
Abstract
Many animals produce vivid colors by reflecting and amplifying light with stacked guanine crystals within membrane-bound organelles called iridosomes. While the presence of guanine crystals in iridosomes is well documented, the mechanisms facilitating the accumulation of water-insoluble guanine and driving its crystallization remain unclear. Here we used cryo-electron microscopy, live-cell pH imaging, pharmacological perturbations and spectroscopy to study iridosome maturation in zebrafish. Cryo-electron and synchrotron-based soft X-ray microscopies revealed that amorphous guanine initially accumulates in early-stage iridosomes in its protonated state. Live-cell imaging with a pH sensor demonstrated that early iridosomes are acidic, with pH gradually neutralizing during development. Inhibiting V-ATPase disrupted this acidification and significantly reduced crystal formation, indicating its role in pH regulation. Our findings reveal insights into the molecular mechanisms facilitating guanine formation within iridosomes, emphasizing the pivotal role of pH alternations in the precise formation of biogenic crystals.

Similar content being viewed by others


Main
Iridophores are specialized cells that contain reflective nanocrystals, primarily composed of guanine, which they use to reflect, scatter or refract light1,2,3. The intracellular assemblies of these nanocrystals have a critical role in producing the vibrant and dynamic coloration observed in a wide range of organisms, including fish, reptiles, amphibians, cephalopods and certain spiders3,4,5,6,7,8,9,10,11,12. These cells contribute to color variation and facilitate adaptive color changes for camouflage, signaling and vision7,10,13,14,15,16. Biogenic guanine crystals are formed within specialized membrane-bound organelles within iridophores known as iridosomes17. The process leading to crystal formation, predominantly taking place during the early developmental stages of iridophores, involves the synthesis and trafficking of guanine precursors to the iridosome, their concentration within the organelle and the eventual crystallization into bioorganic structures with highly controlled properties3,7,18,19. The mechanism by which guanine is transported into the iridosome remains unknown. However, because guanine cannot passively diffuse across the iridosomal membrane, its transport is likely mediated by dedicated transporters or through the fusion of smaller cargo vesicles.
Several factors influence the guanine crystallization process, including supersaturation, temperature, solvent properties, the presence of impurities, agitation and, importantly, pH. pH greatly impacts the ionization and solubility of guanine, which is insoluble under neutral conditions and only dissolves in extremely acidic or alkaline environments20,21. Manipulating the pH of crystallization solutions in vitro has been shown to control guanine’s crystal phases, polymorphism and size, reflecting the stability of different guanine tautomers under varying pH conditions20.
Iridophores, along with melanophores, another pigment cell that also originates from the neural crest1,22, contain organelles that were suggested to be lysosome-related organelles (LROs)18,23,24,25,26. These organelles are distinguished by their specific cargoes and functions24,27,28. A common hallmark of many LROs is the regulation of pH during their maturation. For example, trans-Golgi-derived Weibel–Palade bodies undergo a pH decrease during maturation, while endosome-derived melanosomes experience a pH increase26,29,30,31. While melanosomes have been well studied for their development from endosomal structures and their role in melanin synthesis24,25,32,33, the detailed biochemical dynamics and structural development of iridosomes remain less explored.
Zebrafish (Danio rerio) iridosomes exhibit several unique features that make them a compelling model system for cell biology. Their size varies from less than 100 nm to several microns in length34, providing an excellent framework for studying size regulation and lipid homeostasis. Unlike most biomineralizing organelles, iridosomes form crystals using nucleobases, which are primary metabolites35. This distinctive feature offers a unique opportunity to investigate primary metabolite homeostasis in a highly specialized cellular environment.
While the exact mechanism of crystal formation differs among organisms, biogenic guanine crystals have been shown to form through nonclassical mechanisms34,36,37,38. In this process, guanine initially accumulates within the iridosome until templated crystallization occurs on preassembled scaffolds34,39. However, a fundamental question remains: what mechanisms enable the accumulation of water-insoluble guanine and drive its crystallization?
Here, we use zebrafish iridophores as a model system to investigate the process taking place in the iridosomes before and during crystal formation. By using advanced imaging and spectroscopic techniques, we aim to delineate the mechanisms through which the microenvironment within iridosomes affects guanine crystal formation. We show that the pH variations have an important role in this process and disrupting these changes severely impairs crystal formation. Initially, the early-stage iridosome is highly acidic, potentially promoting the accumulation of protonated amorphous guanine and the assembly of macromolecular scaffolds. As the organelle matures and crystal increases in size, the organelle becomes less acidic. Thus, the neutralization of pH likely promotes subsequent crystal growth.
Results
Zebrafish iridosomes as a model system for crystal formation
To investigate the early stages of guanine accumulation and the key factors that lead to its crystallization, we focused our investigation on the iridophores of the zebrafish larvae as a model system. Zebrafish use guanine crystals for various functions. These crystals, distributed throughout their skin and eyes, are crucial in shaping skin color and patterns, enabling camouflage, functioning as light barriers and improving visual acuity, particularly in dim environments (Fig. 1a,b)15,40,41,42,43,44,45. Notably, the initial appearance of iridophore crystals in zebrafish larvae occurs around 44 hours post fertilization (hpf)46. This timing makes the zebrafish an excellent model for studying guanine accumulation and subsequent crystal formation.

a, Incident light image of zebrafish larva at 5 days post fertilization (dpf), showing guanine crystals in the eyes and skin. Inset, higher-magnification view of the eye region (highlighted in pink). b, Confocal microscopy images of a 96 hpf zebrafish larval eye showing mCherry-labeled iridophores driven by the pnp4a promoter, a known iridophore marker45 (Tg(pnp4a:PALM-mCherry), red). Guanine crystal reflectance is shown in white. Inset, higher-magnification view of a single iridophore (yellow frame); cell boundaries are marked with a cyan dashed line. c, A 3D surface rendering of representative cryo-FIB-SEM data showing a portion of a single eye iridophore at 72 hpf. Nanometer-scale membrane-bound organelles are segmented in yellow and guanine crystals are segmented in blue. ER, endoplasmic reticulum. Field of view: 9.3 × 12.4 μm, with a depth of 2.7 μm. In total, dozens of cells from four different fish were imaged across multiple cryo-FIB-SEM sessions. d, Cryo-TEM micrographs of iridosomes at different maturation stages from zebrafish larvae. Top and inset, TEM images. Middle, reconstructed slice from a cryo-electron tomogram.
Early iridosomes hold amorphous nitrogen-rich material
To capture the initial stages that take place before crystal formation within their natural context, we examined eye iridophores in zebrafish larvae in situ using cryogenic focused ion beam scanning electron microscopy (cryo-FIB-SEM). This approach revealed numerous membrane-bound organelles and vesicles ranging from 50 to 500 nm in diameter, located adjacent to iridosomes at various developmental stages (Fig. 1c, Supplementary Fig. 1 and Supplementary Video 1). To improve visualization of these compartments and their contents, we used cryogenic transmission electron microscopy (cryo-TEM). This was achieved by rapidly plunge-freezing fluorescence-activated cell sorting (FACS)-sorted iridophores (Methods and Supplementary Fig. 2) in liquid ethane, preserving them in near-native conditions and enabling higher-resolution imaging. Through this technique, we identified a population of membrane-bound compartments with sizes similar to those observed by cryo-FIB-SEM, further supporting their presence in iridophores. Notably, some of these membrane-bound compartments exhibited different levels of contrast, suggesting the presence of electron-dense material akin to that found in the crystals (Supplementary Fig. 1). Using this approach, we were also able to capture crystal maturation, observing that crystals first appear in organelles smaller than 100 nm and progressively grow to reach several microns in length (Fig. 1d).
Because of the small size of the electron-dense intracellular membrane compartments, studying them using classical light microscopy-based imaging or light-based spectroscopy is not feasible. Thus, to further characterize these membrane-bound compartments, we used cryogenic scanning transmission electron microscopy coupled with energy-dispersive X-ray spectroscopy (cryo-STEM-EDS) and electron diffraction (cryo-ED). This approach, applied to isolated plunge-frozen cells, enabled high spatial and spectral resolution (Fig. 2a). Our EDS analysis revealed that relatively mature iridosomes, which already contained guanine crystals, were notably rich in nitrogen (Fig. 2c), consistent with the chemical composition of guanine, where nitrogen accounts for five of its 16 atoms (Supplementary Fig. 3a,b). Interestingly, we also observed that some high-contrast vesicular organelles exhibited significant nitrogen enrichment (Fig. 2a,c).

a, Cryo-STEM dark-field image of guanine crystals and organelles from the edge of an isolated iridophore of a 72 hpf larva. In total, 23 cells isolated from dozens of larvae were imaged and analyzed. Right, cryo-STEM-EDS maps of nitrogen (green), carbon (red) and oxygen (purple). b, ED patterns from the corresponding locations (1–3) in the dark-field image, including intracellular organelles (1, pink; 3, orange) and a crystal (2, blue). c, EDS spectra corresponding to the marked ROIs in a: intracellular organelles (pink and orange frames) and a crystal (blue frame). Inset, closer view of the 0.2–0.6 keV region. d, Graph illustrating the N/C ratio, comparing organelles (n = 61 from 23 cells, orange) and crystals (n = 22 from 6 cells, blue; two-sided F-test, F60,21 = 3.4517, P = 0.002618). The mean values ± s.d. are shown on the graph.
To further investigate this, we calculated the nitrogen-to-carbon (N/C) ratio for the different organelles (Fig. 2d). While the N/C ratio of membrane-bound crystals averaged around one (N/C = 1.07 ± 0.12, n = 22 crystals from 6 different cells), as expected, the organelles that did not contain crystals displayed a much broader range, spanning from well below one to approximately one (N/C = 0.50 ± 0. 23, n = 61organelles from 23 different cells; F-test, F60,21 = 3.45, P < 0.001). Lastly, we note that the oxygen signal detected in the EDS spectra originates from the surrounding aqueous environment, as the data were collected under cryogenic conditions, preserving the sample in a near-native hydrated state.
In parallel, to further correlate morphological information with crystallographic features, we investigated the high-contrast organelles using cryo-ED (Fig. 2b and Supplementary Fig. 4). The diffraction pattern of the crystals corresponded to the bc plane of anhydrous β-guanine47 (Supplementary Figs. 3a and 4d–f). On the contrary, the high-contrast, nitrogen-enriched organelles exhibited no diffraction spots, displaying a pattern typical of amorphous compounds, despite the high sensitivity of this method for detecting nanometer-sized crystals (Fig. 2b and Supplementary Fig. a–c). These findings suggest that, like the crystals, these organelles also contain guanine or another nitrogen-rich substrate. However, unlike the more mature membrane-bound crystals, the material within the studied organelles does not exhibit the organized structure characteristic of guanine crystals.
Protonated guanine accumulates in early iridosomes
To elucidate the composition of the nitrogen-rich material in the intracellular membranous organelles and to further investigate the connection between nitrogen accumulation and guanine crystallization, we used a combination of synchrotron-based cryogenic soft X-ray tomography (cryo-SXT) and X-ray absorption near edge structure (XANES). The correlative use of these advanced techniques facilitates the visualization of entire isolated cells while concurrently allowing for chemical characterization with high spatial and spectral resolution using XANES microscopy48,49 (Fig. 3a) of plunge-frozen isolated iridophores at various stages of maturation (Fig. 3b,e). This was achieved by performing tilt series within the ‘water window’ energy range. Within this energy range, carbon and nitrogen exhibit high absorption levels, whereas oxygen and, consequently, water is almost transparent. This results in carbon-rich and nitrogen-rich substances appearing darker in transmission images, whereas the water-abundant cytosol appears lighter (Supplementary Video 2). Moreover, X-ray attenuation properties allow the acquisition of data from samples up to 15 µm thick, permitting the imaging of whole cells that have been isolated from larvae. The reconstructed tomograms reveal various cellular compartments with enhanced contrast compared to the cytoplasm, including the cell membrane, nucleus, guanine crystals and diverse membranous organelles potentially containing nitrogen-rich material (Fig. 3b–g and Supplementary Video 3).

a, Schematic illustration of the experimental design and data collection process using (cryo-SXM), which enables correlated cryo-SXT and XANES. b,e, Cryo-SXM images of single cell from isolated zebrafish larvae iridophores at 1 mpf (b) and 72 hpf (e). Single-cell boundaries are labeled with cyan dashed line. In total, 25 different cells isolated from dozens of larvae were imaged and analyzed. c,f, Two-dimensional volume slices from a reconstructed cryo-SXT of a region within the 1 mpf cell (c) and 72 hpf cell (f), showing multiple crystals and organelles. d,g, XANES 2D heat maps showing the distribution of nitrogen levels within a 1 mpf cell (d) and 72 hpf cell (g).
We proceeded to gather two-dimensional (2D) projection images of iridophores within the nitrogen K-edge energy range of 395–420 eV, where nitrogen absorption is at its peak (Supplementary Video 4). Following data acquisition, the XANES images were refined through flat field normalization and filtering processes (Methods). This treatment resulted in detailed 2D nitrogen maps that highlight areas of concentrated nitrogen. In the analysis of cells from larvae 1 month after fertilization (mpf), where crystallization is predominantly complete, we primarily observed mature, elongated crystals (Fig. 3b–d). In contrast, cells from 72 hpf larvae revealed smaller crystals in close proximity to nitrogen-rich organelles (Fig. 3e–g). The abundance of nitrogen-rich organelles in 72 hpf samples was key in uncovering the chemical identity and characteristics of nitrogen in these organelles. We derived this information from the XANES spectra corresponding to various regions of interest (ROIs) within the nitrogen maps. Notably, the spectrum associated with guanine crystals displayed two distinct peaks at 399 eV (π*1) and 401 eV (π*2)50. These peaks represent the excitation of guanine nitrogen’s 1s electrons to π* orbitals. Additionally, a broader peak around 407 eV was observed, indicative of the 1s to σ* orbital transitions on the different nitrogen sites in the guanine molecule (Fig. 4a).

a, A cryo-XANES spectra of the nitrogen K-edge of a crystal (blue) and organelle (orange). b,c, Swarm graphs showing the ratio between π*2 and π*1 peaks of crystals (blue) and organelles (orange), using normal radiation dose (n = 143 organelles and n = 192 crystals from 25 different cells) (b) and using minimal radiation dose (n = 36 organelles and n = 52 crystals from 5 different cells) (c). Each data point represents an ROI within the XANES measurement field of view. The mean ± s.d. is indicated by horizontal bars overlaid on the swarm plot. Differences in ratios between categories were tested with a two-sided linear mixed-effects model, with category as a fixed factor and sample ID (cell number) as a random factor; P = 2 × 10−16 in b and P = 3.32 × 10−5 in c.
Notably, the darkly contrasted intracellular membranous organelles also displayed an X-ray absorption pattern akin to that of guanine, suggesting guanine as their primary component (Fig. 4a). However, a more detailed analysis of the organelles’ absorption spectra revealed distinct variances when compared to the spectra of guanine crystals (Fig. 4a–c). Specifically, the π*1 absorption peak of the organelles (3.18 ± 0.63, n = 143 organelles from 25 different cells) was significantly broader than the crystals (1.82 ± 0.67, n = 192 crystals from 25 different cells; t-test, P < 0.001) (Supplementary Fig. 5). This suggests a less ordered molecular arrangement of guanine within these organelles, distinct from its ordered arrangement in the crystals (Fig. 4a).
By overlaying the spectra of several hundred crystals and contrasting them with a similar number of organelle spectra, a clear discrepancy in the ratios of the π*1 and π*2 peaks was observed.
After normalizing these spectra, we calculated the ratio of the π*1 and π*2 peaks and found that, on average, this ratio for the crystals was 0.85 ± 0.16 (n = 192 crystals from 25 different cells), whereas, for the organelles, it was significantly higher, at 1.17 ± 0.18 (n = 143 organelles from 25 different cells; t-test, P < 0.001) (Fig. 4b).
Building on the finding that guanine aggregates in the organelles appear less ordered, we hypothesized that the X-ray absorption of the highly anisotropic guanine crystals would be markedly influenced by their orientation relative to the beam, whereas the absorption in organelles would be comparatively unaffected. To investigate this, we conducted absorption measurements on both organelles and crystals during sample tilting. Our results confirmed that the absorption by the crystals was significantly dependent on their alignment with the beam (Supplementary Fig. 6a–d). This effect was especially pronounced when the planar crystals were oriented perpendicular to the beam, when we observed a substantial reduction in their π* peaks in parallel to an increase in their σ* peak (Supplementary Fig. 6c,d). In contrast, the organelles’ absorption remained relatively stable regardless of the tilting angle (Supplementary Fig. 6e–g). These findings further support the idea that the guanine molecules in the organelles are in a less ordered state.
In the course of conducting tilt series experiments, we noticed a notable change; extended radiation exposure seemed to modify the absorption spectrum of the crystals and the organelle in a different manner. While the absorption intensity of the organelle was reduced, the one of the crystals seems to increase with extended radiation exposure, further suggesting that the structural organization of the guanine within the organelle might be different than the crystalline guanine. To further study this observation, we embarked on a more detailed investigation into the impact of radiation. We exposed both crystals and organelles to repeated radiation and carefully observed the subsequent alterations in their absorption properties. For organelles, the impact was straightforward; the amplitude of both π* and σ* absorption peaks diminished with increased radiation exposure (Supplementary Fig. 7a–d). However, the response in crystals was distinctly different. While the σ* peak’s amplitude decreased because of radiation, the amplitudes of the π*1and π*2 peaks in the crystals surprisingly increased (Supplementary Fig. 7a–d). We speculate that this increase in π* absorption could be because of radiation damage that induces bond breaking and an overall reduction in electron density, thus altering the transition probability into antibonding π states in the XANES signal of nitrogen. This may change the XANES fine structure. It is also possible that the breaking of bonds between the molecular sheets allows molecules to rotate more freely thus affecting their orientation to the X-ray beam.
To investigate whether the π*2-to-π*1 peak ratio differences between crystals and organelles arose solely from differential responses to beam damage, we performed experiments under minimal radiation conditions, ensuring that no visible damage occurred during a single scan (Methods and Supplementary Fig. 8a,b). We measured both crystals and organelles using a minimal-dose radiation approach (Supplementary Fig. 8c–e). Under these conditions, the difference in the π*2-to-π*1 ratios between crystals and organelles was less pronounced. Nevertheless, both the π*2-to-π*1 ratios and their spread were significantly larger in the organelles (2.54 ± 1.14, n = 36 organelles from 5 different cells) compared to the crystals (1.75 ± 0.53, n = 52 crystals from 5 different cells; t-test, P < 0.001) (Fig. 4c).
Considering that the organelles’ orientations do not affect their peak ratios, these observations suggest that factors beyond mere molecular ordering influence the π*2-to-π*1 ratio. Previous studies on the XANES characteristics of guanine indicated that different protonation states of guanine show different absorption profiles51. Specifically, it was shown that protonated guanine exhibits an elevated π*2-to-π*1 ratio compared to the neutral form. To further test this hypothesis, and compare aspects of the expected spectra of neutral and protonated forms of guanine, we conducted a series of density functional theory (DFT) calculations for the density of empty states. The results, shown in Supplementary Fig. 9a,b, compare the calculated density of states (DOS), projected onto nitrogen atoms, of a gas-phase guanine molecule and a gas-phase N7 protonated guanine molecule (we restricted our attention to the DOS, as opposed to a full simulation of XANES data, to avoid controversies arising from different levels of theoretical treatment of core holes). Considering the ratio between the first two peaks as a proxy for the π*2-to-π*1 ratio, we find that it was ~1.2 for the isolated neutral guanine molecule but ~1.75 for the protonated form. This supports the experimental findings on the difference between the ratio of π*1 and π*2. Thus, the differences in the π*2-to-π*1 ratio observed for the organelles could indeed suggest that the guanine within the organelle is protonated. Because guanine has a pKa of ~3.3 (refs. 20,52,53) (Supplementary Fig. 3b), protonated guanine should only be present in large amounts in an acidic environment. In Supplementary Fig. 3c, we present a schematic illustration of the lowest unoccupied molecular orbital (LUMO) of the guanine molecule, which helps visualize some of the π orbitals.
Early iridosomes are acidic and neutralize as they mature
To test whether the iridosomes contain an acidic environment, we performed confocal imaging with LysoTracker and LysoSensor dyes54,55 known to selectively stain acidic intracellular compartments such as lysosomes. Our observations during the early developmental stages (~48 hpf), a period characterized by active crystal formation before the crystals reach full maturity, revealed strong LysoTracker and LysoSensor staining of iridosomes (Supplementary Fig. 10), corroborating the acidity of these organelles. Importantly, within the same cell at early developmental stages, iridosomes at varying levels of maturation exhibited differential LysoSensor staining, with smaller, less mature iridosomes displaying more pronounced staining (Fig. 5a and Supplementary Fig. 10).

a, Confocal microscopy images of several iridophores within zebrafish larvae eye at 96 hpf stained with LysoSensor (LysoS), which exhibits increased fluorescence in acidic environments. Top, maximum intensity projection image from a z stack showing iridophore intracellular acidic compartments (green) and crystals (white) within the iridophore. Right, zoomed-in view. Bottom, heat map representation of a single plane of the z stack. b, Box-and-whisker plot showing the fluorescent intensity of ROIs from small <1 μm2 iridosomes (n = 505, green) compared to medium 1–5 μm2 (n = 769, pink) and >5 µm2 iridosomes (n = 601, blue). The mean values ± s.d. are shown. The ROI mean values were compared using a two-sided linear mixed model, with classification group as a fixed factor and fish ID (n = 4) and crystal ID (n = 120) as random factors. P = 0.0231276 for 1–5 μm2 versus <1 μm2; P = 0.0000004 for <1 μm2 versus >5 μm2 and P = 0.0043115 for >5 μm2 versus 1–5 μm2. The horizontal lines mark the maximum, median and minimum values of the data, boxes mark the upper and lower quartiles and data points beyond this range are plotted as outliers. c, Graphs depicting the reflectance coverage area (top) and the Lysotracker fluorescence signal (bottom) in the eyes of larvae (blue) treated with bafilomycin A1 (n = 9) and lansoprazole (n = 10) compared to control larvae (purple) treated with DMSO (n = 10 and 11, respectively). The mean values ± s.d. are shown. Each data point represents an independent biological replicate. Statistical analysis was performed using a two-way ANOVA, with crystal reflectance and fluorescence coverage areas log-transformed before analysis. P = 1.83 × 10−15 for bafilomycin reflectance, P = 3.02 × 10−12 for lansoprazole reflectance, P = 4.2 × 10−14 for bafilomycin fluorescence and P = 6.27 × 10−5 for lansoprazole fluorescence compared to DMSO control. d,e, Confocal microscopy z-stack images of eye iridophores from 72 hpf zebrafish larvae treated with DMSO (control) or treated with 50 nM bafilomycin A1 (d) or 250 μM lansoprazole (e). Insets, higher-magnification views of areas within an iridophore. Lysotracker staining (LysoT, magenta) and crystal reflectance (Ref, white) are shown.
We then systematically quantified the LysoSensor fluorescence of membrane-bound iridosomes containing crystals at different developmental stages across multiple larvae (n = 4 different larvae) and found a consistent trend; smaller iridosomes exhibited stronger signals, which gradually decreased as crystal size increased (Fig. 5b, Supplementary Fig. 11 and Methods). Crystals smaller than 1 μm2 showed the highest average signal (1731.77 ± 671.50; n = 59 crystals, 505 ROIs), which was significantly higher than that of crystals measuring 1–5 μm2 (1,393.14 ± 547.22, n = 37 crystals, 769 ROIs; linear mixed-effects model, P < 0.05), which in turn was significantly higher than crystals larger than 5 μm2 (934.43 ± 432.96, n = 24 crystals, 601 ROIs; linear mixed-effects model, P < 0.01). Data were collected from six different cells from four different larvae. These findings collectively reinforce our earlier results, suggesting that iridosomes are initially acidic, the organelle’s pH gradually neutralizes as it matures and crystal formation ensues.
Disrupting pH variations hinders crystal formation
To further investigate the role of pH regulation in guanine crystal formation, we treated zebrafish larvae with bafilomycin A1, an inhibitor of vacuolar-type ATPase (V-ATPase)44, at early developmental stages (60–72 hpf). Treated larvae showed a marked reduction in LysoTracker staining (2.98% ± 0.56%, n = 12 larvae) compared to controls (12.14% ± 1.76%, n = 14 larvae; two-way analysis of variance (ANOVA), P < 0.001), indicating disrupted pH regulation. In parallel, a significant decrease in crystal quantity was observed in treated larvae (1.93% ± 0.62%, n = 12 larvae) compared to untreated controls (12.71% ± 1.23%, n = 14 larvae; two-way ANOVA, P < 0.001) (Fig. 5c,d).
To determine whether bafilomycin A1 affects only the formation of new crystals or also impacts existing ones, we treated larvae at 132–144 hpf, a stage when many crystals had already formed. We found no significant difference in total crystal quantity between treated larvae (19.58% ± 1.73%, n = 9 larvae) and controls (20.87% ± 1.29%, n = 10 larvae; two-way ANOVA, P > 0.05), suggesting that bafilomycin A1 specifically inhibits the formation and growth of developing crystals while leaving mature crystals largely unaffected (Supplementary Fig. 12a–e).
Although bafilomycin A1-treated larvae remained viable, a systematic analysis of their standard length56 revealed a developmental delay (Supplementary Fig. 13a). To determine whether the observed reduction in crystal growth was specifically because of impaired pH regulation rather than a general developmental delay, we tested lansoprazole, which is known to interfere with organelle pH homeostasis57.
Larvae treated with lansoprazole showed a significant reduction in LysoTracker staining (6.24% ± 1.27%, n = 10 larvae) compared to control larvae (12.99% ± 1.59%, n = 11 larvae; two-way ANOVA, P < 0.001) and a marked decrease in crystal quantity (2.84% ± 0.57%, n = 10 larvae) relative to controls (12.38% ± 2.10%, n = 11 larvae; two-way ANOVA, P < 0.001), without any detectable effect on overall larval development (Fig. 5c,e and Supplementary Fig. 13b).
Similarly, larvae treated with chloroquine, a compound known to raise lysosomal pH and disrupt organelle acidification58,59, exhibited significantly decreased crystal formation (6.56% ± 3.19%, n = 24 larvae) compared to controls (10.91% ± 4.67%, n = 19 larvae; two-way ANOVA, P < 0.001), with a minor impact on the overall larvae developmental progression (Supplementary Fig. 13c,d).
Together, these results indicate that the observed effects on crystal formation are not because of general developmental delays but rather reflect the importance of organelle pH regulation. They further support a model in which pH dynamics within iridosomes have a critical role in enabling guanine crystal formation.
Discussion
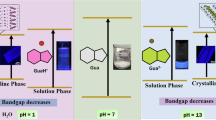
The precise formation of guanine crystals within iridophores depends on the regulation of guanine solubility. In our study, we demonstrated that, in iridophores, this is accomplished by regulating the microenvironment within the iridosome, the organelle in which crystals form. At first, these organelles are highly acidic, which likely allows soluble guanine to accumulate in a protonated form. As iridosomes mature, they transition toward a more neutral pH, prompting the deprotonation of guanine, which may trigger crystallization. Additional gradual increase in pH could promote crystal growth by further shifting the equilibrium toward neutral guanine, supplying the growing crystals with additional building blocks (Fig. 6).

A proposed model for guanine accumulation and crystal formation. Initially, the iridosome is highly acidic, allowing soluble guanine to accumulate in a protonated form. As the iridosome matures, its pH gradually transitions toward neutrality, prompting the deprotonation of guanine, which triggers crystal nucleation. The continued gradual increase in pH promotes crystal growth by shifting the equilibrium toward neutral guanine, providing additional building blocks for the growing crystals.
We first showed that zebrafish larval iridophores are populated with numerous nitrogen-rich organelles, measuring between 100 and 500 nm, alongside small guanine crystals. Cryo-XANES analysis correlated with cryo-SXT three-dimensional (3D) imaging revealed that the nitrogen-rich material within these organelles is guanine. However, the X-ray absorption characteristics of the organelles, namely, their markedly low dependence on orientation and their response to beam damage, suggest that the guanine within them lacks the ordered structure observed in the crystals. This was further substantiated by the absence of ED patterns from the organelles, affirming the noncrystalline nature of the encapsulated guanine.
Guanine is particularly insoluble in aqueous solutions and, under ambient conditions, there is a strong driving force toward crystallization. Thus, the presence of concentrated amorphous guanine within organelles can only be obtained under conditions that increase its solubility and inhibit its crystallization20. The mechanisms that enable the trafficking and accumulation of insoluble guanine within the intracellular organelles and the driving force behind crystallization have yet to be illuminated. By applying a multidisciplinary approach integrating state-of-the-art cryo-electron microscopy, cryo-spectroscopy, live-cell imaging and pharmacological perturbations, we show that the organelle microenvironment is acidic and that, during organelle maturation and crystal formation, the pH of the organelle gradually neutralizes. This suggests that pH variations are important for the overall maturation of the organelle and the formation of the guanine crystals within it.
Firstly, the elevated π*2-to-π*1 ratios we observed for the organelles compared to the crystals suggest that the organelles are acidic. The spread of π*2-to-π*1 ratios exhibited by different organelles even when using a minimal-dose radiation approach suggests that organelles at different maturation stages have different pH values. Using live-cell imaging, we further demonstrated that early crystals containing iridosomes are highly acidic and that, during maturation, they gradually become less acidic, which may further promote the growth of the guanine crystals within them.
It was previously suggested that iridosomes, like other pigment organelles such as melanosomes, are LROs18,23. One of the characteristics of LROs is that pH variations often have a crucial role in their maturation and function24,29,30,31,32.
Our findings that the iridosomes are acidic as well further support this classification, although additional studies looking at characteristic molecular markers are needed to further confirm this point. Recent studies illustrated that guanine crystals develop through the templated nucleation of thin leaflets on preassembled scaffolds34,39. Studies on other functional amyloids related to LROs indicated that pH fluctuations can reversibly influence amyloid assembly60. Thus, the initially acidic environment within the iridosome may facilitate the formation of amyloid fibers34,39, which could template the heterogeneous nucleation of the guanine crystals. The subsequent neutralization of pH as the organelle matures and the crystals grow may lead to the disassembly of these amyloid structures, offering a potential reason for their absence in developed organelles.
Lastly, we found that impeding pH variations, through different pharmacological perturbations, reduced the acidity of the iridosomes and led to a significant decrease in crystal formation. Prior investigations into the iridophores’ proteome and transcriptome revealed the upregulation of several V-ATPase genes, including tcirg1a (ref. 61), tmem179b, atp11a and atp6v1f (ref. 35), suggesting the involvement of multiple proton pumps in regulating iridosome acidity. This indicates a complex mechanism underlying the precise adjustment of the iridosome’s microenvironment. Future research will be essential to pinpoint the specific proton pumps responsible for this regulation in iridophores.
In conclusion, our findings provide insights into the molecular mechanisms that enable guanine formation within iridosomes, highlighting the important role of luminal pH variations in the orchestration of biogenic crystal formation.
Methods
Ethics and safety statement
This study followed the Weizmann Institute of Science Animal Care and Use Committee (protocol number 02000223-1).
Zebrafish husbandry and handling
Zebrafish (D. rerio) were housed at ∼28 °C with a 14-h light and 10-h dark cycle and fed with Artemia and flake food62. Fish were maintained and fed following the standard protocols. Crosses were performed with 3–12-month old wild-type AB strain adults. Embryos were kept in E3 zebrafish embryo medium at 28 °C until reaching the desired developmental stage (48 hpf–1 mpf). The published transgenic line used was Tg(pnp4a:PALM-mCherry)wprt10Tg (RRID: ZDB-FISH-210414-18)45.
Cell dissociation
Cell dissociation was carried out by customizing a protocol described previously34,35. Fish were anesthetized with tricaine and immersed in TrypLE Express (Invitrogen, 12604039). Fish were incubated at 37 °C and shaken at 200 rpm for 1 h, followed by mechanical disruption with a Pasteur pipette to further dissociate the cells. Cells were then strained through a 40 µm cell strainer with HL-15 buffer (Hanks’ balanced salt solution 40% (Sigma H8264) and 60% Leibovitz’s L-15 medium (Gibco 21083-027)) and centrifuged. Dissociated cells were then pelleted at 3,500 rcf for 5 min at 4 °C and then resuspended in 5 ml of fresh HL-15 with 1% FBS.
FACs sorting
Cell isolation was carried out by customizing a protocol described previously34,35. Cells were isolated from dozens of Tg(pnp4a:PALM-mCherry)-positive larvae and sorted using FACS. Following resuspension in 5 ml of cold HL-15 with 1% FBS, the isolated cells were incubated with Hoescht to mark the nuclei for 30 min before FACs. Cells were analyzed and sorted using a BD FACSAria III cell sorter with a 100 µm nozzle. Cells were illuminated using a both 405 nm and 561 nm lasers. Cells were gated on the basis of attributes to separate cells from each other and from cellular debris. Cellular debris were detected using forward scatter, side scatter and Hoescht signals to select against the smallest particles (1 mm or less). Cells were additionally sorted and enriched based on detection using 561 nm filters, corresponding to the pnp4a:PALM-mCherry signal (Supplementary Fig. 2). Cells were collected into ice-cold HL-15 medium with 1% FBS and kept on ice until mounted on grids for downstream imaging.
Live-cell imaging
Live-cell imaging was carried out on an inverted Zeiss LSM 980 and 900 confocal microscopes. Crystals were imaged with reflection. For LysoSensor imaging, live larvae were incubated in 1 µM LysoSensor green DND-189 (Invitrogen, L7535) for 60 min in the dark in 28 °C, washed three times with E3 zebrafish embryo medium and mounted in agarose for imaging. For bafilomycin A1 and lansoprazole, imaging and quantification were performed using 3–4 larvae per condition across three independent experiments. Larvae were incubated with 50 nM bafilomycin A1 (Sigma, B1793), 250 µM lansoprazole (Sigma, L8533) or an equivalent volume of DMSO as a control starting at 60 hpf until imaging at 72 hpf. After bafilomycin A1 or lansoprazole57 treatment, larvae were incubated for 90 min with 10 µM Lysotracker (Invitrogen, L12492) in the dark, washed three times with E3 zebrafish embryo medium and mounted in agarose for imaging. Chloroquine imaging and quantification were performed using 3–6 larvae per condition across three independent experiments. Larvae were exposed to 10 mM chloroquine (Sigma, C6628) in E3 zebrafish embryo medium from 6 hpf to 72 hpf63 (solution was changed daily) and then mounted in agarose for imaging.
For confocal imaging quantifications, z-stack images of the eye area with crystals and LysoTracker staining (1.25 µm each slice) were taken using A ×20 magnification lens. Whole-larva imaging for standard length measurements was performed using a ×2.5 lens.
High-pressure freezing, freeze fracture and cryo-FIB-SEM
High-pressure freezing, freeze fracture and cryo-FIB-SEM were performed by adapting a previously described protocol34. Dozens of cells from four different zebrafish larvae were imaged across multiple cryo-FIB-SEM sessions. Zebrafish larvae at 48–96 hpf were anesthetized with tricaine and cryoimmobilized using a Leica LM ICE high-pressure freezing device (Leica Microsystems).
The samples were then fractured and coated with a 6 nm platinum layer using a freeze-fracture device (BAF 60, Bal-Tec), with the stage temperature maintained at −160 °C under a vacuum of approximately 5 × 10−7 mbar. The coated sample was subsequently transferred to the cryo-FIB-SEM (Crossbeam 550, Zeiss) using a vacuum cryotransfer device (VCT 100, Leica Microsystems).
Rough milling was performed before imaging to expose the ROI, using a 30 kV FIB acceleration voltage and a 1.5 nA FIB probe current. Fine milling was conducted at a 5-mm working distance, a 30 kV FIB acceleration voltage and an FIB probe current ranging from 100 to 700 pA. Throughout the process, the cryostage temperature was maintained below −160 °C and the chamber vacuum remained at approximately 5 × 10−7 mbar.
The dataset shown in Fig. 1c was acquired from a 72 hpf larva. The imaged volume dimensions were 10 µm × 7.5 µm, with a slice thickness of 10 nm. Imaging was performed with a lateral image pixel size of 6 nm, a field of view of 9.3 × 12.4 μm and a 2 kV SEM acceleration voltage.
Cryo-FIB-SEM 3D image processing
Cryo-FIB-SEM 3D image processing was carried out by customizing a protocol described previously34. A wavelet decomposition algorithm as described by Spehner et al.64 was applied to correct vertical stripe artifacts in the image stacks. Specifically, we used a coif wavelet with N = 10 and a vertical coefficient σ = 10. This method was selected because it better preserves the overall image intensity compared to a simple Fourier filtering approach.
Following this, local charge imbalances in the images were corrected using a morphological reconstruction (dilation) approach according to the method described by Spehner et al.64. The processing pipeline was implemented in Python 3.7 to ensure efficient execution of these corrections.
Once preprocessing was complete, the images were automatically aligned using MIB software (Microscopy Image Browser, University of Helsinki Institute of Biotechnology Electron Microscopy Unit)65. To achieve further refinement, manual alignment was subsequently performed using the alignment tool in Amira3D software (Thermo Scientific), ensuring optimal registration of the image stack.
Segmentation
Segmentation and 3D representation of the reconstructed data were conducted using Amira software (Thermo Scientific). Data segmentation was performed on the basis of contrast variations following the unique shape and structure of each component.
Plunge-freezing
Plunge-freezing was carried out by customizing a protocol described previously34. FACS-sorted iridophore cells (3.5 μl) were mixed with 1 μl of gold beads (15 or 150 nm) and applied to glow-discharged holey carbon R2/2 Cu 200-mesh SiO2 grids (Quantifoil) coated with type I collagen (rat tail, EMD Millipore 08-115) to promote cell adherence. The grids were then blotted and vitrified by rapid plunging into liquid ethane using a Leica EM GP automatic plunger, under controlled conditions of 4 °C and 90% humidity. The frozen grids were stored in liquid nitrogen until further use.
Cryo-electron tomography
Cryo-electron tomography data collection was carried out by customizing a protocol described previously34. Data were collected on a Titan Krios TEM G3i (Thermo Fisher Scientific) equipped with a BioQuantum energy filter with a K3 direct electron detector (Gatan). Datasets were collected at 300 kV with the K3 camera (counting mode) using the Thermo Fisher tomography software. The TEM magnification corresponded to a camera pixel size of 1.6 Å (K3 counting mode) and the target defocus was set between 3 and 5 μm. The total dose for a full tilt series was 120 electrons per Å2. Tomogram tilt series were collected using the dose-symmetric scheme, ±60° at 2° steps. The tilt series images alignment and reconstruction were performed in IMOD66.
Cryo-STEM
Cryo-STEM was carried out by customizing a protocol described previously34. STEM images and analytical EDS maps were acquired in a double-aberration-corrected Themis-Z microscope (Thermo Fisher Scientific) at an accelerating voltage of 200 kV. All measurements were completed on fully vitrified plunge-frozen samples in a Gatan 914 cryotransfer holder. STEM images were recorded using a Fischione Model 3000 detector (Thermo Fisher Scientific) and a bright-field detector. EDS hyperspectral data were obtained with a Super-X SDD detector and quantified with the Velox software (Thermo Fisher Scientific) through background subtraction and spectrum deconvolution.
The four-dimensional (4D) STEM67 datasets were obtained on the electron microscope pixel array detector68, which allowed for the rapid collection of unsaturated diffraction patterns with single-electron sensitivity. An electron probe with a convergence angle of 0.2 mrad was adjusted and further defocused to a diameter of typically 50 nm to reduce the electron fluence. The defocused electron beam was rastered with beam overlapping across the sample. A primary beam current of 3 pA resulted in a total fluence of 0.2 e− per Å2 for Supplementary Fig. 4a–e and 1.8 e− per Å2 for Supplementary Fig. 4f. All 4D STEM datasets, acquired from 23 cells isolated from dozens of larvae, were imaged and analyzed using custom-written software.
Cryo-SXT and XANES
X-ray imaging was performed at the MISTRAL beamline (ALBA Synchrotron)69. Initially, a tilt series at 520-eV X-ray energy was collected to allow 3D volume reconstruction of cells and their internal structures. At this energy, nitrogen-rich bodies are highly absorbing. The tilt series typically consisted of 141 images taken at 1° intervals. The exposure time was 2 s to optimize signal-to-noise level while minimizing radiation damage. No radiation damage was observed using this exposure time at the achieved spatial resolution. The datasets were acquired using a 40 nm zone-plate objective lens. The effective pixel size in the projection images was 13 nm. The projection images of the tilt series were normalized using the flat field (average of ten images with no sample, collected at a 1 s exposure time) to take into account the intensity distribution delivered to the sample by the capillary condenser lens. The alignment and reconstruction of the tomographic projections was performed in IMOD66 using gold nanoparticles as fiducial markers.
Energy scan at the nitrogen K-edge was acquired by imaging the same field of view at varying X-ray energies. Specifically, the acquisition was performed in the energy range from 395 to 420 eV with a variable energy step (0.5 and 0.1 eV). Each image was taken with a 2-s exposure time. Low-dose control dataset series were collected as follows: 394–397 eV with a step size of 1 eV, 399.4–403 eV with a step size of 0.3 eV and 404–408 eV with a step size of 2 eV. A total of 17 points were acquired with a 1 s exposure time for each. An energy scan at different tilt angles was performed using the same scheme, with steps of 10° between each tilt angle.
XANES analysis
To obtain the nitrogen 2D distribution maps (Fig. 3d,g), a signal-to-noise ratio criterion was applied on each pixel in the image70. At resonant energies, the nitrogen absorption increases orders of magnitudes while the other elements absorb almost the same, this allows to define the signal s as the absorbance variation between its average value in the energy range of 406–409 eV (absorption maxima) and its average value in the pre-edge energy region (390–396 eV) and the noise n as the s.d. in the pre-edge energy region. Pixels that did not satisfy the condition s/n > 1 were considered free of nitrogen (white pixels the corresponding map) and filtered out before spectrum extraction.
Basic preprocessing to transform data to negative log absorption was performed using ImageJ (version 1.51k) and ROIs were selected manually in the program. Further noise reduction and analysis were conducted using Python 3.11 (ref. 71). Data were smoothed using Gaussian filters to better show trends. Visualizations were performed using Matplotlib72 and Seaborn73. Before π* peak analysis, background noise was calculated as a linear fit at the prepeak region and was reduced from the spectra. Analysis of the peaks in the data was performed either naively or using the SciPy signal module. In some cases, a univariate spline from SciPy was used to obtain specific data points. The Python analysis scripts are available from GitHub (https://github.com/Gur-Lab-WIS). A total of 25 FACS-isolated and imaged cells were analyzed using this workflow.
DFT calculations
All calculations were performed using the Vienna ab initio simulation package (version 6.3.0)74, based on the projector augmented-wave method, with the Perdew–Burke–Ernzerhof (PBE)75 functional and an energy cutoff of 700 eV. The calculations were performed using a supercell (30 × 30 × 30 Å) containing a single molecule. Geometries are available in Supplementary Data 1 and 2. The total energy convergence criterion was set to 10−6 eV and forces were relaxed to below 0.005 eV per Å. The electronic occupancies were determined using Gaussian smearing with a width of 0.5 eV (Sigma) and the DOS calculations were performed without symmetry, using 5,000 bands. The LUMO of a single guanine molecule was calculated using Qchem (version 6.2)76 with the PBE functional and the 6-31G* basis set.
LysoSensor fluorescence intensity quantification
The confocal image stack was imported into Amira software (Thermo Scientific) for analysis. For consistency, only crystals that were properly aligned with the imaging plane were selected. These selected crystals were then segmented to measure and calculate their area.
To assess fluorescence intensity, we defined multiple ROIs along both lumen edges of the labeled vesicle containing the crystal lumen, carefully excluding the crystalline regions (Supplementary Fig. 10). The mean grayscale value was extracted from these ROIs to quantify fluorescence intensity.
Using the label analysis tool in Amira, we obtained two key datasets for each crystal: crystal area, measured from the segmented regions, and a list of grayscale values, representing fluorescence intensity from the labeled lumen. This approach ensured precise quantification of both structural and fluorescence parameters while avoiding interference from the crystalline regions. LysoSensor data were collected from a total of four larvae, 120 crystals and 1,875 ROIs.
Reflectance and fluorescence coverage quantification
For the quantification step done on confocal images, a custom graphical user interface application was developed. The application accepts images in the czi format and creates maximal intensity projections over the images, allowing for further processing and visualization. The application then performs an automatic object detection and segmentation step, followed by optional manual annotation, user-specified thresholding and area to area calculation and output. In every step of the workflow, the application allows visualization of the dataset and of the segmentation. It was implemented in Python using OpenCV, PIL, Tkinter/CustomTkinter, Matplotlib, NumPy and Pandas for image processing, visualization and data management and aicspylibczi for handling czi files. The application was specifically designed for the lab and is available from GitHub (https://github.com/Gur-Lab-WIS).
Statistical analysis
Statistical experimental details can be found in the text and in the relevant figure legends. Results are presented as the mean ± s.d. and differences were considered significant if the P value was <0.05 (*), <0.01 (**) or <0.001 (***). Nonsignificant (NS) results had P > 0.05.
No statistical methods were used to predetermine sample sizes but our sample sizes are similar to those reported in previous publications35,48,55. The data met the assumptions of the statistical tests. Equality of variances was tested with Levene’s test, normality of residuals was tested using a quantile–quantile plot and numerically with a Shapiro–Wilk test. Randomization procedures were used throughout the experimental design. Animals or samples were randomly assigned to experimental groups. When applicable, experimental conditions and stimulus presentations were randomized to minimize order effects and experimental bias. Data collection and analysis were not performed blind to the conditions of the experiments.
Differences between categories were tested using linear mixed-effects models, with category as a fixed factor and sample ID as a random factor. In addition, two-way ANOVA and unpaired two-tailed Student’s t-tests were performed where appropriate to compare group means. When applicable, post hoc tests were used to identify significant differences between specific pairs of conditions. Tests were run using the packages ‘lme4’ and ‘emmeans’ in R (version 4.4.2). All statistical tests were two-sided unless otherwise noted.
Statistics and reproducibility
For Fig. 1a,b, a zebrafish larva and a larval eye are depicted, respectively. These images are representative of dozens of independent experiments performed in our lab, all of which produced consistent results. For Fig. 1d, crystal-containing organelles were imaged from cells isolated from hundreds of larvae over multiple cryo-EM sessions, consistently revealing similar structures during development. For Fig. 5a–e, representative fluorescent images are shown from experiments that were independently repeated at least three times, each yielding similar outcomes.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Processed XANES data are available from GitHub (https://github.com/Gur-Lab-WIS). Data are available from the corresponding authors upon request.
Code availability
All scripts and custom-made applications are available from GitHub (https://github.com/Gur-Lab-WIS).
References
Bagnara, J. T. et al. Common origin of pigment cells. Science 203, 410–415 (1979).
Cunningham, J. & MacMunn, C. A. I. I. On the coloration of the skins of fishes, especially of pleuronectidæ. Proc. R. Soc. Lond. 53, 384–388 (1893).
Gur, D., Palmer, B. A., Weiner, S. & Addadi, L. Light manipulation by guanine crystals in organisms: biogenic scatterers, mirrors, multilayer reflectors and photonic crystals. Adv. Funct. Mater. 27, 1603514 (2017).
Rohrlich, S. T. Fine structural demonstration of ordered arrays of cytoplasmic filaments in vertebrate iridophores. J. Cell Biol. 62, 295–304 (1974).
Denton, E. J. & Land, M. Mechanism of reflexion in silvery layers of fish and cephalopods. Proc. R. Soc. Lond. B Biol. Sci. 178, 43–61 (1971).
Lythgoe, J. N. & Shand, J. Changes in spectral reflexions from the iridophores of the neon tetra. J. Physiol. 325, 23–34 (1982).
Denton, E. J. Review lecture: on the organization of reflecting surfaces in some marine animals. Philos. Trans. R. Soc. Lond. B Biol. Sci. 258, 285–313 (1970).
Teyssier, J., Saenko, S. V., van der Marel, D. & Milinkovitch, M. C. Photonic crystals cause active colour change in chameleons. Nat. Commun. 6, 6368 (2015).
Gur, D. et al. Structural basis for the brilliant colors of the sapphirinid copepods. J. Am. Chem. Soc. 137, 8408–8411 (2015).
Palmer, B. A. et al. The image-forming mirror in the eye of the scallop. Science 358, 1172–1175 (2017).
Skold, H. N., Aspengren, S. & Wallin, M. Rapid color change in fish and amphibians—function, regulation, and emerging applications. Pigment Cell Melanoma Res. 26, 29–38 (2013).
Barzilay, Y. et al. Specialized molecular pathways drive the formation of light-scattering assemblies in leucophores. Proc. Natl Acad. Sci. USA 122, e2424979122 (2025).
Land, M. The physics and biology of animal reflectors. Prog. Biophys. Mol. Biol. 24, 75–106 (1972).
Gur, D. et al. The physical and cellular mechanism of structural color change in zebrafish. Proc. Natl Acad. Sci. USA 121, e2308531121 (2024).
Figon, F. & Casas, J.Morphological and physiological colour changes in the animal kingdom. eLS https://doi.org/10.1002/9780470015902.a0028065 (2018).
Gur, D. et al. The mechanism of color change in the neon tetra fish: a light‐induced tunable photonic crystal array. Angew. Chem. Int. Ed. Engl. 54, 12426–12430 (2015).
Odiorne, J. The occurrence of guanophores in Fundulus. Proc. Natl Acad. Sci. USA 19, 750–754 (1933).
Figon, F., Deravi, L. F. & Casas, J. Barriers and promises of the developing pigment organelle field. Integr. Comp. Biol. 61, 1481–1489 (2021).
Levy-Lior, A. et al. Guanine-based biogenic photonic-crystal arrays in fish and spiders. Adv. Funct. Mater. 20, 320–329 (2010).
Gur, D. et al. Guanine crystallization in aqueous solutions enables control over crystal size and polymorphism. Cryst. Growth Des. 16, 4975–4980 (2016).
Guille, K. & Clegg, W. Anhydrous guanine: a synchrotron study. Acta Crystallogr. C Cryst. Struct. Commun. 62, o515–o517 (2006).
Kelsh, R. N. et al. Zebrafish pigmentation mutations and the processes of neural crest development. Development 123, 369–389 (1996).
Ullate-Agote, A. et al. Genome mapping of a LYST mutation in corn snakes indicates that vertebrate chromatophore vesicles are lysosome-related organelles. Proc. Natl Acad. Sci. USA 117, 26307–26317 (2020).
Dell’Angelica, E. C., Mullins, C., Caplan, S. & Bonifacino, J. S. Lysosome-related organelles. FASEB J. 14, 1265–1278 (2000).
Bowman, S. L., Bi‐Karchin, J., Le, L. & Marks, M. S. The road to lysosome‐related organelles: Insights from Hermansky–Pudlak syndrome and other rare diseases. Traffic 20, 404–435 (2019).
Delevoye, C., Marks, M. S. & Raposo, G. Lysosome-related organelles as functional adaptations of the endolysosomal system. Curr. Opin. Cell Biol. 59, 147–158 (2019).
Marks, M. S. & Seabra, M. C. The melanosome: membrane dynamics in black and white. Nat. Rev. Mol. Cell Biol. 2, 738–748 (2001).
Seiji, M., Fitzpatrick, T. B., Simpson, R. T. & Birbeck, M. S. Chemical composition and terminology of specialized organelles (melanosomes and melanin granules) in mammalian melanocytes. Nature 197, 1082–1084 (1963).
Ambrosio, A. L., Boyle, J. A., Aradi, A. E., Christian, K. A. & Di Pietro, S. M. TPC2 controls pigmentation by regulating melanosome pH and size. Proc. Natl Acad. Sci. USA 113, 5622–5627 (2016).
Valentijn, K. M., Sadler, J. E., Valentijn, J. A., Voorberg, J. & Eikenboom, J. Functional architecture of Weibel–Palade bodies. Blood 117, 5033–5043 (2011).
Terglane, J., Menche, D. & Gerke, V. Acidification of endothelial Weibel–Palade bodies is mediated by the vacuolar-type H+-ATPase. PLoS ONE 17, e0270299 (2022).
Canovas, F. G., Garcia-Carmona, F., Sanchez, J. V., Pastor, J. L. & Teruel, J. A. The role of pH in the melanin biosynthesis pathway. J. Biol. Chem. 257, 8738–8744 (1982).
Klumperman, J. & Raposo, G. The complex ultrastructure of the endolysosomal system. Cold Spring Harb. Perspect. Biol. 6, a016857 (2014).
Eyal, Z. et al. Plate-like guanine biocrystals form via templated nucleation of crystal leaflets on preassembled scaffolds. J. Am. Chem. Soc. 144, 22440–22445 (2022).
Deis, R. et al. Genetic control over biogenic crystal morphogenesis in zebrafish. Nat. Chem. Biol. 21, 383–392 (2025).
Gur, D. et al. Guanine-based photonic crystals in fish scales form from an amorphous precursor. Angew. Chem. Int. Ed. Engl. 52, 388–391 (2013).
Seitz, K. A. Elektronenmikroskopische untersuchungen an den Guanin-Speicherzellen von Araneus diadematus clerck (Araneae, Araneidae). Z. Morphol. Tiere 72, 245–262 (1972).
Wagner, A. et al. The non-classical crystallization mechanism of a composite biogenic guanine crystal. Adv. Mater. 34, e2202242 (2022).
Wagner, A. et al. Macromolecular sheets direct the morphology and orientation of plate-like biogenic guanine crystals. Nat. Commun. 14, 589 (2023).
Oshima, N. & Kasai, A. Iridophores involved in generation of skin color in the zebrafish, Brachydanio rerio. Forma 17, 91–101 (2002).
Hirata, M., Nakamura, K.-i, Kanemaru, T., Shibata, Y. & Kondo, S. Pigment cell organization in the hypodermis of zebrafish. Dev. Dyn. 227, 497–503 (2003).
Singh, A. P., Schach, U. & Nusslein-Volhard, C. Proliferation, dispersal and patterned aggregation of iridophores in the skin prefigure striped colouration of zebrafish. Nat. Cell Biol. 16, 607–614 (2014).
Gur, D. et al. The dual functional reflecting iris of the zebrafish. Adv. Sci. 5, 1800338 (2018).
Salis, P. et al. Developmental and comparative transcriptomic identification of iridophore contribution to white barring in clownfish. Pigment Cell Melanoma Res. 32, 391–402 (2019).
Lewis, V. M. et al. Fate plasticity and reprogramming in genetically distinct populations of Danio leucophores. Proc. Natl Acad. Sci. USA 116, 11806–11811 (2019).
Petratou, K. et al. A systems biology approach uncovers the core gene regulatory network governing iridophore fate choice from the neural crest. PLoS Genet. 14, e1007402 (2018).
Hirsch, A. et al. ‘Guanigma’: the revised structure of biogenic anhydrous guanine. Chem. Mat. 27, 8289–8297 (2015).
Kahil, K. et al. Cellular pathways of calcium transport and concentration toward mineral formation in sea urchin larvae. Proc. Natl Acad. Sci. USA 117, 30957–30965 (2020).
Sviben, S. et al. A vacuole-like compartment concentrates a disordered calcium phase in a key coccolithophorid alga. Nat. Commun. 7, 11228 (2016).
Shaked, N. et al. Guanine crystal formation by the unicellular organism Phacotus lenticularis is part of a cellular stress response. PLoS ONE 20, e0316193 (2025).
Shimada, H. et al. Nitrogen K-edge X-ray absorption near edge structure (XANES) spectra of purine-containing nucleotides in aqueous solution. J. Chem. Phys. 141, 055102 (2014).
Darvishzad, T., Lubera, T. & Kurek, S. S. Puzzling aqueous solubility of guanine obscured by the formation of nanoparticles. J. Phys. Chem. B 122, 7497–7502 (2018).
Mason, S. F. Purine studies. Part II. The ultra-violet absorption spectra of some mono- and poly-substituted purines. J. Am. Chem. Soc. 0, 2071–2081 (1954).
Peri, F. & Nüsslein-Volhard, C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell 133, 916–927 (2008).
Iyer, H., Shen, K., Meireles, A. M. & Talbot, W. S. A lysosomal regulatory circuit essential for the development and function of microglia. Sci. Adv. 8, eabp8321 (2022).
Parichy, D. M., Elizondo, M. R., Mills, M. G., Gordon, T. N. & Engeszer, R. E. Normal table of postembryonic zebrafish development: staging by externally visible anatomy of the living fish. Dev. Dyn. 238, 2975–3015 (2009).
Sasaki, T. et al. Autolysosome biogenesis and developmental senescence are regulated by both Spns1 and v-ATPase. Autophagy 13, 386–403 (2017).
Poole, B. & Ohkuma, S. Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J. cell Biol. 90, 665–669 (1981).
Homewood, C., Warhurst, D., Peters, W. & Baggaley, V. Lysosomes, pH and the anti-malarial action of chloroquine. Nature 235, 50–52 (1972).
McGlinchey, R. P. et al. Repeat domains of melanosome matrix protein Pmel17 orthologs form amyloid fibrils at the acidic melanosomal pH. J. Biol. Chem. 286, 8385–8393 (2011).
Gur, D. et al. In situ differentiation of iridophore crystallotypes underlies zebrafish stripe patterning. Nat. Commun. 11, 6391 (2020).
Aleström, P. et al. Zebrafish: housing and husbandry recommendations. Lab. anim. 54, 213–224 (2020).
Tucker, S. K., Ghosal, R., Swartz, M. E., Zhang, S. & Eberhart, J. K. Zebrafish raptor mutation inhibits the activity of mTORC1, inducing craniofacial defects due to autophagy-induced neural crest cell death. Development 151, dev202216 (2024).
Spehner, D. et al. Cryo-FIB-SEM as a promising tool for localizing proteins in 3D. J. Struct. Biol. 211, 107528 (2020).
Belevich, I., Joensuu, M., Kumar, D., Vihinen, H. & Jokitalo, E. Microscopy image browser: a platform for segmentation and analysis of multidimensional datasets. PLoS Biol. 14, e1002340 (2016).
Kremer, J. R., Mastronarde, D. N. & McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76 (1996).
Ophus, C. Four-dimensional scanning transmission electron microscopy (4D-STEM): from scanning nanodiffraction to ptychography and beyond. Microsc. Microanal. 25, 563–582 (2019).
Tate, M. W. et al. High dynamic range pixel array detector for scanning transmission electron microscopy. Microsc. Microanal. 22, 237–249 (2016).
Sorrentino, A. et al. MISTRAL: a transmission soft X-ray microscopy beamline for cryo nano-tomography of biological samples and magnetic domains imaging. J. Synchrotron Radiat. 22, 1112–1117 (2015).
Meirer, F. et al. Three-dimensional imaging of chemical phase transformations at the nanoscale with full-field transmission X-ray microscopy. J. Synchrotron Radiat. 18, 773–781 (2011).
Van Rossum, G. & Drake, F. L. Python/C API Manual-Python 3 (CreateSpace, 2009).
Hunter, J. D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 9, 90–95 (2007).
Waskom, M. et al. Mwaskom/Seaborn: v0.8.1. Zenodo https://doi.org/10.5281/zenodo.592845 (2017).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Epifanovsky, E. et al. Software for the frontiers of quantum chemistry: an overview of developments in the Q-Chem 5 package. J. Chem. Phys. 155, 084801 (2021).
Acknowledgements
This work was supported by an European Research Council starting grant (grant number 101077470, ‘CRYSTALCELL’) and by the Israel Science Foundation (grant no. 691/22) awarded to D.G. This work benefited from access to ALBA and was supported by iNEXT-Discovery (project number 871037), funded by the Horizon 2020 program of the European Commission. Cryo soft X-ray experiments were performed at the MISTRAL Beamline at ALBA Synchrotron with the collaboration of ALBA staff. We thank E. Pereiro for her support and assistance in coordinating with the synchrotron. Additionally, we thank T. Lerer-Goldshtein for her support in developing the project’s experimental approach. Electron microscopy studies were conducted at the Irving and Cherna Moskowitz Center for Nano and Bio-Nano Imaging at the Weizmann Institute of Science. R.D. is a fellow of the Ariane de Rothschild Women Doctoral Program.
Author information
Authors and Affiliations
Contributions
Conceptualization, Z.E., R.D., A.G.-A. and D.G. Investigation, Z.E., R.D., A.G.-A., Y. Barzilay, Y. Broder, A.P.K., N.V., M.H., A.S., I.K.-A., K.R., L.H., L.K., P.R. and D.G. Formal analysis, Z.E., R.D., A.G.-A., Y. Broder, A.P.K., R.R., R.M., P.R. and D.G. Visualization, Z.E., R.D., A.G.-A., Y. Barzilay, N.V., A.S., I.K.-A., K.R. and L.H. Funding acquisition, D.G. Supervision, D.G. Writing—original draft, Z.E. and D.G. Writing—review and editing, Z.E., R.D., A.G.-A. and D.G.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemical Biology thanks Esther Amstad, Florent Figon, Zhengzhe Liu, Michael Marks, Jordi Poater and the other, anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–13, Videos 1–4 and Data 1 and 2.
Supplementary Video 1 (download MOV )
A 3D reconstruction and animation of a cryo-FIB-SEM volume rendering of an iridophore from a 72-hpf zebrafish larval eye. Guanine crystals are shown in blue, along with vesicles in orange, mitochondria in red and the endoplasmic reticulum in yellow.
Supplementary Video 2 (download MOV )
Tilt series of an iridophore from a 1-mpf zebrafish, acquired at 520 eV using cryo-SXM.
Supplementary Video 3 (download MOV )
A 3D reconstruction and animation of a cryo-SXT dataset of an iridophore isolated from a 1-mpf zebrafish, corresponding to the cell shown in Supplementary Video 2. Guanine crystals are shown in blue, along with vesicles in orange, mitochondria in red and the cell membrane in yellow.
Supplementary Video 4 (download MOV )
Raw energy scan of the same field of view shown in Supplementary Videos 2 and 3, acquired across the range of 395–420 eV, corresponding to the nitrogen K-edge absorption. Cellular compartments appearing bright white indicate regions enriched in nitrogen.
Supplementary Data 1
Coordinates of the guanine molecules used for the DFT calculations.
Supplementary Data 2
Coordinates of the N7-protonated guanine molecules used for the DFT calculations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Eyal, Z., Deis, R., Gorelick-Ashkenazi, A. et al. pH variations enable guanine crystal formation within iridosomes. Nat Chem Biol 22, 19–27 (2026). https://doi.org/10.1038/s41589-025-02020-0
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41589-025-02020-0
This article is cited by
-
Acid enables biogenic crystallization
Nature Chemical Biology (2025)
