
Abstract
Tobacco smoke, alone or combined with alcohol, is the predominant cause of head and neck cancer (HNC). We explore how tobacco exposure contributes to cancer development by mutational signature analysis of 265 whole-genome sequenced HNC samples from eight countries. Six tobacco-associated mutational signatures were detected, including some not previously reported. Differences in HNC incidence between countries corresponded with differences in mutation burdens of tobacco-associated signatures, consistent with the dominant role of tobacco in HNC causation. Differences were found in the burden of tobacco-associated signatures between anatomical subsites, suggesting that tissue-specific factors modulate mutagenesis. We identified an association between tobacco smoking and alcohol-related signatures, indicating a combined effect of these exposures. Tobacco smoking was associated with differences in the mutational spectra, repertoire of driver mutations in cancer genes and patterns of copy number change. Our results demonstrate the multiple pathways by which tobacco smoke can influence the evolution of cancer cell clones.
Similar content being viewed by others

Main
HNC, including malignancies affecting the mouth, pharynx and larynx, represents ~4% of the global cancer burden, with an annual incidence of about 750,000 new cases1. The incidence rate of HNC varies between different countries, largely reflecting the distribution of its main risk factors, including tobacco smoking, alcohol consumption1,2 and infection with high-risk strains of human papillomavirus (HPV) for oropharynx cancer3,4,5. Other proposed risk factors include consumption of hot beverages, obesity and poor oral health, although evidence for their role in HNC is limited6,7,8. In addition, a substantial proportion of HNCs (about 42% for women and 26% for men) cannot be attributed to known lifestyle habits or exposures9.
Epidemiological studies in Europe and America suggest that seven out of ten HNC cancers are caused by preventable behavioral risk factors, with tobacco use, either alone or in combination with alcohol, accounting for most cases9. Conversely, alcohol use on its own is responsible for only ~4% of the disease burden, suggesting a limited effect on HNC burden. This raises the question of whether alcohol acts as an independent carcinogen or simply enhances the known carcinogenic effect of tobacco. Furthermore, the susceptibility to these exposures varies depending on the anatomical region, with smoking posing a higher risk for developing larynx cancer and the risk associated with alcohol being greater for other subsites10.
Considering the dominant role of tobacco in HNC development, risk differences across subsites and potential interactions with other risk factors, HNC offers a particularly interesting opportunity to investigate the effects of tobacco exposure. In this context, the analysis of mutational signatures is an effective tool to track the complex mutagenic patterns linked to this and other exposures over a patient’s lifetime11,12,13. Certain mutational signatures have been related to well-established biological mechanisms and exposures. Signatures SBS4, found predominantly in lung cancer, and SBS92, in bladder cancer, capture two distinct mutagenic processes linked to tobacco use12,14,15. Conversely, signature SBS16 has been attributed to alcohol consumption in esophageal and liver cancer13,16.
Previous studies exploring the genomic landscape of HNC have relied predominantly on exome sequencing data, which have limited power to detect mutational signatures, lacked a diverse geographical and ethnic representation of cases and/or were limited to specific anatomical subsites17,18,19,20. Therefore, the carcinogenic mechanisms underpinning this cancer type in different geographical regions and anatomical subsites remain unclear. To bridge this gap, we performed whole-genome sequencing of 265 HNC samples from individuals exposed to known and suspected risk factors across eight countries with varying incidence rates. By leveraging mutational signature analysis combined with extensive epidemiological data, we shed light on the complexity of tobacco-induced mutagenesis and its interplay with alcohol consumption and other HNC risk factors.
Results
Case-series overview and multicountry study design
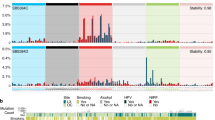
A total of 265 HNC cases were included in the study, comprising retrospective collections from eight countries in Europe and South America6,21 (Fig. 1 and Supplementary Table 1). These provide a broad geographic representation of HNC, including cases from high-incidence regions, with sex-combined age-standardized rates (ASRs) ranging from 9.4 per 100,000 to 18.2 per 100,000 in Romania, Slovakia, Czech Republic and Brazil, as well as moderate-incidence regions, with ASRs from 3.8 to 7.8 per 100,000 in Colombia, Argentina, Greece and Italy1. The study population encompasses diverse ethnic backgrounds, including European, Latin American, African and East Asian descent (Supplementary Table 2 and Supplementary Fig. 1). The dataset contains cases from all HNC anatomical subsites, with 127 oral cavity, 46 oropharynx, 17 hypopharynx and 75 larynx cancers. Epidemiological questionnaire data were available on exposure to known and suspected HNC risk factors, including cases from drinkers and smokers, with both exposed and nonexposed (Supplementary Table 3). DNAs from paired tumor and blood samples were extracted and whole-genome sequenced to average coverage of 55-fold and 27-fold, respectively.

a, Incidence of HNC, sex-combined, ASRs per 100,000, data from GLOBOCAN 2022. Dots indicate countries included in this study and number of participating patients. Panel a adapted from ref. 1, © International Agency for Research on Cancer. Data version: GLOBOCAN 2022-08.02.2024. b, Anatomical subsites of HNC, with number of tumor samples indicated in brackets. Panel b created using BioRender.com. c, Known and suspected risk factors included in the study, based on epidemiological questionnaire data and HPV detection. Frequencies of risk factors in the complete dataset (left) and by anatomical subsite (right) are indicated. OC, oral cavity; OPC, oropharynx; HPX, hypopharynx; LYX, larynx.
Mutation burden
Among the 265 HNC cases, we observed a median of 12,887 single-base substitutions (SBSs; range = 720–244,026), 63 doublet-base substitutions (DBSs; range = 2–7,113) and 757 small insertions and deletions (indels; range = 124–9,898) (Supplementary Table 4). Tumor samples from tobacco users exhibited higher SBS, DBS and indel burdens compared to nonsmokers (Extended Data Fig. 1b and Supplementary Table 5), as previously reported for larynx cancer14. Differences were also found between anatomical subsites, with larynx samples presenting higher mutation burdens, even after correcting for tobacco status (Extended Data Fig. 1a and Supplementary Table 5). No significant differences were found between geographical regions or ancestry profiles (Extended Data Fig. 1c and Supplementary Table 5).
Mutational signatures of exogenous and endogenous exposures
To investigate the mutational processes and carcinogenic exposures that have been operative in HNC development, we extracted SBS, DBS and indel signatures and estimated the contribution of each signature to every sample. We obtained 15 de novo SBS signatures, which were decomposed into 18 reference signatures from the Catalog of Somatic Mutations in Cancer (COSMIC v3.2) database, and two signatures that could not be decomposed into any combination of existing signatures, SBS_I and SBS_L (Fig. 2a,b, Extended Data Fig. 2, Supplementary Tables 6 and 9–11 and Supplementary Note).

a, SBS, DBS and indel signatures extracted in 265 HNC tumors. The size of each dot represents the proportion of samples presenting each mutational signature in the whole HNC dataset and across anatomical subsites. The color represents the mean relative attribution of each signature. Dots filled in white indicate signatures without significantly different relative burdens across subsites. Significance was assessed using a two-sided Kruskal–Wallis test and Bonferroni correction. Top, the mutations per megabase attributed to each signature in samples with counts higher than zero. b, Mutational spectrum of undecomposed signatures extracted from HNC. c, Known SBS signatures of tobacco exposure identified in the HNC dataset. ROS, reactive oxygen species; HR, homologous recombination; DSB, double-strand break.
Among the identified signatures, several have been previously associated with exogenous mutational processes12. The tobacco-related signatures SBS4 and SBS92 were found in 33.6% and 7.6% of HNC samples and, respectively, accounted for 6.3% and 3.5% of the mutational burden on average across all HNC cases. SBS16, attributed to alcohol consumption13,16, was present in 19.2% of the samples with a modest impact on the HNC mutation burden of 1.4% on average. Signatures SBS7a and SBS7b, related to ultraviolet (UV) light exposure, co-occurred in 4.2% of cases.
We also identified signatures associated with endogenous exposures and aberrant cellular processes. Notably, SBS2 and SBS13, which result from cytosine deamination by apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like (APOBEC)12, were present in the majority of HNC cases (92.8% and 91.7%, respectively) (Fig. 2a) and were highly correlated (Supplementary Fig. 2). Combined, these signatures accounted for an average of 20.4% of the total SBS mutation burden. Other prevalent signatures included SBS18, which is caused by reactive oxygen species (77.4% of samples), and clock-like signatures SBS1 (78.1%) and SBS5 (54.7%) (Fig. 2a).
Extraction of DBS signatures identified four de novo signatures, which decomposed into four COSMIC reference signatures (DBS1, DBS2, DBS4 and DBS6) and one nondecomposed signature (DBS_D) (Fig. 2a,b, Extended Data Fig. 3a and Supplementary Tables 7 and 9–11). We also extracted seven de novo indel signatures, all of which were decomposed into 12 COSMIC signatures (Fig. 2a,b, Extended Data Fig. 3b and Supplementary Tables 8–11). DBS and indel signatures of exogenous exposures were positively correlated with their SBS counterparts (Supplementary Fig. 2). For instance, the known tobacco-related signatures DBS2 (59.2% of samples) and ID3 (41.4%), along with DBS6, which has been previously registered as of unknown etiology, correlated with both SBS4 and SBS92. These associations are consistent with the SBS, DBS and indel signatures being generated by the same underlying mutational process. Similarly, ID11 (38.1%), which was associated with alcohol consumption in esophageal cancer13, exhibited a positive correlation with the alcohol signature SBS16, while UV-related DBS1 (16.6%) and ID13 (1.5%) signatures showed the same link with SBS7a–c.
To establish which mutagenic exposures were active earlier or later during the development of HNC, we estimated the molecular timing of each SBS signature (Methods and Supplementary Table 12). Signatures of tobacco and alcohol consumption, as well as the SBS_L signature, were enriched in early clonal mutations (Extended Data Fig. 4a–c), consistent with carcinogenic exposures occurring in normal cells22. Similarly, SBS_I was significantly enriched in early clonal mutations in cases exposed to tobacco and in oral cavity cases, while no significant differences were seen in other subsites (Extended Data Fig. 4d,e). Signatures of APOBEC signaling and SBS39 were enriched in late clonal mutations, suggesting that the corresponding mutational processes increased in activity during the evolution of cancer clones22.
HNC tumors present complex tobacco-related mutation patterns
We then investigated the associations between mutational signatures and epidemiological features using regression analysis (Supplementary Tables 13 and 14). Several signatures were independently associated with tobacco consumption, including the previously recognized tobacco-related signatures SBS4, SBS92, DBS2 and ID3, as well as signature DBS6, reported as of unknown etiology, and the newly discovered SBS_I (Fig. 3a,b, Extended Data Fig. 5a, Supplementary Table 13 and Supplementary Note). The tobacco-associated SBS signatures were composed of three different substitution patterns (predominantly C>A for SBS4, T>C for SBS92 and T>A for SBS_I) (Fig. 2c) and exhibited transcriptional strand bias15,23 (Supplementary Fig. 3). This strand bias often occurs as a result of transcription-coupled DNA repair and is found in mutations owing to bulky adducts, caused by exogenous exposures such as tobacco smoke carcinogens23. Assuming this mechanism is responsible for the strand bias in SBS_I, this is indicative of adduct formation on adenine bases.

a, Mutational burdens of tobacco-related signatures in HNC cases sorted by subsite and tobacco status. The tumor mutational burden (TMB) per sample is also displayed. b, Mutational burdens for SBS, DBS and indel signatures showing significant positive associations with tobacco consumption (n = 265 biologically independent samples). The Kruskal–Wallis test (two-sided) was used to test for global differences. Box-and-whisker plots are in the style of Tukey. The line within the box is plotted at the median, while the upper and lower ends indicate the 25th and 75th percentiles. Whiskers show 1.5× the interquartile range (IQR). The y axes were cut at 1.25× upper whisker for clarity. Bar plots indicate the frequencies of dichotomized signatures. c, Percentage of driver mutations occurring in C>A contexts in LYX and OC HNC from smokers. d, SBS96-mutation spectrum of driver mutations in LYX and OC HNC from smokers, showing enrichment in the frequency of C>A driver mutations in LYX cases.
The distribution of tobacco-associated signatures varied across different anatomical subsites (Fig. 3a, Extended Data Fig. 5c,d and Supplementary Table 13). Previously established tobacco signatures exhibited higher signature burdens and frequencies in larynx cases compared to other subsites. For instance, SBS4 was present in 17.3% of oral cavity, 17.4% of oropharynx, 52.9% of hypopharynx and 66.7% of larynx cases. Similar distributions were observed for SBS92, DBS2 and ID3. Conversely, the previously unknown SBS_I signature was present in smokers across all subsites, with particular enrichment in the oral cavity. The associations between signatures and subsites remained significant after correction for tobacco consumption and other confounding variables (Extended Data Fig. 5c and Supplementary Table 13). The enrichment of SBS_I signature tobacco smokers and oral cavity cases was confirmed in an external dataset (Supplementary Note).
Effects of tobacco exposure on the driver mutation spectra
We explored the driver mutation profile in tobacco-related HNC. This revealed 96 cancer genes with driver mutations in our dataset, including TP53, NOTCH1, CDKN2A, KMT2D and CASP8, which are commonly implicated in HNC24 (Extended Data Fig. 6a,b and Supplementary Tables 15 and 16). TP53 mutations were significantly enriched among smokers compared to nonsmokers (83.2% (164/197) versus 61.8% (42/68), Fisher’s exact test q = 0.0112), while CASP8 mutations were more frequent among nonsmokers (6.09% (12/197) versus 20.6% (14/68), Fisher’s exact test q = 0.0135). A total of 642 driver mutations were identified (Methods), and these showed enrichment of C>A substitutions in smokers compared to nonsmokers (24.9% (114/457) versus 17.3% (32/185), Fisher’s exact test P = 0.0379) (Extended Data Fig. 6c), consistent with the SBS4 mutation profile12. The frequency of C>A driver mutations in tobacco-exposed cases was higher in the larynx subsite compared to oral cavity (31.5% (53/168) versus 19.9% (38/191), Fisher’s exact test P = 0.0148) (Fig. 3c,d). This reflects the lower contribution of SBS4 to mutations in tobacco-exposed oral cavity HNC compared to larynx cases, which has been carried through into the generation of driver mutations. T>A driver mutations were also observed among smokers, albeit in low frequencies (6.6% (11/168) in larynx and 8.4% (16/191) in oral cavity), hinting at a lower presence of SBS_I in driver mutations.
Tobacco-related signatures correlate with HNC incidence
We analyzed the link between tobacco mutagenesis and variations in HNC incidence across different countries, sexes and anatomical subtypes. Our findings support previous epidemiological evidence, which has shown a connection between HNC incidence and smoking habits2 (Fig. 4a,b). Moreover, HNC incidence correlated with tobacco-related signatures (Fig. 4c and Supplementary Fig. 4), showing a higher ASR of HNC incidence in demographic groups presenting higher signature burdens. This further confirms that the geographical and demographic differences in tobacco exposure have a dominant role in driving HNC incidence.

a, Association between ASR of HNC incidence and tobacco smoking per country and sex (n = 16) measured by linear regression analysis. Estimate of ASR of tobacco smoking prevalence was obtained from the WHO Global Health Observatory data repository (2019). b, Association between cigarette quantity smoked per day in the HNC dataset and ASR incidence per country, sex and subsite, adjusted for age (n = 265). c, Association of tobacco-related signatures with ASR incidence per country, sex and subsite, adjusted for age. Data are represented as average mutations attributed to tobacco-related SBS (SBS4, SBS92 and SBS_I), DBS (DBS2 and DBS6) and indel (ID3) mutational signatures per group. The number of cases per group and frequency of positive cases are indicated by size and color, respectively. For a–c, 95% confidence interval is shown in clear blue. The P values shown correspond to ASR incidence in regressions across all data points with ASR of tobacco smoking (a), cigarette quantity (b) or mutation attributions (c) as explanatory variables.
Alcohol-related signatures in drinkers and smokers
Next, we assessed the signature profile in HNC cases with a history of alcohol intake. Regression analysis revealed significant associations between alcohol consumption and the following three specific signatures: SBS16, ID11 and DBS4 (Extended Data Fig. 5b, Supplementary Table 13 and Supplementary Note). Although the etiology of DBS4 is unclear, it has been found prevalent in esophageal cancer cases from countries with high alcohol intake rates13. SBS16, ID11 and DBS4 presented higher signature burdens in cases exposed to both tobacco and alcohol compared to alcohol alone (Fig. 5a,b). In the regression analysis, these signatures showed significant associations with the combined exposure (Fig. 5c and Supplementary Table 13). Models incorporating both tobacco and alcohol showed improved performance over those with alcohol alone (Supplementary Note). In conjunction, this indicates a combined effect of smoking and drinking in shaping the mutation profile of HNC, even after correcting for alcohol quantity and other potential confounding factors (Supplementary Note). The results are, therefore, consistent with SBS16, DBS4 and ID16, all being generated by the same underlying alcohol-related mutational process, and with the mutagenicity of this process being increased with co-exposure to tobacco smoke.

a, Mutational burdens of tobacco-related signatures in HNC cases sorted by subsite, alcohol and tobacco status. TMB per sample is also displayed. b, Mutational burdens for SBS, DBS and indel signatures showing positive associations with the tobacco plus alcohol status (n = 265 biologically independent samples). The Kruskal–Wallis test (two-sided) was used to test for global differences. Pairwise comparisons with the tobacco plus alcohol group were assessed with Dunn’s test (P values are shown in gray). Box-and-whisker plots are in the style of Tukey. The line within the box is plotted at the median, while the upper and lower ends indicate the 25th and 75th percentiles. Whiskers show 1.5× IQR. The y axes were cut at 1.25× upper whisker for clarity. Bar plots indicate the frequencies of dichotomized signatures. c, Associations between alcohol-related mutational signatures and the combined tobacco and alcohol exposures measured by logistic regression analysis. Regressions were corrected for sex, age of diagnosis, anatomical subsite and region. The Bonferroni method was used to adjust P values for multiple hypothesis testing. Effect size (log2(OR), color) and significance level (−log10(adjusted P), size). Dots filled in white indicate nonsignificant associations (Bonferroni-adjusted P < 0.05). OR, odds ratio.
For driver mutations, samples from individuals exposed to both tobacco and alcohol were characterized by a particularly high TP53 frequency of mutations (87.0% (141/162), 71.4% (25/35), 68.8% (22/32) and 55.6% (20/36) in the tobacco plus alcohol, alcohol alone, tobacco alone and unexposed groups, respectively; Fisher’s exact test q = 0.0024) (Extended Data Fig. 6). The driver mutation burden in the SBS16 context was too low to assess differences in the driver spectra between groups. However, TP53 mutations in the SBS16 contexts were exclusively found in samples from individuals exposed to both tobacco and alcohol (n = 5 TP53 variants).
HPV-positive HNC is characterized by APOBEC signatures
HPV infection in oropharynx cases did not elicit a specific mutational signature profile (Supplementary Table 13). However, most of the mutations in HPV-infected cases were driven by APOBEC activity (57.6% of the signature burden on average). This reflects a trend toward higher relative burdens of APOBEC signatures compared to HPV-negative oropharynx (30.0% of signature burden) (Extended Data Fig. 7a–c), consistent with previous reports18. Notably, the presence of APOBEC signatures was nearly ubiquitous across HNC cases (Fig. 2a), suggesting a broader role for APOBEC activation beyond its antiviral function11.
We also observed differences between HPV-positive and HPV-negative oropharynx cases exposed to tobacco. Among smokers, only 1/6 (16.7%) HPV-positive oropharynx cases presented tobacco-related SBS signatures, compared to 7/26 (26.9%) in HPV-negative cases (Fisher’s exact test P = 0.0214). Despite the well-known influence of tobacco smoking on the driver profiles of HNC20,24, the driver alterations in HPV-positive smokers differed from that of HPV-negative smokers and, instead, resembled the profile in HPV-positive cases from nonsmokers. This included PIK3CA mutations, PTEN mutations and deletions, as well as the absence of TP53 mutations and of FADD gains (Extended Data Fig. 7d,e). This, together with the reduced presence of tobacco-related signatures, suggests that oncogenesis in HPV-positive smokers may primarily be driven by viral infection rather than tobacco exposure.
Signature profiles of exposure to putative HNC risk factors
We next investigated the presence of additional environmental exposures beyond the most widely known HNC risk factors. Notably, UV-related signatures, SBS7a–c, DBS1 and ID13, were detected predominantly in oral cavity cases (Fig. 6a, Supplementary Table 13 and Supplementary Note). SBS7 signatures have been previously described in HNC, but the anatomical and epidemiological features of positive cases have not been previously investigated12. Samples with a relative SBS7a–c burden of >10% were categorized as positive for UV exposure, a criterion met by 13 oral cavity cases from the lip, tongue and floor of the mouth (Fig. 6b). All positive cases were either tobacco or alcohol users, with 11/13 presenting both risk factors (Fig. 6b). Thus, our data suggest a potential role of UV light exposure in HNC carcinogenesis23, which could be enhanced by tobacco and/or alcohol.

a, Mutational burdens for mutational signatures related to UV light exposure showing positive associations with the HNC anatomical subsite (n = 265 biologically independent samples). The Kruskal–Wallis test (two-sided) was used to test for global differences. Box-and-whisker plots are in the style of Tukey. The line within the box is plotted at the median, while the upper and lower ends indicate the 25th and 75th percentiles. Whiskers show 1.5× IQR. Frequencies of positive samples in each category are indicated in bar plots. b, SBS, DBS and indel signature burdens in samples positive for UV exposure based on relative SBS7a–c contributions above 10% of relative mutational burdens. Samples are sorted by lip (inner, n = 3 or unspecified, n = 1), tongue and floor of the mouth location within the OC. Positive tobacco and alcohol status are indicated in black.
Our analysis did not show any specific mutational patterns associated with other putative HNC risk factors, including hot drink consumption, poor oral health score and high body mass index6,7 (Supplementary Table 13). This suggests that these agents are likely not causing direct mutagenesis. Finally, the previously unknown DBS_D signature and ID4, with unknown etiology, were enriched among nonsmokers (Extended Data Fig. 5e), suggesting a potential link to unidentified mutational processes in this population.
HNC risk factors elicit distinct copy number profiles
HNC is characterized by complex patterns of copy number aberrations throughout the genome19,20. Unsupervised hierarchical clustering analysis on the copy number counts in HNC samples (n = 242) revealed two main clusters—one displaying diploid genomes (cluster D) and another presenting polyploidy and high burden of copy number gains and losses (cluster P) (Extended Data Fig. 8 and Supplementary Fig. 5). These clusters were further subdivided into four groups (D1, D2, P1 and P2). Notably, subgroup D2 was characterized by a copy-neutral profile, exhibiting substantially lower burdens of copy number events compared to the other groups.
The copy number clusters were associated with distinct epidemiological profiles (Fig. 7c,d). Specifically, tobacco-related HNC was enriched within both the diploid and polyploid copy number-high clusters (that is, D1, P1 and P2), while the copy number-silent cluster D2 was mostly constituted by samples from nonsmokers, including cases with unknown risk factors and alcohol drinkers in the absence of tobacco (Supplementary Table 17). Consistent with this pattern, the D2 cluster was enriched in samples from female patients, oral cavity cases and older age (Supplementary Table 17), aligning with the characteristic features of HNC with undefined risk factor24. Finally, HPV-positive oropharynx cases were enriched in the diploid clusters, predominantly in cluster D1.

a, Copy number signatures extracted in 242 HNC tumors. The size of each dot represents the proportion of samples presenting the signature, and the color represents the mean relative attribution of each signature. b, Copy number spectrum of the newly identified signature CN_G, defined by a 48 context copy number classification incorporating loss-of-heterozygosity status, total copy number state and segment length to categorize segments from allele-specific copy number profiles. c, Copy number profiles of HNC cases classified by copy number cluster. Relative signature burdens, copy number burden and associated epidemiological characteristics are indicated. The displayed epidemiological variables show significant differences by copy number cluster as per Fisher’s exact test and Benjamini–Hochberg procedure. d, Summary of exposures, driver alterations and copy number signatures associated with each cluster. Alluvial diagram depicts the frequency of each etiology in the copy number clusters. WGD, whole-genome duplication; CIN, chromosomal instability; LOH, loss of heterozygosity.
To identify distinct copy number particularities within each cluster and etiology, we conducted copy number signature analysis25 (Fig. 7a,b, Extended Data Fig. 9 and Supplementary Note). Cluster D1 exhibited enrichment in signatures of chromosomal instability within a diploid genome background (signatures CN1, CN9 and CN13) (Extended Data Fig. 10a). By contrast, cluster D2 presented a signature profile related to a diploid copy-neutral background (CN1). Clusters P1 and P2 displayed associations with signatures of whole-genome duplication (CN2 and CN20) along with genomic aberrations (CN5 and CN_G) (Extended Data Fig. 10b,c). Cluster P1 was consistent with double whole-genome duplications (CN18), while P2 showed signatures of chromosomal instability in conjunction with genome doubling (CN12). Collectively, our analysis suggests that HNC risk factors align with different copy number profiles and provides an enhanced characterization of the copy number aberrations in each HNC etiology (Fig. 7d). Specifically, tobacco use, alone or with alcohol, may trigger chromosomal instability and aneuploidy, while HPV infection may confer a copy number-unstable diploid profile. Finally, samples with unknown risk factors exhibit a copy-neutral profile.
We explored whether this difference in the copy number profile could be due to the driver profile that is associated with each risk factor (Fig. 7d and Extended Data Fig. 10d). TP53 mutations and MYC gains, two known promoters of genomic instability25,26,27, as well as gains in the anti-apoptotic FADD gene, were enriched in cluster P. CASP8 and HRAS mutations were enriched in the D2 copy-neutral cluster, in agreement with previous studies in HNC20,24,25. Finally, PTEN and RB1 mutations were enriched in the D1 cluster. Overall, these results indicate that tobacco use in HNC is associated with a distinct copy number-rich profile and driver alterations related to genome instability.
Discussion
The role of tobacco as one of the most avoidable cancer risk factors has been known for over 50 years. Yet, the detailed mechanisms by which tobacco smoke leads to DNA damage and carcinogenesis in different tissues are still not fully understood14,28,29. In this study encompassing HNC cases from eight countries in Europe and South America, we shed light on the effects of tobacco as the main mutagenic exposure in HNC and explored the complex mutational patterns and genomic alterations linked to tobacco exposure in different HNC subsites, as well as its interplay with alcohol consumption and other risk factors.
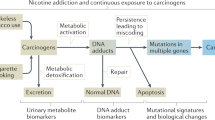
Tobacco smoke contains a mixture of thousands of chemicals, including over 60 carcinogens, among which benzo(a)pyrene (BaP) and nitrosamines are the most widely studied. These carcinogens undergo metabolic activation, generating reactive intermediates that interact with DNA in exposed tissues, resulting in complex mutagenic processes that can lead to cancer development29. In HNC, tobacco exposure resulted in six different signatures, identifying at least three mutational processes due to tobacco in HNC. Signature SBS4, characterized by C>A transversions, has been largely attributed to BaP adducts14,30,31. Exposure to this compound is also consistent with the CC>AA substitutions and C deletions present in DBS2 and ID3 tobacco signatures, respectively31. Conversely, signature SBS92, composed predominantly of T>C transitions, has not been related to specific carcinogens in tobacco smoke15. Finally, the T>A-rich substitution profile captured by the previously unidentified signature SBS_I is compatible with adduct formation on adenines, which have been observed in response to multiple tobacco compounds31,32,33. Among those, exposure to nicotine-derived nitrosamine ketone, one of the main tobacco carcinogens in oral tissues34, also yielded a T>A-rich signature in vitro and in mouse tumors35,36. Notably, a signature exhibiting high T>A frequencies and transcriptional strand bias has been described in normal lung epithelia from patients with a history of smoking37.
Our epidemiological analysis revealed that the mutational effects of tobacco vary among anatomical subsites. The canonical tobacco signatures, SBS4 and SBS92, were found predominantly in larynx cases, along with the tobacco-related DBS and indel signatures. Conversely, SBS_I was extracted in HNC cases from all subsites, with a notable enrichment in oral cavity cases. While previous studies primarily reported SBS4 in laryngeal HNC14,38, suggesting a minimal mutational impact of tobacco in the oral cavity and pharynx, our findings reveal different tobacco-related mutagenic processes occurring across all subsites. Altogether, our observations hint at varying susceptibility, exposure level or clearance of tobacco carcinogens across tissues, leading to different genotoxic effects. A possible explanation for these differences is the tissue-specific pattern of cytochrome P450 function. CYP1A1, the main BaP metabolizer, is primarily expressed in lung and larynx, whereas enzymes responsible for nitrosamine metabolism, such as CYP2E1, are predominant in the upper aerodigestive tract, including the oral cavity34,39,40,41. These differences in the response to tobacco across tissues may partially explain the greater susceptibility to smoking found for larynx cancers compared to other anatomical subsites10. While tobacco use was associated with elevated mutation burdens and BaP-related driver mutations in larynx cancers, this was not observed in oral cavity cases, aligning with a reduced carcinogenic effect. Thus, additional carcinogenic processes may be necessary to aid in the development of oral cavity and oropharynx cancers, including alcohol and HPV infection10.
In this regard, we also identified mutagenic processes linked to alcohol exposure13,16, including signatures SBS16, ID11 and an unreported association with signature DBS4. In HNC, alcohol-related signatures were predominantly observed in patients reporting both alcohol and tobacco consumption, consistent with epidemiological evidence showing a synergistic effect between these two factors on disease risk9,42. Furthermore, a previous study suggested an enrichment of SBS16 in oropharynx cases from tobacco users18. Altogether, our findings indicate that tobacco could enhance the carcinogenic effects of alcohol through shared mutagenic processes. Experimental evidence suggests that salivary concentrations of acetaldehyde, the genotoxic byproduct of alcohol metabolism, are greatly increased by tobacco smoking43, which could result in enhanced alcohol-related mutagenesis in cases with combined exposure.
Our data show that tobacco use, alone or in conjunction with alcohol, is also associated with a distinct copy number-rich profile, characterized by signatures of chromosomal instability, and resembling a previously described subset of copy number-rich HNC19. These genomic profiles are likely due to driver alterations leading to genome instability such as TP53 mutations, which are prevalent among smokers and drinkers24. Although high copy number burdens have been reported in lung adenocarcinoma cases from smokers14, the link between this exposure and specific copy number or driver profiles in HNC was previously unclear44. Cases with unknown etiology, on the other hand, exhibit few copy number alterations, prevalence of CASP8 and HRAS mutations and wild-type TP53. A similar copy-neutral group of samples has been observed in HNC, with an unreported link with HNC etiology19,45.
Regarding the mutagenic potential of other investigated risk factors, HPV infection did not elicit a specific mutational signature profile, but it was associated with distinct driver mutations and a copy number-unstable diploid genome. Poor oral hygiene, high body mass index and consumption of hot drinks did not display a direct effect on the mutation profile of HNC cases and likely contribute to the development of HNC of unknown etiology through mechanisms distinct from direct mutagenesis. This pattern has been proposed for several carcinogens in prior studies13,46. Nevertheless, there may exist additional unidentified mutagens leading to HNC, as hinted by the presence of the previously unidentified signature SBS_L as well as the enrichment of DBS_D and ID4 among nonsmokers.
Furthermore, we provide evidence suggesting that sunlight exposure may contribute to HNC development. Specifically, we identified signatures consistent with pyrimidine dimer formation (SBS7a–c and DBS1) in oral HNC cases, indicative of DNA damage by UV light12,23. UV light has only been described as a risk factor for malignancies in the external lip47, but experimental evidence suggests that oral cavity epithelia are susceptible to this exposure, and its carcinogenic processes could be enhanced by tobacco smoking48,49,50,51,52. While we cannot exclude the possibility of other mutational processes eliciting CC>TT substitutions, such as those driven by reactive oxygen species53, the presence of ID13 signatures, identified in melanoma12, provides additional evidence supporting the role of sunlight exposure in oral HNC.
In summary, through our comprehensive analysis of the mutational, genomic and epidemiological profile of HNC cases from diverse geographical regions, we have uncovered genomic mechanisms by which tobacco smoke and other risk factors contribute to HNC development. These findings enhance our understanding of the complexity and tissue specificity of tobacco mutagenesis, offering additional evidence that may inform prevention strategies aimed at reducing the risk of this disease.
Methods
Recruitment of cases and informed consent
The International Agency for Research on Cancer (IARC)/World Health Organization (WHO) coordinated participant recruitment through the HEADSpAcE and Central European international networks, comprising 13 collaborators from the eight participating countries in Europe and South America (Supplementary Table 18). Inclusion criteria for patients were ≥18 years of age (ranging from 18 to 90 years, with a mean of 60 and s.d. of 12 years), confirmed diagnosis of primary HNC and no prior cancer treatment. Written informed consent was obtained for all participants. Patients were excluded if they had any condition that could interfere with their ability to provide informed consent or if there were no means of obtaining adequate tissues as per protocol requirements. Ethical approvals were first obtained from each local research ethics committee and federal ethics committee when applicable, as well as from the IARC Ethics Committee (project 17-10).
Bio-samples and data collection
Dedicated standard operating procedures, following guidelines from the International Cancer Genome Consortium, were designed by the IARC/WHO to select adequate retrospective case series with complete biological samples and exposure information as described previously11,13 (Supplementary Table 18). In brief, for all case series included, anthropometric measures were taken, together with relevant information regarding medical and familial history. All biological samples from retrospective cohorts were collected using rigorous, standardized protocols and fulfilled the required standards of sample collection defined by the IARC/WHO for sequencing and analysis. Retrospective case series were included after examination of their respective recruitment protocols to ensure the availability of necessary biological samples based on standard operating procedures, following guidelines from the International Cancer Genome Consortium, and also based on the collection of relevant exposure history based on a comparison of validated epidemiological questionnaires from each specific region. Comparable smoking and alcohol history was available from all centers, as well as detailed epidemiological information on oral health, coffee, tea and mate consumption for specific regions2.
Potential limitations of using retrospective clinical data collected using different protocols from different populations were addressed by central data harmonization to ensure a comparable group of exposure variables (Supplementary Table 1). All patient-related data, as well as clinical, demographical, lifestyle, pathological and outcome data, were pseudonymized locally using a dedicated alphanumerical identifier system before being transferred to the IARC/WHO central database.
Expert pathology review
Original diagnostic pathology departments provided diagnostic histological details of contributing cases through standard abstract forms, together with a representative hematoxylin–eosin-stained slide of formalin-fixed paraffin-embedded tumor tissues whenever possible. The IARC/WHO centralized the entire pathology workflow and coordinated a centralized digital pathology examination of frozen tumor tissues collected for the study, as well as formalin-fixed paraffin-embedded sections when available, via a web-based report approach and a dedicated expert panel, following standardized procedures as described previously13. A minimum of 50% viable tumor cells was required for eligibility for whole-genome sequencing.
DNA extraction
Extraction of DNA from fresh frozen tumors and matched blood samples was centrally conducted at IARC/WHO. Of the cases that proceeded to the final analysis (n = 265), germline DNA was extracted from blood samples using previously described protocols and methods13.
HPV infection status and genome detection
The HPV infection status was determined by HPV16 E1, E2, E6 and E7 serology. To assess the HPV status in oropharynx cases with missing serologic information (n = 3), we used two orthogonal next-generation sequencing-based viral integration tools—Virus intEgration sites through iterative Reference SEquence customization (VERSE) and Fast Viral Integration and Fusion Identification (FastViFi)54,55 (Supplementary Table 19 and Supplementary Note). VERSE was used as part of the VirusFinder2.0 package (https://bioinfo.uth.edu/VirusFinder/), and FastViFi was installed using GitHub (https://github.com/sara-javadzadeh/FastViFi). Default parameters were used for running both tools.
Whole-genome sequencing
A total of 618 patients with HNC were enrolled in the study. Out of those, 315 cases were selected based on pathologic review and DNA quality (tumor and germline), and DNA was received at the Wellcome Sanger Institute for whole-genome sequencing. To ensure that the tumor and matched normal sample originated from the same individual, Fluidigm SNP genotyping with a custom panel was performed. Whole-genome sequencing (150 bp paired-end) was performed on the NovaSeq 6000 platform with a target coverage of 40× for tumors and 20× for matched normal tissues. All sequencing reads were aligned to the GRCh38 human reference genome using Burrows-Wheeler-MEM (v.0.7.16a and v.0.7.17). A standard set of postsequencing quality criteria was applied for metrics, including total coverage, evenness of coverage and contamination. Cases were excluded if coverage was below 30× for tumors or 15× for normal tissue. For evenness of coverage, the median over mean coverage (MoM) score was calculated, and tumor samples with MoM scores outside the range of values (0.92–1.09), which were determined by previous studies to be appropriate, were excluded56. Conpair56 (https://github.com/nygenome/Conpair) was used to detect contamination, and any tumor or normal sample with a value above 3% was excluded57. A total of 265 cases passed all criteria and were included in subsequent analysis.
Somatic variant calling
A standard analysis pipeline (https://github.com/cancerit) was used to perform variant calling for copy number variants (CNVs; ASCAT58 and Battenberg59, when tumor purity allowed), SNVs (cgpCaVEMan60), indels (cgpPindel61) and structural rearrangements (BRASS). CaVEMan and BRASS were run using the copy number profile and purity values determined from ASCAT when possible (complete pipeline, n = 242), or using copy number defaults and an estimate of purity obtained from ASCAT–Battenberg when tumor purity was insufficient to determine an accurate copy number profile (partial pipeline, n = 23). For SNVs, additional filters (ASRD ≥ 140 and CLPM = 0) in addition to the standard PASS filter. To further exclude the possibility of caller-specific artifacts being included in the analysis, a second variant caller, Strelka2, was run for SNVs and indels13,62, with variants called by both the Sanger variant-calling pipeline and Strelka2 included in the final analysis.
Generation of mutational matrices
Mutational matrices for SBS, DBS, indels and CNVs were generated using SigProfilerMatrixGenerator (https://github.com/AlexandrovLab/SigProfilerMatrixGenerator) with default options (v1.2.0)63.
Mutational signature analysis
Multiple methods were used to extract mutational signatures. The primary extractions were performed using SigProfilerExtractor (https://github.com/AlexandrovLab/SigProfilerExtractor) with a second method, mSigHdp, used to validate the de novo mutational signatures extracted (https://github.com/steverozen/mSigHdp)15,64. SigProfilerExtractor (v1.1.13) was run using nndsvd_min initialization (NMF_init=‘nndsvd_min’) for 1–20 signature solutions and 500 non-negative matrix factorization (NMF) replicates. For SBS, mutational signatures were extracted in both SBS1536 and SBS288 contexts. Both results were similar (Supplementary Note), with the SBS1536 results taken forward for the final analysis (Supplementary Table 6). Signatures were extracted using SigProfilerExtractor in the following contexts for other variant types: DBS78 for DBS, ID83 for indels and CNV48 for CNVs (Supplementary Tables 7, 8 and 20). The extracted de novo signatures were decomposed to COSMIC reference signatures where possible; this step is important as it allows the detection of de novo signatures that are made up of multiple reference signatures that have not separated during the extraction process (Supplementary Note). mSigHdp extractions were performed using the suggested parameters and using the country of origin to construct the hierarchy for SBS96 and ID83 contexts. A comparison of the SigProfilerExtractor and mSigHdp results can be found in the Supplementary Note.
Attribution of activities of mutational signatures
MSA (v2.0, https://gitlab.com/s.senkin/MSA) was used to attribute both de novo and decomposed mutational signatures65. For decomposed attributions, the panel of signatures included COSMIC reference signatures identified during the decomposition of mutational signatures in addition to newly extracted signatures that were not decomposed. A conservative approach was used for MSA attributions using the (params.no_CI_for_penalties=false) option for the calculation of optimum penalties. Pruned attributions were used for the final analysis, where confidence intervals were applied to each attributed mutational signature, and any signature activity with a lower confidence limit equal to 0 was removed.
Driver mutations
Driver mutations in HNC were identified using the following methods. First, the normalized ratio of non-synonymous to synonymous mutations (dN/dS) was used to identify genes under positive selection in HNC66. Results were calculated both for the whole genome (q < 0.01) and with restricted hypothesis testing for a panel of 369 known cancer genes66. Variants in any gene identified as under positive selection in global dN/dS or in the 369-cancer gene panel were considered as potential driver mutations and were then classified as likely drivers if they met any of the following criteria: (1) truncating mutations in genes annotated as tumor suppressors, (2) mutations annotated as likely or known oncogenic in MutationMapper, (3) truncating variants in genes with selection (q < 0.05) for truncating mutations assumed to be tumor suppressors and thus likely drivers, (4) missense variants in all genes under positive selection and with dN/dS ratios for missense mutations above five (assuming four of every five missense mutations are drivers) labeled as likely drivers or (5) in-frame indels in genes under significant positive selection for in-frame indels. The Cancer Gene Census (https://cancer.sanger.ac.uk/census) and the Cancer Genome Interpreter tool (https://www.cancergenomeinterpreter.org) were used to annotate potential drivers with the mode of action. Missense mutations were assessed using the MutationMapper tool (http://www.cbioportal.org/mutation_mapper).
Copy number profile
The copy number profiles were investigated in a subset of cases with available copy number data (complete pipeline, n = 242). Unsupervised clustering analysis of the copy number counts was performed using Euclidean distance and Ward’s agglomerative procedure. Driver copy number alterations were defined as cancer-related alterations in the COSMIC cancer gene census as follows25,67: (1) homozygous deletion (copy number = (0, 0)) of genes listed as deleted in COSMIC and (2) amplification (copy number > 2 × ploidy + 1) of genes listed as amplified (A) in COSMIC or PIK3CA gains, a commonly reported HNC alteration20,24.
Evolutionary analysis
MutationTimeR22 was run to annotate mutations as either early clonal, late clonal, subclonal or not-assigned clonal (meaning clonality could not be assigned). Samples with at least 256 early clonal mutations and at least 256 late clonal mutations were retained (n = 173), and the early and late clonal mutations for these samples were split into individual VCF files. SigProfilerAssignment68 was run on the resulting VCF files to identify the mutational processes active in the early clonal and late clonal mutations for each sample. Differences between the early and late relative activity of each mutational signature were assessed using a Wilcoxon signed-rank test, and P values were corrected across signatures using the Benjamini–Hochberg procedure (q value).
Genomic ancestry and admixture analyses
The genetic ancestry of individuals within the HNC dataset was inferred using the ADMIXTURE tool (v1.3.0)69. The admixture and principal component analyses were restricted to HapMap SNPs. Germline variants with minor allele frequency <1% within regions of long-range and high linkage disequilibrium in the human genome (GRCh38) were excluded, remaining 1,182,596 variants. After pruning for linkage disequilibrium (r2 < 20% within a 50 kb window), 159,464 independent variants remained in HNC genotype data. The 1000 Genomes reference population genotype data70 (phase 3) for Europeans (n = 489), Africans (n = 661), East Asians (n = 504) and Latin Americans (n = 347) were filtered and merged with the HNC genotype data based on the pruned set of variants present in both datasets. ADMIXTURE analysis was performed on the merged genotype data with k = 4, which would correspond to the four ancestral continental population groups reflecting the participants of our study (Supplementary Table 2). To complement the ADMIXTURE results, principal component analysis was run on the same samples, and HNC cases were visualized in two dimensions in comparison with each reference population included in the 1000 Genomes dataset (Supplementary Fig. 1).
Regressions and associations with signatures
Signature attributions were dichotomized into presence and absence using confidence intervals, with presence defined as both lower and upper limits being positive and absence as the lower limit being zero. If a signature was present in at least 75% of cases (SBS1, SBS2, SBS13, SBS18, SBS_I, ID1 and ID2), it was dichotomized into above and below the median of attributed mutation counts. The binary attributions served as dependent variables in logistic regressions, and relevant risk factors, epidemiological characteristics or ancestry data were used as factorized independent variables. Regressions with variables presenting data separation were performed using Firth’s penalized logistic regression.
For SBS, DBS and indel mutation burden analyses, cases defined as hypermutators (mutation burdens more than 1.5 interquartile range above Q3) were excluded, and associations with epidemiological factors were assessed using linear regression analysis.
To adjust for confounding factors, sex, age of diagnosis, subsite, region, tobacco and alcohol status were added as covariates in all regressions. The region variable was categorized as Europe and South America. The Bonferroni method was used to test for significant P values.
Regressions with HNC incidence (ASRs) were performed as linear regressions with signature attributions for signatures present in at least 75% of cases. Signatures present in less than 75% of cases were dichotomized into presence and absence, as indicated above, and analyzed using the logistic regressions. ASRs were obtained from Global Cancer Observatory (GLOBOCAN 2022)1. Regressions were performed on a sample basis and adjusted for age.
Statistics and reproducibility
Analyses were conducted using R (v.4.1.2)71. Data handling and statistical analysis were conducted using the R packages dplyr 1.1.4, tidyr 1.3.1, stringr 1.5.1, logistf 1.26.0 and yardstick 1.3.1 (refs. 72,73,74,75,76). Figures 1–7 and Extended Data Figs. 1,4,5,7,8 and 10 were created using ggplot2 3.5.1, ggrepel 0.9.5, ggpubr 0.6.0, ggalluvial 0.12.5, viridis 0.6.5, cowplot 1.1.3, patchwork 1.2.0, gridExtra 2.3, circlize 0.4.16, scales 1.3.0, ComplexHeatmap 2.10.0, EnrichedHeatmap 1.24.0, GenomicRanges 1.46.1 and CNAqc 1.0.0 (refs. 77,78,79,80,81,82,83,84,85,86,87,88,89,90). Signature extraction was replicated two times independently at both Wellcome Sanger Institute and University of California San Diego (UCSD). Signature attribution was replicated two times independently at both Wellcome Sanger Institute and IARC. All attempts at replication were successful and provided similar results. No other experiments other than those mentioned here were replicated independently. Additional details relating to the methods used in this study can be found in Supplementary Note.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Whole-genome sequencing data and patient metadata are deposited in the European Genome–Phenome Archive (EGA) associated with the study (EGAS00001005450). Aligned BAM files for all HNC cases included in the final analysis are deposited in dataset EGAD00001015386; consensus SNV and indel variant-calling files are in dataset EGAD00001015388; patient metadata are in dataset EGAD00001015387; structural rearrangement variant-calling files are in dataset EGAD00001015389; CNV calling is in dataset EGAD00001015390. Mutational catalogs for the Pancancer Analysis of Whole Genomes (PCAWG) dataset can be accessed at https://www.synapse.org/Synapse:syn11726616. The human reference genome (GRCh38) is available in the National Center for Biotechnology Information under the BioProject ID PRJNA31257. All other data are provided in the accompanying Supplementary Tables 1–29 and Supplementary Note, including mutation and copy number spectra of the extracted signatures, as well as signature activities, driver mutations and CNV in the HNC dataset.
Code availability
All algorithms used for data analysis are publicly available with repositories noted within the respective method sections and in the accompanying reporting summary. Code used for regression analysis and figures is available at GitLab (https://gitlab.com/Mutographs/mutographs-hnc) and Zenodo (https://doi.org/10.5281/zenodo.14851388)91.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Simard, E. P., Torre, L. A. & Jemal, A. International trends in head and neck cancer incidence rates: differences by country, sex and anatomic site. Oral Oncol. 50, 387–403 (2014).
IARC. Alcohol Consumption and Ethyl Carbamate. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans (IARC Publications, 2010).
IARC. Tobacco Smoke and Involuntary Smoking. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans (IARC Publications, 2004).
IARC. Human Papillomaviruses. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans Vol. 90 (IARC Publications, 2007).
Hashim, D. et al. The role of oral hygiene in head and neck cancer: results from International Head and Neck Cancer Epidemiology (INHANCE) consortium. Ann. Oncol. 27, 1619 (2016).
Gaudet, M. M. et al. Body mass index and risk of head and neck cancer in a pooled analysis of case–control studies in the International Head and Neck Cancer Epidemiology (INHANCE) Consortium. Int. J. Epidemiol. 39, 1091–1102 (2010).
Loomis, D. et al. Carcinogenicity of drinking coffee, mate, and very hot beverages. Lancet Oncol. 17, 877–878 (2016).
Hashibe, M. et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol. Biomarkers Prev. 18, 541–550 (2009).
Lubin, J. H. et al. Total exposure and exposure rate effects for alcohol and smoking and risk of head and neck cancer: a pooled analysis of case–control studies. Am. J. Epidemiol. 170, 937–947 (2009).
Perdomo, S. et al. The Mutographs biorepository: a unique genomic resource to study cancer around the world. Cell Genom. 4, 100500 (2024).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Moody, S. et al. Mutational signatures in esophageal squamous cell carcinoma from eight countries with varying incidence. Nat. Genet. 53, 1553–1563 (2021).
Alexandrov, L. B. et al. Mutational signatures associated with tobacco smoking in human cancer. Science 354, 618–622 (2016).
Islam, S. M. A. et al. Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. Cell Genom. 2, 100179 (2022).
Letouzé, E. et al. Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nat. Commun. 8, 1315 (2017).
Plath, M. et al. Unraveling most abundant mutational signatures in head and neck cancer. Int. J. Cancer 148, 115–127 (2021).
Gillison, M. L. et al. Human papillomavirus and the landscape of secondary genetic alterations in oral cancers. Genome Res. 29, 1–17 (2019).
Yang, J., Chen, Y., Luo, H. & Cai, H. The landscape of somatic copy number alterations in head and neck squamous cell carcinoma. Front. Oncol. 10, 479033 (2020).
Sayáns, M. P. et al. Comprehensive genomic review of TCGA head and neck squamous cell carcinomas (HNSCC). J. Clin. Med. 8, 1896 (2019).
Slot, D. E., Van Der Weijden, F. & Ciancio, S. G. Oral health, dental care and mouthwash associated with upper aerodigestive tract cancer risk in Europe: the ARCAGE study. Oral Oncol. 50, e57 (2014).
Gerstung, M. et al. The evolutionary history of 2,658 cancers. Nature 578, 122–128 (2020).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Leemans, C. R., Snijders, P. J. F. & Brakenhoff, R. H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 18, 269–282 (2018).
Steele, C. D. et al. Signatures of copy number alterations in human cancer. Nature 606, 984–991 (2022).
Meyer, N. & Penn, L. Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 8, 976–990 (2008).
Steele, C. D., Pillay, N. & Alexandrov, L. B. An overview of mutational and copy number signatures in human cancer. J. Pathol. 257, 454 (2022).
Cogliano, V. J. et al. Preventable exposures associated with human cancers. J. Natl Cancer Inst. 103, 1827–1839 (2011).
Hecht, S. S. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat. Rev. Cancer 3, 733–744 (2003).
Nik-Zainal, S. et al. The genome as a record of environmental exposure. Mutagenesis 30, 763 (2015).
Kucab, J. E. et al. A compendium of mutational signatures of environmental agents. Cell 177, 821–836 (2019).
Westcott, P. M. K. et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature 517, 489 (2015).
Pfeifer, G. P. et al. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 21, 7435–7451 (2002).
Hecht, S. S. & Hatsukami, D. K. Smokeless tobacco and cigarette smoking: chemical mechanisms and cancer prevention. Nat. Rev. Cancer 22, 143 (2022).
Peterson, L. A. Context matters: contribution of specific DNA adducts to the genotoxic properties of the tobacco-specific nitrosamine NNK. Chem. Res. Toxicol. 30, 420–433 (2017).
Mingard, C. et al. Dissection of cancer mutational signatures with individual components of cigarette smoking. Chem. Res. Toxicol. 36, 714–723 (2023).
Yoshida, K. et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature 578, 266–272 (2020).
Deneuve, S. et al. Molecular landscapes of oral cancers of unknown etiology. Preprint at medRxiv https://doi.org/10.1101/2023.12.15.23299866 (2023).
Degawa, M. et al. Metabolic activation and carcinogen–DNA adduct detection in human larynx. Cancer Res. 54, 4915–4919 (1994).
Jones, N. J., McGregor, A. D. & Waters, R. Detection of DNA adducts in human oral tissue: correlation of adduct levels with tobacco smoking and differential enhancement of adducts using the butanol extraction and nuclease P1 versions of 32P postlabeling. Cancer Res. 53, 1522–1528 (1993).
Yamazaki, H., Inui, Y., Yun, C. H., Guengerich, F. P. & Shimada, T. Cytochrome P450 2E1 and 2A6 enzymes as major catalysts for metabolic activation of N-nitrosodialkylamines and tobacco-related nitrosamines in human liver microsomes. Carcinogenesis 13, 1789–1794 (1992).
Hoes, L., Dok, R., Verstrepen, K. J. & Nuyts, S. Ethanol-induced cell damage can result in the development of oral tumors. Cancers 13, 3846 (2021).
Gapstur, S. M. et al. The IARC perspective on alcohol reduction or cessation and cancer risk. N. Engl. J. Med. 389, 2486–2494 (2023).
Pickering, C. R. et al. Squamous cell carcinoma of the oral tongue in young non-smokers is genomically similar to tumors in older smokers. Clin. Cancer Res. 20, 3842–3848 (2014).
Lawrence, M. S. et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582 (2015).
Riva, L. et al. The mutational signature profile of known and suspected human carcinogens in mice. Nat. Genet. 52, 1189–1197 (2020).
IARC. Solar and Ultraviolet Radiation. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans (IARC Publications, 1992).
Agrawal, A. et al. UV radiation increases carcinogenic risks for oral tissues compared to skin. Photochem. Photobiol. 89, 1193–1198 (2013).
Von Koschembahr, A. et al. Solar simulated light exposure alters metabolization and genotoxicity induced by benzo[a]pyrene in human skin. Sci. Rep. 8, 14692 (2018).
King, G. N. et al. Increased prevalence of dysplastic and malignant lip lesions in renal-transplant recipients. N. Engl. J. Med. 332, 1052–1057 (1995).
Sugano, N., Minegishi, T., Kawamoto, K. & Ito, K. Nicotine inhibits UV-induced activation of the apoptotic pathway. Toxicol. Lett. 125, 61–65 (2001).
Onoda, N. et al. Nicotine affects the signaling of the death pathway, reducing the response of head and neck cancer cell lines to DNA damaging agents. Head Neck 23, 860–870 (2001).
Reid, T. M. & Loeb, L. A. Tandem double CC–>TT mutations are produced by reactive oxygen species. Proc. Natl Acad. Sci. USA 90, 3904 (1993).
Javadzadeh, S. et al. FastViFi: fast and accurate detection of (hybrid) viral DNA and RNA. NAR Genom. Bioinform. 4, lqac032 (2022).
Wang, Q., Jia, P. & Zhao, Z. VERSE: a novel approach to detect virus integration in host genomes through reference genome customization. Genome Med. 7, 2 (2015).
Whalley, J. P. et al. Framework for quality assessment of whole genome cancer sequences. Nat. Commun. 11, 5040 (2020).
Bergmann, E. A., Chen, B. J., Arora, K., Vacic, V. & Zody, M. C. Conpair: concordance and contamination estimator for matched tumor–normal pairs. Bioinformatics 32, 3196 (2016).
Van Loo, P. et al. Allele-specific copy number analysis of tumors. Proc. Natl Acad. Sci. USA 107, 16910–16915 (2010).
Nik-Zainal, S. et al. The life history of 21 breast cancers. Cell 149, 994–1007 (2012).
Jones, D. et al. cgpCaVEManWrapper: simple execution of CaVEMan in order to detect somatic single nucleotide variants in NGS data. Curr. Protoc. Bioinformatics 56, 15.10.1–15.10.18 (2016).
Raine, K. M. et al. cgpPindel: identifying somatically acquired insertion and deletion events from paired end sequencing. Curr. Protoc. Bioinformatics 52, 15.7.1 (2015).
Kim, S. et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594 (2018).
Bergstrom, E. N. et al. SigProfilerMatrixGenerator: a tool for visualizing and exploring patterns of small mutational events. BMC Genomics 20, 685 (2019).
Liu, M., Wu, Y., Jiang, N., Boot, A. & Rozen, S. G. mSigHdp: hierarchical Dirichlet process mixture modeling for mutational signature discovery. NAR Genom. Bioinform. 5, lqad005 (2023).
Senkin, S. MSA: reproducible mutational signature attribution with confidence based on simulations. BMC Bioinformatics 22, 540 (2021).
Martincorena, I. et al. Universal patterns of selection in cancer and somatic tissues. Cell 171, 1029–1041 (2017).
Tate, J. G. et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019).
Díaz-Gay, M. et al. Assigning mutational signatures to individual samples and individual somatic mutations with SigProfilerAssignment. Bioinformatics 39, btad756 (2023).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Fairley, S., Lowy-Gallego, E., Perry, E. & Flicek, P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 48, D941–D947 (2020).
R Core Team. R: The R Project for Statistical Computing (R Foundation for Statistical Computing, 2022).
Wickham, H., François, R., Henry, L., Müller, K. & Vaughan, D. dplyr: a grammar of data manipulation. dplyr.tidyverse.org/ (2023).
Wickham, H., Vaugan, D. & Girlich, M. tidyr: tidy messy data. tidyr.tidyverse.org/ (2024).
Wickham, H. stringr: simple, consistent wrappers for common string operations. stringr.tidyverse.org/ (2023).
Heinze, G., Ploner, M., Jiricka, L. & Steiner, G. logistf: Firth’s bias-reduced logistic regression. https://doi.org/10.32614/CRAN.package.logistf (2003).
Kuhn, M., Vaughan, D. & Hvitfeldt, E. yardstick: tidy characterizations of model performance. yardstick.tidymodels.org (2024).
Wickham, H. ggplot2: create elegant data visualisations using the grammar of graphics. ggplot2.tidyverse.org/ (2016).
Slowikowski, K. ggrepel: an R package. ggrepel.slowkow.com/ (2024).
Kassambara, A. ggpubr: ggplot2 based publication ready plots. rpkgs.datanovia.com/ggpubr/ (2023).
Brunson, J. C. & Read, Q. D. ggalluvial: alluvial plots in ggplot2. corybrunson.github.io/ggalluvial/ (2023).
Garnier, S. et al. viridis: colorblind-friendly color maps for R. sjmgarnier.github.io/viridis/ (2024).
Wilke, C. O. cowplot: streamlined plot theme and plot annotations for ggplot2. wilkelab.org/cowplot/ (2024).
Pedersen, T. patchwork: the composer of plots. patchwork.data-imaginist.com/ (2024).
Auguie, B. & Antonov, A. gridExtra: miscellaneous functions for ‘Grid’ graphics. cran.r-project.org/web/packages/gridExtra/index.html (2017).
Gu, Z., Gu, L., Eils, R., Schlesner, M. & Brors, B. circlize implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812 (2014).
Wickham, H., Pedersen, T. L. & Seidel, D. scales: scale functions for visualization. scales.r-lib.org/ (2023).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Gu, Z., Eils, R., Schlesner, M. & Ishaque, N. EnrichedHeatmap: an R/Bioconductor package for comprehensive visualization of genomic signal associations. BMC Genomics 19, 234 (2018).
Lawrence, M. et al. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9, e1003118 (2013).
Antonello, A. et al. Computational validation of clonal and subclonal copy number alterations from bulk tumor sequencing using CNAqc. Genome Biol. 25, 38 (2024).
Torrens, L. Mutographs HNC. Zenodo https://doi.org/10.5281/zenodo.14851388 (2025).
Acknowledgements
The authors thank L. O’Neill, K. Roberts, K. Smith, S. Austin-Guest and the staff of Sequencing Operations at the Wellcome Sanger Institute for their contribution to sequencing, and are grateful for the support provided by the IARC General Services, including the Laboratory Services and Biobank team led by Z. Kozlakidis, the Section of Support to Research overseen by T. Landesz. The authors thank M. Blanks and M. McCord for useful discussions, and thank all the patients and their families involved in this study. This work was delivered as part of the Mutographs team supported by the Cancer Grand Challenges partnership funded by Cancer Research UK (C98/A24032). Work at the Wellcome Sanger Institute was also supported by the Wellcome Trust (grants 206194 and 220540/Z/20/A), and work at the IARC/WHO was supported by regular budget funding. This work was also supported by the US National Institute of Health (grants R01ES032547-01, R01CA269919-01 and 1U01CA290479-01 to L.B.A.), as well as by L.B.A.’s Packard Fellowship for Science and Engineering. The research performed in L.B.A.’s lab was supported by UC San Diego Sanford Stem Cell Institute. The HNC collection received funding from the European Union’s Horizon 2020 research and innovation program under grant 825771 and the São Paulo Research Foundation (FAPESP 2018/26297-3). J.P. and F.G. were partially supported by the Italian Ministry of Health—Ricerca Corrente. The funders had no roles in study design, data collection and analysis, decision to publish or preparation of the manuscript. The views expressed in this article by authors identified as IARC/WHO personnel are their own and do not necessarily represent the decisions, policies or views of the IARC/WHO.
Author information
Authors and Affiliations
Contributions
S.P. and P.B. conceived and designed the study. S.P., P.B., M.R.S. and L.B.A. provided supervision. L.T., S.M., A.C.D.C., M.K., S.C., S.S., T.C., R.C.C.P., J.R.A., V.G., A.A., E.N.B., R.V., J.W., S.F., M.D., D.J. and J.T. conducted data analysis. B.A. carried out pathology review. P.C., C. Carreira and C.L. carried out sample manipulation. F.R.P., L.P.K., J.P., F.G., J.C.D.C., P.L., A.L., M.V., D.M., I.N.M., L.M.A., R.R., J.R.V.P., S.V.V.Z., I.H., M.P.C., C. Canova, E.F. and P.A.R. led or facilitated patient and sample recruitment. J.W., S.F. and C.L. performed data generation. A.C.D.C. and L.H. carried out scientific project management. L.T. and S.M. contributed and were responsible for overall scientific coordination. L.T., S.M., A.C.D.C., L.B.A., P.B., M.R.S. and S.P. did the writing of the manuscript, with contributions from all other authors.
Corresponding author
Ethics declarations
Competing interests
L.B.A. is a cofounder, chief scientific officer, scientific advisory member and consultant for io9, has equity and receives income. The terms of this arrangement have been reviewed and approved by the University of California, San Diego, in accordance with its conflict of interest policies. L.B.A. is also a compensated member of the scientific advisory board of Inocras. L.B.A.’s spouse is an employee of Biotheranostics. E.N.B. and L.B.A. declare a US provisional patent application filed with UCSD with serial numbers 63/289,601, 63/269,033 and 63/483,237. A.A. and L.B.A. declare a US provisional patent application filed with UCSD with serial number 63/366,392. L.B.A. also declares US provisional application 63/412,835 and international application PCT/US2023/010679 filed with UCSD and is also an inventor of a US Patent 10,776,718 for source identification by non-negative matrix factorization. All other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks N. Gopalakrishna Iyer and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Mutational burdens in HNC.
a–c, Mutational burdens for SBSs, DBSs and small InDels burdens by anatomical subsite (a), smoking status (b) and country (c). Moreover, b depicts the mutation burdens by smoking status in the whole HNC dataset (left) and across anatomical subsites (right). Kruskal–Wallis test (two-sided) was used to test for global differences. Box-and-whisker plots are in the style of Tukey. The line within the box is plotted at the median, while upper and lower ends indicate 25th and 75th percentiles. Whiskers show 1.5× interquartile range (IQR). Hypermutators defined as samples with mutation burdens above 100,000 for SBS (n = 4), 6,000 for DBS (n = 1) and 5,000 for InDels (n = 1) were removed from the analysis. OC, oral cavity; OPC, oropharynx; HPX, hypopharynx.
Extended Data Fig. 2 SBS signature decomposition.
Decomposed SBS signatures, including reference COSMIC signatures and signatures not decomposed into COSMIC reference signatures.
Extended Data Fig. 3 DBS and InDel signature decomposition.
Decomposed DBS (a) and InDel (b) signatures, including reference COSMIC signatures and signatures not decomposed into COSMIC reference signatures.
Extended Data Fig. 4 Evolutionary analysis of mutational signatures in HNC.
a–c, Comparison of mutational signatures between early and late clonal mutations in HNC (n = 173), including tobacco and alcohol-related signatures enriched in early relative activities (a), signatures enriched in late relative activities (b) and undecomposed signatures (c). d,e, Relative activities of SBS_I in early and late clonal mutations across tobacco exposures (d) and anatomical subsites (e). In a–e, lines show the change in relative activity between the early and late clonal mutations within a positive sample. Colored lines represent an activity change of more than 6% (blue indicates higher in the clonal early mutations; orange indicates higher in the clonal late mutations). The number of positive samples is represented in the title of each plot. Box-and-whisker plots are in the style of Tukey, and show the distribution of activities in samples where the signature was present in the early and/or late clonal mutations. The line within the box is plotted at the median, while upper and lower ends indicate 25th and 75th percentiles. Whiskers show 1.5× IQR. Significance was assessed using a two-sided Wilcoxon signed-rank test, and p values were corrected using the Benjamini–Hochberg procedure (q value). f, Associations between mutational signatures present in early and late mutations and tobacco smoking measured by logistic regression. The Bonferroni method was used to adjust P values for multiple hypothesis testing. Signatures with significant associations (adjusted p-value < 0.05) and odds ratio (OR) > 2 are colored. Regressions were corrected for sex, age of diagnosis, anatomical subsite, region and alcohol consumption. g, Associations between tobacco-related mutational signatures present in early and late mutations and anatomical site, measured by logistic regression. The Bonferroni method was used to adjust P values for multiple hypothesis testing. Effect size (log2(OR), color) and significance level (−log10(adjusted p-value), size). Dots filled in white indicate non-significant associations (adjusted p-value < 0.05). Regressions were corrected for sex, age of diagnosis, region, tobacco and alcohol consumption. LYX, larynx.
Extended Data Fig. 5 Association of mutational signatures with exposures and anatomical subsites.
a,b, Associations between mutational signatures and tobacco smoking (a) or alcohol consumption (b) measured by logistic regression. The Bonferroni method was used to adjust P values for multiple hypothesis testing. SBS, DBS and InDel signatures with significant associations (adjusted p-value < 0.05) and odds ratio (OR) > 2 are colored. Regressions were corrected for sex, age of diagnosis, anatomical subsite, region and alcohol or tobacco consumption. c, Associations between tobacco-related mutational signatures and anatomical subsites measured by logistic regression. Regressions were corrected for sex, age of diagnosis, region, tobacco and alcohol exposure. The Bonferroni method was used to adjust p values for multiple hypothesis testing. Effect size (log2(OR), color) and significance level (−log10(adjusted p-value), size). Dots filled in white indicate non-significant associations (adjusted p-value < 0.05). d, Mutational burdens for tobacco-related mutational signatures by anatomical subsite (n = 265 biologically independent samples). e, Mutational burdens for mutational signatures showing significant negative associations with tobacco consumption. The Kruskal–Wallis test (two-sided) was used to test for global differences. Box-and-whisker plots are in the style of Tukey. The line within the box is plotted at the median, while upper and lower ends indicate 25th and 75th percentiles. Whiskers show 1.5× IQR. Y axes were cut at 1.25× upper whisker for clarity. Bar plots indicate the frequencies of dichotomized signatures.
Extended Data Fig. 6 Driver alterations and driver mutation spectra in HNC.
a, Driver mutations in HNC samples (n = 265) sorted by tobacco and alcohol status. Genes mutated in more than 2% of the cases are shown. b, Driver mutations and copy number events in HNC samples with available copy number data (n = 242). Only driver genes with both copy number gains and losses are included. Top, tumor mutational burden (TMB) per sample. Middle, presence of mutations per sample. Bottom, epidemiological characteristics. Frequency of mutations in the HNC dataset and q values from two-sided Fisher’s exact text are displayed. c, SBS96-mutation spectrum of driver mutations in smokers and non-smoker HNC cases and percentage of driver mutations occurring in C>A contexts.
Extended Data Fig. 7 Mutational signature and driver spectra of oropharynx HNC cases by HPV status.
a,b, Relative mutational burdens for APOBEC-related signatures (SBS2 and SBS13) in oropharyngeal HNC cases sorted by human papillomavirus (HPV) and tobacco status (n = 46 biologically independent samples). The Kruskal–Wallis test (two-sided) was used to test for global differences. Box-and-whisker plots are in the style of Tukey. The line within the box is plotted at the median, while upper and lower ends indicate 25th and 75th percentiles. Whiskers show 1.5× IQR. c, Average relative attributions of SBS signatures by HPV positivity and tobacco status in OPC cancers. d, Driver mutations in OPC HNC samples (n = 46) sorted by HPV and tobacco status. Genes mutated in more than 2% of the samples are shown. e, Driver mutations and copy number events in OPC HNC samples with available copy number data (n = 44). Only driver genes with copy number gains and losses are included. Top, TMB per sample. Middle, presence of mutations per sample. Bottom, HPV status and tobacco smoking. Frequency of mutations in the HNC dataset and q values from two-sided Fisher’s exact text are displayed.
Extended Data Fig. 8 Copy number profile of head and neck cancer clusters.
a, Genome-wide segments showing major and minor allele counts in 10 randomly picked samples per copy number cluster. b, Ploidy, copy number burden and burden of gains, losses and copy-neutral LOH (NLOH) across clusters (n = 242 biologically independent samples). The Kruskal–Wallis test (two-sided) was used to test for global differences. Box-and-whisker plots are in the style of Tukey. The line within the box is plotted at the median, while upper and lower ends indicate 25th and 75th percentiles. Whiskers show 1.5 × IQR.
Extended Data Fig. 9 Copy number signature decomposition.
Decomposed copy number signatures including reference COSMIC signatures and signatures not decomposed into COSMIC reference signatures.
Extended Data Fig. 10 Copy number signature enrichment by HNC clusters and driver profile.
a,b, Signature burdens for copy number signatures by copy number cluster showing associations with clusters D and P. c, Signature burdens for CN5 and CN_G signatures in copy number clusters D and P. The Kruskal–Wallis test (two two-sided) was used to test for global differences. Box-and-whisker plots are in the style of Tukey. The line within the box is plotted at the median, while upper and lower ends indicate 25th and 75th percentiles. Whiskers show 1.5× IQR. Y axis was cut at 1.25× upper whisker for clarity. Bar plots indicate the frequencies of dichotomized signatures. d, Associations between copy number clusters or signatures and driver alterations. Effect size (log2(OR), color) and significance level (−log2(q), size) from two-sided Fisher’s exact tests, corrected using the Benjamini–Hochberg procedure, are displayed. Only significant associations are shown (q < 0.05).
Supplementary information
Supplementary Information (download PDF )
Supplementary Note and Supplementary Figs. 1–17.
Supplementary Tables (download XLSX )
Supplementary Tables 1–29.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Torrens, L., Moody, S., de Carvalho, A.C. et al. The complexity of tobacco smoke-induced mutagenesis in head and neck cancer. Nat Genet 57, 884–896 (2025). https://doi.org/10.1038/s41588-025-02134-0
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41588-025-02134-0
This article is cited by
-
Long-term exposure to the ethanol-derived metabolite acetaldehyde elevates structural genomic alterations but not base substitutions
Communications Biology (2026)
-
Somatic mutation and selection at population scale
Nature (2025)
-
Geographic and age variations in mutational processes in colorectal cancer
Nature (2025)
