
Abstract
The gut microbiota is associated with human health and disease. Here we conducted a genome-wide association study of host genetic factors influencing gut microbiota composition in 12,652 individuals from the Trøndelag Health Study (HUNT), with replication in Nordic cohorts (n = 16,017–21,976). We identified 12 reproducible SNP–species associations across six genomic loci, including known (LCT, ABO) and novel (HLA-DQB1, MUC12, SLC37A2, FUT2) regions. Additionally, we detected genetic signals associated with gut microbiota functional modules at three loci (LCT, ABO, FUT2). Follow-up analyses suggest that these host–microbiota associations are linked to the pathogenesis of celiac disease and hemorrhoidal disease. Mendelian randomization analyses provided evidence supporting a causal effect of body mass index on gut microbiota composition. These findings highlight the interplay between host genetics and gut microbiota for human health and disease.
Similar content being viewed by others


Main
The human gut microbial community is highly diverse and plays an important role in normal gut physiology, including digestion, metabolism and immune regulation1. The gut microbiota has also been associated with a range of diseases, but most causal effects of the microbiota on human health are still to be established2,3,4. The composition and function of the gut microbiota are influenced by multiple factors, including diet, medication, age and host genetics5.
Twin studies have demonstrated that the gut microbiota composition is influenced by host genetics6,7, but previous genome-wide association studies (GWASs) have identified only two genetic loci (the LCT and ABO loci) reproducibly associated with gut microbiota composition2,3,4,8. Earlier studies have been underpowered, lacked replication and/or relied on 16S ribosomal RNA gene sequencing profiles with low taxonomic coverage2,3,4,9,10,11. To our knowledge, no previous GWAS has identified a replicated host genetic signal for gut microbiota functionality.
Several host traits and lifestyle factors have been associated with human gut microbial composition, including body mass index (BMI), physical activity, smoking and different diseases12. However, observational associations may be biased by confounding, and the causal direction for a major factor such as BMI remains unclear12,13. Mendelian randomization (MR) methods have been developed to infer causal relationships from genetic data, and are, under certain assumptions, less influenced by confounding and reverse causality than traditional observational designs. Nevertheless, using MR to evaluate bidirectional causal relationships between gut microbiota composition and host traits requires robust genetic instruments for gut microbial exposures and well-powered GWAS summary statistics for gut microbial outcomes. Unfortunately, previous host GWASs of gut microbiota composition using metagenome sequence data have generally been underpowered (n ≤ 7,738) as sources for exposure and outcome data in MR2,3,4,9,10,11.
To unravel host genetic factors influencing gut microbiota composition, we performed a large-scale GWAS including 12,652 participants with metagenome sequence data available in the Trøndelag Health Study (HUNT). The results were replicated in large Nordic cohorts (n = 16,017–21,976; Fig. 1a). To further understand the host-microbiota interactions, we performed a GWAS of gut microbiota functional potential (Kyoto Encyclopedia of Genes and Genomes (KEGG) modules) in 12,652 participants in HUNT, followed by replication in four Swedish cohorts (n = 16,017; Supplementary Fig. 4a). Our subsequent disease-focused phenome-wide association study (PheWAS) linked the novel findings in the gut microbiota GWAS to plausible diseases. Finally, taking advantage of the summary statistics from our large GWAS in the discovery cohort, we explored possible causal associations between BMI and gut microbiota composition.

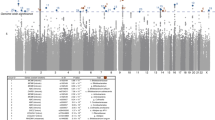
a, Overall design of the discovery GWAS in HUNT and the subsequent replications in four Swedish cohorts (all 13 SNP–species associations for replication) and in the FINRISK cohort (four SNP–species associations available for replication). b, Total SNP-based heritability, estimated as the ratio of genetic variance (Vg) to phenotypic variance (Vp), given as mean with 95% confidence intervals for the 546 evaluated species using genome-wide complex trait analysis (GCTA). c, Manhattan plot summarizing the SNP associations with the 546 gut microbiota species evaluated in the discovery GWAS. Results after replication are shown in Table 1 and Supplementary Table 7. The unadjusted P values are based on two-sided z-tests. Red line indicates genome-wide significance (P < 5.0 × 10−8). Gray dotted line indicates study-wide significance threshold adjusted for number of effective tests (P < 1.3 × 10−10). The identified genetic loci (index SNP ± 500 kb) are annotated with the species with study-wide significant associations. QC, quality control.
Results
GWAS of gut microbiota species
We evaluated associations between the relative abundance of 546 gut microbiota species (prevalence ≥30 %) and 7,971,622 genetic variants (minor allele frequency (MAF) ≥ 0.01) in 12,652 HUNT participants (Fig. 1, Supplementary Tables 1 and 2 and Supplementary Note). We identified genetic signals in seven loci, including 13 SNP-species signals (P < 1.3 × 10−10; Fig. 1c, Table 1 and Supplementary Fig. 3) selected for replication. Among the 13 selected SNP–species associations, 12 were successfully replicated (concordant direction of effect in the replication data and P < 3.8 × 10−3 (Bonferroni correction for 13 comparisons); n = 16,017–21,966; Table 1, Figs. 1a and 2 and Supplementary Fig. 3).

Overview of the gut microbiota species for families with independent genetic signals associated with species. The numbers of independent significant association signals are given within the circles either for those that were study-wide significant (SWS; P < 1.3 × 10−10), given per significant locus (one color per locus) or for all genome-wide significant associations not including the SWS associations (GWS; 1.3 × 10−10 ≤ P < 5.0 × 10−8, given in gray). The total number of independent SWS signals selected for replication are 13, located in 7 different loci. The total number of GWS signals, also including the SWS signals, is 106. For further details of individual associations, see Table 1 and Supplementary Table 6.
The replicated SNP–species associations included two well-known gut-microbiota-associated loci (LCT on Chr2 and ABO on Chr9)2,3,4,8. In addition, the identified FUT2 locus has previously been reported to pass the traditional genome-wide significance level (P < 5.0 × 10−8), but with no replication2. Further, we identified replicated genetic signals in three loci (HLA-DQB1 on Chr6, MUC12 on Chr7 and SLC37A2 on chr 11) not previously associated with the relative abundance of gut microbiota species in any GWAS (Table 1).
We performed several sensitivity analyses (excluding participants on antibiotic treatment, adjusting for bowel motility, only including unrelated individuals, using the centered log-ratio transformation, excluding cohabitation), revealing essentially unchanged effect estimates for the SNP-species associations (Supplementary Tables 8 and 9 and Supplementary Note).
GWAS on α-diversity parameters identified one genome-wide significant genetic signal for Shannon diversity index (rs12140644-G; beta = −0.11, standard error (s.e.) = 0.02, P = 2.6 × 10−8), whereas no significant signal was observed for richness in HUNT.
GCTA revealed SNP heritability estimates (h2) of between 0% and 25% for the 546 evaluated species (mean heritability of 6.8%; Fig. 1b and Supplementary Table 3). The heritability was higher for species with genome-wide significant genetic associations compared to those without (Supplementary Table 4). The SNP heritability for the two α-diversity measures, Shannon diversity index and richness, was 15.6 ± 4.1% and 24.5 ± 4.2%, respectively (Supplementary Note).
GWAS of KEGG functionality modules
To explore the biology underlying host genetics-gut microbiota relationships, we performed GWAS on 461 gut microbiota KEGG functionality modules (prevalence ≥ 30%; Supplementary Fig. 4a and Supplementary Table 24) in HUNT. We identified genetic signals in four loci, including eight SNP-KEGG functionality module associations (P < 4.9 × 10−10; Table 2, Supplementary Fig. 4a,c), which were selected for replication. Among these, six SNP-KEGG functionality module associations were successfully replicated (concordant direction of effect in the replication data and P < 6.25 × 10−3 (Bonferroni correction for eight comparisons)) in the Swedish replication data sets (n = 16,017; Table 2 and Supplementary Fig. 4a), none of which have been previously reported (Supplementary Note). The replicated genetic signals for KEGG functionality modules were found at three loci (LCT, ABO and FUT2) (Table 2). The identified genetic signals for the KEGG functionality modules were either the same SNP or a SNP strongly linked to the SNP identified for the relative abundance of gut microbiota species at the corresponding loci (Tables 1 and 2 and Supplementary Fig. 3g–i). Using GCTA, SNP heritability estimates for KEGG functionality modules varied between 0% and 26% (mean heritability of 7.2%; Supplementary Fig. 4b and Supplementary Table 10).
Genetic signal at the LCT locus
We observed that the index SNP rs182549-T allele in LCT, strongly linked with the lactase persistence rs4988235-A allele (linkage disequilibrium (LD) correlation r2 = 0.996 in the European ancestry 1000 Genomes reference panel), was reproducibly associated with lower relative abundance of Bifidobacterium adolescentis (P = 2.1 × 10−45; Table 1 and Supplementary Fig. 3a) and decreased functional potential of four KEGG functionality modules including SenX3-RegX3 (phosphate starvation response) two-component regulatory system (P = 5.5 × 10−37), glutamate transport system (P = 2.7 × 10−37), putative zinc/manganese transport system (P = 3.3 × 10−33), and crassulacean acid metabolism (P = 2.0 × 10−22; Table 2). The relative abundance of Bifidobacterium adolescentis was strongly correlated with all four identified KEGG functionality modules, especially with the SenX3-RegX3 (phosphate starvation response) two-component regulatory system (60% of the variance explained) and the glutamate transport system (61% of the variance explained; Supplementary Table 35).
Genetic signal at the HLA-DQB1 locus
The Agathobacter sp000434275 index SNP (rs28407950, P = 4.0 × 10−20; Supplementary Table 7) was assigned by the Open Targets platform14 to have functional implications for the nearby HLA-DQB1 gene, with expression quantitative trait loci (eQTLs) in the small intestine and in sigmoid colon (Supplementary Tables 11 and 12). Our disease-focused PheWAS of rs28407950 revealed strong associations for rs28407950-T, which is associated with higher relative abundance of Agathobacter sp000434275 and reduced risk of major autoimmune diseases such as type 1 diabetes (P = 1.2 × 10−183), asthma (P = 4.3 × 10−118) and celiac disease (P = 2.1 × 10−84; Supplementary Table 13).
Observationally, the relative abundance of Agathobacter sp000434275 was inversely associated with the plausible gastrointestinal condition celiac disease in HUNT (n = 240 cases and 12,437 controls; odds ratio = 0.53, 95% confidence interval 0.44-0.63 per standard deviation (s.d.) increase in relative abundance of Agathobacter sp000434275, P = 3.7 × 10−12). This association is also illustrated in a distribution plot (Supplementary Fig. 6a), and the mean relative abundance of Agathobacter sp000434275 was 0.046% in celiac disease cases and 0.120% in controls (Supplementary Note).
MR revealed that having celiac disease was causally associated with reduced relative abundance of Agathobacter sp000434275 (beta = −0.036, s.e. = 0.014, P = 9.9 × 10−3; Supplementary Tables 25 and 26), whereas the possible impact of Agathobacter sp000434275 on celiac disease is unclear (Supplementary Note).
Genetic signal at the MUC12 locus
The Open Targets platform assigned the Coprobacillus cateniformis index SNP (rs4556017, P = 1.1 × 10−37; Supplementary Table 7) to have functional implications for the nearby MUC12 gene (Supplementary Table 14). For rs4556017, there is an eQTL for MUC12 in the rectum (Supplementary Table 15). Colocalization analyses provided strong evidence (posterior probability, PP4 = 99.96%) for a shared causal variant in the MUC12 locus affecting both the relative abundance of C. cateniformis and the expression of MUC12 in rectum (Fig. 3c,d). The rs4556017-T allele was associated with both increased relative abundance of C. cateniformis and increased MUC12 expression in rectum, compared with the reference C allele.

a,b, Bayesian colocalization analyses demonstrating colocalized genetic signals for C. cateniformis and hemorrhoidal disease, with a posterior probability of a single shared causal variant of 99.8%. c,d, Bayesian colocalization analyses demonstrating colocalized genetic signals for C. cateniformis and MUC12 expression in rectum, with a posterior probability of a single shared causal variant of 99.96%. e,f, MUC12 expression in human sigmoid colon. e, MUC12 mRNA is indicated in red, MUC2 protein as a marker of Goblet cells in green, EpCAM (epithelial cell adhesion molecule) protein in yellow as a marker of colonocytes and nuclei in white. f, MUC12 mRNA is indicated in red, MUC12 protein in green, EpCAM protein in yellow as a marker of colonocytes and nuclei in white. The experiment was repeated three times using samples from four different individuals. g,h, Bayesian colocalization analyses providing evidence for colocalization of the genetic signals for the relative abundance of Clostridium sp900540255 and high cholesterol, with a posterior probability of a single shared causal variant of 80.3%.
Our disease-focused PheWAS of the top associated SNP rs4556017 revealed a robust association with hemorrhoidal disease (P = 1.3 × 10−22; Supplementary Table 16). Using Bayesian colocalization analyses, we found strong evidence (PP4 = 99.8%) for a shared causal variant in the MUC12 locus affecting both the relative abundance of C. cateniformis and the risk of hemorrhoidal disease (Fig. 3a,b and Supplementary Note).
We observed that the MUC12 transcript displayed the highest expression in colon among all 54 investigated human tissues available in the Genotype-Tissue Expression (GTEx) portal (Supplementary Fig. 7). To determine the cellular distribution of MUC12 expression in the human sigmoid colon, we performed in situ hybridization (RNAscope). MUC12 mRNA was abundantly expressed in colonocytes, but expression was also observed in MUC2-positive goblet cells (Fig. 3e,f), whereas MUC12 protein was observed at the luminal surface of colonocytes (Fig. 3f and Supplementary Fig. 8).
Genetic signal at the SLC37A2 locus
We observed a novel genetic signal (index SNP rs73024305) for the relative abundance of Dysosmobacter sp001916835 in the SLC37A2 (solute carrier family 37 (glycerol-3-phosphate transporter) member 2) locus at Chr11 (Table 1, Supplementary Fig. 3e and Supplementary Tables 17 and 18). Evidence from the Open Targets platform for SLC37A2 being the underlying gene for this genetic signal includes correlation between rs73024305 and the transcriptional activity of enhancers and transcription start sites of SLC37A2 using the FANTOM5 expression atlas15 (Supplementary Table 17) and an eQTL (Supplementary Table 18). The PheWAS of the top associated SNP rs73024305 did not reveal robust associations with diseases (Supplementary Table 19). Association analyses with circulating metabolites demonstrated that Dysosmobacter sp001916835 was associated with reduced levels of the secondary bile acid isoursodeoxycholate (P = 5.1 × 10−160, rs = −0.29) and increased levels of the metabolite 3-phenylpropionate (P = 1.3 × 10−161, rs = 0.29; Supplementary Table 27).
Interaction between genetic variants at ABO and FUT2 loci
The strongest replicated association in the FUT2 locus was observed between rs2287921 and Clostridium sp900540255 (P = 3.4 × 10−21; Fig. 1c and Table 1). This SNP is in LD with the functional variant rs601338 (r2 = 0.65; D′ = 0.87 in European-ancestry populations, A/A = non-secretor for rs601338 is associated with C/C for rs2287921) that introduces a stop codon in FUT2 (Supplementary Tables 20 and 21)16. The association between the secretor SNP rs601338 and Clostridium sp900540255 was also significant in HUNT (P = 8.8 × 10−13). The other three gut microbiota species index SNPs in this locus were also strongly linked with the functional FUT2 variant rs601338 (Table 1, Supplementary Fig. 3f and Supplementary Table 28).
FUT2 encodes the enzyme alpha-1,2-fucosyltransferase 2, required for synthesizing fucosylated mucin glycans in the intestinal mucosa16. As alpha-1,2-fucosyltransferase 2 is required for the expression of ABO antigens on the intestinal mucosa, we hypothesized that there might be an interaction between the top genetic signal identified in the ABO locus (rs550057) and the top genetic signal in the FUT2 locus (rs2287921). We observed a strong interaction between these two SNPs (P = 5.2 × 10−7 for the SNP × SNP interaction term) for the association with the relative abundance of Mediterraneibacter torques (Supplementary Table 29 and Supplementary Note).
Our disease-focused PheWAS of the index SNP rs2287921 in the FUT2 locus revealed an association with a composite cardiovascular-related outcome parameter (P = 6.9 × 10−18; Supplementary Table 22). The rs2287921-T allele (responsible for the formation of the secretor status) was associated with a decreased risk of cardiovascular-related outcomes. Separate analyses revealed that the observed association was mainly driven by the strong association of rs2287921-T with a reduced risk of high cholesterol and hypertension (Supplementary Tables 22 and 34). Bayesian colocalization analyses provided strong evidence for a shared causal variant in the FUT2 locus for relative abundance of Clostridium sp900540255 and high cholesterol (PP4 = 80%; Fig. 3g,h and Supplementary Note).
Analyses of circulating metabolites known to be associated with poor cardiometabolic health revealed that Mediterraneibacter faecis, linked to FUT2 secretion status, was associated with reduced levels of the metabolites p-cresol sulphate (P = 1.0 × 10−26, rs = −0.12) and phenylacetate (P = 4.6 × 10−33, rs = −0.13), whereas Streptococcus gordonii, linked to FUT2 non-secretors, was associated with increased levels of imidazole propionate (P = 6.3 × 10−40, rs = 0.14; Supplementary Table 27). The KEGG functionality module GWAS demonstrated that rs35866622-T, reflecting FUT2 non-secretor status, was reproducibly associated with enhanced Ihk-Irr (virulence regulation) two-component regulatory system (P = 6.1 × 10−11; Table 2). The relative abundance of S. gordonii explained a major part (85%; Supplementary Table 35) of the variance in the activity of Ihk-Irr (virulence regulation) two-component regulatory system, suggesting that this KEGG functionality module is primarily driven by S. gordonii.
Evidence of an effect of BMI on gut microbiota composition
Observational studies have reported associations between gut microbiota composition and BMI, but the underlying causality is unclear13. To explore the possible causal associations between BMI and overall gut microbiota composition, we used our current large discovery GWAS data set in HUNT (n = 12,652). We first determined the observational association between two α-diversity parameters (Shannon diversity index and species richness) and BMI (Fig. 4a and Supplementary Tables 31 and 32). In models adjusted for age and sex, both Shannon diversity index and richness were inversely associated with BMI. Two-sample MR showed that increased genetically determined BMI reduced both Shannon diversity index and species richness, with similar effect estimates as in the observational linear regression association analyses. Using the single genetic instrument available from the current GWAS for the Shannon diversity index, we did not observe any evidence of reverse causality (Fig. 4a and Supplementary Table 31). However, this analysis is likely underpowered.

a, Observational (linear regression) and causal (MR) associations for α-diversity parameters (Shannon index and richness) and BMI. Effect estimates (mean ± 95% confidence intervals) are given as s.d. change in outcome per s.d. increase in exposure. No genetic instrument was available to test the causal effect of richness on BMI. Observational cross-sectional linear regression analyses in HUNT were adjusted for age, sex and library plate (n = 12,836). Both the exposures and outcomes were inverse rank transformed before analysis. b, Comparison of observational (linear regression analyses) association with causal (MR) association for the effect of BMI on gut microbiota species. The overall observational and causal associations between the 546 evaluated gut microbiota species and BMI were evaluated. Observational associations between gut microbiota species and BMI in HUNT were adjusted for age, sex and library plate and are given as s.d. change in BMI per s.d. change in gut microbiota species (x-axis; n = 12,840). Effect estimates for the causal effect of BMI on gut microbiota species (y-axis) are given as s.d. change in gut microbiota species per s.d. increase in BMI. Both the exposures and outcomes were inverse rank transformed before analyses. The red line represents the correlation of the effect estimates for the observational associations, of all 546 evaluated gut microbiota species, and the effect estimates of the corresponding causal effect of BMI on relative abundance of gut microbiota species. R, Pearson correlation coefficient. P value is given for the correlation analysis. The P value was calculated using a two-sided z-test.
Next, we explored the overall observational and causal associations between BMI and the 546 evaluated gut microbiota species. In models adjusted for age and sex, the relative abundances of 129 species (24%) were nominally (P < 0.05) directly observationally associated with BMI, and 296 species (54%) were inversely associated with BMI (Supplementary Table 32). Two-sample MR revealed evidence supporting that BMI was nominally causally associated with the relative abundance of 104 species (39 species increased and 65 species decreased with increased BMI, P < 0.05; Supplementary Table 32). For these 546 MRs of the effect of BMI on species, the number of nominal significant P values observed (n = 104) was higher than expected by random (n = 27.3; Chi-square, P = 3.3 × 10−49), suggesting that BMI influences relative abundances of species (Supplementary Table 32). Correlation analyses of the betas for the observational associations of all 546 evaluated species and the betas from the MR of the corresponding causal effect of BMI on the relative abundance of species showed a strong positive correlation (Pearson correlation 0.76, P = 7.0 × 10−104; Fig. 4b). This finding provides statistical evidence that BMI exerts an overall effect on the relative abundance of the 546 evaluated species, which contributes to the observational associations between gut microbiota species and BMI (Supplementary Note).
Discussion
The gut microbiota has been associated with human health and disease, but causality is unclear. To unravel host genetics factors influencing gut microbiota composition, we performed a large-scale GWAS including 12,652 participants followed by replication in up to 21,976 participants. We demonstrated a robust contribution of genetic variation to the variability in α-diversity parameters, relative abundance of individual species, and gut microbiota functionality modules in HUNT. We identified 12 reproducible SNP-species associations in six loci, including the known LCT and ABO loci and the novel HLA-DQB1, MUC12, SLC37A2, and FUT2 loci. Reproducible genetic signals for KEGG functionality modules were also identified at the LCT, ABO, and FUT2 loci. Our follow-up analyses suggested that the identified SNP-species associations might contribute to understanding the underlying pathogenesis of celiac disease and hemorrhoidal disease. In addition, we identified BMI as a determinant of gut microbiota composition.
The substantially higher number of reproducible genetic signals in the present study compared to previous GWASs on gut microbiota composition2,3,4,9,10,11 is likely due to our large discovery cohort, the use of metagenome sequencing instead of 16S ribosomal RNA gene sequencing, the use of a state-of-the-art microbiome profiling methodology17, and using the same standardized high-resolution Genome Taxonomy Database (GTDB) species taxonomy for all included participants (Supplementary Note).
In the present study, the lactase-persistence allele in the LCT locus was associated with lower relative abundance of Bifidobacterium adolescentis, replicating previous GWAS findings2,3,4. Extending on the previous findings, we showed that the lactase intolerance genetic variant was also associated with increased activity of certain KEGG functionality modules. A likely explanation is enhanced growth of B. adolescentis due to the presence of lactase not metabolized by the lactose intolerant hosts. Thereby, there is an increase in the functional pathways that are largely represented by B. adolescentis abundance. In addition, these changes in functionality may also be explained by altered dietary habits in combination with altered gut microbiota composition in participants with lactose intolerance.
The SenX3-RegX3 two-component regulatory system was increased in participants with the lactase intolerance genetic variant. This variant is expressed in Bifidobacterium and shown to promote expression of the high-affinity phosphate transporter Pst, involved in energy metabolism, which leads to high inorganic phosphate uptake, for example, for ATP synthesis18. Gut microbiota species that are more abundant in lactose-intolerant subjects, such as B. adolescentis, could have an altered energy metabolism when metabolizing lactose (or other glycans), leading to an increased functionality in the SenX3-RegX3 system (Supplementary Note).
It has been proposed that changes in gut microbiota composition may result in the transition from genetic predisposition to the actual onset of celiac disease characterized by loss of gluten tolerance19. However, no gut microbiota species have been reproducibly associated with celiac disease19. In the present study, the rs28407950-T allele in the HLA-DQB1 locus was associated with higher relative abundance of Agathobacter sp000434275 and reduced risk of celiac disease. As celiac disease is a gastrointestinal condition with partly unclear etiology19,20, we hypothesized that Agathobacter sp000434275 may play a contributory role. Our cross-sectional observational association analyses in HUNT demonstrated that a high relative abundance of Agathobacter sp000434275 was associated with a low prevalence of celiac disease. There was some evidence from MR that that celiac disease reduces the relative abundance of Agathobacter sp000434275, whereas the possible impact of Agathobacter sp000434275 on celiac disease is unclear (Supplementary Note). In summary, these findings clearly demonstrate that low relative abundance of Agathobacter sp000434275 is associated with celiac disease, but further studies are warranted to determine causality.
The gut microbiota composition has also been proposed to be involved in the pathogenesis of hemorrhoidal disease, but no gut microbiota species have been reproducibly linked to the disease21,22. The present study showed that the rs4556017-T in the MUC12 locus was associated with a higher relative abundance of C. cateniformis and reduced risk of hemorrhoidal disease. We also observed that MUC12, a transmembrane mucin23, is highly expressed in colonocytes of the human colon and that the C. cateniformis index SNP rs4556017 has an eQTL for MUC12 in the rectum. A connection between C. cateniformis and MUC12 expression was supported by strong colocalization evidence for a shared causal variant in the MUC12 locus affecting both the relative abundance of C. cateniformis and the expression of MUC12 in rectum, with increased relative abundance of C. cateniformis associated with increased MUC12 expression in rectum (Supplementary Note).
MUC12 has a transmembrane single-pass domain, a cytoplasmic tail, and an enormous extracellular mucin domain densely decorated with glycans. The glycocalyx of enterocytes and colonocytes is built and composed of transmembrane mucins, such as MUC12 and MUC17, that reach about a micrometer out in the lumen from the cell surface23. The small intestinal MUC17-based glycocalyx prevents direct bacterial binding to enterocytes24 and may influence the gut microbiota composition25. Based on these findings, it is possible that MUC12 in the glycocalyx of colon/rectum might regulate the relative abundance of C. cateniformis in feces, which in turn may influence the risk of hemorrhoidal disease. Alternatively, MUC12 might independently affect both the relative abundance of C. cateniformis in feces and the risk of hemorrhoidal disease. Further studies are required to delineate the interactions between C. cateniformis, MUC12, and hemorrhoidal disease.
The present study observed a novel reproducible genetic signal for Dysosmobacter sp001916835 in the SLC37A2 locus. There was some transcriptional support for SLC37A2, which encodes a glucose-6-phosphate transporter located in the endoplasmic reticulum26, being the gene driving this association. We speculate that host cellular sugar transport/metabolism may influence the host-microbiome interaction. We observed that a higher abundance of Dysosmobacter sp001916835 was associated with reduced circulating levels of the secondary bile acid isoursodeoxycholate and increased levels of 3-phenylpropionate. Isoursodeoxycholate has been reported to be a marker of poor cardiometabolic health27 whereas 3-phenylpropionate indicates high fiber intake28, suggesting that the relative abundance of Dysosmobacter sp001916835 might reflect a healthy diet. However, additional studies are required to characterize the underlying biology connecting Dysosmobacter sp001916835 with human host genetics.
We observed an interaction between the identified genetic signals at the ABO and FUT2 loci for the relative abundance of Mediterraneibacter torques, supporting previous findings3,4,8. This interaction is most likely explained by the fact that FUT2 is required for ABO antigen expression on the intestinal mucosa. This impacts the relative abundance of certain gut microbiota species dependent on antigens with accessible glycans in non-O blood-type secretors, providing direct energy sources for these species3. M. torques is a known mucin glycoprotein degrader with strong fucosidase activity29, and it is likely that reduced FUT2 activity, resulting in less fucosylated mucin glycans, leads to a lower abundance of M. torques2.
The index SNP in the FUT2 locus was also associated with a composite cardiovascular-related parameter. Further separate analyses revealed that an association with high cholesterol and hypertension mainly drove this association. A connection between Clostridium sp900540255 and high cholesterol was supported by strong evidence for genetic colocalization (Supplementary Note).
Interestingly, FUT2 non-secretors had increased relative abundance of S. gordonii, a species normally present in the oral cavity but has also been linked to cardiovascular disease30. It was recently demonstrated that the relative abundance of S. gordonii in the gut is linked to subclinical coronary atherosclerosis in the well-powered SCAPIS cohort30. Gut bacteria have been proposed to affect the development and progression of atherosclerosis via secretion of atherogenic metabolites or through infections local or distal to the atherosclerotic plaque31. We observed that high circulating levels of p-cresol sulphate and imidazole propionate, which have been associated with poor cardiometabolic health31, were linked via association with certain gut microbiota species to FUT2 non-secretors. Our gut microbiota functionality studies revealed that FUT2 non-secretors were also associated with enhanced Ihk-Irr (virulence regulation) two-component regulatory system. The Ihk/Irr system influences the expression of genes involved in cell wall synthesis and modification, which are critical for the bacteria’s ability to resist destruction by neutrophils32,33. We also made the interesting observation that the relative abundance of S. gordonii explained a major part of the variance in the activity of the Ihk-Irr (virulence regulation) two-component regulatory system, suggesting that this KEGG functionality module is primarily derived from S. gordonii. Therefore, it is likely that FUT2 non-secretors, via increased relative abundance of S. gordonii, have enhanced functional capacity of the gut microbiota to evade the host innate immune defense. Further studies are warranted to determine the interaction between FUT2 secretor status, gut microbiota composition and functionality, and circulating metabolites in relation to health outcomes.
Previous observational studies have reported associations between gut microbiota composition and BMI, but the causal direction is unclear13. In the present study, two α-diversity parameters (Shannon diversity index and richness) were inversely associated with BMI. MR showed that genetically predicted increased BMI reduced both Shannon diversity index and richness with similar effect sizes as in the cross-sectional linear regression analyses (observational associations). In addition, we demonstrate that BMI exerts an overall effect on the relative abundance of individual gut microbiota species, which contributes to the observational associations between gut microbiota species and BMI. These findings provide evidence of an overall causal effect of BMI, a measure of adiposity, on gut microbiota composition (Supplementary Note).
Strengths of the present study are (i) the large sample size of the discovery cohort, (ii) replication in large independent cohorts, (iii) the use of state-of-the-art metagenome sequencing for gut microbiota analyses, providing high-resolution information on taxonomy and gut microbiota functionality and (iv) the access to large-scale gut microbiota associations with circulating metabolites, which are useful for mechanistic insights. The present study also has limitations. The discovery and replication cohorts mainly included participants of European ancestry living in Nordic countries, and the findings might not be generalizable to populations with other geographical or ancestral origins. The present study provides stronger genetic instruments for a broader panel of species exposures to be used in MR compared with previous GWASs2,3,4,9,10,11, but these genetic instruments are still relatively weak and often include only a single genome-wide significant independent genetic signal, precluding tests of horizontal pleiotropy. Finally, although several novel loci for gut microbiota species were identified in the present large-scale study, larger meta-analyses of cohorts using the same high-resolution taxonomy are required to further disentangle the genetic architecture by which host genetics regulates the gut microbiota composition and functionality.
In conclusion, our findings support important interactions between host genetics and gut microbiota composition in human health and disease and demonstrate that BMI is a determinant of overall gut microbiota composition.
Methods
Discovery cohort: HUNT
The HUNT study is a longitudinal population-based health study conducted in the county of Trøndelag, Norway (Supplementary Note)34,35,36. Among 56,042 participants in the HUNT4 survey (2017-2019), 13,268 participants submitted stool samples for gut microbiome profiling and data from 12,887 of these participants passed the post-metagenome sequencing quality control (Fig. 1a). A total of 12,652 HUNT4 participants of European ancestry had both genetic and gut microbiome data available and were included in the present GWAS (Fig. 1a). The local ethical review board approved the study (regional committee for medical and health research ethics, Midt-Norge; REK-656785), and all participants provided written informed consent.
Replication cohorts
Swedish cohorts
The replication included participants from four population-based Swedish cohorts. Inclusion in the present study was limited to individuals of European ancestry with high-quality metagenomics and genotype data available. The Swedish CArdioPulmonary BioImage Study (SCAPIS)37 includes 8,733 participants of European ancestry, aged 50-65, from Malmö and Uppsala with samples collected between 2014 and 2018. The Swedish Infrastructure for Medical Population-Based Life-Course and Environmental Research (SIMPLER; https://www.simpler4health.se/w/sh/en) combines data from the Cohort of Swedish Men and the Swedish Mammography Cohort38, including 4,515 men and women from the region of Västmanland (SIMPLER-V) and 981 women from the city of Uppsala (SIMPLER-U) with fecal samples collected between 2011 and 2021. The Malmö Offspring Study (MOS) involves 1,788 adult participants, children, and grandchildren of the Malmö Diet and Cancer (MDC) Study cohort39, with samples collected between 2013 and 2017. The association analyses performed in the Swedish cohorts have been approved by the Swedish Ethical Review Authority (DNR 2022-06137-01 and DNR 2024-01992-02). Ethical approval and written informed consent were obtained for the individual Swedish cohorts. Ethical approval for SCAPIS was granted by the Swedish Ethical Review Board (DNR 2010-228-31 M) and all participants gave written informed consent. The SIMPLER studies received approval from the Swedish Ethical Review Board (DNR 2009/2066-32, DNR 2009/1935-32, DNR 2010/0148-32, DNR 2014/892-31/3), and all participants gave written informed consent. The MOS study received approval from the Ethics Review Committee of Lund University (DNR 2012-594), and all participants gave written informed consent.
FINRISK
For four identified SNP-species associations (but no SNP-KEGG functionality module), data for replication was also publicly available from the FINRISK cohort (n = 5,959)4. The participants in the FINRISK cohort were analyzed using shallow metagenome sequencing as previously described4 and the Genome Taxonomy Database (GTDB) was used for annotation of taxa included in the published FINRISK GWAS4.
Gut microbiome profiling - HUNT
Metagenome sequencing of HUNT samples
Stool collection and DNA isolation and quantification have been performed using a standardized procedure, as previously described13 before sequencing and microbiome profiling at Clinical Microbiomics in Denmark (Fig. 1a and Supplementary Note)17.
The enzymatic fragmentation of DNA and library construction was conducted on a Tecan DreamPrep NGS automation system using the Celero EZ DNA-seq Core Module. A DNA sample volume of 10 μl was used. To ensure that the maximum amount of 500 ng input DNA recommended by the manufacturer was not exceeded, all samples with DNA concentrations >50 ng μl−1 were diluted to 30 ng μl−1. The fragmented DNA was amplified using PCR. Short and long DNA fragments were removed using double-sided magnetic bead size selection (AMPure XP, Beckman Coulter, reference A63882). Adapter sequences from Celero 96-Plex Adaptor Plate were added to each sample during library construction. The final concentration for each library was quantified by Tecan Infinite F Nano+ Plate Reader using NuQuant NGS Library Quantification Module. Qubit and TapeStation were used to determine the concentration of the final library before sequencing at 2 × 150 bp on an Illumina NovaSeq 6000. Samples were sequenced to an average depth of 22.9 million read pairs per sample. For each sample, more than 85% (mean ≥ 30 = 93.8%) of the bases had a Phred quality score of ≥30.
Gene catalog and species definitions
Gut microbiome profiling was performed using the Clinical Microbiomics Human Microbiome Profiler (CHAMP) pipeline, which uses the GTDB r214 for taxonomic annotation of prokaryotes17. Clinical Microbiomics gave species missing from the GTDB r214 database a unique species ID (“hMGS”). The HMR05 catalog used in the present study was based on 30,382 samples from nine human body sites, including prokaryotic metagenome-assembled genomes (MAGs) mainly from the Unified Human Gastrointestinal Genome collection40 and the Early-Life Gut Genomes catalog41. In addition, genome assemblies from NCBI and PATRIC were added to capture otherwise missing species of interest (human-associated pathogens, probiotics, food ingredients and species relevant for benchmarking). MAGs were clustered by species using the Genome Taxonomy Database Toolkit (GTDB-Tk release R214), whereas unannotated MAGs were clustered at 95% identity using FastANI. The catalog included 6,809 microorganisms.
Human-relevant eukaryotic species were manually identified from various sources, including an analysis of gut fungal species42, publicly available lists of pathogens, the eukaryotes profiled by MetaPhlAn 4 (ref. 43), and various species relevant for benchmarking. The result was 2,740 genomes representing 244 species.
For MAGs not obtained from publicly available MAG collections, reads were host-filtered, trimmed, and assembled into contigs with Megahit (v.1.2.9)44 or metaSPAdes (v.3.15.5)45 and then binned using VAMB (v.3.0.6)46. MAGs were considered high-quality if they had >90% completeness and < 5% contamination based on CheckM2 (v.2022-07-19) and passed the GUNC chimerism test (v.1.0.5)47. All MAGs were taxonomically annotated using GTDB-Tk (v.2.3.0)48 with GTDB database (v.r214)49. To combine MAGs from multiple VAMB batches and MAG collections, MAGs annotated to the same species were merged into species clusters. MAGs without GTDB-Tk species-level annotations were merged with each other or with existing species clusters at 95% identity (dRep50; FastANI51). This resulted in 6,567 prokaryotic species clusters, 10% of which were unannotated at the species level. We used a three-step clustering approach to derive a pan-genome catalog for each species. First, genes were clustered with MMseqs2 (v.14)52 with 98% identity and 90% bi-directional coverage. Second, the representatives from the first iteration were clustered with MMseqs2 with 95% identity and 90% bi-directional coverage. Representatives of the second iteration were chosen as the ones with highest cardinality from the first iteration. Third, the second iteration representatives were clustered with cd-hit (cd-hit-est, v.4.8.1)53 with 95% identity and 90% coverage of the shorter sequence. Genes shorter than 100 bp or with species prevalence < 1% were discarded. For prokaryotes and eukaryotes separately, the entire set of pangenomes was then clustered with MMseqs2 with 97% identity and 90% bi-directional coverage to obtain between-species clusters. The pan-genomes from prokaryotic (n = 6,567) and eukaryotic (n = 244) species were merged into a final catalog of 25,761,278 genes.
To enable quantification of each species in the database, up to 250 signature genes were selected for each species based on core genes (≥60% prevalence in species MAGs) with a length ≥200 bp and ≤20 kb. Furthermore, signature genes were required to be species unique, with no alignments of 100 bp with >97% sequence-identity to other genes in the catalog. However, if fewer than 20 genes meeting this criterion were available for a species, then genes with segments >200 bp without alignments to other genes were used, and non-unique segments of these genes were masked.
HUNT sequencing data preprocessing
Read pairs mapped to the human reference genome GRCh38.p14 were removed using Bowtie2 (v2.4.2)54. Reads were then trimmed to remove adapters and bases with a Phred score below 30 using AdapterRemoval (v. 2.3.1)55. Host-filtered read pairs with both lengths ≥100 bp, defined as high-quality nonhost (HQNH) reads, were retained.
Mapping HUNT sample reads to the gene catalog
HQNH reads from the HUNT samples were mapped to the gene catalog using BWA mem (v. 0.7.17)56. An individual read was considered uniquely mapped to a gene if the mapping quality (MAPQ) was ≥20 and the read aligned with ≥95% identity over ≥100 bp. However, if >10 bases of the read did not align with the gene or extend beyond the gene, the read was considered unmapped. Reads meeting the alignment length and identity criteria but not the MAPQ threshold were considered multi-mapped. Each read pair was counted as either (1) uniquely mapped to a specific gene, if one or both individual reads were uniquely mapped to a gene, or (2) multi-mapped, if neither read was uniquely mapped, and at least one was multi-mapped, or (3) unmapped, if both individual reads were unmapped. If the two reads were each uniquely mapped to a different gene, the gene mapped by read one was counted but not the gene mapped by read two. A gene count table was created with the number of uniquely mapped read pairs for each gene.
Species relative abundance calculation
The relative abundance of each species (MAGs) was calculated based on the species signature genes with observed read counts within the expected 99% quantile and normalized sample-wise so that the total abundance of all species was summed to 100%. The expected read counts for signature genes in each species in each sample were modelled with a negative binomial distribution as follows. First, if ≥50 of the signature genes for a species had non-zero read counts and ≥99% of genes were expected to have non-zero read counts given the total read count for that species, then signature genes with zero reads were ignored in that sample. Second, the expected 99% quantile (between 0.5% and 99.5%) of read counts was calculated for each gene based on a negative binomial distribution with a mean proportional to the effective gene length (accounting for read length and mapping alignment criteria) and dispersion defined as log2 (effective gene length). The abundance of each species was then calculated as the mean read count normalized by effective gene length based on reads mapping to signature genes with observed read counts within the expected 99% quantile. Species abundances were set to zero if less than five genes with non-zero read counts were within the 99% quantile. Furthermore, species with < 66% of genes with non-zero read count within the 99% quantile were set to zero, unless the median abundance of signature genes was non-zero, in which case the median gene-length-corrected abundance of non-zero genes was used. Abundances were then normalized sample-wise such that all species’ total abundance was 100% (Supplementary Note).
In addition, for the estimation of α-diversity measures, rarefied species abundance profiles were calculated by random sampling, without replacement, of a fixed number of signature gene counts per sample and following the procedure described above. In HUNT, 164,245 signature gene counts were sampled for the rarefied dataset. After profiling the rarefied data set, the α-diversity measures (Shannon diversity index and richness) were calculated using rarefied species relative abundances with the diversity function of the vegan R package.
In the HUNT gut microbiota cohort, on average, 84% of the high-quality microbiome reads from a sample were mapped to the Clinical Microbiomics HMR05 gene catalog, and on average, 459 gut microbiota species were detected per sample. A total of 12,887 high-quality samples passed the post-metagenome sequencing quality control in HUNT, with 4,870 gut microbiota species present in at least one sample (Fig. 1a). For the subsequent association studies, the relative abundance of species and α-diversity measures were inverse rank normal transformed.
Functional annotation and profiling
EggNOG-mapper (v. 2.1.7, Diamond mode)57 was used to map prokaryotic genes in the gene catalog to the EggNOG orthologous groups database (v. 5.0)58 and Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) database. Eukaryotic genes were annotated using KofamScan59. Functional potential profiles based on KOs were calculated as the proportion of the total gene abundance mapped to a given KO.
KEGG modules (v. 78.2)60 were defined as a set of KOs that enable a specific function or pathway. Functional potential profiles based on KEGG modules were generated from the species profiles in HUNT. For this, we identified the set of species associated with each of the KEGG modules by following three criteria: (1) a species was associated with a KEGG module if it included at least 2/3 of the genes encoding the proteins/enzymes needed to complete the functionality of the module; (2) if a module had alternative reaction paths, only one of these was required to be 2/3 complete; and (3) for modules with three or fewer steps, all steps were required to be comprised in the given species. KEGG module profiles based on relative abundances were then calculated by adding the relative abundances of each species fulfilling the criteria for being associated with a given KEGG module.
Gut microbiome profiling: Swedish cohorts
SCAPIS and MOS
DNA extraction, quality control, and library preparation of metagenomic DNA for SCAPIS and MOS have been performed by Clinical Microbiomics A/S and described in detail before30. Libraries from stool DNA were sequenced using the Illumina NovaSeq 6000 instrument using 2 × 150 bp paired-end reads, generating on average 26.0 and 25.3 million read pairs, respectively in SCAPIS and MOS with 97.8% of the sequenced bases having Phred quality score >20.
SIMPLER (SIMPLER-V and SIMPLER-U)
Stool samples were thawed, and a small portion was aliquoted and combined with 800 μl DNA/RNA Shield. These aliquots were sent to the Centre for Translational Microbiome Research at the Karolinska Institutet in Stockholm for metagenomic DNA extraction and sequencing, conducted during 2022 and 2023. The DNA from the samples was extracted using the MagPure Stool kit. The genomic DNA was then fragmented and used to construct libraries using the MGIEasy FS DNA Library Prep Set kit. The prepared DNA libraries were evaluated using a TapeStation D1000 kit, and their quantity was determined by a QuantIT High Sensitivity dsDNA Assay on a Tecan Spark. The pooled libraries were circularized using the MGI Easy Circularization kit and sequenced with 2 × 150 bp paired-end reads on the DNBSEQ G400 or T7 sequencing instrument, following the manufacturer’s instructions, resulting in an average yield of 51 million reads per sample.
Microbial taxonomy profiling was performed for all four Swedish replication cohorts at Clinical Microbiomics using the CHAMP profiler based on the Human Microbiome Reference HMR05 catalog in a similar manner to that described for HUNT above.
Genotyping and imputation: HUNT
HUNT participants were genotyped using Illumina HumanCoreExome arrays and genotype data were imputed to the Human Reference Consortium (HRC) 1.1 panel (Supplementary Note)61.
Genotyping and imputation: Swedish cohorts
The Swedish cohorts were genotyped using Illumina GSA arrays and genotype data were imputed to the HRC 1.1 panel.
GWAS of gut microbiota species
We performed GWAS of the relative abundance of 546 gut microbiota species (prevalence >30%; primary outcomes) in 12,652 HUNT4 participants of European ancestry using linear ridge regression under an additive genetic model for each variant as implemented by REGENIE (v.3.4.1)62. Variants with a MAF < 1% or INFO < 0.3 were excluded from the analyses, leaving 7,971,623 common and low-frequency genetic variants for testing. Before analysis, we applied an inverse rank normal transformation of the relative abundance of each gut microbiota species. Age, sex, genotyping batch, library plate, and the first ten principal components of ancestry were included as covariates in the analyses. To be taken forward to replication efforts, we required a P < 1.3 × 10−10 (genome-wide significance adjusted for number of effective tests, which we estimated to be 391)3. We selected the index SNP for each species in each locus. Among the 13 selected SNP-species associations, 12 passed a conservative threshold of P < 9.2 × 10−11, adjusting for all 546 gut microbiota species analyses, whereas the last one selected passed the significance level adjusting for number of effective tests (P < 1.3 × 10−10) in HUNT (Table 1).
To test for multiple independent association signals within each locus, we performed stepwise conditional regression analyses in each identified locus. We included the same covariates and inverse-rank transformation of the variables as in the main analysis, but we added the index variant in the previous step as a covariate for each consecutive step.
Replication of identified SNP–species signals
We took 13 SNP–species associations discovered in HUNT forward for replication in five Nordic replication cohorts. We tested all SNP-species associations in the four meta-analyzed Swedish replication cohorts (SCAPIS, n = 8,733; SIMPLER-V, n = 4,515; SIMPLER-U, n = 981; MOS, n = 1,788; total sample size, n = 16,017). For four identified SNP-species associations, data for replication was publicly available from the Finnish FINRISK cohort (n = 5,959)4. For successful replication, concordant direction of effect in the combined replication data set (n = 16,017-21,976) and P < 3.8 × 10−3 (Bonferroni correction for 13 comparisons) were required.
Meta-analyses were performed either with fixed-effect inverse-variance weighted or sample size weighted meta-analysis using METAL (v. 2011-03-25)63. For meta-analyses including the FINRISK study, sample size weighted meta-analysis was used because the effect sizes were on a different scale than the other cohorts (HUNT and the Swedish cohorts used standardized inverse rank transformed relative abundance of species, whereas FINRISK used standardized center log-transformed relative abundance of species).
Sensitivity analyses considering antibiotic treatment
In sensitivity analyses, we excluded participants with recent antibiotic use defined as a dispensed prescription (Anatomical Therapeutic Chemical codes J01 and J04; n = 1,046) up to 3 months before the delivery of the fecal sample kit (Supplementary Note).
GWAS for KEGG functionality modules and α-diversity measures
To further follow up on the results from the primary GWAS on gut microbiota species, we used the above described GWAS approach and performed GWAS of 461 gut microbiota KEGG functionality modules (prevalence ≥30%; Supplementary Table 24) and two α -diversity measures (Shannon diversity index and richness (number of observed species) calculated from rarefied data with a rarefication target of 164,245 signature gene counts). Shannon diversity index was calculated using the R package vegan (v.2.6-4).
For KEGG functionality modules, we required a P < 4.9 × 10−10 (genome-wide significant threshold adjusted for the number of effective tests, which was estimated to be 102)3 to be selected for replication in the Swedish cohorts (Supplementary Fig. 4a). We selected the most significant genetic signal for each KEGG functionality module in each locus (Table 2). Tests for replication were performed in the Swedish cohorts for eight SNP-KEGG functionality module associations identified in the HUNT discovery cohort (Supplementary Fig. 4a). For successful replication, concordant direction of effect in the replication data and P < 6.25 × 10−3 (Bonferroni correction for eight comparisons) were required (Supplementary Fig. 4a).
Definition of celiac disease cases in HUNT
Participants with celiac disease were identified through serological screening and linkage to hospital journal records and the Norwegian Patient Registry64. The 240 celiac disease participants with available fecal samples from HUNT4 were included in the present study. The associations between the relative abundance of a gut microbiota species and celiac disease prevalence were determined by logistic regression, adjusting for age, sex and library plate.
SNP heritability
SNP heritability using GCTA
We estimated the narrow-sense (additive) SNP heritability (Vg/Vp ± SE, where Vg is the variance explained by the SNPs and Vp is the total phenotypic variance) of the α-diversity parameters (Shannon diversity index and richness), and of the relative abundances of gut microbiota species and KEGG modules in HUNT, using genome-wide complex trait analysis (GCTA) (v. 1.94.1)65,66. We first created a genetic relationship matrix (GRM) based on 365,943 genotyped autosomal variants in 8,593 unrelated (no first- or second-degree relatives as estimated by KING v.2.3.2)67 HUNT4 participants. Secondly, we used the GRM with GCTA-GREML (genomic relatedness-based restricted maximum-likelihood) to estimate the phenotypic variance explained by the genetic variants for relative abundance of each investigated parameter after inverse rank transformation. For each estimate, we included age, sex, genotyping batch and library plate as covariates in the analysis.
Heritability estimates using LD score regression
As an alternative method to estimate the heritability of α-diversity parameters (Shannon index and richness) and the relative abundance of gut microbiota species, we used LD score regression as implemented in the LD score tool available on Github (https://github.com/bulik/ldsc)68. The LD score regression analyses were restricted to HapMap3 SNPs with MAF > 5% in the 1000 Genomes European reference population. We used precalculated LD scores from the same reference panel (https://data.broadinstitute.org/alkesgroup/LDSCORE/).
MR
As exposures in the two-sample MR, we used genetic instruments for the relative abundance of gut microbiota species and Shannon diversity index, derived from the current discovery GWAS, selected human diseases, identified in our PheWAS, derived from publicly available GWAS data sets (celiac disease69, hemorrhoidal disease70, cardiovascular-related outcomes71), and BMI72. We only selected variants with a MAF > 1% and P < 5 × 10−8. We selected instruments with r2 < 0.01 (based on the European populations in LDlink)73 to ensure little correlation between instruments. The variance explained (R2) and F statistic for the genetic instruments were estimated from the respective GWAS summary statistics (Supplementary Table 25). For exposures with multiple genetic instruments, we applied the inverse variance weighted method using fixed or random effects depending on the Cochran’s Q statistic test of heterogeneity. We then used the MR-Egger regression as a sensitivity analysis to test for possible directional horizontal pleiotropy74. In further sensitivity analyses, we used the weighted median MR method. For exposures with only one genetic instrument, we estimated the Wald ratio. The MR analyses were conducted using the R package MendelianRandomization75.
Colocalization
To assess if any of the identified gut microbiota species loci were consistent with having shared causal variants with selected human diseases or tissue-specific eQTLs of interest, we combined their GWAS summary statistics and performed a Bayesian colocalization analysis as implemented in the R package coloc (Supplementary Note)76.
Associations between gut microbiota species and circulating metabolites in the SCAPIS cohort
For mechanistic insights, we also evaluated the associations for the gut microbiota species with replicated genetic signals with circulating metabolites, analyzed using the Metabolon platform in the Swedish SCAPIS cohort77. We considered the top three annotated circulating metabolites associated with each species with replicated SNP-species GWAS findings (Supplementary Table 27). To investigate the association of genetic variants and species abundance with plasma metabolite levels, we conducted partial Spearman’s rank correlations adjusted for age, sex, place of birth and metabolomics delivery batch.
Dual RNAscope and immunohistochemistry of MUC12 in the human sigmoid colon
Biopsies from the sigmoid colon were collected from patients (>18 years) with normal intestinal macroscopy who were referred for colonoscopy to the Sahlgrenska University Hospital, Gothenburg. The protocol complied with the Declaration of Helsinki and was approved by the Research Ethical Committee in Gothenburg (ethical permission 2020-03196). All patients gave written informed consent. Collected biopsies were fixed in 4% paraformaldehyde and embedded in paraffin.
An anti-MUC12-S2 polyclonal rabbit antibody was raised against the peptide DYTLEYEELFENLAEIVKAKIMNEC. Fluorescent in situ hybridization (FISH) on the tissue described above was performed using the Multiplex Fluorescent Detection Reagent v2 (ACD; 323110), following the manufacturer’s standard RNAscope protocols. The probe Hs-MUC12-O1-C1 (1569341-C1) was used to detect MUC12, with fluorescent signals visualized using the TSA Plus Cyanine 5 system (PerkinElmer, NEL705A001KT). Immediately after the MUC12 RNAscope FISH detection, immunofluorescence was performed on the same tissue section. Blocking serum was applied and incubated for 1 h before the addition of primary antibodies against either EpCAM (1:250, Abcam, ab71916, lot #1076051-3) and MUC2-C3 (1:100, GeneTex, GTX100664, lot #44818) or EpCAM and MUC12-S2 (1:250), which were diluted in blocking serum and left to incubate overnight at 4 °C. Subsequently, secondary antibodies, either Goat anti-Mouse IgG, IgM (H + L) Secondary Antibody, Alexa Fluor 488 (1:400, ThermoFisher, catalog #A10680, lot #1664758) or Cy3 AffiniPure Donkey Anti-Rabbit IgG (H + L) (1:400, Jackson ImmunoResearch, catalog #711-165-152, lot #171768) were applied and incubated for 1 h at room temperature, after which the tissue was counterstained with DAPI and imaged using a Nikon Spinning Disk system.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Individual-level data from HUNT can be accessed by, or in collaboration with, a Norwegian principal investigator. Researchers can apply for HUNT data access from HUNT Research Centre (https://www.ntnu.edu/hunt) if they have obtained project approval from the Regional Committee for Medical and Health Research Ethics (REC). Information on the application and conditions for data access is available at https://www.ntnu.edu/hunt/data. For the replication cohorts, the genetic data used in the SCAPIS, SIMPLER and MOS board are not shared publicly due to confidentiality. Data will be shared upon reasonable request after permission from the Swedish Ethical Review Authority (https://etikprovningsmyndigheten.se) and from the respective cohort boards (https://www.scapis.org/data-access, https://www.simpler4health.se and https://www.malmo-kohorter.lu.se/malmo-offspring-study-mos). Summary statistics of the discovery GWAS are available at the GWAS Catalog under study accession numbers GCST90666541–GCST90667549 (https://www.ebi.ac.uk/gwas). Genome Taxonomy Database Toolkit (GTDB-Tk release R214) can be found at https://gtdb.ecogenomic.org.
Code availability
All analyses have been performed using publicly available software, tools, packages and databases as given in Methods. Most analyses have been performed using R v4.1.1 or higher (https://cran.r-project.org/).
References
Wilmes, P. et al. The gut microbiome molecular complex in human health and disease. Cell Host Microbe 30, 1201–1206 (2022).
Kurilshikov, A. et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 53, 156–165 (2021).
Lopera-Maya, E. A. et al. Effect of host genetics on the gut microbiome in 7,738 participants of the Dutch Microbiome Project. Nat. Genet. 54, 143–151 (2022).
Qin, Y. et al. Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort. Nat. Genet. 54, 134–142 (2022).
Gomaa, E. Z. Human gut microbiota/microbiome in health and diseases: a review. Antonie Van Leeuwenhoek 113, 2019–2040 (2020).
Goodrich, J. K. et al. Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe 19, 731–743 (2016).
Goodrich, J. K. et al. Human genetics shape the gut microbiome. Cell 159, 789–799 (2014).
Ruhlemann, M. C. et al. Genome-wide association study in 8,956 German individuals identifies influence of ABO histo-blood groups on gut microbiome. Nat. Genet. 53, 147–155 (2021).
Bonder, M. J. et al. The effect of host genetics on the gut microbiome. Nat. Genet. 48, 1407–1412 (2016).
Wang, J. et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 48, 1396–1406 (2016).
Turpin, W. et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat. Genet. 48, 1413–1417 (2016).
Vujkovic-Cvijin, I. et al. Host variables confound gut microbiota studies of human disease. Nature 587, 448–454 (2020).
Grahnemo, L. et al. Cross-sectional associations between the gut microbe Ruminococcus gnavus and features of the metabolic syndrome. Lancet Diabetes Endocrinol. 10, 481–483 (2022).
Ochoa, D. et al. The next-generation Open Targets Platform: reimagined, redesigned, rebuilt. Nucleic Acids Res. 51, D1353–D1359 (2023).
Noguchi, S. et al. FANTOM5 CAGE profiles of human and mouse samples. Sci. Data 4, 170112 (2017).
Kashyap, P. C. et al. Genetically dictated change in host mucus carbohydrate landscape exerts a diet-dependent effect on the gut microbiota. Proc. Natl Acad. Sci. USA 110, 17059–17064 (2013).
Pita, S. et al. CHAMP delivers accurate taxonomic profiles of the prokaryotes, eukaryotes, and bacteriophages in the human microbiome. Front. Microbiol. 15, 1425489 (2024).
An, H. et al. Integrated transcriptomic and proteomic analysis of the bile stress response in a centenarian-originated probiotic Bifidobacterium longum BBMN68. Mol. Cell. Proteomics 13, 2558–2572 (2014).
Matera, M. & Guandalini, S. How the microbiota may affect celiac disease and what we can do. Nutrients 16, 1882 (2024).
Caio, G. et al. Celiac disease: a comprehensive current review. BMC Med. 17, 142 (2019).
Palumbo, V. D. et al. Altered gut microbic flora and haemorrhoids: could they have a possible relationship? J. Clin. Med. 12, 2198 (2023).
Yang, F., Lan, Z., Chen, H. & He, R. Causal associations between human gut microbiota and hemorrhoidal disease: A two-sample Mendelian randomization study. Medicine (Baltimore) 103, e37599 (2024).
Pelaseyed, T. et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 260, 8–20 (2014).
Layunta, E., Javerfelt, S., Dolan, B., Arike, L. & Pelaseyed, T. IL-22 promotes the formation of a MUC17 glycocalyx barrier in the postnatal small intestine during weaning. Cell Rep. 34, 108757 (2021).
Layunta, E. et al. MUC17 is an essential small intestinal glycocalyx component that is disrupted in Crohn’s disease. JCI Insight 10, e181481 (2024).
Ng, P. Y. et al. Sugar transporter Slc37a2 regulates bone metabolism in mice via a tubular lysosomal network in osteoclasts. Nat. Commun. 14, 906 (2023).
Louca, P. et al. The secondary bile acid isoursodeoxycholate correlates with post-prandial lipemia, inflammation, and appetite and changes post-bariatric surgery. Cell. Rep. Med. 4, 100993 (2023).
Wang, Z. et al. Gut microbiota and blood metabolites related to fiber intake and type 2 diabetes. Circ. Res. 134, 842–854 (2024).
Schaus, S. R. et al. Ruminococcus torques is a keystone degrader of intestinal mucin glycoprotein, releasing oligosaccharides used by Bacteroides thetaiotaomicron. mBio 15, e0003924 (2024).
Sayols-Baixeras, S. et al. Streptococcus species abundance in the gut is linked to subclinical coronary atherosclerosis in 8973 participants from the SCAPIS cohort. Circulation 148, 459–472 (2023).
Jonsson, A. L. & Backhed, F. Role of gut microbiota in atherosclerosis. Nat. Rev. Cardiol. 14, 79–87 (2017).
Voyich, J. M. et al. Engagement of the pathogen survival response used by group A Streptococcus to avert destruction by innate host defense. J. Immunol. 173, 1194–1201 (2004).
Voyich, J. M., Musser, J. M. & DeLeo, F. R. Streptococcus pyogenes and human neutrophils: a paradigm for evasion of innate host defense by bacterial pathogens. Microbes Infect. 6, 1117–1123 (2004).
Krokstad, S. et al. Cohort profile: the HUNT study, Norway. Int. J. Epidemiol. 42, 968–977 (2013).
Asvold, B. O. et al. Cohort profile update: the HUNT study, Norway. Int. J. Epidemiol. 52, e80–e91 (2023).
Naess, M. et al. Data resource profile: the HUNT Biobank. Int. J. Epidemiol. 53, dyae073 (2024).
Bergstrom, G. et al. The Swedish CArdioPulmonary BioImage Study: objectives and design. J. Intern. Med. 278, 645–659 (2015).
Warensjo Lemming, E. et al. Dietary fatty acids and incident hip fractures in cohorts of women and men. A relative validation and follow-up study. J. Nutr. Health Aging. 28, 100247 (2024).
Brunkwall, L. et al. The Malmo Offspring Study (MOS): design, methods and first results. Eur. J. Epidemiol. 36, 103–116 (2021).
Almeida, A. et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 39, 105–114 (2021).
Zeng, S. et al. A compendium of 32,277 metagenome-assembled genomes and over 80 million genes from the early-life human gut microbiome. Nat. Commun. 13, 5139 (2022).
Nash, A. K. et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 5, 153 (2017).
Blanco-Miguez, A. et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 41, 1633–1644 (2023).
Li, D., Liu, C. M., Luo, R., Sadakane, K. & Lam, T. W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Nissen, J. N. et al. Improved metagenome binning and assembly using deep variational autoencoders. Nat. Biotechnol. 39, 555–560 (2021).
Orakov, A. et al. GUNC: detection of chimerism and contamination in prokaryotic genomes. Genome Biol. 22, 178 (2021).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 38, 5315–5316 (2022).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785–D794 (2022).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Jain, C., Rodriguez, R. L., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90 K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114 (2018).
Steinegger, M. & Soding, J. Clustering huge protein sequence sets in linear time. Nat. Commun. 9, 2542 (2018).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Schubert, M., Lindgreen, S. & Orlando, L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9, 88 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Cantalapiedra, C. P., Hernandez-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Aramaki, T. et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252 (2020).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Brumpton, B. M. et al. The HUNT study: a population-based cohort for genetic research. Cell Genom. 2, 100193 (2022).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 53, 1097–1103 (2021).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Lukina, P. et al. Coeliac disease in the Trondelag Health Study (HUNT), Norway, a population-based cohort of coeliac disease patients. BMJ Open 14, e077131 (2024).
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569 (2010).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Trynka, G. et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet. 43, 1193–1201 (2011).
Zheng, T. et al. Genome-wide analysis of 944 133 individuals provides insights into the etiology of haemorrhoidal disease. Gut 70, 1538–1549 (2021).
Donertas, H. M., Fabian, D. K., Valenzuela, M. F., Partridge, L. & Thornton, J. M. Common genetic associations between age-related diseases. Nat. Aging 1, 400–412 (2021).
Yengo, L. et al. Meta-analysis of genome-wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Hum. Mol. Genet. 27, 3641–3649 (2018).
Machiela, M. J. & Chanock, S. J. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525 (2015).
Yavorska, O. O. & Burgess, S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int. J. Epidemiol. 46, 1734–1739 (2017).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Dekkers, K. F. et al. An online atlas of human plasma metabolite signatures of gut microbiome composition. Nat. Commun. 13, 5370 (2022).
Acknowledgements
The Trøndelag Health Study (HUNT) is a collaboration between HUNT Research Centre (Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology NTNU), Trøndelag County Council, Central Norway Regional Health Authority and the Norwegian Institute of Public Health. The genotyping in HUNT was financed by the National Institutes of Health (NIH) (grant number NIH R35 HL135824-03); Stiftelsen Kristian Gerhardt Jebsen (grant number SKGJ-MED-015); University of Michigan; the Research Council of Norway; the Liaison Committee for Education, Research and Innovation in Central Norway; and the Joint Research Committee between St Olav’s Hospital and the Faculty of Medicine and Health Sciences, NTNU. The genotyping and imputation efforts in HUNT were a collaboration between researchers from the Department of Public Health and Nursing (ISM) (MH, NTNU), and the University of Michigan Medical School and the University of Michigan School of Public Health. The genotyping was performed at the Genomics Core Facility (GCF) (MH, NTNU). The celiac disease study in HUNT was funded by the Research Council of Norway (grant number 288308); the Liaison Committee for Education, Research and Innovation in Central Norway, Samarbeidsorganet (grant numbers 17/38297 and 18/42795); and the Norwegian Coeliac Society (grant date 20180702). Data from the Norwegian Patient Registry has been used in this publication. The interpretation and reporting of these data are the sole responsibility of the authors, and no endorsement by the Norwegian Patient Registry is intended nor should be inferred. C.O. was supported by funding from the Swedish Research Council (2020-01392, 2024-02412); the Swedish state under the agreement between the Swedish government and the county councils, the ALF-agreement (ALFGBG-965235 and ALFGBG-1005227); the Lundberg Foundation (LU2021-0096, LU2024-0110); the Novo Nordisk Foundation (NNF 190C0055250 and 22OC0078421); the Knut and Alice Wallenberg Foundation (KAW 2015.0317; KAW 2020.0230) and the European Union (ERC Advanced Grant-2022, HeMaFA, Project 101096347). Views and opinions expressed are however those of the authors only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them. The funders did not influence the study design, data analysis or manuscript writing. T.P. was supported by the Swedish Society for Medical Research (Svenska Sällskapet för Medicinsk Forskning, S17-0005). Swedish cohorts. Financial support was obtained in the form of grants from the European Research Council (ERC-STG-2018-801965 (T.F.); ERC-CoG-2014-649021 (M.O.-M.)), the Swedish Heart-Lung Foundation (Hjärt-Lungfonden, 2023-0687 (T.F.); 2018-0343 (J.Ä.); 2020-0711 (M.O.-M.); 20200173 (G.E.)), the Swedish Research Council (VR, 2019-01471 (T.F.), 2018-02784 (M.O.-M.), 2018-02837 (M.O.-M.), 2019-01015 (J.Ä.), 2020-00243 (J.Ä.) and EXODIAB 2009-1039 (M.O.-M.); 2019-01236 (G.E.)), the A.L.F. governmental grant 2018-0148 (MO-M), The Novo Nordisk Foundation NNF20OC0063886 (M.O.-M.) and The Swedish Diabetes Foundation DIA 2018-375 (M.O.-M.). The Malmö Offspring Study (MOS) was funded by the Swedish Research Council (VR, 521-2013-2756 (P.M.N.)), the Swedish Heart and Lung Foundation (Hjärt-Lungfonden 20150427 (P.M.N.)) and by A.L.F. from the local Region Skåne County Council (P.M.N.). We acknowledge the Swedish Heart-Lung Foundation, the main funding body of SCAPIS. Funding for the SCAPIS study was also provided by the Knut and Alice Wallenberg Foundation, the Swedish Research Council and VINNOVA (Sweden’s Innovation agency), the University of Gothenburg and Sahlgrenska University Hospital, Karolinska Institutet and Stockholm County council, Linköping University and University Hospital, Lund University and Skåne University Hospital, Umeå University and University Hospital, Uppsala University and University Hospital. We acknowledge the national research infrastructure SIMPLER for generating and making data, computational facilities, and resources available. SIMPLER receives funding through the Swedish Research Council under grants 2017-00644, 2017-06100 and 2021-00160 (to Uppsala University and K. Michaëlsson). The computations and data handling in the Swedish cohorts were enabled by resources in project sens2019512 and simp2023007 provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) and the Swedish National Infrastructure for Computing (SNIC) at Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX), funded by the Swedish Research Council through grant agreement 2022-06725. The T1DGC HLA Reference Panel for Imputation (T1DGC-Special) was conducted by the T1DGC-Special Investigators and supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The data from the T1DGC-Special reported here were supplied by NIDDK Central Repository (NIDDK-CR) and are available for request athttps://repository.niddk.nih.gov. This article was not prepared under the auspices of the T1DGC-Special study and does not necessarily reflect the opinions or views of the T1DGC-Special study, NIDDK-CR or NIDDK.
Funding
Open access funding provided by University of Gothenburg.
Author information
Authors and Affiliations
Contributions
The design of the study was performed in collaboration among M.R.M., E.C., K.H. and C.O. Metagenome sequencing and GWAS in the HUNT discovery cohort were performed by M.R.M., E.C., M.N., L.G., A.E.T., R.M., J.M.M., A.C.E., H.B.N., J.S., A.L., B.B., G.F.G., K.H. and C.O. Mechanistic studies were done by L.L., T.P., R.H. and E.N.-J. Replication in the Swedish cohorts was done by K.D., P.L., K.P., G.B., K.M., M.O.-M., T.F. and G.E. M.R.M., A.E.T. and C.O. wrote the first draft of the manuscript. All authors contributed to subsequent drafts of the manuscript and made the decision to submit the manuscript for publication.
Corresponding authors
Ethics declarations
Competing interests
A.C.E., J.M.M. and H.B.N. are employees of Clinical Microbiomics. C.O. is an applicant on filed patent applications on the effect of probiotics on bone metabolism. The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Andre Franke and Alexander Kurilshikov for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Note and Supplementary Figs. 1–8.
Supplementary Table 1–35 (download XLSX )
Supplementary Tables 1–35.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moksnes, M.R., Coward, E., Nethander, M. et al. The HUNT study identifies host genetic factors reproducibly associated with human gut microbiota composition. Nat Genet 58, 530–539 (2026). https://doi.org/10.1038/s41588-026-02502-4
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41588-026-02502-4
