
Abstract
Surface passivation in perovskite solar cells can enhance device efficiency, yet incomplete interfacial functionality poses challenges to long-term reliability. Here we present a strategic interfacial engineering approach using sodium heptafluorobutyrate to fully functionalize the perovskite surface. Sodium heptafluorobutyrate acts as an ion shield that tunes the perovskite surface work function and increases the defect formation energy, resulting in an improved interface with the electron transport layer that minimizes recombination and boosts electron extraction under operation. We find that a sodium-heptafluorobutyrate-functionalized perovskite surface promotes a uniform, compact C60 layer that effectively blocks ion diffusion and stabilizes the device stack. This approach allows p–i–n perovskite solar cells to achieve a record power conversion efficiency (PCE) of 27.02% (certified 26.96% with a maximum-power-point-tracking PCE of 26.61%). Devices with an active area of 1 cm2 deliver a PCE of 25.95%. Perovskite solar cells retain 100% of their initial efficiency following 1,200 h of continuous 1-sun illumination at the maximum power point. Devices also demonstrate exceptional thermal stability, retaining 92% of the initial PCE when ageing at 85 °C for 1,800 h and 94% after 200 thermal cycles between –40 °C and +85 °C.
Similar content being viewed by others

Main
Metal halide perovskite solar cells (PSCs) have impressive power conversion efficiencies (PCEs)1,2 due to their strong light absorption and appreciably high carrier mobility, and their low cost makes them particularly attractive3,4. Inverted (p–i–n) PSCs have achieved almost 27% PCE, competitive with silicon-based solar cells5,6. Such device architectures possess inherently better compatibility with flexible substrates and low-temperature processing, which could facilitate scale-up and the manufacture of tandem cells7,8,9. To drive forward the industrialization of perovskite photovoltaics, however, device reliability is a critical issue that is increasingly receiving attention10.
In the p–i–n architecture, the upper interface involves contact between the perovskite surface and the electron-selective contact (ESC). The surface of perovskite films often exhibit a high abundance of defects, primarily due to the formation of volatile compounds during the fabrication process, leaving behind defects11,12. These defects, in turn, lead to notable non-radiative recombination losses13,14,15. When illuminated or operated at elevated temperatures, the weaker bonds within the perovskite readily dissociate, generating vacancies and interstitial defects that migrate towards the metal contacts, posing a significant challenge to device stability16,17. Typically, the suppression of ion migration relies on passivating undercoordinated sites within the perovskite lattice. Nonetheless, the ultimate barrier to prevent ion migration within a device often lies at the ESC interface. Fullerene-type materials such as carbon 60 (C60) are commonly used as the ESC, which tend to aggregate when operated at elevated temperatures18. Consequently, not only charge transport is hampered but also the ability of the ESC to block reactions between the metal electrode and the underlying perovskite layer is undermined, thereby compromising device durability16. Therefore, achieving an optimal interface contact requires (1) the passivation of surface defects and stabilization of surface structure in the perovskite, (2) favourable interface charge transport and (3) a compact and stable ESC that blocks ion mobility. By creating a superior upper interface, the operational stability of PSCs should be prolonged.
To address these issues, we consider sodium heptafluorobutyrate (SHF), combining the perfluorous tail and carboxylate head, offering dual functionality as a candidate. SHF has the potential to form a robust hydrophobic barrier, simultaneously providing the effective passivation of surface defects. Using SHF, we present an advanced perovskite surface-to-ESC integrated interfacial engineering strategy. SHF increases the defect formation energy of the perovskite surface, stabilizing undercoordinated Pb(II) and eliminating the non-photoactive phase. Due to suitable dipole characteristics, SHF induces carrier redistribution and tunes the perovskite surface work function (WF) to enhance the open-circuit voltage. During ESC deposition, SHF causes C60 to stack in densely packed layers, which suppresses ion diffusion within the device. The resulting PSCs exhibit a record PCE of 27.02% (certified PCE being 26.96% with a stabilized PCE of 26.61%). Additionally, 1-cm2 devices show a PCE of up to 25.95%. In particular, the SHF-treated device retains its initial PCE (0% efficiency loss) after 1,200 h of maximum power point tracking (MPPT) under 1-sun illumination (International Summit on Organic Photovoltaic Stability ISOS-L-1) and demonstrated over 92% retention when aged at ~85 °C (ISOS-D-2) over 1,800 h. The device cycled between −40 °C and 85 °C showed a retention of 94% of its initial PCE after 200 cycles (ISOS-T-3).
Functionalization of the perovskite surface with SHF
To improve the surface electronic properties of perovskites in a targeted manner, we studied their defect characteristics through first-principles DFT calculations (Fig. 1a). We compared the defect formation energy (VPb and VI) on two types of perovskite surface termination (PbI and formamidinium iodide (FAI)), with the structure models shown in Supplementary Figs. 1–4. The computation revealed that the formation energy of a Pb vacancy is smaller than that of an I vacancy for the PbI-terminated surface, with both being less than that for an FAI-terminated surface (Fig. 1b). This suggests that defects are more easily formed on a pure PbI-terminated surface compared with a pure FAI-terminated surface, corresponding to lower surface stability. In particular, to aid crystal growth and defect passivation, a lead(II) iodide (PbI2)-rich stoichiometry is often used in perovskite fabrication, which also generates a PbI-terminated surface19,20, probably undermining the operational stability of PSCs.

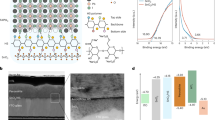
a, Constructed SHF adsorption model on the perovskite structure. b, Defect formation energy of the perovskite surface with and without SHF adsorption. PVK, perovskite. c, Planar Δρ for the PbI2-terminated surface of single SHF-adsorbed perovskite. d,e, KPFM maps of perovskites without (d) and with (e) SHF. The colour scale represents the surface potential (V). Image size, 5 μm × 5 μm. f,g, Integrated GIWAXS patterns on (001) peaks of perovskites without (f) and with (g) SHF for incident angles from 0.05° to 1.0°. h–k, Simulated energy-level diagrams of control (h and j) and SHF-containing (i and k) devices before and after dark equilibration. The SHF layer introduces a vacuum-level shift and enhances the electric field across C60, consistent with an increased built-in potential. WFb, bulk work function; WFs, surface work function; EF is the Fermi level energy; EC and EV are the conduction and valence band edges, respectively, and Evac is the vacuum energy level.
SHF was used to functionalize the perovskite surface via a post-treatment strategy (Fig. 1a and Supplementary Figs. 2–4). DFT calculations show that in the surface adsorption system, the formation energy of vacancies on the PbI surface significantly increases (Fig. 1b), indicating that SHF plays a crucial role in enhancing the surface stability. Mulliken charge population calculations indicate that surface adsorption per SHF will result in 0.22 electron transfer to SHF (Fig. 1c), amplified on the SHF-stacking surface (Supplementary Figs. 5–7).
SHF consists of Na+ and a perfluorocarbon chain terminated with a COO− group, forming a system with a dipole moment. On interacting with the perovskite surface, Na+ introduces localized positive charges, whereas the COO− group generates localized negative charges. Due to their electrostatic interaction and the large size of perfluorocarboxylate anions, Na+ ions potentially remain at the perovskite surface beside the COO− groups21. This combination would establish an interfacial dipole pointing from SHF towards the perovskite. Kelvin probe force microscopy (KPFM) confirms that SHF alters the surface WF of the perovskite, validating the dipole effect. Figure 1d,e shows the potential distribution patterns of control and SHF-treated perovskite films. The average contact potential difference value increases from 0.43 to 0.68 eV after treating the perovskite with SHF (Supplementary Fig. 8). This increase is consistent with the electrostatic potential and WF estimated from DFT (Supplementary Figs. 9–11). We further reveal that the appropriate SHF coverage is a key factor in tuning the surface WF (Supplementary Fig. 12). In this case, the formed positive dipoles at the perovskite–ETL interface increases the PSC’s built-in potential22,23,24 (Fig. 1h,i and Supplementary Fig. 13). To clarify the impact of SHF-induced dipoles under device-relevant conditions, we carried out drift-diffusion simulations on full device stacks (Fig. 1h–k). Although the perovskite band structure remains largely unaffected in the dark equilibrium state, the introduced SHF significantly enhances the internal electric field across the undoped C60 layer. This originates from reduced energetic offsets at the perovskite–C60 interface, consistent with a larger built-in potential. The enhanced interfacial field is expected to promote charge extraction and increase the device’s open-circuit voltage (VOC). To validate the role of molecular dipoles, control experiments were performed using sodium acetate (NaOAc; CH3COONa) under identical processing conditions. As shown in Supplementary Fig. 14, NaOAc treatment led to a minor change in the surface potential, in contrast to the pronounced shift observed with SHF. To understand the molecular origin of the observed dipole difference, we performed DFT calculations on SHF and NaOAc. The structures were optimized using the wB97XD functional and Def2TZVP basis set. The calculated dipole moment of SHF is 8.97 D, significantly larger than that of NaOAc (5.91 D; Supplementary Fig. 15), highlighting the contribution of the fluorinated tail in enhancing the overall molecular polarity.
The interaction between SHF and perovskite was further supported by Fourier transform infrared spectra, X-ray photoelectron spectra and nuclear magnetic resonance spectra25,26 (Supplementary Figs. 16–18). Grazing-incidence wide-angle X-ray scattering (GIWAXS) was used to characterize the surface crystal structure of the perovskite films. At low incidence angles (<1.0°; Fig. 1f), a sharp peak at q ≈ 9.4 nm−1 is observed in the untreated film, indexed as the hexagonal polytype 4H (ref. 27). In particular, SHF treatment eliminates the 4H phase (Fig. 1h). Additionally, a shift in the diffraction peak of the (001) crystal plane for the SHF-treated perovskite is observed, originating from the film surface (Fig. 1g). The shift decreases as the diffraction angle increases, indicative of a surface-specific interaction between SHF and the perovskite28 (Supplementary Fig. 19). According to the Bragg equation, q = 4πsinθ/λ = 2π/d, where d is the Bragg spacing in scattering, a diffraction peak position shifting to higher angles corresponds to a contraction of the lattice29. This helps stabilize the perovskite surface structure due to the improved Goldschmidt tolerance factor30. Note that the interaction of SHF with the perovskite surface is further supported by X-ray diffraction (Supplementary Fig. 20).
Structure and optoelectronic properties of interfacial contact
Since the ESC coats the perovskite surface, its structural characteristics are affected by the underlying perovskite layer, impacting the device performance. We monitored the perovskite film morphology using scanning electron microscopy (SEM) and atomic force microscopy (AFM). The improved perovskite surface due to SHF treatment was confirmed by SEM and AFM images (Fig. 2a and Supplementary Figs. 21 and 22), which facilitated a better interface contact with the ESC. Subsequently, the structural and optoelectronic properties of ESC as a function of the thickness of the C60 layer was studied31,32. In the SHF-treated device, the alignment of the electronic levels is significantly improved, evident from the reduced steepness of the secondary electron cut-off for the perovskites in contact with C60 (Fig. 2b,c). The reduced slope variation indicates that the individual secondary electron cut-offs at different kinetic energies tend to align more consistently. This is attributed to the enhanced perovskite surface uniformity, which promoted more favourable C60 deposition. The valence band scans of the C60 layers indicate their contribution to the total spectrum (Supplementary Fig. 23). The data for the C60 layers were fitted as the sum of the pristine perovskite valence band signal and the signal from 10-nm-thick C60, allowing the perovskite and C60 contributions to be separated. The reduced interfacial energy barrier (Supplementary Figs. 24 and 25) would enhance the PSC performance, as we show below using drift-diffusion simulations.

a, SEM image of SHF-treated perovskite films. b,c, Ultraviolet photoelectron spectroscopy spectra of the cut-off region for C60 with different deposition thicknesses on the perovskite films without (b) and with (c) SHF treatment. d,e, AFM images of perovskite/C60 (d) and perovskite/SHF/C60 (10-nm C60 deposited; e). Image size, 10 μm × 10 μm. f,g, AFM images of perovskite/SHF (f) and perovskite/SHF/C60 (g) samples (the AFM scans were taken from different areas (f) with one-half of the sample covered during deposition and 2-nm C60 deposited only on the other half (g)). Image size, 1 μm × 1 μm. The colour scales in d–g represent the surface height (nm).
The stacking properties of the deposited C60 layer were progressively probed using AFM. At a 10 × 10 μm2 scale, 10-nm C60 deposited on the control film surface exhibited noticeable voids (Fig. 2d). By contrast, C60 on the SHF-treated perovskite surface formed a densely packed layer (Fig. 2e). This difference suggests that the SHF layer mediates C60 growth and provides a favourable interfacial contact between the perovskite and ESC. The SHF probably reduces the surface energy and allows C60 to form a more uniform layer. By contrast, when non-polar molecules such as C60 are deposited directly onto hydrophilic surfaces like perovskites, they tend to aggregate to minimize unfavourable surface interaction energy33,34. As the thickness decreases into 2-nm C60, the differences between control and treated surfaces became more apparent, probably due to the direct effect of the initially deposited thin C60 layer contacting with the underlying perovskite surface (Supplementary Fig. 26). To gain deeper insights, the growth of a 2-nm C60 layer on SHF-treated perovskite surface was monitored at a 1 × 1 μm2 scale, with the perovskite film partially covered as a control (Fig. 2f). We observed that C60 tends to grow in island-like forms consisting of particle aggregates, showing an uneven structure (Fig. 2g). Interestingly, these islands are not isolated, but well interconnected, forming a cohesive layer.
Carrier dynamics and photovoltaic performance
To provide insights into the changes in electronic states, steady-state photoluminescence (PL) measurements were performed, where an enhanced PL intensity indicates the decreased defect-induced non-radiative recombination9 (Fig. 3a). Additionally, we observed an improvement of over two times in the PL quantum yield after SHF treatment (Supplementary Fig. 27), indicating that SHF effectively enhanced passivation. After contacting the C60 layer, the SHF-treated stack (perovskite/SHF/C60) retained a higher PL quantum yield (nearly three times) compared with the control (perovskite/C60), demonstrating reduced interfacial losses and improved optoelectronic quality.

a, Steady-state PL spectra for the untreated and SHF-treated perovskite films. The y axis shows the normalized PL intensity. b, trSPV charge dynamics at the perovskite–ESC interfaces. c, Schematic describing the charge separation mechanisms. d, PCE distribution of control and SHF-treated PSCs, collected from 20 independent devices per condition (n = 20). Box plots show the interquartile range with the median as the centre line; whiskers indicate the 5th–95th percentiles. e,f, J–V curves of untreated and SHF-treated PSCs with working areas of 0.0982 cm2 (e) and 1 cm2 (f).
To evaluate the charge transport properties in PSCs, transient surface photovoltage (trSPV) measurements were conducted, exciting the sample with a 5-ns laser pulse with a photon energy of 1.8 eV and fluences of 0.010 μJ cm−2, inducing a charge carrier density equivalent to 1 sun (refs. 35,36,37). The trSPV signal amplitude is proportional to the concentration of electrons extracted to the ESC, expressed as SPV(t) ≈ ΔnESC(t), where t denotes time37. The untreated perovskite sample without an ESC exhibits a positive trSPV signal due to hole accumulation near the perovskite surface, which can potentially limit electron extraction (Fig. 3b). By contrast, the SHF layer in the treated perovskite induces a negative trSPV signal, highlighting a qualitative change in the charge separation behaviour, with electrons dominating near the ESC side (Fig. 3c). The effect of SHF on the indium tin oxide (ITO)/perovskite/C60 and ITO/perovskite/C60/bathocuproine (BCP) samples was further investigated (Fig. 3b). SHF treatment considerably increases the amplitude and rise speed, with the peak amplitude for perovskite/SHF/C60/BCP being about twice as large as the untreated sample. This increase demonstrates a substantial improvement in electron extraction. The rise in amplitude, particularly in the post-extraction region (1–100 μs), indicates that SHF helps to align the energy-level alignment and passivate the perovskite surface. Additionally, similar trSPV results were observed for the perovskite–ESC interface on quartz, with an even more pronounced boost in rise speed and amplitude due to SHF treatment (Supplementary Figs. 28–30). Dark current and electrochemical impedance spectroscopy further indicate better interfacial contact following SHF treatment, leading to superior charge transport (Supplementary Figs. 31 and 32).
PSCs were fabricated with a configuration comprising transparent conducting oxide substrate/self-assembled monolayers (SAMs) (4-(7H-dibenzo[c,g]carbazol-7-yl)butyl)phosphonic acid (CbzNaph)/perovskite/C60/BCP/silver (Ag; Supplementary Fig. 33). Statistical analysis (Fig. 3d) shows an overall higher PCE distribution for SHF-treated PSCs compared with control (untreated) PSCs. Statistics for VOC, short-circuit current density (Jsc) and fill factor (FF) are presented in Supplementary Fig. 34, confirming the concentrated parameter distributions in PSCs, highlighting the effectiveness of SHF treatment on performance and reproducibility38. The champion control device exhibits a PCE of 24.81%, with a VOC of 1.161 eV and FF of 82.09% (Fig. 3e). In comparison, the SHF-treated PSCs show a champion PCE of 27.02%, along with an increased VOC of 1.197 eV and FF of 86.17% (Supplementary Fig. 35 and Supplementary Table 1), as well as reduced photocurrent–voltage (J–V) hysteresis39 (Supplementary Fig. 36 and Supplementary Table 2). The external quantum efficiency (EQE) spectra (Supplementary Fig. 37) show that the closed EQE onset are essentially the same in the devices. The integrated Jsc values match well with the values acquired from the J–V results (<1.3% discrepancy)40,41. Devices treated with NaOAc exhibited moderate performance improvements compared with the control but remained significantly inferior to SHF-treated devices (Supplementary Fig. 38). The laboratory-tested champion device was evaluated at the Fujian Metrology Institute, an accredited independent PV calibration laboratory, for independent validation. The device achieved a certified PCE of 26.96% under an aperture area of 7.82 mm2, with a certified MPPT efficiency of 26.61% (Supplementary Figs. 39 and 40). Additionally, a device with a 1-cm2 active area achieved a record PCE of 25.95% (Fig. 4f).

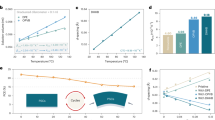
a, MPPT of encapsulated control and SHF-treated devices under simulated 1-sun illumination (ISOS-L-1I) in air (initial PCEs of 23.7% and 25.2%, respectively). b, Thermal stability of control and SHF-treated devices aged at 85 °C (ISOS-D-2I). Initial average PCEs of 24.1% and 25.9% (n = 3 independent devices per condition; error bars represent standard deviation). c, Thermal cycling stability of encapsulated devices aged between −40 °C and 85 °C (ISOS-T-3) (initial PCEs of 24.5% and 26.4%). d,e, ToF-SIMS ion species depth profiles of Ag− (Ag), \({{\rm{C}}}_{60}^{-}\) (C60), \({{\rm{PbI}}}_{2}^{-}\) (perovskite), I− (perovskite), \({{\rm{PO}}}_{2}^{-}\) (SAM), \({{\rm{InO}}}_{2}^{-}\) (ITO), \({{\rm{AgI}}}_{2}^{-}\) (AgI) and F− (SHF) of control (d) and SHF-treated (e) devices after thermal ageing. f,g, Ion diffusion schematics within control (f) and SHF-treated (g) devices.
To gain further insights into the influence of SHF treatment on photovoltaic performance, the devices were simulated using Driftfusion42 (Supplementary Table 3). As shown in Supplementary Fig. 41, the simulation’s parameters increased the steady-state VOC, FF and PCE by amounts comparable with those observed experimentally. In the control device, which has a smaller built-in potential, the direction of the electric field in the perovskite bulk reverses at a lower applied voltage. This has a negative impact on VOC and FF due to hole accumulation at the perovskite–C60 interface43 (Supplementary Fig. 42a). Conversely, when SHF is present, the resulting interfacial dipole improves the energetic alignment between the perovskite and C60, increasing the device’s built-in potential. Consequently, there is less hole accumulation at the perovskite–C60 interface at a given applied voltage, resulting in higher VOC and FF (Supplementary Fig. 42b). To assess the effect of Fermi-level variation, we simulated energy band profiles by adjusting the Fermi levels of the perovskite and C60 layers (Supplementary Figs. 43 and 44). The results suggest that the SHF-induced dipole dominates the interfacial energy alignment.
Operational stability of PSCs
Following the ISOS-L-1I protocol, the device stability was evaluated under simulated 1-sun illumination at the maximum power point44. The SHF-treated device demonstrated stable power output without PCE degradation after 1,200 h of continuous operation (Fig. 4a). In comparison, the control device decayed to ~60% of its initial value within only 800 h. The thermal stability of the devices was also assessed at 85 °C according to the ISOS-D-2I protocol (Fig. 4b), with the SHF-optimized device retaining 92% of its initial PCE after 1,800 h, whereas the control device degraded to approximately 52% of its initial PCE over the same period. Following the ISOS-T-3 protocol, devices were cycled between −40 °C and 85 °C. The SHF-treated device retained 94% of its initial PCE after 200 cycles, significantly outperforming the control device, which retained only 57% of its PCE (Fig. 4c). As expected, the fluorous chain in SHF provides a hydrophobic barrier (Supplementary Fig. 45), and hence, the stability of unencapsulated devices in a humid atmosphere was investigated (Supplementary Fig. 46).
To understand the mechanism behind the enhanced stability, the elemental distribution of aged devices was determined by time-of-flight secondary ion mass spectrometry (ToF-SIMS). From Fig. 4d and Supplementary Fig. 47a, in the control device, a substantial diffusion of I− ions towards the Ag electrode is observed, where they react to form AgI, with Ag+ ions concurrently diffusing into the perovskite layer. For the SHF-treated device, ion migration is considerably suppressed, with the amount of AgI detected being around two orders of magnitude lower than that in the control device (Fig. 4e and Supplementary Fig. 47b). Since ion migration leads to perovskite decomposition45,46, their differences could be related to device stability. Indeed, the diffusion of I− ions through ESC to Ag electrode, where it reacts to form AgI, further accelerates the degradation process (Fig. 4f). The SHF layer stabilizes the perovskite–ETL interface, blocking the passage of I− ions to prolong the operational stability of the device (Fig. 4g). Additionally, SHF inherently possesses high thermal stability, with an initial decomposition temperature reaching up to 260 °C (Supplementary Fig. 48). Comparison with ToF-SIMS data from the pre-aged sample demonstrates that SHF maintains its stability over time, confirming its sustained effectiveness during device operation (Supplementary Fig. 49).
Discussion
We report efficient and stable PSCs by anchoring a perfluorinated ionic barrier between the perovskite and ESC, which delivers PCEs of up to 27.02% and a certified PCE of 26.96% (certified MPPT PCE of 26.61%). SHF treatment increases the defect formation energy, eliminates non-photoactive phases and tunes the perovskite surface WF, effectively reducing the interfacial energetic offset between the perovskite and C60. The favourable surface structure provided by SHF also facilitates the growth of C60 layers, resulting in a denser ESC, which inhibits mobile ion shuttling within the device. Under accelerated ageing test conditions, the devices exhibit enhanced operational stability. These findings pave the way to the next generation of high-efficiency and high-stability perovskite-based optoelectronic devices.
Methods
Materials
FAI (99.99%, trace elements basis), methylammonium iodide (99.99%, trace elements basis) and methylammonium chloride (99.99%, trace elements basis) were purchased from Dyenamo. PbI2 (99.99%, trace metals basis) was purchased from TCI. Self-assembly molecules of CbzNaph (>99%) were purchased from Luminescence Technology. Caesium iodide (99.999%, AB 109298) was purchased from abcr Gute Chemie and used without further purification. Phenethylammonium bromide was obtained from Greatcell Solar Materials. C60 (≥99.99%, OE) was provided by CreaPhys. BCP (99.8%) was acquired from Ossila. Ag shots (2–3 mm, 99.999%) were bought from Alfa Aesar. SHF (C4F7NaO2) was obtained from Santa Cruz Biotechnology. Dimethyl sulfoxide-d6 (99.9 at.% D), N,N-dimethylformamide-d7 (≥99.5 at.% D), isopropyl alcohol, absolute ethanol and anhydrous chlorobenzene were acquired from Sigma-Aldrich.
Solution, film and device fabrication
The patterned ITO, fluorine-doped tin oxide and quartz as substrates were cleaned with deionized water, acetone and isopropyl alcohol in this order for 15 min. The substrates were further exposed to ultraviolet–ozone for 30 min. The self-assembly molecule CbzNaph (0.3 mg ml−1) was dissolved in absolute ethanol and used as a hole-selective contact. The SAM solution was spin coated on the substrate at 3,000 rpm in a two-step process with 3-s acceleration and then kept for 30 s. Then, the substrate–SAM was transferred onto a hotplate and annealed at 100 °C for 10 min in a N2-filled glovebox. The perovskite precursor solution was prepared by dissolving 1.55 M of Cs0.05FA0.9MA0.05PbI3 with 3% excess PbI2 in dimethylformamide/dimethyl sulfoxide with a volume ratio of 4:1. Then, 10 mg ml−1 of methylammonium chloride was added to the precursor solution to improve the film morphology. The mixture was stirred overnight and then filtered through a 0.22-µm polytetrafluoroethylene membrane before use. The precursor solution was spin coated at 1,000 rpm for 10 s, followed by 5,000 rpm for 40 s. In the last 5 s during the spin-coating procedure, 200 μl of chlorobenzene as the antisolvent was dropped onto the substrate. The precast films were then annealed at 100 °C for 30 min to obtain perovskite films. For initial surface passivation, 1 mg ml−1 of phenethylammonium bromide was subsequently used, dissolved in isopropyl alcohol/dimethyl sulfoxide (195:5, v/v), which was spin coated on the prepared perovskite films at 5,000 rpm for 30 s and then annealed at 100 °C for 10 min. The fabricated perovskite encompassing two- and three-dimensional structures was used as a control. SHF was dissolved in isopropyl alcohol (in varying concentrations of 1, 3 and 5 mM) and spin coated onto the film surface, under 3,000 rpm for 30 s at a ramp of 1,000 rpm s−1 and then annealed at 100 °C for 5 min. The samples were then transferred into a thermal evaporator, where 23-nm C60, 7-nm BCP and 100-nm Ag layers were thermally deposited in a sequential manner, where C60 and BCP were evaporated by temperature control and the Ag electrode was evaporated by power control. The evaporation was carried out under a vacuum pressure of around 1.0 × 10−6 mbar at an initial deposition rate of 0.1 nm s−1. For stability tests, 20 nm of SnO2 was deposited as a barrier layer by thermal atomic layer deposition. The device area was determined by a shadow mask during metal evaporation. The active areas of the laboratory-test solar cells are 0.0982 cm2 and 1 cm2, and the aperture area for certification is 0.0782 cm2.
Characterization
X-ray diffraction patterns were recorded using a PANalytical X’Pert Pro X-ray powder diffractometer with Cu Kα radiation (λ = 1.5419 Å). Field-emission SEM images were obtained on a SEM SUPRA 40 electron microscope. Fourier transform infrared spectra were recorded on a JASCO 4100 spectrometer. X-ray photoelectron spectroscopy was conducted on a Shimadzu AXIS SUPRA+ instrument. Water contact angles of the perovskite films were measured using a JC000DI optical contact-measuring system. Solution-state nuclear magnetic resonance spectra were acquired on Bruker Topspin v. 2.1 (AV400) and v. 3.0 (AV500). PL was measured on a laboratory-built PL setup, installed with a reflectance probe and a fibre-optic spectrometer from Ocean Insight, using LabVIEW software. AFM was performed on an NX10 system, Park Systems (XE-70). The AFM was kept in a glovebox with a N2 atmosphere. KPFM measurements were performed on Bruker MultiMode with NanoScope 9.1, Gwyddion, following the lift-mode amplitude-modulated KPFM in a N2 atmosphere in the dark using the following settings: f0 = 300 kHz, k = 40 N m−1 and L = 125 mm (Bruker RTESP-300). Kelvin probe was carried out for WF analysis using a Kelvin probe setup with a vibrating gold mesh driven by a piezoelectric 26 crystal (Kelvin probe S and CPD controller from Besocke Delta Phi).
Photoelectron spectroscopy measurements
Photoemission spectra were recorded using a hemispherical electron analyser (SPECS Phoibos 100) in an ultrahigh-vacuum system equipped with a monochromated He I (hν = 21.218 eV) source for ultraviolet photoelectron spectroscopy and a standard Mg Kα (hν = 1,253.6 eV; anode power, 30 W) X-ray source for X-ray photoelectron spectroscopy. The base pressure of the analysis chamber was maintained at 1 × 10−9 mbar. All spectra were recorded at room temperature and under normal emission to the sample. For measuring the secondary electron cut-off, a sample bias of −10 V was used to overcome the analyser WF. A Solux MR16 4700K white halogen lamp (intensity equivalent to 150 mW cm−2) was used to examine any light-induced energy-level changes at the interfaces.
Photovoltaic performance measurements
The J–V data were recorded under 1-sun-equivalent illumination using a wavelabs SINUS-70 LED class AAA solar simulator in a N2-filled glovebox. The light intensity was calibrated with a filtered KG3 silicon reference cell from the Fraunhofer-Institut für Solare Energiesysteme ISE. J–V scans were performed with a Keithley 2400 source measure unit, controlled by a measurement program written in LabVIEW. The stabilized power outputs under the maximum power points were tracked under illumination, and the PSCs were biased at the voltage at the maximum power point. Thermal cycling tests were conducted with a 10-min dwell and a ramp rate of 100 °C h−1. The EQE spectra were measured for PSCs, where fluorine-doped tin oxide was used in the devices. The EQE was obtained on an Oriel Instruments QEPVSI-b system integrated with a Newport 300-W xenon arc lamp, which was controlled using TracQ-Basic software. The system was equipped with a chopper and monochromatic light of Newport Cornerstone 260 for filtering the white light. The EQE setup was calibrated through a standard silicon photodetector before measurements.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available in the article and its Supplementary Information. Data are also available from the corresponding authors upon reasonable request.
Change history
22 January 2026
A Correction to this paper has been published: https://doi.org/10.1038/s41566-026-01852-z
References
Kim, M. et al. Conformal quantum dot–SnO2 layers as electron transporters for efficient perovskite solar cells. Science 375, 302–306 (2022).
Zhao, Y. et al. Inactive (PbI2)2RbCl stabilizes perovskite films for efficient solar cells. Science 377, 531–534 (2022).
Jeong, J. et al. Pseudo-halide anion engineering for α-FAPbI3 perovskite solar cells. Nature 592, 381–385 (2021).
Xiong, S. et al. Direct observation on p- to n-type transformation of perovskite surface region during defect passivation driving high photovoltaic efficiency. Joule 5, 467–480 (2021).
Best research-cell efficiency chart. NREL https://www.nrel.gov/pv/cell-efficiency.html (2024).
Chen, H. et al. Improved charge extraction in inverted perovskite solar cells with dual-site-binding ligands. Science 384, 189–193 (2024).
Li, G. et al. Structure and performance evolution of perovskite solar cells under extreme temperatures. Adv. Energy Mater. 12, 2202887 (2022).
Wang, Y. et al. Dopant-free small-molecule hole-transporting material for inverted perovskite solar cells with efficiency exceeding 21%. Adv. Mater. 31, 1902781 (2019).
Wu, S. et al. Modulation of defects and interfaces through alkylammonium interlayer for efficient inverted perovskite solar cells. Joule 4, 1248–1262 (2020).
Li, G. et al. Highly efficient p-i-n perovskite solar cells that endure temperature variations. Science 379, 399–403 (2023).
Jiang, Q. et al. Surface reaction for efficient and stable inverted perovskite solar cells. Nature 611, 278–283 (2022).
Jiang, Q. & Zhu, K. Rapid advances enabling high-performance inverted perovskite solar cells. Nat. Rev. Mater. 9, 399–419 (2024).
Azmi, R. et al. Damp heat–stable perovskite solar cells with tailored-dimensionality 2D/3D heterojunctions. Science 376, 73–77 (2022).
Li, X. et al. Constructing heterojunctions by surface sulfidation for efficient inverted perovskite solar cells. Science 375, 434–437 (2022).
Stolterfoht, M. et al. Visualization and suppression of interfacial recombination for high-efficiency large-area pin perovskite solar cells. Nat. Energy 3, 847–854 (2018).
Jiang, Q. et al. Compositional texture engineering for highly stable wide-bandgap perovskite solar cells. Science 378, 1295–1300 (2022).
Azmi, R. et al. Double-side 2D/3D heterojunctions for inverted perovskite solar cells. Nature 628, 93–98 (2024).
Xing, Z. et al. Bowl-assisted ball assembly for solvent-processing the C60 electron transport layer of high-performance inverted perovskite solar cell. Angew. Chem. Int. Ed. 62, e202305357 (2023).
Zhao, L. et al. Chemical polishing of perovskite surface enhances photovoltaic performances. J. Am. Chem. Soc. 144, 1700–1708 (2022).
Li, G. et al. Managing excess lead iodide with functionalized oxo-graphene nanosheets for stable perovskite solar cells. Angew. Chem. Int. Ed. 62, e202307395 (2023).
Sadegh, F. et al. Facile NaF treatment achieves 20% efficient ETL-free perovskite solar cells. ACS Appl. Mater. Interfaces 14, 38631–38641 (2022).
Chen, Q., Wang, C., Li, Y. & Chen, L. Interfacial dipole in organic and perovskite solar cells. J. Am. Chem. Soc. 142, 18281–18292 (2020).
Canil, L. et al. Tuning halide perovskite energy levels. Energy Environ. Sci. 14, 1429–1438 (2021).
Canil, L. et al. Halogen-bonded hole-transport material suppresses charge recombination and enhances stability of perovskite solar cells. Adv. Energy Mater. 11, 2101553 (2021).
Liang, Z. et al. Homogenizing out-of-plane cation composition in perovskite solar cells. Nature 624, 557–563 (2023).
Tan, Q. et al. Inverted perovskite solar cells using dimethylacridine-based dopants. Nature 620, 545–551 (2023).
Qin, M. et al. Manipulating the mixed-perovskite crystallization pathway unveiled by in situ GIWAXS. Adv. Mater. 31, 1901284 (2019).
Lin, Y.-H. et al. Bandgap-universal passivation enables stable perovskite solar cells with low photovoltage loss. Science 384, 767–775 (2024).
Li, G. et al. Ionic liquid stabilizing high-efficiency tin halide perovskite solar cells. Adv. Energy Mater. 11, 2101539 (2021).
Yuan, S. et al. Simultaneous cesium and acetate coalloying improves efficiency and stability of FA0.85MA0.15PbI3 perovskite solar cell with an efficiency of 21.95%. Sol. RRL 3, 1900220 (2019).
Prashanthan, K. et al. Internal electric fields control triplet formation in halide perovskite-sensitized photon upconverters. iScience 26, 106365 (2023).
Agresti, A. et al. Titanium-carbide MXenes for work function and interface engineering in perovskite solar cells. Nat. Mater. 18, 1228–1234 (2019).
Lee, K.-M. et al. Thickness effects of thermally evaporated C60 thin films on regular-type CH3NH3PbI3 based solar cells. Sol. Energy Mater. Sol. Cells 164, 13–18 (2017).
Hoque, M. J. et al. Ultra-resilient multi-layer fluorinated diamond like carbon hydrophobic surfaces. Nat. Commun. 14, 4902 (2023).
Levine, I. et al. Charge transfer rates and electron trapping at buried interfaces of perovskite solar cells. Joule 5, 2915–2933 (2021).
Utomo, D. S. et al. Nonfullerene self-assembled monolayers as electron-selective contacts for n-i-p perovskite solar cells. ACS Energy Lett. 9, 1682–1692 (2024).
Dittrich, T. & Fengler, S. Surface Photovoltage Analysis of Photoactive Materials (World Scientific, 2019).
Jiang, Y. et al. Reducing energy disorder in perovskite solar cells by chelation. J. Am. Chem. Soc. 144, 5400–5410 (2022).
Lu, Y.-N. et al. Constructing an n/n+ homojunction in a monolithic perovskite film for boosting charge collection in inverted perovskite photovoltaics. Energy Environ. Sci. 14, 4048–4058 (2021).
Saliba, M. & Etgar, L. Current density mismatch in perovskite solar cells. ACS Energy Lett. 5, 2886–2888 (2020).
Saliba, M., Unger, E., Etgar, L., Luo, J. & Jacobsson, T. J. A systematic discrepancy between the short circuit current and the integrated quantum efficiency in perovskite solar cells. Nat. Commun. 14, 5445 (2023).
Calado, P. et al. Driftfusion: an open source code for simulating ordered semiconductor devices with mixed ionic-electronic conducting materials in one dimension. J. Comput. Electron. 21, 960–991 (2022).
Hart, L. J. et al. More is different: mobile ions improve the design tolerances of perovskite solar cells. Energy Environ. Sci. 17, 7107–7118 (2024).
Khenkin, M. V. et al. Consensus statement for stability assessment and reporting for perovskite photovoltaics based on ISOS procedures. Nat. Energy 5, 35–49 (2020).
Eames, C. et al. Ionic transport in hybrid lead iodide perovskite solar cells. Nat. Commun. 6, 7497 (2015).
Zhou, J. et al. Modulation of perovskite degradation with multiple-barrier for light-heat stable perovskite solar cells. Nat. Commun. 14, 6120 (2023).
Acknowledgements
This work was supported by Southeast University, China. We acknowledge support from the technicians at the Helmholtz-Zentrum Berlin (HZB). We thank HZB for the allocation of synchrotron radiation beamtime at Myspot beamline at BESSY II. We also acknowledge E. Unger for her support on the GIWAXS measurement at BESSY II; the Harbin Supercomputer Center for the support of computational resources; the Theoretical and Computational Chemistry Team from Shiyanjia’s laboratory for providing invaluable assistance; L. Canil for the Kelvin probe measurement; A. Dallmann for the nuclear magnetic resonance measurement; the facility of the HZB SPV lab; T. Dittrich and I. Levine for their assistance; K.-L. Wang from Soochow University; L.-L. Deng and S.-Y. Xie from Xiamen University; and the École Polytechnique Fédérale de Lausanne (EPFL) for support. T.W.G. acknowledges HyPerCells—a joint graduate school of the University of Potsdam and the HZB. A.M. acknowledges financial support from the German Science Foundation (DFG) SPP 2196 and Horizon Europe Framework Programme, HORIZON-MSCA-2021-PF-01 (HyPerGreen), agreement number 101061809. B.A.-T. acknowledges support from the TWAS-DFG fellowship, African Research Universities Alliance Early Career Research Fellowship (ARUA-ECRF) and BANGA-Africa programme. K.P. acknowledges the Deutscher Akademischer Austauschdienst (DAAD) for funding via the Research Grants—Doctoral Programmes in Germany, 2018/19 (57381412). This work was supported by the Joint Fund of Provincial Science and Technology Research and Development plan of Henan Province (number 232301420004), and the Outstanding Youth Fund of the Natural Science Foundation of Henan Province (number 242300421069). This work has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement number 804519). L.J.F.H., P.R.F.B. and J.N. acknowledge funding from the EPSRC ATIP project (EP/TO28513/1).
Author information
Authors and Affiliations
Contributions
Conceptualization: G.L., M.L. and A.A. Methodology: G.L., L. Wu, Z.L., M.L. and A.A. Investigation: G.L., M.L. and A.A. Film fabrication and characterizations: G.L., Z.Z., B.A.-T. and L. Wu. Device fabrication: G.L., Z.Z., L. Wang and M.L. ToF-SIMS measurement: Z.Z. and L. Wang. KPFM measurement: T.W.G. SPV measurement: A.M. Ultraviolet photoelectron spectroscopy measurement: K.P., L.F. and N.K. AFM measurement: G.L., Z.Z., K.P. T.W.G., L.F. and N.K. PL measurements: G.L. SEM measurement: G.L. and Z.Z. GIWAXS measurement: J.L. Drift-diffusion modelling: L.J.F.H. and P.R.F.B. Discussions and suggestions: J.N. Supervision: G.L., L. Wang, Z.L., M.L. and A.A. Writing—original draft: G.L. Writing—review and editing: G.L., Z.Z., B.A.-T., L. Wu, T.W.G., K.P., A.M., J.L., R.Z., L.J.F.H., L. Wang, Z.L., B.H., M.S., P.R.F.B., J.N., P.J.D., M.K.N., M.L. and A.A.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Photonics thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Notes 1–4, Figs. 1–49, Tables 1–3 and References.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, G., Zhang, Z., Agyei-Tuffour, B. et al. Stabilizing high-efficiency perovskite solar cells via strategic interfacial contact engineering. Nat. Photon. 20, 55–62 (2026). https://doi.org/10.1038/s41566-025-01791-1
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41566-025-01791-1
This article is cited by
-
Highlights of mainstream solar cell efficiencies in 2025
ENGINEERING Energy (2026)
