
Abstract
Protein–protein interactions are at the core of all key biological processes. However, the complexity of the structural features that determine protein–protein interactions makes their design challenging. Here we present BindCraft, an open-source and automated pipeline for de novo protein binder design with experimental success rates of 10–100%. BindCraft leverages the weights of AlphaFold2 (ref. 1) to generate binders with nanomolar affinity without the need for high-throughput screening or experimental optimization, even in the absence of known binding sites. We successfully designed binders against a diverse set of challenging targets, including cell-surface receptors, common allergens, de novo designed proteins and multi-domain nucleases, such as CRISPR–Cas9. We showcase the functional and therapeutic potential of designed binders by reducing IgE binding to birch allergen in patient-derived samples, modulating Cas9 gene editing activity and reducing the cytotoxicity of a foodborne bacterial enterotoxin. Last, we use cell-surface-receptor-specific binders to redirect adeno-associated virus capsids for targeted gene delivery. This work represents a significant advancement towards a ‘one design-one binder’ approach in computational design, with immense potential in therapeutics, diagnostics and biotechnology.
Similar content being viewed by others


Main
Proteins rarely perform their biological functions in isolation but rather rely on protein–protein interactions (PPIs) to execute complex biological processes. Designing protein binders that can specifically target and regulate PPIs therefore holds immense therapeutic and biotechnological potential. However, traditional methods for generating protein binders, such as immunization, antibody library screening or directed evolution, are often laborious, time-consuming and provide limited control over the target site.
Computational protein design offers a powerful alternative, enabling the tailoring of binders to specific targets and binding sites. Physics-based methods such as Rosetta allowed early binder design by means of scaffolding and side-chain optimization2,3. However, such methods suffer from low experimental success rates (less than 0.1%) and require the sampling of many designs2,4. Moreover, they typically require the docking of predefined scaffolds onto a fixed target structure, leading to surface incompatibilities and suboptimal binding, or even precluding the targeting of certain sites.
Recent breakthroughs in deep learning have revolutionized the field of biomolecular modelling. Models such as AlphaFold2 (AF2)1 have demonstrated remarkable capabilities in accurately predicting protein structures and complex PPIs. Indeed, AF2 filtering has been shown to increase the success rates of binder design by evaluating the plausibility of predicted complexes4,5. Deep learning has also been successfully applied for de novo design of proteins and binders. The current state-of-the-art methods involve the use of RFdiffusion5 for backbone generation coupled with ProteinMPNN sequence generation6. When applied to binder design, this approach shows significantly improved success rates compared with previous methods5. However, RFdiffusion relies on sequence design over side-chain-free backbones placed at a rigid target interface, with binder selection ultimately depending on AF2-based complex prediction to identify plausible interactions. This highlights a gap between backbone generation and functional interface design that AF2 filtering helps to bridge.
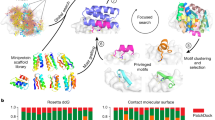
Given the use of AF2 in improving binder filtering success, we proposed that we could harness it directly for the design of protein binders. We present BindCraft, a user-friendly pipeline for de novo design of protein binders that requires minimal user intervention and computational expertise. BindCraft leverages backpropagation through the AF2 network to efficiently hallucinate new binders and interfaces (Fig. 1a). We demonstrate the efficiency of our pipeline on 12 diverse, challenging and therapeutically relevant protein targets (Fig. 1b). We identify several high-affinity binders for each target without the need for high-throughput screening of hundreds to thousands of designs experimentally. This marks an important advancement in the design of protein binders on demand, and makes binder design accessible to research groups without expertise in computational design methods or access to high-throughput screening facilities.

a, Schematic representation of the BindCraft binder design pipeline. Given a target protein structure, a binder backbone and sequence is generated using AF2 multimer, then the surface and core of the binder are optimized using MPNNsol while keeping the interface intact. Finally, designs are filtered based on AF2 monomer model prediction. b, Overview of protein targets for binder design. Parts of the model coloured in green were used during design, grey areas were excluded. Values in the blue box indicate the number of successful designs, where binding was observed on SPR measurement versus the total number of designs tested. Values in the yellow box indicate the measured Kd of the highest affinity binder without experimental sequence optimization, whereas values in orange boxes indicate estimated Kd* values due to poor fit. PD-1 binders were tested as a bivalent Fc fusion.
Accurate design of de novo binders
Our goal was to create an accessible, efficient and automated pipeline leveraging AF2 for accurate binder design with minimal experimental screening. We use the ColabDesign implementation of AF2 to backpropagate hallucinated binder sequences through AF2 weights and calculate an error gradient. This error gradient is used to update and optimize the binder sequence to fit specific design criteria (Methods), as in previous hallucination approaches7,8,9,10. By iterating over the network, we can enable the generation of binder structure, sequence and interface concurrently (Fig. 1a). In contrast to methods such as RFdiffusion5 or RIFdock2,4, which keep the target backbone fixed during design, BindCraft repredicts the binder–target complex at each design iteration. This allows for defined levels of flexibility on the side chain and backbone for both binder and target, resulting in backbones and interfaces that are moulded to the target binding site. The resulting target backbone root mean square deviation (r.m.s.d.Cα) ranges from 0.5 Å to 5.5 Å (Extended Data Fig. 1a). Flexibility can further be increased by masking the sequence of the input template and providing only Cα coordinates (Supplementary Fig. 1a). To mitigate the generation of purely alpha-helical binders, we apply a ‘negative helicity loss’, which enables the design of fully beta-sheeted binders (Extended Data Fig. 1b), albeit with a decrease in in silico success rates (Supplementary Fig. 1b,c).
We use AF2 multimer11 for designing initial binders, as this version of AF2 was trained on protein complexes and would probably be able to more accurately model PPIs compared with AF2 monomer1. We note that AF2 multimer hallucinates on average 20% larger interfaces, with a larger proportion of loops and higher confidence than the AF2 monomer model (Supplementary Fig. 1d). We use all five trained model weights of AF2 multimer to avoid overfitting of sequences to a single model. However, we and others12,13 have previously demonstrated that AF2-hallucinated proteins can show low levels of expression when tested experimentally. We therefore subsequently optimize the sequence of the binder core and surface using a message-passing neural network (MPNNsol)6,12 while keeping the interface intact (Fig. 1a). The optimized binder sequences are repredicted using the AF2 monomer model1. This model was exclusively trained on monomeric proteins, which minimizes prediction bias of PPIs and enables robust filtering for high quality interfaces. Last, as deep learning models have been shown to sporadically produce physically improbable results1,11, we filter the predicted designs based on AF2 confidence metrics, as well as Rosetta physics-based scoring metrics (Methods).
Each target shows varying levels of in silico design success, with 16.8–62.7% of initial AF2 trajectories showing satisfactory confidence metrics and 0.6–65.9% of MPNNsol-optimized designs passing the final computational filters after AF2 monomer complex reprediction (Supplementary Fig. 1e). In silico design success rates are dependent on the target protein and the length of binders being generated (Supplementary Fig. 1f). When compared to the state-of-the-art binder design approach RFdiffusion5, BindCraft yields similar success rates in terms of generation time across several targets and binder lengths (Extended Data Fig. 1c). Notably, we observe a difference in amino acid distribution at the binder interface, with an underrepresentation of bulky amino acids in RFdiffusion-generated designs (Extended Data Fig. 1d).
We benchmarked BindCraft on 12 targets to assess its generalizability (Fig. 1b). The designs show broad sequence and structural diversity (Supplementary Fig. 2a and Supplementary Data 1 and 2), with an average template modelling (TM) score of 0.62 and 14.4% sequence identity to closest Protein Data Bank (PDB) hits (Extended Data Fig. 1e). This suggests that designed proteins recover elements of known structural motifs while also sampling novel folds, consistent with the limited structural diversity expected for compact scaffolds. The high geometric and chemical complementarity of BindCraft binders (Supplementary Data 1) enables the design of new high-affinity interfaces (Extended Data Fig. 1e and Supplementary Fig. 2b). We assessed the novelty of the designed interfaces by comparison to known interactions in the PDB using PPIRef14,15. The designed interfaces showed an average TM score of 0.15 across all targets, indicating that they are distinct from naturally occurring PPIs.
All the described steps are automated into a single workflow, with settings optimized to ensure the design procedure is generalizable across different targets. This allows research groups without protein design expertise to generate binders on demand for any application. By minimizing human intervention needed to generate and sort high quality binder designs, BindCraft democratizes protein binder design and makes it accessible to a broader scientific community.
Binders targeting cell-surface receptors
To test the performance of our pipeline, we designed binders against therapeutically relevant cell-surface receptors and tested for binding activity in vitro. We first targeted the human PD-1 protein, a key immune checkpoint receptor expressed on the surface of T cells16. We purified and screened 53 designs for binding using biolayer interferometry (BLI) in a bivalent Fc-fusion format. We observed a binding signal for 13 binders, with the best binder showing an apparent dissociation constant (Kd*) lower than 1 nM (Fig. 2a,b), although the exact Kd could not be determined due to the extremely slow dissociation rate and avidity effect from the Fc-fusion construct. To confirm the binding site, we performed a competition assay with the well-characterized anti-PD-1 monoclonal antibody, pembrolizumab, which should engage the same binding site. Indeed, our binder could not outcompete the antibody binding (Kd = 27 pM), indicating it is targeting overlapping binding sites (Extended Data Fig. 2a).

a, Design model of binder2 in complex with PD-1. b, Representative BLI sensorgram showing binding kinetics of binder2 (bivalent Fc fusion) to PD-1. c, Design model of binder4 in complex with PD-L1. d, Binding affinity determination by SPR for the PD-L1–binder4 interaction. e, Design model of binder5 in complex with IFNAR2. f, Binding affinity determination by SPR for the IFNAR2–binder5 interaction. g, Design model of binder1 in complex with CD45. h, SPR binding affinity fit for binder1. i, Schematic of CpE-based cytotoxicity and CLDN1 binder inhibition. j, Single cycle kinetic analysis with SPR of CLDN1 binder12 binding to soluble analogues of CLDN1. k, Cell-based assay showing concentration-dependent inhibition of CpE cytotoxicity by CLDN1 binder9, binder12 and CpE inhibitor. Bar plots represent the mean of n = 2 replicates, with standard deviation indicated by error bars. l, MST measurements showing blocking of CpE binding to CLDN1 wild type when preincubated with binder12. MST data were plotted from a single representative measurement. Panel i was created using BioRender (https://biorender.com).
Encouraged by these results, we opted to test fewer designs for all subsequent targets to minimize experimental screening. We next designed binders against PD-L1 (ref. 16) and the interferon 2 receptor (IFNAR2)17, both important modulators of immune signalling. We tested nine designs against PD-L1 out of which seven showed a binding signal (Supplementary Fig. 3a), whereas for IFNAR2 we could detect binding for three out of nine designs (Fig. 1b). The top performing binder4 against PD-L1 showed a Kd* of 615 nM (Fig. 2b,c) as determined by surface plasmon resonance (SPR) and an expected alpha-helical signature as measured by circular dichroism (Supplementary Fig. 3b). Size exclusion chromatography with multi-angle light scattering (SEC–MALS) analysis shows binder4 to be dimeric in solution, but to engage in a 1:1 binding mode with its target (Extended Data Fig. 2b). We then probed the binding of our PD-L1 binder4 using a previously characterized de novo designed binder3. We could confirm they compete for the intended target binding site (Extended Data Fig. 2c), while engaging in a distinct mode of binding compared with PD-1 (Extended Data Fig. 2d).
The top performing binder5 against IFNAR2 showed an affinity of 260 nM by SPR (Fig. 2e,f), a typical alpha-helical signature and high stability (Supplementary Fig. 3c), and monomeric nature in solution (Extended Data Fig. 2e). We tested binder5 against IFNAR2’s native binding partner, the cytokine interferon alpha 2 (IFNA2)17. We observe competition for the IFNA2 binding site, validating our designed binding mode (Extended Data Fig. 2f), while primarily occupying distinct binding sites (Extended Data Fig. 2g). To assess the specificity of our designs, we probed the binding of each top performing binder against other immunoglobulin-like fold receptors. Despite their structural similarity (Supplementary Data 2), we observe no off-target binding (Extended Data Fig. 2h). We observe the AF2 i_pTM metric to effectively discriminate the on-target interactions from off-targets (Extended Data Fig. 2h). These results demonstrate that we are able to efficiently design binders, straight from the computational design pipeline, against known binding sites, without the need for extensive screening to identify hits with nanomolar affinity.
Next, we sought to determine whether our pipeline could design binders against extracellular receptors lacking well-characterized binding sites. We selected CD45 as a target because of the structural complexity of its extracellular domain, comprising four immunoglobulin-like domains d1–d4 with heavy N-glycosylation in the smallest isoform18. We tested 16 binders experimentally, out of which 4 showed binding on SPR (Fig. 1b). The best performing binder1 showed a Kd of 14.7 nM and targeted the junction region of domains d3 and d4 (Fig. 2g,h). We also observed the expected alpha-helical signal in circular dichroism, validating the correct folding of our design (Supplementary Fig. 3d). These results indicate that BindCraft can also effectively design binders against new or previously uncharacterized binding sites.
Targeting unexplored binding sites
Membrane proteins lacking distinct extracellular domains are of critical biological and therapeutic importance, yet pose a significant challenge for binder design due to difficulties in experimental validation and screening. Computationally designed soluble analogues that retain natural epitopes12 offer a promising solution by enabling rapid prescreening of potential binders. To validate this strategy, we targeted claudins, which are critical for maintaining epithelial and endothelial tight junction barrier integrity19. Claudins are naturally targeted by Clostridium perfringens enterotoxin (CpE), which forms a membrane-penetrating pore that leads to cell death20. We proposed that binders competing with CpE for its binding site could mitigate cytotoxicity (Fig. 2i).
Using a soluble analogue of claudin 1 (sCLDN1)12, we designed binders against the extracellular domain and prescreened them for binding using two variants of the soluble analogue (Supplementary Fig. 4a,b). We tested seven binders and found all except binder17 to bind to sCLDN1-14 and sCLDN1-18 (Supplementary Fig. 4a), which both harbour the native CLDN1 extracellular epitope (Supplementary Fig. 4b). We observed the strongest binding signal for binder12, which showed nanomolar affinity for the soluble analogues (Fig. 2j). To assess the binder’s utility, they were tested against wild-type claudin 1 (CLDN1 WT) using a cell-based cytotoxicity assay. Here, binder9 and binder12 effectively inhibited CpE-based cytotoxicity, protecting CLDN1 WT-expressing cells from cell death in a concentration-dependent manner and on the order of a known CpE inhibitor (Fig. 2k and Supplementary Fig. 4c). Notably, both of these binders result from the same initial trajectory and carry the same interface residues.
To validate that the inhibition of cytotoxicity was the result of direct interactions with CLDN1 WT, we used microscale thermophoresis (MST). We found that both CpE and binder12 interacted with CLDN1 WT, and that preincubation of binder12 with CLDN1 WT blocked CpE binding, indicating competition for the same binding site (Fig. 2l and Extended Data Fig. 2i). The binders failed to protect claudin 4 (CLDN4)-expressing cells from CpE-induced toxicity (Supplementary Fig. 4d,e), most probably due to CpE’s roughly 400-fold higher affinity for CLDN4 (ref. 20). Our findings demonstrate that soluble analogues can enable the discovery of binders that effectively modulate membrane protein function.
To assess the generalizability of our pipeline for targeting proteins lacking known binding sites, we designed binders against a protein with no natural sequence homologues. We chose the de novo designed beta-barrel fold 14 (BBF-14)12, as beta-barrels are not commonly regarded as PPI partners. We purified the 11 top-scoring designs from which 6 showed binding (Fig. 1b). The best binder, binder4 (Extended Data Fig. 3a), is composed of a mixed alpha-beta topology, with the interface formed by both the split beta-sheets and a helix motif. The beta-sheet interface is not mediated by backbone hydrogen bonding, but rather by side-chain interactions. Binder4showed a Kd of 20.9 nM for BBF-14, as determined by SPR (Extended Data Fig. 3b). To assess the fidelity of our design procedure, we solved a structure of BBF-14 bound to binder4 (Extended Data Fig. 3c,d, Extended Data Table 1). When aligned on the BBF-14 target, binder4 has a backbone r.m.s.d.Cα of 1.7 Å, confirming both the accuracy of the fold and the designed binding mode (Extended Data Fig. 3c). This result underscores our ability to generate binders purely based on structural information, without relying on existing binding sites or any influence from co-evolutionary data.
Last, we selected the conserved structural protein SAS-6 as a design target. SAS-6 oligomers are essential for centriole biogenesis in eukaryotes21. Using BindCraft, we generated several designs and experimentally tested nine top-scoring binders. Binder4 binds with 5.7 μM affinity to the monomeric form of CrSAS-6 (Extended Data Fig. 3e,f) and 4.2 μM affinity to the dimeric form (Extended Data Fig. 3g), indicating compatibility with its oligomeric form. It targets an overlapping site with the previously reported monobody MBCRS6-15 (Extended Data Fig. 3h), which shifts SAS-6 assembly from a ring to a helical structure22. We speculate that BindCraft enables on-demand binder design to probe biological function, even within higher-order assemblies.
Masking allergenic epitopes
Allergic rhinitis and seasonal allergies affect up to 50% of the population in some countries23. Current treatments primarily target global inflammation with immunosuppressants and monoclonal antibodies. However, neutralizing allergic reactions could potentially offer a more effective strategy for managing allergies. Allergens comprise a diverse group of proteins with different folds, biological functions and highly charged surfaces24. Generally, hydrophobic binding sites are considered more tractable for computational binder design2, making allergens more challenging targets.
To test BindCraft’s ability to target allergens, we designed binders against dust mite allergens Der f7 and Der f21, and the major birch allergen Bet v1, which is responsible for up to 95% of birch-related allergies25. We examined 10 designs against Der f7 experimentally and identified 4 binders (Fig. 1b), with binder2 showing the highest binding affinity with a Kd of 12.8 nM (Fig. 3a). To confirm the binding mode of binder2, we solved crystal structures in complex with Der f7 obtaining two crystal forms with resolutions of 2.2 Å and 3.0 Å (Extended Data Fig. 4a,b and Extended Data Table 1). Aligned on the allergen, binder2 shows a backbone r.m.s.d.Cα of 1.7 Å (Fig. 3b), validating the design’s structural accuracy. Binder2 is monomeric in solution (Extended Data Fig. 4c) and binds the same epitope as mouse monoclonal antibodies raised against Der f7 (ref. 26).

a, Left: design model of binder2 against dust mite allergen Der f7. Right: SPR binding affinity fit for binder2. b, Crystal structure (coloured) of the Der f7–binder2 complex overlaid with the design model (grey). c, Left: design model of binder10 against dust mite allergen Der f21. Right: SPR binding affinity fit for binder10. d, Crystal structure (coloured) of the Der f21–binder10 complex overlaid with the design model (grey). e, Left: design model of binder2 against birch allergen Bet v1. Right: SPR binding affinity fit for binder2. f, SEC–MALS analysis of Bet v1 allergen (blue, expected molecular weight (MW) 18.5 kDa) and Bet v1 mixed with binder2 (orange, expected molecular weight 29.3 kDa). g, Cryo-EM structure (PDB 7MXL) of Bet v1 bound to commercial anti-Bet v1 REGN antibody mix. h, Competition assay on immobilized REGN5713-Bet v1 complex binding of the REGN5714 antibody but not Bet v1 binder2, confirming binding at the designed site. i, Blocking ELISA showing the capacity of the REGN antibody mix (orange) or binder2 (blue) to prevent the binding of Bet v1 to IgE from the sera from three patients allergic to birch. Number suffix represents individual serum from a patient. Data points represent average of two technical replicates with the error bars depicting standard deviation.
Similarly, we evaluated seven binders against Der f21 and could detect binding for four designs by SPR (Fig. 1b). The best performing binder10 showed an apparent affinity of 793 nM (Fig. 3c). Although dimeric in solution (Extended Data Fig. 4d), a 2.6 Å resolution crystal structure validates a 1:1 mode of binding of binder10 against a highly charged helical site of Der f21 (Extended Data Fig. 4e). The binder10 shows a backbone r.m.s.d.Cα of 3.1 Å, caused by an alternative rotamer conformation of an interface tyrosine (Fig. 3d). Mutational analyses of Der f21 indicate that our binders target epitopes distinct from those recognized by IgE in the sera of allergic individuals27.
Last, we identified two successful designs from seven tested binders against the birch allergen Bet v1 (Fig. 1b). Binder2 showed a 120 nM binding affinity by SPR (Fig. 3e), dimerizes in solution (Extended Data Fig. 4f), but in complex with Bet v1 shows a mass of 27.8 kDa, indicative of a 1:1 binding mode (Fig. 3f). The binder2 has a warped helical topology, where its C-terminal helix inserts itself into the ligand binding pocket of Bet v1 (ref. 28). To assess the specificity of allergen-targeting binders, we incubated the top binders with each of the three allergens. Even at 10 μM binder concentration, we observe no off-target binding to other allergens (Extended Data Fig. 4g), indicating high specificity of the designed anti-allergens.
Previously, a cocktail mix of three antibodies binding to three immunogenic epitopes of Bet v1 was developed to prevent allergic response29. Its cryogenic electron microscopy (cryo-EM) structure indicates that our binder targets a known epitope recognized by the REGN5713 antibody (Fig. 3g). To validate, we immobilized REGN5713 on SPR and loaded the Bet v1 allergen on it. We observe a binding signal with REGN5714 as the analyte, but not with binder2, confirming that it targets an overlapping epitope with REGN5713 (Fig. 3h). We further proposed that our binders can compete with Bet v1 specific IgE present in serum samples from patients who are to allergic birch, similar to the REGN antibody mix29. To test the neutralization activity of our anti-Bet v1 binder2, we performed a blocking enzyme-linked immunosorbent assay (ELISA) using the serum of three patients allergic to birch with high titre of anti-Bet v1 IgE. Biotinylated Bet v1 was preincubated with either the REGN antibody cocktail or our designed binder2 (Fig. 3i). The REGN antibody mix blocked up to 90% of Bet v1 binding to IgE, whereas our single binder blocked up to 50% in two out of three donors. This is on par with blocking rates of single antibodies29, indicating that there is therapeutic potential for de novo designed binders in neutralizing allergic responses.
Modulating multi-domain nucleases
Nucleic acid interaction interfaces are widely considered undruggable due to their large, charged and convex surfaces, which are difficult to target with small molecules30. To test our pipeline on such interfaces, we focused on the multi-domain CRISPR–Cas9 nuclease from Streptococcus pyogenes (SpCas9). Although widely used for gene editing due to its programmability31, SpCas9 originally functions as a bacterial immune system protecting against phages32. In response, phages evolved small anti-CRISPR proteins (Acrs) that block CRISPR–Cas by occluding nucleic acid binding sites33. We wondered whether artificial Acrs could be designed to mimic this function.
We designed binders against the bipartite REC1 domain of SpCas9, containing a highly charged guide RNA-binding pocket34 (Fig. 4a). All six tested binders bound the full-length apo SpCas9 enzyme (Supplementary Fig. 5a). The top performing binder3 and 10 showed apparent binding affinities in the range of 300 nM by SPR, although complete titration curves were challenging to obtain. To validate their binding mode, we attempted to solve cryo-EM structures of binder3 and binder10 bound to the full-length SpCas9 apo enzyme. Despite the high quality of the data and clearly observable density for the binders (Extended Data Fig. 5a), we were unable to obtain a satisfactory cryo-EM density to build an atomic model due to poor resolution in the target area (Extended Data Fig. 5b). This observation could be because of the dynamic nature of the apo form of Cas9 (ref. 35). Nevertheless, we observe clear density at the REC1 site and can confidently dock both binders, validating the designed binding mode (Fig. 4b,c and Extended Data Fig. 5c,d).

a, Zoom in on the SpCas9 REC1 domain with bound guide RNA (PDB 4ZT0). A designed binder is overlaid in the binding pocket. b, Cryo-EM structure of binder3 bound to the apo form of SpCas9. The REC1 domain is highlighted in green, the rest of SpCas9 is in grey. Cryo-EM density overlaid in grey. c, Cryo-EM structure of binder10 bound to the apo form of SpCas9. The REC1 domain is highlighted in green, the rest of SpCas9 is coloured in grey. Cryo-EM density overlaid in grey. d, SpCas9-based editing of HEK293T cells in the absence (grey bar, dashed line) or presence of designed binders (green bars) or natural Acrs (blue bars). e, Structural architecture of Clostidium butyricum Argonaute with bound gDNA and tDNA (PDB 6QZK). The PAZ domain and N + PIWI domains used as design targets are highlighted in light and dark blue. f, CbAgo-gDNA-mediated cleavage of target DNA in the absence (grey bar, dashed line) or presence of designed binders (green bars) or designed SpCas9 binders (blue bars). Bar plots represent the mean of n = 3 replicates, with standard deviation indicated by error bars. g, CbAgo-gDNA-mediated cleavage of target DNA in absence of binders (grey line) or in presence of designed binder2 (pink line) or binder3 (purple line). Plotted points represent an average of three measurements with standard deviation indicated by error bars.
To evaluate their function, we cotransfected human embryonic kidney 293T (HEK293T) cells with CRISPR–SpCas9 and either designed binders or natural Acrs36,37,38. We observe a significant reduction of gene editing activity in the presence of our designed binders (Fig. 4d). They outperform AcrIIC2, which inhibits guide RNA loading using a different targeting mechanism37. AcrIIA2 and AcrIIA4, which inhibit target DNA (tDNA) binding (Supplementary Fig. 5b), nearly eliminate gene editing activity, underscoring the differences in inhibitory strategies. These results demonstrate that BindCraft can generate previously unseen inhibitors of nucleic acid-interacting proteins by means of previously unseen mechanisms.
To expand our binder design to other large nucleases, we designed binders against the multi-domain Argonaute (Ago) nuclease from Clostridium butyricum (CbAgo). Akin to Cas9, CbAgo acts as an immune system that uses small oligonucleotide guides to target and cleave invading DNA39,40. So far, no natural inhibitors of Argonaute nucleases have been described. We designed binders targeting either the N-PIWI channel or the PAZ domain of CbAgo (Fig. 4e). We tested the effect of 12 binders on CbAgo-mediated tDNA cleavage and two binders strongly inhibit CbAgo activity (Fig. 4f). Whereas 0.4 μM CbAgo alone has a kcat of 0.004 s−1, in presence of 2 μM binder2 and binder3 the kcat is reduced 80-fold to 5 × 10−5 s−1 and 40-fold to 9.8 × 10−5 s−1, respectively (Fig. 4g). We found that binder2 binds to CbAgo with a Kd of 5 nM, as determined by BLI (Extended Data Fig. 5e). SEC analysis of binder2 with CbAgo validates that it forms a stable complex with CbAgo (Extended Data Fig. 5f). Adding the guide DNA (gDNA) destabilizes the CbAgo–binder2 complex, which confirms that binder2 occupies the gDNA binding channel (Extended Data Fig. 5g,h).
These results demonstrate that we can design protein binders even against challenging nucleic acid binding sites and grooves, potentially opening paths towards new types of protein-based therapeutic, gene editing modulator and molecular biology tool for basic research.
AAV retargeting for gene delivery
Viral vectors, such as those derived from adeno-associated viruses (AAVs), have expanded gene therapy possibilities by leveraging the natural ability of viruses to introduce genetic material into cells and tissues. However, AAVs have poor specificity to cell types, tissues and organs. Achieving specific targeting often requires high doses, raising the risk of off-target effects and immunogenicity. Several efforts have been made to modify the tropism of AAV capsids, by insertion of peptide segments41 or receptor-binding moieties, such as DARPins42. However, such approaches involve library screening or immunization campaigns, usually with limited control over the target site. We proposed that BindCraft could efficiently design miniprotein binders capable of retargeting AAVs to cell-type specific receptors (Fig. 5a). Its high design success rate could enable direct AAV transduction testing in cellulo, bypassing biochemical prescreening, and providing a platform for the rapid development of retargeted AAV vectors to cells and tissues of interest.

a, Schematic representation illustrating AAV-cmv-GFP retargeting on genetic insertion of a cell-type receptor-specific miniprotein binder, replacing the natural primary attachment to cell-surface glycans. b, Chimeric assembly of a retargeted AAV particle, composed of the capsid proteins with (pink) and without (green) inserted binder in a defined stoichiometric ratio. c, Transduction efficiency measured by flow cytometry of different AAV variants targeting HER2 or PD-L1, determined after transfer of packaging cell supernatant onto HEK293 cells stably overexpressing the respective target receptors. The signal-to-noise ratio, defined as target/non-target ratio between the transduction rates measured on each cell line, is indicated as ‘×’ fold change. For comparison, each of the two cell lines is similarly transduced with the wild-type AAV6-cmv-GFP (WT) and the AAV capsid variant carrying knockout (KO) mutations. Transduction efficiencies were measured in triplicates (n = 3) and error bars indicate a 95% confidence interval. d, Design model of binder1 against HER2. e, Design model of binder202 against PD-L1. f, Heatmap of the transduction rates at a normalized multiplicity of infection (MOI) of 1 × 105 vg per cell of the AAV variants carrying the binder1 against HER2 and binder202 against PD-L1, as well as the KO and WT controls, on HEK293 cells stably overexpressing the respective target receptors. g, Transduction with the PD-L1-targeting AAV carrying the binder202. The lower histogram shows that an anti-PD-L1 antibody, which targets the binding site of AAV-binder202, blocks the transduction of HEK293 cells stably overexpressing PD-L1. Panel a was created using BioRender (https://biorender.com).
Traditionally, retargeting molecules are either inserted into the variable regions VR-IV or VR-VIII protruding near the threefold symmetry axis of the AAV capsid, or fused to the N terminus of the viral capsid protein 2 (VP2). Based on a large mutational study on AAV capsid fitness43, we explored an alternative insertion site, located between residues 497 and 498 of the VR-V near the threefold symmetry axis of the AAV capsid (Fig. 5b). We chose AAV6-cmv-GFP as a starting vector and introduced point mutations to deplete its natural primary interactions with heparin and sialic acid (knockout, Fig. 5a). We then designed binders against HER2 and PD-L1 with an extra N-termini and C-termini distance loss to facilitate a direct capsid integration, using a short –(GSG)1– extension on each terminus (Fig. 5b).
To simultaneously screen the designed AAVs for production and transduction efficiency, a small-scale assay was designed that relies on directly transferring the supernatant of AAV-packaging cells onto the targeted cells (Extended Data Fig. 6a,b). This assay led us to identify one reprogrammed AAV to target HER2 and four targeting PD-L1 that showed enhanced specificity for HEK293 cells stably overexpressing their respective target receptor (Fig. 5c and Extended Data Fig. 6c). Characterization of most efficient variants, HER2–b1 and PD-L1–b202 (Fig. 5d,e), showed that both AAVs had enhanced specificity towards cells expressing their target receptor (Fig. 5f). When the interaction was challenged with an antibody targeting the same receptor-binding site, the transduction of PD-L1-expressing cells by the PD-L1-targeting AAV was blocked, suggesting that the designed binder mediates the transduction through the engagement with the target receptor (Fig. 5g).
Conclusions
The computational design of de novo PPIs has been a cornerstone problem in protein design, primarily due to our lack of understanding of the determinants of molecular recognition. The development of deep learning-based structure prediction networks, such as AF2 (refs. 1,11), has revolutionized the field and enabled more accurate filtering of de novo designs with favourable biochemical profiles. Here we introduce a robust pipeline based on backpropagation through the AF2 network, an approach that has been explored in previous studies7,8,9,10,44, and extend its capabilities to the hallucination of protein binders. Unlike most existing methods, BindCraft enables target protein flexibility, which is critical for capturing binding-induced structural changes essential for molecular recognition.
We demonstrate BindCraft’s performance across a diverse set of 12 challenging targets. The binder affinities lie predominantly in the nanomolar range, with success rates from 10% to 100%, with an average success rate of 46.3%, which is remarkable for a purely computational approach. These rates allow for the screening of far fewer designs experimentally to identify functioning binders, when compared with the current state-of the-art RFdiffusion5 and the recently described closed-source AlphaProteo binder design pipeline45. Notably, a binder designed with our pipeline recently ranked first in a community-wide binder design competition, showing 82 nM affinity against the challenging epidermal growth factor receptor (EGFR) target46.
The structural accuracy of our method not only allows us to create proteins that bind to defined surfaces but also enables their functionalization. We demonstrate this by using our designed binders to reduce the binding of birch allergen Bet v1 to specific IgE from patient-derived serum samples. Whereas a single binder showed moderate blocking activity, broader epitope coverage could enhance efficacy. De novo binders therefore offer a promising alternative to antibodies for such treatments, because of their high stability. However, owing to the synthetic nature of our binders and their relatively large size (60–240 amino acids), concerns about immunogenicity and effective delivery persist, although these issues are gradually being addressed in preclinical models47. Furthermore, BindCraft’s high experimental success rates allow direct screening of intended biological function, as exemplified by the retargeting of AAV towards specific cell-surface receptors, enabling precise and customizable transduction profiles. This promises to simplify the development of targeted viral vectors, offering a versatile platform for gene therapy applications, including therapeutic delivery to disease-relevant cells and tissues while minimizing the risk of potential off-target effects.
Despite the successes outlined here, there are limitations to the BindCraft design approach. Backpropagation through AF2 is GPU-intensive, and final design filtering with AF2 monomer in single sequence mode may exclude prospective high-affinity binders2,3,4,5,48 (Extended Data Fig. 7a,b). We assessed the possibility of using the recently released AlphaFold3 (ref. 49) model for filtering, but still found a large proportion of false positive predictions (Extended Data Fig. 7c). Furthermore, AF2 is known to be insensitive to point mutations50, which could be detrimental at PPI interfaces, but can be mitigated by orthogonal physics-based scoring methods, such as Rosetta51. Last, a potential limitation is the use of the AF2 i_pTM metric for the ranking of designs, which has emerged as a powerful binary predictor of binding activity (Extended Data Fig. 7a,b), but does not correlate with the interaction affinity46 (Supplementary Fig. 6). Nevertheless, BindCraft represents a significant leap in the accurate design of binders for direct functional applications. We foresee that through iterative refinement of our pipeline, we will eventually reach a ‘one design, one binder’ stage, omitting the need for screening. This will enable rapid generation of binders for applications in research, biotechnology and therapeutics for a wide range of research groups without protein design expertise.
Methods
BindCraft design protocol
The input and design settings for running the BindCraft pipeline are organized into user-friendly JSON files. To initiate design trajectories, a target PDB format structure needs to be specified, along with the desired minimum and maximum length of the binders, and the desired number of final filtered designs. A target hotspot can be specified as either individual residues or entire chains, or can be omitted completely in which case a binding site is selected according to the combined design loss.
The binder hallucination process is performed using the ColabDesign implementation of AF2. The design process is initialized with a random sequence for the binder, which is predicted in single sequence mode, and a structural input template for the target. This is passed through the AF2 network to obtain a structure prediction and calculate the design loss. The design loss function is composed of several terms, with default weight values indicated in parentheses:
-
(1)
binder confidence pLDDT (weight 0.1)
-
(2)
interface confidence i_pTM (weight 0.05)
-
(3)
normalized predicted alignment error (pAE) within the binder (weight 0.4)
-
(4)
normalized predicted alignment error (pAE) between binder and target (weight 0.1)
-
(5)
residue contact loss within binder (weight 1.0)
-
(6)
residue contact loss between the target and binder: if hotspots are specified, the rest of the target is masked from this loss (weight 1.0)
-
(7)
radius of gyration of binder (weight 0.3)
-
(8)
‘helicity loss’: penalize or promote backbone contacts every one in a three-residue offset to promote the hallucination of helical or non-helical designs (weight −0.3)
-
(9)
optional ‘N&C termini loss’ increases the proximity of the N and C termini of the binder to allow splicing into protein loops (weight 0.1).
The loss function is used to calculate position specific errors, which are then backpropagated through the AF2 network to produce a L × 20 error gradient, where L is the sequence length. Using multiple iterations and stochastic gradient descent optimization, this error gradient is recomputed and used to optimize the input binder sequence for the next iteration to minimize the resulting loss. We backpropagate through the AF2 multimer model weights11 and swap randomly between the five trained models at each iteration to ensure robust sequence generation and reduce the risk of overfitting to a single model.
As our goal is to arrive at a real discrete sequence for the binding interface, the sequence optimization is performed in four stages. The first sequence optimization stage is performed in a continuous sequence space using logit inputs. At each step, the sequence representation is based on linear combination of (1 − λ) × logits + λ × softmax(logits/T), where λ = (step + 1)/iterations and temperature (T) of 1.0. Here, many amino acids are considered per each binder position, which allows the exploration of a larger and less constrained sequence-structure space. After 50 iterations, we terminate trajectories showing poor AF2 confidence scores, as we found that such trajectories rarely converge to high confidence designs. Furthermore, if a beta-sheeted trajectory is detected, we increase the number of recycles during design from one to three to ensure accurate prediction. The continuous sequence space optimization is then continued for a further 25 iterations. During the second optimization stage, the sequence logits are normalized to sequence probabilities using the softmax function for 45 iterations to funnel the design space towards a more realistic sequence representation defined as softmax(logits/T) At each step, the temperature is lowered, where temperature is equal to (1 × 10−2 + (1 − 1 × 10−2) × (1 − (step + 1)/iterations)2). The temperature is also used to scale the learning rate for rate decay. For the third stage, we implement the straight-through estimator, allowing the model to see the one-hot representation, but backpropagate through the softmax representation. This procedure is performed for five iterations. For the final fourth stage, the sequence inputs are converted to a one-hot discrete encoding. At each step, X random mutations are independently sampled and tested from the probability distribution of the softmax representation from the previous stage, and mutations with best loss are fixed. X is defined on the basis of the length of the binder sequence (0.05× binder length). This procedure is performed for 15 iterations. At the end, trajectories with pLDDT below 0.7, fewer than 7 interface contacts or significant backbone clashes are rejected.
Successful binder design trajectories are subjected to MPNNsol sequence optimization to improve stability and solubility12. To this end, we preserve binder residues in a 4 Å radius around the target interface, and design 20 new sequences for the remaining binder core and surface residues using the soluble weights of ProteinMPNN6, with a temperature of 0.1 and 0.0 backbone noise. These optimized sequences are then repredicted using the AF2 monomer model, with three recycles and two template-based models49 in single sequence mode, to ensure robust and unbiased complex assessment. Each of the two resulting models is then energy minimized using Rosetta’s FastRelax protocol52 with 200 iterations, and interface scores are computed using the InterfaceAnalyzer mover53 with side-chain and backbone movement enabled.
Designs are finally filtered using a set of predefined filters to ensure the selection of high quality designs for experimental testing. Filters were initially defined based on experimental observations from previous binder design studies2,3,4,5 and refined over the course of this work. These include:
-
(1)
AF2 confidence pLDDT score of the predicted complex (>0.8)
-
(2)
AF2 interface predicted confidence score (i_pTM) (>0.5)
-
(3)
AF2 interface predicted alignment error (i_pAE) (<0.35)
-
(4)
Rosetta interface shape complementarity (>0.60)
-
(5)
number of hydrogen bonds at the interface (>3)
-
(6)
number of unsaturated hydrogen bonds at the interface (<4)
-
(7)
hydrophobicity of binder surface (<35%)
-
(8)
r.m.s.d. of binder predicted in bound and unbound form (<3.5 Å)
-
(9)
fewer than three lysines and methionines at the binder interface.
We allow only two MPNNsol generated sequences per individual AF2 trajectory to pass filters to promote interface diversity amongst selected binders. This design procedure is set up to loop until a defined number of final desired designs is reached. For optimal results, we recommend running the design pipeline until at least 100 designs pass computational filters. This generally requires the sampling of about 300–3,000 trajectories. We then usually pick 10 designs from the top 20 (ranked by i_pTM) for experimental testing.
To generate designs against targets described in the section ‘Accurate design of de novo binders’, we used the input structures, binder specifications and hotspot designations described in Supplementary Table 1. For AF2 predictions, we used full-length input sequences from UniProt. In all cases, the amino acid cysteine was excluded from sequence design. For AAV targets, the N-termini and C-termini loss is activated with default weight.
Computational benchmarks of BindCraft
To evaluate the flexibility of the target structure post-design, the input PDB structure of the target was aligned to the target chain A of the design trajectory, and r.m.s.d.Cα was calculated using PyRosetta. For increasing target flexibility, the sequence of the input target template was masked by enabling the flag ‘rm_target_seq’ in ColabDesign for trajectory hallucination54, and 200 trajectories were generated.
For the impact of the helicity loss on binder secondary structure composition, the ‘weights_helicity’ flag in BindCraft was set to 1, 0, −0.3, −1, −2 and −3, and 200 trajectories were generated for each instance using otherwise default settings.
To compare the design capabilities of AF2 monomer and multimer weights, we generated 200 trajectories each. For AF2 multimer trajectories, we used the default settings in which AF2 multimer models 1–5 are used for design and AF2 monomer models 1–2 trained with templates are used for reprediction. For AF2 monomer this is inverted, we use AF2 monomer models 1 and 2 for design and AF2 multimer models 1–5 for reprediction.
For benchmarks involving design and trajectory success rates, we run the design pipeline either for 200 trajectories or until 100 designs passing in silico filters are accumulated (where indicated). We then designate trajectories with pLDDT above 0.7 as ‘passing’, whereas trajectories that have a pLDDT below 0.7, more than 1 Cα backbone clash between chains or fewer than 3 contacts between the binder and target are designated ‘low confidence’.
RFdiffusion benchmarks were performed as described in the original publication5, with the exception of running the pipeline in deterministic mode for tracking purposes. Briefly, backbones of designated lengths were sampled using RFdiffusion against selected targets and sequences were designed using original ProteinMPNN weights with a temperature of 0.0001 and 8 sequences per backbone. Each complex was predicted using AF2 monomer model 1 and two MPNN designed sequences for each backbone were allowed to pass filters as defined in the original publication (pLDDT > 0.8, i_pAE < 0.32, binder r.m.s.d. < 1.0 Å). The pipeline was run until 100 designs passed filters. The computational time was calculated as backbone generation time + ProteinMPNN sequence generation + AF2 complex prediction for each design. Notably, although single model prediction was used in the case of RFdiffusion, we used prediction using two template-based AF2 models in the case of BindCraft.
Pairwise structural similarities and sequence identities across targets and binders in Supplementary Data 2 were extracted using Foldseek55 exhaustive search and TMalign alignment type.
To determine fold and interface novelty of designed binder complexes, we searched the binder chain against the PDB using Foldseek in TMalign mode. Hits with the highest template modelling score (qtmscore) and their sequence identities (fident) for each binder were plotted. Owing to the low resolution structural representations in Foldseek, an alternative strategy was used to assess interface novelty. Residues were extracted using PPIRef in a 6 Å radius around the designed interface, then searched against the precomputed PDB interaction pairs using the iDist method, with a default threshold of 0.04 (ref. 14). The closest interface hit is then aligned using USalign to calculate the template modelling score and sequence identity15.
Benchmarking of designs from other design pipelines was performed using the BindCraft prediction method of either AF2 monomer or multimer in single sequence, with templates provided for the target according to the specifications in their respective publications.
AlphaFold3 predictions of designed BindCraft complexes were performed using the AlphaFold3 server49 with multiple-sequence alignments and templates enabled.
Pairwise Pearson correlation coefficients (r) among experimental binding (yes, 1, no, 0), Affinity (nanomolar, length and all AF2 and Rosetta-derived features were computed and visualized as a heatmap to assess linear relationships and correlation across all pairs of values. Coefficient values outlined in the cells are considered significant at |r| ≥ 0.7.
Protein expression, purification and characterization
DNA sequences of designed proteins, as well as BBF-14, Der f7, Der f21 and Bet v1 targets were ordered from Twist Biosciences with Gibson cloning adaptors for cloning into bacterial expression vectors pET21b or pET11. Proteins were expressed in Escherichia coli BL21 Codon Plus (DE3) cells (Novagen) by inducing with 0.5 mM isopropyl-β-d-thiogalactoside for 6 h at 18 °C. Pellets were resuspended and lysed in lysis buffer (50 mM Tris-HCl pH 7.5, 500 mM NaCl, 5% glycerol, 1 mg ml−1 lysozyme, 1 mg ml−1 phenylmethylsulfonyl fluoride and 1 µg ml−1 DNase) using sonication. Cell lysates were clarified using ultracentrifugation, loaded on a 1 ml Ni-NTA Superflow column (Qiagen) and washed with 7 column volumes of 50 mM Tris-HCl pH 7.5, 500 mM NaCl and 10 mM imidazole. Proteins were eluted with 10 column volumes of 50 mM Tris-HCl pH 7.5, 500 mM NaCl, 500 mM imidazole. Claudin binders were dialysed against 20 mM HEPES pH 8.0, 150 mM NaCl, 4% glycerol and directly frozen.
The Fc-fused PD-L1 target3, IFNAR2 target, IFNA2 cytokine and antibodies were expressed using a mammalian Expi293 secreted expression system (Thermo Fisher Scientific, A14635). Six days posttransfection, the supernatants were collected, cleared and purified either using a 1 ml Ni-NTA Superflow column (Qiagen) or protein A affinity column (Qiagen). SAS-6 (ref. 22), SpCas9 (ref. 56), CbAgo and the catalytic mutant of CbAgo (D541A, D611A)40 have been purified as described previously.
Remaining bacterial and mammalian expressed proteins were then concentrated and injected onto a Superdex 75 16/600 or Superdex 75 10/300 gel filtration column (GE Healthcare) in 50 mM Tris-HCl pH 7.5, 250 mM KCl or PBS. Proteins after size exclusion were concentrated, frozen in liquid nitrogen and stored at −80 °C. Molar mass, sample homogeneity and multimeric state were confirmed using SEC–MALS (miniDAWN TREOS, Wyatt) by injecting 100 µg of protein in PBS (Column, Superdex 75 10/300 or Superdex 200 10/300, GE Healthcare). Folding, secondary structure content and melting temperatures were assessed using circular dichroism in a Chirascan V100 instrument from Applied Photophysics in PBS at a concentration of 0.1–0.3 mg ml−1.
Expression and purification of PD-1 target and binders
DNA sequences were synthesized in the pcDNA3.4 vector with an osteonectin secretion signal at the N terminus (Twist Biosciences). De novo designs were fused to the N terminus of human IgG1 Fc. The extracellular domain (25–167) of human PD-1 (UniProtKB Q15116) was fused to a C-terminal AviTag and His tag. Plasmid DNA was prepared from glycerol stocks (Twist Biosciences) using Cowin Biosciences GoldVac EndoFree plasmid maxi kit. Plasmids were transfected into 3 ml or 50 ml cultures of Expi293F (Gibco) cells as per the manufacturer’s recommendations. Cells incubated at 37 °C for 4–5 days before collection. Following protein expression, the cell culture supernatant was filtered through a 0.22-µM filter and purified using MabSelect protein A affinity chromatography resin (Cytiva). The column was washed with PBS and the protein was eluted in Tris glycine buffer pH 2.5. Following elution, proteins were dialysed into PBS using a 10-kDa molecular weight cut-off dialysis cassette. For production of biotinylated PD-1 protein, the PD-1 plasmid was cotransfected with BirA plasmid (2:1 ratio). The BirA plasmid contains the BirA sequence (UniProtKB P06709) with a C-terminal Flag tag in the pcDNA3.4 vector.
Binding characterization of PD-1
Designs were initially screened for binding to biotinylated human PD-1 or a random protein using BLI (Sartorius OctetRED384). Biotinylated human PD-1 protein and biotinylated lysozyme (GeneTex) were prepared at 500 nM in PBS containing 0.1% bovine serum albumin (BSA) (PBSA). The designs were diluted to 5 µM in PBSA. Streptavidin-labelled biosensors were saturated with either biotinylated human PD-1 or biotinylated chicken lysozyme. The designs were then allowed to associate with the immobilized ligand for 60 s, followed by a dissociation step in PBSA. The baseline subtracted signal (nanometres) was calculated and used to prioritize human PD-1 specific binders for further characterization.
To determine the affinity of selected designs, 100 nM biotinylated human PD-1 prepared in PBSA was immobilized onto a streptavidin-labelled biosensor for 15 s. Serial dilutions of the designs (from 2.5 µM to 5 nM) were then allowed to associate with the immobilized ligand for 180 s, followed by a dissociation step in PBSA for 300 s. Following background subtraction of the BLI binding curves using the buffer only (PBSA) curve, the Kd was determined using the 1:1 model in the Data Analysis HT v.11.1 curve fitting module.
To determine whether the designed protein competed with pembrolizumab for binding to PD-1, 100 nM biotinylated human PD-1 in PBSA was immobilized onto streptavidin coated biosensors for 15 s. An initial association with 200 nM pembrolizumab prepared in PBSA was performed for 180 s, followed by a second association with 200 nM design prepared in PBSA for 180 s.
SPR binding and competition assays
SPR measurements were performed using the Biacore 8 K system (Cytiva) in HBS-EP + buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% (v/v) Surfactant P20 GE Healthcare). Target proteins were immobilized on a CM5 chip (GE Healthcare) through amide coupling in 10 mM NaOAc pH 4.5 for 130–250 s at a flow rate of 10 µl min−1 aiming for 100 relative response units. Designed binders or control proteins were injected as analytes in either a single 10 µM concentration during binder prescreening or in serial dilutions to assess binding kinetics. These were injected at a flow rate of 30 µl min−1 for a varying contact time, followed by dissociation. If necessary, the chip surface was regenerated after each injection using 10 mM Glycine-HCl pH 2.5 for 30 s at a flow rate of 30 µl min−1. Binding curves were fitted with a 1:1 Langmuir binding model in the Biacore 8K analysis software. Steady-state response units were plotted against analyte concentration and a sigmoid function was fitted to the experimental data in Python v.3.9 to derive the Kd.
Competition assays were performed as follows. For PD-L1 and IFNAR2, target receptors were immobilized, and binders and competitors were injected as analytes. Two subsequent injections were performed with only competitor (A,1 µM), only design (B,1 µM) or first competitor (1 µM, A) and then design + competitor (both 1 µM, A + B). For Bet v1, REGN5713 (Antibody format) was immobilized on the SPR chip and in a first injection (1) loaded with Bet v1 allergen (1 µM), before either REGN5714 (Fab format) or Birch–binder2 were injected (both 1 µM) (2).
Cell-surface specificity measurements
For specificity measurements, PD-1–b4 was expressed and purified as a His-tagged protein. PD-1-Fc was produced with mutations at glycosylation sites (N → D) and free cysteine residues (C → S). All other proteins were purified as previously described.
BLI experiments were performed using a Gator BLI system and GatorOne software (Gator Bio, v.2.7.3.0728). Assays were conducted in a running buffer containing 10 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA and 0.005% (v/v) Surfactant P20 (GE Healthcare).
For immobilization, Fc-tagged target proteins (PD-L1, PD-1 and IFNAR2) were diluted to 5 µg ml−1 and captured onto protein A biosensor tips (Gator Bio). After immobilization, the biosensor tips were dipped into 1 µM solutions of purified binder.
Protein crystallization and structure determination
The BBF-14–binder4 complex was crystallized at a concentration of 5 mg ml−1 using sitting drop vapour diffusion at 16 °C in 0.1 M MES pH 6.0, 0.2 M sodium acetate trihydrate, 20% w/v polyethylene glycol (PEG) 8000 buffer (SG1-Eco Screen, Molecular Dimensions). The Der f7–binder2 complex in P21 crystal form was crystallized at a concentration of 15 mg ml−1 using sitting drop vapour diffusion at 16 °C in 0.1 M MES pH 6.5, 0.2 M KSCN, 25% w/v PEG 2000 MME buffer (Clear Strategy Screen I, Molecular Dimensions). The Der f7–binder2 complex in C121 crystal form was crystallized at a concentration of 15 mg ml−1 using sitting drop vapour diffusion at 16 °C in 0.1 M MES pH 6.5 and 20% v/v PEG smear high BCS (BCS Screen, Molecular Dimensions). The Der f21–binder10 complex was crystallized at a concentration of 30 mg ml−1 using sitting drop vapour diffusion at 16 °C in 0.1 M sodium citrate pH 5.6, 1.0 M LiSO4, 0.5 M NH4SO4 buffer (SG1-Eco Screen, Molecular Dimensions). Crystals were cryoprotected in 25% glycerol and flash-cooled in liquid nitrogen. Diffraction data were collected at the European Synchrotron Radiation Facility MASSIF-3 and ID30B beamlines, Grenoble, France at a temperature of 100 K. Crystallographic data were processed using the autoPROC package57. Phases were obtained by molecular replacement using Phaser58. Atomic model refinement was completed using COOT59 and Phenix.refine58. The quality of refined models was assessed using MolProbity60. Structural figures were generated using ChimeraX61.
Cryo-EM structure determination
SpCas9 was mixed with a threefold excess of either binder3 or binder10, and the complex was purified using S200 10/300 gel filtration column (GE Healthcare) in 20 mM Tris-HC pH 7.5, 250 mM KCl. The purified complex was applied to a glow discharged 300-mesh holey carbon grid 300-mesh holey carbon grid (Au 1.2/1.3 QuantifoilMicro Tools), blotted for 4 s at 95% humidity, 10 °C, plunge frozen in liquid ethane (Vitrobot Mark IV, FEI) and stored in liquid nitrogen. Data collection was performed on a 300 kV Titan Krios G4 microscope equipped with a FEI Falcon IV detector and SelectrisX energy filter. Micrographs were recorded at a magnification of ×165,000, pixel size of 0.726 Å and a nominal defocus ranging from −0.8 mm to −2.2 mm.
Acquired cryo-EM data were processed using cryoSPARC v.4.6.0 (ref. 62). Micrographs were patch motion corrected, and micrographs with a resolution estimation worse than 5 Å were discarded after patch contrast transfer function estimation. Initial particles were picked using blob picker at 90–135 Å. Particles were extracted with a box size of 360 × 360 pixels, downsampled to 220 × 220 pixels. After two-dimensional classification, clean particles were used for ab initio three-dimensional (3D) reconstruction and initial non-uniform 3D reconstruction63. This model was used for extra template-based picking of particles. Following several rounds of 3D classification, in which classes containing unbound Cas9 were excluded, the class with the most detailed binder features was re-extracted using full box size and subjected to non-uniform and local refinement to generate final reconstructions. The local resolution was calculated and visualized using ChimeraX61. The in silico models were docked into density using ChimeraX61.
Birch allergen blocking assay
Anti-Bet v1 binder blocking capacity was assessed by first coating NuncSorp (Thermo Fisher) plates with 2 μg ml−1 of anti-human IgE monoclonal antibody (NBS-C BioScience; clone Le27; 0908-1-010) in coating buffer (15 mM Na2CO3, 34.87 mM NaHCO3) and incubating overnight at 4 °C. The plates were washed with PBS + 0.05% Tween and blocked using PBS + 1% BSA for 2 h at room temperature. Then, sera from patients allergic to birch were added at a concentration of 4 ng ml−1 of anti-Bet v1 IgE. Biotinylated Bet v1 allergen at 1 nM concentration was preincubated for 2 h at room temperature with fourfold serial dilutions of the Bet v1–binder2 starting at 2 μM or with fivefold serial dilutions of the cocktail of REGN5713, REGN5714 and REGN5715 (starting at 50 nM each) and then added to the IgE coated plate. After 2 h of incubation at room temperature, the plates were washed with PBS + 0.05% Tween and streptavidin horseradish peroxidase (BD Pharmigen; 554066; 1:1,000 dilution) was added and incubated for 1 h. Plates were washed and tetramethylbenzidine substrate (BD Biosciences; 555214) was added and incubated for another 20 min. The reaction was stopped with 2 M sulfuric acid. Absorbance was measured on a spectrophotometer at 450 nm with a 630-nm reference, and blocking percentage was measured by subtracting the absorbance of the sample in the absence of the binder.
MST
CLDN1 WT was labelled with Cy5 by adding a 1:5 molar excess of dye and incubating for 2 h on ice. The excess dye was removed by passing through a PD-10 column. The labelled protein was collected and stored in small aliquots at −80 °C after flash freezing in liquid nitrogen.
For MST-based interaction studies, the Monolith (Nanotemper) instrument was used. Serial dilutions of the ligand (CLDN1–b12/CpE–Nd33) were made in buffer B (25 mM HEPES pH 8.0, 200 mM NaCl, 5% glycerol 0.03% DDM) and mixed with 10 nM labelled CLDN1 WT. After 10 min of incubation, samples were transferred to capillaries (Monolith standard capillary) and readings were initiated. The spectral shift data were plotted and fitted into a Kd model, and estimated Kds were obtained. When data were not fitted using the Kd model, the Hill model was used to fit data. For studying the competitive binding of CpE–Nd33 and CLDN1–b12 to the target CLDN1 WT, a second set of experiments was performed. CLDN1 WT was incubated with CLDN1–b12 (2 × Kd) and subsequently challenged with CpE–Nd33.
Cytotoxicity assay
To study whether claudin binders were able to inhibit pore formation in Sf9 cells expressing claudins, adherent Sf9 cells in a 24-well plate were infected with baculovirus containing either CLDN1 or CLDN4. The assay was performed as shown previously20. Briefly, for each claudin, a 12-well experiment was performed. Six wells were used to test the effect of binders on the pore-forming capacity of CpE–Nd33 and the other six wells were used as controls. After 36 h of infection, 4 µM of each binder were added into six different wells and the plate was then gently mixed by swirling and incubated for 5 min. After that, 300 nM of CpE–Nd33 was added to each of the six wells. The following controls were used in experiment 1. Sf9 without baculovirus infection, 2. Sf9 infected with claudin but not treated with CpE–Nd33, 3. Sf9 infected with Claudin and treated with CpE–Nd33 4. Sf9 infected with Claudin and treated with COP4 Fab (referred to as CpE inhibitor) 5. Sf9 infected with Claudin and treated by COP4 followed by addition of CpE–Nd33 after incubation for 5 min. The number of cells dead or alive were then measured after 18 h of incubation by staining the cells with trypan blue and measuring the number of cells using an automated cell counter (Invitrogen Countess).
SpCas9 gene editing
For SpCas9-single-guide RNA (sgRNA) plasmid cloning, lentiCRISPR v2 (Addgene no. 52961, a gift from F. Zhang) was digested with BsmBI (NEB). Oligonucleotides encoding for the sgRNA targeting the NSD2 gene were annealed and ligated into the digested lentiCRISPR v2 plasmid. All binders were human-codon optimized using the GenSmart Codon Optimization tool and ordered as inserts with homology overhangs for cloning from Twist bioscience. Final binder plasmids were generated by isothermal assembly (NEBuilder HiFi DNA Assembly Cloning Kit, NEB).
HEK293T (ATCC CRL-3216) cells were maintained in DMEM plus GlutaMax (Thermo Fisher Scientific), supplemented with 10% (vol/vol) fetal bovine serum (Sigma-Aldrich) and 1 × penicillin-streptomycin (Thermo Fisher Scientific) at 37 °C and 5% CO2. Cells were maintained at confluency below 90% and passaged every 2–3 days. For testing inhibitor efficiency, HEK293T cells were seeded in 48-well cell culture plates (Greiner) and transfected at 70% confluency using 300 ng Cas9 + sgRNA plasmid, 500 ng of inhibitor plasmid and 5 µl of Lipofectamine 2000 according to the manufacturer’s instructions (Thermo Fisher Scientific). The next day, cells were split and selected with Puromycin, Blasticidin or both. Three days posttransfection, cells were gathered and genomic DNA was isolated by direct lysis.
The DNA from the cell lysate was prepped for next-generation sequencing as previously described64. In the first PCR round, genomic regions of interest were amplified using GoTaq Green Master Mix (Promega) and primers that included Illumina forward and reverse adaptor sequences. A second PCR round, also using GoTaq Green Master Mix (Promega), introduced p5–p7 barcodes into the products from the first round. The resulting amplified amplicons were pooled and quantified using a Qubit 3.0 fluorometer (Invitrogen). The libraries were then sequenced using a MiSeq platform (Illumina, 150 bp, paired-end). Sequencing data and resulting gene editing insertion-deletion rates were analysed using CRISPResso2 (ref. 65).
CbAgo in vitro cleavage assay
For in vitro cleavage assays, binders, CbAgo, 5′-phosphorylated 16-nt single-stranded DNA (ssDNA) guide (oDS423) and Cy5-labelled 45-nt ssDNA target (oDS401) were mixed to final concentrations of 2:0.4:0.4:0.2 μM in 10 mM HEPES pH 7.5, 125 mM KCl and 2 mM MgCl2. To this end, first the binder protein and CbAgo were mixed and incubated at 37 °C for 15 min, after which the mixture was incubated on ice and guide ssDNA and target ssDNA were added. Subsequently, reaction mixtures were incubated at 37 °C, and samples were taken at 0-min, 4-min, 10-min, 30-min and 60-min timepoints. Samples taken at each timepoint were directly quenched by adding 2× RNA loading dye (25 mM EDTA, 5% v/v glycerol, 90% v/v formamide) and heating for 5 min at 95 °C. Cleavage products were resolved using denaturing (7 M urea) 20% polyacrylamide gel electrophoresis, and gels were imaged using a Amersham Typhoon gel scanner (Cytiva Life Sciences). Cleavage reactions were performed in triplicates for each binder protein. CbAgo target cleavage was quantified using ImageQuant TL 1D v.8.2.0 (Cytiva Life Sciences), and fitted with nonlinear least squares fit (nlsLM from R package minpack.lm) to a double-exponential decay model to model initial (fast) and turnover (slow) cleavage:
If fitting to a double-exponential decay model yielded no fit after 1,024 iterations with residuals and gradient convergence tolerance of 1 × 10−9, the turnover cleavage (slow) was considered negligible and a single-exponential decay model (that is, B = 0) was used.
For all samples Kcat was calculated from the fit constants for the initial rate (A and K1):
The mean and standard deviation of Kcat was calculated using the three experimental replicates.
CbAgo BLI binding kinetics
BLI measurements were conducted using the Gator BLI system and GatorOne software (Gator Bio, v.2.7.3.0728). The running buffer consisted of 150 mM KCl, 20 mM HEPES (pH 7.5) and 0.5% BSA. His-tagged CbAgo binders were immobilized on the sensor tips at a concentration of 10 µg ml−1. After immobilization, the tips were transferred into serial dilutions of CbAgo. Binding curves were globally fitted using a 1:1 interaction model in the Gator software.
CbAgo SEC binding verification
Purified CbAgo was diluted to 0.8 mg ml−1 (9.3 µM) and mixed with 0.2 mg ml−1 binder protein and 9.3 µM 5′-phosphorylated 16-nt ssDNA guide (oDS423) in SEC buffer (20 mM HEPES pH 7.5, 250 mM KCl and 2 mM MgCl2). The mixture was incubated for 15 min at room temperature. After incubation samples were resolved at room temperature on a Superdex 200 Increase 10/300 GL column (Cytiva Life Sciences) connected to a 1260 Infinity II high-performance liquid chromatography system (Agilent) using SEC buffer with a flow rate of 0.75 ml min−1. The elution was measured using a Agilent 1260 Infinity II Multiple Wavelength Detector at 280 nm. The data were analysed using Astra v.8.1 (Wyatt Technology).
AAV engineering
HEK293 cells adapted to culture in orbitally shaken bioreactors (HEKExpress, ExcellGene SA) were maintained in Serum-free BalanCD HEK293 Medium (Irvine Scientific) supplemented with l-alanyl-l-glutamine (Gibco GlutaMax) at 37 °C, 80% humidity, 5% CO2, under constant shaking at 180 rpm (shaking diameter 5 cm). Cells were passaged every 3–4 days to a concentration of 0.2 × 106 cells per ml. For the generation of cell lines stably expressing the target receptors used in the AAV transduction experiments, the receptor complementary DNAs (cDNAs) were obtained from an open reading frame collection (HER2, ORFeome Collaboration cDNA Clone, PD-L1, Addgene no. 121142) and cloned into a pRRLSIN lentiviral shuttle construct (Addgene no. 12252) with expression under the control of the human phosphoglycerate kinase (hPGK) promoter. Lentiviral particles were generated using standard procedures for calcium phosphate transfection of HEK293T cells with the pRRLSIN-hPGK-WPRE, p8.92, pMD2G and pAdVAntage plasmids. At 48 h, the vector-containing supernatant was harvested, filtered and concentrated by ultracentrifugation. The number of lentiviral particles present in the obtained vector suspension was quantified using a p24 antigen ELISA kit (ZeptoMetrix). HEKExpress cells were transduced in a six-well plate at a density of 3.0 × 106 cells per well using a multiplicity of infection (MOI) of 100 vg per cell (conversion factor 1 pg p24 = 1 × 104 vg). After 5 days, the cells were stained for the presence of the respective target receptor using an APC-conjugated antibody (0.8 µg ml−1, BioLegend, 329707 (PD-L1), 324407 (HER2)) in staining buffer (PBS containing 0.5% BSA (Merck)) and sorted by flow cytometry using a Sony SH800 cell sorter. After expansion, the cells were aliquoted and frozen at −80 °C until further use.
The pRepCap plasmids for the AAV production by transient transfection of HEKExpress cells, encoding the rep (AAV2) and cap (varying) genes, were chosen according to the different variants as indicated. For serotype 6 wild-type AAV, an AAV6 plasmid was ordered from the manufacturer (Aldevron, pALD-AAV6). For the variant carrying the knockout mutations to deplete the primary interactions with heparin (K459S and K531E) and sialic acid (V473D, N500E and T502S), a corresponding gene fragment was ordered as an insert with homology overhangs (Twist Biosciences) and cloned into pALD-AAV6 by BspEI/MscI yielding the pRepCap knockout. To introduce the sequence encoding the designed miniprotein binders, an intermediate plasmid was created that, in addition to the five mutations to deplete the primary interactions, carries two silent mutations yielding MluI/NheI restriction sites in proximity to the chosen site of binder insertion between amino acid positions 497 and 498 of the serotype 6 VP3. The DNA sequences for the designed miniprotein binders, flanked by a single –(GSG)1– linker at both termini, were human-codon optimized using the GenSmart Codon Optimization tool and ordered as inserts with homology overhangs (Twist Biosciences) for subcloning.
For the AAV production for screening at small scale, the cells were seeded in 24-well cell culture plates at a density of 0.4 × 106 cells per ml in a volume of 500 µl and transfected with 520 ng of pHelper (Aldevron, pALD-HELP), 250 ng of shuttle plasmid (Aldevron, pALD-ITR-GFP), 270 ng of pRepCap (varying) and 1.5 µg of polyethyleneimine (Polysciences). If applicable, the variants’ pRepCap plasmids were respectively mixed in a ratio of 1:2 with the pRepCap knockout plasmid. Then 12 h after transfection, the cells’ media was exchanged and supplemented with 4 mM valproic acid (Sigma). The cell culture was incubated at the standard conditions described above but without shaking, and the AAV containing medium was harvested on day 5 by collecting the supernatant using centrifugation at 400g for 5 min at room temperature to remove cells.
For the AAV production for validation at a normalized MOI, the cells were seeded at a density of 1.0 × 106 cells per ml in a volume of 300 ml in a TubeSpin 600 bioreactor tube (TPP) and transfected with 231 µg pHelper (Aldevron, pALD-HELP), 105 µg of shuttle plasmid (Aldevron, pALD-ITR-GFP), 105 ng pRepCap (varying) and 900 µg polyethyleneimine (Polysciences). If applicable, the pRepCap plasmids were obtained as described above, and mixed in a ratio of 1:2 with the pRepCap knockout plasmid. Then 6 h after transfection, the cell culture medium was supplemented with 4 mM valproic acid (Sigma). The cell culture was incubated at the standard conditions described above for 7 days, and the vector was harvested on day 3–4 and on day 7 by collecting the supernatant after centrifugation of the bioreactor tube at 800g for 10 min at room temperature. The supernatant was filtered (Stericup Quick Release, Millipore Express PLUS 0.22 μm PES, 1,000 ml, Merck Millipore). The particles were concentrated from the cell culture supernatant to a concentration of at least 3.0 × 1010 vg ml−1 by using Amicon Ultra-15 centrifugal filter units at a molecular weight cut off of 100 kDa (Merck). The wild-type and knockout variant particles were processed alternatively according to Gaudry et al.66. In short, the particles were purified from the cell culture supernatant using the POROS CaptureSelect AAVX resin (Thermo Fisher Scientific) on an ÄKTA Pure chromatography system followed by buffer exchange to PBS, 0.001% Pluronic F-68 (10% stock solution, Gibco) through Amicon Ultra-15 centrifugal filter units, at a molecular weight cut off of 100 kDa (Merck). The number of genome-containing AAV particles was determined after treatment with DNase I (Thermo Fisher) by digital PCR using the QIAcuity system and PCR kit (Qiagen).
For transduction, the target cells were seeded in 96-well cell culture plates at a density of 3 × 105 cells per ml in a volume of 100 µl. After 6 h, the cells’ media was replaced with 100 µl of AAV containing medium from the production in 24-well cell culture plates, or a 100-µl dilution to 3 × 1010 vg ml−1 of the material from the production in 300 ml of culture to yield a MOI of 1 × 105 vg per cell. If applicable, 0.8 µg ml−1 target receptor blocking antibody (BioLegend, 329707) was added. After 48 h, the cells were washed twice with 100 µl of PBS containing 0.5% BSA (Merck), and the transduction signal (GFP) was measured by flow cytometry on an Attune NxT analyser (Thermo Fisher) equipped with an automated plate reader. The results were analysed using FlowJo v.10.8 Software (BD Life Sciences).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are available in the main text or as Supplementary Information. Structural models PDB 2LAG (IFNAR2), 5FMV (CD45), 9HAG (BBF-14), 3UV1 (Der f7), 5YNY (Der f21), 6QZK (CbAgo), 1N8Z (HER2) and 4ZT0 (SpCas9) were used to generate target structures for binder design using AF2. Atomic coordinates and structure factors of the reported X-ray structures have been deposited in the PDB under accession numbers 9HAC (BBF-14–b4), 9HAD (DerF21–b10), 9HAE (DerF7–b2, crystal form P21) and 9HAF (DerF7–b2, crystal form C121). The cryo-EM maps for SpCas9-binder3 and SpCas9-binder10 were deposited in the Electron Microscopy Data Bank under accession codes EMD-53510 and EMD-53511, respectively. Structural models of binders are available at Zenodo (https://doi.org/10.5281/zenodo.14249738)67.
Code availability
The full BindCraft code along with installation instructions and binder design protocols are available on GitHub under MIT license at https://github.com/martinpacesa/BindCraft. A Google Colab notebook for running BindCraft is available through GitHub at https://github.com/martinpacesa/BindCraft/blob/main/notebooks/BindCraft.ipynb.
References
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Cao, L. et al. Design of protein-binding proteins from the target structure alone. Nature 605, 551–560 (2022).
Gainza, P. et al. De novo design of protein interactions with learned surface fingerprints. Nature 617, 176–184 (2023).
Bennett, N. R. et al. Improving de novo protein binder design with deep learning. Nat. Commun. 14, 2625 (2023).
Watson, J. L. et al. De novo design of protein structure and function with RFdiffusion. Nature 620, 1089–1100 (2023).
Dauparas, J. et al. Robust deep learning–based protein sequence design using ProteinMPNN. Science 378, eadd2187 (2022).
Anishchenko, I. et al. De novo protein design by deep network hallucination. Nature 600, 547–552 (2021).
Goverde, C. A., Wolf, B., Khakzad, H., Rosset, S. & Correia, B. E. De novo protein design by inversion of the AlphaFold structure prediction network. Protein Sci. 32, e4653 (2023).
Frank, C. et al. Scalable protein design using optimization in a relaxed sequence space. Science 386, 439–445 (2024).
Jendrusch, M. A. et al. AlphaDesign: a de novo protein design framework based on AlphaFold. Mol. Syst. Biol. https://doi.org/10.1038/s44320-025-00119-z (2025).
Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. Preprint at bioRxiv https://doi.org/10.1101/2021.10.04.463034 (2022).
Goverde, C. A. et al. Computational design of soluble and functional membrane protein analogues. Nature 631, 449–458 (2024).
Wicky, B. I. M. et al. Hallucinating symmetric protein assemblies. Science 0, eadd1964 (2022).
Bushuiev, A. et al. Learning to design protein-protein interactions with enhanced generalization. in Twelfth Intl Conf. Learning Representations (ICLR, Vienna, 2024).
Zhang, C., Shine, M., Pyle, A. M. & Zhang, Y. US-align: universal structure alignments of proteins, nucleic acids, and macromolecular complexes. Nat. Methods 19, 1109–1115 (2022).
Ribas, A. & Wolchok, J. D. Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018).
Walter, M. R. The role of structure in the biology of interferon signaling. Front. Immunol. 11, 606489 (2020).
Hermiston, M. L., Xu, Z. & Weiss, A. CD45: a critical regulator of signaling thresholds in immune cells. Annu. Rev. Immunol. 21, 107–137 (2003).
Günzel, D. & Yu, A. S. L. Claudins and the modulation of tight junction permeability. Physiol. Rev. 93, 525–569 (2013).
Vecchio, A. J., Rathnayake, S. S. & Stroud, R. M. Structural basis for Clostridium perfringens enterotoxin targeting of claudins at tight junctions in mammalian gut. Proc. Natl Acad. Sci. USA 118, e2024651118 (2021).
Gönczy, P. & Hatzopoulos, G. N. Centriole assembly at a glance. J. Cell Sci. 132, jcs228833 (2019).
Hatzopoulos, G. N. et al. Tuning SAS-6 architecture with monobodies impairs distinct steps of centriole assembly. Nat. Commun. 12, 3805 (2021).
Bousquet, J. et al. Allergic rhinitis. Nat. Rev. Dis. Prim. 6, 95 (2020).
Dall’Antonia, F., Pavkov-Keller, T., Zangger, K. & Keller, W. Structure of allergens and structure based epitope predictions. Methods 66, 3–21 (2014).
Ipsen, H. & Løwenstein, H. Isolation and immunochemical characterization of the major allergen of birch pollen (Betula verrucosa). J. Allergy Clin. Immunol. 72, 150–159 (1983).
Tai, H.-Y. et al. The different modes of binding of the dust mite allergens, Der f 7 and Der p 7, on a monoclonal antibody WH9 contribute to the differential reactivity. J. Microbiol. Immunol. Infect. 51, 478–484 (2018).
Pang, S. L. et al. Crystal structure and epitope analysis of house dust mite allergen Der f 21. Sci. Rep. 9, 4933 (2019).
Kofler, S. et al. Crystallographically mapped ligand binding differs in high and low IgE binding isoforms of birch pollen allergen Bet v 1. J. Mol. Biol. 422, 109–123 (2012).
Atanasio, A. et al. Targeting immunodominant Bet v 1 epitopes with monoclonal antibodies prevents the birch allergic response. J. Allergy Clin. Immunol. 149, 200–211 (2022).
Bushweller, J. H. Targeting transcription factors in cancer—from undruggable to reality. Nat. Rev. Cancer 19, 611–624 (2019).
Pacesa, M., Pelea, O. & Jinek, M. Past, present, and future of CRISPR genome editing technologies. Cell 187, 1076–1100 (2023).
Koonin, E. V. & Makarova, K. S. Origins and evolution of CRISPR-Cas systems. Philos. Trans. R. Soc. Lond. B 374, 20180087 (2019).
Wiegand, T., Karambelkar, S., Bondy-Denomy, J. & Wiedenheft, B. Structures and strategies of anti-CRISPR-mediated immune suppression. Annu. Rev. Microbiol. 74, 21–37 (2020).
Jiang, F., Zhou, K., Ma, L., Gressel, S. & Doudna, J. A. A Cas9–guide RNA complex preorganized for target DNA recognition. Science 348, 1477–1481 (2015).
Shibata, M. et al. Real-space and real-time dynamics of CRISPR-Cas9 visualized by high-speed atomic force microscopy. Nat. Commun. 8, 1430 (2017).
Dong, D. et al. Structural basis of CRISPR–SpyCas9 inhibition by an anti-CRISPR protein. Nature 546, 436–439 (2017).
Zhu, Y. et al. Diverse mechanisms of CRISPR-Cas9 inhibition by Type IIC anti-CRISPR proteins. Mol. Cell 74, 296–309 (2019).
Liu, L., Yin, M., Wang, M. & Wang, Y. Phage AcrIIA2 DNA mimicry: structural basis of the CRISPR and Anti-CRISPR arms race. Mol. Cell 73, 611–620 (2019).
Kuzmenko, A. et al. DNA targeting and interference by a bacterial Argonaute nuclease. Nature 587, 632–637 (2020).
Hegge, J. W. et al. DNA-guided DNA cleavage at moderate temperatures by Clostridium butyricum Argonaute. Nucleic Acids Res. 47, 5809–5821 (2019).
Goertsen, D. et al. AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nat. Neurosci. 25, 106–115 (2022).
Münch, R. C. et al. Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol. Ther. 21, 109–118 (2013).
Ogden, P. J., Kelsic, E. D., Sinai, S. & Church, G. M. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science 366, 1139–1143 (2019).
Wang, J. et al. Scaffolding protein functional sites using deep learning. Science 377, 387–394 (2022).
Zambaldi, V. et al. De novo design of high-affinity protein binders with AlphaProteo. Preprint at https://arxiv.org/abs/2409.08022 (2024).
Cotet, T.-S. et al. Crowdsourced protein design: lessons from the adaptyv EGFR binder competition. Preprint at bioRxiv https://doi.org/10.1101/2025.04.17.648362 (2025).
Berger, S. et al. Preclinical proof of principle for orally delivered Th17 antagonist miniproteins. Cell 187, 4305–4317 (2024).
Goudy, O. J., Nallathambi, A., Kinjo, T., Randolph, N. Z. & Kuhlman, B. In silico evolution of autoinhibitory domains for a PD-L1 antagonist using deep learning models. Proc. Natl Acad. Sci. USA 120, e2307371120 (2023).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Pak, M. A. et al. Using AlphaFold to predict the impact of single mutations on protein stability and function. PLoS ONE 18, e0282689 (2023).
Baryshev, A. et al. Massively parallel measurement of protein–protein interactions by sequencing using MP3-seq. Nat. Chem. Biol. 20, 1514–1523 (2024).
Leman, J. K. et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods 17, 665–680 (2020).
Coventry, B. & Baker, D. Protein sequence optimization with a pairwise decomposable penalty for buried unsatisfied hydrogen bonds. PLoS Comput. Biol. 17, e1008061 (2021).
Roney, J. P. & Ovchinnikov, S. State-of-the-art estimation of protein model accuracy using AlphaFold. Phys. Rev. Lett. 129, 238101 (2022).
van Kempen, M. et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 42, 243–246 (2023).
Pacesa, M. et al. R-loop formation and conformational activation mechanisms of Cas9. Nature 609, 191–196 (2022).
Vonrhein, C. et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. Sect. D 67, 293–302 (2011).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. Sect. D 75, 861–877 (2019).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D 66, 486–501 (2010).
Williams, C. J. et al. MolProbity: more and better reference data for improved all‐atom structure validation. Protein Sci. 27, 293–315 (2018).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Marquart, K. F. et al. Predicting base editing outcomes with an attention-based deep learning algorithm trained on high-throughput target library screens. Nat. Commun. 12, 5114 (2021).
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
Gaudry, J.-P., Aebi, A., Valdés, P. & Schneider, B. L. Production and purification of adeno-associated viral vectors (AAVs) using orbitally shaken HEK293 cells. Methods Mol. Biol. 2810, 55–74 (2024).
Pacesa, M., Nickel, L. & Correia, B. Structural models of BindCraft designed binders. Zenodo https://doi.org/10.5281/zenodo.14249738 (2024).
Acknowledgements
We thank SCITAS at EPFL for support in running design trajectories. We thank A. Marchand, P. E. M. Balbi, A. Sadek and S. Mauro for support with protein purification. We thank F. Pojer, K. Lau and A. Larabi (Protein Production and Structure Characterization Core Facility, EPFL, Switzerland) for help with crystallization, biochemical characterization and providing SpCas9 protein. We thank A. Aebi and J. P. Gaudry (Bertarelli Foundation Gene Therapy Platform, EPFL, Switzerland) for their assistance with AAV production and purification. We thank D. Nurizzo and M. Nanao (European Synchrotron Radiation Facility, MASSIF-3 and ID30B beamline, Grenoble, France) for assistance with crystallographic data collection. We thank A. Myasnikov, B. Beckert and S. Nazarov (Dubochet Center for Imaging, EPFL-UNIL-UNIGE, Switzerland) for assistance with cryo-EM data collection. We thank M. Garcia and Francesco Palumbo (Flow Cytometry Core Facility, EPFL, Switzerland) for their help with flow cytometry measurements. We thank R. Fernandes’ group for generously providing purified CD45 extracellular domain protein. Funding: M.P. was supported by the Peter und Traudl Engelhorn Stiftung. B.E.C. and G.N.H. were supported by the Swiss National Science Foundation, the NCCR in Molecular Systems Engineering. L.N., C.S., B.L.S. and B.E.C were supported by the Novartis Foundation for Medical-Biological Research. S.O. and Y.C. were supported by National Institutes of Health (NIH) grant nos. DP5OD026389 and NSF MCB2032259 and by Amgen. Y.D.M. was funded by the Gabriella Giorgi-Cavaglieri Foundation. A.A.-S. was funded by Fondation Machaon. D.C.S. was supported by a grant from the European Research Council (grant no. ERC-2020-STG 948783). G.S. was supported by the Swiss National Science Foundation grant no. 214936. L.K. was funded by the University of Zurich Research Priority Program ITINERARE. C.S. was directly supported by grants from the Foundation Teofilo Rossi di Montelera e di Premuda advised by CARIGEST, and the interdisciplinary SV iPhD program at EPFL. S.K., J.C. and A.J.V. are supported by NIH NIGMS grant no. R35GM138368.
Author information
Authors and Affiliations
Contributions
M.P., L.N., C.S. and B.E.C. conceived of the study and designed experiments. M.P., L.N., Y.C., C.A.G. and S.O. developed the code base. M.P., L.N. and C.S. generated protein designs. M.P., L.N., C.S., J.S. and S.G. purified proteins. L.N. and K.H.G. performed protein binding assays. E.P. and S.B. performed CD45 binder characterization. K.H.G. and L.V. developed and performed PD-1 binder characterization assays. G.N.H. purified SAS-6. L.K. performed gene editing assays. A.A.-S. performed blocking assays for birch allergen. M.P. and L.N. solved crystal and cryo-EM structures. P.B., A.H.W. and S.L. performed and analysed CbAgo activity assays and SEC analysis. S.K., J.C. and A.J.V. performed and analysed Claudin experiments. C.S., L.N., B.L.S. and B.E.C. designed and performed AAV experiments. B.J.Y., A.M.W., P.G., Y.D.M., G.S., D.C.S., A.J.V., S.O., B.L.S. and B.E.C. supervised the work and acquired core funding. M.P., L.N., C.S. and B.E.C. wrote the initial paper. All authors read and contributed to the paper. M.P., L.N. and C.S. agree to rearrange the order of their respective names according to their individual interests.
Corresponding authors
Ethics declarations
Competing interests
K.H.G., L.V., B.J.Y. and A.M.W. are employees of Visterra Inc., USA. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature thanks Patrick Aloy, Qiangfeng Cliff Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 In silico analysis of BindCraft designs.
a, Graphs plotting the RMSDCα of the target structure after initial AF2 binder hallucination, compared to the input target structure for different design targets. b, Effect of the helicity loss on secondary structure content of binders designed against PD-L1. Negative values that discourage the formation of alpha-helices can result in purely beta-sheeted binders. c, Number of GPU hours elapsed to generate 100 binder designs passing computational filters across different binder lengths and targets. Numbers above bars indicate the number of designs that needed to be sampled. Filtering conditions for BindCraft and RFdiffusion are described in Methods. d, Amino acid type distribution at the designed binder interfaces generated by BindCraft and RFdiffusion averaged across four different targets. e, Comparison of maximum TM-scores and sequence identities of designed binder folds and designed binder–target interfaces against their closest matches in the PDB. Binder folds (green) were assessed using Foldseek55, and interfaces (pink) were evaluated using PPIref14 and USalign15. For depicted box plots, the centre line represents the median of the data (50th percentile) and the box spans the 25th and 75th percentiles of the data. The whiskers show the minimum and maximum values of the distribution. Outliers (circles) are data points that fall outside the 1.5 interquartile range.
Extended Data Fig. 2 Experimental characterization of binding modes of binders targeting cellular receptors.
a, Competition assay showing that the anti-PD-1 antibody, pembrolizumab, occupies the same binding site on PD1 as binder2. b, SEC-MALS analysis of PD-L1_b4 alone (left) and in complex with PD-L1-Fc (right). c, Competition assay showing previously published de novo binder DBL2_04 occupying the same binding site as PD-L1 binder4. d, Structural overlay highlighting the binding mode of PD1 (grey) versus PD-L1_b4 (salmon). e, SEC-MALS analysis of IFNAR2_b5. f, Competition assay displaying the natural IFNA2 binding partner occupies an overlapping binding site with IFNAR2 binder5. g, Structural overlay highlighting the binding of mode of IFNA2 (grey) versus IFNAR2_b5 (salmon). h, Specificity of three selected binders against PD-L1, IFNAR2, and PD-1, as assessed by BLI at 1 µM. i_pTM values are shown for each binder-target interaction (green: on-target; black: off-target). i Structural overview of the binding mode of CLDN1 binder12 (salmon) and the native CpE toxin interaction partner (grey).
Extended Data Fig. 3 Targeting natural and de novo binding epitopes.
a, Design model of binder4 in complex with de novo designed beta-barrel BBF-14. b, Single cycle kinetic analysis with SPR of binder4 binding to BBF-14. c, Comparison of crystal structure (coloured) of the BBF-14_binder4 complex overlaid with the design model (grey). d, Refined 2mFo − mFc electron density map of the BBF-14_binder4 complex rendered in gray and contoured at 1.0σ. The model complex refined against the map is shown as cartoon representation with BBF-14 coloured green and binder4 in salmon. e, Design model of binder4 binding to the challenging structural protein target SAS-6. f, SPR binding traces of binder4 to CrSAS-6 monomeric form. g, SPR binding traces of binder4 to CrSAS-6 dimeric form. h, Structural model of the oligomeric form of SAS-6 with binder4 (salmon) overlaid with the previously characterized monobody MBCRS6-122.
Extended Data Fig. 4 Biophysical and structural properties of binders targeting allergens.
Refined 2mFo − mFc electron density map of the Der f7_binder2 complex rendered in grey and contoured at 1.0σ for crystal form P21 in a and the crystal form C121 in b. The model complex refined against the map is shown as cartoon representation with Der f7 coloured green and binder2 in salmon. c, SEC-MALS analysis of Der f7_binder2. d, SEC-MALS analysis of Der f21_binder10 e, Refined 2mFo − mFc electron density map of the Der f21_binder10 complex rendered in grey and contoured at 1.0σ. The model complex refined against the map is shown as cartoon representation with Der f21 coloured green and binder10 in salmon. f, SEC-MALS analysis of Bet v1_binder2. g, Specificity of three selected binders against Der f7, Der f21, and Bet v1, as assessed by SPR with increasing concentrations (4.6–10 µM). i_pTM values are shown for each binder-target interaction (green: on-target; black: off-target).
Extended Data Fig. 5 Biophysical and structural analysis of binders against nucleic acid-guided nucleases.
a, Representative 2D class averages of apo SpCas9 (left), SpCas9 bound to binder3 (centre) and binder10 (right). b, Views of the unsharpened cryo-EM density maps coloured by local resolution. Predicted model of the apo conformation of SpCas9 with bound c, binder3 or d, binder10 docked into its respective cryoEM density. e, Representative BLI sensorgram displaying binding kinetics of CbAgo and binder2. f, Size exclusion chromatography (SEC) analysis of CbAgo only (grey line) or binder2 only (orange line) or combined (green line). g, SEC analysis of CbAgo only (grey line) or in presence of gDNA (orange line) or in presence of both gDNA and binder2 (blue line). h, Structural comparison of the binder2 overlaid with the target DNA-bound structure of CbAgo, indicating overlapping binding sites.
Extended Data Fig. 6 Screening of functional cell-type specific AAVs.
a, Schematic illustrating the small-scale screening assay. Both the production cell line as well as the target cells overexpressing the target receptors are derived from the same parent cell line, allowing to directly transfer the supernatant of AAV-packaging cells onto the targeted cells for transduction. The right scatter plot illustrates the transduction signal measured by flow cytometry (GFP expression) b, Supernatant viral titres (log-scale) of the different AAV variants screened (Fig. 5c), as indicated. Titres were measured in duplicates (n = 2) and error bars indicate 95% confidence interval. c, Receptor expression levels of the created stable cell lines for screening, stained by APC-conjugated antibodies. Panel a was created using BioRender (https://biorender.com).
Extended Data Fig. 7 Benchmarking prediction accuracy across design pipelines.
Experimentally validated binders and non-binders from previously published binder design pipelines have been repredicted using the BindCraft prediction pipeline with either AF2 a, monomer (default) or b, multimer models. Of note, EvoPro48 and RFdiff5 designs have been already prefiltered by AF2 monomer in their respective publications, and indicate the presence of false positives. RIFdock4 and Masif-seed3,4 designs were not prefiltered by AF2. The centre line in box plots represents the median of the data (50th percentile), the box spans the 25th and 75th percentiles of the data. The whiskers show the minimum and maximum values of the distribution. Outliers (circles) are data points that fall outside the 1.5 interquartile range. c, The i_pTM values of AlphaFold2 and AlphaFold3 predictions of experimentally characterized BindCraft designs.
Supplementary information
Supplementary Information (download PDF )
Supplementary Text, Figs. 1–6, Table 1 and References.
Supplementary Data 1 (download CSV )
List of binder designs for each target selected for experimental validation with associated in silico design metrics.
Supplementary Data 2 (download XLSX )
Pairwise template modelling score structural comparisons and sequence identity fractions for target proteins and binders against different targets.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pacesa, M., Nickel, L., Schellhaas, C. et al. One-shot design of functional protein binders with BindCraft. Nature 646, 483–492 (2025). https://doi.org/10.1038/s41586-025-09429-6
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09429-6
This article is cited by
-
De novo design of potent CRISPR–Cas13 inhibitors
Nature Chemical Biology (2026)
-
De novo design of GPCR exoframe modulators
Nature (2026)
-
Computation and deep-learning-driven advances in CRISPR genome editing
Nature Structural & Molecular Biology (2026)
-
Semantic design of functional de novo genes from a genomic language model
Nature (2026)
-
The computationally designed TRI2-2 miniprotein inhibitor protects against multiple SARS-CoV-2 Omicron variants
Communications Biology (2026)
