
Abstract
Mitochondrial electron transport chain (ETC) function is linked to the generation of ATP, signaling molecules including reactive oxygen species (ROS), pyrimidines and tricarboxylic acid cycle metabolites1. Mitochondrial electron transport is required for T cell proliferation2,3,4. However, which mitochondrial ETC functions are necessary for each dynamic state of CD8+ T cell responses is unknown. Here we report that impairing mitochondrial complex III function, which diminishes respiration, proton pumping linked to ATP production and superoxide production, decreases peripheral naive numbers, antigen-induced CD8+ T cell proliferation and memory formation. Acute stimulation of mitochondrial complex III-deficient CD8+ T cells induced an exhausted-like phenotype. Expression of Ciona intestinalis alternative oxidase (AOX) in mitochondrial complex III-deficient CD8+ T cells restores respiration without generating ROS or proton pumping, and rescues proliferation and the exhausted phenotype but not naive or memory formation. Thus, T cell development, proliferation and memory formation have distinct requirements for mitochondrial complex III ROS.
Similar content being viewed by others


Main
Harnessing CD8+ T cells against cancer and chronic infection has drawn attention to improving their metabolic fitness across their dynamic state changes. Initially, research on T cell metabolic reprogramming focused on how antigen stimulation triggers increased glycolytic flux to support the growth and ATP needs of these highly proliferative cells5,6,7. Subsequent studies revealed that mitochondrial metabolism also increases after T cell activation2,8,9,10,11,12. Indeed, activated T cells require mitochondrial electron transport chain (ETC) complexes II, III and IV for proliferation2,3,4. The mitochondrial ETC performs multiple functions beyond ATP production, including superoxide production, apoptosis regulation via cytochrome c release and the maintenance of tricarboxylic acid (TCA) cycle flux1. However, which of those functions is essential for T cell proliferation and differentiation is not well understood. Previously, we proposed that mitochondrial complex III-generated superoxide, a type of reactive oxygen species (ROS) and signaling molecule, is necessary for T cell activation and proliferation2. Recent work suggests that chronic stimulation-induced T cell exhaustion is attributed to dysfunctional mitochondrially generated ROS13,14,15,16,17. Whether dysfunctional mitochondrial metabolism, in the absence of chronic stimulation, is sufficient to push CD8+ T cells down an exhausted phenotypic trajectory, is not understood. Using genetic modifications of mitochondrial complex III in T cells to isolate the mitochondrial contributions of ROS production from ATP and TCA cycle flux, we found that T cells do not require complex III-derived ROS for activation or proliferation, but it is necessary for establishment and maintenance of naive and memory CD8+ T cell populations. Importantly, in the absence of mitochondrial complex III, acute stimulation resulted in an exhausted-like T cell phenotype, which did not require mitochondrial complex III ROS production.
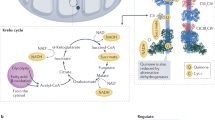
To directly test the hypothesis that mitochondrial complex III-generated ROS are necessary for T cell activation and proliferation, we generated mice lacking mitochondrial complex III ROS production with intact cellular respiration and TCA cycle flux2. Under normal conditions, mitochondrial complexes I and II transfer electrons to ubiquinone (CoQ), converting it into ubiquinol (CoQH2). The process regenerates the NAD+ and FAD essential for the oxidative TCA cycle. Mitochondrial complex III transfers the electrons from ubiquinol to the downstream electron carrier cytochrome c (Fig. 1a). Mitochondrial complex III also pumps protons and serves as a major site for superoxide production (ROS). Dihydroorotate dehydrogenase (DHODH), the rate-limiting enzyme in pyrimidine synthesis, also donates electrons to ubiquinone and consequently requires mitochondrial complex III function. An alternative oxidase from Ciona intestinalis (AOX) can transfer electrons from CoQH2 directly to oxygen, regenerating CoQ, without proton pumping or superoxide production18. Thus, in the absence of complex III, AOX expression allows the mitochondrial complexes I and II, and DHODH, to continue electron transfer to CoQ, to sustain anabolism, without ROS production from complex III19,20,21,22,23. Expression of AOX also prevents CoQ pool over-reduction thus decreasing superoxide production from reverse electron transport at mitochondrial complex I24,25. AOX also bypasses functions linked to cytochrome c and complex IV. Mitochondrial complex III-deficient, AOX-expressing cells rely on mitochondrial complex I for mitochondrial ATP production via proton motive force (Fig. 1a). Therefore, the combination of mitochondrial complex III knockout and AOX expression effectively dampens mitochondrial superoxide production while enabling cellular respiration, nucleotide synthesis and the TCA cycle to proceed as usual. These properties of AOX have been validated by us and others in several contexts18,19,21,26,27.

a, Schematic of WT (top) or RISP KO + AOX (bottom) ETC as in mice with Cre-driven recombination of floxed Uqcrfs1 (RISPfl/fl) and lox-stop-lox AOX (AOXLSL) expression. b–e, Isolated CD8+ T cells of the indicated genotypes were activated in vitro for 48 h then collected for RISP protein normalized to vinculin (b), AOX mRNA expression by qPCR (c), antimycin- and piericidin-corrected basal and ATP-coupled OCR (d), and antimycin, piericidin and SHAM-corrected OCR tracing (e). f, Isolated CD8+ T cells from indicated genotypes were labeled with Cell Trace Violet and anti-CD3ε/CD28 bead + IL-2, stimulated for 96 h then analyzed for the division index. g–j, Isolated CD8+ T cells of the indicated genotypes were activated in vitro, then collected for NAD+/NADH measurements (g), or metabolomics (h–j) measuring dihydroorotate (h), 2-hydroxygluterate (2HG) over α-ketogluterate (α-KG) ratio (i), or succinate over α-KG ratio (j). All data points represent individual mice as biological replicates: n = 7 WT, n = 5 KO, n = 8 KO + AOX (b); n = 5 WT, n = 4 KO, n = 5 KO + AOX (c); n = 6 WT, n = 5 KO, n = 6 KO + AOX (d,e); n = 5 WT, n = 4 KO, n = 3 KO + AOX (f); n = 4 WT, n = 3 KO, n = 6 KO + AOX (g); n = 6 WT, n = 7 KO, n = 5 KO + AOX (h–j). Data are means ± s.e.m., *P < 0.05. Statistical tests for b, c and f–j are one-way ANOVA with Tukey’s multiple-comparison test and for d are two-way ANOVA with Tukey’s multiple-comparison test. P values are: *P = 0.0018 WT/KO, *P = 0.001 WT/KO + AOX (b); *P = 0.0007 WT/KO + AOX, *P = 0.001 KO/KO + AOX (c); *P = < 0.0001 basal WT/KO, *P = < 0.0001, KO/KO + AOX, *P = < 0.0001, coupled WT/KO, *P = 0.0005, WT/KO + AOX, *P = 0.0149, KO/KO + AOX (d); *P = < 0.0001 WT/KO, *P = < 0.0001 KO/KO + AOX (f); *P = 0.0255 WT/KO, *P = 0.0319 KO/KO + AOX (g); *P = 0.0026 WT/KO, *P = 0.0052 KO/KO + AOX (h); *P = 0.0407 WT/KO (i). Panel a created using BioRender.com.
To investigate the necessity of individual mitochondrial ETC functions for T cell proliferation and cell fate, we crossed CD4-Cre mice with mice conditionally lacking the mitochondrial complex III subunit RISP (Uqcrfs1fl/fl), inducing T cell-specific loss of mitochondrial complex III function, herein referred to as RISP KO28. Next, we crossed those with mice conditionally expressing C. intestinalis alternative oxidase (AOXLSL)18. From the CD4-Cre RISPfl/fl AOXLSL mice we used three genotypes: RISP wild-type (WT) (Cre deficient), RISP KO and RISP KO + AOX. To validate the model, we confirmed RISP protein depletion in CD8+ T cells from RISP KO and RISP KO + AOX mice and AOX RNA transcript detection in CD8+ T cells from RISP KO + AOX mice (Fig. 1b–c). Measurement of the oxygen consumption rate (OCR) showed that AOX expression in the absence of mitochondrial complex III function restores basal, but only partially restores ATP-coupled cellular respiration (Fig. 1d). RISP KO cells show elevated basal extracellular acidification rate (ECAR) compared with RISP WT CD8+ T cells, which was not reduced in RISP KO + AOX cells (Extended Data Fig. 1a). RISP KO + AOX OCR is insensitive to mitochondrial complex III inhibition by antimycin but sensitive to AOX inhibition by salicylhydroxamic acid (SHAM) (Fig. 1e). AOX expression in RISP-deficient CD8+ T cells rescued the RISP KO in vitro proliferation defect (Fig. 1f), suggesting that slightly reduced capacity to generate ATP does not limit proliferation. RISP depletion decreased the NAD+/NADH ratio and increased dihydroorotate and the TCA cycle metabolite 2-hydroxyglutarate and, to a lesser extent, succinate (Fig. 1g–j). These data demonstrate that in the absence of complex III function, complex I (NAD+/NADH) and complex II (succinate), as well as oxidative TCA cycle activity, are also inhibited, all of which is restored by AOX expression. However, mitochondrial membrane potential (MMP) displayed no disruption in RISP KO cells (Extended Data Fig. 1b). Notably, cells can reverse ETC complex V whereby ATP is consumed to generate a proton gradient, allowing maintenance of the MMP in the absence of normal ETC function29.
RISP KO mice are able to establish equal number of T cells in the thymus but have fewer naive T cells in the spleen and lymph nodes, which are not rescued by AOX expression (Fig. 2a,b). This suggests that mitochondrial complex III ROS is necessary for T cell population establishment or maintenance in the periphery. To gain additional insight, naive CD8+ T cells were sorted for RNA sequencing (RNA-seq). Compared with RISP WT and RISP KO + AOX, RISP KO showed enrichment of pathways relevant to proliferation and activation, including metabolic, G2M checkpoint, JAK-STAT signaling and integrated stress response (ISR) (Extended Data Fig. 1c,d). The naive population defects were not explained by overt thymic development failures or increased apoptosis as measured by annexin V staining, although apoptotic cells are quickly cleared and this method may not capture minor differences in cell death. A higher percentage of CD8+ T cells in RISP KO mice express CD44, suggesting some dysregulation in the peripheral naive population (Extended Data Fig. 2a–f).

a,b, CD4-Cre RISPfl/fl AOXLSL mice of indicated genotypes were harvested for direct ex vivo quantification from pooled peripheral lymph nodes (a) or spleen (b). c–k, CD4-Cre RISPfl/fl AOXLSL mice of indicated genotypes were infected with LCMV Armstrong and gp33 tetramer-positive cells were tracked over time in peripheral blood (c) then isolated at >40 days post-infection (d.p.i.) for quantification from pooled peripheral lymph nodes (d), quantification from spleen (e) and sorted along with a polyclonal naive CD8+ T cell control population from the same mice from pooled spleen and lymph nodes for RNA-seq (f–k). f, PCA of gp33 tetramer-positive cells from each genotype. g, Heatmap of selected significantly differentially regulated exhaustion genes. h–k, Sorted gp33 tetramer-positive cells analyzed for enrichment of published exhaustion gene expression profiles KO/WT (h) and KO + AOX/WT (i) from Utzschneider et al.37 and KO/WT (j) and KO + AOX/WT (k) from Wherry et. al.36 (j,k). NES, Normalized enrichment score. All data points represent individual mice as biological replicates: n = 21 WT, n = 14 KO, n = 13 KO + AOX (a); n = 21 WT, n = 14 KO, n = 12 KO + AOX (b); n = 9 WT, n = 6 KO, n = 6 KO + AOX (c); n = 7 WT, n = 7 KO, n = 7 KO + AOX (d); n = 7 WT, n = 7 KO, n = 7 KO + AOX (e); n = 5 WT, n = 5 KO, n = 3 KO + AOX (f–k). Data are means ± s.e.m. *P < 0.05, statistical tests are one-way ANOVA with Tukey’s multiple comparisons test (LN CD4+ *P = 0.0205 WT/KO, LN CD8+ *P = < 0.0001 WT/KO, *P = < 0.0001 WT/KO + AOX (a); spleen CD4+ *P = 0.0022 WT/KO, spleen CD8+ *P = < 0.0001 WT/KO, spleen CD8+ *P = 0.0001 WT/KO + AOX (b); *P = 0.0154 WT/KO (d); *P = 0.0002 WT/KO, *P = 0.002 WT/KO + AOX (e)).
To determine whether AOX expression restores activation and proliferation of RISP-deficient CD8+ T cells in vivo, we infected CD4-Cre RISPfl/fl AOXLSL mice of each genotype with LCMV Armstrong and tracked the H-2D(b) LCMV gp33-41 tetramer-positive population (Extended Data Fig. 3a). After infection, the gp33+ population in RISP KO mice failed to expand whereas those in RISP KO + AOX mice proliferated to a similar extent as the RISP WT (Fig. 2c). We quantified memory gp33+CD8+ T cells from lymph nodes and spleen, and sorted CD44higp33+ and naive polyclonal CD8+ T cell populations for RNA-seq (Fig. 2d–k). RISP KO and RISP KO + AOX mice had fewer gp33+CD8+ T cells in secondary lymphoid organs (Fig. 2d–e), but this is difficult to interpret because of the observed naive precursor defect in those mice. Nevertheless, RNA-seq principal component analyses (PCAs) of gp33+ memory CD8+ T cells demonstrated that much of what distinguishes RISP KO from RISP WT cells is restored by AOX expression (Fig. 2f). Among significantly differentially expressed genes in RISP KO, many are associated with CD8+ T cell exhaustion, including Pdcd1 (encoding PD-1), Lag3, Tox, Tigit and tcf7 (refs. 30,31,32,33,34,35) (Fig. 2g and Extended Data Fig. 3b–d). Compared with RISP WT, RISP KO, but not RISP KO + AOX, gp33+ memory CD8+ T cells were significantly enriched in two exhaustion gene signatures from previous studies36,37 (Fig. 2h–k).
To investigate whether the proliferation defect is the result of impaired activation in the absence of mitochondrial complex III, we crossed congenically marked transgenic P14 mice, wherein nearly all CD8+ T cells are specific for the gp33-41 epitope of LCMV, to Gzmb-CreERT2 mice combined with conditional deletion of mitochondrial complex III subunit QPC (Uqcrqfl/fl) and conditional AOX expression. This generated Gzmb-CreERT2QPCfl/flAOXLSL P14 CD8+ T cell donors of three genotypes: QPC KO, QPC KO + AOX and QPC WT38 (Extended Data Fig. 4a–h). QPC WT P14 cells are Gzmb-CreERT2+, QPCf/w, AOXw/w and all recipients were pre-fed tamoxifen chow. In this system all genotypes are wild type during initial activation and gene recombination occurs after activation when CD8+ T cells begin expressing Gzmb, approximately day 4.5 after LCMV infection39 (Extended Data Fig. 4b). Tracking the P14 donor cells in peripheral blood revealed that, even when activation occurs before mitochondrial complex III depletion, QPC KO P14 cells are unable to expand in response to LCMV infection (Extended Data Fig. 4c–f). Normal QPC KO + AOX P14 proliferation suggests that CD8+ T cell proliferation requires mitochondrial complex III functions independent of ROS production.
To evaluate memory differentiation in mitochondrial complex III-deficient cells with normalized precursor frequency, we crossed the CD4-Cre RISPfl/fl AOXLSL mice to congenically marked P14 mice and performed adoptive transfer experiments (Fig. 3a–j). In this context, the RISP KO P14 CD8+ T cells display reduced proliferation and RISP KO + AOX P14 CD8+ T cells expanded to WT levels in peripheral blood at day 7 post-infection (Fig. 3a,b). At memory timepoints we quantified P14 CD8+ T cells in blood and secondary lymphoid organs and found a trend toward reduced memory formation of RISP KO + AOX P14 CD8+ T cells (Fig. 3c–e). Temporal evaluation of subset differentiation revealed that RISP KO and RISP KO + AOX P14 CD8+ T cells form a higher proportion of effectors and fail to generate a substantive memory population (Fig. 3f–g). At day 7 post-infection RISP KO P14 CD8+ T cells form significantly more KLRG1+CD127− short-lived effectors (SLECs) than RISP WT or RISP KO + AOX P14 CD8+ T cells, which is sustained through day 30 post-infection. RISP KO P14 CD8+ T cells form concomitantly fewer KLRG1−CD127+ memory precursor cells (MPECs). It is interesting that RISP KO + AOX P14 CD8+ T cells form normal proportions of SLECs and MPECs on days 7–21, but by day 30 the memory population is lost, indicating that they can initiate memory formation but are unable to terminally differentiate and persist (Fig. 3g). At day 30 post-infection, a lower proportion of RISP KO P14 CD8+ T cells produced all three cytokines measured (Fig. 3h). Despite functional deficits in the RISP KO P14 CD8+ T cells, the LCMV Armstrong virus was cleared as quickly as in RISP WT P14 CD8+ T cell recipients, ensuring that prolonged antigen dynamics do not cause the observed phenotype (Extended Data Fig. 5a). Notably, RISP P14 CD8+ T cell-recipient mice were Cre−/−AOXLSL/WT to enable central tolerance and prevent AOX rejection. Furthermore, experiments using SOX2-Cre AOXLSL/WT recipients, which globally express AOX, phenocopied the KO + AOX memory formation defect, mitigating concerns regarding AOX immunogenicity or rejection (Extended Data Fig. 5b).

a–j, Congenically marked P14 CD8+ T cells from CD4-Cre RISPfl/fl AOXLSL mice of the indicated genotype were adoptively transferred into naive mice which were then infected with LCMV Armstrong, evaluated by flow cytometry over time in peripheral blood (PBL) (a). Results after 7 d.p.i. in PBL (b), 45 d.p.i. in PBL (c) or isolated for quantification 65 d.p.i. from spleen (d) or pooled peripheral lymph nodes (LNs) (e). P14 phenotype and function were further evaluated by flow cytometry, gated on P14 representative flow plots from PBL 30 d.p.i. (f), over time from PBL (g), by restimulating splenocytes 30 d.p.i. with gp33 peptide for 4 h (h), or from spleen 30 d.p.i. (i) and from spleen 7 d.p.i. (j). All data points represent individual mice as biological replicates: n = 7 WT, n = 6 KO, n = 6 KO + AOX (a–d); n = 7 WT, n = 5 KO, n = 6 KO + AOX (e); n = 5 WT, n = 3 KO, n = 4 KO + AOX (f,g); n = 5 WT, n = 3 KO, n = 5 KO + AOX (h,i); n = 3 WT, n = 3 KO, n = 3 KO + AOX (j). Data are means ± s.e.m., *P < 0.05. The statistical tests for b–g, i and j are one-way ANOVA with Tukey’s multiple-comparison test and for h two-way ANOVA with Tukey’s multiple-comparison test (*P = 0.0007 WT/KO, *P = 0.0011 KO/KO + AOX (b); *P = 0.0262 WT/KO (c); *P = 0.0382 WT/KO (d); top % KLRG1+CD127− day 7, *P = 0.0018 WT/KO, *P = < 0.0001 KO/KO + AOX, *P = 0.04 WT/KO + AOX, day 15 *P = 0.0023 WT/KO, *P = 0.0194 KO/KO + AOX, day 21 *P = 0.0122 WT/KO, day 30 *P = < 0.0001 WT/KO, *P = 0.0175 KO/KO + AOX, *P = 0.0011 WT/KO + AOX; bottom % KLRG1−CD127+ day 15 *P = 0.0006 WT/KO, *P = 0.0107 KO/KO + AOX, day 21 *P = 0.0053 WT/KO, *P = 0.0293 KO/KO + AOX, day 30 *P = < 0.0001 WT/KO, *P = 0.0028 KO/KO + AOX, *P = 0.0003 WT/KO + AOX (g); *P = 0.0058 WT/KO, 0.0439 KO/KO + AOX (h); day 30 PD-1 *P = 0.0361 WT/KO, LAG3 *P = 0.0361 WT/KO, TCF1 *P = 0.0251 WT/KO (i); day 7 PD-1 *P = 0.0255 WT/KO, *P = 0.0306 WT/KO + AOX, LAG3 *P = 0.0121 WT/KO and TIM3 *P = 0.0208) (j). gMFI, geometric mean fluorescence intensity.
At the 30-d timepoint, we found that RISP KO P14 CD8+ T cells expressed higher levels of exhaustion markers including PD-1, LAG3 and lower TCF1 (Fig. 3i), consistent with the RNA-seq data of memory gp33+ CD8+ T cells from endogenously infected mice (Fig. 2g). At day 7 post-LCMV Armstrong, markers of exhaustion are more highly expressed by RISP KO P14 CD8+ T cells and in some cases RISP KO + AOX P14 CD8+ T cells (Fig. 3j). To investigate whether the exhaustion signature results from impaired T cell activation in the absence of mitochondrial complex III, Gzmb-CreERT2QPCfl/flAOXLSL P14 CD8+ T cells of each genotype were sorted for RNA-seq 7 d after adoptive transfer and LCMV infection (Extended Data Fig. 4a). We found that QPC KO P14 CD8+ T cells do not express a normal effector signature36 and demonstrate significant upregulation of exhaustion genes, including Pdcd1 and Tox (Extended Data Fig. 4g–h). QPC KO + AOX P14 CD8+ T cells do not deviate significantly from QPC WT, indicating that the exhaustion phenotype is not dependent on impaired T cell activation or mitochondrial ROS. To elucidate how RISP KO P14 CD8+ T cells respond to chronic stimulation, we transferred RISP WT or RISP KO P14 CD8+ T cells before infection with LCMV clone 13. RISP KO P14 CD8+ T cells developed higher proportions of terminally exhausted (TCF1−,CX3CR1−) cells at later timepoints and expressed more Tim3 and PD-1 than RISP WT as early as 8 d post-infection (d.p.i.) (Extended Data Fig. 6a–c).
To observe the rapidity of the differentiation defects after activation, CD8+ T cells from CD4-Cre RISPfl/fl AOXLSL mice were analyzed by RNA-seq after overnight in vitro activation (Fig. 4a). RISP KO CD8+ T cells were enriched for published exhaustion profiles and Tox expression was increased36,37 (Fig. 4a,b). Collectively, these data, combined with the RNA-seq from LCMV-infected mice, suggest that mitochondrial complex III-deficient CD8+ T cells are susceptible to rapid induction of exhaustion-like phenotypes on acute stimulation.

a,b, Splenic CD8+ T cells from CD4-Cre RISPfl/fl AOXLSL mice of indicated genotypes activated overnight with anti-CD3ε/CD28 beads + IL-2, then analyzed for pathway enrichment from gene set enrichment analysis (GSEA). Expression profiles were compared against the Molecular Signatures Database (MSigDb) Hallmark dataset v.0.3 with the addition of published exhaustion gene expression profiles from Utzschneider et al.37 and Wherry et al.36, as well as the ISR from Wong et al.44 (a); TOX expression (b). c–e, Splenic CD8+ T cells from CD4-Cre RISPfl/fl AOXLSL mice of indicated genotypes stimulated in vitro for memory formation, experimental schematic (c), then analyzed by flow cytometry representative histograms (d) and mean fluorescence intensity of indicated markers (e). f, Splenic P14 CD8+ T cells enriched from CD4-Cre RISPfl/fl AOXLSL mice of indicated genotypes and stimulated in vitro before adoptive transfer into MC38-gp33 tumor-bearing mice 5 d after subcutaneous tumor administration, with tumors measured over time. All data points represent individual mice as biological replicates: n = 7 WT, n = 6 KO, n = 7 KO + AOX (a,b); n = 4 WT, n = 5 KO, n = 5 KO + AOX (e); n = 5 WT, n = 5 KO, n = 5 KO + AOX (f). Data in e and f are means ± s.e.m. *P < 0.05. The statistical test in e is one-way ANOVA with Tukey’s multiple-comparison test. In b, the boxplot limits represent the interquartile range (IQR) with a center line at the median and the statistical test is Wald’s test with false recovery rate correction. The whiskers represent the largest point within 1.5× the IQR. All points are overlaid with jittering (TOX *P = 0.0036 WT/KO, *P = 0.036 KO/KO + AOX, PD1 *P = 0.0001 WT/KO, *P = 0.0012 KO/KO + AOX, TIM3 *P = 0.0176 WT/KO, TCF7 *P = < 0.0001 WT/KO, *P = < 0.0001 KO/KO + AOX, Gzmb *P = 0.0139 WT/KO, *P = 0.0224 KO/KO + AOX, % CD62L *P = < 0.0001 WT/KO, *P = < 0.0001 WT/KO + AOX, *P = 0.0112 KO/KO + AOX (e)). max., maximum. Illustration in c created using BioRender.com.
To test whether the RISP KO + AOX memory differentiation defect and RISP KO exhaustion-like phenotype are cell autonomous, we used a standard in vitro memory formation assay (Fig. 4c). After 6 d total in culture, RISP KO CD8+ T cells expressed significantly increased TOX, PD1, TIM3 and GZMB and decreased TCF1 (Fig. 4d–e). It is interesting that RISP KO and RISP KO + AOX CD8+ T cells are unable to undergo memory differentiation, as defined by re-expression of L-selectin (CD62L) (Fig. 4d–e). These data are consistent with our in vivo findings that, upon acute activation, RISP KO CD8+ T cells developed an exhausted-like state, whereas RISP KO + AOX CD8+ T cells were mostly protected from that phenotype. Importantly, in the absence of mitochondrial complex III ROS, CD8+ T cells could not fully differentiate into the canonical memory phenotype. To further test the function of RISP-deficient cells, we transferred in vitro activated P14 CD8+ T cells from RISP WT, RISP KO or RISP KO + AOX mice into mice bearing subcutaneous MC38-gp33 tumors (Fig. 4f). Although RISP WT P14 CD8+ T cells efficiently eliminated gp33-expressing tumors, RISP KO and RISP KO + AOX P14 CD8+ T cells did not. These data indicate that mitochondrial complex III-derived ROS are important for in vivo T cell function, although this may be the result of failures of migration, persistence or effector function.
Mitochondrial metabolism and ROS generation, as well as structure and dynamics, have been linked to T cell proliferation, migration, cytokine production and memory formation40,41,42,43. Elevated ROS levels are associated with CD8+ T cell exhaustion after chronic stimulation13. Here we tested whether mitochondrial complex III-dependent ROS are necessary for distinct T cell stages (for example, naive maintenance, activation, proliferation and differentiation). First, naive T cell population defects seen in RISP KO mice were not rescued by AOX expression, indicating that mitochondrial ROS are required for naive T cell development and/or maintenance. Future work will determine whether that is because of undetected defects in development, thymic emigration or survival. Next, using reductionist genetic systems, we discovered that CD8+ T cells require mitochondrial respiration, but not mitochondrial complex III-derived ROS, for proliferation.
Interestingly, across multiple model systems, we uncovered that acute stimulation is sufficient to give an exhausted phenotype in the absence of mitochondrial complex III function, a condition not typically known to drive CD8+ T cell exhaustion. AOX expression in mitochondrial complex III-deficient CD8+ T cells normalized the expression of most exhaustion markers; however, a clear defect in memory formation remained30,31,32,33,34,35. Tracking T cell differentiation over time revealed that RISP KO + AOX CD8+ T cells initiated a memory precursor population that was lost at later timepoints, indicating that mitochondrial complex III-derived ROS is required for terminal differentiation or persistence of memory CD8+ T cells. The minor reduction in mitochondrial ATP in RISP KO + AOX CD8+ T cells could influence memory formation; however, proliferative capacity was unaffected despite being an ATP-intensive process. Together these data show that mitochondrial respiration is required to prevent an exhaustion-like state after activation independent of ROS, but dependent on respiration-linked metabolism. By contrast, mitochondrial complex III ROS are necessary for optimal memory CD8+ T cell differentiation and persistence. Thus, T cell development, proliferation and memory formation have distinct requirements for mitochondrial complex III ROS.
Methods
Mice, adoptive transfers, infections and tumors
All mice were housed and used in accordance with the Institutional Animal Care and Use Committee (IACUC) at Northwestern University. RISPfl/fl (Uqcrfsfl/fl) mice2, QPCfl/fl (Uqcrqfl/fl) mice45 and AOXLSL(ref. 18) mice have been previously described. Gzmb-CreERT2 mice, generated by D. Fearon38, were a gift from K. Khanna at the University of Connecticut. CD4-Cre (stock no. 022071), P14 transgenic (stock no. 004694), Thy1.1 (stock no. 000406) and Sox2-Cre (stock no. 008454) mice were purchased from the Jackson Laboratory, backcrossed in-house when necessary and bred to relevant gene combinations. All mouse lines were screened for C57BL/6J genetic background purity by Illumina SNP chip (DartMouse). For all experiments, male and female mice aged between 1.5 months and 6 months were age matched and assigned to experimental groups by pre-experimental genotyping. Sex matching was used when required for adoptive transfer experiments. For inducible Cre recombination, tamoxifen was administered via chow (Envigo).
P14 immune chimeras were generated by transferring 5 × 104 congenically marked P14 CD8+ T cells from donors bearing the appropriate genotypes into naive recipient mice by tail vein injection. At least 12 h later, recipients were infected with 2 × 105 plaque-forming units (p.f.u.) of LCMV Armstrong via intraperitoneal injection (intraperitoneally (i.p.)). For endogenous studies, naive recipient mice were infected with 2 × 105 p.f.u. of LCMV Armstrong i.p. LCMV Armstrong was a gift from D. Masopust at the University of Minnesota. For acute infections and in vivo tumor-killing assays, adoptive transfer recipients of CD4-Cre RISP AOX P14 CD8+ T cells were WT mice carrying an unexpressed AOX gene (for example, Cre−/−, AOXL/W), to enable central tolerance to the AOX protein. One group of such recipients was also used for Gzmb-CreERT2 QPC AOX P14 CD8+ T cell recipients as noted.
For LCMV clone 13 (Cl13) studies, naive CD8+ T cells were isolated from the spleens of donor CD90.1+ P14 mice of the indicated genotypes using the EasySep Mouse Naive CD8+ T Cell Isolation Kit (STEMCELL, cat. no. 19858). A total of 2,500 naive P14 CD8+ T cells were intravenously transferred into naive C57BL/6 recipient mice, followed by LCMV Cl13 infection. To establish chronic infection, each mouse received an intravenous injection of 2 × 106 p.f.u. of LCMV Cl13. The virus was propagated on BHK21 cells and viral titers were quantified using a plaque formation assay on Vero cells.
For in vivo cytotoxicity assays, MC38-gp33 tumor cells were a gift from A. Goldrath at the University of California, San Diego and, on arrival, were independently confirmed as being Mycoplasma negative and gp33 expressing. For tumor administration, 5 × 105 MC38-gp33 tumor cells were injected subcutaneously into the recipient mouse’s shaved flank. Isolated P14 CD8+ T cells from donors bearing the indicated genotypes were activated in vitro for 3 d using 1:1 anti-CD3ε/anti-CD28 T cell expansion beads (Miltenyi, cat. no. 130-093-627) and 50 U ml−1 of interleukin-2 (IL-2; National Institutes of Health (NIH) repository) before adoptive transfer of 106 P14 intravenously (i.v.), into mice with established palpable tumors 5 d after tumor cell administration. Tumor-bearing mice were monitored and tumors were measured with calipers in accordance with the relevant approved IACUC protocols.
Restimulation of P14 CD8+ T cells was performed in 96-well, flat-bottomed plates (MIDSCI, cat. no. TP92096). A total of 2 × 106 splenocytes was seeded in complete Roswell Park Memorial Institute (RPMI) medium and incubated at 37 °C for 4 h in the presence of 0.133 µg ml−1 of WT GP33 peptide (KAVYNFATC; GenScript, cat. no. RP20257), 5.0 µg ml−1 of Brefeldin A solution (BioLegend, cat. no. 420601) and 0.2 µg ml−1 of anti-CD107a antibody.
Cell isolations, in vitro restimulations, flow cytometry and sorting
To obtain a single-cell suspension, spleen, lymph nodes or thymus was mechanically dissociated using the back of a syringe against a scored polystyrene Petri dish in phosphate-buffered saline (PBS) containing 2% serum and filtered through a 70-μm filter. Peripheral blood and splenocytes were treated with ACK lysis buffer. Total spleen, lymph node and thymus counts were obtained using the Cellometer K2 Counter with AOPI stain (Nexcelom). CD8+ T cells were purified from a single-cell suspension using the EasySep Mouse CD8+ T Cell Isolation Kit (STEMCELL, cat. no. 19853A) according to the manufacturer’s instructions. Single-cell suspensions were stained with antibodies (Supplementary Table 4) and Live/Dead Fixable Near-IR 780 (Invitrogen, cat. no. L34994A) or annexin V (Invitrogen, cat. no. V13246) and propidium iodide and either acquired on a FACSSymphony or Fortessa flow cytometer (BD Biosciences) or sorted on a FACS ARIA II (BD Bioscience) running FACSDiva software or Tyto (Miltenyi) Sorter, using regular speed cartridges. The H-2D(b) LCMV gp33-41-biotinylated monomer was obtained from the NIH Tetramer Core, tetramerized and conjugated to allophycocyanin (APC) fluorochrome and used at 1:100–1:200 dilution. Flow cytometry analysis was performed using FlowJo v.10. The gating strategy is shown in Extended Data Fig. 7.
In vitro activation for proliferation and memory formation
Isolated CD8+ T cells were labeled with Cell Trace Violet for proliferation assays according to the manufacturer’s instructions (Thermo Fisher Scientific, cat. no. C34557) and loaded into a round-bottomed, 96-well plate with anti-CD3ε/anti-CD28 beads (Miltenyi, cat. no. 130-093-627) at a ratio of three beads per cell with 50 U ml−1 of IL-2 for 96 h in adapted mouse plasma-like medium for T cells (Supplementary Table 1)46 or complete RPMI medium (Supplementary Table 2), then stained and assessed by flow cytometry. For in vitro memory formation assays, CD8+ T cells were similarly isolated and activated in vitro with anti-CD3ε/anti-CD28 beads at a 1:1 ratio and 50 U ml−1 of IL-2 for 3 d. On the third day, the cells were removed from the beads and cultured with IL-15 and IL-7 for another 3 d. All in vitro memory formation assays were performed in complete RPMI medium. On day 6, cells were stained directly on the surface, with intracellular (BD cytofix/cytoperm, cat. no. BDB554714) or intranuclear (BioLegend True Nuclear Kit, cat. no. 424401) antibodies (Supplementary Table 4) before acquisition on the flow cytometer.
FFA for mouse serum virus titer
To determine viral titers in mouse serum samples, a focus-forming assay (FFA) was performed using Vero cells. Mouse serum was collected, kept on ice 1 h and centrifuged to collect the supernatant. Samples were frozen and stored at −80 °C until use. Vero cells were seeded in 96-well plates at a density of 30,000 cells per well and incubated overnight at 37 °C with 5% CO2. The following day, serum samples were serially diluted in Dulbecco’s modified Eagle’s medium supplemented with 2% fetal bovine serum and added to the Vero cell monolayer. After a 20-h incubation, infected cells were fixed and stained using a rat anti-LCMV nucleoprotein monoclonal antibody (clone VL-4, BioXCell, cat. no. BED106), followed by a goat anti-rat immunoglobulin G2a–FITC secondary antibody (Bethyl, cat. no. A110-109F). Clusters of infected cells (foci) were imaged and quantified using the IncuCyte S3 Live-Cell Analysis System. Viral titers were expressed as focus-forming units per milliliter of serum. Day 4 post-infection serum from LCMV Armstrong-infected mice was used as a positive control.
Protein quantification by capillary electrophoresis immunoassay
For RISP protein quantification, isolated CD8+ T cells were lysed in NP40 cell lysis buffer (Thermo Fisher Scientific, cat. no. FNN0021) with 1× Halt protease inhibitor cocktail (Thermo Fisher Scientific, cat. no. 78430) and protein concentration was determined by Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, cat. no. 23225). Protein was quantified using the ProteinSimple WES (Bio-Techne) platform and Compass software following the manufacturer’s instructions. Primary antibodies used were: anti-RISP (Abcam, cat. no. ab14746, 1:50) and anti-vinculin (CST, cat. no. 13901, 1:100,000).
OCR and ECAR measurements by Seahorse
For OCR and ECAR measurements, a Seahorse XF HS Mini Analyzer (Agilent) was used. CD8+ T cells were isolated and enriched (see above) from lymph nodes and spleen, then activated in vitro with anti-CD3ε/anti-CD28 beads at a ratio of 3 beads per cell for 48 h in a round-bottomed plate. Cells were then seeded, centrifuged and rested to adhere to a Cell Tak-coated mini-seahorse cell plate (following the manufacturer’s instructions) at a density of 100,000 cells per well in Seahorse RPMI medium without serum (Supplementary Table 3). Drugs were prepared in the same medium as the cells at concentrations appropriate to make the following final concentrations in the well on addition: oligomycin 1 μM (Sigma-Aldrich, cat. no. 75351), antimycin A 500 nM (Sigma-Aldrich, cat. no. A8674), SHAM 2 mM (Sigma-Aldrich, cat. no. S607), piericidin 1 μM (Cayman, cat. no. 15379) and 10 mM 2-deoxy-d-glucose (Sigma-Aldrich, cat. no. D8375). Seahorse XF Wave software (Agilent Technologies) and GraphPad Prism were used to analyze the data.
Metabolomics
Naive CD8+ T cells from CD4-Cre RISP AOX mice were sorted at 4 °C on the MACS Tyto, activated with anti-CD3ε/anti-CD28 T cell expansion beads (Miltenyi, cat. no. 130-093-627) at a 3:1 ratio overnight then collected, washed in PBS, counted and centrifuged at 500g, before being resuspended at 10,000 cells per μl in 80% acetonitrile in high-performance liquid chromatography (HPLC)-grade H2O, vortexed vigorously and then frozen. The samples were stored at −80 °C for a minimum of 12 h before performing three additional freeze–thaw cycles with wet ice and dry ice. Samples were then centrifuged at 16,000g for 30 min at 4 °C. Supernatants were transferred to a fresh tube for analysis.
A 10-μl aliquot of the sample was used for high-resolution HPLC–tandem mass spectrometry (MS/MS) and high-resolution HPLC–MS/MS was performed as previously described26.
NAD/NADH ratio measurement
The NAD/NADH ratio was measured using the NAD/NADH-Glo Assay (Promega, cat. no. G9071) following the manufacturer’s instructions. Data were acquired and analyzed 60 min after starting incubation.
RNA isolation, RT–PCR
RNA was isolated using the AllPrep DNA/RNA Mini Kit (QIAGEN) following the manufacturer’s instructions. Real-time (RT)–PCR was performed on the CFX Opus 384 Real-Time PCR System (BioRad) using CYBRFast 1-Step RT-qPCR Lo-ROX Kit (Tonbo Biosciences) and 10 μM (final concentration) of the following primers: Uqcrfs1 (RISP)-F: 5′-CTGCTTCTGTCCGTTTTTCC-3′, Uqcrfs1 (RISP)-R: 5′-GCATAAGCAACACCCACAGT-3′, AOX-F: CGAGACGTTATCCTGGCAAT; AOX-R: CTCGATCCCAAGTCATGGTT. Data were analyzed using the comparative Ct (ΔΔCt) method.
Bulk RNA-seq
For 40-d.p.i. samples, RNA quality and quantity were assessed using TapeStation 4200 RNA tapes (Agilent) and RNA-seq libraries were prepared from 0.2–6.0 ng of total RNA using the SMART-seq v.4 Ultra Low Input RNA Kit (Takara, cat. no. 634773). For all other samples, libraries were prepared from 15 ng of total RNA using the NEBNext Ultra DNA Library Prep kit for Illumina (NEB, cat. no. E7370L). Library quality control was then performed using TapeStation 4200 High-Sensitivity DNA tapes (Agilent). Dual-indexed libraries were pooled and sequenced on a NextSeq500 instrument (Illumina) for 40-d.p.i. samples or a NextSeq2000 instrument for all other samples (Illumina), 75 or 100 cycles, respectively, single end, to an average sequencing depth of 14.1 million reads per sample. FASTQ files were generated using bcl2fastq v.2.20 (Illumina) for NextSeq500 output or bcl-convert v.4.0.3 (Illumina) for NextSeq2000 output using default parameters. To facilitate reproducible analysis, samples were processed using the publicly available nf-core/RNA-seq pipeline v.3.12.0 implemented in Nextflow v.23.10.0 using Singularity v.3.8.1 with the minimal command nextflow run nf-core/rnaseq \-r ‘3.12.0’ \-profile nu_genomics \–additional_fasta ‘transgenes.fa’ \–star_index false \–three_prime_clip_r2 3 \–genome ‘GRCm38’ for SMART-seq samples. For NEBnext samples, 3′-clipping was not performed. In brief, lane-level reads were trimmed using trimGalore! v.0.6.7 and aligned to the hybrid genome described above using STAR v.2.6.1d. Gene-level assignment was then performed using salmon v.1.10.1.
Bulk differential expression analysis
All analysis was performed using customized scripts in R v.4.1.1 with the DESeq2 v.1.34.0 framework. A ‘local’ model of gene dispersion was employed because this fit dispersion trends better without obvious overfitting and pairwise comparisons were performed on a combined factor of age, treatment and genotype. α was set at 0.05 for all differential expression analyses, otherwise default settings were used. High-level analysis was performed using customized scripts available in the N GitHub repository.
RNA-seq statistical analysis
For all analyses, normality was first determined using the Shapiro–Wilk test and visual inspection of histograms. In cases where distributions were clearly non-normal, nonparametric statistics were employed. For transcriptomics quantification, all analysis was performed using customized scripts in R v.4.1.1, all of which are publicly available on GitHub at N. Plotting was performed using ggplot2 v.3.4.2 unless otherwise noted. Comparisons for these figures were added using ggsignif v.0.6.4. Heatmaps were generated using heatmap v.1.0.12 with Euclidean distance as the distance metric and the Ward D2 clustering method. In all boxplots, box limits represent the IQR with a center line at the median. The whiskers represent the largest point within 1.5× the IQR. All points are overlaid with jittering.
Gene set enrichment analysis
For gene set enrichment analysis, the fgsea v.1.20.0 package was used. Hallmark gene set lists were downloaded from the Molecular Signatures Database v.7.5.1 at http://www.gsea-msigdb.org/gsea/downloads.jsp33. Additional gene lists were included as follows: the ‘ISR’ list was a gift from C. Sidrauski, Calico Life Sciences44. Enrichment analysis was performed for all gene sets simultaneously, using the ‘fgseaMultilevel’ method with gene-level Wald’s statistics as rankings and default parameters.
Raw FASTQ files from Utzschneider et al.37 were downloaded from the Gene Expression Omnibus (GEO) using accession no. GSE142686 and processed using the nf-core/rnaseq pipeline as outlined above with the addition of the gene sequence for green fluorescent protein (AY151052.1_GFP) using the --additional_fasta command. The ‘core exhaustion signature’ was then re-derived as described in Utzschneider et al.37 by identifying the intersection of gene hits, split by up- and downregulation, of significantly differentially expressed genes (q < 0.05; Wald’s tests in DESeq2 with false recovery rate correction) when comparing LCMV Armstrong versus LCMV docile 21 d.p.i. in (1) ID3-GFP+ and (2) ID3-GFP− datasets. All comparisons were made as described in the RNA-seq methods, with α set to 0.05 using a local model of gene dispersion. For Wherry et al.36 profiles of exhausted and effector CD8+ T cells from LCMV-infected mice, Supplementary Table 1 clusters were used for phenotype-exclusive gene lists36.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Code availability
All code and materials used in the analysis are available via GitHub at https://github.com/Chandel-Lab/Steinert_2025 for the purposes of reproducing or extending the analysis.
References
Chakrabarty, R. P. & Chandel, N. S. Beyond ATP, new roles of mitochondria. Biochemist 44, 2–8 (2022).
Sena, L. A. et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236 (2013).
Tarasenko, T. N. et al. Cytochrome c oxidase activity is a metabolic checkpoint that regulates cell fate decisions during T cell activation and differentiation. Cell Metab. 25, 1254–1268.e7 (2017).
Chen, X. et al. Succinate dehydrogenase/complex II is critical for metabolic and epigenetic regulation of T cell proliferation and inflammation. Sci. Immunol. 7, eabm8161 (2022).
Brand, K., Williams, J. F. & Weidemann, M. J. Glucose and glutamine metabolism in rat thymocytes. Biochem. J. 221, 471–475 (1984).
Frauwirth, K. A. et al. The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777 (2002).
Peng, M. et al. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484 (2016).
Wang, R. et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011).
Chang, C.-H. et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 (2013).
Ron-Harel, N. et al. Mitochondrial biogenesis and proteome remodeling promote one-Carbon metabolism for T cell activation. Cell Metab. 24, 104–117 (2016).
Tan, H. et al. Integrative proteomics and phosphoproteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity 46, 488–503 (2017).
Buck, M. D. et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76 (2016).
Scharping, N. E. et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 22, 205–215 (2021).
Vardhana, S. A. et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol. 21, 1022–1033 (2020).
Yu, Y.-R. et al. Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nat. Immunol. 21, 1540–1551 (2020).
Guo, Y. et al. Metabolic reprogramming of terminally exhausted CD8+ T cells by IL-10 enhances anti-tumor immunity. Nat. Immunol. 22, 746–756 (2021).
Bengsch, B. et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity 45, 358–373 (2016).
Dhandapani, P. K. et al. Hyperoxia but not AOX expression mitigates pathological cardiac remodeling in a mouse model of inflammatory cardiomyopathy. Sci. Rep. 9, 12741 (2019).
Fernandez-Ayala, D. J. M. et al. Expression of the Ciona intestinalis alternative oxidase (AOX) in Drosophila complements defects in mitochondrial oxidative phosphorylation. Cell Metab. 9, 449–460 (2009).
Sommer, N. et al. Bypassing mitochondrial complex III using alternative oxidase inhibits acute pulmonary oxygen sensing. Sci. Adv. 6, eaba0694 (2020).
Billingham, L. K. et al. Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat. Immunol. 23, 692–704 (2022).
Hakkaart, G. A. J., Dassa, E. P., Jacobs, H. T. & Rustin, P. Allotopic expression of a mitochondrial alternative oxidase confers cyanide resistance to human cell respiration. EMBO Rep. 7, 341–345 (2006).
Martínez-Reyes, I. et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 585, 288–292 (2020).
Robb, E. L. et al. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 293, 9869–9879 (2018).
Scialò, F. et al. Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 23, 725–734 (2016).
Stoolman, J. S. et al. Mitochondria complex III–generated superoxide is essential for IL-10 secretion in macrophages. Sci. Adv. 11, eadu4369 (2025).
Szibor, M. et al. Broad AOX expression in a genetically tractable mouse model does not disturb normal physiology. Dis. Model. Mech. 10, 163–171 (2017).
Waypa, G. B. et al. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am. J. Respir. Crit. Care Med. 187, 424–432 (2013).
Martínez-Reyes, I. et al. TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol. Cell 61, 199–209 (2016).
Utzschneider, D. T. et al. T cell factor 1-expressing memory-like CD8+ T cells sustain the immune response to chronic viral infections. Immunity 45, 415–427 (2016).
Yao, C. et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat. Immunol. 20, 890–901 (2019).
Khan, O. et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571, 211–218 (2019).
Seo, H. et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl Acad. Sci. USA 116, 12410–12415 (2019).
Alfei, F. et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269 (2019).
Scott, A. C. et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274 (2019).
Wherry, E. J. et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684 (2007).
Utzschneider, D. T. et al. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat. Immunol. 21, 1256–1266 (2020).
Bannard, O., Kraman, M. & Fearon, D. T. Secondary replicative function of CD8+ T cells that had developed an effector phenotype. Science 323, 505–509 (2009).
Masopust, D., Vezys, V., Wherry, E. J., Barber, D. L. & Ahmed, R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J. Immunol. 176, 2079–2083 (2006).
Steinert, E. M., Vasan, K. & Chandel, N. S. Mitochondrial metabolism regulation of T cell-mediated immunity. Annu. Rev. Immunol. 39, 395–416 (2021).
Franchina, D. G., Dostert, C. & Brenner, D. Reactive oxygen species: involvement in T cell signaling and metabolism. Trends Immunol. 39, 489–502 (2018).
Wilfahrt, D. & Delgoffe, G. M. Metabolic waypoints during T cell differentiation. Nat. Immunol. 25, 206–217 (2024).
Simula, L. et al. Mitochondrial metabolism sustains CD8+ T cell migration for an efficient infiltration into solid tumors. Nat. Commun. 15, 2203 (2024).
Wong, Y. L. et al. eIF2B activator prevents neurological defects caused by a chronic integrated stress response. eLife 8, e42940 (2019).
Weinberg, S. E. et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 565, 495–499 (2019).
Leney-Greene, M. A., Boddapati, A. K., Su, H. C., Cantor, J. R. & Lenardo, M. J. Human plasma-like medium Improves T lymphocyte activation. iScience 23, 100759 (2020).
Acknowledgements
We thank the NIH Tetramer Core Facility (contract no. 75N93020D00005) for providing the H-2D(b) LCMV gp33-41 biotinylated monomer. We also thank Northwestern University Robert H. Lurie Comprehensive Cancer Center Metabolomics Core (H. Abdala-Valencia) and Flow Cytometry Core Facilities (S. Swaminathan) for their expertise and support. We thank K. M. Khanna at University of Connecticut (currently at NYU) for sending the GzmbCreERT2 mice made by D. Fearon, H. T. Jacob for sharing the AOX mice (Faculty of Medicine and Health Technology, Tampere University), A. Goldrath at University of California San Diego (currently at the Allen Institute) for providing the MC38-gp33 tumor cell line and D. Masopust (University of Minnesota) for providing the LCMV Armstrong virus. This work was supported by NIH grant (nos. R01AI148190 and 5P01HL154998 to N.S.C.) and E.M.S. was a Cancer Research Institute Fellow supported by the Cancer Research Institute during much of this work. R.A.G. is supported by Schmidt Science Fellows, in partnership with the Rhodes Trust.
Author information
Authors and Affiliations
Contributions
E.M.S. and N.S.C. conceptualized the study. Experimentation was done by E.M.S., B.F.B., V.D.D. and A.K. Technical expertise was provided by A.K., Y.Z., M.S. and S.E.W. E.M.S., R.A.G, Y.Z. and K.V. were responsible for data analysis. E.M.S., R.A.G., Y.Z., K.V., W.C., S.E.W. and N.S.C. interpreted the data. E.M.S. and N.S.C. wrote the manuscript. E.M.S., B.F.B., V.D.D., R.A.G, K.V., A.K., W.C., M.S., S.E.W. and N.S.C. edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
M.S. is a shareholder in a start-up company founded to develop therapeutics based on AOX. The other authors declare no competing interests.
Peer review
Peer review information
Nature Immunology thanks Dirk Brenner and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: S. Houston in collaboration with the Nature Immunology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Characterization of complex III deficient CD8+ T cells from CD4-Cre RISPfl/fl AOXLSL mice.
Isolated CD8+ T cells from CD4-Cre RISPfl/fl AOXLSL mice of indicated genotypes were activated in vitro for 48 hr then collected for (a) ECAR or (b) TMRE and MitoTracker Green staining for flow cytometry. GSEA of naïve CD8 T cells from (c) RISP KO/WT genotypes or (d) RISP KO + AOX/WT genotypes. Expression profiles were compared against the Molecular Signatures Database (MSigDb) ‘Hallmark’ dataset v0.3 with the addition of published exhaustion gene expression profiles from Utzschneider NatImmuno 2020 (ref. 37) and Wherry Immunity 2007 (ref. 36), as well as the integrated stress response from Wong eLife 2019 (ref. 44) (a gift from C. Sidrauski at the Calico Life Sciences). All data points represent individual mice as biological replicates. a. n = 2 WT, n = 2 KO, n = 2 KO + AOX; b. n = 4 WT, n = 4 KO, n = 4 KO + ; c-d. n = 4 WT, n = 5 KO, n = 3 KO + AOX. For a-b. data are means ± s.e.m, *P < 0.05 and statistical test in b. is two-way ANOVA with Tukey’s multiple comparisons test.
Extended Data Fig. 2 T cell defects in naïve CD4-Cre RISPfl/fl AOXLSL mice.
Naïve CD4-Cre RISPfl/fl AOXLSL mice were used for direct ex vivo quantification by flow cytometry (a) of indicated populations from thymus, (b) percent of indicated naïve T cells that are apoptotic from pooled peripheral lymph nodes or (c) spleen (d) total indicated T cells from lymph nodes or spleen, (e) Percent of total T cell populations that express CD44 from lymph nodes or spleen, (f) percent of total T cell populations that are apoptotic by annexin V staining from lymph node or spleen. All data points represent individual mice as biological replicates, a. n = 20 WT, n = 13 KO, n = 10 KO + AOX; b-c. n = 8 WT, n = 7 KO, n = 6 KO + AOX; d. Lymph nodes n = 25 WT, n = 15 KO, n = 11 KO + AOX; Spleen n = 24 WT, n = 15 KO, n = 10 KO + AOX; e. Lymph nodes n = 21 WT, n = 15 KO, n = 13 KO + AOX; Spleen n = 21 WT, n = 14 KO, n = 12 KO + AOX; f. n = 8 WT, n = 7 KO, n = 6 KO + AOX. Data are means ± s.e.m, *P < 0.05, statistical tests are one-way ANOVA with Tukey’s multiple comparisons test, (d. CD4 LN *P = < 0.0376 WT/KO; CD8 LN *P = < 0.0001 WT/KO, *P = 0.0001 WT/KO + AOX; CD4+ Spleen *P = 0.0095 WT/KO, CD8+ Spleen *P = < 0.0001 WT/KO, *P = 0.0009 WT/KO + AOX; e. CD8+ LN *P = 0.0006 WT/KO; CD8+ Spleen *P = 0.163 KO/KO + AOX, *P = < 0.0001 WT/KO).
Extended Data Fig. 3 Gene expression of gp33+ memory CD8 T cells from CD4-Cre RISPfl/fl AOXLSL mice > 40 days after LCMV infection.
CD4-Cre RISPfl/fl AOXLSL mice of RISP WT, KO and KO + AOX genotypes were infected with LCMV Armstrong, and gp33 tetramer positive cells were sorted from spleen and lymph node for RNAseq along with a naïve CD8+ T cell control population from the same mice, (a) experimental strategy (b) isolated gp33 tetramer positive cells and polyclonal naïve cells from the same mice, heatmaps of all significantly differentially expressed genes among expression of all genes measured (c) GSEA of memory CD8 T cells from RISP KO/WT genotypes. Expression profiles were compared against the Molecular Signatures Database (MSigDb) ‘Hallmark’ dataset v0.3 with the addition of published exhaustion gene expression profiles from Utzschneider et. al.37 and Wherry et. al.36, as well as the integrated stress response from Wong eLife 2019 (ref. 44) (a gift from C. Sidrauski at the Calico Life Sciences). (d) boxplots of selected genes from RNA-seq analysis. All data points represent individual mice as biological replicates, b-d. n = 4 WT, n = 5 KO, n = 3 KO + AOX. In d box plots, box limits represent the interquartile range with a center line at the median. Whiskers represent the largest point within 1.5× interquartile range. All points are overlaid with jittering. Statistical tests on boxplots are Wald test with FDR correction. Illustrations in a created using BioRender.com.
Extended Data Fig. 4 Gzmb-CreERT2 enables post-activation deletion of mitochondrial complex III (QPC) and AOX expression.
Gzmb-CreERT2 QPCfl/fl AOXLSL (a) experimental strategy, (b) diagram of gene recombination timing (c–h) Naïve mice were fed tamoxifen chow for 2 weeks before congenically marked P14 CD8+ T cells from Gzmb-CreERT2 QPCfl/fl AOXLSL mice of indicated genotypes were adoptively transferred by i.v. injection the day before infection with LCMV. P14 were (c) analyzed overtime in PBL or analyzed at day 7 post infection (d) from PBL, (e) for quantification from spleen, or (f) for quantification from pooled peripheral lymph nodes. P14 CD8+ T cells were sorted from pooled lymph nodes and spleen on day 7 post infection for RNAseq (g) and compared to established effector T cell profiles from Wherry et. al.36 (h) boxplots of gene expression for selected genes. All data points represent individual mice as biological replicates. c. n = 11 WT, n = 15 KO, n = 15 KO + AOX; d. n = 11 WT, n = 15 KO, n = 15 KO + AOX; e-f. n = 4 WT, n = 5 KO, n = 4 KO + AOX; g-h. n = 5 WT, n = 4 KO, n = 4 KO + AOX. Data in c-f are means ± s.e.m, *P < 0.05, and statistical tests for d-f are one-way ANOVA with Tukey’s multiple comparisons test. In h box plots, box limits represent the interquartile range with a center line at the median. Whiskers represent the largest point within 1.5× interquartile range. All points are overlaid with jittering. Statistical tests on boxplots are Wald test with FDR correction. (d. *P = < 0.0001 WT/KO, *P = < 0.0001 KO/KO + AOX, *P = < 0.0001 WT/KO + AOX; e. *P = 0.0038 WT/KO). Illustrations in a and b created using BioRender.com.
Extended Data Fig. 5 Gp33-specific CD8 T cell (P14) adoptive transfer and LCMV infection viral titers and Sox2Cre AOX recipients.
(a) Congenically marked P14 CD8+ T cells from CD4-Cre RISPfl/fl AOXLSL mice of indicated genotypes were adoptively transferred by i.v. injection the day before infection with LCMV Armstrong infection, and serum was collected on days 7, 15, 21 and 30 post infection for focus forming assay to determine viral titer. Serum from a mouse infected with LCMV Armstrong 4 days prior was used as a positive control. A horizontal dotted line is placed at 10^4 to indicate PFU limit of detection of the focus forming assay as previously described56. (b) Sox2-Cre AOXLSL mice were used as AOX globally expressing naïve recipients, which were fed tamoxifen chow for 2 weeks before congenically marked P14 CD8+ T cells from Gzmb-CreERT2 QPCfl/fl AOXLSL mice of indicated genotypes were adoptively transferred by i.v. injection the day before infection with LCMV and the P14 were followed in blood to determine if complex III deficient AOX expressing P14 were able to form normal memory populations in an AOX expressing mouse. All data points represent individual mice as biological replicates. a. day 4 endogenous control n = 5; day 7 n = 5 WT, n = 5 KO, n = 5 KO + AOX; day 15 n = 3 WT, n = 5 KO, n = 5 KO + AOX; day 21 n = 3 WT, n = 4 KO, n = 4 KO + AOX,;day 30 n = 5 WT, n = 5 KO, n = 5 KO + AOX; b. n = 5 WT, n = 4 KO, n = 5 KO + AOX. Data are means ± s.e.m, *P < 0.05, statistical test in a. is two-way ANOVA with Tukey’s multiple comparisons test. (a. D7*P = 0.0002 WT/KO, *P = 0.0015 WT/KO + AOX.).
Extended Data Fig. 6 CD4-Cre RISPfl/fl AOXLSL P14 adoptive transfer and LCMV Clone 13 infection.
CD4-Cre RISPfl/fl AOXLSL P14 were adoptively transferred into naïve C57bl6/J mice prior to infection with LCMV Clone 13 then isolated for phenotypic evaluation by flow cytometry (a) from spleen 15 days post infection, representative flow plots (b) quantification of indicted populations from 15 or 24 days post infection spleens or (c) gMFI of indicated markers at the indicated timepoints post infection from spleen. control All data points represent individual mice as biological replicates. b. n = 5 WT, n = 6 KO,; c. n = 3 WT, n = 3 KO. Data are means ± s.e.m, *P < 0.05, statistical test is one-way ANOVA (b. %TEX *P = 0.0118 WT/KO; c. D8 PD1 *P = 0.0142 WT/KO, D24 Gzmb *P = 0.0119 WT/KO).
Extended Data Fig. 7 Basic Gating Strategy Used in Flow Cytometry Analysis.
a, Basic flow cytometric gating strategy for the identification of CD8+ T cells for quantification and phenotypic analysis.
Supplementary information
Supplementary Information (download PDF )
Supplementary Information Methods Tables 1–4.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Steinert, E.M., Furtado Bruza, B., Danchine, V.D. et al. Mitochondrial respiration is necessary for CD8+ T cell proliferation and cell fate. Nat Immunol 26, 1267–1274 (2025). https://doi.org/10.1038/s41590-025-02202-x
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41590-025-02202-x
This article is cited by
-
Targeting organelle function in T cells for cancer immunotherapy
Nature Reviews Immunology (2025)
