
Abstract
Cancer metastasis is a major contributor to patient morbidity and mortality1, yet the factors that determine the organs where cancers can metastasize are incompletely understood. Here we quantify the absolute levels of 124 metabolites in multiple tissues in mice and investigate how this relates to the ability of breast cancer cells to grow in different organs. We engineered breast cancer cells with broad metastatic potential to be auxotrophic for specific nutrients and assessed their ability to colonize different tissue sites. We then asked how tumour growth in different tissues relates to nutrient availability and tumour biosynthetic activity. We find that single nutrients alone do not define the sites where breast cancer cells can grow as metastases. In addition, we identify purine synthesis as a requirement for tumour growth and metastasis across many tissues and find that this phenotype is independent of tissue nucleotide availability or tumour de novo nucleotide synthesis activity. These data suggest that a complex interplay between multiple nutrients within the microenvironment dictates potential sites of metastatic cancer growth, and highlights the interdependence between extrinsic environmental factors and intrinsic cellular properties in influencing where breast cancer cells can grow as metastases.
Similar content being viewed by others

Main
Understanding the factors that govern tumour growth in metastatic sites could lead to more effective therapies for advanced cancer. Among the tumour microenvironment factors that contribute to where tumours can grow as metastases, nutrient availability is a key component2,3,4,5,6,7,8,9. Because nutrient levels vary across tissues, they can limit where cancers grow as metastases3,4,10,11, forcing cancer cells to undergo metabolic adaptations that create new dependencies for colonization of different tissues. For instance, limited lipid availability in the brain results in a site-specific dependency on fatty acid synthesis or desaturation for brain metastasis of breast tumours12,13 or leukaemias14. Similarly, reduced brain serine levels sensitize breast cancer brain metastases to inhibition of the serine synthesis enzyme PHGDH15. Both pyruvate and asparagine availability can influence breast cancer metastasis to the lungs16,17. These studies raise the question of whether reduced availability of specific nutrients in certain tissues broadly predicts where metastatic cancer cells are likely to colonize and grow.
Here we sought to explore the relationship between tissue nutrient availability and metastatic potential in triple-negative breast cancer (TNBC). We quantified the absolute levels of 124 metabolites present in plasma and across six mouse tissues and constructed a series of nutrient auxotrophs to assess their metastatic potential to multiple tissues following intracardiac injections. Our analysis revealed that although levels of some metabolites correlate with metastatic potential, the levels of individual nutrients in isolation are insufficient to determine metastatic preference or the ability of specific nutrient auxotrophs to grow in a tissue site. Instead, our findings suggest that metastatic preference is driven by a combination of multiple nutrient levels and cell-intrinsic factors.
Nutrient availability across tissues
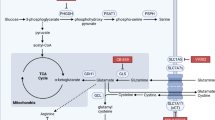
To assess nutrient availability across multiple tissue sites, including where TNBC cells can grow as primary tumours or metastases, we isolated plasma and interstitial fluid from the mammary fat pad (MFP), liver, lung, kidney and pancreas of female NOD-SCID-γ (NSG) and female C57BL/6J (B6) mice. We also collected cerebrospinal fluid (CSF) as a surrogate for the brain extracellular environment because acquiring sufficient interstitial fluid for analysis of multiple metabolites from normal brain tissue was not feasible. Quantitative mass spectrometry determined the absolute levels of 124 metabolites in the interstitial fluid from these different tissue sites, as well as in plasma (Fig. 1a and Extended Data Fig. 1a–j). We confirmed high correlations across biological replicates, stability of most metabolites during handling and consistency between plasma collection methods and across NSG and B6 strains (Extended Data Figs. 1k–3). Only a small subset of metabolites showed method-specific or strain-specific variation. For example, hypoxanthine increased after prolonged incubation on ice that exceeded collection time (Extended Data Fig. 2e), and some metabolites differed between NSG and B6 in specific tissues (Extended Data Fig. 3).

a, Schematic of plasma, CSF and tissue interstitial fluid (IF) isolation from female NSG or B6 mice. Metabolites were quantified by liquid chromatography–mass spectrometry alongside a dilution series of chemical standards; 124 metabolites were quantified. TCA, tricarboxylic acid. Created in BioRender. Abbott, K. (2025) https://BioRender.com/tp4l6fd. b,c, PCA (b) and hierarchical clustering (c) of metabolites measured in plasma, CSF or tissue IF samples from NSG mice. Data represent n = 10 (plasma, liver IF, lung IF and pancreas IF), n = 6 (kidney IF and MFP IF) or n = 4 (CSF) biologically independent samples. Metabolite measurements were performed twice for plasma, liver, lung and pancreas IF samples and once for the remaining tissues; data from repeated measurements were pooled for analysis. Columns of the heatmap were z-score normalized. d, Bar plot showing the number of metabolites with significantly lower (depleted) or higher (elevated) levels in IF or CSF relative to plasma. Significance was determined by Welch’s t-test (two-sided, unequal variance) with fold change > 2 and P < 0.05. e, Loadings plot presenting the contribution of individual metabolite classes to the PCA components in b. The colours indicate pathway assignment in a. f–k, log2 fold change of selected metabolites in tissue IF or CSF relative to plasma. Data are mean ± s.e.m., with n values as in b,c. l–n, Heatmaps of average log2 fold change (FC) in metabolite concentrations relative to plasma. Scale bars indicate value ranges. o, Area under the curve (AUC) values from proliferation assays of MDA-MB-231 control (Ctrl) or indicated knockout (KO) cells cultured ± relevant rescue metabolites (Extended Data Fig. 6a–f). AUC values were normalized to control + rescue. Data are mean ± s.d.; n = 3 biologically independent samples. Representative plots are shown from one of two independent experiments with similar results. Statistical analysis was done by a Kruskal–Wallis test with Dunn’s multiple comparisons (two-sided). Arg, arginine; Cit, citrulline; Hx, hypoxanthine; Pro, proline; Ser, serine; Urd, uridine. PYCR denotes PYCR1/2/3 triple knockout.
Principal component analysis (PCA) and hierarchical clustering revealed that metabolites measured in tissue interstitial fluid samples cluster distinctly from those measured in plasma and the CSF (Fig. 1b,c and Extended Data Fig. 4a,b). When considering the differences in metabolite concentrations between tissue interstitial fluid and plasma, we found that although some metabolites were depleted in tissue interstitial fluid relative to plasma, numerous metabolites were present at higher concentration in interstitial fluid than in plasma (Fig. 1d and Extended Data Fig. 4c). By contrast, CSF showed lower levels of many metabolites than plasma, a pattern probably attributable to the selective permeability of the blood–brain barrier and that is consistent with previous studies examining CSF from both humans and mice18,19. Of note, we found that nucleotide-related metabolites, but not amino acids, were greater contributors to the PCA components separating the fluid samples in both mouse strains (Fig. 1e and Extended Data Fig. 4d). This suggests that levels of nucleotides and nucleotide precursors or salvage products are an important contributor to the differences in nutrient availability across tissue environments.
Many of the differences in metabolite concentrations across tissues align with known metabolic characteristics of each tissue. Most tissue interstitial fluid displayed lower glucose and higher lactate levels than plasma (Fig. 1f,g and Extended Data Fig. 4e,f), consistent with active glucose metabolism20. The liver stood out with higher levels of both glucose and lactate in interstitial fluid than plasma, consistent with its glycogenolytic and gluconeogenic capabilities, as well as its role in recycling lactate back to glucose21. In liver interstitial fluid, we also observed lower arginine and higher ornithine levels than plasma (Fig. 1h,i and Extended Data Fig. 4g,h), probably reflecting active arginase activity in this tissue22. Conversely, citrulline was depleted in kidney interstitial fluid (Extended Data Fig. 4i), aligning with human data23 and perhaps reflective of an active urea cycle in this tissue. Liver interstitial fluid contained relatively higher concentrations of branched-chain amino acids (isoleucine, leucine and valine) than other tissues (Fig. 1j,k and Extended Data Fig. 4j–l), which may reflect known low branched-chain amino acid aminotransferase activity in this organ relative to other tissues24.
Consistent with previous measurements18,19, the levels of many amino acids were lower in CSF than in circulation, with notable variation in amino acid levels measured in other tissues (Fig. 1l and Extended Data Fig. 1a). Unlike previous reports18,19, serine was not depleted in the CSF relative to plasma, although it was lower than other tissue interstitial fluid. This lack of serine depletion in CSF was consistent across both NSG and B6 mouse strains (Extended Data Fig. 4m), and may be influenced by other factors such as circadian rhythm25 or diet26. In addition, although many nucleotide species were elevated in tissue interstitial fluid and low in CSF compared with plasma (Fig. 1m,n and Extended Data Figs. 1d and 4n), the purine hypoxanthine was consistently high across all tissue interstitial fluid as well as in CSF, although some of this elevation may be due to a post-collection artefact (Extended Data Fig. 2e). Collectively, these findings denote many variances in nutrient availability across different tissues, which could constrain the ability of cancer cells to metastasize to particular organs.
Generation of nutrient auxotrophs
Cancer cells exposed to nutrient-limited microenvironments often adapt by upregulating metabolic pathways to synthesize the deficient metabolite10. To eliminate this adaptive response and isolate how nutrient availability constrains metastatic growth, we engineered widely metastatic TNBC cell lines to be auxotrophic for specific metabolites that vary across tissues. We selected the human lines MDA-MB-231 and HCC1806 (ref. 12) and the B6 mouse cell line EO771 (ref. 27), then used CRISPR–Cas9 to knock out genes essential for the synthesis of specific metabolites with known nutrient rescues (Extended Data Fig. 5a). Specifically, we targeted ASNS (asparagine)28,29, ASS1 (arginine)30,31, PHGDH (serine)15,32, PYCR1, PYCR2 and PYCR3 (proline)33, DHODH (pyrimidines, for example, uridine)34,35 and GART (purines, for example, inosine or hypoxanthine)36. We confirmed variable expression of each of these proteins in parental MDA-MB-231, HCC1806 and EO771 cells by western blot (Extended Data Fig. 5b,c); of note, MDA-MB-231 cells expressed lower levels of ASS1 and PHGDH, consistent with previous observations32,37. We also confirmed loss of relevant enzyme expression in each knockout line by western blot (Extended Data Fig. 5d–i), and further confirmed that each knockout line exhibited impaired proliferation in the absence of the relevant nutrient that corresponded to the intended auxotrophy (Fig. 1o and Extended Data Fig. 6a–f). Supplementation of the specific metabolite rescued proliferation of each auxotroph cell line to comparable levels as the parental line. EO771 cells were noted to occasionally stop proliferating when cultured in RPMI, which we attribute to glucose depletion, as refreshing the medium or supplementing glucose fully rescued sustained proliferation (Extended Data Fig. 6g).
To generate proline auxotrophs, deletion of the mitochondrial (PYCR1 and PYCR2) and the cytoplasmic (PYCR3) genes encoding pyrroline-5-carboxylate reductase was required, as the presence of any of these genes has been reported to enable proline synthesis33 and could support proliferation following proline withdrawal (Extended Data Figs. 5g and 6d). As arginine starvation in parental cells arrests proliferation via mTOR inactivation38, addition of citrulline was required for control cells to proliferate without arginine (Fig. 1o and Extended Data Fig. 6b); however, ASS1-null cells could not grow without arginine even if citrulline was added, validating that ASS1-knockout cells lost the ability to generate arginine from citrulline. For GART, despite the presence of some species detected at 110 kDa (ref. 39) in MDA-MB-231 and HCC1806 using an anti-GART antibody (Extended Data Fig. 5i), depletion of the 50-kDa monomeric form was sufficient to result in hypoxanthine auxotrophy in all three cell lines (Extended Data Fig. 6f). To assess the specificity of these auxotrophies, we also tested HCC1806 ASNS-knockout, ASS1-knockout, PHGDH-knockout and PYCR1, PYCR2, PYCR3 triple knockout lines in media lacking other amino acids targeted in this study. Each knockout line maintained robust proliferation when deprived of amino acids unrelated to its engineered enzyme deficiency, supporting the specificity of the observed auxotrophies (Extended Data Fig. 6h–k). One exception was that ASNS-knockout cells showed reduced proliferation in citrulline-depleted media, suggesting a potential interaction with arginine synthesis pathways. Overall, these data confirm that the genetic modifications render cells dependent on external supplementation of the expected metabolites for proliferation and survival in standard culture conditions.
In constructing the nutrient auxotrophs, we identified three distinct categories of metabolite requirements. First, removing asparagine or proline from the culture medium reduced proliferation of all cell lines when the relevant synthesis genes were knocked out (Extended Data Fig. 6a,d). Second, withdrawal of arginine in all cell lines, or serine in MDA-MB-231 cells, reduced proliferation even when the synthesis genes were intact (Extended Data Fig. 6b,c). Third, nucleotides, which are typically absent from standard culture media, could be supplemented to rescue cell dependency on DHODH or GART (Extended Data Fig. 6e,f). These findings indicate that for some nutrients, cellular synthesis capacity is important for maximal proliferation regardless of environmental availability. Nevertheless, this panel of metabolite auxotrophs with varying metabolite synthesis requirements enable testing of how metabolite synthesis influences the ability of cells to colonize different tissues with different nutrient levels.
Single nutrients do not predict metastasis
Before testing the auxotrophs in vivo, we confirmed that MDA-MB-231, HCC1806 and EO771 cells tagged with firefly luciferase (Fluc) metastasize to multiple organs after intracardiac injection, consistent with previous reports12,40 (Extended Data Fig. 7). We then injected Fluc-tagged auxotrophs and control cells and compared their ability to form tissue-specific metastases (Fig. 2a–c and Extended Data Fig. 8). Nucleotide auxotrophs showed the strongest defects, as DHODH loss impaired metastasis in all sites for MDA-MB-231 and HCC1806, and in the liver and kidney or adrenal gland for EO771, whereas GART was essential across all tissues in all three lines (Extended Data Fig. 8a,b). Despite abundant hypoxanthine in tissue fluids and CSF (Fig. 1n and Extended Data Figs. 1d and 4n), GART remained essential, arguing that individual nutrient requirements are not sufficient to define the tissues to which these cancer cells can metastasize.

a, Schematic of intracardiac injection of control and auxotroph cells expressing Fluc into the left ventricle, enabling metastatic spread to the brain, liver, lung, ovary, bone, and kidney or adrenal glands. Colonization was quantified by bioluminescence imaging of harvested tissues at end point. MDA-MB-231–Fluc and HCC1806–Fluc cells were injected into NSG mice; EO771–Fluc cells were injected into B6 mice. Created in BioRender. Abbott, K. (2025) https://BioRender.com/73th1x3. b, Petal plots used to display metastatic patterns, where each petal represents a tissue and its length indicates growth of auxotrophs relative to controls. c, Petal plots showing the metastatic distributions of different metabolite auxotroph cells relative to control cells. Data are mean ± 95% confidence interval; n = 3–7 biologically independent mice per group, with exact numbers reported in the Source Data. Plots were derived from Extended Data Fig. 8. Bo, bone; Br, brain; Ki, kidney; Li, liver; Lu, lung; Ov, ovary. d, Scatter plots of average metabolite concentrations in tissue IF versus auxotroph dependency (log2 fold depletion in tumour growth of knockout relative to control). MDA-MB-231 (black) and HCC1806 (blue) were compared with NSG tissue metabolite levels; EO771 (red) was compared with B6 levels. Brain values reflect CSF. Data are mean ± s.e.m.; n = 3–7 biologically independent mice per group with exact numbers reported in the Source Data. Pearson correlation coefficients (r) and exact P values are provided in the Source Data (two-sided tests; *P < 0.05 and **P < 0.01). PYCR denotes PYCR1/2/3 triple knockout. Experiments were performed once.
In contrast to nucleotides, amino acid dependencies were heterogeneous and cell line specific (Fig. 2c and Extended Data Fig. 8c–f). ASNS was required for metastasis to bone and kidney or adrenal gland in MDA-MB-231 and HCC1806 but not in EO771, whereas effects on brain metastasis were modest, with only slight impairment in MDA-MB-231 and EO771 and no effect in HCC1806. Loss of ASS1 most consistently impaired bone metastasis across lines, although the magnitude was modest, whereas effects in other tissues varied. PHGDH dependency also differed, as MDA-MB-231 required PHGDH broadly, consistent with previous reports of serine synthesis dependence in brain metastasis15, but HCC1806 showed dependency only in lung and bone, and EO771 displayed minimal reliance. These findings demonstrate that low serine levels in the brain18,19 do not universally confer PHGDH dependence. Finally, proline synthesis was broadly required in MDA-MB-231 but variably so in HCC1806 and EO771, although all three lines showed some dependency on PYCR1, PYCR2 and PYCR3 (PYCR1/2/3) for brain metastasis. Collectively, these results indicate that, unlike nucleotide synthesis, amino acid auxotrophy does not reliably predict tissue-specific metastatic capacity. Instead, dependencies differ across cell lines and tissues, highlighting the combined influence of cell-intrinsic traits and local environments.
To assess variability and reproducibility, we repeated the in vivo metastasis experiment for four HCC1806 auxotroph lines (ASNS, ASS1, PHGDH and PYCR1/2/3), which had shown variable tissue-specific growth. Metastatic burden was quantified by bioluminescence imaging, and auxotroph-to-control fold changes were compared across experiments (Extended Data Fig. 9a–e). Of 24 tissue comparisons, 22 were consistent, supporting the reproducibility of the assay. The only differences were modestly better growth of ASS1-knockout cells in the brain and ovary in the repeat experiment. These results indicate that the heterogeneous metastatic phenotypes reflect biological differences rather than experimental variability.
To test whether auxotrophs retained their phenotype after metastasis formation, we isolated HCC1806 ASNS-knockout brain metastases and ASS1-knockout liver metastases following intracardiac injection. Western blotting confirmed that neither enzyme was re-expressed (Extended Data Fig. 9f,g). In culture, ASNS-knockout cells proliferated only with asparagine, and ASS1-knockout cells failed to grow without arginine, even with citrulline added (Extended Data Fig. 9h–k), both consistent with their original in vitro phenotypes. Thus, auxotrophy is preserved in vivo, and growth in nutrient-depleted tissues is not due to re-expression of the deleted enzyme.
To evaluate whether nutrient availability predicts tissue-specific auxotroph dependencies, we correlated metabolite levels with metastatic outgrowth (Fig. 2d). We hypothesized that auxotrophs would struggle to grow in tissues lacking the corresponding nutrient but would grow normally in tissues where it was abundant: for example, PHGDH-null cells in serine-poor versus serine-rich sites. Only a few significant correlations were observed: proline concentration correlated positively with PYCR1/2/3 dependency in HCC1806 cells, whereas arginine showed a negative correlation with ASS1 dependency in MDA-MB-231 cells. Broader analysis revealed few associations, most unrelated to the engineered auxotrophy (Extended Data Fig. 10a–d). Thus, single-nutrient levels alone do not reliably predict metabolic dependencies in metastasis.
We also considered whether the low but non-zero levels of amino acids measured in vivo might partially sustain auxotrophs. For example, arginine is present at 3 µM in liver interstitial fluid and asparagine at 4 µM in CSF (Extended Data Fig. 10e,f). In titration assays, ASS1-null cells showed a half-maximal inhibitory concentration (IC50) of 2.5 µM arginine (with citrulline present), and ASNS-null cells showed an IC50 of 10 µM asparagine (Extended Data Fig. 10g–j). These concentrations are at or below those measured in vivo, suggesting that residual nutrient availability could allow some proliferation but does not fully support proliferation of the auxotrophs in culture. Thus, additional support from the tumour microenvironment, such as stromal exchange or protein scavenging, must further enable cancer cell proliferation in nutrient-limited tissues.
Auxotroph tumour growth in the MFP or brain
Because nucleotide synthesis auxotrophs fail to grow in multiple tissues, it might be required for survival in circulation. To test this possibility, and to ask whether nutrient availability more directly constrains tumour growth at the implantation site itself, we implanted auxotrophs into the brain or MFP, bypassing dissemination through the circulation (Fig. 3a–c and Extended Data Fig. 11). We chose these sites because the levels of many nutrients, especially amino acids, are lower in the brain18,19, and previous work has suggested that tumours preferentially grow in their primary-site tissue environment41. To monitor tumour growth, we engineered auxotrophs to express Gaussia luciferase (Gluc)42, enabling noninvasive tracking by blood luminescence. We also implanted EO771 auxotrophs into the brain to compare metabolic dependencies in that site across all three lines.

a, Schematic of direct implantation of control or auxotroph cells expressing Gluc into the brain or MFP of mice, with tumour growth monitored over time via blood luminescence. MDA-MB-231–Gluc and HCC1806–Gluc cells were injected into NSG mice; EO771–Gluc cells were injected into B6 mice. Created in BioRender. Abbott, K. (2025) https://BioRender.com/e78ptc1. b, Petal plots illustrating auxotroph tumour growth relative to controls. Each petal represents a cell line and tumour site; petal length reflects relative tumour growth of auxotrophs versus controls. c, Petal plots showing growth distributions of auxotroph versus control tumours. Data are mean ± 95% confidence interval; n = 2–10 biologically independent mice per group with exact numbers reported in the Source Data. Plots were derived from Extended Data Fig. 11. Cell lines and tumour sites are as in b. d, Scatter plot correlating auxotroph dependency for brain growth based on route of cell delivery. Axes show log2 fold change in tumour growth of knockout versus control cells following intracranial (x axis) or intracardiac (y axis) injections. Data are mean ± s.e.m.; n = 2–10 biologically independent mice per group (exact numbers are in Extended Data Figs. 8 and 11). Two-sided Pearson correlation coefficient and exact P value are provided in figure panel. e, Scatter plots of average metabolite concentrations in MFP IF and CSF (proxy for brain) versus auxotroph dependency for growth in each site (log2 fold depletion of knockout relative to control). Symbols represent tissue metabolite concentrations. Data are mean ± s.e.m.; n = 3–7 biologically independent mice per group, with exact numbers reported in the Source Data. PYCR denotes PYCR1/2/3 triple knockout. Experiments were performed once.
We first compared how the route of cell delivery to the brain influenced gene dependency for tumour growth in that site. Our analysis revealed a statistically significant but modest correlation between gene dependencies observed with intracranial and intracardiac injections (Fig. 3d and Extended Data Fig. 12a), indicating that the overall patterns of gene dependency are independent of the method of cell delivery to the brain. However, we found that dependencies were generally stronger when cells were directly implanted via intracranial injection. In addition, we observed some notable outliers in which the route of injection affected gene dependency. For instance, HCC1806 cells demonstrated a strong dependency on ASS1 when injected intracranially, but this dependency was reduced following intracardiac injection. EO771 cells lacking DHODH or PHGDH were able to form metastatic brain tumours when injected in the heart but not when directly implanted in the brain.
We found that nucleotide synthesis was invariably required, as loss of DHODH or GART impaired brain and MFP tumour growth in all three lines and extended survival (Fig. 3c and Extended Data Fig. 11a–f). These data are also consistent with a broad requirement for de novo nucleotide synthesis to grow in tissues despite high extracellular nucleotide levels (Fig. 1m,n). By contrast, amino acid dependencies were more variable, as was observed in intracardiac experiments. MDA-MB-231 required ASNS in both the brain and MFP, whereas HCC1806 showed no ASNS dependence in the brain and EO771 only showed a modest effect (Fig. 3c and Extended Data Fig. 11g–i). ASS1 was required for HCC1806 tumours in both the brain and the MFP, for MDA-MB-231 only in the MFP, and was dispensable in EO771 (Extended Data Fig. 11j–l). PHGDH was essential in MDA-MB-231 across sites but variably required in HCC1806 and EO771 (Extended Data Fig. 11m–o). Proline synthesis (PYCR1/2/3) was required in the brain for HCC1806, modestly in MDA-MB-231, but not for EO771 (Extended Data Fig. 11p–r). A consistent link between tissue nutrient availability and tumour growth was not observed (Fig. 3e). Together, these data show that direct implantation recapitulates the heterogeneous auxotroph dependencies observed with intracardiac injection and reinforces that single-nutrient levels do not reliably predict tissue tumour growth requirements.
MFP and brain tumour metabolic activity
To further study the relationship between nutrient levels in tissues and the ability to synthesize specific nutrients to allow tumour growth in a given tissue, we traced the fate of 13C-labelled glucose in MDA-MB-231-derived brain or MFP tumours in mice (Fig. 4a). We monitored the fraction of m+6-labelled glucose in plasma over the course of the infusion and observed that labelling approached steady state by the final time point (Fig. 4b and Extended Data Fig. 12b). We observed that plasma glucose labelling was slightly lower in mice bearing brain tumours than in those with MFP tumours, whereas 13C-labelling of pyruvate, lactate and amino acids in plasma was largely similar between cohorts (Extended Data Fig. 12c–e). These findings suggest that tumour location may modestly influence systemic glucose metabolism.

a, Schematic of [U-13C]-glucose infusion to trace metabolite fate in female NSG mice bearing MDA-MB-231 tumours in the MFP or brain. Created in BioRender. Abbott, K. (2025) https://BioRender.com/os441ve. b, Fractional labelling of plasma glucose (m+0 and m+6) following [U-13C]-glucose infusion (0.4 mg min−1, 10 h) in mice with MFP or brain tumours. Data are mean ± s.e.m.; n = 5 (MFP tumours) or n = 4 (brain tumours) biologically independent mice. c–i, Fractional labelling of the indicated metabolites measured by liquid chromatography–mass spectrometry. Separate cohorts of infused mice were implanted with tumours in either the MFP or brain, and both tumour and noncancerous tissues were collected from the same mice in each cohort. Data are mean ± s.e.m.; n = 5 (MFP tumour and noncancerous MFP), n = 4 (brain tumour) and n = 3 (noncancerous brain) biologically independent samples. Statistical analysis was performed using one-way analysis of variance with Holm–Sidak multiple comparisons test (two-sided). Experiments were performed once.
Within tissues, lactate and tricarboxylic acid cycle metabolite labelling was higher in the brain and brain tumours than in the MFP (Extended Data Fig. 13a,b), consistent with high glucose utilization in the brain43 and greater biosynthetic demands imposed by low amino acid availability behind the blood–brain barrier15,19. Accordingly, labelling of asparagine, glycine, serine and proline was higher in brain tumours than in MFP tumours (Fig. 4c–f and Extended Data Fig. 13c,d). However, growth of auxotrophs lacking ASNS, PHGDH or PYCR1/2/3 was similarly affected in both sites (Fig. 3c), arguing that elevated amino acid synthesis in brain tumours does not necessarily indicate increased dependency.
For nucleotides, MFP tumours synthesized more purine and pyrimidine nucleotides than normal tissue, whereas brain and brain tumours showed lower synthesis, closer to normal MFP (Fig. 4g–i and Extended Data Fig. 13e,f). Regardless, DHODH and GART were required for tumour growth in both sites (Fig. 3c), again indicating that synthesis activity does not predict pathway dependency. Although glucose-derived labelling was reduced in brain tumours, total nucleotide levels were largely similar between brain and MFP tumours (Extended Data Fig. 13e), suggesting compensation through uptake or salvage. Supporting this, DHODH-knockout MDA-MB-231 cells cultured with uridine showed reduced pyrimidine labelling from 13C-glucose but maintained overall nucleotide levels (Extended Data Fig. 13g–k), providing evidence that salvage can sustain nucleotide pools when de novo synthesis is impaired.
Together, these results demonstrate that although metabolic activity differs between brain and MFP tumours, neither individual nutrient levels nor pathway activity reliably predict auxotroph dependencies or metastatic potential. Rather, it implies that tumour cells differentially leverage existing nutrient resources or engage alternative pathways such as nucleotide salvage within a tissue microenvironment to meet the metabolic demands for tumour growth.
Dietary nutrient depletion and metastasis
To test whether systemic nutrient manipulation alters metastatic colonization, we fed NSG mice a diet deficient in both serine and glycine before intracardiac injection of HCC1806 cells44,45. The diet robustly depleted serine and glycine in plasma and multiple tissues (Extended Data Fig. 14a–f), but effects on metastasis were site specific: tumour burden in the brain, liver and lung was unchanged, whereas bone, kidney and ovary metastases were reduced (Extended Data Fig. 14g–i). Thus, although nutrient depletion can influence metastatic outgrowth, single nutrients do not consistently predict tissue-specific colonization.
Genetic correlates of metastasis
To probe links between metabolic requirements and metastasis, we analysed Dependency Map (DepMap) data. CRISPR dependency of the amino acid synthesis genes studied here did not correlate with tissue-specific metastatic potential (Extended Data Fig. 15a). By contrast, CRISPR dependencies for DHODH and GART correlated with metastatic potential in several tissues, particularly the lung (Fig. 5a and Extended Data Fig. 15b), consistent with our experimental findings (Fig. 2c). Thus, CRISPR dependencies may better predict metastatic potential than gene expression or metabolite levels in select cases.

a, Scatter plots correlating the metastatic potential of breast cancer cells to the lung with in vitro CRISPR dependencies of the indicated genes (left). Each dot represents one cell line. P values from two-sided Pearson correlation tests are shown in the panels. Heat map showing −log10(P) values from correlations between metastatic potential to the indicated tissues and CRISPR dependency scores of the indicated genes (right). Data are from the DepMap portal. b, Scatter plots correlating metabolite concentrations in tissue IF with the metastatic potential of control cells following intracardiac injection. MDA-MB-231 (black) and HCC1806 (blue) values were compared with NSG tissue metabolite data; EO771 (red) was compared with B6 data. Brain values reflect CSF. Data are mean ± s.e.m.; n = 3–7 biologically independent mice per group, with exact numbers provided in the Source Data. Summary data used to derive metastatic potential values are presented in Extended Data Fig. 8. Pearson correlation coefficients (r) and exact P values are provided in the Source Data (two-sided tests). c, Volcano plots depicting Pearson correlation coefficients and P values for metabolites correlated with metastatic potential of control cell lines following intracardiac injection. The black circles indicate metabolites significantly correlated in all three cell lines. Significance was defined as |r| > 0.5 and P < 0.05 (two-sided). G1P, glucose-1-phosphate; GPC, glycerophosphocholine; 3M2OP, 3-methyl-2-oxopentanoic acid; PEtn, O-phosphoethanolamine.
Multiple metabolites influence metastasis
Because single nutrients did not reliably predict metastatic site tropism, we next asked whether broader tissue metabolite levels correlate with metastatic outgrowth of control cell lines. Several metabolites, including serine, glycine, tyrosine, glutamate, inosine and thymidine, correlated positively with metastatic potential, with 23 metabolites consistently significant across all three lines (Fig. 5b,c). Many correlations were reproducible in both NSG and B6 datasets, although some strain-specific differences (for example, carnosine) were observed (Extended Data Fig. 15c,d). These findings suggest that metastatic potential reflects the combined influence of multiple metabolites rather than any single nutrient.
Discussion
A consistent correlation between tissue-specific, single-nutrient levels and dependency on specific metabolic genes was not observed across three breast cancer cell lines. Although extensive data support the notion that nutrient availability influences metabolic behaviour5,46, these findings suggest that metabolic dependencies in cancer are not solely determined by the availability of metabolites synthesized downstream of a given metabolic enzyme. Rather, the data indicate that metabolic dependencies and tissue preference of metastases are shaped by a complex interplay between multiple nutrients in tissues and cell-intrinsic factors that influence tumour metabolic requirements. For example, despite reports of low levels of serine in the brain microenvironment18,19, our measurements indicate that serine levels in CSF are not depleted relative to plasma, although they are lower than in other tissue interstitial fluid. Nevertheless, we observed that serine synthesis via PHGDH is not universally required across cells for brain tumour growth or brain metastasis. One possibility is that different cell lines can differentially salvage products of serine synthesis, influencing their dependence on de novo synthesis. Consistent with this, cancer cells vary in their response to serine starvation even within the same tumour type47. Serine synthesis activity can also be shaped by availability of other nutrients, reflecting a metabolic ‘budget’ in which cells prioritize the uptake of some metabolites to allocate resources towards others47. Moreover, dependency on pathways such as PHGDH may also shift across stages of metastasis: low expression of PHGDH promotes early dissemination, whereas re-expression supports established lesions48. Thus, reliance on serine synthesis or other metabolic pathways reflects a dynamic interaction between environmental supply and intrinsic demands, complicating prediction and targeting of metabolic vulnerabilities. Consistent with this heterogeneity, even among TNBC models, we noted that HCC1806 behaved differently from MDA-MB-231 and EO771 in some conditions.
The observation that auxotrophic cancer cells can grow in tissues where the corresponding nutrient is depleted raises important questions about what factors enable tumour outgrowth in tissues with low levels of the rescue nutrient. Although the levels of metabolites such as arginine and asparagine are lower in sites such as the liver or CSF, they are not entirely absent. Defining whether such concentrations are limiting requires functional data. Regardless, residual nutrient availability is unlikely to fully explain tumour growth, as buffering mechanisms—including upregulation of high-affinity transporters, support from non-cancer cell types in the tumour microenvironment2,49 or scavenging of macromolecules50,51—may collectively reduce demand for the rescue nutrient and sustain proliferation.
Quantification of metabolites available across tissues challenges the conventional view that tumour microenvironments are generally nutrient deprived. Supporting this, tumour interstitial fluid is not depleted of many nutrients23,52. Of note, tumour interstitial fluid is similar to normal kidney interstitial fluid in human patients with renal cell carcinoma, suggesting that tumours adapt to existing environmental conditions rather than reshaping them to suit their needs23. This adaptation is particularly critical in the brain, where the blood–brain barrier restricts nutrient access53. Despite lower amino acid levels, we did not find a universal dependency on amino acid synthesis, indicating that cell-intrinsic metabolic traits and differential ability to salvage protein from the microenvironment probably drive these varied dependencies.
Although heightened synthesis activity measured by labelled glucose incorporation often correlates with pathway dependency, such as for serine or lipid synthesis in the brain12,13,15, our results show that this is not always the case. Bulk tumour measurements may obscure cell-specific metabolism, or genetic knockouts may trigger compensatory adaptations such as nutrient salvage. For example, pancreatic cancer cells increase uridine salvage following DHODH loss54. Thus, tumour metabolic dependencies cannot be inferred solely from synthesis activity.
Despite the heterogeneity in metabolic dependencies that we observed in auxotrophs, we found a strong dependency on GART for breast cancer cells to form tumours in multiple tissues. This robust dependency underscores the importance of purine synthesis in this cancer and is corroborated by evidence that breast cancer can be treated with antifolates55. Of note, we did not observe elevated purine synthesis in brain compared with MFP tumours, which is consistent with two studies showing lower purine synthesis activity in the brain than in other tissues56,57. Although those data were interpreted as a reliance of brain tissue on nucleoside or nucleobase salvage58, synthesis also remains important for tumour growth.
Our use of different models to explore gene–nutrient interactions highlights how methodological approaches can lead to different interpretations of metabolic dependencies. For example, although DHODH was found to be essential for MDA-MB-231 and HCC1806 cells across all experimental approaches, DHODH was required by EO771 cells for brain and MFP tumour growth but was minimally essential for metastasis when introduced into mice via intracardiac injections. Differences in metabolic dependencies between intracardiac and intracranial injections probably reflect the distinct microenvironments and stages of metastasis encountered with each method. We selected intracardiac injection to assess nutrient levels and metastatic outgrowth because it allows for simultaneous evaluation of multiple organ sites, providing a broad view of tissue-specific constraints on metastasis. To distinguish these effects from local tumour growth capacity, we also used direct intracranial and MFP injections, enabling a focused analysis of how metabolic dependencies vary by tissue independent of dissemination. Although no single metastasis model fully recapitulates the clinical distribution of metastases, each has inherent strengths and limitations59 and offer insights into the metabolic requirements of cancer cells for metastatic tumour growth. Studies using alternative systems—such as genetically engineered mouse models—could further build on the findings.
This study only examined breast cancer, and whether the relationship between single-nutrient levels and metabolic requirements for metastasis extends to other cancer types is not known. Moreover, tumours arising from clonal knockout cells necessary to create auxotrophs may not capture intratumour metabolic heterogeneity that could contribute to nutrient sharing among cancer cells within tumours48,60. Nonetheless, this study provides insight into TNBC cell metabolic dependencies and their relationship to tissue nutrient environments, showing that the interplay between levels of multiple microenvironmental nutrients and cell-intrinsic wiring shapes metastatic growth. More broadly, these results illustrate the challenge of defining tumour metabolic vulnerabilities. Despite the extensive scope of this study, many dependencies could not be explained by single nutrients or pathways. Future progress will require approaches that integrate nutrient availability, genetics, stromal interactions and adaptive pathways to predict and ultimately exploit metabolic vulnerabilities in diverse tumour contexts.
Methods
Mice
All experiments conducted in this study were approved by the MIT Committee on Animal Care or the MGH Institutional Animal Care and Use Committee. Female B6 and female NSG mice 6–10 weeks of age were used. Animals were housed at ambient temperature and humidity (18–23 °C, 40–60% humidity) on a 12-h light–12-h dark cycle and co-housed with littermates with ad libitum access to food and water. All experimental groups were age matched, numbered and randomly assigned to treatment or control groups to minimize bias. Randomization was performed using a randomized numbering sequence at the time of injection or treatment. Investigators were not blinded during animal injections or treatments; however, researchers were blinded during data acquisition and quantification. No statistical methods were used to pre-determine sample size. Animal studies were performed once per condition unless otherwise specified in the figure legends. Data were collected from distinct animals, where n represents biologically independent samples. Mice were euthanized when total tumour burden reached 2 cm in diameter or earlier if humane end point criteria (for example, more than 20% weight loss, body condition score of less than 2 or signs of distress) were observed. These institutional limits were not exceeded in any experiment.
Cell lines and culture
Human cell lines used in this study were obtained from the American Type Culture Collection (HCC1806: CRL-2335, from a female patient; MDA-MB-231: HTB-26, from a female patient). Mouse EO771 breast cancer cells were obtained from CH3 BioSystems (94A001, from B6 mice). Cell lines obtained from ATCC and CH3 BioSystems are authenticated by the suppliers; no additional authentication was performed by the authors. Lines were regularly tested for mycoplasma contamination using the MycoAlert PLUS Mycoplasma Detection Kit (LT07-710, Lonza BioSciences). All cells were cultured in a Heracell humidified incubator (Thermo Fisher Scientific) at 37 °C and 5% CO2. Cell lines were routinely maintained in RPMI-1640 (10-040-CV, Corning Life Sciences) supplemented with 10% heat-inactivated fetal bovine serum (10437-028, Gibco), and for cell culture experiments, 10% dialysed fetal bovine serum (26400-044, Gibco) was supplemented.
RPMI-1640 medium lacking serine or arginine was made using a previous method61. In brief, enough of all amino acids and sodium phosphate dibasic (S5136, Sigma-Aldrich) of RPMI-1640 media except for serine or arginine were weighed out to make 25 l of media, then the resulting powders were homogenized using an electric blade coffee grinder (80365, Hamilton Beach) that had been washed with methanol then water. The resulting powders were resuspended in water along with sodium bicarbonate (S5761, Sigma-Aldrich) and RPMI-1640 medium without amino acids and sodium phosphate (R8999-04A, US Biological) to make RPMI-1640 medium lacking serine or arginine. Serine or arginine was reconstituted in water to generate stock solutions and added back as needed to reach 286 µM serine or 1.15 mM arginine, reflecting standard RPMI-1640 concentrations.
DMEM medium lacking serine or arginine was prepared using a base powder formulation lacking all amino acids and pyruvate (D9800-13, US Biological). To this base, glucose, sodium pyruvate and sodium bicarbonate were added to match standard DMEM composition. Amino acids were weighed out and homogenized as described above for RPMI, then dissolved into the reconstituted DMEM base. Serine or arginine was added back as needed to reach 400 µM serine or 398 µM arginine, reflecting standard DMEM concentrations.
Isolation of interstitial fluid, CSF and plasma
Tissue interstitial fluid was collected from mouse tissues using an adapted protocol based on previous published work52. Female NSG or female B6 mice on ad libitum diet 6–9 weeks of age were used for all tissue interstitial fluid isolations. Organs from five mice were combined per interstitial fluid sample, and each pooled sample was treated as an individual data point for the liquid chromatography–mass spectrometry (LC–MS) measurements and analysis. All mice were euthanized at the same time of day to account for any effects of time of day on metabolism, and were euthanized by cervical dislocation to minimize metabolic artefacts associated with CO2 exposure62. Tissues were kept on a chilled metal block on ice throughout the harvest and when ready to pool, were briefly rinsed in ice-cold saline (16005, Azer Scientific), excess liquid removed by blotting on filter paper (28298-020, VWR), then placed in a 50-ml conical vial lined with a 20-μm nylon mesh filter (148134, Spectrum Labs) and 0.25 μl of 0.5 M EDTA, pH 8.0 at the bottom. The tissues were centrifuged at 400g for 10 min at 4 °C. The flow through was collected and centrifuged again at 400g for 10 min at 4 °C before flash freezing and storage in −80 °C until further analysis. Before tissue harvest, plasma from each live mouse was collected by facial cheek bleed into EDTA-coated tubes (41.1395.105, Sarstedt), then centrifuged at 800g for 10 min at 4 °C. Supernatant containing plasma was flash frozen and stored at −80 °C until further analysis. An equal volume of each plasma sample was pooled from each mouse cohort before analysis by LC–MS.
CSF was isolated from the mouse brain as previously described63. In brief, female NSG or female B6 mice were anaesthetized with ketamine (90 mg kg−1) and xylazine (9 mg kg−1) via intramuscular injection, and CSF was collected by gently inserting a sharpened capillary (inner diameter of 0.75 mm and outer diameter of 1.0 mm) in the cisterna magna. Collected fluid was visually monitored for blood contamination. CSF was centrifuged at 800g for 10 min at 4 °C, and the supernatant was flash frozen and stored at −80 °C until further analysis. Following CSF collection, animals were euthanized via cervical dislocation and blood was collected via cardiac puncture and immediately placed in EDTA tubes (41.1395.105, Sarstedt), then centrifuged at 800g for 10 min at 4 °C. Plasma was flash frozen in liquid nitrogen and stored at −80 °C until further analysis.
To evaluate the stability of metabolite levels during sample handling, we performed a time-course experiment in female B6 mice. For plasma, blood was collected via cheek bleed directly into EDTA-coated tubes and either processed immediately as described above or held on ice for 5, 10 or 30 min before processing. For liver interstitial fluid, one liver was collected from each mouse and either processed immediately or placed on a chilled metal block on ice for 5, 10 or 30 min before interstitial fluid isolation using the standard centrifugation protocol described above. All samples were analysed by LC–MS to quantify time-dependent changes in metabolite levels.
MFP and brain tumour model generation
MDA-MB-231, HCC1806 and EO771 cell lines were transduced with a lentiviral vector expressing Gluc and GFP separated by an internal ribosomal entry site42, and the top 10% of GFP+ cells were isolated by FACS. To generate MFP tumours, mice were first anaesthetized with ketamine (90 mg kg−1) and xylazine (9 mg kg−1) via intraperitoneal injection, and 1 × 105 MDA-MB-231–Gluc or HCC1806–Gluc cells were injected into the MFP in 30 or 50 µl volume of PBS. To generate brain tumours, mice were first anaesthetized with ketamine (90 mg kg−1) and xylazine (9 mg kg−1) via intramuscular injection. Then, 1 × 105 MDA-MB-231–Gluc, 1 × 105 HCC1806–Gluc or 5 × 104 EO771–Gluc cells were diluted in 1 µl PBS and stereotactically injected into the left frontal lobe of the mouse brain. Tumour volumes were assessed 1–2 times per week by mixing 7 µl tail vein blood with 7 µl 0.5 mM EDTA (pH 8.0) and quantifying luminescence using Promega GloMax Plate Reader (Promega) and the substrate coelenterazine (303, NanoLight Technology).
Intracardiac injection and metastasis quantification
MDA-MB-231, HCC1806 and EO771 cell lines were transduced with a lentiviral vector expressing Fluc and GFP12, and infected cells were sorted by FACS using a fixed gate for GFP. For intracardiac injections, 1 × 105 cancer cells were suspended in 100 µl PBS and injected into the left ventricle of mice anaesthetized with inhaled isoflurane using ultrasound guidance64. In vivo metastasis progression was monitored via real-time bioluminescence imaging (BLI) using the IVIS Spectrum Imaging System (PerkinElmer). Mice were anaesthetized with inhaled isoflurane, injected intraperitoneally with D-luciferin (150 mg kg−1), then imaged every 3 min with the auto-exposure setting in supine position for a total time of 15 min. The time at which luminescence reached its maximum was used for total photon flux values (photons per second).
For end point ex vivo tissue BLI quantification, mice were intraperitoneally injected with D-luciferin (150 mg kg−1), anaesthetized with inhaled isoflurane and whole-body BLI was recorded 10 min post-luciferin injection. Following immediate euthanasia by cervical dislocation, tissues were rapidly dissected and imaged with the auto-exposure setting. BLI analysis was conducted using Living Image software (v4.7.2, PerkinElmer).
Generation of metastatic cell lines
Following intracardiac injection of GFP-labelled or Fluc-labelled cancer cells and end point BLI, GFP+ metastases in the brain and/or liver were identified and microdissected using a fluorescent dissecting microscope. Brain metastases were placed directly into 15-ml conical tubes containing 5 ml of digestion buffer (1 mg ml−1 collagenase I (LS004194, Worthington Biochemical), 3 mg ml−1 dispase II (04942078001, Roche) and 0.1 mg ml−1 DNase I (D4527, Sigma) in PBS). Liver tissue bearing GFP+ metastases were placed into digestion buffer and finely minced using sterile surgical tools. Tissues were then digested at 37 °C for 1 h with gentle rotation. After digestion, samples were triturated using a flamed glass Pasteur pipette to further dissociate tissue. EDTA was added to a final concentration of 10 mM, and samples were incubated at room temperature for 5 min. Cell suspensions were filtered through a 70-µm cell strainer into 50-ml conical tubes, centrifuged, washed twice with PBS and resuspended in RPMI medium supplemented with 10% heat-inactivated fetal bovine serum and penicillin–streptomycin. Cells were plated and expanded in culture, and GFP+ populations were subsequently isolated by flow cytometry for downstream analyses.
Petal plot generation
To account for baseline activity and inter-tissue variability in our luciferase assay data, we normalized the readings against control cell lines expressing non-targeting control single guide RNA. This normalization involved dividing the raw data for each gene by the average non-targeting control values corresponding to each tissue type. We used bootstrap methods for statistical reliability, resampling the normalized data to estimate mean value distributions and calculate 95% confidence intervals, thereby capturing the inherent variability in the biological data. Values indicating fold changes above 1, suggesting normal or enhanced cell growth, were capped at 1 to focus on growth dependencies. Data visualization, inspired by Jin et al.12, was achieved through a petal plot (a radial variant of a bar plot), succinctly depicting luciferase activities along with 95% confidence intervals. Replicates within the dataset were averaged to provide a single representative measure per tissue type. The analysis was conducted using R studio (v4.3.1), using the dplyr (v1.1.2) and ggplot2 (v3.4.3) packages for data processing and visualization, the boot package (v2019.6.0) for bootstrap confidence interval calculations, and the reshape2 package (v1.4.4) for data reshaping.
Quantification of metabolite levels in biological fluids
Metabolite quantification in mouse fluid samples was performed as previously described52. In brief, 5 μl of sample or external chemical standard pool (ranging from approximately 5 mM to about 1 μM) was mixed with 45 μl of acetonitrile:methanol:formic acid (75:25:0.1) extraction mix including isotopically labelled internal standards. All solvents used in the extraction mix were high-performance LC grade (HPLC). Samples were vortexed for 15 min at 4 °C, and insoluble material was sedimented by centrifugation at 16,000g for 10 min at 4 °C. Of the soluble polar metabolite extract, 20 µl was taken for LC–MS analysis. Following analysis by LC–MS, metabolite identification was performed with XCalibur 2.2 software (Thermo Fisher Scientific) using a 5 ppm mass accuracy and a 0.5-min retention time window. For metabolite identification, external standard pools were used for assignment of metabolites to peaks at given m/z and retention time (see Supplementary Table 1 for the m/z and retention time for each metabolite analysed). Absolute metabolite concentrations were determined as previously published52.
13C-glucose tracing in brain and MFP tumours
Infusion of [U-13C]-glucose (CLM-1396-1, Cambridge Isotope Laboratories) into tumour-bearing mice was performed as previously described13,65,66. MDA-MB-231-Gluc cells (1 × 105) were intracranially injected into the brain or injected into the MFP of mice and tumours were permitted to grow for 14 or 17 days, respectively. Catheters were surgically implanted into the jugular vein of tumour-bearing animals 3 days before the infusion. Mice were fasted for 4 h before the start of the infusion, then conscious, free-moving animals were infused with [U-13C]-glucose at a constant rate of 0.4 mg min−1 for 10 h. Mice remained fasted throughout the infusion period. Following the 10-h infusion, animals were terminally anaesthetized with an infusion of Fatal Plus. Blood was collected immediately by cardiac puncture, and tumours and non-cancerous tissue were collected within 5 min of death; for brain tumours, GFP+ tumour tissue was isolated from non-GFP+ tissue under a fluorescent dissecting microscope. Tissues were flash frozen using the BioSqueezer (1210, BioSpec Products) and stored at −80 °C for subsequent metabolite extraction and analysis. Blood was placed into EDTA tubes (41.1395.105, Sarstedt) and centrifuged at 800g at 4 °C to separate plasma, which was flash frozen and stored at −80 °C. All isotope labelling experiments in mice were performed at the same time of day.
Snap frozen tissues were ground into powder using a mortar and pestle, then weighed into glass vials (C4010-1 and C4010-60BLK, Thermo Fisher Scientific). Metabolites were extracted in 1.5 ml of dichloromethane:methanol:0.88% KCl (w/v) (8:4:3 v/v/v), vortexed for 15 min at 4 °C and centrifuged at maximum speed for 10 min at 4 °C. Polar metabolites (aqueous fraction) were transferred to Eppendorf tubes, dried under nitrogen gas and stored at −80 °C until further analysis. Before analysis by LC–MS, samples were resuspended in water, vortexed for 10 min, centrifuged at maximum speed for 10 min and supernatant transferred to vials. For plasma, 5 µl of fluid was extracted in 45 µl of 80% methanol containing 500 nM 13C and 15N amino acid standards (MSK-A2-1.2, Cambridge Isotope Laboratory). Samples were vortexed for 15 min, centrifuged at maximum speed for 10 min, and supernatant transferred to vials and analysed by LC–MS. Following analysis by LC–MS, metabolite peak areas were called using XCalibur (v.2.2; Thermo Fisher Scientific) or Compound Discoverer (v.3.3; Thermo Fisher Scientific) software. Ion counts for each metabolite were normalized to the weight of the tissue sample; for plasma samples, total ion counts were reported. Isotope corrections were applied using IsoCorrector (v.3.18)67.
Dietary serine or glycine restriction and metastasis assay
To assess the effect of systemic nutrient availability on metastasis, female NSG mice (6–8 weeks of age) were fed either a control diet (TD.110839, Envigo) or a high-serine diet (TD.160575, Envigo). Mice were maintained on the assigned diets for 2 weeks before intracardiac injection of GFP-lablled or Fluc-labelled HCC1806 cells. Mice were maintained on each diet until end point. For metabolomics analysis of tissues of mice on the diets, separate mice were placed on the two diets for 2 weeks at which point plasma and tissues were collected and analysed by LC–MS. Mice were randomly assigned to diets, and all tissue and plasma samples were collected at the same time of day to control for circadian effects. Before tissue harvest, plasma from each live mouse was collected by facial cheek bleed into EDTA-coated tubes (41.1395.105, Sarstedt), then centrifuged at 800g for 10 min at 4 °C. Supernatant containing plasma was flash frozen and stored at −80 °C until further analysis. Tissues were rinsed in cold saline, blotted dry, then snap frozen and stored at −80 °C. Frozen tissues were ground into powder using a mortar and pestle and metabolites were extracted in 50 µl extraction mix (80% methanol + containing 500 nM 13C and 15N amino acid standards (MSK-A2-1.2, Cambridge Isotope Laboratory)) per mg of tissue. Samples were vortexed for 15 min, centrifuged at maximum speed for 10 min, dried down under nitrogen gas and then stored at −80 °C. For analysis, samples were resuspended in HPLC-grade water and analysed by LC–MS.
For plasma, 5 µl of fluid was extracted in 45 µl of 80% methanol containing 500 nM 13C and 15N amino acid standards (MSK-A2-1.2, Cambridge Isotope Laboratory). Samples were analysed by quantitative metabolomics as described below. Samples were vortexed for 15 min, centrifuged at maximum speed for 10 min, and supernatant was transferred to vials and analysed by LC–MS. Following analysis by LC–MS, metabolite peak areas were called using XCalibur (v.2.2; Thermo Fisher Scientific).
In vitro [U-¹³C]-glucose tracing
MDA-MB-231 control or DHODH-knockout cells were seeded in six-well plates in RPMI supplemented with 100 µM uridine. After attachment, cells were washed and incubated for 24 h in RPMI (R8999, US Biological) containing 10 mM [U-13C6]-glucose (CLM-1396-1, Cambridge Isotope Laboratories) and 100 µM uridine. Following incubation, cells were washed, extracted in 80% methanol containing 500 nM 13C and 15N amino acid standards (MSK-A2-1.2, Cambridge Isotope Laboratory), vortexed, clarified by centrifugation, dried and stored at −80 °C until analysis by LC–MS. Replicate plates were counted in parallel, and metabolite peak areas were normalized to internal standards and to cell number.
LC–MS analysis
Metabolite profiling was conducted on a QExactive benchtop orbitrap mass spectrometer equipped with an Ion Max source and a Heated Electrospray Ionization (HESI-II) probe, which was coupled to a Dionex UltiMate 3000 HPLC system (Thermo Fisher Scientific). External mass calibration was performed using the standard calibration mixture every 7 days. An additional custom mass calibration was performed weekly alongside standard mass calibrations to calibrate the lower end of the spectrum (m/z 70–1,050 positive mode and m/z 60–900 negative mode) using the standard calibration mixtures spiked with glycine (positive mode) and aspartate (negative mode). Of each sample, 2 µl was injected onto a SeQuant ZIC-pHILIC 150 × 2.1 mm analytical column equipped with a 2.1 × 20 mm guard column (both particle size of 5 mm; EMD Millipore). Buffer A was 20 mM ammonium carbonate and 0.1% ammonium hydroxide; and buffer B was acetonitrile. The column oven and autosampler tray were held at 25 °C and 4 °C, respectively. The chromatographic gradient was run at a flow rate of 0.150 ml min−1 as follows: for 0–20 min, linear gradient from 80% to 20% B; for 20–20.5 min, linear gradient from 20% to 80% B; and from 20.5–28 min, hold at 80% B. The mass spectrometer was operated in full-scan, polarity-switching mode, with the spray voltage set to 3.0 kV, the heated capillary held at 275 °C and the HESI probe held at 350 °C. The sheath gas flow was set to 40 units, the auxiliary gas flow was set to 15 units and the sweep gas flow was set to 1 unit. MS data acquisition was performed in a range of m/z = 70–1,000, with the resolution set at 70,000, the automatic gain control target at 1 × 106 and the maximum injection time at 20 ms.
Cell proliferation
For assessment of proliferation by continuous live-cell imaging, cells were trypsinized, pelleted and washed with PBS, counted, resuspended in the appropriate medium and then plated directly into clear 96-well plates. Cells were then treated with or without the relevant rescue metabolite, and plates were placed into an IncuCyte Live Cell Analysis Imaging System S3 (Sartorius) in a humidified incubator at 37 °C and 5% CO2. Images were acquired every 3 h using the 10x objective. Cell confluence was determined from a mask generated by the standard settings in the IncuCyte Zoom Analysis S3 v2018B software. Cells were plated in either RPMI or DMEM medium lacking the metabolite corresponding to their engineered auxotrophy: asparagine (ASNS knockout), arginine (ASS1 knockout), proline (PYCR knockout) or serine (PHGDH knockout). DHODH-knockout and GART-knockout experiments were performed in complete RPMI medium. The metabolite concentrations used for rescues unless otherwise stated were: 1.15 mM arginine, 379 µM asparagine, 1 mM citrulline, 100 µM hypoxanthine, 174 µM proline, 286 µM serine and 100 µM uridine.
Western blot
Cells were washed once in PBS and scraped into RIPA lysis buffer containing protease and phosphatase inhibitors (5871, Cell Signaling Technology), rocked for 15 min at 4 °C, and insoluble material was sedimented by centrifugation at 21,000g for 10 min at 4 °C. Protein concentration was determined via Bradford Assay, and samples were mixed with LDS sample buffer (NP0008, Thermo Fisher Scientific) and 2.5% 2-mercaptoethanol and then incubated at 95 °C for 5 min. Proteins were resolved by SDS–PAGE and then transferred onto nitrocellulose membranes using a wet tank transfer system (Bio-Rad). Membranes were blocked in 5% milk in TBST and probed overnight at 4 °C with the appropriate antibody diluted in 5% BSA in TBST. For detection, membranes were incubated with horseradish peroxidase-linked anti-rabbit (1:5,000; 7074, Cell Signaling Technology) or anti-mouse (1:5,000; 7076, Cell Signaling Technology) IgG secondary antibody diluted in 5% milk in TBST, and chemiluminescent signal was detected using a digital imager (LAS 4000, GE Healthcare). The primary antibodies used were: ASNS (1:1,000; 14681-1-AP, Proteintech), ASS1 (1:1,000; clone D4O4B, 70720, Cell Signaling Technology), PHGDH (1:1,000; HPA021241, Sigma-Aldrich), PYCR1 (1:1,000; 13108-1-AP, Proteintech), PYCR2 (1:1,000; 17146-1-AP, Proteintech), PYCRL (1:1,000; clone OTI1B12, NBP2-03337, Novus Biologicals), PYCRL (1:1,000; A17763, ABclonal), DHODH (1:100; clone E-8, sc-166348, Santa Cruz Biotechnology), GART (1:1,000; 13659-1-AP, Proteintech), β-actin (1:5,000; clone D6A8, 8457, Cell Signaling Technology) and vinculin (1:1,000; clone E1E9V, 13901, Cell Signaling Technology). In cases in which re-probing of the same membrane was required, the membrane was incubated with hydrogen peroxide solution 30% (w/w in water; H1009, Sigma-Aldrich) for 1 h at 37 °C to inactivate horseradish peroxidase. Western blots were quantified by densitometry using Fiji (v2.0.0)68; data are normalized to the loading control from the same gel. Uncropped, unprocessed scans for all key blots are provided in Supplementary Fig. 1.
Lentivirus generation
LentiX cells were transfected with lentiviral packaging plasmids pMD2.G (12259, Addgene) pMDLg/pRRE (12251, Addgene) and pRSV-Rev (12253, Addgene) using TransIT-293 Transfection Reagent (MIR 2700, Mirus). Forty-eight hours post-transfection, medium was collected and filtered through a 0.45-µm low-protein-binding membrane. Lentivirus-containing supernatants were used immediately or stored at −80 °C. Cell lines were transduced with virus and 10 µg ml−1 polybrene for 24 h.
CRISPR–Cas9-mediated gene knockout
Cas9-expressing cell lines were generated by infecting parental cell lines with viral particles containing lentiCas9/blast (52962, Addgene) followed by selection with 10 µg ml−1 blasticidin. Two guide RNAs (gRNAs) per gene of interest were designed by CRISPick (https://portals.broadinstitute.org/gppx/crispick/public) and cloned into lentiGuide-Puro (52963, Addgene). Primer sequences used in this study are listed in Supplementary Table 3. Cas9+ cell lines were transduced with lentiGuide-Puro-gRNA viral particles, then selected with 1 µg ml−1 puromycin. Single cells were plated into individual wells of 96-well plates in RPMI medium; following expansion, western blotting confirmed complete knockout. To generate and maintain DHODH-knockout or GART-knockout cells, 100 µM uridine or 100 µM hypoxanthine were supplemented in RPMI medium, respectively. For the generation of PYCR1, PYCR2, PYCR3 (PYCRL) triple knockout MDA-MB-231 or HCC1806 cells, two gRNAs targeting both PYCR1 (exons 4 and 5) and PYCR2 (exons 2 and 3), and two gRNAs targeting PYCR3 (exon 3), were used.
To knock out Pycr1, Pycr2 and Pycr3 (PycrI) in EO771, we used ribonucleoprotein (RNP) complexes with Cas9–GFP and Cas9–RFP. The Cas9 protein and gRNAs specific to each target gene were ordered from Integrated DNA Technologies (IDT) as RNA oligonucleotides and used to assemble RNP complexes. EO771 cells were nucleofected with the RNP complexes using a 4D-Nucleofector (Lonza Bioscience) and the SF Cell Line 4D-Nucleofector X Kit L (V4XC2024, Lonza Walkersville) with Pulse Code CM-150. Post-nucleofection, cells were cultured and monitored for GFP and RFP expression. Single-cell sorting was performed 24 h after nucleofection to isolate successfully nucleofected cells.
DepMap and metastatic potential data
Gene expression, CRISPR-gene dependency and metastatic potential values used for correlation analyses were sourced from the Broad Institute DepMap portal website, 24Q2 release (https://depmap.org/portal). Metastatic potential values were derived from Jin et al.12, which used a standardized in vivo barcoding approach to assess tissue-specific metastatic behaviour across a large panel of human cancer cell lines.
Statistics and reproducibility
All in vitro experiments were independently repeated at least twice with similar results. Mouse experiments were performed once for each condition unless otherwise specified. All n values represent biologically independent samples. Sample sizes, reproducibility and statistical tests used for each figure are denoted in the figure legends or Source Data. All graphs were generated using GraphPad Prism 10.3.1 (GraphPad Software). Relative fold changes were determined by normalizing every data point to the mean value of the control group.
After determining the concentration of each metabolite in each plasma or tissue interstitial fluid sample, all multivariate statistical analysis on the data was performed using Metaboanalyst (v6.0)69. Metabolite concentrations were log transformed, and metabolites that contained greater than 50% missing values were removed before analysis. For the remainder of the metabolites, missing values were replaced using one-fifth of the lowest positive value. Hierarchical clustering was performed with Euclidean distance measurement and clustering by the Ward algorithm. Univariate analysis was performed comparing metabolite levels between groups in which metabolite differences of interest were defined by a fold change greater than 2 and significance as a raw P value of less than 0.05 assuming unequal group variance. Using the fold change and adjusted P value cut-offs, the number of differentially expressed metabolites was determined.
Illustrations
Experimental schema and illustrative models were generated using BioRender.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Datasets can be found in Supplementary Tables 1–3. The metabolomics data have been deposited to MetaboLights70 with the study identifiers: MTBLS13151 (https://www.ebi.ac.uk/metabolights/MTBLS13151), MTBLS13152 (https://www.ebi.ac.uk/metabolights/MTBLS13152), MTBLS13164 (https://www.ebi.ac.uk/metabolights/MTBLS13164) and MTBLS13165 (https://www.ebi.ac.uk/metabolights/MTBLS13165). Unprocessed western blots can be found in Supplementary Fig. 1. Any additional information required to reanalyse the data reported in this paper is available from the lead contact, M.G.V.H. (mvh@mit.edu). Source data are provided with this paper.
References
Boire, A. et al. Why do patients with cancer die? Nat. Rev. Cancer 24, 578–589 (2024).
Lyssiotis, C. A. & Kimmelman, A. C. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 27, 863–875 (2017).
Elia, I., Doglioni, G. & Fendt, S.-M. Metabolic hallmarks of metastasis formation. Trends Cell Biol. 28, 673–684 (2018).
Teoh, S. T. & Lunt, S. Y. Metabolism in cancer metastasis: bioenergetics, biosynthesis, and beyond. Wiley Interdiscip. Rev. Syst. Biol. Med. https://doi.org/10.1002/wsbm.1406 (2018).
Faubert, B., Solmonson, A. & DeBerardinis, R. J. Metabolic reprogramming and cancer progression. Science 368, eaaw5473 (2020).
Bergers, G. & Fendt, S.-M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 21, 162–180 (2021).
Mosier, J. A., Schwager, S. C., Boyajian, D. A. & Reinhart-King, C. A. Cancer cell metabolic plasticity in migration and metastasis. Clin. Exp. Metastasis 38, 343–359 (2021).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Tardito, S. & MacKay, C. Rethinking our approach to cancer metabolism to deliver patient benefit. Br. J. Cancer 129, 406–415 (2023).
Sullivan, M. R. & Vander Heiden, M. G. Determinants of nutrient limitation in cancer. Crit. Rev. Biochem. Mol. Biol. https://doi.org/10.1080/10409238.2019.1611733 (2019).
Garcia-Bermudez, J., Williams, R. T., Guarecuco, R. & Birsoy, K. Targeting extracellular nutrient dependencies of cancer cells. Mol. Metab. 33, 67–82 (2020).
Jin, X. et al. A metastasis map of human cancer cell lines. Nature 588, 331–336 (2020).
Ferraro, G. B. et al. Fatty acid synthesis is required for breast cancer brain metastasis. Nat. Cancer 2, 414–428 (2021).
Savino, A. M. et al. Metabolic adaptation of acute lymphoblastic leukemia to the central nervous system microenvironment is dependent on stearoyl CoA desaturase. Nat. Cancer 1, 998–1009 (2020).
Ngo, B. et al. Limited environmental serine and glycine confer brain metastasis sensitivity to PHGDH inhibition. Cancer Discov. 10, 1352–1373 (2020).
Elia, I. et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 568, 117–121 (2019).
Knott, S. R. V. et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 554, 378–381 (2018).
Kruse, T., Reiber, H. & Neuhoff, V. Amino acid transport across the human blood-CSF barrier. An evaluation graph for amino acid concentrations in cerebrospinal fluid. J. Neurol. Sci. 70, 129–138 (1985).
Dolgodilina, E. et al. Brain interstitial fluid glutamine homeostasis is controlled by blood-brain barrier SLC7A5/LAT1 amino acid transporter. J. Cereb. Blood Flow Metab. 36, 1929–1941 (2016).
Jang, C. et al. Metabolite exchange between mammalian organs quantified in pigs. Cell Metab. 30, 594–606.e3 (2019).
Rui, L. Energy metabolism in the liver. Comp. Physiol. 4, 177–197 (2014).
Morris, S. M. Regulation of enzymes of the urea cycle and arginine metabolism. Annu. Rev. Nutr. 22, 87–105 (2002).
Abbott, K. L. et al. Metabolite profiling of human renal cell carcinoma reveals tissue-origin dominance in nutrient availability. eLife 13, RP95652 (2024).
Bifari, F. & Nisoli, E. Branched-chain amino acids differently modulate catabolic and anabolic states in mammals: a pharmacological point of view. Br. J. Pharmacol. 174, 1366–1377 (2017).
Panda, S. Circadian physiology of metabolism. Science 354, 1008–1015 (2016).
Lien, E. C. & Vander Heiden, M. G. A framework for examining how diet impacts tumour metabolism. Nat. Rev. Cancer 19, 651–661 (2019).
Le Naour, A., Rossary, A. & Vasson, M.-P. EO771, is it a well-characterized cell line for mouse mammary cancer model? Limit and uncertainty. Cancer Med. 9, 8074–8085 (2020).
Hettmer, S. et al. Functional genomic screening reveals asparagine dependence as a metabolic vulnerability in sarcoma. eLife 4, e09436 (2015).
Krall, A. S., Xu, S., Graeber, T. G., Braas, D. & Christofk, H. R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 7, 11457 (2016).
Delage, B. et al. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int. J. Cancer 126, 2762–2772 (2010).
Crump, N. T. et al. Chromatin accessibility governs the differential response of cancer and T cells to arginine starvation. Cell Rep. 35, 109101 (2021).
Possemato, R. et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350 (2011).
D’Aniello, C., Patriarca, E. J., Phang, J. M. & Minchiotti, G. Proline metabolism in tumor growth and metastatic progression. Front. Oncol. 10, 776 (2020).
Sykes, D. B. et al. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell 167, 171–186.e15 (2016).
Do, B. T. et al. Nucleotide depletion promotes cell fate transitions by inducing DNA replication stress. Dev. Cell https://doi.org/10.1016/j.devcel.2024.05.010 (2024).
Soflaee, M. H. et al. Purine nucleotide depletion prompts cell migration by stimulating the serine synthesis pathway. Nat. Commun. 13, 2698 (2022).
Qiu, F. et al. Arginine starvation impairs mitochondrial respiratory function in ASS1-deficient breast cancer cells. Sci. Signal. 7, ra31 (2014).
Carroll, B. et al. Control of TSC2-Rheb signaling axis by arginine regulates mTORC1 activity. eLife 5, e11058 (2016).
Welin, M. et al. Structural studies of tri-functional human GART. Nucleic Acids Res. 38, 7308–7319 (2010).
Di Micco, R. et al. Rare sites of breast cancer metastasis: a review. Transl. Cancer Res. 8, S518–S552 (2019).
Sivanand, S. et al. Cancer tissue of origin constrains the growth and metabolism of metastases. Nat. Metab. 6, 1668–1681 (2024).
Wurdinger, T. et al. A secreted luciferase for ex vivo monitoring of in vivo processes. Nat. Methods 5, 171–173 (2008).
Berti, V., Mosconi, L. & Pupi, A. Brain: normal variations and benign findings in fluorodeoxyglucose-PET/computed tomography imaging. PET Clin. 9, 129–140 (2014).
Sullivan, M. R. et al. Increased serine synthesis provides an advantage for tumors arising in tissues where serine levels are limiting. Cell Metab. 29, 1410–1421.e4 (2019).
Maddocks, O. D. K. et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542–546 (2013).
Muir, A. & Vander Heiden, M. G. The nutrient environment affects therapy. Science 360, 962–963 (2018).
Chang, S. M. et al. Cancer cells differentially modulate mitochondrial respiration to alter redox state and enable biomass synthesis in nutrient-limited environments. eLife https://doi.org/10.7554/eLife.107123.1 (2025).
Rossi, M. et al. PHGDH heterogeneity potentiates cancer cell dissemination and metastasis. Nature 605, 747–753 (2022).
Kimmelman, A. C. & Sherman, M. H. The role of stroma in cancer metabolism. Cold Spring Harb. Perspect. Med. 14, a041540 (2024).
Garcia-Bermudez, J. et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 20, 775–781 (2018).
Recouvreux, M. V. & Commisso, C. Macropinocytosis: a metabolic adaptation to nutrient stress in cancer. Front. Endocrinol. 8, 261 (2017).
Sullivan, M. R. et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 8, e44235 (2019).
Daneman, R. & Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 7, a020412 (2015).
Mullen, N. J. et al. ENT1 blockade by CNX-774 overcomes resistance to DHODH inhibition in pancreatic cancer. Cancer Lett. 552, 215981 (2023).
Gonen, N. & Assaraf, Y. G. Antifolates in cancer therapy: structure, activity and mechanisms of drug resistance. Drug Resist. Update 15, 183–210 (2012).
Xu, X. et al. One-carbon unit supplementation fuels purine synthesis in tumor-infiltrating T cells and augments checkpoint blockade. Cell Chem. Biol. 31, 932–943.e8 (2024).
Tran, D. H. et al. De novo and salvage purine synthesis pathways across tissues and tumors. Cell https://doi.org/10.1016/j.cell.2024.05.011 (2024).
Ipata, P. L., Camici, M., Micheli, V. & Tozz, M. G. Metabolic network of nucleosides in the brain. Curr. Top. Med. Chem. 11, 909–922 (2011).
Giacobbe, A. & Abate-Shen, C. Modeling metastasis in mice: a closer look. Trends Cancer 7, 916–929 (2021).
Demicco, M., Liu, X.-Z., Leithner, K. & Fendt, S.-M. Metabolic heterogeneity in cancer. Nat. Metab. 6, 18–38 (2024).
Muir, A. et al. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. eLife 6, e27713 (2017).
Brooks, S. P. J., Lampi, B. J. & Bihun, C. G. The influence of euthanasia methods on rat liver metabolism. Contemp. Top. Lab. Anim. Sci. 38, 19–24 (1999).
Lim, N. K.-H. et al. An improved method for collection of cerebrospinal fluid from anesthetized mice. J. Vis. Exp. https://doi.org/10.3791/56774 (2018).
Henning, N., Roberts, F., Fowler, C. & Spanoudaki, V. Ultrasound guided injection into the left ventricle of the mouse. protocols.io https://doi.org/10.21203/rs.3.pex-2336/v2 (2024).
Davidson, S. M. et al. Environment impacts the metabolic dependencies of ras-driven non-small cell lung cancer. Cell Metab. 23, 517–528 (2016).
Lau, A. N. et al. Dissecting cell-type-specific metabolism in pancreatic ductal adenocarcinoma. eLife 9, e56782 (2020).
Heinrich, P. et al. Correcting for natural isotope abundance and tracer impurity in MS-, MS/MS- and high-resolution-multiple-tracer-data from stable isotope labeling experiments with IsoCorrectoR. Sci. Rep. 8, 17910 (2018).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Pang, Z. et al. MetaboAnalyst 6.0: towards a unified platform for metabolomics data processing, analysis and interpretation. Nucleic Acids Res. 52, W398–W406 (2024).
Yurekten, O. et al. MetaboLights: open data repository for metabolomics. Nucleic Acids Res. 52, D640–D646 (2024).
Acknowledgements
We thank members of the Vander Heiden laboratory for helpful discussions; the Koch Institute’s Robert A. Swanson (1969) Biotechnology Center for technical support, specifically the Preclinical Imaging & Testing, Flow Cytometry, Barbara K. Ostrom (1978) Integrated Genomics and Bioinformatics, and the Hope Babette Tang (1983) Histology facilities; the MIT Division of Comparative Medicine staff and the Massachusetts General Hospital Center for Comparative Medicine staff for help with colony maintenance and animal care; and C. S. Nabel for comments on the manuscript. This work was supported in part by R01CA259253 to R.K.J. and M.G.V.H., as well as the Koch Institute Cancer Center Support grant P30CA014051 and the Koch Institute–Dana-Farber Harvard Cancer Center Bridge Project. K.L.A. was supported by the National Science Foundation (DGE-1122374) and US National Institutes of Health (NIH; F31CA271787 and T32GM007287). S. Subudhi was supported by the MGH ECoR FMD fellowship grant (2022A018897). R.F. was supported by the Knut and Alice Wallenberg Foundation (KAW 2019.0581). Y.G. was supported by a postdoctoral fellowship from the Ludwig Center at MIT’s Koch Institute for Integrative Cancer Research. S.E.H. acknowledges support from the Marietta Blau-Grant by the Austrian Agency for Education and Internationalisation. S. Sivanand acknowledges support from the Damon Runyon Cancer Research Foundation (DRG-2367-19). K.M.E. was supported by a Boehringer Ingelheim Fonds MD fellowship. R.F., L.M.R. and G.M.C. were supported by the Aging and Longevity-Related Research Fund and EGL Charitable Foundation. A.A. received support as a Howard Hughes Medical Institute Medical Research Fellow. X.J. acknowledges support from the Key R&D Program of Zhejiang (2024SSYS0034). R.K.J. was supported by grants from the NIH (U01CA224348, R01CA208205, R01NS118929 and U01CA261842), the Ludwig Cancer Center at Harvard, the Nile Albright Research Foundation, the National Foundation for Cancer Research and the Jane’s Trust Foundation. M.G.V.H. acknowledges support from the MIT Center for Precision Cancer Medicine, the Ludwig Center at MIT and the NIH (R35CA242379).
Author information
Authors and Affiliations
Contributions
K.L.A., S. Subudhi, R.F., A.A., X.J., R.K.J. and M.G.V.H. conceived the project. K.L.A. performed metabolite profiling with assistance from S. Subudhi, Y.G., D.L.R., A.S.-Z., E.H.R., S. Sivanand, A.M.B., M.W., T.K. and F.G. K.L.A., R.F., S.C.S., D.L.R., S.E.H., M.W., J.A.H. and L.M.R. generated and validated the nutrient auxotroph cell lines. K.L.A., S.C.S., D.L.R. and S.E.H. performed the proliferation experiments with assistance from M.D. and M.A.N. K.L.A. performed the intracardiac experiments with assistance from S. Subudhi, Y.G., E.H.R. and N.H. K.L.A., S. Subudhi, S.C.S., A.S.K. and G.B.F. performed the orthotopic implantation experiments. K.L.A. and Y.G. performed in vivo isotope tracing with assistance from S. Subudhi, M.W. and T.K. K.L.A. performed in vitro isotope tracing with assistance from S.E.T., M.W. and T.K. K.L.A., S. Subudhi, R.F., M.B.M. and B.T.D. conducted the bioinformatic and statistical analyses. K.L.A. performed the in vivo diet experiment with assistance from K.M.E., M.W. and T.K. V.S., F.J.S.-R. and X.J. provided input on methodology and study design. G.M.C., R.K.J. and M.G.V.H. supervised the study and acquired funding. K.L.A. assembled the paper and prepared the figures. K.L.A. and M.G.V.H. wrote the manuscript with input from all authors. Y.G., S.C.S. and M.B.M. contributed equally as co-second authors.
Corresponding authors
Ethics declarations
Competing interests
R.F. consulted for Lime Therapeutics during this study, unrelated to the work presented. G.M.C. is a co-founder of Editas Medicine and has other financial interests listed in Supplementary Table 4. R.K.J. received consultant or scientific advisory board fees from DynamiCure, SPARC and SynDevRx; owns equity in Accurius, Enlight and SynDevRx; served on the Board of Trustees of Tekla Healthcare Investors, Tekla Life Sciences Investors, Tekla Healthcare Opportunities Fund and Tekla World Healthcare Fund; and received research grants from Boehringer Ingelheim and Sanofi; no funding or reagents from these organizations were used in the study. M.G.V.H. discloses that he is or was a scientific advisor for Agios Pharmaceuticals, iTeos Therapeutics, Sage Therapeutics, Pretzel Therapeutics, Lime Therapeutics, Faeth Therapeutics, Droia Ventures, MPM Capital and Auron Therapeutics. All other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Ralph DeBerardinis, Tara Teslaa and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Metabolite concentrations in mouse plasma, CSF, and tissue interstitial fluid.
a-j, Heatmaps of metabolite concentrations in plasma, cerebrospinal fluid (CSF), and the indicated tissue interstitial fluids (IF) from female NSG mice measured by LC/MS. Metabolites are grouped by pathway; scale bars indicate concentration ranges. Data represent n = 10 (plasma, liver IF, lung IF, pancreas IF), n = 6 (kidney IF, MFP IF), or n = 4 (CSF) biologically independent samples. Metabolite measurements were performed twice for plasma, liver, lung and pancreas IF samples and once for the remaining tissues; data from repeated measurements were pooled for analysis. Metabolite concentrations below the limit of detection are shown in grey and labeled “nd” (not determined). k, Scatter plots comparing LC/MS metabolite concentrations (µM) in plasma or tissue IF from female NSG mice across two independent experiments to assess reproducibility. Data represent mean values from n = 4 (experiment 1) and n = 6 (experiment 2) biologically independent samples. Pearson correlation coefficients (r) and P values from two-sided tests, and the number of metabolites measured, are indicated in each panel.
Extended Data Fig. 2 Analysis of post-collection metabolite stability in mouse plasma and liver interstitial fluid.
a, Heatmap showing log2 fold change in metabolite levels over time relative to t = 0 in plasma and liver interstitial fluid (IF) samples incubated on ice for up to 30 min. Each value represents the mean of n = 4 biologically independent samples per time point from female C57BL/6 J (B6) mice. Experiments were performed once. b-c, Volcano plots of metabolites altered at 5, 10, or 30 min post-collection in plasma (b) or liver IF (c). Significance was defined as fold change ≥ 2 with P < 0.05 by two-sided Welch’s t-test with unequal variance. d, Bar plot showing the percentage of significantly altered metabolites at each time point in (b-c). e, Concentrations of hypoxanthine in plasma and liver IF over time. Data are mean ± SEM; n = 4 biologically independent samples per time point. Statistical analysis was performed using a Kruskal-Wallis test with Dunn’s multiple comparisons (two-sided). f, Scatter plots comparing plasma metabolite concentrations from cardiac puncture versus cheek bleed in female B6 and NSG mice. For B6, n = 7 (cardiac puncture) and n = 5 (cheek bleed); for NSG, n = 4 (cardiac puncture) and n = 6 (cheek bleed). Experiments were performed once. Each point represents the mean concentration of one metabolite across the indicated biological replicates. Pearson correlation coefficients (r) and P values from two-sided tests are shown in the panels. g, Volcano plots of plasma metabolite differences between collection methods in female B6 and NSG mice. Significance was defined as fold change ≥ 2 with P < 0.05 by two-sided Welch’s t-test with unequal variance. Sample sizes are as in (f).
Extended Data Fig. 3 Comparison of tissue metabolite levels between C57BL/6 J and NSG mice.
a, Scatter plots comparing metabolite concentrations across plasma, CSF and tissue interstitial fluid (IF) from female C57BL/6 J (B6) and female NSG mice. For B6 mice: n = 15 (CSF) and n = 5 (plasma, all tissue IF). For NSG mice: n = 10 (plasma, liver IF, lung IF, pancreas IF), n = 6 (kidney IF, MFP IF), and n = 4 (CSF). Metabolite measurements for B6 mice were performed twice for CSF and once for plasma and all tissue IF samples; data from repeated measurements were pooled for analysis. Metabolite measurements for NSG mice were performed twice for plasma, liver IF, lung IF and pancreas IF and once for kidney IF, MFP IF and CSF; data from repeated measurements were pooled for analysis. Each point represents the mean concentration of one metabolite in the indicated tissue. The number of metabolites measured is indicated in each panel. Pearson correlation coefficients (r) and P values from two-sided tests are shown in the panels. b, Volcano plots of metabolites differing between female B6 and NSG mice in plasma, CSF, or tissue IF. Significance was defined as fold change ≥ 2 with P < 0.05 by two-sided Welch’s t-test with unequal variance. Sample sizes are as in (a). 2HB, 2-hydroxybutyrate; 2HG, 2-hydroxyglutarate; GAA, guanidinoacetic acid.
Extended Data Fig. 4 Nutrient levels in plasma, CSF, and tissue interstitial fluid from C57BL/6 J mice.
a-b, Principal component analysis (PCA) (a) and hierarchical clustering (b) of metabolites measured in plasma, cerebrospinal fluid (CSF), and tissue interstitial fluid (IF) from female C57BL/6 J (B6) mice. Data represent n = 15 (CSF) or n = 5 (plasma and all tissue IF) biologically independent samples. Metabolite measurements for B6 mice were performed twice for CSF and once for plasma and all tissue IF samples; data from repeated measurements were pooled for analysis. Columns of the heatmap were z-score normalized. c, Bar plot showing the number of metabolites with significantly lower (depleted) or higher (elevated) levels in IF or CSF relative to plasma. Significance was determined by Welch’s t-test (two-sided, unequal variance) with fold change >2 and P < 0.05. d, Loadings plot presenting the contribution of individual metabolite classes to the PCA components in (a). Colors indicate pathway assignment. e-l, Log2 fold change of selected metabolites in tissue IF or CSF relative to plasma in female B6 or NSG mice. For B6: n = 15 (CSF) or n = 5 (plasma and all tissue IF). For NSG: n = 10 (plasma, liver IF, lung IF, pancreas IF), n = 6 (kidney IF, MFP IF), or n = 4 (CSF). Data are mean ± SEM. m-n, Log2 fold change in amino acid (m) or nucleotide (n) concentrations between paired CSF and plasma samples from B6 (n = 14) or NSG (n = 4) mice. Data are mean ± SEM. Metabolite measurements for B6 mice were performed twice, and data from repeated measurements were pooled for analysis. Metabolite measurements for NSG mice were performed once. Statistical analysis by one-sample Student’s t-test (two-sided); exact P values provided in Source Data (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Extended Data Fig. 5 Validation of amino acid and nucleotide synthesis gene knockouts in breast cancer cells.
a, Schematic of amino acid and nucleotide synthesis pathways, highlighting enzymes targeted to generate auxotrophs. Genes knocked out in breast cancer cells are shown in red; relevant metabolites synthesized are shown in blue. Asp: aspartate; DHO: dihydroorotate; Glu: glutamate; Gln: glutamine; IMP: inosine monophosphate; P5C: 1-pyrroline-5-carboxylic acid; PRPP: phosphoribosyl diphosphate; UMP: uridine monophosphate. Created in BioRender. Abbott, K. (2025) https://BioRender.com/lz9ywec. b, Western blot analysis of parental MDA-MB-231, EO771, and HCC1806 breast cancer cells for expression of the indicated proteins. c, Quantification of western blots in (b). Signal intensity of each protein was normalized to vinculin loading control. Data are mean ± SD; n = 3 biologically independent samples. Statistical analysis was performed using one-way ANOVA with Holm–Sidak multiple comparisons test (two-sided). d-i, Western blot analysis of non-targeting control (Ctrl) or knockout (KO) auxotroph clonal cells for expression of the indicated proteins in MDA-MB-231, HCC1806, or EO771 cells. PYCR KO represents PYCR1/2/3 triple KO. For gel source data, see Supplementary Fig. 1. Representative blots are shown from one of two independent experiments with similar results.
Extended Data Fig. 6 Validation of auxotroph cells by assessing proliferation with or without rescue metabolites.
a-f, Percent confluence of control (Ctrl) or knockout (KO) clonal cells cultured in medium with or without the relevant rescue metabolites. ASNS KO (a) and PYCR KO (d) were cultured in DMEM; ASS1 KO (b) and PHGDH KO (c) in RPMI lacking arginine or serine, respectively; DHODH KO (e) and GART KO (f) in RPMI. Rescue metabolite concentrations: 1.15 mM arginine (Arg), 379 µM asparagine (Asn), 1 mM citrulline (Cit), 100 µM hypoxanthine (Hx), 174 µM proline (Pro), 286 µM serine (Ser), 100 µM uridine (Urd). Data are mean ± SD; n = 3 biologically independent samples. g, Percent confluence of EO771 cells in standard RPMI (11 mM glucose), RPMI containing 25 mM glucose (DMEM-equivalent), or standard RPMI refreshed every 48 h. Data are mean ± SD; n = 3 biologically independent samples. h-k, Percent confluence of HCC1806 Ctrl or auxotroph KO cells (ASNS KO, ASS1 KO, PHGDH KO, PYCR KO) cultured in DMEM lacking the indicated metabolites. h, ± Asn. i, Without Arg, ± Cit. j, ± Ser. k, ± Pro. Rescue metabolite concentrations: 398 µM Arg, 379 µM Asn, 1 mM Cit, 174 µM Pro, 400 µM Ser. Data are mean ± SD; n = 3 biologically independent samples. Asn, asparagine; Arg, arginine; Cit, citrulline; Ser, serine; Pro, proline; Urd, uridine; Hx, hypoxanthine; PYCR KO, PYCR1/2/3 triple KO. Representative plots are shown from one of two independent experiments with similar results.
Extended Data Fig. 7 Assessment of breast cancer cell metastasis.
a, Whole-body bioluminescence imaging (BLI, photons/sec) of MDA-MB-231-Fluc, HCC1806-Fluc, and EO771-Fluc control cells over time following intracardiac injection into female NSG (MDA-MB-231, HCC1806) or C57BL/6 J (EO771) mice. Data are mean ± SEM; n = 7 (MDA-MB-231, NSG), n = 5 (HCC1806, NSG), and n = 7 (EO771, B6) biologically independent mice. Experiments were performed once. Data are from the same experiments shown in Extended Data Fig. 8c. b, BLI and quantification of metastatic burden in organs of mice injected as in (a). Data are mean ± SEM. c, Representative images of mice and tissues showing tumor burden after intracardiac injection as in (a). Color scales reflect BLI radiance; minimum and maximum values are shown next to each set of images.
Extended Data Fig. 8 Metastasis of breast cancer auxotrophs.
Whole-body bioluminescence imaging (BLI, photons/sec) and tissue-specific BLI of female NSG (MDA-MB-231, HCC1806) or C57BL/6 J (EO771) mice injected with control (Ctrl) or auxotroph cells. Each panel corresponds to one gene knockout: a, DHODH KO; b, GART KO; c, ASNS KO; d, ASS1 KO; e, PHGDH KO; f, PYCR KO (PYCR1/2/3 triple KO). Within each panel, data are shown for all three cell lines, where left plots track whole-body BLI over time and right plots show quantitative analysis of tissue-specific metastatic burden. The final BLI time point corresponds to the experimental endpoint when tissues were harvested. Depletion values indicate fold reduction in metastatic load relative to Ctrl: ○, <2-fold; +, 2–5 fold; ++, 5–10 fold; +++, >10-fold. Two bones, kidneys, or ovaries were analyzed per mouse. Data are mean ± SEM; exact n values are shown in each panel (biologically independent mice). Experiments were performed once. Statistical analysis was performed using two-sided unpaired Welch’s t-test with Holm-Sidak multiple comparisons correction (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). Exact P values are provided in the Source Data. The same control mice were used as follows: MDA-MB-231 in (b, c; d, e); HCC1806 in (a, d, f; b, c, e); EO771 in (a, b; d, e). Groups separated by a semicolon denote distinct cohorts of control mice.
Extended Data Fig. 9 Reproducibility of tissue-specific depletion patterns and validation of HCC1806 auxotrophs.
a-d, Upper panels, whole-body bioluminescence imaging (BLI, photons/sec) of female NSG mice injected with HCC1806-Fluc control (Ctrl) or auxotroph cells. Final BLI values correspond to the endpoint when tissues were harvested. Lower panels, tissue-specific BLI showing metastatic burden of auxotrophs. Depletion values indicate fold reduction versus Ctrl: ○, <2-fold; +, 2–5 fold; ++, 5–10 fold; +++, >10-fold. Data are mean ± SEM; exact n values are shown in each panel (biologically independent mice). Two bones, kidneys, or ovaries were analyzed per mouse. Experiments were performed once. Statistical analysis by two-sided unpaired Welch’s t-test with Holm-Sidak multiple comparisons correction. PYCR KO denotes PYCR1/2/3 triple KO. e, Comparison of ASNS, ASS1, PHGDH, or PYCR KO depletion across two independent experiments. Depletion values represent tissue-specific BLI of KO relative to Ctrl. Experiment 1 corresponds to Extended Data Fig. 8; experiment 2 corresponds to (a-d). Data are mean ± SEM; P values from two-sided unpaired Welch’s t-tests with Benjamini–Krieger–Yekutieli correction for multiple comparisons; ns: not significant. f-g, Western blots confirming loss of ASNS in brain (f) and ASS1 in liver (g) metastases from KO versus Ctrl cells. Cells were isolated from biologically independent metastases in (a-d). For gel source data, see Supplementary Fig. 1. h-k, Proliferation of metastasis-derived auxotrophs ex vivo. h-i, Percent confluence of ASNS KO brain lines in RPMI lacking (-Asn) or supplemented (+ Asn, 379 µM). j-k, Percent confluence of ASS1 KO liver lines in RPMI lacking (-Arg), with (+Cit, 1 mM), or (+Arg, 1.15 mM). Data are mean ± SD; n = 3 biologically independent samples per condition. Each line was derived from an independent metastatic outgrowth from different mice in (a-d). In f-k, representative plots are shown from one of two independent experiments with similar results.
Extended Data Fig. 10 Correlations of auxotroph metastatic potential with tissue metabolite levels.
a-b, Scatter plots correlating tissue metabolite concentrations with MDA-MB-231 dependency on GART for metastatic growth. a, Creatine. b, Glucose-6-phosphate (Glucose-6P). The x-axis shows tissue metabolite concentrations; the y-axis shows log2 fold change of knockout (KO) versus control (Ctrl). Symbols denote metabolite concentrations in specific tissues; brain values reflect CSF. Data are mean ± SEM; n = biologically independent mice as in Extended Data Fig. 8. Pearson correlation coefficients (r) and exact P values (two-sided) are shown in figure panels. c, Volcano plots of Pearson correlation coefficients and P values for correlations between metabolite levels and metastatic potential of auxotroph cell lines following intracardiac injection, calculated as in (a-b). d, Fraction of metabolites showing significant positive (pos) or negative (neg) correlations with metastatic potential of the indicated auxotroph cell lines from (c). e-f, Absolute concentrations of arginine (e) or asparagine (f) in liver IF and CSF replotted from Extended Data Fig. 1a. Data are mean ± SD; n = 6 biologically independent mice (liver IF) and n = 4 biologically independent mice (CSF). Dotted lines mark in vitro IC50 values from (i-j). g-h, Percent confluence of HCC1806 Ctrl or KO cells cultured across a titration series of rescue metabolites. g, Ctrl and ASS1 KO cells in RPMI-Arg with 1 mM citrulline plus increasing arginine. h, Ctrl and ASNS KO cells in DMEM with increasing asparagine. Data are mean ± SD; n = 3 biologically independent samples. i-j, Area under the curve (AUC) from proliferation assays in (g-h), plotted against log10-transformed arginine (i) or asparagine (j) concentrations. Dotted lines indicate IC50 values. Data are mean ± SD; n = 3 biologically independent samples. In g-j, representative plots are shown from one of two independent experiments with similar results.
Extended Data Fig. 11 Tumor growth of auxotroph versus control cells following intracranial or MFP implantations.
Tumor growth and survival were measured in NSG mice implanted with MDA-MB-231-Gluc or HCC1806-Gluc cells, or in C57BL/6 J mice implanted with EO771-Gluc cells. Control (Ctrl) and knockout (KO) auxotroph lines were implanted either in the brain or mammary fat pad (MFP), and tumors were monitored over time by secreted Gaussia luciferase (Gluc). Survival in intracranial injections was assessed in the same cohorts. EO771 experiments were performed for brain tumors only. a-c, DHODH KO; d-f, GART KO; g-i, ASNS KO; j-l, ASS1 KO; m-o, PHGDH KO; p-r, PYCR KO (PYCR1/2/3 triple KO). For each gene set, the three panels show MDA-MB-231, HCC1806, and EO771. Within each panel, subplots are arranged (left, middle, right) as brain tumor growth, brain survival, and MFP tumor growth (MDA-MB-231 and HCC only). Experiments were performed once. Data are mean ± SEM for tumor growth and Kaplan-Meier survival curves for survival analysis; n = biologically independent mice indicated in each panel. Tumor growth was analyzed by two-way ANOVA across timepoints and groups, and survival by log-rank (Mantel–Cox) test. Exact P values (two-sided) are provided in figure panels. The same control mice were used as follows: for MDA-MB-231, brain (a, d, g, j, m, p) and MFP (a, d, g, j, m, p); for HCC1806, brain (b, h, k, q; e, n) and MFP (b, h, k, q; e, n); for EO771, brain (c, i, l; f, o, r). Groups separated by a semicolon denote distinct cohorts of control mice.
Extended Data Fig. 12 Auxotroph dependency in brain tumors; plasma metabolite labeling in mice bearing brain versus MFP tumors.
a, Relative dependency of metabolite auxotroph cells on specific metabolic genes for growth in the brain, expressed as log2 fold change of knockout (KO) relative to control (Ctrl) following intracranial or intracardiac injection. Data are mean ± SEM. Group sizes and instances where the same control mice were used across panels are indicated in Extended Data Figs. 8 and 11. PYCR denotes PYCR1/2/3 triple KO. b, Tumor growth in NSG mice implanted with MDA-MB-231-Gluc cells in the mammary fat pad (MFP) or brain. Tumor burden was monitored over time using secreted Gaussia luciferase (Gluc). Data are mean ± SEM; n = 5 biologically independent mice (MFP) or n = 4 biologically independent mice (brain). At the experimental endpoint, following [U-13C]-glucose infusion, tumors and matched noncancerous tissues were harvested from the same mice (MFP tumor with non-tumor MFP tissue; brain tumor with adjacent brain). c-e, Plasma metabolite analyses from the mice in (b) following [U-13C]-glucose infusion. c, Total ion counts for the indicated metabolites at endpoint (white bars, MFP tumor–bearing mice; black bars, brain tumor-bearing mice). d-e, Isotopolog distributions showing fractional labeling of the indicated metabolites. In (b-e), data are mean ± SEM; n = 5 biologically independent mice (MFP tumor-bearing mice) and n = 4 (brain tumor-bearing mice). Experiments were performed once.
Extended Data Fig. 13 Metabolite labeling in MDA-MB-231 brain and MFP tumors.
a, Normalized peak areas for glucose metabolism metabolites in MFP, MFP tumor, brain, or brain tumor tissues isolated at endpoint from the mice shown in Fig. 4 at endpoint. b, Isotopolog distributions of glucose metabolism metabolites measured by LC/MS in MDA-MB-231 tumors (brain or MFP) and matched noncancerous tissues from NSG mice infused with [U-13C]-glucose. c, Normalized peak areas for amino acids in MFP, MFP tumor, brain, or brain tumor tissues isolated as in (a). d, Isotopolog distributions of amino acids in MDA-MB-231 tumors (brain or MFP) and matched noncancerous tissues from NSG mice infused with [U-13C]-glucose. e, Normalized peak areas for nucleotides in MFP, MFP tumor, brain, or brain tumor tissues isolated from the mice shown in Fig. 4 at endpoint. f, Isotopolog distributions of nucleotides in MDA-MB-231 tumors (brain or MFP) and matched noncancerous tissues from NSG mice infused with [U-13C6]-glucose. In (a-f), data are mean ± SEM; n = 5 biologically independent mice (MFP tumor, MFP), 3 (noncancerous brain), and 4 (brain tumor). g, Isotopolog distributions of the indicated metabolites in MDA-MB-231 control (Ctrl) or DHODH knockout (KO) cells cultured 24 h in medium containing [U-13C]-glucose and 100 µM uridine. h, Normalized peak areas for metabolites shown in (g). i-j, Isotopolog distributions of pyrimidines (i) and purines (j) in MDA-MB-231 Ctrl or DHODH KO cells cultured as in (g). k, Normalized peak areas for metabolites shown in (i-j). In (g-k), data are mean ± SD; n = 3 biologically independent samples. Experiments were performed once.
Extended Data Fig. 14 Dietary depletion of serine and glycine alters tissue metabolite levels and site-specific metastasis.
a, Volcano plot of plasma metabolite changes in female NSG mice fed a control (Ctrl) or serine/glycine-deficient (-SG) diet for 15 days. Significance was defined as fold change ≥ 2 with P < 0.05 by two-sided Welch’s t-test with unequal variance; n = 5 mice per group. b, Plasma concentrations of serine and glycine from (a). Data are mean ± SD; n = 5 per group. c-d, Principal component analysis (PCA) (c) and hierarchical clustering (d) of metabolites in brain, kidney, liver, and lung from Ctrl or -SG diet-fed mice. n = 5 per tissue per diet. Heatmap columns were z-score normalized. e, Volcano plots showing tissue metabolite differences between diets, thresholds as in (a). n = 5 per group. f, Normalized peak areas for serine and glycine in tissues from (e). Data are mean ± SEM. g, Body weight of mice maintained on Ctrl or -SG diet. HCC1806-Fluc cells were injected after 15 days, and mice remained on their respective diets until endpoint. Data are mean ± SEM; n = 10 per group. h, Whole-body bioluminescence imaging (BLI, photons/sec) of the same mice shown in (g) tracking metastatic progression. Final values correspond to endpoint when tissues were harvested. Data are mean ± SEM; n = 10 per group. i, Tissue-specific BLI at endpoint following intracardiac injection of HCC1806-Fluc cells in Ctrl or -SG diet-fed mice. Data are mean ± SEM; n = 10 per group (two bones, kidneys, or ovaries analyzed per mouse). Statistical significance for (b, f, i) was assessed by two-sided unpaired Welch’s t-test with Holm-Sidak multiple comparisons correction. *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant. Exact P values (two-sided) are provided in Source Data. Experiments were performed once.
Extended Data Fig. 15 Correlation of metastatic potential with gene expression, CRISPR dependency, and metabolite levels.
a-b, Scatter plots correlating breast cancer cell line metastatic potential with (a) RNA expression (log2 transcripts per million, TPM) or (b) in vitro CRISPR dependency of the indicated genes, from the Dependency Map portal. Each point represents a cell line. c, Scatter plot comparing Pearson correlation (r) values for EO771 metastatic potential across tissues with corresponding metabolite concentrations measured in interstitial fluid from female NSG mice (x-axis) versus C57BL/6 J mice (B6, y-axis). Each point is a metabolite–tissue pair. Points in red were significant in B6 only, points in blue in NSG only, and points in black in both cohorts (significance defined as |r | > 0.5, P < 0.05). d, Scatter plot correlating tissue carnosine concentrations with metastatic potential of control (Ctrl) cells following intracardiac injection (black, MDA-MB-231; blue, HCC1806; red, EO771). Symbols denote metabolite concentrations in specific tissues; brain values reflect CSF. Data are mean ± SEM; n = biologically independent mice as in Extended Data Fig. 8. Pearson correlation coefficients (r) and exact P values (two-sided) are provided in Source Data.
Supplementary information
Supplementary Figure 1 (download PDF )
The uncropped gel blots.
Supplementary Table 1 (download XLSX )
Metabolites detected by LC-MS and their corresponding identification data.
Supplementary Table 2 (download XLSX )
Absolute metabolite concentrations measured by LC-MS across plasma, cerebrospinal fluid, and tissue interstitial fluids from mice.
Supplementary Table 3 (download XLSX )
sgRNA and primer sequences used to generate nutrient auxotroph cell lines via CRISPR-Cas9.
Supplementary Table 4 (download XLSX )
Additional competing interests information for G.M.C.’
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Abbott, K.L., Subudhi, S., Ferreira, R. et al. Nutrient requirements of organ-specific metastasis in breast cancer. Nature 649, 1292–1301 (2026). https://doi.org/10.1038/s41586-025-09898-9
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09898-9
This article is cited by
-
The complex role of nutrients in cancer spread
Nature (2026)
