
Abstract
Cancer is generally thought to be caused by expansion of a single mutant cell1. However, analyses of early colorectal cancer lesions indicate that tumours may instead originate from several genetically distinct cell populations2,3. Detecting polyclonal tumour initiation is challenging in patients, as it requires profiling early-stage lesions before clonal sweeps obscure diversity. To investigate this, we analysed normal colorectal mucosa, benign and dysplastic premalignant polyps and malignant adenocarcinomas (123 samples) from six individuals with familial adenomatous polyposis. Individuals with familial adenomatous polyposis have a germline heterozygous APC mutation, predisposing them to colorectal cancer and numerous premalignant polyps by early adulthood4. Whole-genome and/or whole-exome sequencing showed that many premalignant polyps—40% with benign histology and 28% with dysplasia—were composed of several genetic lineages that diverged early, consistent with polyclonal origins. This conclusion was reinforced by whole-genome sequencing of single crypts from polyps in further patients that showed limited sharing of mutations among crypts within the same lesion. In one case, several distinct APC mutations co-existed in different lineages of a single polyp, consistent with polyclonality. These findings reshape our understanding of early neoplastic events, demonstrating that tumour initiation can arise from the convergence of diverse mutant clones. They also indicate that cell-intrinsic growth advantages alone may not fully explain tumour initiation, highlighting the importance of microenvironmental and tissue-level factors in early cancer evolution.
Similar content being viewed by others


Main
Cancer begins when cells acquire new malignant traits that increase cell growth, survival and invasion5. Investigating the earliest steps in tumour initiation has important implications for understanding cancer biology, improving risk stratification and prevention6,7. However, studying this process in human tissues is challenging, because phenotypic changes and evolutionary progression make it difficult to characterize the cells that originate the lesion when sampling only malignant tumours. Premalignant lesions are therefore a crucial window into these earlier initiating events, because they reflect an important, often long, period of molecular and evolutionary change where cells acquire the necessary traits for full-blown malignancy8,9.
Therefore, to study the initiation of colorectal tumours, we collected and profiled numerous premalignant colon polyps from several individuals with familial adenomatous polyposis (FAP), a hereditary cancer predisposition syndrome caused by a germline heterozygous loss-of-function mutation in the APC gene4. Starting in adolescence, individuals with FAP develop many colorectal polyps, some of which will inevitably progress to colorectal cancer (CRC) without prophylactic colectomy4. The abundant polyps in these individuals are an excellent model for studying early tumour progression, because they arise at different times and retain information about the initial state of the lesion that is easily obscured by later selective sweeps. We sampled the entire process of CRC development, in which normal mucosal epithelium can form premalignant polyps, some of which later may develop dysplastic features and then transform into malignant adenocarcinomas (AdCas).
Colorectal polyps are canonically assumed to develop after an epithelial cell acquires the required oncogenic mutation(s) to clonally expand and create a detectable lesion composed of the descendants of the original initiating mutant cell10,11,12. However, some studies of human premalignant polyps challenge this hypothesis. First, others have found that key early driver mutations (APC and KRAS) are sometimes not found in all cells in a polyp, as would be expected if they initiated a clonal expansion13. Instead, there can be several unique subclonal mutations in APC or KRAS in a single polyp13. Second, limited in situ hybridization and single-crypt sequencing data have indicated that polyps can be composed of epithelial cells from different genetic lineages that diverged early in the patient’s lifetime, before the polyp initiated2,3. Both observations indicate that polyp initiation is not always monoclonal or the result of clonal expansion of a single cell. Instead, colorectal polyp initiation may often be polyclonal or the result of expansion of a group of genetically diverse cells, possibly driven by cell-extrinsic effects.
To evaluate whether polyclonal initiation is evident in FAP patient colorectal premalignant colon polyps, to determine its prevalence and to investigate the role of somatic driver mutations in CRC development, we performed an evolutionary analysis using whole-genome sequencing (WGS) and/or whole-exome sequencing (WES) data from 123 total samples taken either from normal mucosa, benign polyps without evidence of dysplasia or dysplastic premalignant polyps, or malignant AdCas from six patients with FAP. The profiled lesions were of varied age, size and physical location in the colon and represent an important resource in the NIH Human Tumour Atlas Network (HTAN)14 for understanding premalignant progression in CRC. This bulk genomic profiling was used to detect spontaneously occurring somatic mutations15,16,17,18, which are a record of past mutational processes and evolutionary dynamics19,20,21,22. We computationally analysed these bulk sequencing data to infer clonal architecture and dynamics without longitudinal sampling.
We found that 40% of benign and 28% of dysplastic polyps had evidence of polyclonal initiation. Polyclonal samples were less likely to have clonal APC or KRAS driver mutations, indicating that these oncogenic mutations do not always drive monoclonal expansions that lead to tumour initiation. Single-crypt WGS of three polyps and one AdCa from two further individuals with FAP indicates the polyps are often polyclonal. Two of these polyps show early genetic divergence of individual crypts, including one polyp with several unique APC second-hit mutations acquired after lesion initiation, consistent with polyclonal initiation. Taken together, our results indicate that polyclonal initiation of premalignant polyps is common, challenging the classic model of monoclonal initiation.
FAP polyps have canonical CRC drivers
To investigate early events in CRC lesion initiation, we performed WGS on fresh frozen histologically normal mucosal tissue and several benign and dysplastic polyps from six individuals with FAP (Fig. 1a, Supplementary Tables 1–3, Supplementary Methods and Supplementary Fig. 1). Five of these individuals had a germline truncating heterozygous mutation in APC detected in all tissue samples and blood (Supplementary Figs. 2–6), whereas A014 had no detectable germline mutation in APC as assessed by WGS or clinical diagnostic genetic testing. Because this individual has no family history of FAP (Supplementary Table 1), this finding is consistent with reports that roughly half of index cases have no pathogenic variants detectable in the blood and up to 20% of index cases with a detectable APC mutation have a somatic mosaic variant that might not be detected in all tissues23,24.

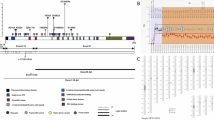
a, Overview of the FAP cohort (six patients, n = 123 samples, WGS and/or WES). b, Oncoplot summarizing the landscape of non-silent SNVs, small insertions/deletions, copy-number gains/amplifications (Gain/Amp), deep deletions (Loss/Del) and copy-neutral LOH (cnLOH) in CRC driver genes based on WGS when available or WES. Only somatic mutations are shown. WGD, whole-genome doubling. c, UpSet plot showing the combination of somatic mutations in APC, KRAS, FBXW7 and TP53. d, ppVAF distributions for all mutations (left) and APC second-hit driver mutations (right) in mucosa (green), benign polyps (orange) and dysplastic polyps (blue) from WGS data. e,f, ppVAF posterior distributions of APC mutations in samples with a single APC second-hit mutation (e, separated into clonal (left) or subclonal (right) APC mutations) or several APC second-hit driver mutations (f), ordered by ascending ppVAF point estimates. Asterisks in f denote clustered APC second-hit mutations (Supplementary Fig. 7). APC second-hit mutation ppVAF distributions are shown in blue, and KRAS driver mutation ppVAF posterior distributions are shown in pink. WGS was used in samples for which it was available; otherwise WES data were used to estimate ppVAFs. g, Clonal driver mutation fractions (Supplementary Methods) from WGS (when available) or WES data for benign and dysplastic polyps showing mutations in APC (benign, n = 31; dysplastic, n = 53 mutations), KRAS (benign, n = 6; dysplastic, n = 25 mutations) and FBXW7 (benign, n = 3; dysplastic, n = 5 mutations). Error bars are 95% Bayesian credible intervals (Supplementary Methods). Illustrations in a were created using BioRender. Ma, Z. (2025) https://BioRender.com/4v5zpca.
Somatic alterations, including single-nucleotide variants (SNVs) and copy-number alterations (CNAs), in driver genes associated with CRC were detected in nearly all polyp samples (91% of benign and 95% of dysplastic polyps) but rarely in normal mucosa (10%) (Fig. 1b). Driver alterations in APC and KRAS were common in polyps; 83% of benign polyps and 82% of dysplastic polyps (adenomas) had second-hit APC driver mutations or APC loss of heterozygosity (LOH), whereas only 7% of normal mucosal samples had biallelic APC inactivation. Furthermore, 17% of benign and 35% of dysplastic polyps had both APC and KRAS somatic mutations (Fig. 1c). The high prevalence of APC and KRAS somatic mutations in polyp samples is consistent with previous reports in both FAP and sporadic adenomas25,26 (Extended Data Figs. 1 and 2) and indicates that these mutations are associated with polyp initiation. Further CRC driver mutations were found in FBXW7, TP53 and other proliferative signalling associated genes in some lesions (Fig. 1b). Although recurrent gains of chromosomes 7, 13 and 20 were noted, as observed in other premalignant colorectal lesions25,26, polyps were generally less aneuploid than malignant CRC samples (Extended Data Fig. 3). The fraction of genome altered was low in both benign (median 0.003) and dysplastic (median 0.03) polyps and increased with disease stage in both our FAP cohort and previously published FAP25 and sporadic cohorts26 (Extended Data Fig. 3). Thus, FAP polyps harbour early oncogenic events, most frequently somatic SNVs and more rarely CNAs.
Clonal drivers are often absent in FAP polyps
To better assess the role of driver mutations in polyp initiation and growth, we computed the purity- and ploidy-adjusted variant allele frequencies (ppVAFs) of all somatic mutations detected (Supplementary Methods). The ppVAF is the estimated fraction of epithelial cells that have a mutation and is analogous to the cancer cell fraction in malignant samples27. The cancer cell fraction is usually calculated using tumour purity values estimated from CNA frequencies28, but our normal and polyp samples have relatively few CNAs compared to CRCs (Extended Data Fig. 3), making copy-number-based purity estimation difficult. Instead, we used the epithelial cell fractions previously measured and reported in further samples from this patient cohort using single-cell assay for transposase-accessible chromatin using sequencing (scATAC-seq)29 to estimate the distribution of sample purities for normal mucosa, polyps and AdCas (Supplementary Methods, Supplementary Notes 1 and 2 and Extended Data Fig. 4). The ppVAF point estimates and posterior probability distributions indicate that somatic APC mutations are found at higher allele frequencies than other somatic mutations, reinforcing the idea that they occur early in polyp development and/or experience positive selection (Fig. 1d).
However, many polyp samples in our cohort lacked clonal driver mutations (Fig. 1e–g). This observation is not consistent with the hypothesis that APC second-hit mutations or other driver mutations are required for monoclonal expansions leading to polyp formation. Furthermore, 8 of the 69 polyps harboured more than one APC somatic driver mutation (Fig. 1f). Although five represent clustered mutational events that occurred in the same subclone (Supplementary Fig. 7) or probable biallelic loss in a single subclone in the patient without a germline APC mutation (A014), two samples (A001C107 and G055) show evidence of two subclonal APC mutations. Because a strong selective advantage for several truncating APC mutations to appear in the same subclone with a germline heterozygous mutation is unlikely, this observation indicates that several unique APC mutations existed in the epithelial cell population that initiated the lesion and/or further APC mutations were acquired by cells in the lesion that did not have an APC second hit at the time of lesion initiation. Together, these results indicate that polyps are not always caused by a monoclonal expansion of an epithelial cell with a driver mutation.
Polyps have subclones that diverged early
Because we observed that polyp initiation is not always associated with monoclonal expansion of one or more driver mutations, we wondered how lesion initiation occurred in samples without clonal driver mutations. We hypothesized that several colonic crypts (clonal units of epithelial homeostasis derived from a small number of stem cells30) might collectively initiate each polyp, rather than a single mutant crypt. This would lead to a polyp composed of several distinct genetic lineages that diverged early. We defined initiation of a single polyp from several crypts as ‘polyclonal’ and used the WGS data to determine whether polyp initiation was probably monoclonal or polyclonal (Fig. 2a). In monoclonal samples, clonal expansion of a single crypt leads to all somatic mutations in that founding crypt being present in all epithelial cells in the resulting lesion. This sweep leads to many detectable clonal mutations in the sequenced polyp. By contrast, polyclonal lesions will probably have fewer clonal mutations, because any clonal mutations in the lesion must be present in all founding crypts in a polyp (Fig. 2b).

a, Schematic showing that polyclonal polyps are initiated from several colorectal crypts in the intestinal mucosa (green crypts), whereas monoclonal polyps are the result of a clonal expansion of a single crypt (pink). b, Schematic showing that monoclonal lesions have an MRCA occurring after lesion initiation (dotted line) because they initiate from a single crypt, leading to many detected clonal mutations (purple Xs on tree). Bottom parts show hypothetical ppVAF distribution shapes for monoclonal and polyclonal samples, with clonal mutations marked in purple. c, Number of expected clonal SNVs detected in WGS data from samples from patients with FAP. Samples with fewer than 63 clonal SNVs (dashed line, corresponding to an MRCA at less than 1 year old) were classified as having an early MRCA and are probably polyclonal. d, ppVAF distributions for example monoclonal (left) and polyclonal (right) lesions. e, ppVAF distributions for all monoclonal (left) and polyclonal (right) lesions combined. f, Fraction of WGS samples classified as polyclonal based on estimated clonal SNV count. Fractions were estimated from n = 25 mucosal samples, n = 15 benign polyp samples and n = 54 dysplastic polyp samples. Error bars are 95% Bayesian credible intervals (Supplementary Methods). Illustrations in a were created using BioRender. Ma, Z. (2025) https://BioRender.com/4v5zpca.
We estimated the number of clonal SNVs in each sample using the ppVAF posterior probability distributions computed for each somatic mutation from the sequencing data. We classified each somatic SNV as clonal or subclonal based on its ppVAF posterior probability distribution and counted the number of clonal mutations in each sample (Supplementary Methods and Supplementary Note 1). We used these estimated clonal SNV counts to determine whether lesions were probably monoclonal or polyclonal. Because most SNVs in these samples have clock-like mutational signatures31 (Extended Data Fig. 5), the number of clonal mutations can be used to estimate an upper bound on the age of the most recent common ancestor (MRCA) for the sample (Supplementary Methods). Samples with an MRCA earlier than the time at which the lesion initiated are by definition polyclonal. In polyclonal samples, several genetic lineages must have existed during lesion initiation, so these genetic lineages must therefore have diverged from the MRCA before initiation. If the MRCA occurred at or after lesion initiation, the lesion is composed of cells that originated from a single genetic lineage at the time of initiation and is monoclonal. We observed that, whereas normal mucosal samples had few (fewer than 15) clonal SNVs, benign and dysplastic polyps had a much wider range of expected clonal SNV counts (0–3,882 SNVs) (Fig. 2c). The low clonal mutation count in the normal samples is consistent with previous observations indicating that normal intestinal mucosa is highly polyclonal, often having few mutations shared between nearby crypts32.
To investigate the prevalence of polyclonal polyps, we estimated the frequency of polyps with an MRCA that existed before one year of age (fewer than 63 clonal SNVs, using WGS data) (Supplementary Methods), which is far earlier than polyps appear in most individuals with FAP33 and is similar to the estimated clonal mutation counts of the polyclonal normal mucosa. We found that 6 out of 15 benign polyps (40%) had an early MRCA, indicating that they are polyclonal, and 15 out of 54 dysplastic polyps (28%) had an early MRCA (Fig. 2d–f). Samples referred to as polyclonal generally do not have a clonal mutation peak visible in the raw VAF or ppVAF distributions (Fig. 2d,e and Supplementary Figs. 8–17), consistent with their early divergence time. WES data from some samples with WGS data as well as 27 further samples from our cohort were also subjected to clonal SNV count thresholding to estimate the prevalence of early-diverging samples and showed a similar fraction of these lesions (Extended Data Fig. 6). Additionally, clonal SNV counts indicate that polyclonality is prevalent in premalignant and malignant multiregion WES samples from a different published FAP cohort25 (Extended Data Fig. 7 and Supplementary Methods). A similar analysis using published multiregion WES data from individuals without a hereditary CRC predisposition syndrome26 indicates that polyclonal initiation also occurs in sporadic premalignant adenomas but not AdCas (Extended Data Fig. 7).
These analyses indicate that polyclonal initiation is probably present in at least 25–40% of polyps in individuals with FAP. Selective sweeps occurring after lesion initiation can obscure a polyclonal population structure, so the true fraction of lesions that initiate polyclonally may be higher. This is particularly true in older lesions or lesions containing driver mutations with a substantial selective advantage, highlighting the importance of studying early premalignant adenomas. Amongst dysplastic polyps, those of monoclonal origin had shorter telomeres than polyclonal lesions (P = 0.0015; Fisher’s combined P value from per-patient Wilcoxon rank-sum tests) (Supplementary Methods and Extended Data Fig. 8), consistent with a higher number of total cell divisions due to earlier lesion initiation and/or increased proliferation rate. Early, proliferative lesions would have shorter telomeres when sampled and would be more likely to seem monoclonal, particularly if there were expansion of an advantageous clone.
Polyclonal polyps have subclonal drivers
We used these monoclonal/polyclonal classifications to further investigate the role of driver mutations in polyp initiation and progression. Most monoclonal and polyclonal polyps have an APC second-hit driver mutation (Fig. 3a). As expected, the ppVAFs of APC somatic driver mutations are lower in polyclonal samples than monoclonal samples (Fig. 3b; P = 0.003 for benign, P = 0.005 for dysplastic polyps, Wilcoxon rank-sum test), indicating that these mutations are not the sole drivers of lesion initiation and growth and have not caused a selective sweep in the polyclonal polyps. Similarly, KRAS mutations are more likely to be found at lower subclonal frequencies in dysplastic polyclonal polyps than dysplastic monoclonal polyps (P = 0.074 for benign, P = 0.012 for dysplastic polyps, Wilcoxon rank-sum test; Fig. 3c,d). These data are consistent with the notion that premalignant lesions are not always the product of monoclonal expansions associated with common early CRC driver mutations. However, such driver mutations probably have a selective advantage that often increases their frequency and can lead to selective sweeps in more advanced lesions.

a, Fraction of monoclonal and polyclonal WGS samples with second-hit APC somatic driver mutations. b, ppVAFs of APC second-hit mutations in benign (left) and dysplastic (right) polyps. c, Fraction of monoclonal and polyclonal WGS samples with KRAS driver mutations. d, ppVAFs of KRAS driver mutations in benign (left) and dysplastic (right) polyps. Error bars in a and c are 95% Bayesian credible intervals (Supplementary Methods), and fractions in a and c were estimated from n = 25 mucosal, n = 6 polyclonal benign, n = 9 monoclonal benign, n = 15 polyclonal dysplastic and n = 39 monoclonal dysplastic samples.
Single-crypt WGS shows polyclonality
We performed single-crypt isolation and WGS from polyps and AdCas from two further patients with FAP (Fig. 4, Extended Data Figs. 9 and 10, Supplementary Methods and Supplementary Table 4) to examine the clonal relationships between cell populations within each lesion. Lesions from patients with FAP were dissected into several regions before single-crypt isolation to better determine the association between spatial proximity and genomic similarity. After filtering out low-quality crypts and polyps/tumours, we built phylogenies using single-crypt WGS data from the remaining three polyps and one AdCa sample (Supplementary Methods and Supplementary Note 3). We first focused on the polyp for which the most crypts were sequenced (FAP03_P2; 20 single crypts included after filtering). We found that crypts within the polyp diverged early in life, with no mutations shared by all crypts sequenced in the polyp (Fig. 4c). This finding indicates that several distinct genetic lineages with a very early common ancestor combined to form the polyp. The lack of clonal somatic mutations in this polyp is reflected in the structure of its single-crypt phylogeny (Fig. 4c), with long terminal branches and no trunk, as hypothesized in the polyclonal schematic shown in Fig. 2b. This high degree of polyclonality seems spatially structured, with one polyp region (R4) seeming to be monophyletic (Fig. 4c), with 1,025 mutations shared between and exclusive to all 5 crypts in the region (Extended Data Fig. 10b). This implies that the crypts in R4 share a more recent common ancestor than the rest of the polyp, which may have expanded clonally to create that region. By contrast, the other regions are polyclonal mixtures of crypts from different genetic lineages.

a, Schematic showing collection of polyp P2 from patient FAP03, regional dissection, single-crypt isolation and WGS procedure (Supplementary Methods and Supplementary Information). b, Images of individual isolated crypts from FAP03_P2. c, Single-crypt phylogeny reconstructed from single-crypt WGS data from polyp P2, patient FAP03 and adjacent normal mucosa (blue). Putative CRC-associated mutations are highlighted. The spatial region from which each crypt originated is indicated by the tip label colours (Supplementary Table 4). d, Schematic showing collection of CRC lesion T3 from patient FAP01 as well as regional dissection of the lesion. e, Images of individual isolated crypts from FAP01_T3. f, Single-crypt phylogeny reconstructed from lesion T3 from patient FAP01 and normal mucosa (blue). Putative CRC-associated mutations are highlighted, with mutations denoted in grey if they were filtered out of some samples but not others or if the sequencing depth at the mutated site was too low to detect the variant (Supplementary Methods and Supplementary Note 3, section 5). The spatial region from which each crypt originated is indicated by the tip label colours. Illustrations in a and d were created using Biorender (Ma, Z. (2025) https://BioRender.com/4v5zpca) and FigDraw (https://www.figdraw.com).
The single-crypt WGS analysis also showed putative cancer driver mutations in distinct subpopulations within the polyp. Importantly, no driver mutations are shared by all crypts in the polyp. Instead, there were three truncating frameshift mutations in APC occurring in independent subclones (Fig. 4 and Extended Data Fig. 9c) that either arose after lesion initiation or were present at subclonal frequencies in the initiating cell population. One of these APC second-hit mutations is found in all crypts in region R4, raising the possibility that it caused a clonal expansion that dominated that part of the polyp. These observations indicate that this polyp did not initiate from a clonal expansion driven by a single APC second hit but rather is a mixture of epithelial cell clones with independent growth advantages.
By contrast, single-crypt WGS of the malignant AdCa tumour sample FAP01_T3 showed a monoclonal expansion, with 479 mutations shared by all 10 crypts in the sample (Fig. 4d–f). This expanded clone includes a second-hit stopgain mutation in APC, a missense driver mutation in SMAD4 (R361C) and a truncating mutation in CTNNB1 (beta-catenin) (Fig. 4f). Lower sequencing depth in some loci in some samples makes it more difficult to determine which driver mutation(s) are found in all crypts and possibly responsible for the initial expansion; in particular, low coverage (less than or equal to 3×) at the APC second-hit mutation locus in crypts R6_G9, R5_G4 and R1_G7 limited our ability to detect the mutation in these samples (Supplementary Note 3). However, the phylogenetic structure of this tumour (long trunk from which all tumour crypts originate) clearly indicates that it is monoclonal (compare phylogeny to schematics in Fig. 2c) and provides a contrasting example to the phylogenetic pattern in the polyclonal polyp FAP03_P2.
Two further polyps from these patients with single-crypt sequencing data (FAP01_P6 and FAP03_P1) show different patterns (Extended Data Fig. 9). The 908 clonal mutations in the 7 crypts from polyp FAP01_P6 indicate that it is monoclonal. The lesion has a clonal expansion, possibly resulting from the APC LOH event found in all crypts from this polyp and further fuelled by a subclonal KRAS mutation (Q61R), consistent with most of the APC second-hit driven monoclonal polyps in our bulk sequencing dataset. By contrast, the single-crypt WGS data from polyp FAP03_P1 are more difficult to interpret. Although very few clonal mutations are present (21 shared by all 7 crypts), indicating early genetic divergence of the crypts within it, the somatic APC truncating mutation present in 6 out the 7 crypts (which share only 57 other mutations) raises the possibility that this APC second hit occurred very early in life and may have contributed to lesion initiation. In summary, this single-crypt sequencing dataset provides high-resolution orthogonal validation of polyclonal initiation in FAP and highlights the mutational heterogeneity in polyclonal lesions.
Discussion
Although it is widely assumed that malignancies are the product of clonal expansions from single mutant cells of origin10, limited case studies in CRC and other cancers indicate that premalignant lesions can be polyclonal2,3,34,35. However, systematic assessment of the prevalence of polyclonality in patient samples is still lacking, despite the utility of sequencing to detect this phenomenon. To address this, we used WGS data from 69 colorectal polyps and found 40% with benign histology and 28% with dysplasia from individuals with FAP originated from several colon crypts. Furthermore, analysis of single-crypt WGS data supported our conclusion that premalignant colorectal lesions in individuals with FAP can initiate polyclonally and showed local expansion of subpopulations within a polyp with unique APC second-hit mutations. These findings point towards a possible role for cell-extrinsic mechanisms in tumour initiation and indicate that this process may involve cell–cell interactions in a premalignant multicellular ecosystem as well as cell-intrinsic effects of driver mutations.
The finding that many polyps are polyclonal has implications for understanding the molecular and microenvironmental determinants and dynamics of tumour initiation and progression. Polyclonal initiation provides a genetically diverse starting point for premalignant evolution. This intralesion heterogeneity can persist because sweeps are relatively rare, with long periods of stasis between such events. The finding that premalignant polyps may experience several selective sweeps before accruing genetic alterations and transforming to CRC is consistent with our previous findings that subsequent evolution is often effectively neutral19,20,21. Common drivers (APC, KRAS) may not directly initiate the lesion by causing a clonal expansion but instead may be present at subclonal frequencies in the initiating population or be acquired after lesion initiation. This process can lead to monoclonal conversion of a previously polyclonal lesion and may lead to underestimation of the frequency of polyclonal initiation in our dataset, indicating that the extent of this phenomenon may be substantially higher than reported here.
Our study has several limitations. First, our bulk sequencing analysis does not directly estimate the purity of each sample individually, but rather uses the distribution of epithelial cell fractions estimated from scATAC-seq data from the HTAN FAP patient cohort as a measure of purity. Although this procedure avoids the pitfalls of copy-number-based algorithms that render them inappropriate for non-malignant tissue samples (Supplementary Methods, Extended Data Fig. 4 and Supplementary Note 2), samples with lower purity due to stromal or immune cell inclusion may be falsely called polyclonal. Although this cannot account for all polyclonal samples we identify by means of bulk genomic sequencing (Supplementary Note 2), further profiling with strategies that either isolate epithelial cells for sequencing (such as single-crypt WGS) or directly measure epithelial cell fraction in the same sample will be instrumental to accurately estimate the fraction of polyclonal samples. Additionally, we focused on CRC initiation in the context of a hereditary cancer predisposition syndrome (FAP). Focus on this patient population also introduces the possibility that some of our polyclonal polyps could be caused by stochastic collision of independently initiated lesions due to the increased density of colorectal polyps in patients with FAP. However, recently published studies indicate that polyclonality is a general phenomenon in colorectal lesions. Our findings are consistent with studies in the analogous APC mutant murine model, which demonstrate cellular cooperativity during colorectal tumorigenesis35 and polyclonal tumour origins36,37,38,39. Moreover, analysis of clonal SNV counts in sporadic CRC adenomas similarly indicates that 29% are polyclonal36,38. Thus, polyclonality is unlikely to be restricted to hereditary colon cancers or exclusively caused by random polyp collisions.
Our findings raise questions about the events necessary and/or sufficient for cancer development. Here, we did not identify what mechanistic role these further epithelial cell clones are playing in tumorigenesis, and follow-up studies will be required to determine how these clones function and interact. Evidence for the role of cell–cell interactions in tumour development between epithelial cells indicates that both cooperation between premalignant clones40 and recruitment of neighbouring non-malignant epithelium36,41 may contribute to polyclonal initiation, although other interactions may also be involved. Furthermore, signals from non-epithelial cells in the microenvironment, including fibroblasts and immune cells42,43,44, may lead to malignant phenotypic changes in several colonic crypts at once, resulting in a polyclonal lesion. These findings also raise questions about the role of canonical driver mutations in tumour initiation: how does acquisition of an oncogenic mutation in an individual cell lead to expansion of a diverse group of cells, not all of which have the mutation? More generally, polyclonal initiation may be common across diverse tissue types34,35. Indeed, a recent study in premalignant pancreatic cancer lesions demonstrated extreme multifocality, consistent with polyclonal origins34. The conceptual and analytic framework we outline for assessing polyclonality from sequencing data can be extended to other premalignant lesions to systematically investigate this underappreciated phenomenon in cancer initiation.
Methods
See Supplementary Information.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
DNA sequencing data and metadata have been deposited at the HTAN portal: https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002371.v8.p2. The raw single-crypt WGS data have been deposited in NGDC’s Genome Sequence Archive (GSA) under the accession number HRA009036. Processed source data and resources needed for reproducing key analyses are available at Zenodo (https://doi.org/10.5281/zenodo.13228021)45. The WES data for the sporadic CRC cohort are available through the European Genome-Phenome Archive (EGA) under accession number EGAS00001003066. The WES data for the FAP multiregion cohort are available through the Genome Sequence Archive (GSA) under accession number HRA000127. To facilitate comparisons, both of these published cohorts were uniformly reprocessed using the same software versions in the Isabl platform, as was used for the HTAN Pre-cancer Atlas bulk WGS FAP cohort. Data to generate the figures are available at GitHub (https://github.com/cancersysbio/HTAN-FAP).
Code availability
Source code to generate the figures are available at GitHub (https://github.com/cancersysbio/HTAN-FAP) and an archived copy of the code is available at Zenodo (https://doi.org/10.5281/zenodo.17372231)46.
References
Greaves, M. & Maley, C. C. Clonal evolution in cancer. Nature 481, 306–313 (2012).
Novelli, M. R. et al. Polyclonal origin of colonic adenomas in an XO/XY patient with FAP. Science 272, 1187–1190 (1996).
Thirlwell, C. et al. Clonality assessment and clonal ordering of individual neoplastic crypts shows polyclonality of colorectal adenomas. Gastroenterology 138, 1441–1454 (2010).
Galiatsatos, P. & Foulkes, W. D. Familial adenomatous polyposis. Am. J. Gastroenterol. 101, 385–398 (2006).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Spira, A. et al. Precancer atlas to drive precision prevention trials. Cancer Res. 77, 1510–1541 (2017).
Derks, L. L. M. & van Boxtel, R. Stem cell mutations, associated cancer risk, and consequences for regenerative medicine. Cell Stem Cell 30, 1421–1433 (2023).
Gerstung, M. et al. The evolutionary history of 2,658 cancers. Nature 578, 122–128 (2020).
Karlsson, K. et al. Deterministic evolution and stringent selection during preneoplasia. Nature 618, 383–393 (2023).
Nowell, P. C. The clonal evolution of tumor cell populations. Science 194, 23–28 (1976).
Fearon, E. R. & Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 61, 759–767 (1990).
Lamlum, H. et al. APC mutations are sufficient for the growth of early colorectal adenomas. Proc. Natl Acad. Sci. USA 97, 2225–2228 (2000).
Gausachs, M. et al. Mutational heterogeneity in APC and KRAS arises at the crypt level and leads to polyclonality in early colorectal tumorigenesis. Clin. Cancer Res. 23, 5936–5947 (2017).
Rozenblatt-Rosen, O. et al. The Human Tumor Atlas Network: charting tumor transitions across space and time at single-cell resolution. Cell 181, 236–249 (2020).
Martincorena, I. et al. Universal patterns of selection in cancer and somatic tissues. Cell 171, 1029–1041 (2017).
Behjati, S. et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature 513, 422–425 (2014).
Lee-Six, H. et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 574, 532–537 (2019).
Ogawa, H., Horitani, K., Izumiya, Y. & Sano, S. Somatic mosaicism in biology and disease. Annu. Rev. Physiol. 84, 113–133 (2022).
Sottoriva, A. et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 47, 209–216 (2015).
Williams, M. J., Werner, B., Barnes, C. P., Graham, T. A. & Sottoriva, A. Identification of neutral tumor evolution across cancer types. Nat. Genet. 48, 238–244 (2016).
Sun, R. et al. Between-region genetic divergence reflects the mode and tempo of tumor evolution. Nat. Genet. 49, 1015–1024 (2017).
Haughey, M. J. et al. First passage time analysis of spatial mutation patterns reveals sub-clonal evolutionary dynamics in colorectal cancer. PLoS Comput. Biol. 19, e1010952 (2023).
Hes, F. J. et al. Somatic APC mosaicism: an underestimated cause of polyposis coli. Gut 57, 71–76 (2008).
Aretz, S. et al. Somatic APC mosaicism: a frequent cause of familial adenomatous polyposis (FAP). Hum. Mutat. 28, 985–992 (2007).
Li, J. et al. Genomic and transcriptomic profiling of carcinogenesis in patients with familial adenomatous polyposis. Gut 69, 1283–1293 (2020).
Cross, W. et al. The evolutionary landscape of colorectal tumorigenesis. Nat. Ecol. Evol. 2, 1661–1672 (2018).
McGranahan, N. et al. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 7, 283ra54 (2015).
Tarabichi, M. et al. A practical guide to cancer subclonal reconstruction from DNA sequencing. Nat. Methods 18, 144–155 (2021).
Becker, W. R. et al. Single-cell analyses define a continuum of cell state and composition changes in the malignant transformation of polyps to colorectal cancer. Nat. Genet. 54, 985–995 (2022).
Clevers, H. The intestinal crypt, a prototype stem cell compartment. Cell 154, 274–284 (2013).
Alexandrov, L. B. et al. Clock-like mutational processes in human somatic cells. Nat. Genet. 47, 1402–1407 (2015).
Coorens, T. H. H. et al. Extensive phylogenies of human development inferred from somatic mutations. Nature 597, 387–392 (2021).
Kennedy, R. D., Potter, D. D., Moir, C. R. & El-Youssef, M. The natural history of familial adenomatous polyposis syndrome: a 24 year review of a single center experience in screening, diagnosis, and outcomes. J. Pediatr. Surg. 49, 82–86 (2014).
Braxton, A. M. et al. 3D genomic mapping reveals multifocality of human pancreatic precancers. Nature 629, 679–687 (2024).
Parsons, B. L. Multiclonal tumor origin: evidence and implications. Mutat. Res Rev. Mutat. Res 777, 1–18 (2018).
Gaynor, L. et al. Crypt density and recruited enhancers underlie intestinal tumour initiation. Nature 640, 231–239 (2025).
Sadien, I. D. et al. Polyclonality overcomes fitness barriers in Apc-driven tumorigenesis. Nature 634, 1196–1203 (2024).
Lu, Z. et al. Polyclonal-to-monoclonal transition in colorectal precancerous evolution. Nature 636, 233–240 (2024).
Zahm, C. D. et al. Advanced intestinal cancers often maintain a multi-ancestral architecture. PLoS ONE 11, e0150170 (2016).
Islam, M. et al. Temporal recording of mammalian development and precancer. Nature 634, 1187–1195 (2024).
Marusyk, A. et al. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 514, 54–58 (2014).
Thliveris, A. T. et al. Transformation of epithelial cells through recruitment leads to polyclonal intestinal tumors. Proc. Natl Acad. Sci. USA 110, 11523–11528 (2013).
Vermeulen, L. et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 12, 468–476 (2010).
Zhou, R. W., Harpaz, N., Itzkowitz, S. H. & Parsons, R. E. Molecular mechanisms in colitis-associated colorectal cancer. Oncogenesis 12, 48 (2023).
Khan, A. et al. Processed source data and resources for ‘Polyclonal origins of human premalignant colorectal lesions’. Zenodo https://doi.org/10.5281/zenodo.13228021 (2025).
Van Egeren, D. et al. Source code for ‘Polyclonal origins of human premalignant colorectal lesions’. Zenodo https://doi.org/10.5281/zenodo.17372231 (2025).
Acknowledgements
This work was supported by NCI grant no. U2CCA233311 to M.P.S., J.M.F. and C.C. A. Khan, C.L.W., D.V.E., R.O.S. and C.C. were additionally supported by NIH grant no. DP1CA238296 to C.C. R.O.S. is supported by a Stanford Dean’s Fellowship. A.M.H. was funded in part by the Cancer Systems Biology Scholars Postdoctoral Fellowship Program (grant no. 5R25CA180993). D.V.E. is supported by the A. P. Giannini Postdoctoral Fellowship. S.A.N. was supported in part by the Stanford Graduate Fellowship in Science and Engineering and the NHGRI Stanford Genome Training Program (grant no. 5 T32 000044). This work is also supported by NIH grant no. S10OD025212. We thank A. Adimoelja, K. Houlahan, S. Park and other members of the Curtis Lab for feedback on the manuscript.
Author information
Authors and Affiliations
Contributions
A.M.H., A. Khan, C.C., D.V.E. and R.O.S. conceptualized the study. A.M.H., A. Khan, B.B., C.L.W., D.V.E., R.O.S., S.M. and Z. Hu. performed bioinformatic analysis. A. Khan, C.L.W., D.V.E. and R.O.S. performed statistical analyses. A.M.H., A.K.W., C.H., E.D.E., E.M., H.Z., J.M.F., M.A.M., R.C., R.L., S.A.N., T.K.G., U.L., W.R.B. and Z. He collected samples for the FAP Pre-cancer Atlas. A.M.H., E.D.E., H.Z., Z.M., Z. Hu, Z. He and S.M. generated genomic data and designed experimental methods. A.M.H., B.M., C.H., E.D.E., H.C., H.Z., H.L., L.J., J.C., K.C., M.E., R.L., N.B., R.J., S.L., S.W., S.M., T.K.G., T.V.K., Z. He, Z. Hu and Z.M. designed experimental methods and generated data for the FAP Pre-cancer Atlas. D.G.E., J.C., J.S. and T.A.L. reviewed pathology. A. Kundaje, C.C., J.M.F., M.P.S. and W.J.G. provided guidance on methods and study design. A.M.H., A.K.W. and E.M. managed the FAP Pre-cancer Atlas project. C.C., J.M.F. and M.P.S. supervised and are co-senior authors. A.M.H., A. Khan, C.C., D.V.E. and R.O.S. wrote the original draft. C.C. and D.V.E. wrote the final version.
Corresponding author
Ethics declarations
Competing interests
E.D.E. is an employee and stockholder of Invitae, scientific advisory board member and stockholder of Taproot Health, and advisor and stockholder of Exir Bio. M.P.S. is cofounder and scientific advisor of Personalis, Qbio, SensOmics, January AI, Mirvie, Protos, NiMo and Onza and is on the advisory board of Genapsys. A. Kundaje has affiliations with Biogen (consultant), SerImmune (SAB), RavelBio (scientific cofounder and SAB) and PatchBio (SAB). W.J.G. is a consultant for Guardant Health, scientific cofounder of Protillion Biosciences and 10x Genomics and has licensed patents related to ATAC-seq. C.C. is a stockholder in Illumina, DeepCell and 3T Biosciences and has served as an advisor/consultant to AstraZeneca, Bristol Myers Squibb, DeepCell, Genentech, Pfizer and 3T Biosciences. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Richard Halberg, Carla Robles-Espinoza and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Somatic mutations in FAP and sporadic CRC samples.
a. Oncoplot summarizing the landscape of non-silent exonic single nucleotide variants (SNVs), small insertions/deletions, copy number amplifications (Amp), deletions (Del), and copy neutral loss of heterozygosity (cnLOH) within CRC driver genes based on WES data from the previously published FAP multi-region cohort25 (left) and the sporadic multi-region sequencing cohort26 (right). Only somatic mutations are shown. b-d. Comparison of non-silent exonic mutations across polyp stages between the two FAP cohorts and the CRC cohort (p-values are estimated using two-sided Wilcoxon rank sum tests). In box-and-whisker plots, the box represents the interquartile range (IQR) with the center line representing the median, and the whiskers are the largest and smallest data values within 1.5 times the IQR from the box edges. Each point represents one sequenced normal or polyp sample, with n = 29 mucosal, n = 35 benign, and n = 57 dysplastic samples in the multi-polyp HTAN FAP cohort (b), n = 7 mucosal, n = 13 benign, and n = 22 dysplastic samples in the multi-region FAP cohort (c), and n = 27 adenoma and n = 30 carcinoma samples in the sporadic CRC cohort (d). e. Lollipop plots showing the distribution and classes of mutations in APC across two FAP cohorts and one sporadic CRC cohort.
Extended Data Fig. 2 Distribution of APC and KRAS somatic mutations in FAP polyps and normal mucosa.
a. Number of normal mucosa, benign and dysplastic polyps with 0, 1, or 2 somatic APC mutations (‘hits’). b. Number of normal mucosa, benign and dysplastic polyps with 0, 1, or 2 somatic KRAS mutations.
Extended Data Fig. 3 Landscape of somatic copy-number alterations in FAP lesions and sporadic colorectal tumors.
a-c. Fraction of samples with copy-number gains (red, positive y-axis) or losses (blue, negative y-axis) in the HTAN FAP multi-polyp cohort (a), FAP multi-region cohort25 (b), and sporadic CRC multi-region cohort26 (c). Centromeres are shown as light yellow vertical regions. Only samples without whole-genome doubling (WGD) inferred by FACETS are shown, and the HTAN AdCas were not shown since only one sample was in this group. d-f. Comparison of the fraction genome altered (FGA) across disease stages for the HTAN FAP cohort (d), multi-region FAP cohort (e), and the sporadic CRC cohort (f). In e-f, the points of the same color and at the same x-axis position within each disease stage indicate multiple regions sequenced from the same lesion. WGD samples were included in these plots. In total there were n = 29 mucosal, n = 35 benign, and n = 57 dysplastic samples in the multi-polyp HTAN FAP cohort (d), n = 7 mucosal, n = 13 benign, n = 22 dysplastic, and n = 17 AdCa samples in the multi-region FAP cohort (e), and n = 27 adenoma and n = 30 carcinoma samples in the sporadic CRC cohort (f). P-values were computed using two-sided Wilcoxon tests. In box-and-whisker plots, the box represents the IQR with the center line representing the median, and the whiskers are the largest and smallest data values within 1.5 times the IQR from the box edges.
Extended Data Fig. 4 Purity estimation and ppVAF transformation in normal and premalignant samples.
a-b. Comparison between sample purities estimated by two copy-number based algorithms (FACETS and Sequenza) in the HTAN WGS (a) and WES (b) samples. Colors indicate the type of sample (green: mucosa, orange: benign polyp, blue: dysplastic polyp) and shape indicates the inferred clonal origin of the sample from the bulk sequencing data (circle: monoclonal, triangle: polyclonal). c-d. Comparison between WES and WGS sample purities estimated by FACETS (c) or Sequenza (d) for samples profiled with both modalities. As before, colors indicate the type of sample (green: mucosa, orange: benign polyp, blue: dysplastic polyp) and shape indicates the inferred clonal origin of the sample from the bulk sequencing data (circle: monoclonal, triangle: polyclonal). e-f. The distributions of purities inferred by Sequenza (dashed colored lines; mucosa shown in green in e, benign (orange) and dysplastic (blue) polyps are shown in f) or FACETS (solid colored lines) are very different from the epithelial cell fractions measured using scATAC-seq (grey filled distributions). g-h. Toy examples showing estimation of ppVAFs using uncertain sample purity values, for a mutation with 20 mutant and 80 wild-type reads. Using a known sample purity with no uncertainty (vertical line in sample purity distributions in g), the posterior distribution for the ppVAF has the narrowest possible width (corresponding posterior distribution in h). As the sample purity distribution gets wider (orange and red distributions in g), the ppVAF posterior distributions for a mutation with the same reference and alternate allele sequencing counts get wider as well (corresponding orange and red distributions in h). The true ppVAF of the mutation given an 80% pure sample is noted by the dashed vertical line in g. i-l. Raw VAF distributions (grey) and corresponding ppVAF distributions (blue), computed using the scATAC-seq measured polyp sample purity distribution (shown in f) from four example samples where computational purity estimation using copy-number based algorithms (FACETS and Sequenza) produced poor results. In these examples, many mutations, including the bulk of the clonal mutation peak, have substantially higher VAFs than the expected clonal heterozygous VAF calculated from the FACETS purity (vertical solid line) and/or the Sequenza purity (vertical dashed line).
Extended Data Fig. 5 SNV signatures using WGS data and known COSMIC signatures.
a. Original and reconstructed mutation profiles using COSMIC (v.3.2) signatures (SBS1,5,18 and 40) for mutations in normal mucosa, benign, and dysplastic polyps based on WGS. Each pair of profiles shows the original (top) and reconstructed (bottom) mutation profiles categorized by nucleotide substitution type (C > A, C > G, C > T, T > A, T > C, T > G) across 96 trinucleotide contexts. Cosine similarity and residual sum of squares (RSS) between original and reconstructed profiles are shown for each stage. b. Relative contributions of SBS1, SBS5, SBS18, and SBS40 signatures in each stage. The color gradient indicates the level of contribution, with darker shades representing higher contributions. c. Cosine similarity of signature contributions across the three stages. Darker shades represent higher cosine similarity between the stage and signature. d. Cosine similarity between the original and reconstructed mutation profiles using the combined signatures including the clock-like signatures (SBS1, SBS5) for normal mucosa, benign, and dysplastic polyps.
Extended Data Fig. 6 Detecting polyclonality using clonal SNV counts in WES FAP samples.
a. Comparison of the number of total SNVs detected with WES and WGS in samples with data from both modalities. The diagonal line indicates the approximate expected relationship between WES and WGS (approximately 2% of the human genome is in the exome). b. Comparison between early/late MRCA classifications using WES and WGS data for samples with both modalities. Classification thresholds shown as dashed lines and regions in light red indicate samples that were classified differently using the two modalities. c. Comparison between counting clonal SNVs using our expected clonal counting procedure (Supplementary Methods) and counting mutations with ppVAF upper bounds (defined as the upper ppVAF value where the posterior probability is half the maximum) equal to 1, as was used in38. Classification thresholds are shown as dashed lines and regions in light red indicate samples that were classified differently using the two procedures. The diagonal line shows the expected relationship if the counting procedures were equivalent (line is at y = x). In panels a-c, green points indicate mucosal samples, orange points indicate benign polyps, and blue points indicate dysplastic polyps (coloring as in d-e). d. Number of estimated clonal SNVs detected in WES data from FAP polyps. Samples with fewer than 0.63 clonal SNVs (dashed line, corresponding to an MRCA at <1 year old) were classified as having an early MRCA and are likely polyclonal. e. Fraction of WES samples classified as early MRCA (putative polyclonal) based on estimated clonal SNV count, calculated using n = 10 mucosal, n = 25 benign, and n = 16 dysplastic samples. Error bars are 95% Bayesian credible intervals (Supplementary Methods).
Extended Data Fig. 7 Clonal SNV counts and monoclonal/polyclonal classifications in published multi-region WES FAP and sporadic CRC cohorts.
a. Estimated clonal SNV counts in an additional published FAP cohort with multi-region WES data24. Each lesion (polyp or adenocarcinoma tumor) is at an x-axis location, and sequencing data from each region in the lesion are shown as individual points. Point colors indicate sample type; green: mucosa, orange: benign, blue: dysplastic, and pink: AdCa. X’s indicate the number of SNVs shared between all regions of each lesion, and the dashed horizontal line is the polyclonal cutoff. Samples marked in green text on the x-axis were classified as polyclonal, while samples marked in purple text are monoclonal. b. Estimated clonal SNV counts in a published sporadic CRC cohort with WES data from adenomas and adenocarcinomas26. Each lesion (adenoma or adenocarcinoma tumor) is at an x-axis location, and sequencing data from each region in the lesion are shown as individual points. Point colors indicate sample type; blue is adenomas and pink is adenocarcinomas. X’s indicate the number of SNVs that are shared between all regions of each lesion, regardless of VAF, and the dashed horizontal line is the polyclonal cutoff. Samples marked in green text on the x-axis were classified as polyclonal, while samples marked in purple text are monoclonal. c-d. Fraction of polyclonal samples (where each sample is a lesion or normal mucosal region, not an individual sequenced region within a lesion/region) in the multi-region datasets (FAP cohort data in c with n = 5 mucosal, n = 10 benign, n = 9 dysplastic, and n = 5 AdCa samples, and sporadic CRC cohort data in d with n = 8 adenomas and n = 4 carcinomas). Colors indicate sample type as in a and b. Error bars are 95% Bayesian credible intervals (Supplementary Methods).
Extended Data Fig. 8 Telomere length in monoclonal and polyclonal polyps.
Boxplot showing computationally estimated telomere length (TL) (Supplementary Methods) from WGS data for normal mucosa and benign and dysplastic polyps, separated by patient. Polyclonal and monoclonal lesions are plotted separately for benign and dysplastic samples. The age at which colectomy was performed for each patient is noted under the patient IDs and patients are ordered by age. Each point is one sequenced sample from one polyp or normal mucosal region, with n = 9 mucosal, n = 6 polyclonal dysplastic, and n = 10 monoclonal dysplastic samples for A002, n = 4 mucosal and n = 1 monoclonal dysplastic samples for A014, n = 2 mucosal, n = 2 polyclonal benign, n = 2 monoclonal benign, n = 4 polyclonal dysplastic and n = 16 monoclonal dysplastic samples for F001, n = 2 mucosal, n = 6 monoclonal benign, n = 2 polyclonal dysplastic, and n = 11 monoclonal dysplastic samples for G001, n = 4 mucosal and n = 2 polyclonal dysplastic samples from A015, and n = 4 mucosal, n = 4 polyclonal benign, n = 1 monoclonal benign, n = 1 polyclonal dysplastic, and n = 1 monoclonal dysplastic samples from A001. P-values were computed to compare polyclonal and monoclonal TL in dysplastic polyps with at least two data points within individual patients using two-sided Wilcoxon rank-sum tests. The combined p-value was computed using Fisher’s method. In box-and-whisker plots, the box represents the IQR with the center line representing the median, and the whiskers are the largest and smallest data values within 1.5 times the IQR from the box edges.
Extended Data Fig. 9 FAP sgWGS sampling distribution and additional phylogenetic trees.
a-b. Distribution of sampled lesions for patient FAP01 (a) and FAP03 (b). Samples taken from the normal mucosa are shown in blue. Lesions that passed our filters are highlighted in green. Each patient’s germline APC mutation is annotated as well. c. Oncoplot heatmap of mutations in a list of 20 known CRC drivers across the four sgWGS-sampled lesions that passed our filters. For lesion FAP01_T3, this includes mutations originally filtered out (Figure 4f; Supplementary Note 3, Section 5). CNVs are grouped into loss-of-heterogeneity (LOH), amplification (AMP) and deletion (DEL) variants. SNVs are grouped into frame-shift (FS), premature stop-codon (STOP) and miss-sense (MISS) variants. For APC SNVs, the amino-acid position affected by the mutation is indicated as well. d-e. Phylogenetic trees from lesions FAP01_P6 (d) and FAP03_P1 (e). Trees were reconstructed from all SNVs and 1-base INDELs at VAF ≥ 0.2, using the neighbor-joining algorithm. Colors indicate the spatial region of the polyp or lesion from which glands were extracted, or glands from normal mucosa (in blue). Terminal branches are annotated with missense mutations, nonsense mutations, frameshift mutations and CNVs in a list of 20 known CRC drivers, if the sample below that branch carries such a mutation. R, region; P, polyp; N, normal. Illustrations in a and b were created using BioRender (https://biorender.com), Figdraw (https://www.figdraw.com) and Servier Medical Art under a CC BY 4.0 licence.
Extended Data Fig. 10 Mutation sharing across sgWGS samples within lesions.
a. Mutation sharing across the 20 samples from FAP03_P2. The y-axis was split to be able to faithfully represent the extent of higher-level mutation sharing, while still capturing (on a log10 scale) the mutations shared by 5 or fewer samples. b. Mutation sharing within the six regions sampled from lesion FAP03_P2. c. Mutation sharing across the 10 samples from FAP01_T3. d. Mutation sharing across the 7 samples from FAP01_P6. d. Mutation sharing across the 7 samples from FAP03_P1. a-e. In all plots, the colored bars indicate the counts of mutations that passed our coverage and VAF filters, while the grey bar indicates the counts of mutations that got filtered out (Supplementary Note 3).
Supplementary information
Supplementary Information (download PDF )
Methods, Supplementary Figs. 1–17 and legends and Supplementary Notes 1–3.
Supplementary Table 1 (download XLSX )
Information including gender, race/ethnicity, colectomy age and germline mutations of the six FAP donors.
Supplementary Table 2 (download CSV )
Metadata about the normal mucosa and polyp information including stage and location and sequencing data type.
Supplementary Table 3 (download CSV )
Sequencing coverage of whole-genome and -exome sequencing data.
Supplementary Table 4 (download CSV )
Metadata about the single-crypt whole-genome sequencing data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Van Egeren, D., Schenck, R.O., Khan, A. et al. Polyclonal origins of human premalignant colorectal lesions. Nature 650, 1017–1024 (2026). https://doi.org/10.1038/s41586-025-09930-y
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09930-y
