Caco2-N cells. Data are means ± s.d. from n = 3 independent experiments. P values from two-tailed Student’s t-test. Images were representative of three independent biological repeats. Scale bars are indicative of 200 μm. e, A schematic shows the S810/813I and S810/813 A of SARS-CoV-2 recombinant viruses evaded GALNT2 antiviral activity, while reducing the viral fitness due to reduced utilization of TMPRSS2 for viral entry.
Supplementary information
Supplementary Information (download PDF )
Supplementary Tables 1–7 and Supplementary Methods.
Source data
Source Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Fig. 2 (download PDF )
Unprocessed western blots and/or gels.
Source Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Fig. 4 (download PDF )
Unprocessed western blots and/or gels.
Source Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Fig. 5 (download PDF )
Unprocessed western blots and/or gels.
Source Data Fig. 6 (download XLSX )
Statistical source data.
Source Data Fig. 6 (download PDF )
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 1 (download PDF )
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 2 (download PDF )
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 4 (download PDF )
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 5 (download PDF )
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 6 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 6 (download PDF )
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 7 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 7 (download PDF )
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 9 (download PDF )
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 10 (download XLSX )
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ran, W., Yang, J., Yu, S. et al. Interferon-stimulated gene GALNT2 restricts respiratory virus infections. Nat Microbiol 11, 256–270 (2026). https://doi.org/10.1038/s41564-025-02200-7
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41564-025-02200-7
