
Abstract
Genome-wide association studies (GWAS) have identified >80 Alzheimer’s disease and related dementias (ADRD)-associated genetic loci. However, the clinical outcomes used in most previous studies belie the complex nature of underlying neuropathologies. Here we performed GWAS on 11 ADRD-related neuropathology endophenotypes with participants drawn from the following three sources: the National Alzheimer’s Coordinating Center, the Religious Orders Study and Rush Memory and Aging Project, and the Adult Changes in Thought study (n = 7,804 total autopsied participants). We identified seven independent significantly associated loci, of which three were new (COL4A1, LZTS1 and APOC2). Separately testing known ADRD loci, 19 loci were significantly associated with at least one neuropathology after false-discovery rate adjustment. Genetic colocalization analyses identified pleiotropic effects and quantitative trait loci. Methylation in the cerebral cortex at two sites near APOC2 was associated with cerebral amyloid angiopathy. Studies that include neuropathology endophenotypes are an important step in understanding the mechanisms underlying genetic ADRD risk.
Similar content being viewed by others

Main
Amnestic dementia, often diagnosed as late-onset Alzheimer’s disease (LOAD), is increasingly recognized to be a heterogeneous clinical syndrome that may reflect multiple underlying heritable pathological processes1,2,3. LOAD genome-wide association studies (GWAS) have primarily used clinical diagnosis or proxy phenotypes based on family history of dementia4,5,6,7. While these GWAS have been immensely successful, identifying over 80 disease-associated genetic loci6, the use of clinical phenotypes complicates interpretation and partly obscures the complex and common reality of mixed neuropathologies in aged individuals3,8. To complement the successes from previous studies, GWAS using neuropathology endophenotypes (NPEs) is an essential next step to identify loci that drive specific Alzheimer’s disease and related dementias (ADRD)-associated pathologic mechanisms9.
Amyloid plaques and neurofibrillary tangles (NFT), together known as Alzheimer’s disease (AD) neuropathologic changes (ADNC), are present at autopsy in most brains from patients diagnosed with clinical LOAD, but ~20% of clinically diagnosed patients do not have ADNC, and >50% of those with ADNC have comorbid non-AD pathologies3,10. For example, transactive response (TAR) DNA-binding protein 43-kDa (TDP-43) pathology was found in >50% of elderly autopsied individuals11 in a community-based cohort study. Limbic-predominant age-related TDP-43 encephalopathy (LATE) is an amnestic dementia syndrome defined by a distinguishing pattern of LATE-neuropathological change (LATE-NC) characterized by TDP-43 proteinopathy that is most severe in the medial temporal lobes3,12,13. Hippocampal sclerosis of aging is characterized by neuronal death, gliosis and atrophy of the hippocampus beyond normal ranges based on levels of ADNC, commonly co-occurs with LATE-NC, and is associated with severe cognitive impairment12,14.
Cerebrovascular pathologies also contribute to cognitive decline and dementia and are prevalent among elderly autopsied research participants15. Cerebral amyloid angiopathy (CAA) is characterized by amyloid-β deposition in cerebral blood vessels16. CAA often co-occurs with ADNC but can independently contribute to cerebral injury15,16. Infarcts of both grossly visible arteries and microscopically examined vessels (the latter referred to as microinfarcts) are also common contributors to cognitive decline15,17. Cerebral large-vessel atherosclerosis and small-vessel/arteriolar thickening (arteriolosclerosis) are associated with infarcts, white matter rarefaction and hippocampal sclerosis18,19 and contribute to cognitive decline20,21. Collectively, these factors reveal a complex web of pathologies that contribute to cognitive impairment and dementia.
Examining the genetic risk factors of each subtype of neuropathology can provide an important and complementary approach to large GWAS of clinical- and family history-based outcomes for studying LOAD/ADRD risk. Previous GWAS of NPEs have confirmed known LOAD risk loci and have identified new neuropathology risk loci22,23,24,25,26,27. Some NPEs, particularly LATE-NC, have yet to be studied systematically using GWAS. Here we performed GWAS on 11 NPEs using three high-quality data sources with both autopsy and genotype data. We also performed downstream functional analyses to explore potential biological functional mechanisms of newly identified risk loci and provide insight into previously identified putative AD risk loci.
Results
Participant and NPE characteristics
Genotype and neuropathology data were analyzed from the following three autopsy data sources: (1) the National Alzheimer’s Coordinating Center (NACC; n = 5,940), (2) the Religious Orders Study and Rush Memory and Aging Project (ROSMAP; n = 1,183) and (3) the Adult Changes in Thought (ACT; n = 681) study (Fig. 1). In total, 7,804 unique participants were included in our analyses. The number of participants included in each GWAS ranged from 6,363 for amyloid-β plaques to 7,786 for Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) neuritic amyloid plaque score, except for LATE-NC, which had a smaller sample due to the more recent discovery and evaluation of TDP-43 pathology (n = 3,112; Table 1).

We performed GWAS meta-analyses of 11 NPEs across three data sources. White boxes represent data sources or summary statistics used in this study. Purple boxes represent individual steps throughout the genetic association analysis, and green boxes represent downstream functional analyses. The first stage of this analysis involved independent GWAS performed in parallel across the NACC neuropathology dataset, the ACT study and the combined ROSMAP. We then performed a meta-analysis using results from each individual GWAS using METAL. Variants reaching a suggestive threshold of association (P ≤ 1 × 10−5) in the meta-analysis were then carried forward for downstream analyses, including functional and colocalization analyses. Variants reaching the genome-wide significant threshold (P ≤ 5 × 10−8) and exhibiting ≥80% colocalization between two NPEs were followed up using existing methylation data to assess the association. All variants reaching genome-wide significance were considered associated with the respective NPE. We also report variants that reached a suggestive threshold (P ≤ 5 × 10−7) or reached the lower suggestive threshold (P ≤ 1 × 10−5) and were in a previously known disease-associated locus. GTEx, Genotype-Tissue Expression Project; QTL, quantitative trait locus; eQTL, expression QTL; sQTL, splicing QTL; mQTL, methylation QTL; AD, Alzheimer’s disease.
The 11 studied NPEs included AD-related pathologies (CERAD score for neuritic amyloid plaques, amyloid-β plaques including diffuse plaques and Braak NFT staging28,29,30), non-AD neurodegenerative proteinopathies (LATE-NC and Lewy bodies12,31), cerebrovascular pathologies (CAA, gross infarcts, microinfarcts, circle of Willis atherosclerosis and arteriolosclerosis30,32) and hippocampal sclerosis32. NPEs commonly co-occurred, forming the following four identifiable clusters of pathologies: vascular, AD, LATE and Lewy body (Extended Data Fig. 1). Methods, Supplementary Methods and Supplementary Table 1 describe the applied phenotype definitions and harmonization approach.
GWAS meta-analysis of NPEs
We first performed GWAS on the 11 NPEs for the NACC, ROSMAP and ACT studies separately (Fig. 1). Genetic association analyses were performed with logistic or proportional-odds logistic regression mixed-effects models as appropriate (Methods). We then performed fixed-effects meta-analyses using METAL 2011-03-25 (ref. 33) on variants with minor allele frequencies ≥1% in at least one study33. Quantile–quantile plots and corresponding estimates of genomic inflation (λ values) did not suggest systematic bias (Extended Data Fig. 2).
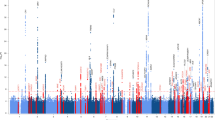
In total, the meta-analysis revealed six loci with at least one variant meeting genome-wide significance (P < 5 × 10−8) across eight NPEs (amyloid-β plaques, arteriolosclerosis, atherosclerosis, Braak NFT stage, CAA, CERAD plaque score, hippocampal sclerosis and LATE-NC), with a total of 12 associations between genomic loci and NPEs (Fig. 2). Four of the six loci were from genes previously associated with late-onset ADRD (the broader APOE region, TMEM106B, GRN and BIN1; Fig. 2a–c,f,i,k), while two loci were new, where the lead variant was in or closest to LZTS1 and COL4A1, respectively (Fig. 2d,e). Although most meta-analyses had no significantly different effect size estimates across the three data sources, there were three with significant tests for heterogeneity (Table 2), all of which were associations with APOE.

a–k, Manhattan plots are shown for amyloid-β plaques (a), Braak NFT stage (b), CERAD score for neuritic plaques (c), arteriolosclerosis (d), atherosclerosis in the circle of Willis (e), CAA (f), gross infarcts (g), microinfarcts (h), LATE-NC (i), Lewy body (j) and hippocampal sclerosis (k). The y axes denote the −log10(P value of meta-analysis two-sided z test) of the variant–phenotype association, and the x axes outline the chromosomal position, with alternate chromosomes represented in black and blue. Labels indicate the nearest gene at a locus. The horizontal lines define the genome-wide significance level (solid black, P = 5 × 10−8), near genome-wide significance level (dotted gold, P = 5 × 10−7), and suggestive significance level (P < 5 × 10−5) in loci with evidence of AD association from a previous study (for example, ref. 6; dotted purple). Points and gene symbols are coded with the same colors. Gray gene symbols indicate genome-wide significant hits within the APOE region that did not survive conditional analysis. All GWAS are in cohorts of European ancestry and adjusted for age at death, sex, genotyping cohort and top ten genetic PCs. We identified seven genome-wide significant loci and 30 near genome-wide significant or suggestive loci. The genome-wide significant loci resulted in 12 associations with eight NPEs (amyloid-β plaques, arteriolosclerosis, atherosclerosis, Braak NFT stage, CAA, CERAD score, hippocampal sclerosis and LATE-NC). Four genes were previously associated with ADRD (APOE, BIN1, TMEM106B, GRN; a–c,f,i,k), while the three new loci were in or closest to LZTS1, COL4A1 and APOC2 (d–f). APOC2 is within the broader APOE region but remained significantly associated with CAA after adjusting for APOE \(\epsilon\) diplotypes (f). Three NPEs (gross infarcts, microinfarcts and Lewy bodies) had zero genome-wide significant hits, but all three had near genome-wide significant and/or suggestive hits from either new or known loci. APOE was associated with a range of NPEs, including LATE-NC, which is not pathognomonic of AD. On the other hand, neither GRN nor TMEM106B (recently identified in ADRD GWAS) was associated with the AD pathognomonic NPEs but were specific to gross infarcts, LATE-NC and hippocampal sclerosis at either genome-wide or suggestive significance. sig., significant; assocs, associations; sugg., suggestive.
We subsequently discovered a new locus near APOC2 within the broader APOE region that is associated with CAA after adjusting for APOE \(\epsilon\) diplotypes (Fig. 2f). No loci reached genome-wide significance with gross infarcts, microinfarcts or Lewy body pathology (Fig. 2g–j).
Known ADRD-associated loci
As expected, the APOE region (rs429358) is associated with multiple NPEs at genome-wide significance. Specifically, the APOE region (rs429358) is associated with (1) amyloid-β plaques (odds ratio (OR) = 1.98; P = 2.3 × 10−55; Table 2 and Fig. 2a), (2) Braak NFT stage (OR = 2.06, P = 9.7 × 10−89; Fig. 2b), (3) CERAD score (OR = 2.42, P = 4.7 × 10−103; Fig. 2c), (4) CAA (OR = 2.49, P = 4.4 × 10−138; Fig. 2f) and (5) LATE-NC (OR = 1.70, P = 1.7 × 10−14; Fig. 2i). Our results corroborate previous studies22,34 that described an association between APOE and CAA.
BIN1 was associated with Braak NFT stage (rs6733839; OR = 1.21; P = 1.6 × 10−9; Table 2 and Fig. 2b), and variants within TMEM106B were associated with both LATE-NC (rs2043539; OR = 0.70, P = 5.8 × 10−11; Fig. 2i) and hippocampal sclerosis (rs7805419; OR = 0.65; P = 3. 2× 10−13; Fig. 2k). A locus in GRN was also associated with hippocampal sclerosis (rs5848; OR = 1.40, P = 3.2 × 10−8; Fig. 2k).
New loci outside the APOE region
We also discovered two new loci outside the broader APOE region that are associated with three NPEs. Associations identified in the NPE GWAS meta-analysis included a suggestive PIK3R5 intronic locus associated with Braak NFT stage (rs72807981; OR = 0.71, P = 1.3 × 10−7; Supplementary Table 8 and Figs. 2b and 3a,b; see Supplementary Results for details on suggestive loci), an intronic LZTS1 locus associated with arteriolosclerosis (rs78909048; OR = 0.44, P = 5.8 × 10−10; Table 2 and Figs. 2d and 3d,e) and a variant 12 kilobase pairs (kbp) upstream of COL4A1 associated with the circle of Willis atherosclerosis (rs2000660; OR = 0.73, P = 2.7 × 10−8; Figs. 2e and 3g,h).

a, Braak stage association plot from NPE GWAS meta-analysis (n = 7,776) for the region around PIK3R5. Colored dots represent the chromosomal position (x axis, Mb) in hg38 coordinates and −log10(P value from meta-analysis two-sided z test; y axis) of each variant in the region. Dots are colored to represent the LD r2 with the lead variant (purple diamond) estimated with PLINK–r2 using 1000 Genomes phase 3 European-descended participants. The recombination rate was calculated using GRCh38 genetic map files downloaded from https://bochet.gcc.biostat.washington.edu/beagle/genetic_maps/ and taking the ratio of difference of CM and Mb between positions. Boxes below data indicate the location of genes in the region. (Plot generated using LocusZoom73.) b, Association of PIK3R5 lead variant (rs72807981) with Braak stage for individual cohorts (NACC, n = 5,927 and ACT, n = 677; this variant was not present in ROSMAP) and meta-analysis (n = 6,604) using METAL (y axis). Points along the x axis represent OR of association, and error bars indicate 95% CI. c, Human brain cell-type expression profile of PIK3R5 in ref. 35. Columns represent mean FPKM. Error bars indicate the s.e. of measurement for each cell type based on the number of human samples sequenced for each type (fetal astrocytes, n = 6; mature astrocytes, n = 12; endothelial, n = 2; microglia, n = 3; neurons, n = 1 and oligodendrocytes, n = 5). PIK3R5 is primarily expressed in microglia. d, Arteriolosclerosis association plot from NPE GWAS meta-analysis (n = 6,668) for the region around LZTS1 (see a for interpretation). e, Association of LZTS1 lead variant (rs78909048) with arteriolosclerosis for individual cohorts (NACC, n = 4,930; ROSMAP, n = 1,163 and ACT, n = 575) and meta-analysis (n = 6,668) using METAL (y axis; see b for interpretation). f, Human brain cell-type expression profile of LZTS1 in ref. 35. LZTS1 is primarily expressed in fetal astrocytes and endothelial cells (see c for interpretation). g, Cerebral atherosclerosis association plot from NPE GWAS meta-analysis (n = 7,340) for the region around COL4A1 (see a for interpretation). h, Association of COL4A1 lead variant (rs2000660) with cerebral atherosclerosis for individual cohorts (NACC, n = 5,496; ROSMAP, n = 1,175 and ACT, n = 669) and meta-analysis (n = 7,340) using METAL (y axis; see b for interpretation). i, Human brain cell-type expression profile of COL4A1 in ref. 35. COL4A1 is preferentially expressed in fetal astrocytes and endothelial cells with lower expression in neurons (see c for interpretation). Mb, megabase.
We next characterized which cell type(s) in the human brain express the new genes identified. According to brainrnaseq.org35,36, PIK3R5 is most highly expressed in microglia (Fig. 3c), LZTS1 is most highly expressed in fetal astrocytes and endothelial cells (Fig. 3f) and COL4A1 is most highly expressed in fetal astrocytes, endothelial cells and neurons (Fig. 3i).
New locus association within the APOE region
Based on the meta-analysis, we observed 12 genetic locus-phenotype associations within the broader APOE region (defined as less than 500 kbp from the start or end site of APOE transcription) across five NPEs (amyloid plaques, Braak stage, CAA, CERAD score and LATE-NC), where APOE itself (rs429358) was the top variant in the region for all five NPEs (Fig. 2a–c,f,i). We performed additional analyses in this region adjusting for APOE \(\epsilon\) diplotypes to determine whether any of the genome-wide significant signals within the broader APOE region remained significant.
In the APOE-adjusted analysis, the lead variant from the nonadjusted analysis (rs429358), which tags the APOE ε4 allele (the well-known common variant with the strongest association with LOAD), was no longer associated with any of the five phenotypes. One locus with lead variant rs7247551 remained significantly associated with CAA (OR = 0.81; P = 8.0 × 10−12; Table 2 and Fig. 2f). rs7247551 is located between APOC2 and CLPTM1. No variants remained genome-wide significantly associated with any other APOE-associated NPE. Sensitivity analyses showed that the effect size of rs7247551 did not significantly differ based on APOE diplotype in NACC, ROSMAP or ACT (Supplementary Results and Extended Data Fig. 3).
APOC2 replicates in an independent cohort
We obtained data from a recent GWAS of CAA in 815 participants with dementia in the Mayo Clinic Brain Bank. Using their data, we replicated the association between rs7247551 and CAA while adjusting for APOE \(\epsilon\) diplotypes (P = 0.0012). We also confirmed that rs7247551 was indeed new and not in linkage disequilibrium (LD) with the variant previously reported in ref. 26 (rs5117; r2 < 0.01). Together, these results provide evidence for a new locus within the broader APOE region that is independent of the APOE \(\epsilon\) diplotypes and is associated with CAA pathology burden. It further suggests that the genetic risk for CAA in the broader APOE region may differ from the AD-specific neuropathologies (neuritic amyloid plaques and NFT).
Associations of clinical and proxy AD risk loci with NPE
We further tested whether LOAD-associated loci identified in a recent ADRD GWAS were associated with any evaluated NPEs6. Reference 6 identified a total of 83 distinct non-APOE loci (39 previously identified and 44 new) associated with ADRD (hereafter, ‘ADRD loci’), between 76 and 78 of which had lead variants that met inclusion criteria in our study for each NPE. In total, 26 NPE-locus associations from 19 loci had adjusted P (Q values) ≤0.05 across the 11 NPEs. Of the 26 associations, 24 had concordant directions of effect with ref. 6 (Table 3 and Supplementary Table 2).
Notably, the pathognomonic AD pathologies, operationalized using Braak NFT stage and CERAD score, had concordant directions of effect with 68 and 59 of 77 ADRD loci, respectively, regardless of statistical significance. Five ADRD loci (BIN1, MME, HLA-DQA1, TMEM106B and CELF1/SPI1; Table 3) were significantly associated with the Braak NFT stage after multiple testing corrections. Two ADRD loci (PICALM and TPCN1) were associated with CAA. Eleven ADRD loci (CR1, BIN1, INPP5D, ZCWPW1/NYAP1, PTK2B, CELF1/SPI1, PICALM, SORL1, FERMT2, SNX1 and ABCA7) were significantly associated with the CERAD score after multiple testing corrections. Four ADRD loci (TMEM106B, IL34, GRN and MAPT) were significantly associated with hippocampal sclerosis, all of which except for IL34 (hippocampal sclerosis—OR = 0.74, P = 0.0019; AD—OR = 1.06, P = 5.6 × 10−6) were concordant in effect direction. Two ADRD loci (TMEM106B and GRN) were significantly associated with LATE-NC. Finally, two ADRD loci (TPCN1 and PLCG2) were significantly associated with microinfarcts. These results indicate that NPE studies largely corroborate the findings of large AD GWAS based on clinical and proxy phenotypes. Several ADRD loci, particularly MAPT (hippocampal sclerosis), TMEM106B and GRN (hippocampal sclerosis and LATE-NC), were associated only with non-AD pathology.
Identifying potential genetic mechanisms of NPE development
We also assessed possible mechanisms through which identified NPE-associated variants may be involved in disease risk.
Gene-prioritization and enrichment analyses
Using gene-based, pathway, and enrichment analyses, we identified APOE as significantly associated with NFT, diffuse plaques, CAA, neuritic plaques and LATE-NC (Supplementary Table 3)37. TMEM106B was associated with both hippocampal sclerosis and LATE-NC. We further found that rs2000660 (associated with the circle of Willis atherosclerosis) is located within an enhancer region 13 kbp upstream of COL4A1 transcription start site (Extended Data Fig. 4). Annotations from FAVOR identified a synonymous, exonic single-nucleotide polymorphism (SNP), rs650724, in high LD (r2 = 0.86) with this lead variant that is highly conserved across mammals (mamPhCons = 0.987; Supplementary Table 4)38.
Genetic colocalization analysis
We investigated whether loci associated with multiple NPEs show evidence for genetic colocalization39. We identified two NPE pairs exhibiting genetic colocalization, including (1) Braak NFT stage and CERAD score (BIN1; probability of colocalization (PrC) > 99%; Extended Data Fig. 5) and (2) hippocampal sclerosis and LATE-NC (TMEM106B; PrC = 90%; Fig. 4a–c).

We investigated whether loci associated with multiple NPEs show evidence for genetic colocalization using a Bayesian colocalization analysis approach implemented in the coloc R package72. a, The TMEM106B lead variant (rs2043539) reached genome-wide significance with LATE-NC. b,c, Hippocampal sclerosis colocalized with LATE-NC on TMEM106B (PrC = 90%). d,e, TMEM106B expression colocalized with LATE-NC (PrC = 91%). f–i, Two methylation QTL (mQTLs), cg09613507 (PrC = 89%; f,g) and cg23422036 (PrC = 95%; h,i), also colocalized with LATE-NC. a, b, d, f and h show regional LocusZoom73 plots for each trait. Purple diamonds represent lead variants. c, e, g and i compare −log10(P) values between each trait compared to LATE-NC −log10(P) values across the TMEM106B rs2043539 locus (color legend same as in a). The TMEM106B expression and the methylation data were obtained from ROSMAP. j, Decreased TMEM106B expression was associated with more severe LATE-NC pathology (P = 0.043). Unless otherwise specified, for all boxplots, boxes outline the first quartile, median and third quartile. Whiskers extend up to 1.5× the distance between the first and third quartiles. k, Hypermethylation of cg09613507 was associated with more severe LATE-NC pathology (P = 0.0093). l, Methylation at cg23422036 was not significantly associated (P = 0.10). m, Unrelated to TMEM106B, BIN1 expresses eight distinct RNA isoforms simultaneously in the frontal cortex from six AD cases and six controls. To understand the complexities and nuances of ADRDs, we also need to understand the nuances of the genes purported to be driving disease. CDS, coding sequence; CTRL, control; CPM, counts per million; F, female; M, male; hipp. scler., hippocampal sclerosis; expr. cer., expression in cerebellum.
We also tested for colocalization between NPE loci and quantitative trait loci (QTL). The TMEM106B locus (associated with hippocampal sclerosis and LATE-NC) colocalized with TMEM106B expression in multiple tissues, including the cerebellar hemisphere (PrC = 91%; Fig. 4d,e). Two CpG sites located either within TMEM106B (cg09613507; Fig. 4f) or upstream (cg23422036; Fig. 4h) colocalized with both hippocampal sclerosis (cg09613507-hippocampal sclerosis PrC = 94%, cg23422036-hippocampal sclerosis PrC = 94%; Fig. 4b,f,g) and LATE-NC (cg09613507-LATE-NC PrC = 89% and cg23422036-LATE-NC PrC = 95%; Fig. 4a,f–i). A hippocampal sclerosis-associated locus within GRN strongly colocalized with both LATE-NC and GRN expression in multiple tissues (PrC > 99.9%; Extended Data Fig. 6).
APOC2 colocalized with several traits, including methylation QTL (mQTL) for four CpG sites in ROSMAP (cg04401876, cg10169327, cg13119609 and cg09555818; PrC = 96–99%; Fig. 5a–i). The APOC2 locus also colocalized with an expression of multiple genes in Genotype-Tissue Expression Project (GTEx), including APOE expression in the wall of the aorta (PrC = 94%), CLPTM1 expression in the skin of the leg and suprapubic region and APOC2 expression in 17 different tissues (for example, brain cortex, caudate, nucleus accumbens and cerebellum; PrC = 89–96%).

Using the same Bayesian colocalization analysis approach from Fig. 4 (coloc 5.2.2 R package72), we tested for colocalization between CAA and methylation sites using existing data from ROSMAP. a, Lead SNP rs7247551, near APOC2, reached genome-wide significance with CAA. b,d,f,h, The rs7247551 was also significantly associated with four mQTL. b–i, cg04401876 (PrC = 96%; b,c), cg10169327 (PrC = 96%; d,e), cg13119609 (PrC = 99%; f,g) and cg09555818 (PrC = 97%; h,i) all colocalized with CAA. a, b, d, f and h show regional LocusZoom73 plots for each trait. c, e, g and i compare −log10(P) values between each trait compared to CAA −log10(P) values across the APOC2 rs7247551 locus. Variants in LD with the lead variant (purple diamond in a–i) are shaded in a–i according to the color legend on the left-hand side of a. j, Plots of normalized methylation level versus CAA pathology severity. Hypomethylation at cg09555818 (OR = 0.82, P = 0.003) and cg13119609 (OR = 0.78, P = 0.0006) were significantly associated with more severe CAA pathology. Unless otherwise specified, for all boxplots, boxes outline the first quartile, median and third quartile. Whiskers extend up to 1.5× the distance between the first and third quartiles. k, Both cg09555818 (P = 0.0063; k) and cg13119609 (P = 0.0069; not shown) were significantly associated with APOC2 expression. l,m, The rs7247551 G allele was significantly associated with increased APOC2 expression in the frontal cortex in ROSMAP (β = 0.072, P = 0.00013; l); however, the direction of effect was opposite of that found in brain tissues in GTEx (P = 7.2 × 10−7; m). n,o, The rs7247551 was not associated with APOE (P = 0.81; n) or APOC2 (P = 0.89; o) expression in frontal cortex in ROSMAP. p, APOC2 is highly expressed, especially in microglia and oligodendrocytes. Columns represent mean FPKM. Error bars indicate the s.e. of measurement for each cell type based on the number of human samples sequenced for each type (fetal astrocytes, n = 6; mature astrocytes, n = 12; neurons, n = 1; oligodendrocytes, n = 5; endothelial, n = 2 and microglia, n = 3). expr., expression; norm., normalized.
Multiple suggestive NPE loci showed evidence of colocalization with expression QTL (eQTL) in GTEx. In total, 50 NPE loci (lead variant P < 1 × 10−5) colocalized with various QTL types (that is, expression, methylation or splicing QTL [sQTL]; PrC ≥ 80%). A total of 407 NPE–QTL pairs colocalized across 47 tissues (Supplementary Table 5), many giving credence to previously discovered associations and insight into potential mechanisms. For example, rs1643235 (ABCC9) colocalized with hippocampal sclerosis and gene expression in multiple tissues, including the cortex (PrC = 80%), corroborating previous studies27,40,41 because the ABCC9 SNP rs4148674 (r2 = 0.96 with rs1643235) was a robust eQTL for ABCC9 and the strongest ABCC9 region signal for association with hippocampal sclerosis (Extended Data Fig. 7).
Functional studies in ROSMAP
Using data from ROSMAP participants with DNA methylation and/or RNA-sequencing (RNA-seq) data available from the dorsolateral prefrontal cortex (DLPFC), we found that neither TMEM106B nor GRN expression was associated with hippocampal sclerosis (P > 0.05), while decreased TMEM106B expression was associated with more severe LATE-NC pathology (P = 0.043; Fig. 4j). Of the two CpG sites that colocalized with hippocampal sclerosis and LATE-NC, hypermethylation of cg09613507 was associated with more severe LATE-NC pathology (P = 0.0093; Fig. 4k), while cg23422036 was not significantly associated (P = 0.10; Fig. 4l).
Additionally, we tested for association between CAA pathology and methylation levels at four CpG sites (cg09555818, cg04401876, cg10169327 and cg13119609) that colocalized with the chromosome 19 (chr19) CAA risk locus (rs7247551). We first confirmed that all four CpG sites were significantly associated with rs7247551 (P < 0.0001) and had directions of effect consistent with those previously reported for ROSMAP42. Hypomethylation at cg09555818 (OR = 0.82, P = 0.003) and cg13119609 (OR = 0.78, P = 0.0006) were significantly associated with more severe CAA pathology (Fig. 5j). Both cg09555818 (P = 0.0063; Fig. 5k) and cg13119609 (P = 0.0069) were significantly associated with APOC2 expression.
Next, as APOC2 expression in multiple brain tissues colocalized with CAA in GTEx but not ROSMAP, we investigated whether there was a nominal association between APOC2 expression in the DLPFC and rs7247551. We found that the G allele of rs7247551 was nominally associated with increased APOC2 expression in the DLPFC (β = 0.072, P = 0.00013; Fig. 5l); however, the direction of effect was opposite of that found in brain tissues in GTEx (that is, the G allele of rs7247551 was associated with decreased APOC2 expression in GTEx; P = 7.2 × 10−7; Fig. 5m). Expression of APOC2 in the DLPFC was not associated with CAA in ROSMAP (OR = 0.98, P = 0.89). We performed an additional post hoc analysis for nominal APOE eQTL activity of rs7247551 in ROSMAP. We confirmed that rs7247551 was not associated with APOE expression in the DLPFC in ROSMAP (P = 0.81; Fig. 5n). APOC2 expression was also not significantly associated with the severity of CAA pathology in ROSMAP (P = 0.089; Fig. 5o). Notably, APOC2 is highly expressed, especially in microglia and oligodendrocytes (Fig. 5p).
Potential effects via differential RNA splicing
Many human genes undergo alternative splicing43,44,45. Recent work in ref. 46 demonstrated that many medically relevant genes express multiple RNA isoforms that result in unique proteins, including genes involved in ADRD. Specific examples include APP, MAPT and BIN1, which express five, five and eight distinct RNA isoforms above noise levels, respectively, in the human frontal cortex. Thus, we explored whether any genes associated with NPEs in this work express multiple RNA isoforms in the human frontal cortex.
While all loci that were associated with NPE have multiple annotated RNA isoforms, ranging from 4 (LZTS1) to 24 (PICALM) per Ensembl, most of the genes exhibited expression for only a single isoform above noise levels, per data from ref. 46. Some expressed multiple RNA isoforms, but the isoforms were not predicted to result in distinct proteins (for example, LZTS1)—although recent data suggest that alternative untranslated regions have direct functional consequences47,48,49. BIN1, however, actively transcribes eight distinct RNA isoforms in the frontal cortex (Fig. 4m). BIN1 is also expressed in multiple brain cell types in humans, according to brainrnaseq.org35,36. Given the diversity of BIN1 isoforms simultaneously expressed, we need to understand whether specific isoforms are involved in disease, and we propose differential RNA isoform expression as a potential mechanism through which disease genes may be affecting disease.
Discussion
The present study of 11 ADRD-related NPEs comprised 7,804 participants. These results provide an autopsy-based complement to previous studies based on clinical diagnoses and expand on the findings of previous genetic studies of dementias and neuropathologies2,6,22,23,24,26,41. In this work, we confirm previous findings that several loci associated with AD (including APOE) are also associated with non-ADNC pathogenesis. Additionally, some of the known ADRD loci (that is, TMEM106B and GRN) did not associate with any of the classical AD-defining NPEs in this study. Thus, our results improve our collective understanding of the complex nature of ADRD and its genetic bases.
Sharpening the endophenotypes enabled the discovery of new hit genes that did not reach genome-wide significance in previous studies oriented toward studying AD clinical phenotypes. We identified clear genetic associations between specific neuropathologies and loci already associated with ADRD and three new NPE-associated loci. Yet, many questions remain and will require deeper investigation. For example, three of the NPEs studied (gross infarcts, microinfarcts and Lewy body pathology) did not have any locus reach genome-wide significance in our study. Likely explanations for the lack of genome-wide associations for these three pathologies include (1) insufficient sample size, (2) the cohorts not being specifically designed to study these phenotypes, and (3) variability in the collection and scoring of these phenotypes over time and between research centers. Nonetheless, these three NPEs each had suggestive associations, including with known loci (Supplementary Results). Specifically, TMEM106B and APOE had suggestive associations with gross infarcts and Lewy body pathology, respectively. APOE ?4 was previously associated with Lewy body pathology by a study discussed in ref. 50. As study cohorts grow larger, researchers will be able to re-assess these suggestive associations.
As expected, APOE variation was associated with ADNC, CAA and LATE-NC. Although LATE-NC is diagnostically characterized by TDP-43 proteinopathy3,12,13, brains with a greater burden of neuritic amyloid plaques are more likely to have TDP-43 proteinopathy (and vice versa)12. Similarly, others have previously shown an association between APOE ?4 status and TDP-43 pathology in ROSMAP11. Unraveling the complex interaction between APOE and proteinopathies (including the suggestive association to Lewy bodies) may provide a crucial insight into properly treating patients with these often comorbid pathologies. A study using a mouse model suggests that TDP-43 directly interacts with amyloid-β and promotes worse pathology51, but further studies are needed to better understand this interaction.
By adjusting for APOE ? diplotypes, we identified a new locus near APOC2 associated with CAA. Like APOE, APOC2 is part of the apolipoprotein family and is involved in lipoprotein metabolism. Both genes directly bind fat droplets (chylomicrons)52,53 and are implicated in heart disease54—a known link to APOE and dementia. APOC2 and APOE expression is also coregulated through the same mechanisms in liver52,53. We replicated the association between APOC2 and CAA while adjusting for APOE in an independent sample of 815 Mayo Clinic Brain Bank participants used in ref. 26, providing additional evidence for the APOC2 locus being important for CAA pathology beyond the known effects of APOE \(\epsilon\) haplotypes.
Previously, a study discussed in ref. 55 reported that a variant proximal to rs7247551 (rs10413089; 822 bp away) showed evidence of association with clinical AD independently of APOE ? status in their cohort but determined their results were inconclusive. Their original association did not survive multiple testing corrections, but the association replicated in an independent cohort. Summary statistics from ref. 6 reflect that rs7247551 was significantly associated with ADRD but did not report results of an independent analysis within the broader APOE region. Thus, the present study is the first to confirm that this association is independent of the known effects of APOE alleles. Both the potential association with clinical AD status found by ref. 55 and the association with CAA we report herein should be followed up in future studies.
Several variants in the APOC2 locus were lead eQTLs for APOC2 brain expression in both GTEx and ROSMAP. Colocalization analysis confirmed that the new CAA risk locus shares a functional variant with both APOC2 eQTL and nearby brain cortex mQTL. We confirmed that two of the CpG sites affected by the CAA risk locus, cg09555818 and cg13119609, were, in turn, significantly associated with CAA pathology. Both CpG sites are located within the APOC4–APOC2 readthrough transcript region, overlapping APOC4 and APOC2. Our results are consistent with the hypothesis that the association between rs7247551 and CAA risk may be driven by hypomethylation of CpG sites in the APOC2 region. Previous studies in other human cohorts also implicate hypomethylation at cg09555818 and cg13119609 in AD56,57,58. Collectively, these results provide evidence that APOC2 may be the target gene of the rs7247551 CAA risk locus, although more research must be done for verification.
We also identified associations between known loci and multiple NPEs, including BIN1, APOE and TMEM106B. These associations provide context regarding their involvement in disease pathogenesis. One intronic locus of TMEM106B was significantly associated with both hippocampal sclerosis and LATE-NC, while a locus within GRN was associated with hippocampal sclerosis. Both genes are associated with frontotemporal lobar degeneration with TDP-43 inclusions59,60, hippocampal sclerosis61,62, and were recently associated with clinical AD5,6. We found that hippocampal sclerosis, LATE-NC, and clinical AD all colocalize at these two loci, suggesting that hippocampal sclerosis, LATE-NC and clinical AD likely share causal loci for these genes. Furthermore, hippocampal sclerosis (GRN and TMEM106B) and LATE-NC (TMEM106B) colocalized with brain eQTL and mQTL, and TMEM106B expression and methylation were associated with LATE-NC in downstream analyses. Notably, lead GRN and TMEM106B variants identified in GWAS of ADRD were not associated with the ADNC NPEs. Given that a substantial fraction of individuals diagnosed with dementia have LATE-NC or hippocampal sclerosis pathology (with estimates as high as ~50%)3, it is plausible that the associations found between these genes and clinical AD in recent GWAS are due to individuals with these non-AD pathologies who were diagnosed clinically with AD.
A locus ~30 kbp downstream of BIN1 on chr2q14 was significantly associated with Braak stage and suggestively associated with CERAD score for neuritic plaques. In previous GWAS, this locus was second only to APOE for strength of association with LOAD6. We verified through colocalization analysis that the same locus drives association signals with the Braak NFT stage and CERAD score. Interestingly, the lead variant in this locus, rs6733839, was not associated with nonneuritic amyloid plaques nor CAA, neither of which include tau deposits. Previous research supports the hypothesis that BIN1 is associated with LOAD through its effect on NFT rather than amyloid pathology63,64.
We also discovered intriguing new loci mapped to COL4A1 and LZTS1 associated with atherosclerosis in the circle of Willis and brain arteriolosclerosis, respectively.
One locus on chr13q34 with lead variant rs2000660 located 12 kbp upstream of COL4A1 was significantly associated with atherosclerosis in the circle of Willis. Previous research found that the COL4A1/COL4A2 locus is associated with numerous vascular disease phenotypes, including peripheral artery disease, coronary artery disease, stroke, arteriolar stiffness, rare familial cerebrovascular diseases and stroke65,66,67 In a recent GWAS, rs2000660 was a lead risk variant for migraines68. The relevance of the COL4A1 locus to cerebral vascular traits is thus highly supported by previous research, and the biological role of collagen IV in vascular disease is possibly related to the disruption of the extracellular matrix65. COL4A1 is preferentially expressed in astrocytes and endothelial cells and codes for a component of collagen IV, an important component of basal lamina. Endothelial cells are strongly implicated in atherosclerosis, and in recent years, researchers have suggested that astrocytes may also be directly involved in cerebrovascular disease69. The rs2000660 was not nominally associated with any other vascular NPE in our study, and a previous GWAS of circle of Willis atherosclerosis using ROSMAP participants did not identify the COL4A1 as a risk locus24. The rs650724, a variant in high LD with rs2000660 (r2 = 0.84), is a synonymous coding variant (p.Ser1600Ser in ENST00000375820.10; p.Ser319Ser in ENST00000650424.1) within COL4A1. The rs2000660 is also located within an enhancer for COL4A1, presenting possible molecular functional mechanisms driving association in this locus.
An intronic variant within PIK3R5 (rs72807981; chr17p13) met our suggestive threshold of association with the Braak NFT stage. PIK3R5 codes for a phosphatidylinositol 3-kinase involved in cell growth, motility and survival. There is previous research suggesting that PIK3R5 is more highly expressed in aged adults with Braak NFT stages V and VI versus nondemented controls70. PIK3R5 is expressed preferentially in microglial cells in humans35, suggesting that its association with neurofibrillary pathology may be immune-mediated, although future work is needed to validate the association between PIK3R5 and NFT.
One new intronic locus in LZTS1 was found to be protective against brain arteriolosclerosis. The relatively modest literature regarding LZTS1 has focused mostly on cancers; however, LZTS1 is primarily expressed in endothelial cells and astrocytes, cell types relevant to vascular function and dysfunction. One paper suggests that LZTS1 is involved in neuronal delamination and development of glial-like cells during mammalian neocortical development71, but additional work related to LZTS1 and its function in the cerebrovasculature and brain parenchyma is needed.
In conclusion, we identified promising new loci associated with NPEs and replicated multiple known risk loci for ADRD using NPE-based GWAS. Our study demonstrates the importance of studying genetic risk factors of NPEs as a complement to studies of clinical and proxy phenotypes of LOAD.
Methods
Participants
An overview of our study design is presented in Fig. 1. Each participating study previously obtained informed consent from participants or caregivers for participants with substantial cognitive impairment. Parent study protocols were approved by the local institutional review boards. This study was approved by the University of Kentucky Office of Research Integrity Institutional Review Board.
NACC
The present study used NACC data from 36 National Institute on Aging (NIA)-funded Alzheimer’s Disease Research Centers (ADRCs). Individual ADRCs use different recruitment strategies and perform autopsies on-site, but neuropathology data at each ADRC are collected using a standard form (https://files.alz.washington.edu/documentation/np11-form.pdf) and submitted to NACC where they are aggregated and anonymized. The NACC Neuropathology dataset based on the first version of this form was originally implemented in 2001 (ref. 30), and this analysis uses data from then through the March 2023 freeze. Participants were excluded if they did not have autopsy data available or if they were noted in the NACC Neuropathology dataset to have at least one of 19 conditions that could potentially bias results. These conditions include brain tumors, severe head trauma and frontotemporal lobar degeneration (see Supplementary Table 6 for the full list of variables used for exclusion criteria).
ROSMAP
ROSMAP consists of harmonized data from the following two longitudinal cohort studies: the Religious Orders Study (ROS) and the Rush Memory and Aging Project (MAP)74. ROS and MAP were both approved by the Institutional Review Board of Rush University Medical Center. All participants signed the Uniform Anatomic Gift Act, as well as informed and repository consents. ROS began in 1994 and has recruited over 1,500 Catholic priests, nuns and brothers across the United States. MAP started in 1997 and has enrolled more than 2,300 community members in the greater Chicago area of northeastern Illinois. The ROSMAP NP data used in this study were received from Rush University Medical Center in January 2020. Using KING 2.2.7 (ref. 75), we found that several participants in ROSMAP also had neuropathology and genotype data available in NACC. In these cases, records in the NACC were preferentially kept.
ACT
The ACT study began in 1994 and recruited residents in the greater Seattle area aged 65 years and older without dementia at the time of enrollment76,77,78,79. The study has expanded to include three cohorts with continued enrollment using the original enrollment criteria and has a current total of 4,960 participants across all three cohorts. The ACT NP data used in this study were obtained from Kaiser Permanente in May 2023.
Genotype data and quality control
Genotype data for all cohorts underwent imputation using the Trans-Omics for Precision Medicine (TOPMed) Imputation Server 1.7.3 and the TOPMed reference panel using Minimac 4 (refs. 80,81,82). Postprocessing was performed with BCFtools 1.10.2 (ref. 83) and SAMtools 1.10 (ref. 84), PLINK 1.9 and 2.0 (ref. 85), R 4.2.1 and 4.2.2 (https://cran.r-project.org/), and R packages data.table 1.14.10 (https://CRAN.R-project.org/package=data.table) and stringi 1.803 (ref. 86). The 3.4.2 NACC and ACT raw genotype data were obtained from the September 2020 freeze of the Alzheimer’s Disease Genetics Consortium (ADGC) in March 2021 and subsequently imputed. Pre-imputed ROSMAP genotype data were received from collaborators in the Hohman Lab at Vanderbilt University in December 2021. Genetic variants with minor allele frequency (MAF) < 0.1% and imputation quality scores of <0.8 were removed before further quality control measures. Due to the small sample sizes of participants with substantial non-European ancestry (based on proximity to 1000 Genomes ‘EUR’ superpopulation cluster in principal component (PC) analysis), especially in replication cohorts, these participants were excluded from the analysis. Standard GWAS quality control procedures were followed for variant and participant inclusion (Supplementary Methods).
Defining and harmonizing NPEs for analysis
In total, we combined and/or harmonized 11 NPEs for analysis across the four studies. We note that there are differences in the way that some neuropathological data were collected across studies, and our strategy for harmonizing was informed by practical considerations for maximizing available sample sizes given the available endophenotypes. Thus, several synthetic NPEs were created by merging existing NPEs within a cohort or by harmonizing categorical variables from one cohort and continuous variables from another. Hippocampal sclerosis, microinfarcts and gross infarcts were recorded as binary case–control phenotypes. Arteriosclerosis, atherosclerosis, CAA, CERAD score for neuritic plaques, amyloid plaques, LATE-NC and Lewy body pathology were recorded as four-stage ordinal variables that either measured progressive severity of pathology (‘none’ < ‘mild’ < ‘moderate’ < ‘severe’) or progressing anatomical distribution of pathology. Braak NFT was recorded as a seven-stage ordinal variable that followed the anatomical distributional stages originally characterized in ref. 87. We provide a deeper description of our harmonization approach in the Supplementary Methods, and a detailed listing of variables harmonized across data sources to construct NPEs for analysis is available in Supplementary Table 1.
To assess the co-occurrence of NPEs in our data, we estimated polychoric correlations (an approach that assumes that observed ordinal or binary variables reflect latent normally distributed variables) between NPE pairs, followed by hierarchical clustering using the polycor 0.8-1 (ref. 88), psych89 and pheatmap 1.0.12 (ref. 90) R packages (Extended Data Fig. 1).
DNA methylation data
Preprocessed and quality-controlled DNA methylation data for 740 ROSMAP participants were downloaded from Synapse.org (Synapse IDs: syn3157275 and syn3191087). DNA methylation preparation and quality control measures have been previously described91,92. Briefly, approximately 50 mg of frozen gray matter tissue from the DLPFC was sampled from each participant. DNA was then extracted and processed using the Illumina Infinium HumanMethylation450 BeadChip. Quality control measures included removing low-quality probes, removing participants with poor bisulfite-conversion efficiency and adjusting methylation levels by age, sex and batch, which adequately controlled for batch effects92. Missing methylation levels were imputed using 100-nearest neighbors91,92.
RNA-seq data
Preprocessed and quality-controlled bulk-tissue RNA-seq data from the DLPFC of ROSMAP participants were downloaded from Synapse.org (Synapse IDs: syn21088596, syn21323366, syn3505732 and syn3505724). As previously described, samples were prepared by sectioning approximately 100 mg of gray matter tissue from the DLPFC and RNA extracted using the Qiagen MiRNeasy Mini (217004) protocol and then submitted for transcriptome library construction using the dUTP protocol and Illumina sequencing92. A total of 634 participants in seven batches were sequenced with an average sequencing depth of 50 million paired reads per sample92. Subsequent quality control and batch corrections were performed, and the final output of the RNA-Seq pipeline was fragments per kilobase of transcript per million mapped reads (FPKM)92.
Statistical analyses
Single-variant GWAS
We analyzed ordinal endophenotypes using proportional-odds logistic mixed-effects models implemented in the POLMM 0.2.3 (refs. 93,94) and GRAB 0.1.1 R packages93 and analyzed binary variables similarly with logistic mixed-effects models implemented in the SAIGE R package95. Fixed-effect covariates included age at death, sex, cohort and the first ten genetic PCs created using the PCA in Related Samples (PC-AiR) method in the GENESIS 2.26.0 R package96. We included a genetic relationship matrix (GRM) as a random effect to account for relatedness between participants. An additive mode of inheritance was assumed in all analyses.
Analysis of individual data sources proceeded in two stages. In stage one, GRM was constructed using a pruned set of independent variants, defined as having a pairwise r2 < 0.2 within moving windows of 15 kbp. Null models, which included fixed covariates and the GRM, were then fitted using the GWASTools 1.42.1 (ref. 97), SNPRelate 1.30.1 (ref. 98), POLMM 0.2.3 (refs. 93,94), GRAB 0.1.1 (refs. 93,94) and/or SAIGE 1.1.3 (ref. 95) R packages. In stage two, score tests were performed on each variant with a saddle-point approximation used to calculate P values. We considered all variants with a P <5 × 10−8 to be genome-wide significant. To identify independent risk loci, we clumped results using the ‘--clump’ flag in PLINK 1.9 with the pairwise LD threshold set to r2 ≤ 0.05 (https://www.cog-genomics.org/plink/1.9/)85. Following analyses of individual cohorts, we performed fixed-effects meta-analyses using METAL software using inverse-variance weighting on variants with MAF > 1% in at least one cohort33. Variants with a total minor allele count <20 after meta-analysis were then excluded.
Conditional analysis of the APOE region
The region surrounding the APOE gene on chr19 is consistently the single strongest genetic risk factor for LOAD in GWAS. Three common forms of the APOE gene—\(\epsilon\)2, \(\epsilon\)3 and \(\epsilon\)4—are present in our study populations (see Table 1 for distribution of APOE alleles in participants), and the \(\epsilon\)2 and \(\epsilon\)4 alleles are associated with lower and higher risk of LOAD, respectively, relative to the \(\epsilon\)3 allele99. We therefore expected that variants in the APOE region, defined as the region within 200 kbp from the start and end transcription sites of APOE, would be associated with multiple NPEs in our study. Moreover, we hypothesized that genetic variants in the APOE region may influence neuropathology risk independently of the effects of APOE \(\epsilon\) alleles. To test this hypothesis, we re-analyzed variants in chr19 while adjusting for APOE \(\epsilon\) diplotype. We limited re-analysis to endophenotypes with at least one genome-wide significant association signal within the APOE locus in the final meta-analysis of the three independent GWAS. APOE diplotypes were determined either using the rs7412 and rs429358 variants according to the SNPedia online ref. 100 or taken directly from study data if available. Both rs7412 and rs429358 variants had high imputation quality scores (r2 = 0.997 and 0.975, respectively). The \(\epsilon 3/\epsilon 3\) diplotype was used as a reference, and we included fixed-effect indicator variables to adjust for \(\epsilon 2/\epsilon 2\), \(\epsilon 2/\epsilon 3\), \(\epsilon 3/\epsilon 4\) and \(\epsilon 4/\epsilon 4\) diplotypes. We chose this approach rather than adjusting for counts of \(\epsilon\)2 and \(\epsilon\)4 alleles because it is robust to potential nonlinear effects of genotypes. We performed additional sensitivity analyses for loci identified through this approach (Supplementary Information).
Replication of known AD risk loci in NPE
We used the 83 ADRD loci presented in a recent large GWAS to investigate whether AD-associated loci were associated with NPE6. We restricted our comparison to AD loci with lead variants with MAF ≥ 0.01, leaving 76–78 loci for comparison for each NPE. LD for variants near the top-known AD-associated variants was evaluated using the R package LDLinkR 1.2.3 (ref. 101). We controlled the false-discovery rate for each NPE using the Benjamini–Hochberg procedure102. Variants with an adjusted Q value ≤ 0.05 were considered significant.
FUMA and FAVOR annotation, gene-prioritization and functional enrichment pipeline
We mapped variants to genes and performed subsequent gene and gene-set analyses using the FUMA and FAVOR pipelines37,103. Variants were mapped to genes if they had P ≤ 1 × 10−5 in the GWAS meta-analysis and were located within 10 kbp of a protein-coding gene’s transcription start or end sites. Gene-based analyses were performed using MAGMA 1.10. The top variant PCs that accounted for 99.9% of the variance in a gene’s region were used to test for significance using an F test. We considered genes with resulting P ≤ 2.5 × 10−6 to be significantly associated with NPE. Gene-set enrichment analyses were performed using MAGMA104 gene-set analysis of Gene Ontology and curated gene sets from MSigDB105. Bonferroni P value corrections were made for each NPE individually.
Colocalization analyses
We used multiple sources of publicly available summary statistics from external studies as data sources for Bayesian colocalization analyses. First, we downloaded Genotype-Tissue Expression Project (GTEx) v8 European ancestry QTL analysis summary statistics, which contains summary statistics for significant gene expression and splicing QTL variants (eQTL and sQTL, respectively) in 48 body tissues106. We also used gene expression and DNA mQTL analysis summary statistics from studies using tissue from the DLPFC of ROSMAP participants42. These studies examined the associations of genetic variants with molecular traits and provided curated lists of significant QTL variants. Finally, we downloaded the summary statistics from a recent GWAS of LOAD for a targeted post hoc colocalization analysis in TMEM106B and GRN6.
For each NPE outcome in our study, we first created a list of genetic variants with P ≤ 1 × 10−5 in the GWAS meta-analysis. We then queried the lists of significant QTL variants in GTEx and ROSMAP using R (https://cran.r-project.org/) and Python 3.8.16 and 3.10.8 (https://www.python.org/) to identify neuropathology-associated QTL variants. For each genetic locus associated with NPEs that had at least one significant QTL in either GTEx or ROSMAP, we performed colocalization analysis using the ‘coloc.abf’ function in the coloc 5.2.2R package39. For ordinal variables, we chose dichotomizing cut points to determine case–control proportions. We used coloc’s default prior PrC of PrC = 1 × 10−5 and considered a posterior PrC > 80% as a threshold for evidence of colocalization.
To investigate whether shared GWAS signals drive association among multiple NPEs, we also performed colocalization analysis on loci with variants satisfying P < 1 × 10−4 and concordant effect direction for at least two NPEs in the GWAS meta-analysis. Due to the absence of associations in the region in APOE-adjusted analyses for NPEs other than CAA, we excluded that region for NPE–NPE colocalization analyses.
Association analyses using DLPFC DNA methylation and bulk RNA-seq data from ROSMAP
ROSMAP participants had postmortem bulk-tissue samples collected from the DLPFC, which underwent DNA methylation quantification using the Illumina DNAMethylation450 chip and gene expression and RNA-seq using the Illumina HiSeq 2000 (ref. 92). In total, 708 ROSMAP participants had DNA methylation data available for analysis. We restricted analyses involving DNA methylation or RNA-seq data to NPE-associated loci that reached the genome-wide significance threshold in the meta-analysis and also colocalized with mQTL or eQTL in brain tissue in either GTEx or ROSMAP.
In our APOE \(\epsilon\)-adjusted genetic association analysis, one locus near APOE remained significantly associated with CAA. This locus colocalized with DNA methylation levels at four CpG sites in ROSMAP. To investigate whether these CpG sites were in turn associated with CAA pathology, we combined individual-level DNA methylation and neuropathological data in ROSMAP for analysis. We used cumulative logit models using the ‘clm’ function implemented in the R package ordinal 2023.13.12-04 (ref. 107) with the semi-quantitative CAA variable described above as the outcome for analysis. We performed four analyses, with one of each of the four CpG sites tested as the independent variable of interest in each analysis. We adjusted for age, sex, ROS versus MAP study, bisulfite-conversion efficiency, postmortem interval and APOE \(\epsilon\) diplotype in each analysis. Similar models were used to test associations between hippocampal sclerosis and LATE-NC and methylation levels at CpG sites cg09613507 and cg23422036. Wald tests were performed on the resulting parameter estimates to test for statistical significance. We also performed post hoc analyses examining the association between these CpG sites and APOC2 expression in ROSMAP.
For genes with significant eQTL in GTEx or ROSMAP that colocalized with NPE, we performed additional targeted analyses to assess the association between gene expression and NPE. We first assessed the association between NPE lead variants and gene expression in ROSMAP to confirm nominal eQTL status. We then performed generalized linear regression models between square-root or log-transformed mRNA expression and NPE outcomes adjusting for age at death, sex, PMI and RNA integrity number.
Plots from these analyses were generated using the R package ggplot2 (ref. 108).
Replication of CAA locus using Mayo Clinic neuropathology GWAS
We used data from Mayo Clinic Brain Bank participants available from ref. 26 study of the genetic risk factors of CAA (dataset heretofore referred to as MC-CAA) to attempt to replicate a new CAA locus in the present study in an independent sample26. Neuropathology and genetic variant data were downloaded from Synapse (Synapse IDs: syn10930250, syn21499318, syn21522653 and syn21547862). Eight participants were identified as duplicates between batches or with NACC participants and removed. While CAA is graded on a four-level ordinal scale in the present study, CAA in MC-CAA is graded as an average of CAA burden across five brain regions26. We therefore used linear regression with the outcome variable as sqrt(CAA) with the independent variable of interest being the number of G alleles of variant rs7247551. Covariates included APOE diplotype (\(\epsilon 3/\epsilon 3\), \(\epsilon 2/\epsilon 4\), \(\epsilon 2/\epsilon 3\), \(\epsilon 3/\epsilon 4\) or \(\epsilon 4/\epsilon 4\)), sex, age at death (truncated at 90 years) and the first three genetic PCs.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Meta-analysis summary statistics for each NPE studied will be made available through NIAGADS upon publication at https://dss.niagads.org/. The authors are unable to share genotype or phenotype data from NACC, ADGC, ROSMAP or ACT due to data use restrictions. While these data were de-identified for study authors, these studies contain identifiable information on participants. ROSMAP data can be requested at https://www.radc.rush.edu and https://www.synapse.org. ADGC data can be requested from NIAGADS at https://www.niagads.org/resources/related-projects/alzheimers-disease-genetics-consortium-adgc-collection. NACC neuropathology data can be requested at https://naccdata.org/. ACT data can be requested at https://actagingresearch.org/. Harmonized neuropathology data are available through NIAGADS at https://dss.niagads.org/datasets/ng00067/. The results published here are in whole or in part based on data obtained from the AD Knowledge Portal. Raw long-read RNA-seq data generated and used in this manuscript are publicly available in both Synapse (https://www.synapse.org/#!Synapse:syn52047893) and NIH SRA (accession: SRP456327). Processed long-read RNA-seq data can be easily downloaded or viewed at https://ebbertlab.com/brain_rna_isoform_seq.html.
Code availability
All code used for data preparation and analysis is available at https://doi.org/10.5281/zenodo.11089995 (ref. 109).
Change history
17 June 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41588-024-02046-5
17 June 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41588-024-02045-6
References
Farfel, J. M. et al. Relation of genomic variants for Alzheimer disease dementia to common neuropathologies. Neurology 87, 489–496 (2016).
Katsumata, Y. et al. Multiple gene variants linked to Alzheimer’s-type clinical dementia via GWAS are also associated with non-Alzheimer’s neuropathologic entities. Neurobiol. Dis. 174, 105880 (2022).
Karanth, S. et al. Prevalence and clinical phenotype of quadruple misfolded proteins in older adults. JAMA Neurol. 77, 1299–1307 (2020).
Jansen, I. E. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413 (2019).
Wightman, D. P. et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282 (2021).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Escott-Price, V. & Hardy, J. Genome-wide association studies for Alzheimer’s disease: bigger is not always better. Brain Commun. 4, fcac125 (2022).
Karran, E. & De Strooper, B. The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat. Rev. Drug Discov. 21, 306–318 (2022).
Nelson, P. T., Braak, H. & Markesbery, W. R. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J. Neuropathol. Exp. Neurol. 68, 1–14 (2009).
Yang, H. S. et al. Evaluation of TDP-43 proteinopathy and hippocampal sclerosis in relation to APOE ε4 haplotype status: a community-based cohort study. Lancet Neurol. 17, 773–781 (2018).
Nelson, P. T. et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142, 1503–1527 (2019).
Nelson, P. T. et al. Frequency of LATE neuropathologic change across the spectrum of Alzheimer’s disease neuropathology: combined data from 13 community-based or population-based autopsy cohorts. Acta Neuropathol. 144, 27–44 (2022).
Brenowitz, W. D. et al. Hippocampal sclerosis of aging is a key Alzheimer’s disease mimic: clinical-pathologic correlations and comparisons with both Alzheimer’s disease and non-tauopathic frontotemporal lobar degeneration. J. Alzheimers Dis. 39, 691–702 (2014).
Skrobot, O. A. et al. Vascular cognitive impairment neuropathology guidelines (VCING): the contribution of cerebrovascular pathology to cognitive impairment. Brain 139, 2957–2969 (2016).
Weber, S. A., Patel, R. K. & Lutsep, H. L. Cerebral amyloid angiopathy: diagnosis and potential therapies. Expert Rev. Neurother. 18, 503–513 (2018).
Smith, E. E. et al. Cerebral microinfarcts: the invisible lesions. Lancet Neurol. 11, 272–282 (2012).
Arvanitakis, Z. et al. The relationship of cerebral vessel pathology to brain microinfarcts. Brain Pathol. 27, 77–85 (2017).
Neltner, J. H. et al. Arteriolosclerosis that affects multiple brain regions is linked to hippocampal sclerosis of ageing. Brain 137, 255–267 (2014).
Arvanitakis, Z. et al. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol. 15, 934–943 (2016).
Ighodaro, E. T. et al. Risk factors and global cognitive status related to brain arteriolosclerosis in elderly individuals. J. Cereb. Blood Flow Metab. 37, 201–216 (2017).
Beecham, G. W. et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet. 10, e1004606 (2014).
Shade, L. M. et al. Genome-wide association study of brain arteriolosclerosis. J. Cereb. Blood Flow Metab. 42, 1437–1450 (2022).
Vattathil, S. M. et al. A genetic study of cerebral atherosclerosis reveals novel associations with NTNG1 and CNOT3. Genes (Basel) 12, 815 (2021).
Farrell, K. et al. Genome-wide association study and functional validation implicates JADE1 in tauopathy. Acta Neuropathol. 143, 33–53 (2022).
Reddy, J. S. et al. Genome-wide analysis identifies a novel LINC-PINT splice variant associated with vascular amyloid pathology in Alzheimer’s disease. Acta Neuropathol. Commun. 9, 93 (2021).
Nelson, P. T. et al. ABCC9 gene polymorphism is associated with hippocampal sclerosis of aging pathology. Acta Neuropathol. 127, 825–843 (2014).
Rüb, U. et al. The evolution of Alzheimer’s disease-related cytoskeletal pathology in the human raphe nuclei. Neuropathol. Appl. Neurobiol. 26, 553–567 (2000).
Mirra, S. S. et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486 (1991).
Besser, L. M. et al. The revised National Alzheimer’s Coordinating Center’s neuropathology form-available data and new analyses. J. Neuropathol. Exp. Neurol. 77, 717–726 (2018).
Attems, J. et al. Neuropathological consensus criteria for the evaluation of Lewy pathology in post-mortem brains: a multi-centre study. Acta Neuropathol. 141, 159–172 (2021).
Montine, T. J. et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123, 1–11 (2012).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Shinohara, M. et al. Impact of sex and APOE4 on cerebral amyloid angiopathy in Alzheimer’s disease. Acta Neuropathol. 132, 225–234 (2016).
Zhang, Y. et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53 (2016).
Zhang, Y. et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947 (2014).
Watanabe, K. et al. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Pollard, K. S. et al. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121 (2010).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Katsumata, Y. et al. LATE-NC risk alleles (in TMEM106B, GRN, and ABCC9 genes) among persons with African ancestry. J. Neuropathol. Exp. Neurol. 82, 760–768 (2023).
Dugan, A. J. et al. Analysis of genes (TMEM106B, GRN, ABCC9, KCNMB2, and APOE) implicated in risk for LATE-NC and hippocampal sclerosis provides pathogenetic insights: a retrospective genetic association study. Acta Neuropathol. Commun. 9, 152 (2021).
Ng, B. et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat. Neurosci. 20, 1418–1426 (2017).
Kelemen, O. et al. Function of alternative splicing. Gene 514, 1–30 (2013).
Keren, H., Lev-Maor, G. & Ast, G. Alternative splicing and evolution: diversification, exon definition and function. Nat. Rev. Genet. 11, 345–355 (2010).
Kim, E., Magen, A. & Ast, G. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 35, 125–131 (2007).
Aguzzoli-Heberle, B. et al. Mapping medically relevant RNA isoform diversity in the aged human frontal cortex with deep long-read RNA-seq. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02245-9 (2024).
Ciolli Mattioli, C. et al. Alternative 3′ UTRs direct localization of functionally diverse protein isoforms in neuronal compartments. Nucleic Acids Res. 47, 2560–2573 (2019).
Weber, R. et al. Monitoring the 5′UTR landscape reveals isoform switches to drive translational efficiencies in cancer. Oncogene 42, 638–650 (2023).
Zhang, Q. & Tian, B. The emerging theme of 3′UTR mRNA isoform regulation in reprogramming of cell metabolism. Biochem. Soc. Trans. 51, 1111–1119 (2023).
Dickson, D. W. et al. APOE ε4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology 91, e1182–e1195 (2018).
Shih, Y. H. et al. TDP-43 interacts with amyloid-β, inhibits fibrillization, and worsens pathology in a model of Alzheimer’s disease. Nat. Commun. 11, 5950 (2020).
Wolska, A. et al. Apolipoprotein C-II: new findings related to genetics, biochemistry, and role in triglyceride metabolism. Atherosclerosis 267, 49–60 (2017).
Jong, M. C., Hofker, M. H. & Havekes, L. M. Role of ApoCs in lipoprotein metabolism: functional differences between ApoC1, ApoC2, and ApoC3. Arterioscler. Thromb. Vasc. Biol. 19, 472–484 (1999).
Ben Braiek, A. et al. Identification of biomarker panels as predictors of severity in coronary artery disease. J. Cell. Mol. Med. 25, 1518–1530 (2021).
Cervantes, S. et al. Genetic variation in APOE cluster region and Alzheimer’s disease risk. Neurobiol. Aging 32, 2107.e7–2107.e17 (2011).
Walker, R. M. et al. Identification of epigenome-wide DNA methylation differences between carriers of APOE ε4 and APOE ε2 alleles. Genome Med. 13, 1 (2021).
Walker, R. M. et al. Epigenome-wide analyses identify DNA methylation signatures of dementia risk. Alzheimers Dement. (Amst.) 12, e12078 (2020).
Shao, Y. et al. DNA methylation of TOMM40–APOE–APOC2 in Alzheimer’s disease. J. Hum. Genet. 63, 459–471 (2018).
Rollinson, S. et al. Frontotemporal lobar degeneration genome wide association study replication confirms a risk locus shared with amyotrophic lateral sclerosis. Neurobiol. Aging 32, 758.e1–758.e7 (2011).
Ciani, M. et al. Genome wide association study and next generation sequencing: a glimmer of light toward new possible horizons in frontotemporal dementia research. Front. Neurosci. 13, 506 (2019).
Katsumata, Y. et al. Gene-based association study of genes linked to hippocampal sclerosis of aging neuropathology: GRN, TMEM106B, ABCC9, and KCNMB2. Neurobiol. Aging 53, 193.e17–193.e25 (2017).
Nelson, P. T. et al. Reassessment of risk genotypes (GRN, TMEM106B, and ABCC9 variants) associated with hippocampal sclerosis of aging pathology. J. Neuropathol. Exp. Neurol. 74, 75–84 (2015).
Holler, C. J. et al. Bridging integrator 1 (BIN1) protein expression increases in the Alzheimer’s disease brain and correlates with neurofibrillary tangle pathology. J. Alzheimers Dis. 42, 1221–1227 (2014).
Franzmeier, N. et al. The BIN1 rs744373 SNP is associated with increased tau-PET levels and impaired memory. Nat. Commun. 10, 1766 (2019).
Steffensen, L. B. & Rasmussen, L. M. A role for collagen type IV in cardiovascular disease? Am. J. Physiol. Heart Circ. Physiol. 315, H610–H625 (2018).
Blevins, B. L. et al. Brain arteriolosclerosis. Acta Neuropathol. 141, 1–24 (2021).
Rannikmäe, K. et al. COL4A2 is associated with lacunar ischemic stroke and deep ICH: meta-analyses among 21,500 cases and 40,600 controls. Neurology 89, 1829–1839 (2017).
Hautakangas, H. et al. Genome-wide analysis of 102,084 migraine cases identifies 123 risk loci and subtype-specific risk alleles. Nat. Genet. 54, 152–160 (2022).
Price, B. R. et al. An emerging role of astrocytes in vascular contributions to cognitive impairment and dementia. J. Neurochem. 144, 644–650 (2018).
Guennewig, B. et al. Defining early changes in Alzheimer’s disease from RNA sequencing of brain regions differentially affected by pathology. Sci. Rep. 11, 4865 (2021).
Kawaue, T. et al. Lzts1 controls both neuronal delamination and outer radial glial-like cell generation during mammalian cerebral development. Nat. Commun. 10, 2780 (2019).
Wang, G. et al. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. Ser. B Stat. Methodol. 82, 1273–1300 (2020).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Bennett, D. A. et al. Religious Orders Study and Rush Memory and Aging Project. J. Alzheimers Dis. 64, S161–s189 (2018).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Kukull, W. A. et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch. Neurol. 59, 1737–1746 (2002).
Postupna, N. et al. The delayed neuropathological consequences of traumatic brain injury in a community-based sample. Front. Neurol. 12, 624696 (2021).
Sonnen, J. A. et al. Neuropathology in the adult changes in thought study: a review. J. Alzheimers Dis. 18, 703–711 (2009).
Crane, P. K. et al. Alzheimer’s disease sequencing project discovery and replication criteria for cases and controls: data from a community-based prospective cohort study with autopsy follow-up. Alzheimers Dement. 13, 1410–1413 (2017).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature 590, 290–299 (2021).
Howie, B. et al. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 44, 955–959 (2012).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015).
Gagolewski, M. stringi: fast and portable character string processing in R. J. Stat. Softw. 103, 1–59 (2022).
Braak, H. & Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 (1991).
Fox, J. polycor: polychoric and polyserial correlations. R package version 0.8-1 https://r-forge.r-project.org/projects/polycor (2022).
Revelle, W. psych: procedures for psychological, psychometric, and personality research. R package version 2.3.3 https://CRAN.R-project.org/package=psych (2023).
Kolde, R. et al. raivokolde/pheatmap. GitHub https://github.com/raivokolde/pheatmap (2018).
Yu, L. et al. Association of brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol. 72, 15–24 (2015).
De Jager, P. L. et al. A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Sci. Data 5, 180142 (2018).
Bi, W. et al. Efficient mixed model approach for large-scale genome-wide association studies of ordinal categorical phenotypes. Am. J. Hum. Genet. 108, 825–839 (2021).
Bi, W. et al. Scalable mixed model methods for set-based association studies on large-scale categorical data analysis and its application to exome-sequencing data in UK Biobank. Am. J. Hum. Genet. 110, 762–773 (2023).
Zhou, W. et al. Efficiently controlling for case–control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341 (2018).
Conomos, M. P., Miller, M. B. & Thornton, T. A. Robust inference of population structure for ancestry prediction and correction of stratification in the presence of relatedness. Genet. Epidemiol. 39, 276–293 (2015).
Gogarten, S. M. et al. GWASTools: an R/Bioconductor package for quality control and analysis of genome-wide association studies. Bioinformatics 28, 3329–3331 (2012).
Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328 (2012).
Reiman, E. M. et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat. Commun. 11, 667 (2020).
Cariaso, M. & Lennon, G. SNPedia: a wiki supporting personal genome annotation, interpretation and analysis. Nucleic Acids Res. 40, D1308–D1312 (2012).
Myers, T. A., Chanock, S. J. & Machiela, M. J. LDlinkR: an R package for rapidly calculating linkage disequilibrium statistics in diverse populations. Front. Genet. 11, 157 (2020).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 57, 289–300 (1995).
Zhou, H. et al. FAVOR: functional annotation of variants online resource and annotator for variation across the human genome. Nucleic Acids Res. 51, D1300–D1311 (2023).
De Leeuw, C. A. et al. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Battle, A. et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Christensen, R. H. B. Ordinal: regression models for ordinal data. R package version 2023.12-04 https://CRAN.R-project.org/package=ordinal (2023).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, 2016).
Shade, L. M. Code for Genome-wide association study of multiple neuropathology endophenotypes identifies novel risk loci and provides insights into genetic risk of dementia. Zenodo https://doi.org/10.5281/zenodo.11089995 (2024).
Acknowledgements
We disclosed receipt of the following financial support for the research, authorship and/or publication of this article: RF1AG082339 (to Y.K., S.A.C., M.T.W.E., P.T.N. and D.W.F.), R56AG057191 (to Y.K., S.A.C. and D.W.F.), F30NS124136 (to L.M.P.S.), P30 AG028383 (to P.T.N.), R35GM138636 (to B.A.H., J.A.B., M.L.P. and M.T.W.E.), R01AG068331 (to B.A.H., J.A.B., M.L.P. and M.T.W.E.), U01AG058654 (to J.L.H.), P01AG078116 (to Y.K. and K.Z.A.), K25AG055620 (to S.M.), R01AG082730 (to S.M. and D.W.F.), R01LM012535 (to K.N.), U19AG024904 (to A.J.S.), U01AG068057 (to A.J.S.), U01AG072177 (to A.J.S.), U19AG074879 (to K.N. and A.J.S.), U24AG072122 (to W.A.K.), P30AG072976 (to A.J.S.), the BrightFocus Foundation (A2020161S to M.T.W.E.), Alzheimer’s Association (2019-AARG-644082 to M.T.W.E.), the University of Kentucky Center for Clinical and Translational Science TL-1 Fellowship (TL1TR0019970), the National Center for Advancing Translational Sciences (UL1TR001998) and the Dean of the College of Medicine, University of Kentucky. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health (NIH), the University of Kentucky or other participating institutions. The NACC database is funded by NIA/NIH under grant U01AG016976. NACC data are contributed by the NIA-funded ADCs—P30 AG019610 (E. Reiman), P30 AG013846 (N. Kowall), P50 AG008702 (S. Small), P50 AG025688 (A. Levey), P50 AG047266 (T. Golde), P30 AG010133 (A. Saykin), P50 AG005146 (M. Albert), P50 AG005134 (B. Hyman), P50 AG016574 (R. Petersen), P50 AG005138 (M. Sano), P30 AG008051 (T. Wisniewski), P30 AG013854 (R. Vassar), P30 AG008017 (J. Kaye), P30 AG010161 (D. Bennett), P50 AG047366 (V. Henderson), P30 AG010129 (C. DeCarli), P50 AG016573 (F. LaFerla), P50 AG005131 (J. Brewer), P50 AG023501 (B. Miller), P30 AG035982 (R. Swerdlow), P30 AG028383 (PI. L. Van Eldik), P30 AG053760 (H. Paulson), P30 AG010124 (J. Trojanowski), P50 AG005133 (O. Lopez), P50 AG005142 (H. Chui), P30 AG012300 (R. Rosenberg), P30 AG049638 (S. Craft), P50 AG005136 (T. Grabowski), P50 AG033514 (S. Asthana), P50 AG005681 (J. Morris) and P50 AG047270 (S. Strittmatter). The results published here are in part based on data obtained from the AD Knowledge Portal. Genotyping was supported by the ADGC through the National Institute of Aging (U01AG032984 and RC2AG036528). See Supplementary Note for full ADGC acknowledgments. Samples from the National Cell Repository for Alzheimer’s Disease, which receives government support under a cooperative agreement grant (U24AG21886) awarded by the NIA, were used in this study. We thank contributors who collected samples used in this study, as well as patients and their families whose help and participation made this work possible. Data for this study were prepared, archived and distributed by the National Institute on Aging Alzheimer’s Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24AG041689-01). We thank the study participants and staff of the Rush Alzheimer’s Disease Center. The ROS and the Rush MAP are supported by grants from the NIH (P30AG10161, P30AG72975, R01AG15819, R01AG17917, R01AG22018, R01AG33678, R01AG34374, R01AG36042, R01AG40039, R01AG042210, U01AG46152, U01AG61356, R01AG47976, R01AG43379, RF1AG54057, R01AG56352, R01NS78009 and UH2NS100599) and the Illinois Department of Public Health. The ACT study was funded by the NIA (U19AG066567). Data collection for this work was additionally supported, in part, by previous funding from the NIA (U01AG006781). All statements in this report, including its findings and conclusions, are solely those of the authors and do not necessarily represent the views of the NIA or the NIH. We thank the participants of the ACT study for the data they have provided and the many ACT investigators and staff who steward that data. You can learn more about ACT at https://actagingstudy.org/.
Author information
Authors and Affiliations
Consortia
Contributions
L.M.P.S. conceptualized study design, prepared data, performed analyses and contributed to manuscript preparation. Y.K. and S.M. provided feedback on analyses and contributed to the manuscript. Y.K. also provided software for data preparation. K.Z.A. assisted with creating figures. Q.Q. assisted with gene-based and gene-prioritization analyses and assisted with creating figures. S.A.C. and M.T.W.E. helped with data interpretation and preparing figures, and contributed to manuscript preparation. E.L.A. provided guidance on the interpretation of BIN1 results and provided extensive feedback on manuscript preparation. B.A.H., J.A.B. and M.L.P. performed RNA isoform sequencing and analyses. T.J.H. performed imputation and quality control on ROSMAP genotype data. K.N. and A.J.S. provided imputed and quality-controlled ADNI genotype data used in an earlier version of the manuscript and provided feedback on manuscript preparation. D.A.B. and J.A.S. provided ROSMAP neuropathology data and made critical revisions to the manuscript. P.T.N. provided guidance on defining NPEs and contributed to the manuscript. D.W.F. conceptualized the study design and contributed to manuscript preparation. P.T.N., M.T.W.E. and D.W.F. jointly supervised the study. NACC and ADGC consortia provided data for their respective studies. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
J.A.S. reported personal fees from the Observational Study Monitoring Board Framingham, Observational Study Monitoring Board Discovery (National Institute of Neurological Disorders and Stroke) and Takeda Pharma. A.J.S. reported support from Avid Radiopharmaceuticals, a subsidiary of Eli Lilly (in kind contribution of positron emission tomography tracer precursor), and participated in Scientific Advisory Boards (Bayer Oncology, Eisai, Novo Nordisk and Siemens Medical Solutions) and an Observational Study Monitoring Board (MESA, NIH NHLBI), as well as several other NIA External Advisory Committees. He also serves as editor-in-chief of Brain Imaging and Behavior, a Springer Nature Journal. He was not involved in the editorial handling of this Nature Genetics paper (journals within the Springer Nature Portfolio are editorially independent). The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Lasse Pihlstrom and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Heatmap of the polychoric correlations of 11 neuropathology endophenotypes.
The y-axis (rows) and x-axis (columns) refer to the neuropathology endophenotype pairs with the hierarchical clustering generated by the polychoric correlations calculation. The red and blue color refers to high and low correlations between the neuropathology endophenotype pairs. The three positively correlated clusters of endophenotypes that match general expectations are highlighted by the black solid lines: a ‘vascular’ cluster consisting of gross infarcts, microinfarcts, arteriolosclerosis and atherosclerosis; an ‘Alzheimer’s disease’ cluster consisting of Braak NFT stage, neuritic plaques, amyloid-beta plaques and CAA; and a ‘LATE’ cluster consisting of LATE-NC and HS, respectively.
Extended Data Fig. 2 Quantile–quantile (QQ) plots for the 11 neuropathology endophenotype.
The y-axis refers to the experimental −log10(P) from two-sided z test of the genome-wide association study (GWAS) meta-analysis. The x-axis refers to the theoretical −log10(P) based on percentile. Each point represents a single-nucleotide polymorphism (SNP). The line of identity (y = x) is shown in a black dashed line, indicating the expected alignment under the null hypothesis. Deviations from this line suggest possible inflation due to population structure or polygenic effects. The genomic inflation factor lambda (λ) is calculated for each phenotype indicating minimal inflation of test statistics. The λ estimates ranged from 0.9879 to 1.0047, and visual inspection of the QQ plots did not suggest any systematic bias in the data.
Extended Data Fig. 3 Forest plots of associations between CAA and lead variant (rs7247551) on chromosome 19 stratified by study and APOE ? diplotype.
For each of the data sources (NACC n = 5,927, ROSMAP n = 1,172 and ACT n = 677), we re-analyzed the association between CAA and lead variant rs7247551 from the meta-analysis while stratifying by APOE ? diplotype and visually compared effect sizes across groups. Due to low sample sizes preventing model convergence, APOE ?4 carriers (diplotypes ?2/?4, ?3/?4, ?4/?4) were merged in analyses for ROSMAP and ACT. Points along the x-axis represent the estimated odds ratios, and error bars indicate 95% CI. Results demonstrate a consistent pattern of association between rs7247551 and CAA within each of the data sources used in our study.
Extended Data Fig. 4 Regional plot and the chromatin interaction plot of rs2000660 that is associated with cerebral atherosclerosis.
Atherosclerosis association plot from NPE GWAS meta-analysis (n = 7,340) for the SNP of rs2000660. The x-axis refers to the position of the genome. In the top plot, the y-axis refers to the −log10(P) from meta-analysis two-sided Z test. The lead variant, rs2000660, is circled in black and colored in dark purple. Variants meeting the threshold of P < 1 × 10-5 were colored coded according to linkage disequilibrium r2 to rs200060. Other variants are colored in gray. The figures are generated by FUMA pipelines (https://fuma.ctglab.nl). In the bottom plot, the x-axis refers to the genome position, and the y-axis refers to the type of regulatory elements in the chromatin interaction plot for rs2000660.
Extended Data Fig. 5 CERAD score and Braak NFT stage colocalize on BIN1.
Braak stage and CERAD association plot from NPE GWAS meta-analysis (n = 7,776–7,786) for the region around BIN1. Colored dots represent the chromosomal position (x-axis, Mb, megabase) in hg38 coordinates and −log10(P from meta-analysis two-sided z test; y-axis) of each variant in the region. Dots are colored to represent the linkage disequilibrium r2 with the lead variant (purple dot) estimated with PLINK–r2 using 1000 Genomes Phase 3 European-descended participants. The recombination rate was calculated using GRCh38 genetic map files downloaded from https://bochet.gcc.biostat.washington.edu/beagle/genetic_maps/. Boxes below data indicate the location of genes in the region (plot generated using LocusZoom73).
Extended Data Fig. 6 GRN expression, hippocampal sclerosis and LATE-NC all colocalize on GRN.
GRN gene expression, hippocampal sclerosis and LATE-NC association plot from NPE GWAS meta-analysis (n = 3,112–7,164) for the region around GRN. Colored dots represent the chromosomal position (x-axis, Mb, megabase) in hg38 coordinates and −log10(P from meta-analysis two-sided z test; y-axis) of each variant in the region. Dots are colored to represent the linkage disequilibrium r2 with the lead variant (purple dot) estimated with PLINK–r2 using 1000 Genomes Phase 3 European-descended participants. The recombination rate was calculated using GRCh38 genetic map files downloaded from https://bochet.gcc.biostat.washington.edu/beagle/genetic_maps/. Boxes below data indicate the location of genes in the region (plot generated using LocusZoom73).
Extended Data Fig. 7 Hippocampal sclerosis and ABCC9 expression colocalize on ABCC9.
ABCC9 gene expression and hippocampal sclerosis association plot from NPE GWAS meta-analysis (n = 7,164) for the region around ABCC9. Colored dots represent the chromosomal position (x-axis, Mb, megabase) in hg38 coordinates and −log10(P from meta-analysis two-sided Z test; y-axis) of each variant in the region. Dots are colored to represent the linkage disequilibrium r2 with the lead variant (purple dot) estimated with PLINK–r2 using 1000 Genomes Phase 3 European-descended participants. The recombination rate was calculated using GRCh38 genetic map files downloaded from https://bochet.gcc.biostat.washington.edu/beagle/genetic_maps/. Boxes below data indicate the location of genes in the region (plot generated using LocusZoom73).
Supplementary information
Supplementary Information (download PDF )
Supplementary Methods, Results, consortium authors and affiliations, and ADGC full acknowledgments.
Supplementary Tables 1–8 (download XLSX )
Supplementary Table 1: NPE harmonization across data sources. Supplementary Table 2: Associations between NPE and known AD loci (meta-analysis P value from two-sided t tests). Supplementary Table 3: MAGMA gene confirmation (meta-analysis P value from one-sided t tests). Supplementary Table 4: FAVOR annotation table. Supplementary Table 5: Colocalization analysis results (meta-analysis P value from two-sided t tests). Supplementary Table 6: NACC exclusion criteria. Supplementary Table 7: Suggestive GWAS hits. Supplementary Table 8: Suggestive NPE-associated loci from GWAS meta-analysis of NACC, ROSMAP and ACT datasets (meta-analysis P value from two-sided t tests).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shade, L.M.P., Katsumata, Y., Abner, E.L. et al. GWAS of multiple neuropathology endophenotypes identifies new risk loci and provides insights into the genetic risk of dementia. Nat Genet 56, 2407–2421 (2024). https://doi.org/10.1038/s41588-024-01939-9
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41588-024-01939-9
This article is cited by
-
Reply to: a quantitative trait locus for reduced microglial APOE expression associates with reduced cerebral amyloid angiopathy
Nature Genetics (2026)
-
Therapeutic targeting of neuroimmune mechanisms in neurodegeneration
Nature Reviews Drug Discovery (2026)
-
High throughput identification of genetic regulators of microglial inflammatory processes in Alzheimer's disease
Journal of Neuroinflammation (2025)
-
Shared and disease-specific pathways in frontotemporal dementia and Alzheimer’s and Parkinson’s diseases
Nature Medicine (2025)
-
The genetics of neurodegenerative diseases is the genetics of age-related damage clearance failure
Molecular Psychiatry (2025)
