
Abstract
Asparagine-linked glycans are essential for the maturation and function of most eukaryotic secretory proteins. The biosynthesis and transfer of dolichylpyrophosphate-anchored GlcNAc2Man9Glc3 glycan is a highly conserved process occurring in the endoplasmic reticulum (ER) membrane and involving over a dozen membrane proteins whose dysfunction is linked to congenital disorders of glycosylation (CDGs). Three membrane-integral mannosyltransferases, ALG3, ALG9 and ALG12, mediate four consecutive mannosylation reactions that convert GlcNAc2Man5 to GlcNAc2Man9. Here, using chemoenzymatically synthesized lipid-linked glycan donor and acceptor analogs, we recapitulated this biosynthetic pathway in vitro. High-resolution cryo-electron microscopy structures of pseudo-Michaelis complexes of each step revealed how the branched glycan is accurately synthesized and unwanted side products are averted. Molecular dynamics simulations and mutagenesis studies uncovered a subtle but precise mechanism selecting the dolichylphosphomannose donor substrate over dolichylphosphoglucose, which is also present in the ER membrane. Our results also provide mechanistic explanations for enzyme dysfunction in CDGs and offer opportunities for N-glycan engineering.

Similar content being viewed by others


Main
The addition of glycans to protein asparagine residues, termed N-linked glycosylation, is found throughout the three domains of life and is one of the most common post-translational modifications1,2. Having primarily structural functions at the cell surface in archaea and bacteria, the functional and structural diversity of N-linked glycans exploded during eukaryotic evolution. In the endoplasmic reticulum (ER), N-linked glycans have pivotal roles in protein folding and stabilization, as well as in directing protein localization, degradation and sorting3,4,5. These processes are highly conserved in eukaryotes and involve unique and precisely defined N-glycan structures that serve as covalently attached signals, providing the basis of the generation of highly diverse secretory proteins6. Starting from the uniform oligomannose structures exiting the ER, hydrolases and glycosyltransferases in the Golgi compartments generate a large array of distinct glycan structures that are species, protein and glycosylation site specific. Thus, a kinetically controlled pathway that is based on the differential localization of differentially expressed processing enzymes yields a large diversity of N-glycan structures that mediate cell–cell and cell–molecule interactions, many of which are essential for immune function and host–pathogen interactions or have roles in cancer metastasis, nervous system development and function and Alzheimer disease7,8,9,10,11,12. In humans, the severe phenotypes associated with the more than 100 recorded congenital errors in this pathway demonstrate the biomedical importance of these glycan-mediated quality control steps13,14.
Before acceptor protein glycosylation, N-glycans are synthesized as lipid-linked oligosaccharides (LLOs) covalently bound to dolichylpyrophosphate (Dol-PP) and, thus, anchored in the ER membrane1. The sequential addition of sugars is catalyzed by the asparagine-linked glycosylation (ALG) enzymes (Fig. 1a). Synthesis begins on the cytosolic face of the ER membrane, where DPAGT1, ALG13 and ALG14 convert dolichylphosphate (Dol-P) to Dol-PP-GlcNAc2 (refs. 15,16). Subsequently, ALG1, ALG2 and ALG11 sequentially transfer mannoses c to g (labeling as previously published6; legend in Fig. 1a) from GDP-mannose to the growing LLO. The resulting Dol-PP-GlcNAc2Man5 is then flipped to the ER lumen by RFT1 (refs. 17,18). Extension of the LLO in the ER lumen involves the transfer of four mannoses and three glucoses from dolichylphosphomannose (Dol-P-Man) and dolichylphosphoglucose (Dol-P-Glc) donors. The mannosyltransferases ALG3, ALG9 and ALG12 transfer four mannoses, initiating and completing the B and C branches of the growing the N-glycan (Fig. 1a)19. ALG3 initiates the B branch by adding the h mannose in an α1,3 linkage20,21. The B branch is then completed with an α1,2-linked addition of the i mannose by the activity of ALG9 (refs. 22,23). Only when the B branch is complete does ALG12 initiate the C branch with the addition of the α1,6-linked j mannose24. The C branch is completed by ALG9, which adds the α1,2-linked k mannose22. The GlcNAc2Man9 glycan is subsequently processed by the enzymes ALG6, ALG8 and ALG10, which sequentially transfer three glucoses to the A branch25,26,27. The completed GlcNAc2Man9Glc3 oligosaccharide is transferred to target proteins containing an N-glycosylation sequon, Asn-Xaa-Ser/Thr, by the oligosaccharyltransferase (OST) complex1,28,29,30,31.

a, Schematic of N-glycan synthesis in the human ER. b, Strategy for the chemoenzymatic synthesis of the substrates for the luminal ER mannosyltransferases. Structures of the chemically synthesized starting compounds, Dol25-PP-GlcNAc2 and Dol25-P-Man, are shown. The obtained glycans were transferred to a fluorescent peptide using TbSTT3B and separated on a tricine gel. Structures above the gel lanes are indicated according to the Symbol Nomenclature for Glycans. c–f, Time courses of enzymatic activity using WT ScALG3 and Dol25-PP-GlcNAc2Man5 (c), WT HsALG9 and Dol25-PP-GlcNAc2Man6 (d), WT GgALG12 and Dol25-PP-GlcNAc2Man7 (e) and WT HsALG9 and Dol25-PP-GlcNAc2Man8 (f). Incubation times were as indicated. Lanes with glycopeptide cartoons over them serve as glycopeptide standards for the indicated glycan. Activity assays in b –f were performed at least twice for each enzyme–substrate combination with similar results.
Here, we recapitulate oligomannose N-glycan synthesis in vitro and report structures of ALG3, ALG9 and ALG12 bound to chemoenzymatically generated substrate analogs. In combination with mutagenesis studies and molecular dynamics (MD) simulations, these structures reveal the mechanism and biosynthetic logic of ER luminal N-glycan B-branch and C-branch synthesis. Our results also foster a molecular understanding of associated congenital disorders of glycosylation (CDGs) and advance efforts to engineer therapeutics and vaccines bearing N-glycans.
Results and discussion
Biochemical characterization of ALG3, ALG9 and ALG12
Gene homologs encoding ALG3, ALG9 and ALG12 from several species were expressed in HEK293 cells and screened using florescence size-exclusion chromatography (SEC) to assess protein expression and monodispersity32. On the basis of these results, we selected ALG3 from Saccharomyces cerevisiae (ScALG3), ALG9 from Homo sapiens (HsALG9) and ALG12 from Gallus gallus (GgALG12) for structural and functional studies (Extended Data Fig. 1a,b). Because the native dolichol contains 14–19 isoprenoid units, Dol-P-linked or Dol-PP-linked monosaccharides or glycans are poorly soluble and challenging to synthesize19,33. We, therefore, used synthetic or chemoenzymatically generated substrate analogs with a 25-carbon farnesylcitronellyl moiety, here referred to as Dol25, which increases their solubility34,35(Fig. 1b). The various acceptor substrates were chemoenzymatically generated using synthetic Dol25-PP-GlcNAc2, which was enzymatically extended to GlcNAc2Man5 using purified ALG1, ALG2 and ALG11 enzymes and GDP-Man as the mannose donor34. The chemoenzymatically generated LLOs could be transferred to a fluorescently labeled peptide using purified STT3B from Trypanosoma brucei (TbSTT3B), a single-subunit OST36. The resulting glycopeptides were analyzed using tricine–SDS–PAGE (Fig. 1b). In vitro activity assays were performed by allowing purified enzyme to transfer mannose from Dol25-P-Man to the provided acceptor LLO before enzyme denaturation. These assays demonstrated that detergent-solubilized ScALG3, HsALG9 and GgALG12 were catalytically active (Fig. 1c–f). Assays with Dol25-PP-GlcNAc2Man6 or Dol25-PP-GlcNAc2Man8 acceptor substrates confirmed the dual activity of ALG9 in vitro (Fig. 1d,f). Prolonged incubation times with each of these enzymes showed that they specifically add a single mannose, without further extension (Fig. 1c–f).
High-affinity antigen-binding fragment (Fab) selection using a synthetic library
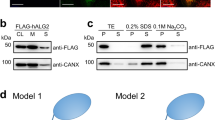
To provide insight into the reaction mechanisms, high-resolution structures of each of the enzymes in complex with donor and acceptor substrates were required. Because they are small integral membrane proteins (ScALG3, 52.9 kDa; HsALG9, 69.9 kDa; GgALG12, 55.8 kDa), particle alignment during cryo-electron microscopy (cryo-EM) data processing is challenging37. We, therefore, generated Avi-tagged constructs of ScALG3, HsALG9 and GgALG12, which allowed in vitro biotinylation and subsequent screening of phage library E for specific, high-affinity synthetic Fabs38,39,40. This yielded the Fabs Sc3-16, Hs9-8 and Gg12-11, which formed high-affinity complexes with ScALG3, HsALG9 and GgALG12 respectively, as shown by SEC, SDS–PAGE and multipoint protein ELISA (Extended Data Fig. 2a–d). At a tenfold molar excess of each Fab, the mannosyltransferase activities of ScALG3, HsALG9 and GgALG12 were unaltered, demonstrating that the Fabs did not interfere with enzyme function (Extended Data Fig. 2e). The sequences of the complementarity-determining regions (CDRs) of Sc3-16, Hs9-8 and Gg12-11 are shown in Extended Data Fig. 2f.
Structures of substrate-bound ScALG3, HsALG9 and GgALG12
To facilitate the trapping of pseudo-Michaelis complexes, we substituted the residues providing the putative catalytic base in ScALG3 (D71N), HsALG9 (D82A) and GgALG12 (E35Q) to either an alanine or the corresponding carboxamide side chain. The catalytic residues were predicted on the basis of sequence homology with other glycosyltransferase class CA (GT-CA) enzymes41 and confirmed by activity assays, where the mutant enzymes showed strongly decreased mannosyltransferase activity (Extended Data Fig. 1c–e and Supplementary Figs. 1–3). Single-particle cryo-EM data of ScALG3-D71N, HsALG9-D82A and GgALG12-E35Q (referred to below as ScALG3, HsALG9 and GgALG12) in complex with Fab fragments and an anti-Fab nanobody targeting the κ light chain at the Fab hinge42 were collected and processed to resolutions ranging from 2.4 to 3.1 Å (Extended Data Table 1 and Extended Data Fig. 3). The Fabs targeting HsALG9 and GgALG12 bound the cytoplasmic face, while the Fab for ScALG3 bound the luminal face of the mannosyltransferases (Extended Data Fig. 2g–j). In each case, the high-resolution maps allowed de novo model building of the proteins and the bound substrate analogs.
Previous phylogenetic studies have suggested that enzymes involved in N-linked glycan or glycosylphosphatidylinositol anchor synthesis and using Dol-P-Man as a donor substrate have a common ancestor, with each enzyme arising through gene duplication and divergent evolution43. In line with this hypothesis, our structures revealed that ScALG3, HsALG9 and GgALG12 feature similar folds and belong to the GT-CA class (Figs. 2b,c and 3b and Extended Data Fig. 4). The GT-CA class includes integral membrane enzymes that use isoprenoid-bound sugars as donor substrates and feature an N-terminal conserved module consisting of seven transmembrane (TM) helices, followed by a C-terminal module of varied length and structure41. The first luminal loop (LL1), located within the conserved domain, comprises two α-helices, the first of which contains the catalytic base. The variable module of ScALG3 is composed of five TM helices, whereas HsALG9 and GgALG12 feature only four TM helices with a C-terminal domain that extends into the ER lumen (Figs. 2b,c and 3b and Extended Data Fig. 4).

a, Schematic showing the initiation and termination of the N-glycan B branch. ScALG3 and HsALG9 structures are displayed as ribbon diagrams, while the acceptor and donor substrates are shown as sticks in pink and gold, respectively. b,c, Cross-section schematic view of ScALG3 (b) and HsALG9 (c) viewed from the ER luminal side, indicating the arrangement of TMs and the substrate-binding pockets. The conserved and variable domains of the GT-CA fold are indicated with gray and colored shading, respectively. The locations of catalytic base residues are indicated by magenta spheres. d, Structure of ternary complex of ScALG3-D71N with bound Dol25-PP-GlcNAc2Man5 (acceptor) and Dol25-P-Man (donor) substrates. e, Close-up view of the ScALG3 active site in stick representation. Carbons of protein residues are colored light blue, acceptor substrate in pink and donor substrate in gold. Selected distances are shown by dashed lines. f, Structure of the ternary complex of HsALG9-D82A with bound Dol25-PP-GlcNAc2Man6 acceptor substrate. Insets in d,f show cartoons of bound substrates, with monosaccharides resolved in the cryo-EM density colored according to the Symbol Nomenclature for Glycans, while unresolved monosaccharides are depicted in gray. In d,f, cryo-EM densities for bound substrates are shown as transparent gray surfaces. Acceptor and donor substrates are depicted in pink and gold stick representation. Key residues are highlighted as sticks and hydrogen-bonding interactions are shown as dashed lines.

a, Schematic showing synthesis of the N-glycan C branch. GgALG12 and HsALG9 structures are displayed as ribbon diagrams, with acceptor and donor substrates shown as sticks in pink and gold, respectively. b, Cross-section schematic view of GgALG12 viewed from the ER luminal side, indicating the arrangement of TMs and the substrate-binding pockets. The conserved and variable domains of the GT-CA fold are indicated with gray and purple shading, respectively. The location of the catalytic base side chain is indicated with a magenta sphere. c, Structure of ternary complex of GgALG12-E35Q with bound Dol25-PP-GlcNAc2Man7 (acceptor) and Dol25-P-Man (donor) substrates. d, Structure of the ternary complex of HsALG9-D82A with bound Dol25-PP-GlcNAc2Man8 (acceptor) and Dol25-P-Man (donor) substrates. Insets in c,d show cartoons of bound substrates, with monosaccharides resolved in the cryo-EM density colored according to the Symbol Nomenclature for Glycans, while unresolved monosaccharides are depicted in gray. In c,d, cryo-EM densities for bound substrates are shown as transparent gray surfaces. Acceptor and donor substrates are depicted in pink and gold stick representation. Key residues are highlighted as sticks and hydrogen-bonding interactions are shown as dashed lines.
ALG3 is classified within the GT-58 family according to the CAZy nomenclature44. It contains 12 TM helices, with cytosolic N and C termini, and extends only minimally outside the membrane. ALG9 and ALG12 are classified as GT-22 enzymes. They contain 11 TM helices and a cytosolic N terminus. N-glycosylation of ScALG3 or GgALG12 was not observed but the variable domain of HsALG9 is N-glycosylated at N593, as indicated by cryo-EM density for the first GlcNAc entity of an N-glycan.
Structural basis of B-branch initiation
To capture the ternary complex representing initiation of the B branch, detergent-solubilized ScALG3 was mixed with chemoenzymatically synthesized acceptor and donor substrates (Dol25-PP-GlcNAc2Man5 and Dol25-P-Man, respectively) before cryo-EM grid vitrification. The resulting structure revealed the acceptor and donor substrate-binding pockets, which flank the catalytic cleft on the luminal face of ALG3 (Fig. 2a,b,d). The acceptor Dol25 moiety is located in a hydrophobic groove formed by TM1 and TM3, where it is exposed to the lipid bilayer (Fig. 2b,d). An electropositive patch formed by R163 and R171 of TM3 is located at the membrane boundary, facing the ER lumen and forming ionic interactions with the pyrophosphate moiety. The two GlcNAc entities are bound to the protein surface adjacent to the catalytic cleft, positioning the c mannose such that the A branch projects toward bulk solvent, whereas the e mannose reaches into the active site (Fig. 2d,e). The A branch is likely mobile because its cryo-EM density weakens as it extends away from the protein surface. Strong CH–π contacts between the b GlcNAc and Y410, as well between the c mannose and W286, help position the substrate for catalysis (Supplementary Fig. 4a). Similar CH–π interactions have been shown to have an important role in other carbohydrate–protein interactions45,46. Within the active site, a hydrogen bond between the e mannose 4-OH and the side-chain nitrogen of Q373 helps orient the 3-OH for attack on the anomeric carbon atom of the donor substrate (Fig. 2e). The side chain of the catalytic D71, here substituted to asparagine, forms a hydrogen bond to the C3-OH of the e mannose, supporting its role as the catalytic base. The distance between the attacking C3-OH of the acceptor and the donor anomeric carbon is ~3.2 Å. Our structure, therefore, represents a pseudo-Michaelis state primed for mannose transfer and the formation of the α1,3-linkage.
The Dol25-P-Man donor substrate enters the active site from the opposite side of ScALG3 relative to the acceptor substrate pyrophosphate group. The fourth luminal loop (LL4) forms a bridge over the donor substrate-binding groove, an architectural feature that was previously observed for GT-CA enzymes including ALG6, ArnT, EmbA-C and AftD35,47,48,49 (Extended Data Fig. 4a). We observed cryo-EM density for the entire Dol25-P-Man molecule, which revealed that the Dol25 moiety interacts with TM6 and TM8 (Fig. 2b). The phosphate group interacts with R280 from LL4, with H371 from LL5 connecting TM9 and TM10, and with the hydroxyl group of Y75 through a water molecule (Fig. 2e). Dol25-P-Man adopts a bent-back conformation, with the axial C2-OH projecting away from the acceptor substrate (Fig. 2e). Bent-back conformations have been observed for the Dol-P-Man donor substrate in the C-mannosyltransferase DPY19 (ref. 50) and in several structures containing nucleotide-activated donor sugars51,52,53,54,55,56. While energetically less favorable, the bent-back conformation is thought to prime the anomeric carbon for attack by the incoming nucleophile of the enzyme-bound acceptor substrate and subsequent release of the leaving group, Dol-P.
Ternary complexes reveal completion of B and C branches
To uncover how ALG9 completes the B and C branches with the addition of α1,2-linked mannoses, we collected cryo-EM data of nanodisc-reconstituted enzyme in the presence of Dol25-P-Man donor substrate and either Dol25-PP-GlcNAc2Man6 (containing an initiated B branch) or Dol25-PP-GlcNAc2Man8 (containing a completed B branch and an initiated C branch) acceptor substrates (Figs. 2a and 3a). Our structures reveal how these distinct substrates are accommodated in the active site of the enzyme (Figs. 2a,c,f and 3a,d). In both cases, the acceptor substrate pyrophosphate group binds to a cluster of electropositive residues (R428 and R577) at the ER luminal membrane boundary while the Dol25 moiety binds to a hydrophobic patch formed by TM8 and TM11. The N-acetyl group of the a GlcNAc points out of the active-site cleft, whereas, in ScALG3 and GgALG12, the GlcNAc sugars are rotated 180° (Figs. 2d,f and 3c,d). A hydrogen bond between the a GlcNAc N-acetyl group and the side-chain nitrogen of N309 and a CH–π interaction between the Y473 side chain and the c mannose anchor the acceptor substrates (Supplementary Fig. 4b). As with the ScALG3 ternary structure, the A branch of bound acceptor LLO projects out of the catalytic cleft. The catalytic base, D82, was substituted to alanine to capture these ternary complexes. The ALG9 structures show the Cβ carbon of A82 within 5 Å of the C2-OH of the acceptor mannose, supporting the notion that the acceptor substrate represents a pretransfer pose.
In both ternary structures of HsALG9, we observed strong cryo-EM density for the Dol25 and phosphate moieties of bound Dol25-P-Man, which was bound in a hydrophobic groove formed by TM6 and TM9 and in a tunnel formed by LL4. The phosphate group interacts with two arginine residues, R112 and R370, which likely facilitate the transfer reaction through stabilization of the leaving group (Figs. 2f and 3d). In contrast to the ScALG3 structure, cryo-EM density for the mannose of bound Dol25-P-Man was not observed in HsALG9. To exclude that futile donor hydrolysis was responsible, we carried out activity assays with substoichiometric amounts of substrate relative to enzyme (Methods) and found no evidence of donor substrate hydrolysis (Supplementary Fig. 5a). This suggested that the donor substrate was chemically intact in our cryo-EM samples but that the mannose group was too mobile to allow visualization. We further investigated this conclusion using MD simulations, as discussed below.
Structural basis of C-branch initiation
The structure of GgALG12 was determined in the presence of the chemoenzymatically generated acceptor substrate Dol25-PP-GlcNAc2Man7 and the donor substrate Dol25-P-Man. The Dol25 moiety of the acceptor is lodged in a groove formed by TM8 and TM11, facing the lipid bilayer (Fig. 3b). The pyrophosphate group is coordinated by H140 of TM3 and R271 of TM8 and located at the ER luminal membrane boundary (Fig. 3c). The side-chain nitrogen of N370 interacts with the C3-OH of the a GlcNAc, positioning the c mannose of the glycan such that the e mannose, to which the B and C branches are connected, projects toward the active site, while the A branch points away from the active site and interacts with the luminal domain of ALG12. The c mannose is further stabilized by a CH–π stacking interaction to the side chain of W256 (Supplementary Fig. 4c). ALG12 catalyzes an α1,6-linked mannose addition and we indeed found the C6-OH of the oligosaccharide e mannose at a distance of ~3.6 Å from the ε-oxygen of the catalytic base, E35, here substituted to glutamine. Notably, the acceptor B branch projects into a cavity created by LL1 and the C-terminal luminal domain of ALG12, where it interacts with several residues lining the pocket.
The donor substrate, Dol25-P-Man, accesses the active site through a tunnel formed by TM6, TM9 and LL4 (Fig. 3b). The phosphate group is coordinated by the side chains of R67 from LL1, R185 from the loop between TM5 and TM6 and R312 located in the loop between TM9 and TM10. Given this coordination of the leaving group phosphate, we believe this structure captures the state before mannose transfer. As with the HsALG9 ternary complexes, density for the donor mannose sugar was not observed despite the use of the catalytic mutant and the addition of Dol25-P-Man before vitrification. We, therefore, performed a similar assay as detailed for ALG9 to detect donor hydrolysis but none was observed (Supplementary Fig. 5b).
Molecular logic of oligomannose core assembly
The correct composition and structure of the GlcNAc2Man9Glc3 glycan transferred to nascent glycoproteins is essential for quality control mechanisms in the ER and for the generation of hybrid and complex glycans in the Golgi. In vivo studies have shown that ALG3, ALG9 and ALG12 synthesize the N-glycan in a defined order20,22,23,24. Our structural and functional data reveal the basis for ordered assembly of the mannose core.
The binding pockets of ALG3, ALG9 and ALG12 are precisely tailored to allow accommodation of acceptor glycans in an extended pose from the pyrophosphate-binding site to the active site (Figs. 2d,f and 3c,d). This suggests that the distance from the pyrophosphate-binding site to the active site functions as a molecular ruler, preventing mannose transfer to incorrect acceptor substrates (Fig. 4a). The observed interactions between the acceptor pyrophosphate and the protein constrain the substrate and enforce a binding pose that presents the correct hydroxyl for activation by the catalytic base. Such activity control likely prevents ER luminal mannosyltransferases from acting as polymerases (Extended Data Fig. 5d), which would result in overly long B and C branches. An analogous ruler concept was suggested for the bacterial glycosyltransferase PglH of Campylobacter jejuni57.

a, The distance between the pyrophosphate-binding site of ALG enzymes and the active sites acts as a molecular ruler to select the correct acceptor substrate. b, Superimposed structures of HsALG9 containing either Dol25-PP-GlcNAc2Man6 (pink sticks) or Dol25-PP-GlcNAc2Man8 (white sticks) substrates reveal two distinct binding modes. c, Acceptor B-branch-binding pocket in GgALG12 structure underpins the requirement of a completed B branch in the acceptor substrate. d, Activity assays with GgALG12 and Dol25-PP-GlcNAc2Man5 (no B branch) or Dol25-PP-GlcNAc2Man6 (initiated but incomplete B branch) acceptor substrates. GgALG12 processes substrates without a complete B branch very slowly compared to the native acceptor substrate. e, Functional studies of ScALG3 with Dol25-PP-GlcNAc2Man6 bearing an initiated C branch showed no turnover even after extended incubation. In d,e, reactions were incubated as indicated above the lanes; for ‘heated’ samples, the enzyme was replaced with heat-inactivated enzyme. The obtained glycans were transferred to a fluorescent peptide (TAMRA-YANATS-NH2) using TbSTT3B and separated on a tricine gel. Lanes with glycopeptide cartoons over them serve as glycopeptide standards to allow comparison. Time-course assays in d,e were performed at least twice with similar results. A schematic for reactions in d,e is shown in Extended Data Fig. 5d. f, Zoomed-in view of ScALG3 catalytic cleft with bound donor and acceptor substrates. The ternary complex shows insufficient room in the active-site cleft to accommodate an initiated acceptor C branch at the indicated C6 hydroxyl.
The two HsALG9 ternary complexes illuminate how two different acceptor substrates are accommodated by a single enzyme. The dual specificity relies on rotation of the α1,6-linkage between the c and e mannoses, allowing presentation of either the h or the j mannose to the active site (Fig. 4b). For the first acceptor substrate (Dol-PP-GlcNAc2Man6), the C branch has not been initiated; thus, only the B-branch-initiating mannose of the acceptor substrate is able to reach the active site. For the second acceptor substrate (Dol-PP-GlcNAc2Man8), the B branch has been already completed and projects toward bulk solvent.
Whereas ALG9 evidently requires ALG3 or ALG12 to initiate the B and C branches before they can be completed, it was less obvious how ALG12 activity was prevented until ALG3 and ALG9 could complete the B branch. The physiological importance is that a GlcNAc2Man7 intermediate containing h and j mannoses must be avoided by the biosynthetic pathway because it could interfere with protein quality assessment processes mediated by the lectin OS9, which depends on such a GlcNAc2Man7 structure58. The ALG12 ternary structure reveals that this is implemented by a deep binding cavity that accommodates the B branch of the acceptor glycan (Fig. 4c). The h mannose interacts with the side chain of E419 through its C4-OH, while the i mannose forms hydrogen bonds to the side chains of S37 and Q41, as well as to the backbone carbonyl of A399 (Fig. 4c). Tight binding and immobilization of the completed B branch in ALG12 likely helps orient the acceptor e mannose to receive the α1,6-linked j mannose, providing a mechanism to strongly favor the GlcNAc2Man7 glycan and prevent premature C-branch initiation. Assays with wild-type (WT) GgALG12 and acceptor substrates either lacking a B branch or featuring a B branch containing a single mannose demonstrated that mannosyl transfer activity was drastically reduced but not impossible, in line with in vivo observations24 (Fig. 4d and Extended Data Fig. 5d). Furthermore, yeast cells lacking ALG9 accumulate a GlcNAc2Man6 intermediate through ALG3 activity, providing evidence that ALG12 does not proceed with C-branch initiation unless the B branch is first completed24. Only with overexpression of ALG12 do ALG9-knockout cells produce a certain amount of GlcNAc2Man7 (ref. 24). Should ALG12 occasionally generate a nonnative Dol-PP-GlcNAc2Man6 intermediate containing an initiated C branch, this acceptor substrate is not processed by ALG3 (Fig. 4e and Extended Data Fig. 5d). The ScALG3 ternary complex structure revealed that an acceptor substrate with an initiated C branch would cause a steric clash in the binding pocket (Fig. 4f). This ensures that no GlcNAc2Man7 intermediates containing initiated B and C branches accumulate.
Our ALG3 structure revealed that the A-branch mannoses project away from the enzyme toward the bulk solvent (Figs. 2d and 4f) and are not required for activity. Hence, ALG3 does not ensure completeness of the GlcNAc2Man5 moiety. However, there is evidence that the flippase RFT1 favors Dol-PP-GlcNAc2Man5 over Dol-PP-GlcNAc2Man3, which may prevent immature glycans from being flipped to the ER lumen18. The OST complex has a specific pocket for the Glc3 moiety of the oligosaccharide but the B and C branches are not recognized29. Following glycan transfer and the removal of the two terminal glucoses, the remaining glucose is cleaved or reattached, depending on the folding status of the linked protein, during the folding process based on calnexin/calreticulin and UDP-Glc:glycoprotein glucosyltransferase (UGGT)59. However, the glucosidases and UGGT require a fully mannosylated glycan with complete B and C branches for efficient removal or addition of glucose and, therefore, for proper functioning of this pathway60,61. Given the importance of the mannoses in these branches for subsequent signaling steps in the ER and Golgi, it is essential that the B and C branches are completed before the glucosyltransferases complete the A branch to prevent transfer of a premature glycan. In vivo evidence suggests that one or more of the ER luminal glucosyltransferases provide this specificity23,62,63.
Catalytic mechanism
Our structural and functional studies define the geometry of the active site for ScALG3, HsALG9 and GgALG12 and allow us to propose structure-based reaction mechanisms (Extended Data Fig. 5a–c). The three mannosyltransferases facilitate an inverting mechanism by accepting a β-linked donor mannose and transferring it to the acceptor LLO with an α-linkage (α1,3 for ALG3, α1,2 for ALG9 and α1,6 for ALG12). We propose a bimolecular nucleophilic substitution reaction, whereby the attacking hydroxyl group of the acceptor mannose is activated by deprotonation by a catalytic base (side chains of D71 in ScALG3, D82 in HsALG9 and E35 in GgALG12). This is followed by an attack of the donor mannose anomeric carbon, causing inversion of configuration and departure of the Dol-P leaving group. In each enzyme, electropositive residues coordinate the donor phosphate, thus stabilizing the negative charge of the leaving group. Notably, the acceptor substrates bind ALG3, ALG9 and ALG12 at the domain boundary of the conserved and variable module, with specific contacts predominantly formed with residues of the variable module (Figs. 2b,c and 3b and Extended Data Fig. 4). We hypothesize that the GT-CA variable modules also facilitate the specific binding of diverse acceptor substrates in other GT-CA enzymes.
Mechanism of donor substrate specificity
The donor substrates Dol-P-Man and Dol-P-Glc are both present in the ER membrane, making it essential for ER luminal mannosyltransferase and glucosyltransferase enzymes to discriminate between these C2 epimers. To explore the molecular basis for donor selectivity, we conducted MD simulations with HsALG9 using the cryo-EM structure of the second ternary complex (capturing the state just before completion of the C branch) as the starting model. In a first set of five replicates over 800 ns, we observed that, while the lipid tail of Dol25-P-Man was highly mobile, the phosphate and sugar moieties were more stably bound (Extended Data Fig. 6a). During the simulations, the axial C2-OH of the donor mannose often formed hydrogen bonds with the side chains of N288 and H366, suggesting that these residues might have a role in donor selection (Extended Data Fig. 6b). However, the Dol25-P-Man molecule shifted further into the active site by about 2 Å in one of the five simulations, with the side chains of R112 and R370 moving apart to accommodate this motion and better coordinate the donor phosphate leaving group (Extended Data Fig. 6c,d). In this new ‘engaged’ pose, the distance between the C2-OH of the acceptor mannose and the anomeric carbon of the donor mannose was shortened, suggesting that this represented a conformation closer to the transition state.
To test the relevance of these simulated donor sugar–protein interactions, we conducted further MD simulations using the engaged Dol25-P-Man pose as the starting structure. We first compared distinct donor molecule analogs and performed simulations with Dol25-P-Man, Dol25-P-Glc or Dol25-P-Man-2F, where the C2-OH group was replaced by a fluorine atom, which cannot serve as a hydrogen-bond donor (Fig. 5a–d and Extended Data Fig. 7a–c). We plotted the relative propensity of different donor sugar poses (‘probability’) during the MD simulations, defined by the observed distances between the donor anomeric carbon and the attacking acceptor oxygen and the angle between the C–H bond of the anomeric carbon and a line between the anomeric carbon and the acceptor C2 oxygen (Fig. 5a). The simulations showed that Dol25-P-Man predominantly adopted a catalytically favorable pose, whereas Dol25-P-Glc rarely adopted a similar pose (Fig. 5b,c and Extended Data Fig. 7a,b). This is likely because, in the engaged pose, the donor mannose C2 is within van der Waals distance of the enzyme and space exists only for an axial hydroxyl group (Fig. 5c). An equatorial hydroxyl group, such as that in Dol-P-Glc, would cause a steric clash, offering a molecular explanation for the donor substrate specificity. When simulating with the nonreactive donor analog Dol25-P-Man-2F, a maximum was observed in a similar position to the Dol25-P-Man simulation (Fig. 5b,d and Extended Data Fig. 7a,c). However, the peak was lower, suggesting that the fluorinated Dol-P-Man analog not only is inactive but may also be less likely to adopt a potentially reactive pose in the active site.

a, Definition of donor pose metrics used for analysis of MD results including distance (r) and angle (θ). b, Probability of different distance (r) and angle (θ) values observed during MD simulations for the donor analogs, Dol25-P-Man or Dol25-P-Glc. In b,d, the plots are colored by the relative probability of observed donor poses. c, Representative pose of Dol25-P-Man in the catalytically favorable conformation in the active site of WT HsALG9. Top, hydrogen bonds between the attacking hydroxyl of the acceptor substrate and the catalytic base (D82) and between the donor C2-OH and the phosphate oxygen are shown with dotted lines. The distance (r) between the acceptor C2 hydroxyl oxygen and donor anomeric carbon is indicated with a dashed line. Bottom, Dol25-P-Man shown as sticks in the active site, with HsALG9 displayed as a partially transparent surface. d, Probability density of different distance (r) and angle (θ) values observed during MD simulations with WT HsALG9 and Dol25-P-Man-2F or with HsALG9 mutants and Dol25-P-Man. e, Relative amount of GlcNAc2Man6 (acceptor) and GlcNAc2Man7 (product) after reaction for 30 s with different HsALG9 mutants. The resulting glycans were transferred to excess fluorescent peptide (TAMRA-YANATS-HN2) with TbSTT3B and measured using high-resolution MS. f, Structure of HsALG9 active site, with protein residues colored green, acceptor substrate colored pink and donor substrate colored gold. The shown structure represents an initial frame of an MD simulation with WT HsALG9 and Dol25-P-Man. Key residues are shown as sticks and labeled, including the substitutions introduced. The color of the label (red, deleterious; green, minimal effect) indicates the effect of the substitution on ALG9 mannosyltransferase activity.
The simulations also revealed an intramolecular hydrogen bond between the mannose C2-OH and the phosphate group of Dol25-P-Man, resulting in a bent-back pose similar to that observed in the experimental ScALG3 structure (Figs. 2d,e and 5c). Notably, we observed that a loss of this hydrogen bond correlated with a loss of the engaged donor pose (Extended Data Fig. 7d,e). The simulations with Dol25-P-Glc revealed an analogous hydrogen bond between the C2-OH and the phosphate oxygen but a catalytically active pose was not favored (Fig. 5b and Extended Data Fig. 7d,e). Dol25-P-Man-2F does not permit similar hydrogen bonding because of the fluorine substitution. Our results suggests that, while Dol-P-Glc can enter the active site of ALG9 in vivo, it cannot proceed to adopt a catalytically active pose.
The hydrogen-bonding interactions between the donor mannose C2-OH and the side chain of N288 are eliminated in the engaged pose. However, N288 may have a role in orientating the donor substrate as it moves into the active site. To probe the role of N288 and H366, we performed additional MD simulations of HsALG9 after replacing these residues individually with alanine. The resulting Dol25-P-Man pose distributions were analogous to those with Dol25-P-Glc, supporting the role of these two residues in correctly orientating the donor substrate (Fig. 5b,d and Extended Data Fig. 7f,g). Given the lack of density for the donor mannose in our ALG9 cryo-EM structures, we also investigated donor mannose mobility by simulating the components vitrified during cryo-EM grid preparation. We carried out MD simulations of HsALG9-D82A with bound Dol25-P-Man and Dol25-PP-GlcNAc2Man8 and observed that the donor sugar was highly mobile (Fig. 5d and Extended Data Fig. 7h). This may be because of the added space or lack of charge in the active site as a result of the aspartate to alanine substitution, preventing the donor mannose from adopting a single favored pose.
Given the close structural similarity of HsALG9 to GgALG12, we expect analogous mechanisms of donor substrate selection (Extended Data Fig. 8a). HsALG9 residues N288 and H366 are conserved in GgALG12 as N247 and H308. Substitution of N247 or H308 to alanine caused a reduction in transfer activity in GgALG12 as with HsALG9, as discussed below, supporting the importance of these residues in GgALG12 (Extended Data Fig. 8b). ScALG3 also needs to select the correct donor substate but has a clearly distinct architecture compared to ALG9 and ALG12 (Extended Data Fig. 4). Among the residues located within 4 Å of the mannose donor C2-OH, we found Y102 and R280, both of which also coordinate the donor phosphate group (Fig. 2e). The side chains of these residues could form hydrogen bonds with the axial 2-OH of the mannose donor (distance to hydroxyl group of Y102 of 3.7 Å and to η-N of R280 of 2.5 Å). These residues might, therefore, have a role in donor substrate selection in addition to the R280-mediated stabilization of the phosphate leaving group. Functional assays with ScALG3 variants containing either Y102A, Y102F or R280A substitutions indeed showed reduced transfer activity, supporting the importance of these residues in facilitating mannose transfer (Extended Data Fig. 8c)
Functional analysis of HsALG9
To validate our structural-based and simulation-based findings, we expressed and purified HsALG9 variants, probed their in vitro function (Fig. 5e and Extended Data Fig. 8d) and mapped the observed activities onto the HsALG9 structure (Fig. 5f). To assay the activity of ALG9 mutants, we transferred the resulting LLOs to a fluorescently labeled peptide and separated them by tricine gel electrophoresis or analyzed the glycopeptide composition by high-resolution mass spectrometry (MS) (Fig. 5e and Supplementary Fig. 6). We found that substitution of arginine residues coordinating the acceptor pyrophosphate (R428A and R577A) reduced ALG9 activity, indicating that these residues have a crucial role in substrate binding. Interestingly HsALG9 mutants R428A and R577A still had some residual activity, which supports the idea that, while the pyrophosphate binding is necessary, binding is partially supported by either one of these residues. Substitution of K574, which is nearby but does not directly coordinate the pyrophosphate group in either of our structures, did not appear to substantially effect activity. In contrast, substitution of either R112 or R370 appeared to abolish activity supporting their essential function in stabilizing the donor leaving group. The HsALG9 structures further revealed that E104 forms a salt bridge with R112 in the active site (Figs. 2f and 3d). When the negative charge of this side chain was removed (E104Q or E104A substitutions), ALG9 activity was strongly reduced, suggesting that E104 may be important for correct orientation of R112. Additionally, when W103 was substituted to a phenylalanine, ALG9 activity was again abolished, suggesting that the observed hydrogen bond between the tryptophan side-chain nitrogen and the C4-OH of the acceptor mannose is required for substrate binding or orientation (Figs. 2f and 3d). We also probed the functional role of residues N288 and H366 that were identified in the MD studies. We found that the N288A and H366A variants both abolished HsALG9 function, supporting their importance for catalysis and validating the MD simulations (Fig. 5d–f and Extended Data Fig. 7f,g).
Structural basis for CDG involving ALG3, ALG9 and ALG12
Our structural data provide mechanistic explanations for many of the ALG3, ALG9 and ALG12 variants associated with CDGs in humans (Extended Data Table 2 and Supplementary Figs. 1–3). All highlighted substitutions affect residues that are conserved between the human and yeast homologs of ALG3 and the human and chicken homologs of ALG12 (Fig. 6). Of note, the ALG3-R171Q mutant has been associated with hyperinsulinemic hypoglycemia, hypotonia, facial dysmorphisms and death within the first 3 weeks of life64. Our structural data reveal that R171 is involved in an electrostatic interaction with the pyrophosphate moiety of the acceptor substrate, suggesting that substrate recruitment would be disrupted in a R171Q mutant (Fig. 6a,d). In ALG9, the Y287C mutant has been linked to severe symptoms including hypotonia, recurrent seizures and psychomotor retardation65,66. Y287 lines the binding site of the donor mannose. We purified the HsALG9 variants Y287C and Y287F and observed that both showed strongly reduced catalytic activity, suggesting that the side-chain hydroxyl group of Y287 might be indirectly involved in binding and orientating the donor substrate (Figs. 5e,f and 6b,e and Extended Data Fig. 8d). In human ALG12, substitution of R311 (equivalent to R312 in GgALG12) to cysteine was reported to cause a CDG characterized by psychomotor retardation, severe cerebellar hypoplasia and hypotonia67. This residue is in direct vicinity of the donor phosphate group and, therefore, likely stabilizes the leaving group of the substitution reaction (Fig. 6f), which can explain why substitution to cysteine results in a loss of function. Additionally, several persons with CDG displaying debilitating symptoms leading to death in the first 2 years of life were found to have substitutions of either F142 or R146 in human ALG12 (F143 or R147 in GgALG12)62,68,69. These residues form a π–cation stacking interaction in our GgALG12 ternary structure (Fig. 6f) and line the acceptor substrate-binding cleft. They do not directly contact the nearby GlcNAc moieties or the pyrophosphate moiety (closest distance: 4.6 Å away) but this π–cation interaction is likely important for the structural integrity of the acceptor substrate-binding cavity.

a–c, Locations of selected substitutions associated with CDGs mapped onto the structures of ScALG3 (a), HsALG9 (b) and GgALG12 (c). Substituted residues are shown as yellow spheres. Equivalent residues in the human constructs are shown in brackets. Superimposed AlphaFold2 models of human homologs of ScALG3 and GgALG12 are shown as light-gray cartoons. d–f, Close-up views of ScALG3 (d), HsALG9 (e) and GgALG12 (f), with selected residues associated with CDGs shown as yellow sticks.
Conclusions
Our studies reveal the mechanism and logic of the assembly of the branched oligomannose glycan GlcNAc2Man9, which underpins essential quality control and folding mechanisms of N-glycoproteins in the ER and provides the oligomannose structure required for Golgi glycan remodeling. Furthermore, we show that the discrimination of the correct donor substrate, Dol-P-Man from its C2 epimer, Dol-P-Glc, is mediated by residues within the active site and the adopted pose of the Dol-P-Man donor when it engages in an intramolecular hydrogen bond. In addition to fundamental understanding in an essential biosynthetic pathway, the molecular insights governing LLO substrate selection contribute a basis for future biotechnological engineering, including the production of viral oligomannose epitopes or structure-guided mutagenesis to modify ALG substrate preferences (for example, bump-and-hole methods70), facilitating new tools for probing the N-linked glycome and glycoproteome. Our results provide a framework for understanding the mechanisms of other GT-C enzymes, particularly those involved in branched-glycan biosynthesis. Lastly, we provide a molecular basis for understanding how ALG3, ALG9 and ALG12 enzyme variants lead to lethal CDGs.
Methods
Cloning, expression and purification of ScALG3, HsALG9 and GgALG12
Gene sequences coding for ScALG3 (UniProt P38179), isoform 1 of HsALG9 (UniProt Q9H6U8) and GgALG12 (UniProt F1P077) were codon-optimized for expression in human cells using GeneArt (Thermo Fisher Scientific) and cloned into a modified pUC57 vector using restriction free cloning71. ScALG3 was cloned with a C-terminal HRV 3C cleaveage sequence followed by eYFP and rho-1D4 sequences72. HsALG9 and GgALG12 where cloned with an N-terminal FLAG tag followed by eYFP and HRV 3C cleavage sequences. All three proteins were transiently expressed in suspension 293 c18 (American Type Culture Collection, CRL-10852) cells maintained at 37 °C in humidified incubators with supplemental carbon dioxide. Protein expression was induced by transient transfection with linear polyethylenimine. Cells expressing protein for 2 days were collected by centrifugation, washed with PBS and flash-frozen before storage at −80 °C.
Cells were thawed in lysis buffer (150 mM sodium chloride, 50 mM HEPES pH 7.5 and 10% (v/v) glycerol) in a 1:5 (w/v) ratio. All subsequent steps were either performed on ice or at 4 °C. Before lysis by Dounce homogenization, 1 mM PMSF, 20 µg ml−1 DNAse I (Roche) and a 1:100 (v/v) dilution of protease inhibitor cocktail (Sigma) were added. Membrane extraction was performed for 1 h in 1% (w/v) lauryl maltose neopentyl glycol (LMNG), 0.1% (w/v) cholesteryl hemisuccinate (CHS) or 1% (w/v) n-dodecyl-β-D-maltopyranoside (DDM) and 0.2% (w/v) CHS for samples destined for nanodisc reconstitution or substrate synthesis. All detergents used were purchased from Anatrace. The lysate was then centrifuged at 186,000g for 30 min in a Type-45Ti (Beckman Coulter) rotor to pellet insolubilized membrane. The supernatant was then incubated with either M2 Flag antibody resin (Sigma) or 1D4 antibody resin (made in house) for 1 h with rotation. The flowthrough was discarded and the resin was washed twice with 15 column volumes of wash buffer (150 mM sodium chloride, 20 mM HEPES pH 7.5, 10% (v/v) glycerol and 0.01% LMNG, 0.001% CHS or 0.017% DDM and 0.0034% CHS). HRV 3C protease was added to the column and incubated for 1 h before the protein of interest was eluted from the column with wash buffer. The protein was concentrated on an Amicon Ultra centrifugal filter (50-kDa molecular mass cutoff; Merck Millipore). For cryo-EM samples, the appropriate Fab, expressed and purified as described below, was added followed by anti-κ light chain nanobody, expressed and purified as described below, in a 1:2:4 molar ratio of protein, Fab and nanobody, respectively. The complex was allowed to assemble for at least 10 min on ice before the next steps were carried out.
For LMNG–CHS-reconstituted samples, SEC was performed using a Superdex 200 increase 10/300 column (GE Healthcare) equilibrated in SEC buffer without detergent (150 mM sodium chloride and 20 mM HEPES pH 7.5) and run at 0.5 ml min−1. For DDM–CHS-reconstituted samples, SEC was performed using a Superdex 200 increase 10/300 column (GE Healthcare) equilibrated in SEC buffer with detergent (150 mM sodium chloride, 20 mM HEPES pH 7.5, 0.017% DDM and 0.0034% CHS) and run at 0.5 ml min−1. Fractions containing the protein of interest were concentrated as before and either directly used or flash-frozen in liquid nitrogen and stored at −80 °C. For nanodisc-reconstituted samples, DDM–CHS-reconstituted HsALG9 in complex with Fab and elbow nanobody was mixed with membrane scaffold protein 1D1 (ref. 73) and porcine liver polar lipid extract (Avanti Polar Lipids) mixed 4:1 (w/w) with cholesterol (Avanti Polar Lipids) in a 1:5:120 molar ratio. The lipids were mixed in chloroform, dried, washed in diethyl ether, dried, resuspended in nanodisc buffer (20 mM HEPES pH 7.5 and 150 mM sodium chloride) and stored at −80 °C until needed74. Before nanodisc assembly, the lipid suspension was thawed and 1% DDM and 0.2 % CHS were added; the mixture was sonicated for approximately 1 h. The lipids were mixed with protein and incubated for 5 min at room temperature with rotation before adding the membrane scaffold protein and incubating for a further 20 min at room temperature. Activated Bio-Beads SM-2 polystyrene beads (Bio-Rad) were added to 0.8 g (wet weight) of Bio-Beads per ml of sample and incubated with rotation at room temperature for 10 min before being moved to 4 °C overnight74. Following nanodisc reconstitution, the sample was further purified using a Superdex 200 increase 10/300 column (GE Healthcare) equilibrated in nanodisc buffer and run at 0.5 ml min−1. Fractions containing the protein of interest were concentrated as before and used to prepare cryo-EM grids.
Chemoenzymatic substrate synthesis
Acceptor substrates were enzymatically extended from chemically synthesized Dol25-PP-GlcNAc2 (refs. 34,35,75). Dol25-PP-GlcNAc2 was extended to Dol25-PP-GlcNAc2Man5 in a one-pot reaction with ScALG1, HsALG2 and ScALG11 (ref. 34). ScALG3, HsALG9 and GgALG12 were then used to further extend the acceptor substrate as desired. All enzymes were extracted and purified in DDM–CHS detergents.
A 1:400 molar ratio of enzyme to acceptor substrate was used for ScALG3, HsALG9 and GgALG12 with an excess of chemically synthesized Dol25-P-Man donor substrate or with submolar amounts of donor substrate regenerated in situ35,36. Donor recycling was accomplished using DPMS from Pyrococcus furiosus, GDP-mannose and 10 mM MgCl2 (ref. 36). DPMS was used at a 1:100 molar ratio of enzyme to acceptor substrate. Reactions were incubated at room temperature overnight in 150 mM sodium chloride, 20 mM HEPES pH 7.5, 0.017% DDM and 0.0034% CHS. Donor product Dol25-P was removed after synthesis, as needed, by the transfer of GlcNAc-1-phosphate from UDP-GlcNAc to Dol25-P using AglH (UniProt P39465) from Sulfolobus acidocaldarius at 45 °C overnight36. AglH was included at a concentration of ~2–3 µM with excess UDP-GlcNAc36. LLOs were purified as needed with solvent extraction of lyophilized product using a 2:1 volumetric ratio of chloroform to methanol, followed by a 10:10:2 volumetric ratio of chloroform to methanol to water34.
Activity assays with ScALG3, HsALG9 and GgALG12
Mutants were prepared using site-directed mutagenesis and enzyme variants were purified as for the WT enzyme. Activity assays were performed with 100 nM WT or enzyme variant, 10 µM acceptor substrate and 20 µM donor substrate. Reactions were carried out at room temperature in reaction buffer (150 mM sodium chloride, 20 mM HEPES pH 7.5, 0.01% LMNG and 0.001% CHS) for 30 s before being heated to 95 °C for 10 min unless otherwise noted. For activity assays with Fabs, the protein was preincubated with a tenfold molar excess of Fab before the addition of substrates and reacted for 60 s before heating. The resulting LLO samples were then transferred to a peptide fluorescently labeled with carboxytetramethylrhodamine (TAMRA) using TbSTT3B (ref. 36). The peptide had the sequence: TAMRA-YANATS-NH2 (Genscript), with the glycosylated asparagine residue shown in bold. Briefly, 200 nM TbSTT3B was incubated with 5 µM LLO and 10 µM peptide in transfer buffer (150 mM sodium chloride, 20 mM HEPES pH 7.5, 10 mM MnCl2, 0.035% DDM and 0.007% CHS) at 30 °C for at least 4 h. The glycopeptides were then separated using tricine–SDS–PAGE and imaged with a fluorescence scanner76.
Liquid chromatography (LC)–MS/MS glycopeptide analysis was performed at the Functional Genomics Center Zürich. The samples were diluted 20-fold with 0.1% formic acid before injection of 2 µl on a calibrated Fusion Lumos MS instrument (Thermo Fischer Scientific) coupled to a nano-Acquity ultrahigh-performance LC system (Waters). Peptides were loaded onto a nanoEase M/Z Symmetry C18 trap column (180 µm × 20 mm, 100 Å, particle size: 5 μm) and separated on a nanoEase M/Z HSS C18 T3 column (75 μm × 150 mm, 100 Å, particle size: 1.8 μm), at a constant flow rate of 300 nl min−1, with a column temperature of 50 °C and a linear gradient of 2% to 35% acetonitrile with 0.1% formic acid in 20 min and then 35% to 98% acetonitrile with 0.1% formic acid in 5 min, before being held isocratically for another 5 min. The MS instrument was operated in data-dependent acquisition mode; one scan cycle comprised a full-scan MS survey spectrum, followed by up to 12 sequential higher-energy collision-induced dissociation (HCD) MS/MS runs on the most intense signals above a threshold of 1 × 104. Full-scan MS spectra (300–1,800 m/z) were acquired in the FT-Orbitrap at a resolution of 120,000 at 400 m/z, while HCD MS/MS spectra were recorded in the FT-Orbitrap at a resolution of 60,000 at 400 m/z. HCD was performed with a target value of 1 × 105 and a normalization collision energy of 32 was applied. Automatic gain control target values were 5 × 105 for full Fourier-transform MS. For all experiments, dynamic exclusion was used with a single repeat count, repeat duration of 15 s and exclusion duration of 30 s.
Assays to detect donor hydrolysis by HsALG9-D82A and GgALG12-E35Q
First, the Dol25-P-Man donor substrate was titrated to determine the minimum concentration needed to completely extend a given concentration of acceptor substrate. This concentration was then used to determine whether donor hydrolysis was occurring. HsALG9-D82A was incubated at 16 µM with 8 µM Dol25-PP-GlcNAc2Man6 and 12 µM Dol25-P-Man for 1 h on ice, whereas GgALG12-E35Q was incubated at 10 µM with 5 µM Dol25-PP-GlcNAc2Man7 and 10 µM Dol-P-Man for 1 h on ice. By incubating with an excess of enzyme, we reasoned that our system could be used to detect donor hydrolysis even in the absence of substrate turnover. The various substrate combinations and concentrations were also incubated without enzyme. The timeframe of 1 h on ice was chosen to simulate the maximum time the proteins would be mixed with substates before vitrification for cryo-EM. Experiments with HsALG9-D82A were performed with protein purified in reaction buffer (150 mM sodium chloride and 20 mM HEPES pH 7.5) with DDM–CHS while GgALG12-E35Q was performed using protein purified with LMNG–CHS. Following incubation on ice, the samples were heated to 95 °C for 10 min and then cooled. The samples with and without enzyme were then divided into three tubes to determine whether donor hydrolysis had occurred. To the first tube, nothing was added. To the second tube, 1 µM HsALG9 WT was added to samples with Dol25-PP-GlcNAc2Man6 or 1 µM GgALG12 WT was added to samples with Dol25-PP-GlcNAc2Man7. The third tube was treated as the second tube but with the addition of 20 µM Dol-P-Man. These tubes were incubated for 10 min at room temperature for mannose transfer to occur. The resulting LLOs were transferred to a fluorescently labeled peptide (5-FAM-GSDANYTYTQ-NH2) using TbSTT3A (ref. 77). Briefly, 200 nM TbSTT3A was incubated with 5 µM LLO and 10 µM peptide in transfer buffer (150 mM sodium chloride, 20 mM HEPES pH 7.5, 10 mM MnCl2, 0.035% DDM and 0.007% CHS). The glycopeptides were then separated using tricine–SDS–PAGE and imaged with a fluorescence scanner76. If we observed full conversion of acceptor substrate, we interpreted this as indicating that hydrolysis of donor substrate did not occur during the incubation with the inactive enzyme variant.
Generation of biotinylated ScALG3, HsALG9 and GgALG12 for phage display
ScALG3, HsALG9 and GgALG12 constructs were modified using restriction-free cloning to include the 15-aa AviTag sequence78. Proteins were expressed and purified as without the AviTag in LMNG–CHS detergent. Purified protein was biotinylated overnight at 4 °C with Escherichia coli BirA (protein-to-BirA molar ratio ranging from 1:1 to 5:1) in biotinylation buffer (150 mM sodium chloride, 50 mM bicine pH 8.3, 10 mM magnesium acetate, 10 mM ATP, 250 µM biotin, 0.01% (w/v) LMNG and 0.001% (w/v) CHS). Following biotinylation, SEC was performed on a Superdex 200 increase column (300/10) equilibrated in SEC buffer (150 mM sodium chloride, 20 mM HEPES pH 7.5, 0.01% (w/v) LMNG and 0.001% (w/v) CHS). The level of biotinylation was evaluated by pulldown using Streptavidin MagneSphere paramagnetic particles (Promega). Biotinylated protein was flash-frozen in liquid nitrogen and stored at −80 °C until needed.
Phage display selection
AviTagged ScALG3, HsALG9 and GgALG12, biotinylated through the AviTag, were used for phage display selection. Before the campaign, the labeling (biotinylation) efficiency of the protein was verified by a pulldown assay on Streptavidin magnetic beads. Phage display selection was performed at 4 °C according to published protocols39. The selection buffer was 25 mM HEPES pH 7.5, 150 mM NaCl, 0.02% LMNG, 0.004% CHS and 0.5% BSA. In the first round, 250 nM of target was immobilized on 250 µl of Streptavidin magnetic beads. Then, 100 μl of a phage library E40 was added to the target bound to the Streptavidin beads and incubated for 30 min. The resuspended beads containing bound phages were washed extensively and then used to infect log phase E. coli XL1-Blue cells. Phages were amplified overnight in 2×YT medium with 50 µg ml−1 ampicillin and 109 plaque-forming units per ml of M13-KO7 helper phage. To obtain binders of high affinity and specificity, four additional rounds of selection were performed while decreasing the target concentration in each round, with the final concentration of the target being 10 nM in the fifth round of selection for all three target proteins. In each round, the amplified pool of phages from the preceding round was used as the input. From the second round onward, the bound phages were eluted using 100 mM glycine pH 2.7. This elution technique often results in the elution of nonspecific and Streptavidin binders. To eliminate them, the precipitated phage pool from the second round onward was negatively selected against 50 µl of Streptavidin magnetic beads before adding to the target. The precleared phage pool was then used as input for the selection.
Single-point phage ELISA
The ELISA experiments were performed at 4 °C in 96-well plates coated with 50 µl of 2 µg ml−1 neutravidin in Na2CO3 buffer (pH 9.6) and subsequently blocked by 1.0% BSA in PBS. A single-point phage ELISA was used to rapidly screen the binding of the obtained Fab fragments displayed on phage. Colonies of E. coli XL1-Blue harboring phagemids from the fifth round of selection were inoculated directly into 500 μl of 2×YT broth supplemented with 100 μg ml−1 ampicillin and M13-KO7 helper phage. The cultures were grown overnight at 37 °C in a 96-deep-well block plate. The ELISA buffer was identical to that used in selection. The experimental wells in the ELISA plates were incubated with 25 nM of respective biotinylated target proteins (ALG3, ALG9 and ALG12) in ELISA buffer for 20 min. Only buffer was added to the control wells. Overnight culture supernatants containing Fab phage were diluted tenfold in ELISA buffer. The diluted phage supernatants were then transferred to ELISA plates that were preincubated with biotinylated target and washed with ELISA buffer. The ELISA plates were incubated with the phage for another 20 min and then washed with ELISA buffer. The washed ELISA plates were incubated with a 1:1 mixture of mouse anti-M13 monoclonal antibody (GE, 27-9420-01; 1:5,000 dilution in ELISA buffer) and peroxidase-conjugated goat anti-mouse IgG (Jackson Immunoresearch, 115-035-003; 1:5,000 dilution in ELISA buffer) for 30 min. The plates were washed again, developed with TMB substrate and then quenched with 1.0 M HCl; the absorbance at 450 nm was determined. The background binding of the phage was monitored by the absorbance from the control wells. The sequences of the clones exhibiting high binding over background were verified by DNA sequencing.
Expression and purification of Fabs
Fabs were expressed in E. coli (BL12 (DE3) or C43) in Luria–Bertani or ZYP-5052 autoinduction medium at 30 °C overnight79. Fabs were purified using a 5-ml protein-G column and dialyzed into buffer (150 mM sodium chloride and 20 mM HEPES pH 7.5)79.
Multipoint protein ELISA for determination of half-maximal effective concentration (EC50)
Multipoint ELISA was performed at 4 °C to estimate the affinity of the Fabs to ALG3, ALG9 and ALG12. The ELISA buffer consisted of 25 mM HEPES pH 7.4, 150 mM NaCl, 0.02% LMNG and 0.004% CHS supplemented with 0.5% BSA. First, 25 nM of target immobilized on a neutravidin-coated ELISA plate was incubated with threefold serial dilutions of the purified Fabs starting from 4 μM for 20 min. The plates were washed and the bound target-Fab complexes were incubated with a secondary horseradish-peroxidase-conjugated Pierce recombinant protein L (Thermo Fisher, 32420; 1:5,000 dilution in ELISA buffer) for 30 min. The plates were again washed, developed with TMB substrate and quenched with 1.0 M HCl; the absorbance at 450 nm was determined. To determine the affinities, the data were fitted to a dose–response sigmoidal function in GraphPad Prism and EC50 values were calculated.
Expression and purification of anti-κ light chain nanobody
The anti-κ light chain nanobody42 was expressed using E. coli BL21 (DE3) cells with ZYP-5052 autoinduction medium80 and was purified by nickel affinity chromatography81.
Cryo-EM
For preparation of cryo-EM grids, detergent-reconstituted ScALG3 and GgALG12 were concentrated to 21 or 33 µM, respectively. For the ScALG3 sample, 50 µM Dol25-PP-GlcNAc2Man5 and 100 µM Dol25-P-Man were added before vitrification. For GgALG12, 150 µM Dol25-PP-GlcNAc2Man7 and 300 µM Dol25-P-Man were included before plunging the grids. For the detergent-reconstituted samples, 100 µM fluorinated octyl-maltoside (Anatrace) was added directly before plunging. HsALG9, reconstituted in nanodiscs, was concentrated to 12 µM before the addition of 50 µM Dol25-PP-GlcNAc2Man6 or Dol25-PP-GlcNAc2Man8 acceptor substrate and 100 µM Dol25-P-Man donor substrate. For all datasets collected, Quantifoil holey carbon film (R1.2/1.3) 300-mesh grids with gold support were glow-discharged before the addition of 3 µl of sample. Vitrification was performed using a Vitrobot Mark IV (Thermo Fisher) set to 4 °C and 100% relative humidity. Grids were blotted for 2.5–4.5 s before plunging into liquid propane and ethane.
Datasets were collected at the ScopeM facility at ETH Zürich using a 300-kV Titan Krios (Thermo Fischer Scientific) transmission EM instrument equipped with a K3 Summit direct electron detector (Gatan) and a GIF Quantum energy filter (Gatan) operated at a slit width of 12 or 20 keV. Fully automated collection of movies was performed in EPU2 (Thermo Fischer Scientific) with a defocus range set from −0.6 to −2.8 µm or from −0.6 to −2.2 µm. The HsALG9 second ternary complex and the ALG12 datasets were collected in correlated double-sampling mode. Data collection parameters for each dataset are listed in Extended Data table 1.
Cryo-EM data processing
Cryo-EM data were processed using cryoSPARC version 4 (ref. 82) as shown in Extended Data Fig. 3. Similar processing strategies were used for each dataset. Beam-inducted motion correction and contrast transfer function (CTF) estimation were performed using patch motion correction and patch CTF. Micrographs with a CTF fit resolution worse than 4.5 Å for the ScALG3 and HsALG9 datasets or 3.5 Å for the GgALG12 dataset were discarded. Particles were initially picked using the blob picker on a subset of micrographs before two-dimensional (2D) classification and ab inito reconstruction was used to create an initial model for template-based particle picking of the full dataset. Particles were initially extracted with a box size of 432 or 512 pixels and 2× or 4× binning. Iterative heterogeneous refinement with two or three classes was used with or without 2D classification to select the best particles with bound Fab. The best particles were reextracted without binning and taken forward for nonuniform refinement. Three-dimensional classification, reference-based per-particle motion correction, per-particle and per-group CTF optimization, masking and local refinement were used as needed to improve resolution. Refined maps were either sharpened in cryoSPARC or using DeepEMhancer83.
Model building and refinement
Models were initially generated de novo using models for the ALG enzymes predicted by ModelAngelo84 or AlphaFold2 (ref. 85), while an anti-κ light chain nanobody–Fab complex model (PDB 6WW2)86 was used for the Fab and nanobody regions of the complex. The models were rigid-body fitted into the map using ChimeraX87 and manually build as required in Coot88. Models were initially refined using Rosetta-based refinement and local density-guided optimization implemented in StarMap89,90,91. Ligand restraints were prepared using AceDRG92 and models were iteratively refined in the PHENIX software suite93 using phenix.real_space_refine94, followed by manual manipulation in Coot. Models were evaluated using MolProbity95 and validation tools implemented in PHENIX96. Figures were prepared using ChimeraX and Adobe Illustrator. Sequences were aligned using Clustal Omega97 within the EMBL-EBI Job Dispatcher98 and prepared for publication using ESPript 3.0 (ref. 99) and Adobe Illustrator.
MD simulation and analysis
The initial model for the HsALG9 MD simulations with donor substrate (Dol25-P-Man) and the second acceptor substrate (Dol25-PP-GlcNAc2Man8) was taken from the EM-refined model. The complex was embedded in a lipid bilayer comprising 400 phosphatidylcholine (POPC) molecules and solvated in a water box (distance from the top of HsALG9 to the wall = 22.5 Å) with 0.15 M sodium chloride using CHARMM-GUI100 and the PPM server101, leading to a box size of 125.86 Å × 126.20 Å × 103.07 Å. The D82A substitution was restored to WT and the disulfide bond between C414 and C480 was built using tleap from AmberTools22 (ref. 102). The atomic structure of the unresolved mannose was manually added onto the donor product with the donor anomeric carbon facing toward the oxygen of the acceptor C2 hydroxyl in VMD103. The energy of the system was first minimized by 70,000 steps with positional restraints on the solute atoms with a force constant of 0.418 kJ mol−1 nm−2. The same minimized system was equilibrated five times with different random number seeds for the generation of the initial velocities consisting of a 75-ps NVT simulation and a 650-ps NPT simulation at 303.15 K using Langevin dynamics104 with a friction coefficient of 1 ps−1 and a gradually decreasing force constant for the positional restraints on the solute atoms (from 0.418 kJ mol−1 nm−2 to 0.004 kJ mol−1 nm−2). An 800-ns-long NPT production run was performed starting from each equilibrated system. The printout frequency for analysis was 0.2 ns. For the NPT simulations, an anisotropic Monte Carlo barostat105,106 was used to keep the pressure at 1 bar with a coupling constant of 0.5 ps and a volume change attempted every 0.2 ps. Nonbonded interactions were treated with a cutoff distance of 9 Å and long-range electrostatic interactions were taken into account with the particle mesh Ewald method107,108. The simulations were integrated with a time step of 2 fs and SHAKE109 bond-length constraints on hydrogen with a geometrical tolerance of 10−6. The translational and rotational center-of-mass motion was removed every 2 ps. Atomic interactions were described by the classical force fields AMBER ff19SB (ref. 110) for the proteins, lipid21 (ref. 111) for POPC, GAFF2 (ref. 112) for donor and acceptor substrates and OPC113 for water molecules, with Joung/Cheatham114 ion parameters for ions. Partial charges of the donor and acceptor substrates were assigned with the AM1-BCC115 method using antechamber116. Energy minimization and MD simulations were conducted with the pmemd.MPI and pmemd.cuda engines in Amber22 (University of California, San Francisco), respectively.
Among the five simulations from the initial guess of the donor mannose position, one of them exhibited a stable geometry at the catalytic center. The final frame from this simulation was taken as the initial structure for the subsequent five systems, including Dol25-P-Man, Dol25-P-Glc and Dol25-P-Man-2F with WT HsALG9 and Dol25-P-Man with HsALG9 mutants N288A, H366A and D82A. Five repeats of 800-ns-long simulations were performed following the same procedure as described above. The changes from Dol25-P-Man to Dol25-P-Glc and Dol25-P-2F were performed with PyMol Builder (Schrödinger) and the N288A and H366A substitutions were realized using tleap from AmberTools22 (ref. 102). For MD simulation analysis, hydrogen bonds were defined as being present with default settings in pytraj (acceptor–hydrogen–receptor angle cutoff of 135° and a donor–acceptor cutoff of 3 Å)117.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Models and EM maps for the ScALG3 ternary complex, the first ternary complex of HsALG9, the GgALG12 ternary complex and the second ternary complex of HsALG9 were deposited to the Protein Data Bank with the accession codes 9S6R, 9S6S, 9S6T and 9S6U, respectively, and the EM Data Bank with accession codes EMD-54629, EMD-54630, EMD-54631 and EMD-54632, respectively. The MS glycoproteomics data generated in this study were deposited to the ProteomeXchange Consortium through the PRIDE118 partner repository with the dataset identifier PXD072144. Source data are provided with this paper.
Code availability
The MD topology files, input files, simulation scripts and analysis scripts are freely available from GitHub (https://github.com/rinikerlab/ALG_mannosyltransferase). Because of the large file size (>20 GB without solvent), the tMD trajectories used in this work can be provided upon requests to sriniker@ethz.ch. The solvent-stripped MD trajectories used in this work are provided on Zenodo (https://doi.org/10.5281/zenodo.17973165)119.
References
Aebi, M. N-linked protein glycosylation in the ER. Biochim. Biophys. Acta 1833, 2430–2437 (2013).
Apweiler, R., Hermjakob, H. & Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1473, 4–8 (1999).
Breitling, J. & Aebi, M. N-linked protein glycosylation in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 5, a013359 (2013).
Nakagawa, H. et al. Disruption of N-linked glycosylation enhances ubiquitin-mediated proteasomal degradation of the human ATP-binding cassette transporter ABCG2. FEBS J. 276, 7237–7252 (2009).
Cherepanova, N., Shrimal, S. & Gilmore, R. N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 41, 57–65 (2016).
Aebi, M., Bernasconi, R., Clerc, S. & Molinari, M. N-glycan structures: recognition and processing in the ER. Trends Biochem. Sci. 35, 74–82 (2010).
Varki, A. Biological roles of glycans. Glycobiology 27, 3–49 (2017).
Zhang, Q., Ma, C., Chin, L. S. & Li, L. Integrative glycoproteomics reveals protein N-glycosylation aberrations and glycoproteomic network alterations in Alzheimer’s disease. Sci. Adv. 6, eabc5802 (2020).
Paprocka, J., Jezela-Stanek, A., Tylki-Szymańska, A. & Grunewald, S. Congenital disorders of glycosylation from a neurological perspective. Brain Sci. 11, 88 (2021).
Williams, S. E., Mealer, R. G., Scolnick, E. M., Smoller, J. W. & Cummings, R. D. Aberrant glycosylation in schizophrenia: a review of 25 years of post-mortem brain studies. Mol. Psychiatry 25, 3198–3207 (2020).
Cai, G. et al. The role of N-glycosylation in the stability, trafficking and GABA-uptake of GABA-transporter 1. FEBS J. 272, 1625–1638 (2005).
Wesener, D. A., Dugan, A. & Kiessling, L. L. Recognition of microbial glycans by soluble human lectins. Curr. Opin. Struct. Biol. 44, 168–178 (2017).
Chang, I. J., He, M. & Lam, C. T. Congenital disorders of glycosylation. Ann. Transl. Med. 6, 477 (2018).
Ng, B. G., Freeze, H. H., Himmelreich, N., Blau, N. & Ferreira, C. R. Clinical and biochemical footprints of congenital disorders of glycosylation: proposed nosology. Mol. Genet. Metab. 142, 108476 (2024).
Gao, X. D., Moriyama, S., Miura, N., Dean, N. & Nishimura, S. I. Interaction between the C termini of ALG13 and ALG14 mediates formation of the active UDP-N-acetylglucosamine transferase complex. J. Biol. Chem. 283, 32534–32541 (2008).
Dong, Y. Y. et al. Structures of DPAGT1 explain glycosylation disease mechanisms and advance TB antibiotic design. Cell 175, 1045–1058 (2018).
Helenius, J. et al. Translocation of lipid-linked oligosaccharides across the ER membrane requires Rft1 protein. Nature 415, 447–450 (2002).
Chen, S. et al. Rft1 catalyzes lipid-linked oligosaccharide translocation across the ER membrane. Nat. Commun. 15, 5157 (2024).
Ramirez, A. S. & Locher, K. P. Structural and mechanistic studies of the N-glycosylation machinery: from lipid-linked oligosaccharide biosynthesis to glycan transfer. Glycobiology 33, 861–872 (2023).
Aebi, M., Gassenhuber, J., Domdey, H. & Te Heesen, S. Cloning and characterization of the ALG3 gene of Saccharomyces cerevisiae. Glycobiology 6, 439–444 (1996).
Huffaker, T. C. & Robbins, P. W. Yeast mutants deficient in protein glycosylation. Proc. Natl Acad. Sci. USA 80, 7466–7470 (1983).
Frank, C. G. & Aebi, M. ALG9 mannosyltransferase is involved in two different steps of lipid-linked oligosaccharide biosynthesis. Glycobiology 15, 1156–1163 (2005).
Burda, P. et al. Stepwise assembly of the lipid-linked oligosaccharide in the endoplasmic reticulum of Saccharomyces cerevisiae: identification of the ALG9 gene encoding a putative mannosyl transferase. Proc. Natl Acad. Sci. USA 93, 7160–7165 (1996).
Burda, P., Jakob, C. A., Beinhauer, J., Hegemann, J. H. & Aebi, M. Ordered assembly of the asymmetrically branched lipid-linked oligosaccharide in the endoplasmic reticulum is ensured by the substrate specificity of the individual glycosyltransferases. Glycobiology 9, 617–625 (1999).
Reiss, G., Te Heesen, S., Zimmerman, J., Robbins, P. W. & Aebi, M. Isolation of the ALG6 locus of Saccharomyces cerevisiae required for glucosylation in the N-linked glycosylation pathway. Glycobiology 6, 493–498 (1996).
Stagljar, I., Te Heesen, S. & Aebi, M. New phenotype of mutations deficient in glucosylation of the lipid-linked oligosaccharide: cloning of the ALG8 locus. Proc. Natl Acad. Sci. USA 91, 5977–5981 (1994).
Burda, P. & Aebi, M. The ALG10 locus of Saccharomyces cerevisiae encodes the α-1,2 glucosyltransferase of the endoplasmic reticulum: the terminal glucose of the lipid-linked oligosaccharide is required for efficient N-linked glycosylation. Glycobiology 8, 455–462 (1998).
Kornfeld, R. & Kornfeld, S. Assembly of asparagine-linked oligosaccharides. Ann. Rev. Biochem. 54, 631–664 (1985).
Ramírez, A. S. et al. Molecular basis for glycan recognition and reaction priming of eukaryotic oligosaccharyltransferase. Nat. Commun. 13, 7296 (2022).
Ramírez, A. S., Kowal, J. & Locher, K. P. Cryo-electron microscopy structures of human oligosaccharyltransferase complexes OST-A and OST-B. Science 366, 1372–1375 (2019).
Wild, R. et al. Structure of the yeast oligosaccharyltransferase complex gives insight into eukaryotic N-glycosylation. Science 359, 545–550 (2018).
Kawate, T. & Gouaux, E. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 14, 673–681 (2006).
Eichler, J. & Imperiali, B. Stereochemical divergence of polyprenol phosphate glycosyltransferases. Trends Biochem. Sci. 43, 10–17 (2018).
Ramírez, A. S. et al. Chemo-enzymatic synthesis of lipid-linked GlcNAc2Man5 oligosaccharides using recombinant ALG1, ALG2 and ALG11 proteins. Glycobiology 27, 726–733 (2017).
Bloch, J. S. et al. Structure and mechanism of the ER-based glucosyltransferase ALG6. Nature 579, 443–447 (2020).
Rossi, L., Alexander, J. A. N., Ramirez, A. S. & Locher, K. P. GLYCO-BUILD: an enzymatic pipeline for the synthesis of peptides carrying eukaryotic N-glycans. Nat. Commun. 17, 359 (2026).
Nygaard, R., Kim, J. & Mancia, F. Cryo-electron microscopy analysis of small membrane proteins. Curr. Opin. Struct. Biol. 64, 26–33 (2020).
Fairhead, M. & Howarth, M. Site-specific biotinylation of purified proteins using BirA. Methods Mol. Biol. 1266, 171–184 (2015).
Paduch, M. et al. Generating conformation-specific synthetic antibodies to trap proteins in selected functional states. Methods 60, 3–14 (2013).
Miller, K. R. et al. T cell receptor-like recognition of tumor in vivo by synthetic antibody fragment. PLoS ONE 7, e43746 (2012).
Alexander, J. A. N. & Locher, K. P. Emerging structural insights into C-type glycosyltransferases. Curr. Opin. Struct. Biol. 79, 102547 (2023).
Ereño-Orbea, J. et al. Structural basis of enhanced crystallizability induced by a molecular chaperone for antibody antigen-binding fragments. J. Mol. Biol. 430, 322–336 (2018).
Oriol, R., Martinez-Duncker, I., Chantret, I., Mollicone, R. & Codogno, P. Common origin and evolution of glycosyltransferases using Dol-P-monosaccharides as donor substrate. Mol. Biol. Evol. 19, 1451–1463 (2002).
Drula, E. et al. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 50, D571–D577 (2022).
Hudson, K. L. et al. Carbohydrate–aromatic interactions in proteins. J. Am. Chem. Soc. 137, 15152–15160 (2015).
Stankovic, I. M., Blagojevic Filipovic, J. P. & Zaric, S. D. Carbohydrate–protein aromatic ring interactions beyond CH/π interactions: a Protein Data Bank survey and quantum chemical calculations. Int. J. Biol. Macromol. 157, 1–9 (2020).
Petrou, V. I. et al. Structures of aminoarabinose transferase ArnT suggest a molecular basis for lipid A glycosylation. Science 351, 608–612 (2016).
Zhang, L. et al. Structures of cell wall arabinosyltransferases with the anti-tuberculosis drug ethambutol. Science 368, 1211–1219 (2020).
Tan, Y. Z. et al. Cryo-EM structures and regulation of arabinofuranosyltransferase AftD from mycobacteria. Mol. Cell 78, 683–699 (2020).
Bloch, J. S. et al. Structure, sequon recognition and mechanism of tryptophan C-mannosyltransferase. Nat. Chem. Biol. 19, 575–584 (2023).
Chiu, C. P. et al. Structural analysis of the sialyltransferase CstII from Campylobacter jejuni in complex with a substrate analog. Nat. Struct. Mol. Biol. 11, 163–170 (2004).
Persson, K. et al. Crystal structure of the retaining galactosyltransferase LgtC from Neisseria meningitidis in complex with donor and acceptor sugar analogs. Nat. Struct. Biol. 8, 166–175 (2001).
Pedersen, L. C., Darden, T. A. & Negishi, M. Crystal structure of β1,3-glucuronyltransferase I in complex with active donor substrate UDP-GlcUA. J. Biol. Chem. 277, 21869–21873 (2002).
Pedersen, L. C. et al. Crystal structure of an α1,4-N-acetylhexosaminyltransferase (EXTL2), a member of the exostosin gene family involved in heparan sulfate biosynthesis. J. Biol. Chem. 278, 14420–14428 (2003).
Gagnon, S. M. L. et al. High resolution structures of the human ABO(H) blood group enzymes in complex with donor analogs reveal that the enzymes utilize multiple donor conformations to bind substrates in a stepwise manner. J. Biol. Chem. 290, 27040–27052 (2015).
Moremen, K. W. & Haltiwanger, R. S. Emerging structural insights into glycosyltransferase-mediated synthesis of glycans. Nat. Chem. Biol. 15, 853–864 (2019).
Ramirez, A. S. et al. Structural basis of the molecular ruler mechanism of a bacterial glycosyltransferase. Nat. Commun. 9, 445 (2018).
Hosokawa, N., Kamiya, Y., Kamiya, D., Kato, K. & Nagata, K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem. 284, 17061–17068 (2009).
Helenius, A. & Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 (2004).
Sousa, M. C., Ferrerogarcia, M. A. & Parodi, A. J. Recognition of the oligosaccharide and protein moieties of glycoproteins by the UDP-Glc-glycoprotein glucosyltransferase. Biochemistry 31, 97–105 (1992).
Grinna, L. S. & Robbins, P. W. Substrate specificities of rat-liver microsomal glucosidases which process glycoproteins. J. Biol. Chem. 255, 2255–2258 (1980).
Grubenmann, C. E. et al. ALG12 mannosyltransferase defect in congenital disorder of glycosylation type lg. Hum. Mol. Genet. 11, 2331–2339 (2002).
Frank, C. G. et al. Identification and functional analysis of a defect in the human ALG9 gene: definition of congenital disorder of glycosylation type IL. Am. J. Hum. Genet. 75, 146–150 (2004).
Sun, L. et al. Congenital disorder of glycosylation Id presenting with hyperinsulinemic hypoglycemia and islet cell hyperplasia. J. Clin. Endocrinol. Metab. 90, 4371–4375 (2005).
Weinstein, M. et al. CDG-IL: an infant with a novel mutation in the ALG9 gene and additional phenotypic features. Am. J. Med. Genet. 136, 194–197 (2005).
Vleugels, W. et al. Quality control of glycoproteins bearing truncated glycans in an ALG9-defective (CDG-IL) patient. Glycobiology 19, 910–917 (2009).
Eklund, E. A. et al. Molecular and clinical description of the first US patients with congenital disorder of glycosylation Ig. Mol. Genet. Metab. 84, 25–31 (2005).
Chantret, I. et al. Congenital disorders of glycosylation type Ig is defined by a deficiency in dolichyl-P-mannose:Man7GlcNAc2-PP-dolichyl mannosyltransferase. J. Biol. Chem. 277, 25815–25822 (2002).
Kranz, C. et al. Expanding spectrum of congenital disorder of glycosylation Ig (CDG-Ig): sibs with a unique skeletal dysplasia, hypogammaglobulinemia, cardiomyopathy, genital malformations, and early lethality. Am. J. Med. Genet. A 143A, 1371–1378 (2007).
Schumann, B. et al. Bump-and-hole engineering identifies specific substrates of glycosyltransferases in living cells. Mol. Cell 78, 824–834 (2020).
van den Ent, F. & Löwe, J. RF cloning: a restriction-free method for inserting target genes into plasmids. J. Biochem. Biophys. Methods 67, 67–74 (2006).
Molday, L. L. & Molday, R. S. 1D4: a versatile epitope tag for the purification and characterization of expressed membrane and soluble proteins. Methods Mol. Biol. 1177, 1–15 (2014).
Bayburt, T. H., Grinkova, Y. V. & Sligar, S. G. Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett. 2, 853–856 (2002).
Geertsma, E. R., Nik Mahmood, N. A., Schuurman-Wolters, G. K. & Poolman, B. Membrane reconstitution of ABC transporters and assays of translocator function. Nat. Protoc. 3, 256–266 (2008).
Meirelles, M. A. & Reymond, J.-L. Optimizing phosphate couplings for dolichyl diphosphochitobiose to enable protein N-glycosylation studies. Helv. Chim. Acta 106, e202300171 (2023).
Schagger, H. Tricine–SDS–PAGE. Nat. Protoc. 1, 16–22 (2006).
Ramírez, A. S. et al. Characterization of the single-subunit oligosaccharyltransferase STT3A from Trypanosoma brucei using synthetic peptides and lipid-linked oligosaccharide analogs. Glycobiology 27, 525–535 (2017).
Beckett, D., Kovaleva, E. & Schatz, P. J. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 8, 921–929 (1999).
Bloch, J. S. et al. Development of a universal nanobody-binding Fab module for fiducial-assisted cryo-EM studies of membrane proteins. Proc. Natl Acad. Sci. USA 118, e2115435118 (2021).
Studier, F. W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 (2005).
Pardon, E. et al. A general protocol for the generation of nanobodies for structural biology. Nat. Protoc. 9, 674–693 (2014).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Sanchez-Garcia, R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 4, 1–8 (2021).
Jamali, K. et al. Automated model building and protein identification in cryo-EM maps. Nature 628, 450–457 (2024).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Tsutsumi, N. et al. Structure of human Frizzled5 by fiducial-assisted cryo-EM supports a heterodimeric mechanism of canonical Wnt signaling. Elife 9, e58464 (2020).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Casañal, A., Lohkamp, B. & Emsley, P. Current developments in Coot for macromolecular model building of electron cryo-microscopy and crystallographic data. Protein Sci. 29, 1069–1078 (2020).
Lugmayr, W. et al. StarMap: a user-friendly workflow for Rosetta-driven molecular structure refinement. Nat. Protoc. 18, 239–264 (2023).
DiMaio, F. et al. Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement. Nat. Methods 12, 361–365 (2015).
Reggiano, G., Lugmayr, W., Farrell, D., Marlovits, T. C. & DiMaio, F. Residue-level error detection in cryoelectron microscopy models. Structure 31, 860–869 (2023).
Long, F. et al. AceDRG: a stereochemical description generator for ligands. Acta Crystallogr. D Struct. Biol. 73, 112–122 (2017).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in PHENIX. Acta Crystallogr. D Struct. Biol. 75, 861–877 (2019).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 74, 531–531 (2018).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–293 (2018).
Afonine, P. V. et al. New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr. D Struct. Biol. 74, 814–840 (2018).
Sievers, F. & Higgins, D. G. Clustal Omega. Curr. Protoc. Bioinformatics 48, 3.13.1–3.13.16 (2014).
Madeira, F. et al. The EMBL-EBI Job Dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 52, W521–W525 (2024).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I. & Lomize, A. L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 (2012).
Case, D. A. et al. AmberTools. J. Chem. Inf. Model. 63, 6183–6191 (2023).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 27–38 (1996).
Goga, N., Rzepiela, A. J., de Vries, A. H., Marrink, S. J. & Berendsen, H. J. Efficient algorithms for Langevin and DPD dynamics. J. Chem. Theory Comput. 8, 3637–3649 (2012).
Zhang, Y., Feller, S. E., Brooks, B. R. & Pastor, R. W. Computer simulation of liquid/liquid interfaces. I. Theory and application to octane/water. J. Chem. Phys. 103, 10252 (1995).
Åqvist, J., Wennerström, P., Nervall, M., Bjelic, S. & Brandsdal, B. O. Molecular dynamics simulations of water and biomolecules with a Monte Carlo constant pressure algorithm. Chem. Phys. Lett. 384, 288–294 (2004).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald—an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Anderson, H. C. Rattle: A ‘velocity’ version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 52, 24–34 (1983).
Tian, C. et al. ff19SB: amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 16, 528–552 (2020).
Dickson, C. J., Walker, R. C. & Gould, I. R. Lipid21: complex lipid membrane simulations with AMBER. J. Chem. Theory Comput. 18, 1726–1736 (2022).
Wang, J. et al. Development and testing of a general amber force field. J. Comput. Chem. 25, 1157 (2004).
Izadi, S., Anandakrishnan, R. & Onufriev, A. V. Building water models: a different approach. J. Phys. Chem. Lett. 5, 3863–3871 (2014).
Joung, I. S. & Cheatham, T. E. 3rd Molecular dynamics simulations of the dynamic and energetic properties of alkali and halide ions using water-model-specific ion parameters. J. Phys. Chem. B 113, 13279–13290 (2009).
Jakalian, A., Jack, D. B. & Bayly, C. I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 23, 1623–1641 (2002).
Wang, J., Wang, W., Kollman, P. A. & Case, D. A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 25, 247–260 (2006).
Roe, D. R. & Cheatham, T. E. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 (2013).
Perez-Riverol, Y. et al. The PRIDE database at 20 years: 2025 update. Nucleic Acids Res. 53, D543–D553 (2025).
Chen, S.-Y. Structures of ALG3, ALG9, and ALG12 reveal the assembly logic of the N-glycan oligomannose core. Zenodo https://doi.org/10.5281/zenodo.17973165 (2026).
Wu, T. T. & Kabat, E. A. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J. Exp. Med. 132, 211–250 (1970).
Korner, C. et al. Carbohydrate deficient glycoprotein syndrome type IV: deficiency of dolichyl-P-Mans:Man5GlcNAc2-PP-dolichyl mannosyltransferase. EMBO J. 18, 6816–6822 (1999).
Thiel, C. et al. Deficiency of dolichyl-P-Man:Man7GlcNAc2-PP-dolichyl mannosyltransferase causes congenital disorder of glycosylation type Ig. Biochem. J. 367, 195–201 (2002).
Acknowledgements
We thank M. Mikolin for technical assistance and C.-W. Lin and the Functional Genomic Center Zürich for MS analysis. We are appreciative to the ScopeM facility (ETH Zürich) staff for technical support. J.A.N.A. is grateful for salary funding from a Natural Science and Engineering Research Council of Canada postdoctoral fellowship during this work. Research in the K.P.L. group is supported by Swiss National Science Foundation (310030_196862 and 315230_220007 to K.P.L and J.-L.R.). Research in the A.A.K. group is supported by the National Institutes of Health (GM117372 to A.A.K.).
Author information
Authors and Affiliations
Contributions
J.A.N.A. performed the cloning, mutagenesis, homolog screening and selection, protein purification, cryo-EM sample and grid preparation, cryo-EM data processing and model building and refinement. S.Y.C. performed the MD simulations under the supervision of S.R. S.M. performed the Fab library screening, Fab expression and initial Fab characterization with assistance from P.A., under the supervision of A.A.K. J.A.N.A. and L.R. synthesized the substrates used for structural and functional assays and J.A.N.A. performed the enzyme assays. R.N.I. and J.K. collected the cryo-EM data. J.L.R. supervised the chemical synthesis of substrates by M.d.C. and M.A.M. M.A. provided advice and contributed to writing the manuscript. J.A.N.A. and K.P.L. conceptualized the project, analyzed the data and wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemical Biology thanks Daisuke Fujinami and the other, anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Biochemical and functional characterisation of ScALG3, HsALG9 and GgALG12.
a, Size exclusion chromatography of ScALG3, HsALG9 and GgALG12 solubilized in LMNG-CHS detergent. b, SDS-PAGE analysis of ScALG3, HsALG9 and GgALG12. Representative gel shown from multiple (n > 10) independent purifications of ALG3, ALG9 and ALG12. Molecular weight markers are indicated in kDa. c-e, Assays with WT and catalytic mutants of c, ScALG3, d, HsALG9 and e, GgALG12 in LMNG-CHS. The reaction products were transferred to a fluorescent peptide (TAMRA-YANATS-NH2) using TbSTT3B and separated by tricine gel electrophoresis. The reaction times for each sample are indicated above the lanes and are coloured red for inactive, green for active and black for partially active. Lanes with glycopeptide cartoons over them serve as glycopeptide standards for the indicated glycan. Activity assays for each construct were performed at least twice with similar results.
Extended Data Fig. 2 Biochemical characterisation of ScALG3, HsALG9 and GgALG12 Fabs.
a-c, Size exclusion chromatography and SDS-PAGE analysis of ScALG3 D71N with Sc3-16 Fab (a), HsALG9 WT with Hs9-8 Fab (b) and GgALG12 E35Q with Gg12-11 Fab (c). In panels a-c, molecular weight markers are indicated in kDa. All samples in panels a-c included the anti-κ-light-chain nanobody. Purification of ScALG3, HsALG9 and GgALG12 were performed multiple times (n > 10) with similar results. Fractions taken for the SDS-PAGE gels are indicated with red lines. d, Multi-point ELISA binding curves for the indicated Fabs. Data (n = 3 biological replicates for Fab each concentration) are presented as mean values ± SD. e, Activity of ScALG3, HsALG9 and GgALG12 in the presence and absence of the Fabs in panels a-c. The reaction products were transferred to a fluorescent peptide (TAMRA-YANATS-NH2) using TbSTT3B and separated by tricine gel electrophoresis. Lanes with glycopeptide cartoons over them serve as glycopeptide standards for the indicated glycan. Activity assays with Fabs were performed at least three times with similar results. f, Sequences of the complementarity determining regions (CDRs) of each Fab used for structural studies. The Kabat numbering system is used for the Fab CDR residues120. g-i, Cryo-EM density maps of g, ScALG3 ternary, h, HsALG9 first ternary, i, GgALG12 ternary and j, HsALG9 second ternary complexes. The cryo-EM density maps are coloured according to the different components as labelled.
Extended Data Fig. 3 Single particle cryo-EM data collection and processing for ScALG3, HsALG9 and GgALG12 structures.
Panels a-f pertain to the ALG3-ternary complex. a, Representative micrograph. b, Reference-free 2D class averages. c, Processing strategy in cryoSPARC82. d, Gold standard half-map and model-map FSC curves shown in black and blue lines, respectively. The dashed lines are shown at 0.143 and 0.5 FSC. e, Final map coloured by local resolution. f, Particle orientation distribution plot. Panels g-l pertain to the ALG9-first-ternary complex and are listed in order as panels a-f. Panels m-r pertain to the ALG12-ternary complex and are listed in order as panels a-f. The scale bar in panels a, g and m represents 100 nm.
Extended Data Fig. 4 ER luminal mannosyltransferase architecture.
Topology, cartoons in rainbow colouring and surface representation for ScALG3 (a), HsALG9 (b) and GgALG12 (c). The GT-CA conserved module is shown in grey shading while the variable module is shaded in colour. The surface representations show the acceptor substrate binding cavity forms at the interface of the GT-CA variable and conserved modules in CAZy families GT-22 (ALG9 and ALG12) and GT-58 (ALG3). The acceptor and donor substrates are shown as pink or gold spheres, respectively.
Extended Data Fig. 5 Catalytic mechanisms of ALG3, ALG9 and ALG12 and schematic of N-glycan assembly.
Proposed reaction mechanisms for a, B branch initiation by ALG3, b, B branch completion by ALG9 and c, C branch initiation by ALG12. Monosaccharides are labelled according to the nomenclature in Fig. 1a. The acceptor carbon bound to the attacking hydroxyl is indicated in each panel. d, Schematic of eukaryotic oligomannose N-glycan assembly. Bold arrows indicate in vivo reactions, while dashed arrows indicate slow reactions that can be forced in vitro. Reactions that do not occur are indicated with a red cross.
Extended Data Fig. 6 Molecular dynamics (MD) simulations of HsALG9 with Dol25-PP-GlcNAc2Man8 acceptor and Dol25-P-Man donor substrates.
a, Structure of donor substrate Dol25-P-Man, with the hydrophobic tail highlighted in grey, the phosphate group in orange, and the mannose in light green. Root mean square fluctuation (RMSF) of the donor substrate atoms was calculated by aligning all MD frames to the averaged conformation. b, Plots of hydrogen bond formation between the side chains of Asn288 (upper panel) or His366 (lower panel) and the donor substrate C2-OH over the duration of 5 simulations. Data are shown in running average of 1 ns for the clearer visibility. c, Comparison of the active site geometry between the representative frames of the first simulation (blue curve, t = 796.4 ns) and the fifth simulation (purple curve, t = 780 ns). Dol25-P-Man and selected side chains are shown in sticks and labelled. Selected interatomic distances are indicated by the dashed lines with colours corresponding to the respective simulations. d, Distance between the acceptor C2 hydroxyl oxygen and donor anomeric carbon (upper panel) and between the acceptor C2 hydroxyl hydrogen and D82 γ-carbon over the course of the simulation. Results of the five independently generated trajectories are shown by different colours as indicated in panel a. The last frame of the fifth (purple curve) trajectory was taken as the initial template for the subsequent simulations. MD simulations were performed with 5 replicates.
Extended Data Fig. 7 Time plots of molecular dynamics (MD) simulations of WT or mutant HsALG9.
a-c, Donor distance (r) and angle (θ) in the simulations of wild type HsALG9 in complex with acceptor substrate and donor analogues, as defined in Fig. 5a. Shown are the plots for Dol25-P-Man (a), Dol25-P-Glc (b), or Dol25-P-Man-2F (c) over the course of the simulation. d,e Plot of probability for distance (r) (d) and angle (θ) (e) values observed during MD simulations in panels a and b, here filtered by the presence or absence of the intramolecular hydrogen bond between the donor C2-OH and the donor phosphate. Here a hydrogen bond is defined by an acceptor-hydrogen-donor angle cutoff of less than 135° and a donor-acceptor distance of less than 3 Å. f-h Distance (r) and angle (θ) in the simulations of HsALG9 mutant N288A (f), H366A (g) or D82A (h) in complex with acceptor substrate and Dol25-P-Man. In each example, MD simulations were performed with 5 replicates.
Extended Data Fig. 8 Residues involved in donor substrate selection and catalysis in ScALG3, HsALG9 and GgALG12.
a, Superposition of HsALG9 and GgALG12 structures. The zoomed insert shows the active site, with Dol25-P shown as sticks. b, Tricine gel electrophoresis of glycopeptides generated by WT or mutant GgALG12, with conditions indicated above the lanes. c, Tricine gel electrophoresis of glycopeptides generated by WT or mutant ScALG3 WT, with conditions indicated above the lanes. d, Tricine gel electrophoresis of glycopeptides generated using WT or mutant HsALG9. In b-d, following the reaction with ScALG3, HsALG9 or GgALG12, the glycans were transferred to a fluorescently labelled peptide (TAMRA-YANATS-NH2) with TbSTT3B. Labels above the gel are coloured to indicate whether mannose transfer has occurred (green) or has not proceeded or is reduced (red). Lanes with glycopeptide cartoons over them serve as glycopeptide standards for the indicated glycan. Activity assays were performed at least three times with similar results.
Supplementary information
Source data
Source Data Fig. 1 (download PDF )
Unprocessed gels.
Source Data Fig. 4 (download PDF )
Unprocessed gels.
Source Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 1 (download PDF )
Unprocessed gels.
Source Data Extended Data Fig. 2 (download PDF )
Unprocessed gels.
Source Data Extended Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 8 (download PDF )
Unprocessed gels.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alexander, J.A.N., Chen, SY., Mukherjee, S. et al. Structures of ALG3/9/12 reveal the assembly logic of the N-glycan oligomannose core. Nat Chem Biol (2026). https://doi.org/10.1038/s41589-026-02164-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41589-026-02164-7
