
Abstract
Most RNA-binding small molecules have limited solubility, weak affinity and/or lack of specificity, restricting the medicinal chemistry often required for lead compound discovery. We reasoned that conjugation of these unfavorable ligands to a suitable ‘host’ molecule can solubilize the ‘guest’ and deliver it site-specifically to an RNA of interest to resolve these issues. Using this framework, we designed a small-molecule library that was hosted by cobalamin (Cbl) to interact with the Cbl riboswitch through a common base displacement mechanism. Combining in vitro binding, cell-based assays, chemoinformatic modeling and structure-based design, we unmasked a cryptic binding site within the riboswitch that was exploited to discover compounds that have affinity exceeding the native ligand, antagonize riboswitch function or bear no resemblance to Cbl. These data demonstrate how a privileged biphenyl-like scaffold effectively targets RNA by optimizing π-stacking interactions within the binding pocket.

Similar content being viewed by others

Main
The design and discovery of small molecules that selectively target RNA is a longstanding problem in chemical biology. This emerging field has the potential to develop chemical probes of RNA function and therapeutics to treat RNA-mediated disease1,2,3. Despite ongoing efforts, there is only one small molecule approved by the US Food and Drug Administration that targets RNA outside the ribosome, risdiplam4. This reinforces the notion that targeting RNA is difficult and often limited by the lack of druggable RNA targets and/or quantitative structure–activity relationship (QSAR) studies1,2,3. Compared with proteins, which are routinely targeted with small-molecule therapeutics5, much less is known about the principles governing RNA–ligand interactions to guide discovery efforts.
One approach to address this knowledge gap is the application of natural RNA–small molecule interactions such as those observed in riboswitches. These bacterial mRNA elements have evolved tertiary structures to selectively bind metabolites and control gene expression6,7,8, making them ideal models to explore RNA–ligand interactions. For example, BioRelix9,10 and Merck11,12 have reported promising lead compounds against the FMN riboswitch that demonstrate efficacy in animal models. Another system of therapeutic interest13 is the cobalamin (Cbl) riboswitch14, which is broadly distributed across bacteria15. Different classes of Cbl riboswitches exhibit distinct binding preferences to forms of Cbl that differ only at their β-axial position (Fig. 1a)16. The biological forms of Cbl are 5′-deoxyadenosylCbl (AdoCbl) and methylCbl (MeCbl, 1)13 but standard forms include the photolysis17 product hydroxoCbl (OHCbl, 2) and the photostable18 cyanoCbl (CNCbl, 3) (Fig. 1a).

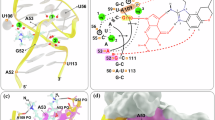
a, Chemical structure of standard Cbls with their corresponding β-axial groups shown. b, Cocrystal structure of the env8–OHCbl complex (PDB 4FRG)19. Left: global RNA architecture displayed as a cartoon, with nucleotides critical for recognition of the variable β-axial group shown in cyan (A68), green (A20) and yellow (G19); OHCbl is represented by magenta van der Waals spheres. Right: the ligand-binding pocket consists of a π-stacking network involving nucleotides G19–A20 and A20–A68 that is unimpeded by the small (–OH) β-axial group. c, Simplified chemical structures of previously characterized20 β-axial-modified Cbls 4–7 tested against env8. d, Reduction-free synthetic route25,26 to make Cbl derivatives from CNCbl using a variable alkyne. e, Overview of our expanded β-axial-modified Cbl library.
We recently identified several β-axial derivatives 4–7 that bind the env8 MeCbl-selective riboswitch19 and regulate in-cell function (Fig. 1b,c)20. This was a surprising result given that these derivatives host bulky β-axial moieties (Fig. 1c) that present a substantial steric problem within the binding pocket (Supplementary Fig. 1a). Chemical probing suggests that recognition of the higher affinity 4 and 7 involves the displacement of an adenosine (A20) from the RNA core (Supplementary Fig. 1b), which is likely replaced by the β-axial group20. Displacement of A20 would yield a cryptic binding site distinct from the displacement of an adjacent adenosine by the 5′-deoxyadenosyl moiety observed in AdoCbl-selective riboswitches (Supplementary Fig. 1c)19. Our preliminary data suggest that the cryptic binding of 4 and 7 may explain their increased affinity and that chemical modifications to the β-axial group have robust effects on RNA binding20.
In this work, we synthesized an expanded library of Cbl derivatives that host systematically varied β-axial moieties (Fig. 1d,e) and used a set of biochemical and cell-based assays to quantify their RNA-binding affinities and regulatory activities. These data were used in a predictive modeling platform to determine the ligand properties associated with tight affinity and strong regulatory activity. As a complementary approach, we used structure-based design to unmask the cryptic β-axial binding pocket and discover lead compounds with affinity exceeding the native ligand. This structure-informed approach also enabled the identification of novel cryptic binders that are chemically distinct from Cbl. Collectively, our work outlines the molecular determinants of specific and high-affinity base-displacement RNA-binding modes, which can guide future efforts to target these common RNA interactions21,22,23,24.
Results
Small-molecule library design and synthesis
Our Cbl derivative library was synthesized using a previously established reduction-free method25,26 where the cyano group at the β-axial position of CNCbl is replaced by the R-group of a variable alkyne (Fig. 1d). While this approach enabled the synthesis of a diverse assortment of photostable β-axial-modified Cbls 8–44 (Fig. 1e), reactions with alkynes containing amines or protonated nitrogen atoms were unsuccessful (Supplementary Fig. 2). The logic of our library construction was to diversify around 4 and 7 to address specific medicinal chemistry questions with QSAR studies. Within this framework, we can determine which chemical features modulate RNA binding and function.
Given that the majority of RNA-binding ligands are aromatic systems capable of π-stacking27,28, many RNA-targeting lead compounds suffer from limited solubility, low affinity and/or lack of specificity, limiting the scope and utility of the downstream medicinal chemistry that is required for successful hit-to-lead discovery campaigns1,2,3. In our approach (Supplementary Fig. 3a), Cbl functions as a soluble ‘host’ of a variable chemical ‘guest’ that interacts with the env8 RNA target in the minor groove. This target–guest interaction is anchored by contacts from the host corrin ring and α-axial group (Fig. 1a) to project the variable β-axial moiety to the same spatial location within the cryptic site. Thus, our model system enables a detailed and systematic approach to determine which chemical features confer high-affinity base-displacement RNA-binding modes, which is common throughout RNA biology21,22,23,24 (Supplementary Fig. 3b).
Cbl derivatives productively bind env8
To quantify the binding of our library to env8, we used a fluorophore-conjugated CNCbl probe (CNCbl–5×PEG-ATTO590)29 that undergoes fluorescence induction upon env8 binding and competed it off with ligands in our derivative library20. Titration of env8 into the probe gave a KD of 3.2 ± 0.1 nM (Fig. 2a) and competitive titrations of MeCbl and Cbl 4 into the env8–CNCbl–5×PEG-ATTO590 complex yielded KD values of 1.4 ± 0.8 nM and 10 ± 5 nM, respectively (Fig. 2b). Using this fluorescence displacement assay, we measured the KD values for Cbls 1–44. All ligands bound env8 with submicromolar affinity (Fig. 2c). The tightest-binding derivative was 29 (7 ± 7 nM) while 12 (800 ± 700 nM) was the weakest. Importantly, 29 functions as a binding lead with an affinity that exceeds 4 and 7 and is comparable (P = 0.19) to the native MeCbl.

a, Binding curve of the fluorescence induction of CNCbl–5×PEG-ATTO590 binding to env8. b, Representative displacement binding curve for MeCbl and Cbl 4. Binding curves are shown as the mean and s.e.m. from independent experiments (n = 3). KD values are reported as the mean and s.d. from independent experiments (n = 3). c, The log-transformed KD values for 1–44 shown as the mean and s.d. from independent experiments (n = 4, except for data from 1, 2, 4–7, 10, 14, 19, 41 and 42, where n = 3, and data from 21, 36 and 37, where n = 5). d, Locations of the training and test sets from the Q2-focused modeling in two-dimensional chemical space constructed from PC1 and PC2 of the whole dataset. e, Measured ln (KD) values plotted with the value predicted by the Q2-focused model. f, Overview of our lead compound screen from a 513-compound alkyne library using our binding-based models. g, Simplified chemical structures of representative derivatives that our binding-based models predict to be tight (45–48), moderate (49 and 50) and weak (51–53) binders. h, Measured ln(KD) values plotted with the value predicted by our binding-based models. The experimental data are presented as the mean and s.d. from independent experiments (n = 3, except for data from 45, 48 and 53, where n = 4) and the predicted data are shown as the mean and s.d. from independent predictions (n = 3) from our three models (baseline, R2-focused and Q2-focused).
QSAR analysis of RNA-binding data
To reveal chemical trends within our binding data, we used a QSAR analysis. We quantified the ‘distance from the alkyne’ of our ortho-substituted, meta-substituted and para-substituted phenyl-F (9–11) and phenyl–CH3 (12–14) derivatives and compared this parameter against their KD values. We observed a strong relationship (R2 = 0.97) for the smaller substituents and an even stronger relationship (R2 = 0.99) for the larger groups (Extended Data Fig. 1a,b), suggesting steric limitations within the cryptic site. We also quantified the electron-donating and electron-withdrawing strength or van der Waals volume of the chemical groups in the para-substituted phenyls (4, 5, 8, 11, 14 and 17–20) and compared these parameters against their KD values. This analysis revealed that phenyl ring electronics (R2 = 0.10) and para-position sterics (R2 = 0.02) both fail to explain env8 binding (Extended Data Fig. 1c–e).
Additional QSAR trends emerge from our heterocyclic Cbls (21–26) (Extended Data Fig. 1f). Comparing the affinities of phenyl 8 (70 ± 20 nM) and pyrimidine 24 (70 ± 20 nM) suggest that heterocycles have no advantage over their hydrocarbon counterpart. Data from thiophene 21 (40 ± 30 nM), thiazole 22 (50 ± 40 nM) and furan 23 (50 ± 10 nM) demonstrate that five-membered heterocycles bind just as well as six-membered ones. Lastly, the increased affinity for our two-ring heterocycles 25 (16 ± 8 nM) and 26 (12 ± 5 nM) suggests that two rings are preferred, which is consistent with our hypothesis that the β-axial group π-stacks within the cryptic site. This notion is supported by the binding data of our two-ring-containing 27–29, which all have affinities under 20 nM (Extended Data Fig. 1g). Even the three-ring-containing (31, 30 ± 20 nM) and four-ring-containing (32, 30 ± 10 nM) derivatives bind with high affinity (Extended Data Fig. 1g).
To analyze these chemical trends in greater detail, we used a multivariate analysis. Binding data were clustered into tight, moderate and weak binders (Extended Data Fig. 2a) and 20 standard chemoinformatic parameters30,31 (Supplementary Tables 1 and 2) were calculated for the β-axial groups of Cbls 4, 5 and 7–44. These descriptors were used in a linear discriminant analysis (LDA)31 with our binding data. When viewed along the first two principal components (PC1 and PC2), each binding cluster occupies unique regions in chemical space (Extended Data Fig. 2b). The LDA loading plot provides qualitative trends for the molecular determinants of each cluster. For example, tight binders have more aromatic rings, moderate binders have more hydrogen-bond donors and weak binders have more accessible surface area (Extended Data Fig. 2c).
QSAR model-based screening of potential binding leads
Motivated by the LDA-identified trends, we sought to determine whether the chemical identity of the β-axial group could be used to predict env8-binding affinity. An expanded set of 347 physiochemical descriptors were calculated for the β-axial group of Cbls 4, 5 and 7–44 and modeled against their natural-log-transformed KD data using a least absolute shrinkage and selection operator (lasso) multiple linear regression (MLR) strategy32. When using all data in training, we obtained a baseline model that predicted our binding data with good accuracy (root-mean-square error (r.m.s.e.) = 0.53) using six descriptors (Extended Data Fig. 3a,b and Supplementary Table 3). When using a stratified data split (Fig. 2d and Extended Data Fig. 3c) and implementing our lasso-MLR strategy, we constructed new models that prioritized either the predictive ability on the test set (Q2-focused) or the overall fit of the training set (R2-focused). The Q2-focused (r.m.s.e.Test = 0.35) and R2-focused (r.m.s.e.Test = 0.65) models showed a similar level of performance (Fig. 2e and Extended Data Fig. 3d).
We then used our binding-based models to screen for high-affinity β-axial groups among a 513-compound alkyne library, which included modified phenyl (phenyl-X) and aliphatic (amide-X) alkynes (Fig. 2f) compatible with our Cbl synthesis (Fig. 1d) and representative of our initial library (Fig. 1e). Among these compounds, 184 were predicted to have tighter affinities than binding lead 29 (Extended Data Fig. 3e). However, the majority of these small molecules were synthetically inaccessible using the nonreductive method25,26 (Extended Data Fig. 3f). These considerations left us with four remaining leads, which were synthesized into the corresponding Cbls 45–48 along with compounds that were predicted to be moderate (49 and 50) and weak (51–53) binders (Fig. 2g). We then used our fluorescence displacement assay to quantify the binding of these derivatives to env8. The experimentally derived ln(KD) data agreed well with our model-based predictions for the moderate and weak binders but deviated greatly for the predicted leads 45 and 46 (Fig. 2h). One explanation for this observation is the sparseness of tight binders in our dataset (8 of 44, 18%). Synthetic routes that access chemical groups facilitating high-affinity RNA-binding interactions could address this issue.
Cbl derivatives promote RNA-regulatory activity
Given that all the derivatives in our library bind env8, we wanted to determine whether they drive RNA-regulatory activity in a cellular environment. We used a previously established33 cell-based assay in which env8 was placed upstream of GFPuv, whose expression is repressed by Cbl-dependent occlusion of the ribosome-binding site. In the absence of Cbl, we detected high fluorescence, indicative of GFPuv expression (Fig. 3a). Conversely, in the presence of 10 nM of MeCbl or Cbl 4, we observed attenuated fluorescence, demonstrating that these ligands promote env8 repression of GFPuv expression in Escherichia coli (Fig. 3a).

a, Box-and-whisker plot of the OD600-normalized relative fluorescence units (RFU) of E. coli cells with and without reporter plasmid (env8–GFPuv) and ligand (MeCbl and Cbl 4) added. Data are shown from independent experiments (n = 4). b, Fold repression (defined as the ratio of (−)-Cbl RFU and (+)-Cbl RFU values)33 for our env8 reporter system in the presence of Cbls 1–44 shown as the mean and s.d. from biological replicates (n = 4 except for data from 6, where n = 3). c, Plot comparing the log-transformed fold repression and KD data. d, Locations of the training and test set from the Q2-focused modeling in two-dimensional chemical space constructed from PC1 and PC2 of the whole dataset. e, Measured fold repression values plotted with the value predicted by the Q2-focused model. f, Overview of our lead compound screen from a 513-compound alkyne library using our function-based models. g, Simplified chemical structures of representative derivatives that our function-based models predict to be strong (54–57), moderate (58 and 59) and weak (60–62) repressors. h, Measured fold repression values plotted with the value predicted by our function-based models. The experimental data are presented as the mean and s.d. from biological replicates (n = 4, except for data from 58, 59, 61 and 62, where n = 3) and the predicted data are shown as the mean and s.d. from independent predictions (n = 3) from our three models (that is, baseline, R2-focused and Q2-focused).
To quantify the extent to which each derivative regulates env8 function, we calculated the fold repression33 for Cbls 1–44. While all ligands were able to regulate env8 function to some extent, the repression was highly variable. The strongest repressors were 36 (8 ± 1) and 41 (7 ± 2) while 43 (1.4 ± 0.1) and 44 (2.0 ± 0.1) were the weakest (Fig. 3b). Importantly, 36 and 41 are new functional leads with fold repression values that exceed 4 and 7 and are comparable (36, P = 0.31; 41, P = 0.19) to the native MeCbl (9.0 ± 0.2).
These data enable the exploration of the relationship between derivative binding and regulatory activity. Derivatives 29 and 36 are among the tightest binders and strongest regulators, making them attractive leads. However, in general, binding affinity and regulatory activity were weakly correlated (R2 = 0.34) (Fig. 3c). This fact reflects the complex nature of promoting a ligand-induced regulatory response, which involves Cbl cellular import, β-axial repair by endogenous enzymes and env8–Cbl binding within the appropriate timescale of a cotranscriptional process33. To investigate this further, we carried out growth experiments in ΔMetE cells, which lack the Cbl-independent methionine synthase MetE34. In the absence of methionine, these cells must use Cbl for one carbon metabolism, making MeCbl essential for growth34. In the presence of 10 nM derivative, both weak (44) and strong (36) repressors support ΔMetE growth with the same doubling time as CNCbl, suggesting that these ligands are imported and repaired to some extent (Extended Data Fig. 4). However, the fact that repression is variable among our derivatives in wild-type E. coli cells suggests that these compounds exist predominantly in their unrepaired, β-axial derivatized state. A confounding factor in comparing the binding and functional data is potentially variable levels of derivative metabolism within the cell.
QSAR model-based screening of potential functional leads
While individual QSAR trends were less pronounced within our functional data (Supplementary Fig. 4), an LDA using the same set of chemoinformatic parameters30,31 (Supplementary Tables 1 and 4) demonstrated that functional clusters (Extended Data Fig. 5a) occupy unique regions in chemical space (Extended Data Fig. 5b) and that strong repressors have more hydrogen-bond donors, moderate repressors have more aromatic rings and weak repressors have more sp3 centers (Extended Data Fig. 5c). These trends motivated us to build QSAR models using the same lasso-MLR workflow32 described above, except using our repression data as the dependent variable. We obtained baseline, Q2-focused and R2-focused models that predict the functional data reasonably well with four to six descriptors (Fig. 3d,e, Extended Data Fig. 6a–d and Supplementary Table 5).
We then used our function-based models to screen for strong repressive β-axial groups among the same alkyne library (Fig. 3f). Although 151 small molecules were predicted to have stronger repression than functional lead 36 (Extended Data Fig. 6e), many of these compounds were unavailable for the same reasons described above (Extended Data Fig. 6f). We identified four available predicted leads, which were synthesized into the corresponding Cbls 54–57 along with compounds that were predicted to be moderate (58 and 59) and weak (60–62) repressors (Fig. 3g). The fold repression of 54–62 was then measured using our reporter assay. Much like our binding data, the experimentally derived repression values agreed well with our model-based predictions for the moderate and weak repressors but deviated greatly for the predicted functional leads 54–56 (Fig. 3h), which is likely explained by the confounding role of derivative metabolism on regulatory activity.
env8–Cbl cocrystal structures unmask cryptic binding site
As a complementary approach to QSAR modeling, we used structure-based design to investigate env8–ligand interactions. We carried out three independent 1-µs molecular dynamics (MD) simulations of env8–CNCbl and env8–Cbl 4 and measured the A20(C6)–G19(C6) and A20(C6)–A68(N7) distances to assess A20 π-stacking with neighboring nucleotides (Fig. 4a). Over the course of the MD trajectories, the A20(C6)–G19(C6) distances were identical across both complexes, whereas 4 induced slight increases in A20(C6)–A68(N7) distances (Fig. 4b). However, these changes were not a result of A20 displacement but were caused by the β-axial phenyl-pNO2 group intercalating between A20 and A68 (Supplementary Fig. 5), which is inconsistent with previous chemical probing data (Supplementary Fig. 1b)20.

a, Representative MD starting states of the env8–CNCbl and env8–Cbl 4 complexes from one (of three) independent 1-µs trajectory. The A20(C6)–G19(C6) and A20(C6)–A68(N7) distances are shown as double arrows. In all structural representations, the binding-pocket nucleotides G19 (yellow), A20 (green) and A68 (cyan) are numbered in reference to full-length env8 and colored, the β-axial group is shown in magenta and dashed lines represent proposed hydrogen bonds. b, The average A20(C6)–G19(C6) (left) and A20(C6)–A68(N7) (right) distances measured over the course of three independent MD trajectories for both env8–CNCbl and env8–Cbl 4 complexes. c, Cocrystal structure of env2–CNCbl showing only the binding-pocket nucleotides and the β-axial group. All mesh representations correspond to a simulated annealing 2Fo − Fc map where A20 and the ligand were omitted from the model and are shown at 1σ contour. d, Same as c but for env2–Cbl 4. e, Same as c but for env2–Cbl 32 (left), along with a van der Waals sphere representation of the binding pocket (right). f, Same as e but for env2–Cbl 29. g, Same as c but for env2–Cbl 42. h, Schematic representation of design hypotheses that emerge from our 11 RNA–ligand cocrystal structures.
The limitations of our MD analysis motivated us to use X-ray crystallography. We determined cocrystal structures of the Cbl riboswitch aptamer domain in complex with CNCbl and 4. However, we only obtained diffraction-quality crystals using the closely related env2 RNA, which shares 95% sequence identity with env8 (Extended Data Fig. 7a). Given that env2 shows near-identical ligand binding and regulatory activity to env8 (Extended Data Fig. 7b,c), all structural information can be translated to env8. The env2–CNCbl structure shows A20 π-stacked with G19 and A68 (Fig. 4c) with strong agreement to the env8–OHCbl structure19 (root-mean-square deviation (r.m.s.d.) = 0.45 Å) (Extended Data Fig. 7d). The env2–Cbl 4 structure, on the other hand, reveals that A20 undergoes base displacement from the RNA core toward the major groove and that the β-axial phenyl ring π-stacks with A68 (Fig. 4d), unmasking the cryptic binding pocket in agreement with our hypothesis20. We also determined the structure of env2 in the apo state, which shows a near-identical (r.m.s.d. = 0.39 Å) binding pocket to our env2–CNCbl structure (Extended Data Fig. 8a), demonstrating that CNCbl interacts with a preorganized binding pocket.
To gain further insight into cryptic β-axial pocket, we determined nine additional env2–ligand cocrystal structures (Extended Data Fig. 8b–j). The structure of env2–Cbl 32 with its bulky pyrene β-axial group showcases that base displacement provides a lot of room (>340 Å3) in the binding pocket (Fig. 4e). With its four phenyl rings, pyrene π-stacks well with A68 but is unable to optimize π-stacking with G19 (Fig. 4e). In contrast, the structure of env2–Cbl 29 demonstrates how the biphenyl β-axial group maximizes π-stacking with both G19 and A68 through rotation of the two rings relative to one another (Fig. 4f). Interestingly, our data support the fitting of two conformations of A20, with the more populated (55%) conformer partially engaged within the RNA core (Fig. 4f), providing a means to offset the energetic penalty of A20 displacement. It is important to note that π-stacking is not the only binding mode available within the cryptic site. The structure of env2–Cbl 42 suggests that two hydrogen bonds to A68 provide comparable affinity (50 ± 10 nM) with a π-stacking interaction (for example, 8, 70 ± 20 nM) (Fig. 4g). Together, our structural data suggest a set of design hypotheses to develop new lead compounds: (1) hydrogen bonding to the phosphate backbone; (2) cation–π stacking; and (3) engagement of A20 (Fig. 4h).
Structure-based design identifies Cbls with affinity exceeding the native ligand
To explore design hypotheses (1) and (2), we adopted new chemistry using a click reaction of Cbl 58 with a variable azide25 to form a biphenyl-like scaffold where the second ring is an R-group harboring triazole (Fig. 5a). We synthesized Cbls 63–65 to install amino-substituted, guanidinium-substituted and hydroxyl-substituted triazoles, respectively (Fig. 5b). We then used our fluorescence displacement assay to quantify the binding of these derivatives to env8 and discovered that 63 (1.0 ± 0.6 nM, P = 0.62) and 64 (1.3 ± 0.7 nM, P = 0.92) had comparable affinity to CNCbl (Fig. 5c). However, these interactions approach the accuracy limit of our binding assay. To confirm these results, we used isothermal titration calorimetry (ITC) with the env8 aptamer domain, which lacks an element of RNA structure that interacts with the Cbl α-axial face, thereby lowering the observed affinity. These data revealed significantly tighter affinity to 63 (32 ± 6 nM, P = 0.02) and 64 (40 ± 3 nM, P = 0.02) as compared with CNCbl (260 ± 60 nM) (Fig. 5d).

a, Modified click reaction25 to make Cbl derivatives from 58 using a variable azide. b, Simplified chemical structures of Cbls 63–65 that were made to systematically test our design hypotheses. c, Fluorescence displacement binding curve for CNCbl and 63–65 shown as the mean and s.e.m. from independent experiments (n = 4, except for data from CNCbl, where n = 3). KD values are reported as the mean and s.d. from independent experiments (n = 4, except for data from CNCbl, where n = 3). d, To verify the near-picomolar binding of 63 and 64, we used ITC with the env8 aptamer domain. Given that this RNA shows weaker ligand binding than the full-length env8 (ref. 19), it is in the perfect regime for ITC. Representative ITC binding isotherm for the env8 aptamer domain with CNCbl, 63 and 64. e, Cocrystal structure of env2–Cbl 63 showing only the binding-pocket nucleotides and the β-axial group. Proposed hydrogen bonds, which are weakly supported by the electron density, are shown as double arrows. All mesh representations correspond to a simulated annealing 2Fo − Fc map where A20 and the ligand were omitted from the model and are shown at 1σ contour. f, Same as e but for env2–Cbl 64. g, Fold repression for our reporter system in the presence of CNCbl and 63–65 and for cells with CNCbl and excess amounts of 63–65 added. The mean and s.d. from biological replicates (n = 4) are shown. For comparison, the mean fold repression of 10 nM CNCbl is shown as a dashed line and its s.d. is represented as a shaded box. h, Simplified chemical structure of the titratable pyridine Cbl 66. i, Representative ITC binding isotherms for env8 aptamer domain with CNCbl, 29 and 66 at pH 8 and pH 5. For all ITC data, KD values are reported as the mean and s.d. from independent experiments (n = 3).
To understand how these new lead compounds bind env8, we determined cocrystal structures of env2–Cbl 63 and env2–Cbl 64. While weakly supported by the electron density, both structures suggest that the amino (Fig. 5e) or guanidinium (Fig. 5f) groups likely electrostatically interact with backbone phosphate of G19 (env2–Cbl 64) or A68 (env2–Cbl 63). However, our env2–Cbl 63 structure unambiguously demonstrates that the terminal triazole ring twists to π-stack with A20 (Fig. 5e), further emphasizing that rotatable bonds are beneficial for optimizing the π-stacking network. This is the first evidence of strong engagement of A20 in any of our structures. Notably, when π-stacked with the ligand, A20 adopts a perpendicular orientation relative to G19, a highly unusual mode of base–base interaction in nucleic acids.
Despite their high affinity, when used in our reporter assay, 63–65 confer minimal repression (Fig. 5g). To investigate this further, we repeated our growth experiments in ΔMetE cells. In the presence of 10 nM derivative, 63–65 support ΔMetE growth with the same doubling time as CNCbl, suggesting that these ligands are imported and repaired to some extent (Extended Data Fig. 9a,b). However, unlike 36 and 44, elevated concentrations of 63–65 prevented the growth of ΔMetE cells (Extended Data Fig. 9c,d), suggesting that these derivatives antagonize some essential aspect of Cbl metabolism in E. coli35. To fully assess how 63–65 affect regulatory activity, we repeated our reporter assay in wild-type E. coli, which grows normally in the presence of high Cbl concentrations (Extended Data Fig. 9e). Adding excess amounts of 63–65 to cells containing 10 nM CNCbl resulted in a loss of CNCbl-induced repression, confirming that these derivatives are antagonists of env8 function (Fig. 5g).
To further explore design hypothesis (2), we synthesized the pyridine Cbl 66 to introduce a conditional positive charge (Fig. 5h). Under the conditions of our binding experiments (pH 8), 66 should behave exactly like its nonpyridine counterpart 29. However, at a pH below its predicted pKa (~5.25), 66 should be charged and we would expect an increase in affinity that is absent in 29. To test this hypothesis, we carried out ITC measurements at pH 8 and 5 with the env8 aptamer domain and CNCbl, 29 and 66. The nontitratable CNCbl and 29 showed a ~2-fold reduction in binding affinity at reduced pH, whereas the affinity of 66 increased 1.7-fold (Fig. 5i), in agreement with our hypothesis. These data suggest that installing pyridines, whose pKa can be lowered by interaction with RNA, can leverage the increased affinity of cation–π interactions without violating Lipinski’s rules36,37.
The discovery of 63–66 motivated us to refine our QSAR models to explore whether additional high-affinity training data improve model performance. When including the data of 49–66, our binding-based models predicted the affinity of the unseen leads 45–48 with improved accuracy (r.m.s.e.original = 1.42, r.m.s.e.new = 0.83) (Extended Data Fig. 10a–c and Supplementary Table 6). Inclusion of the data of 45–48 for training further improved these predictions (r.m.s.e.new,2 = 0.23) (Extended Data Fig. 10d–f). We observed a similar improvement in performance when refining our function-based models (Extended Data Fig. 10g–l and Supplementary Table 7).
Structure-informed screen identifies cryptic binders of env8 divorced from Cbl
Our structural biology efforts unmasked the cryptic β-axial binding pocket within env8, which allows us to explore whether small molecules divorced from a Cbl host can also target this site. To address this issue, we carried out a high-throughput computational screen using a 28,000-compound RNA-focused library. These compounds were docked against env2 in two conformations: one in which A20 was displaced toward the major groove (A20-out, for example, env2–Cbl 29) and one where A20 was engaged in the RNA core (A20-in, for example, env2–Cbl 63) (Supplementary Fig. 6). To leverage our structural data, we specified that docking hits contain the biphenyl-like scaffold and identified eight potential lead compounds (Fig. 6a).

a, Chemical structure of the eight hits that emerged from our structure-informed docking, with their biphenyl-like scaffold highlighted in gray. b, TO-based displacement assay to identify env8-binding compounds that induce fluorescence attenuation. This was used as an orthogonal binding experiment because we observed ligand-induced fluorescence induction of CNCbl–5×PEG-ATTO590. c, MST trace of fluorescently labeled env8 alone and in the presence of CNCbl, 68 and 71. All ligands induced an observable change to the MST trace of env8, suggestive of binding. d, TO displacement binding curve for CNCbl, 68 and 71 shown as the mean and s.d. from independent experiments (n = 3). KD values are reported as the mean and s.d. from independent experiments (n = 3). e, Native polyacrylamide gel showing a gradual shift from a slower-migrating, extended conformation to a faster-migrating, kissing-loop conformation of env8 upon titration of CNCbl (0.01–10 µM). Data are shown from an individual experiment. f, Same as e but after addition of CNCbl, 68 and/or 71 to demonstrate competitive binding of 68 and 71. Similar results were obtained from repeated experiments (n = 8 for CNCbl, n = 3 for 68 and n = 2 for 71). g, MST trace from a competition experiment where env8–CNCbl was monitored with and without the addition of excess amounts of 68 or 71. Both ligands induced an observable change to the MST trace of env8–CNCbl, suggestive of competitive binding. All MST data are shown as representative traces from multiple independent experiments (n = 4). h, Top-ranked docking poses of 68 and 71 to env2 in the A20-out and A20-in states.
To survey the binding of these ligands to env8, we adopted a thiazole orange (TO) displacement assay (Supplementary Fig. 7a)38. Upon the addition of 67–74, 68 and 71 led to sufficient (>25%) TO displacement indicative of binding (Fig. 6b). This conclusion was confirmed with microscale thermophoresis (MST) (Fig. 6c). Competitive titrations of CNCbl, 68 and 71 into the env8–TO complex yielded KD values of 38 ± 5 nM, 34 ± 9 µM and 50 ± 10 µM, respectively (Fig. 6d). The affinity of CNCbl agrees well with data obtained from ITC (38 ± 8 nM) (Supplementary Fig. 7b,c), supporting the use of this method to quantify ligand binding to env8.
To verify that 68 and 71 target the Cbl-binding pocket, we used an electrophoresis mobility shift assay (EMSA) that monitors the ability of env8 to adopt a kissing-loop conformation in the presence of Cbl33, which migrates faster than the unbound RNA (Fig. 6e). Given that the kissing loop is facilitated by interactions with the Cbl α-axial face33, 68 and 71 are not expected to induce this conformational change. Thus, if these ligands competitively bind the same site within env8, then adding 68 or 71 to env8 in the presence of CNCbl should revert the RNA to the slower-migrating species. When excess amounts of 68 or 71 were added to an env8–CNCbl complex, both ligands prevented kissing-loop formation, suggestive of competitive binding (Fig. 6f). This phenomenon was again confirmed with MST (Fig. 6g). Consistent with these observations, 68 and 71 were unable to facilitate env8-mediated repression (Supplementary Fig. 8), which requires kissing-loop formation19. These data support the top-ranked docking poses of 68 and 71, which show their biphenyl-like scaffolds maximizing π-stacking to G19 and A68 within the cryptic β-axial binding pocket and making potential hydrogen bonds to the phosphate backbone (Fig. 6h).
Discussion
Targeting RNA with small molecules is an emerging field hindered by an incomplete understanding of the basic principles governing RNA–ligand interactions1,2,3. To address this knowledge gap, we explored the chemical features promoting specific and high-affinity base-displacement RNA-binding interactions using the Cbl riboswitch. Using a wide range of experimental approaches, we successfully identified lead compounds with affinities exceeding the native ligand. All leads share a biphenyl-like structure, which was previously identified as a privileged RNA-binding scaffold27,28,39. Our data demonstrate that twisting around a rotatable bond confers the ability to optimize the π-stacking network within the binding pocket. Our results not only reinforce our understanding of RNA chemical space but yield new structural insights into how these scaffolds interact with RNA.
Across all newly reported RNA–ligand cocrystal structures, binding-pocket nucleotides G19 and A68 are in near-identical positions, indicating that structural plasticity is conferred solely by the displacement of A20, providing a large (>340 Å3) cryptic site that accommodates the β-axial group of all derivatives. One mechanism to explain this phenomenon is ligand docking into a rigid RNA to induce A20 displacement; however, large, rigid β-axial groups likely prevent the corrin ring from productively engaging the RNA. A more likely explanation is that A20 undergoes transient excursions from the RNA core to form a binding-competent state, a motion that is well documented in nucleic acids and consistent with the timescale of ligand binding40. Considerations of local binding-pocket dynamics are essential to unmask additional RNA cryptic sites41,42, which will facilitate the development of new chemical probes of RNA function and therapeutics to treat RNA-mediated disease. However, our work cautions against using a purely computational approach, as MD simulations could not account for the nuances of base displacement within env8. Advances in machine learning43 and artificial intelligence44 methods may address these limitations in the future.
Our work also showcases that bifunctional host–guest systems are a valuable approach to targeting RNA. Exploration of general host–small molecule conjugates is, therefore, of critical importance. Nucleic-acid-based hosts are attractive candidates because they can solubilize the attached ligand, deliver it to RNA in a sequence-specific manner and contribute binding energy that can turn modest and/or nonspecific binders into ligands with high affinity and specificity. For example, TO was conjugated to a peptide nucleic acid host to monitor double-stranded RNA formation45. Peptide nucleic acid hosts have also been shown to rescue the binding of small molecules that have no affinity for RNA by themselves46. This conceptual framework is similar to the widespread use of bifunctional and multivalent small molecules in protein targeting47, which has parallels in covalent RNA-targeting strategies39,48. We believe that a host–guest approach is generalizable and will enable detailed chemical and structural analysis of this underexplored chemical space to better understand RNA–small molecule interactions.
Methods
Synthesis of Cbl derivatives
Cbl derivatives 4–66 were synthesized using established methodology25,26 outlined in Supplementary Information.
RNA preparation
All RNAs (full sequences shown in Supplementary Table 8) were prepared using DNA templates amplified by PCR, transcribed with T7 RNA polymerase and purified using preparative denaturing PAGE49. Purified RNA was buffer-exchanged and concentrated into Milli-Q H2O using centrifugal concentrators (Millipore-Sigma). Final RNA concentrations were calculated using absorbance at 260 nm and extinction coefficients determined from the summation of the individual bases. Before all binding experiments, RNA was heated at 95 °C for 2 min, incubated on ice for 10 min and then allowed to equilibrate to room temperature.
Cbl fluorescence binding assays
For fluorescence induction titrations, 120-μl reactions containing CNCbl–5×PEG-ATTO590 (ref. 29) (1 nM for full-length env8 or 10 nM for env8 aptamer domain and env2 crystal construct), 1× RNA-binding buffer (50 mM HEPES pH 8.0, 100 mM KCl, 10 mM NaCl and 1 mM MgCl2), 0.01% nonidet P40 and increasing amounts of env8 were incubated for 1 h at room temperature. From each reaction, 50 μl was added in technical duplicates to wells in a 384-well plate (Corning) and ATTO590 fluorescence was monitored (excitation, 594 nm; emission, 620–670 nm) at room temperature using a CLARIOstar Plus microplate reader (BMG Labtech). Fluorescence values were background-corrected by subtracting buffer fluorescence values, integrated over all wavelengths and normalized to reactions containing no RNA. Data were fit to the quadratic binding equation using matlab (v2019a)20. The KD of CNCbl–5×PEG-ATTO590 for env8 is reported as the mean and s.d. of independent experiments (n = 4). For competitive fluorescence displacement titrations, the same general procedure was carried out except with a fixed amount of 0.1 μM RNA, 1 μM CNCbl–5×PEG-ATTO590 (in sufficient excess of its KD to ensure RNA saturation) and increasing amounts of competing ligand. Data were fit to a single-site binding model with a Hill slope parameter (set equal to 1.0) using matlab and the KD from the induction experiment was used to convert the half-maximal inhibitory concentration (IC50) from the competitive titrations to KD values20. The KD values for Cbl derivatives to env8 are reported as the mean and s.d. from independent experiments (n = 3–5 depending on the ligand; Source Data) and can be found in Supplementary Table 9. All P values used to compare affinities were derived from a two-tailed t-test calculated in Microsoft Excel (16.43) assuming unequal variance (because of irregular variance among derivatives).
Cell-based reporter assay
Plasmids harboring env8–GFPuv (Addgene, 99831)33 or pBR322 empty vector control were transformed into E. coli cells (Keio collection, JW3805). For each assay, 1 µl of a saturated overnight culture was added to 1 ml of CSB medium supplemented with 100 µg ml−1 carbenicillin and 10 nM Cbl (unless otherwise noted) and grown to mid-log phase at 37 °C. From each biological replicate, 200 µl of cells were added in technical triplicate to wells in a 96-well plate (Costar) and GFP expression was monitored (excitation, 395 nm; emission, 510 nm) using a microplate reader (Tecan Infinite 200 Pro). The fluorescence values were normalized by the optical density at 600 nm (OD600) and background-corrected by subtracting the OD600-normalized fluorescence from the pBR322 empty vector control. Fold repression values were calculated by dividing the median (across all technical replicates) normalized and background-corrected fluorescence values in the absence of Cbl (unrepressed) by the same values but in the presence of Cbl (repressed)33. Fold repression values for all Cbl derivatives are reported as the mean and s.d. from biological replicates (n = 3–4 depending on the ligand, see source data) and can be found in Supplementary Table 10. All P values used to compare fold repression values were derived from a two-tailed paired t-test calculated in Microsoft Excel.
ΔMetE growth assays
For growth experiments, 5 µl of a saturated overnight culture of ΔMetE E. coli cells (Keio collection, JW3805) was added to 5 ml of methionine-dropout CSB medium supplemented with either 335 µM methionine or 10 nM Cbl and grown to mid-log phase at 37 °C. From each biological replicate, 200 µl of cells were added in technical triplicate to wells in a 96-well plate (Costar) and growth was monitored by measuring the OD600 using a microplate reader (Tecan Infinite 200 Pro). For growth rate experiments, the same procedure was used except that the cells were grown for 24 h and the OD600 was measured at various time intervals. For titration experiments, the same procedure was used except that variable amounts of methionine or Cbl were added to the medium. The doubling time (t1/2) and concentrations at which the growth was at its half-maximum for all ligands are reported as the mean and s.d. from biological replicates (n = 3).
Chemoinformatic LDA
A set 20 standard chemoinformatic parameters30,31 (Supplementary Table 1) were calculated for the β-axial group of Cbls 4, 5 and 7–44 using the molecular operating environment (MOE, v2022.02, Chemical Computing Group)31. All descriptors that were nonconstant were then used in an LDA with our clustered binding or functional data (Extended Data Figs. 2 and 5) using the discriminatory analysis plugin from XLSTAT (2023.2.1414)31.
QSAR modeling
The β-axial group of Cbls 4, 5 and 7–44 (and then later for 45–66; Extended Data Fig. 10) were tuned to the correct protonation and tautomerization states using MOE and each state was sent to a conformational search32 using the default settings in MOE. A total of 347 descriptors (all but x3D and protein descriptors) were calculated for each conformation in MOE, averaged using the Boltzmann-weighted equation and filtered to remove multicollinear features32. Then, a lasso-MLR strategy was used to model these descriptors (independent variable) against either the natural-log-transformed KD or fold repression data (dependent variables) in matlab. Lasso was implemented with fivefold cross-validation to find an optimal lambda value in descriptor selection. MLR exhaustively searched all possible models from the lasso-selected descriptors (with a maximum number of descriptors set by Topliss rule50) with fivefold cross-validation to obtain the best model as defined by specified criteria. We constructed a baseline model without a data split that maximized the R2. We also used a stratified 80:20 data split to ensure that training and test sets had equivalent proportions of tight, moderate and weak binders or strong, moderate and weak repressors and constructed models that prioritized either the predictive ability on the test set (Q2-focused) or the fit of the training set (R2-focused).
Lead compound prediction
We used our binding-based and function-based models to screen for high-affinity β-axial groups among a curated Enamine alkyne library. These small molecules were selected by a substructure search (https://enaminestore.com/search) of modified phenyl (phenyl-X) and aliphatic (amide-X) alkynes (precise structures used for this search in Fig. 2f). This left us with a 513-compound library (469 phenyl-X and 44 amide-X). The binding-based and function-based model-selected descriptors (Extended Data Figs. 3a and 6a) were calculated for the 513-compound library in MOE as described above. The predicted ln(KD) and fold repression values from baseline, Q2-focused and R2-focused models were averaged and used to select tight, moderate and weak binders (Cbls 45–53) (Fig. 2g) and strong, moderate and weak repressors (Cbls 54–62) (Fig. 3g).
MD simulations
All-atom MD simulations of env8–CNCbl and env8–Cbl 4 complexes were performed using the PMEMD module51 in AMBER22 (ref. 52). CNCbl was manually built from OHCbl (Protein Data Bank (PDB) 4FRG)19 in PyMol (2.5.7, Schrödinger) and a structure from the Cambridge Structural Database (961228)26 was used for Cbl 4. Ligands were docked into env8 by alignment to OHCbl from the env8–OHCbl structure (PDB 4FRG)19. Each system was neutralized and solvated in a cubic simulation box with a 25-Å buffer of OPC water molecules53 and 150 mM NaCl to mimic physiological conditions. RNA was parameterized using the OL3 force field54, incorporating the RNA.LJbb correction55 for improved backbone accuracy. Force-field parameters for each ligand were generated using Antechamber with the general Amber force field. The metal center, including the cobalt ion, was parameterized using MCPB.py (7.0) following established guidelines56. Quantum mechanical calculations were performed with Gaussian16 to optimize the geometry and derive charges. The cobalt–carbon bond was explicitly defined in the MCPB.py input to ensure accurate representation of the ligand coordination environment56. To improve simulation efficiency, hydrogen mass repartitioning was applied, allowing a 4-fs time step57. Following system preparation, energy minimization, gradual heating to 300 K and equilibration under constant pressure were conducted. Production simulations were performed in triplicate for each system under constant temperature (300 K) and pressure (1 atm) using the NPT ensemble, with each replicate running for at least 1 µs. Trajectory analysis and postprocessing A20(C6)–G19(C6) and A20(C6)–A68(N7) distance calculations were carried out using Cpptraj (6.24.0)58.
Crystallization of env2–ligand complexes
The env2 aptamer domain RNA in complex with various Cbl derivatives was crystallized at 30 °C using hanging drop vapor diffusion. The RNA–ligand complexes were prepared as solutions containing 250 µM RNA and 375–500 µM ligand in 1× TE buffer. For crystallization, drops containing 2 µl of RNA–ligand solution and 2 µl of precipitant solution (40 mM sodium cacodylate pH 7, 10% (w/v) 2-methyl-2,4-pentanediol (MPD), 12 mM spermine tetrahydrochloride, 80 mM KCl and 20 mM MgCl2) were suspended above a reservoir solution of 500 μl of 35% MPD and grown for 24–96 h. For cryoprotection, the crystals were soaked in a cryoprotectant (precipitant solution with MPD increased to 25%) for 2 min and flash-frozen in liquid nitrogen. Some data were collected on a home-source Rigaku MicroMax-003 X-ray source with a Dectris Pilatus 200K detector and then indexed, integrated and scaled with HKL3000 (ref. 59) (Supplementary Tables 11–14). Other data were collected at the Advanced Light Source on a Dectris on the ALS Beamline 8.2.2. with a Dectris Pilatus3 2M detector and then indexed and integrated with XDS (June 30, 2023)60 and scaled with Aimless (0.7.4)61 (Supplementary Tables 11–14). The crystals that yielded the env2 apo structure were grown in the same solution described above but in the presence of compound 71.
Structure determination and model refinement
For all structures, an initial electron density map was calculated using molecular replacement with Phaser62 in PHENIX (1.19.2)63 using env8 as a starting model (PDB 4FRG)19 but with OHCbl and A20 removed to minimize model bias. All ligands were manually built from Cbl 4 (Cambridge Structural Database 961228)26 in PyMol (2.5.7, Schrödinger) and restraint files were generated with eLBOW64. After initial refinement, the ligand density was unambiguous and ligands were added to the model with LigandFit65. After another round of refinement, A20 was manually built into the mode using Coot (0.9.8.93)66. The map and model were refined and the solvent was built until a maximum agreement between the map and the model was reached. During this process, to mitigate the effect of model bias, multiple rounds of combined high temperature (5,000 K) simulated annealing in torsion space and maximum-likelihood refinement were performed67. At the end of refinement, RNA geometry was corrected with ERRASER (https://rosie.rosettacommons.org)68. A subset of the RNA–ligand cocrystal structures had sufficient resolution to warrant simulated annealing in Cartesian space and individual ADP refinement. A simulated annealing 2Fo − Fc map where A20 and the ligand were omitted from the model was calculated for all structures to support the final placement of the ligand and A20. Models and associated structure factors were deposited to the PDB.
ITC
RNA was exchanged into a 1× RNA-binding buffer by successive washing in centrifugal concentrators (Millipore-Sigma). RNA was diluted up to a final concentration of 10 μM and titrated with ligand (dissolved in the flowthrough of the final wash from buffer exchange) at 100 μM. All titrations were performed at 25 °C using a MicroCal ITC200. Data were fit to a single-site binding model using Origin 7 ITC software (MicroCal). The KD values for all Cbl derivatives to env8 RNA constructs are reported as the mean and s.d. from independent experiments (n = 3).
Computational docking
The 28,000-compound RNA library from Enamine (https://enamine.net/compound-libraries/targeted-libraries/rna) was downloaded as an SDF file and imported into MOE as a database file. The small molecules were tuned to the correct protonation and tautomerization states and each state was sent to a conformational search32 using the default settings in MOE. RNA receptor preparation was carried out using the default Quick Prep protocol in MOE. Our env2–Cbl 29 (A20-out) and env2–Cbl 63 (A20-in) structures were used as representative receptors where A20 was displaced toward the major groove or engaged in the RNA core, respectively. To ensure that A20 was not accessible for A20-out docking, all A20 atoms were removed from the RNA receptor in MOE. Rigid receptor docking was carried out in a templated manner, where the ligand search space started from the biphenyl-like scaffold present in the cocrystal structures with 29 and 63. For A20-in docking, the terminal triazole ring was rebuilt into a phenyl group, such that the scaffolds were identical in both RNA receptors. With this approach, only the 319 ligands with the required scaffold were docked and scored. The Enamine catalog numbers for the eight docking hits are Z4539256213 (67), Z3515426302 (68), Z4144805645 (69), Z8153471773 (70), Z2466243364 (71), Z1209546900 (72), Z7621749448 (73) and Z4116044018 (74).
TO binding assay
For fluorescence induction titrations, 60-μl reactions containing TO (5 nM), 1× RNA-binding buffer and increasing amounts of env8 were incubated in 384-well plates (Corning). Reactions were incubated for 15 min and TO fluorescence was monitored (excitation, 490 ± 10 nm; emission, 530 ± 10 nm) at room temperature using a CLARIOstar Plus microplate reader (BMG Labtech). Fluorescence values were normalized to reactions containing no RNA and data were fit to a single-site binding model with a Hill slope parameter using matlab. The KD of TO for env8 is reported as the mean and s.d. of independent experiments (n = 3). For competitive fluorescence displacement assays, the same procedure was carried out except with a fixed amount of 2 μM RNA, 20 μM TO (in sufficient excess of its KD to ensure RNA saturation) and 2 mM competing ligand. Displacement titrations were carried out in the same manner except using increasing amounts of 68 or 71. Data were fit to a single-site binding model with a Hill slope parameter using matlab and the KD from the induction experiment was used to convert the IC50 from the competitive titrations to KD values20 as described above. The KD values for all ligands to env8 are reported as the mean and s.d. from independent experiments (n = 3).
EMSA
To validate the CNCbl-induced env8 conformational change, 20-μl reactions containing RNA (0.5 μM), 1× RNA-binding buffer and increasing amounts of CNCbl (0.1–10 μM) were incubated at 37 °C for 15 min and then at 4 °C for another 15 min. Once equilibrated to 4 °C, 15 μl of the samples were loaded onto a native 10% (19:1) polyacrylamide gel in 0.5× THE buffer and 1 mM MgCl2 and run at constant 7 W for 3 h at 4 °C in 0.5× THE buffer with 1 mM MgCl2. For the competitive EMSA experiment, the same procedure was used except using a prebound env8–CNCbl complex (0.5 μM CNCbl) and adding 5 mM 68 or 71. All gels were stained with ethidium bromide, imaged on an ultraviolet illuminator (Alpha Innotech) and processed with AlphaView software (AlphaImagerHP). EMSA experiments to establish the competitive binding of 68 and 71 were repeated either two (68) or three (71) times. Unmodified images of the gels shown in Fig. 6e,f and their replicates can be found in Supplementary Fig. 9 and are included as Source Data.
MST
For MST binding experiments, env8 was 3′-end labeled with pCp-AF488 (NU-1706-AF488, Jena Bioscience). The 50-μl labeling reaction comprised 200–500 nmol of RNA, 1 mM ATP, 10% (w/v) DMSO, 1× T4 RNA ligase 1 buffer (B0204S, New England Biolabs), 15% (w/v) PEG 8000, 48 μM pCp-AF488 and 4 μl of T4 RNA ligase 1 (M0204L, New England Biolabs) and was incubated at 16 °C for 18 h. To monitor RNA–ligand binding, labeled env8 (300 nmol) was incubated in 1× RNA-binding buffer and 250 μM ligand for 15 min and transferred to capillaries (NanoTemper). AlexaFluor488 fluorescence was read on a Monolith NT.115 (NanoTemper) instrument on its default blue setting. Competitive MST experiments were carried out in the same manner except using a prebound env8–CNCbl complex (300 nmol of RNA and 10 μM ligand) and adding 1 mM 68 or 71. MST data are shown as representative traces from independent experiments (n = 4).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Atomic coordinates and structure factors were deposited to the PDB under accession numbers 9MFH (env2 apo), 9E5H (env2–CNCbl), 9E5I (env2–Cbl 4), 9E5J (env2–Cbl 5), 9E5K (env2–Cbl 13), 9E5L (env2–Cbl 26), 9E5M (env2–Cbl 29), 9E5O (env2–Cbl 32), 9E5P (env2–Cbl 33), 9E5Q (env2–Cbl 36), 9ELR (env2–Cbl 37), 9E5R (env2–Cbl 42), 9E5S (env2–Cbl 63) and 9E5T (env2–Cbl 64). A structure from the Cambridge Structural Database (961228)26 was used to model Cbl 4. Additional data can be provided upon request. Source data are provided with this paper.
Code availability
Code and data files for carrying out QSAR modeling and MD simulations can be accessed from GitHub (https://github.com/bateyLab/2025_NCB_paper).
References
Childs-Disney, J. L. et al. Targeting RNA structures with small molecules. Nat. Rev. Drug Discov. 21, 736–762 (2022).
Warner, K. D., Hajdin, C. E. & Weeks, K. M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 17, 547–558 (2018).
Umuhire Juru, A. & Hargrove, A. E. Frameworks for targeting RNA with small molecules. J. Biol. Chem. 296, 100191 (2021).
Ratni, H. et al. Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J. Med. Chem. 61, 6501–6517 (2018).
Santos, R. et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34 (2017).
Olenginski, L. T., Spradlin, S. F. & Batey, R. T. Flipping the script: understanding riboswitches from an alternative perspective. J. Biol. Chem. 300, 105730 (2024).
Breaker, R. R. The biochemical landscape of riboswitch ligands. Biochemistry 61, 137–149 (2022).
Serganov, A. & Nudler, E. A decade of riboswitches. Cell 152, 17–24 (2013).
Blount, K. F. et al. Novel riboswitch-binding flavin analog that protects mice against Clostridium difficile infection without inhibiting cecal flora. Antimicrob. Agents Chemother. 59, 5736 (2015).
Vicens, Q. et al. Structureactivity relationship of flavin analogues that target the flavin mononucleotide riboswitch. ACS Chem. Biol. 13, 2908–2919 (2018).
Rizvi, N. F. et al. Discovery of selective RNA-binding small molecules by affinity-selection mass spectrometry. ACS Chem. Biol. 13, 820–831 (2018).
Howe, J. A. et al. Selective small-molecule inhibition of an RNA structural element. Nature 526, 672–677 (2015).
Kräutler, B. Antivitamins B12—some inaugural milestones. Chemistry 26, 15438–15445 (2020).
Nahvi, A. et al. Genetic control by a metabolite binding mRNA. Chem. Biol. 9, 1043–1049 (2002).
Mccown, P. J., Corbino, K. A., Stav, S., Sherlock, M. E. & Breaker, R. R. Riboswitch diversity and distribution. RNA 23, 995–1011 (2017).
Polaski, J. T., Webster, S. M., Johnson, J. E. & Batey, R. T. Cobalamin riboswitches exhibit a broad range of ability to discriminate between methylcobalamin and adenosylcobalamin. J. Biol. Chem. 292, 11650–11658 (2017).
Demerre, L. J. & Wilson, C. Photolysis of vitamin B12. J. Am. Pharm. Assoc. 45, 129–134 (1956).
Juzeniene, A. & Nizauskaite, Z. Photodegradation of cobalamins in aqueous solutions and in human blood. J. Photochem. Photobiol. B 122, 7–14 (2013).
Johnson, J. E., Reyes, F. E., Polaski, J. T. & Batey, R. T. B12 cofactors directly stabilize an mRNA regulatory switch. Nature 492, 133–137 (2012).
Lennon, S. R. et al. Targeting riboswitches with β-axial-substituted cobalamins. ACS Chem. Biol. 18, 1136–1147 (2023).
Peselis, A. & Serganov, A. Structural insights into ligand binding and gene expression control by an adenosylcobalamin riboswitch. Nat. Struct. Mol. Biol. 19, 1182–1184 (2012).
Davila-Calderon, J. et al. IRES-targeting small molecule inhibits enterovirus 71 replication via allosteric stabilization of a ternary complex. Nat. Commun. 11, 1–13 (2020).
Chen, J. L. et al. Design, optimization, and study of small molecules that target tau pre-mRNA and affect splicing. J. Am. Chem. Soc. 142, 8706–8727 (2020).
Matyjasik, M. M., Hall, S. D. & Batey, R. T. High affinity binding of N2-modified guanine derivatives significantly disrupts the ligand binding pocket of the guanine riboswitch. Molecules 25, 2295 (2020).
Chromiński, M., Lewalska, A., Karczewski, M. & Gryko, D. Vitamin B12 derivatives for orthogonal functionalization. J. Org. Chem. 79, 7532–7542 (2014).
Chromiński, M., Lewalska, A. & Gryko, D. Reduction-free synthesis of stable acetylide cobalamins. Chem. Commun. 49, 11406–11408 (2013).
Donlic, A. et al. R-BIND 2.0: an updated database of bioactive RNA-targeting small molecules and associated RNA secondary structures. ACS Chem. Biol. 17, 1556–1566 (2022).
Morgan, B. S., Forte, J. E., Culver, R. N., Zhang, Y. & Hargrove, A. E. Discovery of key physicochemical, structural, and spatial properties of RNA-targeted bioactive ligands. Angew. Chem. Int. Ed. Engl. 56, 13498–13502 (2017).
Braselmann, E. et al. A multicolor riboswitch-based platform for imaging of RNA in live mammalian cells. Nat. Chem. Biol. 14, 964–971 (2018).
Stratton, C. F., Newman, D. J. & Tan, D. S. Cheminformatic comparison of approved drugs from natural product versus synthetic origins. Bioorg. Med. Chem. Lett. 25, 4802–4807 (2015).
Patwardhan, N. N., Cai, Z., Umuhire Juru, A. & Hargrove, A. E. Driving factors in amiloride recognition of HIV RNA targets. Org. Biomol. Chem. 17, 9313–9320 (2019).
Cai, Z., Zafferani, M., Akande, O. M. & Hargrove, A. E. Quantitative structure–activity relationship (QSAR) study predicts small-molecule binding to RNA structure. J. Med. Chem. 65, 7262–7277 (2022).
Polaski, J. T., Holmstrom, E. D., Nesbitt, D. J. & Batey, R. T. Mechanistic insights into cofactor-dependent coupling of RNA folding and mRNA transcription/translation by a cobalamin riboswitch. Cell Rep. 15, 1100–1110 (2016).
Neidhardt, F. C. et al. (eds) Escherichia coli and Salmonella Typhimurium: Cellular and Molecular Biology (American Society for Microbiology, 1987).
Figge, R. M. Methionine biosynthesis in Escherichia coli and Corynebacterium glutamicum. in Amino Acid Biosynthesis—Pathways, Regulation and Metabolic Engineering (ed. Wendisch, V. F.) (Springer, 2006).
Lipinski, C. A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol. 1, 337–341 (2004).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46, 3–26 (2001).
Wicks, S. L. & Hargrove, A. E. Fluorescent indicator displacement assays to identify and characterize small molecule interactions with RNA. Methods 167, 3–14 (2019).
Tong, Y. et al. Programming inactive RNA-binding small molecules into bioactive degraders. Nature 618, 169–179 (2023).
Geng, A., Roy, R. & Al-Hashimi, H. M. Conformational penalties: new insights into nucleic acid recognition. Curr. Opin. Struct. Biol. 89, 102949 (2024).
Stelzer, A. C. et al. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol. 7, 553–559 (2011).
Panei, F. P., Gkeka, P. & Bonomi, M. Identifying small-molecules binding sites in RNA conformational ensembles with SHAMAN. Nat. Commun. 15, 1–15 (2024).
Zhou, Y. & Chen, S.-J. Advances in machine-learning approaches to RNA-targeted drug design. Artif. Intell. Chem. 2, 100053 (2024).
Morishita, E. C. & Nakamura, S. Recent applications of artificial intelligence in RNA-targeted small molecule drug discovery. Expert Opin. Drug Discov. 19, 415–431 (2024).
Sonar, M. V. et al. Fluorescence detection of KRAS2 mRNA hybridization in lung cancer cells with PNA-peptides containing an internal thiazole orange. Bioconjug. Chem. 25, 1697–1708 (2014).
Wierzba, A. J., Richards, E. M., Lennon, S. R., Batey, R. T. & Palmer, A. E.Unveiling the promise of peptide nucleic acids as functional linkers for an RNA imaging platform. RSC Chem. Biol. 6, 249–262 (2024).
Corson, T. W., Aberle, N. & Crews, C. M. Design and applications of bifunctional small molecules: why two heads are better than one. ACS Chem. Biol. 3, 677–692 (2008).
Bereiter, R. et al. Engineering covalent small molecule–RNA complexes in living cells. Nat. Chem. Biol. 21, 843–854 (2025).
Edwards, A. L., Garst, A. D. & Batey, R. T. Determining structures of RNA aptamers and riboswitches by X-ray crystallography. Methods Mol. Biol. 535, 135–163 (2009).
Topliss, J. G. & Edwards, R. P. Chance factors in studies of quantitative structure–activity relationships. J. Med. Chem. 22, 1238–1244 (1979).
Cheatham, T. E., Miller, J. L., Fox, T., Darden, T. A. & Kollman, P. A. Molecular dynamics simulations on solvated biomolecular systems: the particle mesh Ewald method leads to stable trajectories of DNA, RNA, and proteins. J. Am. Chem. Soc. 117, 4193–4194 (1995).
Case, D. A. et al. The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 (2005).
Izadi, S., Anandakrishnan, R. & Onufriev, A. V. Building water models: a different approach. J. Phys. Chem. Lett. 5, 3863–3871 (2014).
Zgarbová, M. et al. Refinement of the Cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J. Chem. Theory Comput. 7, 2886–2902 (2011).
Steinbrecher, T., Latzer, J. & Case, D. A. Revised AMBER parameters for bioorganic phosphates. J. Chem. Theory Comput. 8, 4405–4412 (2012).
Li, P. & Merz, K. M. MCPB.py: a Python based metal center parameter builder. J. Chem. Inf. Model. 56, 599–604 (2016).
Hopkins, C. W., Le Grand, S., Walker, R. C. & Roitberg, A. E. Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 11, 1864–1874 (2015).
Roe, D. R. & Cheatham, T. E. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 (2013).
Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 (2006).
Kabsch, W. XDS. Acta Crystallogr. D Struct. Biol. 66, 125–132 (2010).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 (2013).
Bunkóczi, G. et al. Phaser.MRage: automated molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 69, 2276–2286 (2013).
Adams, P. D. et al. The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106 (2011).
Moriarty, N. W., Grosse-Kunstleve, R. W. & Adams, P. D. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 65, 1074–1080 (2009).
Terwilliger, T. C., Klei, H., Adams, P. D., Moriarty, N. W. & Cohn, J. D. Automated ligand fitting by core-fragment fitting and extension into density. Acta Crystallogr. D Biol. Crystallogr. 62, 915–922 (2006).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D., Pannu, N. S., Read, R. J. & Brunger, A. T. Extending the limits of molecular replacement through combined simulated annealing and maximum-likelihood refinement. Acta Crystallogr. D Biol. Crystallogr. 55, 181–190 (1999).
Chou, F. C., Sripakdeevong, P., Dibrov, S. M., Hermann, T. & Das, R. Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nat. Methods 10, 74–76 (2013).
Acknowledgements
This work is supported by the National Institutes of Health (R35 GM152029 to R.T.B. and R35 GM139644 to A.J.W.) and the Howard Hughes Medical Institute (S.P.L). We thank J. Nix and the staff of beamline 8.2.2 of the Advanced Light Source, Lawrence Berkeley National Laboratory for their support with remote crystallographic data collection. Beamline 8.2.2 of the Advanced Light Source, a Department of Energy Office of Science User Facility under contract no. DE-AC02-05CH11231, is supported in part by the ALS-ENABLE program funded by the National Institutes of Health (P30 GM124169-01). We thank the Macromolecular X-ray Crystallography Core (RRID:SCR_019310) at the University of Colorado Boulder for crystallographic data collection and the Shared Instruments Pool (RRID: SCR_018986) of the Department of Biochemistry at the University of Colorado Boulder for the use of the Monolith NT.115 NanoTemper (S10OD021603) and the MicroCal ITC200 (S10RR026516) instruments. We acknowledge A. Erbse for her assistance with the resources used at the University of Colorado Boulder. We thank F. Longshore-Neate and D. Patel for help with derivative synthesis and N. Zeps and T. Wolters for help with RNA–ligand cocrystallization. We are grateful to Q. Vicens for comments on the manuscript.
Author information
Authors and Affiliations
Contributions
R.T.B. conceptualized the project. L.T.O. designed the small-molecule library and carried out its synthesis, purification and characterization. A.J.W. helped supervise the synthesis and characterization. S.P.L. conducted the MD simulations. L.T.O. performed the in vitro binding and cell-based reporter assays, carried out the chemoinformatic modeling, conducted the RNA crystallization and structure determination, implemented the computational docking and analyzed all the data. L.T.O. wrote the paper with input from all authors.
Corresponding author
Ethics declarations
Competing interests
R.T.B. serves on the scientific advisory boards of SomaLogic and MeiraGTx. The other authors declare no competing interests.
Peer review
Peer review information
Nature Chemical Biology thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Quantitative structure-activity relationship (QSAR) analysis of RNA binding data.
a, Schematic representation of the “distance from alkyne”. b, Plot of KD values and “distance from alkyne” for the phenyl-F (9-11) and -CH3 (12-14) series. c, Chemical structures of the para-substituted phenyls (4, 5, 8, 11, 14, and 17-20) with their respective Hammett constant (σp) and van der Waals (VdW) volume values listed. d, Plot of KD values and the σp of the chemical group in the para-substituted phenyls. e, Same as in d but using the VdW volume of the same chemical groups. f, Plot of KD values for the heterocyclic derivatives (21-26) and 8 for comparison. g, Same as in f but for the multi-ring derivatives (27-32). In f and g, mean and s.d. from independent experiments (n = 4 except for data from 21 where n = 5) are shown.
Extended Data Fig. 2 Linear discriminant analysis (LDA) of RNA binding data.
a, Plot of the KD values for Cbls 1-44, where the data have been normalized to the value for MeCbl. On this basis, we clustered our derivatives into three groups: tight (KD,rel(MeCbl) < 15), moderate (KD,rel(MeCbl) = 15-70), and weak (KD,rel(MeCbl) > 70) binders. Given that standard Cbls 1-3 do not have a chemically diverse β-axial group, they were excluded from the LDA (and downstream QSAR modeling). Cbl 6 was also excluded due to its a sp3 carbon adjacent to the alkyne, a chemical space that was unique within the library and one that we did not wish to pursue. b, LDA plot for principal components 1 and 2 (PC1 and PC2) depicting the clustering of the three binding groups and their centroids using chemoinformatic parameters (Supplementary Table 1). c, LDA loading plot of the contributions of each chemoinformatic parameter on PC1 and PC2, with the most obvious descriptor colored by the binding cluster that they best explain (for example, number of aromatic rings, ArRings; number of hydrogen bond donors, HBD; and accessible solvent area, ASA).
Extended Data Fig. 3 QSAR modeling framework to predict binding lead compounds.
a, Written out multiple linear regression (MLR) baseline, R2-focused, and Q2-focused models. The physical meaning of each physiochemical descriptor is listed in Supplementary Table 2. b, Measured ln [KD] values plotted with the value predicted by the baseline model that did not include a data split. c, Locations of the training and test set from the R2-focused modeling in two-dimensional chemical space constructed from PC1 and PC2 of the whole data set. d, Same as in b but for the R2-focused model. e, Average ln [KD] values predicted by our three models for each compound in the alkyne library. The magenta shaded box represents the 184 compounds that have a predicted affinity tighter than binding lead 29 (ln [KD] < −18.0). f, Rank-ordered plot the average KD values predicted by our binding models for the 184 potential lead compounds that were identified from the alkyne library, separated by type (phenyl-X and amide-X). These compounds are classified as either “out of stock”, “already have”, “counterevidence”, “difficult synthesis”, and “will test”. Here, “out of stock” refers to alkynes that were not purchasable from Enamine. “Counterevidence” refers to compounds that are chemically similar to derivatives in our library that are known to have weak affinity. “Difficult synthesis” refers to compounds that contain amino groups or protonated nitrogen atoms, which were not compatible with the reduction-free synthesis25,26 (Supplementary Fig. 2). The vast majority of “out of stock” compounds also contained amino groups or protonated nitrogen atoms. Thus, even if they become purchasable from Enamine, they are still synthetically inaccessible. Finally, “will test” refers to the four purchasable alkynes that were predicted to yield binding lead Cbls 45-48.
Extended Data Fig. 4 Growth experiments in ΔMetE cells.
a, Growth (as measured by OD600) curve of ΔMetE cells (which lack the Cbl-independent methionine synthase MetE) that were grown in methionine (Met)-dropout CSB media and supplemented with either Met or CNCbl. b, Plot of ln [OD600] against time from the data in a where cell growth is exponential. The doubling time (t1/2) is obtained from the slope and is reported as mean and s.d. from biological replicates (n = 3). c, Titration of ΔMetE cells grown for a variable duration in increasing amounts Met. The concentration at which the growth is at its half maximum value is reported as mean and s.d. from biological replicates (n = 3). d, Same as in c but for titration of CNCbl. e, Same as in a but for CNCbl and Cbls 36 (strong repressor) and 44 (weak repressor). f, Same as in b but for the data in e. g, Same as in d but for cells grown for 8 h in increasing amounts of CNCbl, 36, or 44. All t1/2values and concentrations at which growth is at its half maximum are reported as mean and s.d. from biological replicates (n = 3).
Extended Data Fig. 5 LDA of RNA functional data.
a, Plot of the fold repression (FR) values for Cbls 1-44, where the data have been normalized to the value for MeCbl. On this basis, we clustered our derivatives into three groups: strong (FRrel(MeCbl) < 0.3), moderate (FRrel(MeCbl) = 0.3-0.7), and weak (KD,rel(MeCbl) > 0.7) repressors. b, LDA plot for PC1 and PC2 depicting the clustering of the three functional groups and their centroids using chemoinformatic parameters (Supplementary Table 1). c, LDA loading plot of the contributions of each chemoinformatic parameter on PC1 and PC2, with the most obvious descriptor colored by the functional cluster that they best explain (for example, number of aromatic rings, ArRings; number of hydrogen bond donors, HBD; and fraction of sp3 centers, Fsp3).
Extended Data Fig. 6 QSAR modeling framework to predict functional lead compounds.
a, Written out MLR baseline, Q2-focused, and R2-focused models. The physical meaning of each physiochemical descriptor is listed in Supplementary Table 3. b, Measured fold repression values plotted with the value predicted by the baseline model that did not include a data split. c, Locations of the training and test set from the R2-focused modeling in two-dimensional chemical space constructed from PC1 and PC2 of the whole data set. d, Same as in b but for the R2-focused model. e, Average fold repression values predicted by our three models for each compound in the alkyne library. The magenta shaded box represents the 151 compounds that have a predicted fold repression greater than functional lead 36 (fold repression > 6.7). f, Rank-ordered plot the average fold repression values predicted by functional models for the 151 potential lead compounds that were identified from the alkyne library, separated by type (phenyl-X and amide-X). These compounds are classified as either “out of stock”, “difficult synthesis”, and “will test” as described in the legend of Extended Data Fig. 3.
Extended Data Fig. 7 Validation of the env2 crystal construct.
a, Schematic representation of the secondary structures of full-length env8, env8 aptamer domain, and the env8 and env2 RNA crystallography constructs. The binding pocket nucleotides G19 (yellow), A20 (green), and A68 (cyan) are colored and the sites of variation between the env8 and env2 RNA sequences are indicated in magenta. Other changes to the env8 wild-type RNA sequence are the insertion of a gGAAAc tetraloop at L5, the variable lengths of helices P1 and P5 (either four or six base pairs, bp), and a 3′-end adenosine, which were all incorporated to aid crystallization. b, Binding curve of the fluorescence induction of CNCbl-5×PEG-ATTO590 binding to env8 aptamer domain and env2 crystal construct shown as mean and s.e.m. from independent experiments (n = 3). The KD values are reported as mean and s.d. from independent experiments (n = 3). c, Fold repression values for our env8 and env2 reporter systems in the presence of 10 µM CNCbl, reported as mean and s.d. from biological replicates (n = 4). d, Co-crystal structure of env2-CNCbl aligned to the env8-OHCbl complex (PDB 4FRG ref. 19). Global RNA architecture (left) is displayed as cartoon, with sites of variation between the env8 and env2 shown in magenta. The ligand binding pocket (right) is shown in sticks and shows very strong agreement between the two RNA-ligand complexes (RMSD = 0.45 Å). Given that env2 shows near-identical ligand binding, regulatory activity, and local and global RNA architecture, all structural information from env2 can be translated to env8.
Extended Data Fig. 8 Additional env2-Cbl co-crystal structures.
a, Crystal structure of env2 in the apo state showing only the binding pocket nucleotides (left). Alignment of the binding pockets of env2-CNCbl (protomer A) and the apo state (right). In all structural representations, the binding pocket nucleotides G19 (yellow), A20 (green), and A68 (cyan) are colored and the β-axial is shown in magenta. All mesh representations correspond to a simulated annealing 2Fo-Fc map where A20 and the ligand (if present) were omitted from the model and are shown at 1 σ contour. b, Our env2-CNCbl co-crystal structure has two molecules in the asymmetric unit (ASU). Protomer A is already shown in Fig. 4c. Here, we are showing an alignment of the binding pockets of both protomers, which are nearly identical. c, Our env2-Cbl 4 co-crystal structure also has two molecules in the ASU. Protomer B is already shown in Fig. 4d. Here, we are showing an alignment of the binding pockets of both protomers, which reveals a different orientation of A20. This was a common feature of all but one of our co-crystal structures. In each protomer A, A20 is locked in a crystal lattice contact. In each protomer B, we get a variable conformation of A20, suggesting that A20 is dynamic. This also explains why the electron density of A20 is well defined in protomer A but less so in protomer B. We will therefore focus on the more solution-representative protomer B in all analyses. d, Co-crystal structure of env2-Cbl 5 showing only the binding pocket nucleotides and the β-axial group. e, Same as in d but for env2-Cbl 13. This structure had four molecules in the ASU. In all four protomers, there is well defined density for A20 and minimal conformational variety. f, Same as in d but for env2-Cbl 26. g, Same as in d but for env2-Cbl 33. h, Same as in d but for env2-Cbl 36. i, Same as in d but for env2-Cbl 37. j, Alignment of the binding pockets of protomers A (left) and B (right) for all the co-crystal structures shown here and in Fig. 4.
Extended Data Fig. 9 High concentrations of click Cbls inhibit ΔMetE cell growth.
a, Growth (as measured by OD600) curve of ΔMetE cells grown in Met-dropout CSB media and supplemented with either CNCbl or Cbls 63-65. b, Plot of ln [OD600] against time from the data in a where cell growth is exponential. The t1/2 is obtained from the slope and is reported as mean and s.d. from biological replicates (n = 3). c, Growth of ΔMetE cells grown for 5.5 h and supplemented with different Cbls. Here, (–) refers to cells without Met or Cbl supplemented to the media. d, Titration of ΔMetE cells grown for 8 h in increasing amounts of CNCbl or Cbls 63-65. The concentration at which the growth is at its half maximum value is reported as mean and s.d. from biological replicates (n = 3). e, Growth of Keio E. coli cells from the fold repression experiment shown in Fig. 5g. In this experiment, 10 nM CNCbl was added to all cells and excess amount of Cbls 36, 44, or 63-65 (5 µM) were also added to the growth media. Here, (–) refers to cells without any ligand added and 3 refers to cells with just 10 nM CNCbl added. Data are reported as mean and s.d. from biological replicates (n = 4). In contrast to ΔMetE cells, the growth of Keio cells is unaffected by high concentrations of Cbls.
Extended Data Fig. 10 Refined QSAR modeling.
a, Written out MLR baseline, R2-focused, and Q2-focused models for refined binding-based modeling using data from Cbls 49-66 (binding leads 45-48 excluded). The physical meaning of each physiochemical descriptor is listed in Supplementary Table 6. b, Measured ln [KD] values plotted with the value predicted by the baseline, R2-focused, or Q2-focused models. c, Measured ln [KD] values plotted with the value predicted by our new binding-based models. d, Same as in a but from models including data from 45-48. e, Same as in b but for the refined modeling described in d. f, Same as in c but for the models shown in d. g, Written out MLR baseline, R2-focused, and Q2-focused models for refined function-based modeling using data from Cbls 45-53 and 58-66 (functional leads 54-57 excluded). The physical meaning of each descriptor is listed in Supplementary Table 7. h, Measured fold repression values plotted with the value predicted by the baseline, R2-focused, or Q2-focused models. i, Measured fold repression values plotted with the value predicted by our new function-based models. j, Same as in g but from models including data from 54-57. k, Same as in h but for the refined modeling described in j. l, Same as in c but for the models shown in j. In c, f, i, and l, all experimental data are represented as mean and s.d. from independent experiments (n = 3 except for data from 45 and 48 where n = 4) and all predicted data are shown as the mean and s.d. from independent predictions (n = 3) from our three models (baseline, R2-focused, and Q2-focused).
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–9, Tables 1–15, Note 1 and References.
Supplementary Data 1 (download XLSX )
Statistical source data for Supplementary Fig. 4.
Supplementary Data 2 (download XLSX )
Statistical source data for Supplementary Fig. 6.
Supplementary Data 3 (download XLSX )
Statistical source data for Supplementary Fig. 7.
Supplementary Data 4 (download XLSX )
Statistical source data for Supplementary Fig. 8.
Supplementary Data 5 (download ZIP )
Unprocessed gels for Supplementary Fig. 9.
Source data
Source Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Fig. 6 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 6 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 7 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 9 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 10 (download XLSX )
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Olenginski, L.T., Wierzba, A.J., Laursen, S.P. et al. Designing small molecules targeting a cryptic RNA binding site through base displacement. Nat Chem Biol (2025). https://doi.org/10.1038/s41589-025-02018-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41589-025-02018-8
This article is cited by
-
Seeing the hidden pocket
Nature Chemical Biology (2025)
