
Abstract
Transcranial direct current stimulation (tDCS) has been proposed as a new treatment in major depressive disorder (MDD). This is a fully remote, multisite, double-blind, placebo-controlled, randomized superiority trial of 10-week home-based tDCS in MDD. Participants were 18 years or older, with MDD in current depressive episode of at least moderate severity as measured using the Hamilton Depression Rating Scale (mean = 19.07 ± 2.73). A total of 174 participants (120 women, 54 men) were randomized to active (n = 87, mean age = 37.09 ± 11.14 years) or sham (n = 87, mean age = 38.32 ± 10.92 years) treatment. tDCS consisted of five sessions per week for 3 weeks then three sessions per week for 7 weeks in a 10-week trial, followed by a 10-week open-label phase. Each session lasted 30 min; the anode was placed over the left dorsolateral prefrontal cortex and the cathode over the right dorsolateral prefrontal cortex (active tDCS 2 mA and sham tDCS 0 mA, with brief ramp up and down to mimic active stimulation). As the primary outcome, depressive symptoms showed significant improvement when measured using the Hamilton Depression Rating Scale: active 9.41 ± 6.25 point improvement (10-week mean = 9.58 ± 6.02) and sham 7.14 ± 6.10 point improvement (10-week mean = 11.66 ± 5.96) (95% confidence interval = 0.51–4.01, P = 0.012). There were no differences in discontinuation rates. In summary, a 10-week home-based tDCS treatment with remote supervision in MDD showed high efficacy, acceptability and safety. ClinicalTrials.gov registration: NCT05202119
Similar content being viewed by others


Main
Major depressive disorder (MDD) is common and it is a leading cause of disability worldwide; it is the most notable precursor in suicide1. MDD is characterized by a prolonged low mood or inability to experience usual feelings of pleasure, which is accompanied by disturbances in sleep, appetite, psychomotor functioning and energy levels, and in cognitive functioning. First-line treatments are antidepressant medications and psychological therapies. However, more than a third of individuals with MDD do not achieve full clinical remission despite full treatment trials2,3.
Transcranial direct current stimulation (tDCS) is a form of noninvasive brain stimulation that applies a weak (0.5–2 mA) direct current via scalp electrodes4. Anodal stimulation shifts membrane potentials toward depolarization and increasing cortical excitability, whereas cathodal stimulation tends to shift membrane potentials toward hyperpolarization, decreasing potential cell firing and inhibiting cortical excitability5. tDCS modulates the resting state potential, thereby modulating cortical tissue excitability, rather than directly triggering an action potential that is in contrast to repetitive transcranial magnetic stimulation6. Neurophysiological effects typically persist beyond the immediate stimulation period7. Anodal tDCS can enhance cortical excitability, which is dependent on the N-methyl-d-aspartate receptor and calcium channel activity, demonstrating a sustained increase in synaptic transmission that is long-term potentiation-like, whereas cathodal tDCS decreases excitability and facilitates long-term depression-like changes8. Neural recordings demonstrate measurables effects on cortical electric fields9. Neurophysiological measures reveal network-level modulatory effects, in which anodal tDCS applied to left dorsolateral prefrontal cortex (DLPFC) is associated with significant changes in connectivity in default mode, self-referential and frontoparietal networks compared with sham tDCS10; it can extend into the deeper limbic brain regions, including the amygdala11, which are key regions in MDD neurocircuitry and reflect potential mechanisms of effect4.
tDCS is applied through a flexible cap or band that is worn over the forehead. The anode electrode is typically placed over the left DLPFC and the cathode is placed over the right DLPFC, in the suborbital or frontotemporal region6. In an individual-patient data meta-analysis, active tDCS relative to sham tDCS was associated with a significantly greater rate of clinical response (30.9% versus 18.9%; number needed to treat (NNT) = 9) and remission (19.9% versus 11.7%; NNT = 13) from 572 participants with MDD in nine studies12. tDCS is safe and well tolerated with no significant differences in attrition rate and adverse events between active and sham stimulation, offering a potential new first-line treatment for MDD4. However, a course of tDCS treatment involves daily sessions for several weeks; most studies have been conducted in a research clinic and have required daily visits6,12.
As it is portable and safe, tDCS can be provided for home use4. We developed a protocol that provides home-based tDCS with real-time remote supervision using videoconferencing13. In MDD, we found significant improvements in depressive symptoms, high acceptability and feasibility13, as also observed in additional open-label trials14,15. However, in our protocol, all participants had both the active tDCS device and real-time visits using videoconferencing, which were associated with meaningful experiences of support and containment16. Three randomized controlled trials (RCTs) of home-based tDCS in MDD have taken place17,18,19; however, none were fully remote because all included in-person study appointments, two trials were probably underpowered because of small sample sizes (n = 11 (ref. 18) and n = 58 (ref. 19)), and all were limited to a 6-week duration; they found no significant effects of active relative to sham tDCS17,18,19. However, the recent meta-analysis by Nikolin et al.20 reported that the active tDCS effects continue to increase for up to 10 weeks compared to sham stimulation.
In the present study, we sought to investigate the clinical efficacy and safety of a 10-week course of home-based tDCS for MDD in a large, double-blind, randomized superiority trial conducted in both the UK and USA. All participants had MDD as determined by a structured diagnostic interview; all were in a current depressive episode of at least moderate severity. Participants in our study might be taking stable antidepressant medication for at least 6 weeks, might be in psychotherapy for at least 6 weeks or might be treatment-free, reflecting the range of forms of MDD from first-episode and recurrent MDD to treatment-resistant depression. All study visits were remote and we were able to monitor participants’ tDCS use in real time. The primary objective was to investigate clinical efficacy at the 10-week end point of treatment between active and sham tDCS treatment arms.
Results
Participant data
Recruitment was from 12 May 2022 to 10 March 2023 (ClinicalTrials.gov registration: NCT05202119). From 2,234 individuals who had an initial telephone screen, 368 individuals provided written informed consent and had an assessment using Microsoft Teams videoconferencing. In total, 174 participants with MDD (120 women, 69%) with a mean age of 37.63 years (s.d. = 11.00) were enrolled. One hundred and forty-five (83.3%) had white ethnicity. All had an MDD diagnosis based on the criteria of the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition21, were assessed using a structured clinical interview22 and were in a current depressive episode of at least moderate severity as measured by a minimum score of 16 in 17-item Hamilton Depression Rating Scale (HDRS)23. The mean HDRS was 19.07 (s.d. = 2.73); the median number of depressive episodes was three (interquartile range (IQR) = 1–5) (Table 1). The sex of participants was based on self-report. There were no exclusions of participants based on either sex or gender.
Inclusion criteria included being treatment-free or taking stable antidepressant medication or undergoing psychotherapy for at least 6 weeks before enrollment. Having persistent depressive symptoms of at least moderate severity and meeting the MDD criteria while taking antidepressant medication for at least 6 weeks have been clinical criteria for treatment-resistant depression in previous trials24,25. The composition of the participant cohort was as follows: treatment-free = 57 (32.8%); taking antidepressant medication = 109 (62.6%); undergoing psychotherapy = 26 (14.9%); taking medication and undergoing psychotherapy = 18 (10.3%) participants.
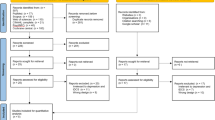
Participants were randomly allocated to active tDCS treatment (87 with MDD, mean age = 37.09 years (s.d. = 11.14)) or sham tDCS (87 with MDD, mean age = 38.32 years (s.d. = 10.92)) (Fig. 1, Table 1 and Supplementary Tables 2–5). One participant did not continue and had not started any treatment; therefore, the modified intention-to-treat (ITT) sample was 173 participants. There were no significant differences in discontinuation rates between groups (total = 25 participants, 14.3%: 13 (14.9%) in the active group and 12 (13.7%) in the sham group (P = 0.99)) (Supplementary Table 6). Based on a priori blinded interim analysis, recruitment ended early (Supplementary Notes—Interim Analysis).

Enrollment, group allocation, follow-up and analysis.
Primary outcome
A significant improvement was observed with regard to a change in depressive symptomatology as measured by the HDRS score from baseline to week 10 (the end of treatment) in the active tDCS treatment arm: HDRS decrease of 9.41 points (s.d. = 6.25) (estimated week 10 mean = 9.58 (s.d. = 6.02)), compared to the sham tDCS treatment arm: HDRS decrease of 7.14 points (s.d. = 6.10) (estimated week 10 mean = 11.66 (s.d. = 5.96)) (95% confidence interval (CI) = 0.51–4.01, P = 0.012) (Fig. 2).

Estimated mean 17-item HDRS rating scores from baseline to week 10 in the modified ITT analysis sample (n = 173) for the active and sham tDCS treatment arms. The error bars represent ± 1 s.e. The HDRS scores range from 0 to 52, with higher values indicating more severe depressive symptoms. A significant improvement was observed in the change in HDRS ratings from baseline to week 10 in the active tDCS treatment arm, that is, an HDRS decrease of 9.41 ± 6.25 (s.d.) (mean HDRS at week 10 = 9.58 ± 0.70 (s.e.)), compared to the sham tDCS treatment arm (HDRS decrease = 7.14 ± 6.10 (s.d.)) (mean HDRS at week 10 = 11.66 ± 0.69 (s.e.)) (95% CI = 0.5–4.0, P = 0.012). The difference in change scores was also significant at week 4 (95% CI = 0.2–3.4, P = 0.03), with a greater score decrease in the active treatment arm. A fully conditional specification (FCS) approach was used to produce 20 multiply imputed complete datasets. The FCS approach accommodates nonmonotonicity in the pattern of missing data and requires regression models to be specified for each variable, with missing values needing imputation. All models included age, sex, undergoing psychotherapy at baseline, use of any antidepressants at baseline and treatment group. The resulting complete datasets were combined using Rubin’s rules. *P < 0.05.
Secondary outcomes
Based on the HDRS ratings, the active tDCS treatment arm was associated with a significantly greater clinical response of 58.3% compared to the sham treatment arm (37.8%; P = 0.017) (post hoc odds ratio (OR) = 2.31 (lower bound = 1.17, upper bound = 4.55); the active treatment arm was associated with a significantly greater remission rate of 44.9% relative to the sham treatment arm (21.8%; P = 0.004) (post hoc OR = 2.93, lower bound = 1.41, upper bound = 6.09).
Based on the Montgomery–Åsberg Depression Rating Scale (MADRS)26 ratings, the active tDCS treatment arm showed a significant improvement from baseline to week 10, with a mean improvement of 11.31 (s.d. = 8.81) (estimated mean at week 10 = 12.46 (s.d. = 9.40)) compared to sham treatment (mean improvement = 7.74; s.d. = 8.47; P = 0.006) (estimated mean at week 10 = 15.30 (s.d. = 9.28)). Regarding clinical response, the active treatment arm was associated with a significantly greater response rate of 64.2% compared to sham treatment (32.3%; P < 0.001) (post hoc OR = 3.76, lower bound = 1.83, upper bound = 7.74). Regarding clinical remission, the active treatment arm was associated with a significantly greater remission rate of 57.5% relative to sham treatment (29.4%; P = 0.002) (post hoc OR = 3.26, lower bound = 1.53, upper bound = 6.94).
Based on the MADRS self-report scale (MADRS-s)27, the active tDCS treatment arm was associated with a significant improvement from baseline to week 10, with a mean improvement of 9.90 (s.d. = 8.94) (estimated mean at week 10 = 16.60 (s.d. = 9.33)) compared to sham treatment (mean improvement = 6.23 (s.d. = 9.13), P = 0.009) (estimated mean at week 10 = 19.55 (s.d. = 9.62)). Regarding clinical response, the active treatment arm was associated with a significantly greater response rate of 51.8% compared to sham (25.1%; P = 0.002) (post hoc OR = 3.22, lower bound = 1.15, upper bound = 6.94). Regarding clinical remission, the active treatment arm was associated with a significantly greater remission rate of 53.8% compared to sham (23.4%; P = 0.002) (post hoc OR = 3.83, lower bound = 1.61, upper bound = 9.13) (Table 2 and Extended Data Figs. 1 and 2). There were no significant differences in quality of life between treatment arms as measured by EQ-5D-3L28,29 (P = 0.326).
Exploratory outcomes
Regarding anxiety symptoms, there were no significant differences between an active mean Hamilton Anxiety Rating Scale (HAM-A)28 score improvement of 6.62 (s.d. = 6.09) (mean = 8.24 (s.d. = 5.65)), compared to a sham improvement of 4.88 (s.d. = 5.88) (mean = 9.29 (s.d. = 4.90)) (P = 0.08). Regarding hypomanic symptoms, the Young Mania Rating Scale (YMRS)29 mean score was 1.27 (s.d. = 1.40) in the active treatment arm at week 10 and 1.84 (s.d. = 1.69) in the sham treatment arm; this was statistically significant (P = 0.03) (Supplementary Tables 12 and 13).
In the neuropsychological assessments, there were no significant differences in Rey Auditory Verbal Learning Test (RAVLT)30 total learning or Symbol Digit Modalities Test (SDMT)31 between treatment arms (Supplementary Table 14).
Per-protocol and sensitivity analyses in participants with persistent depressive symptoms who had been taking antidepressant medication at study enrollment showed significant improvements in depressive symptoms, clinical response and remission (Supplementary Tables 15 and 18).
Analysis of study blinding and unblinding
Before unblinding at week 10 (end of trial), participants were asked to guess whether they thought they were receiving the active or sham tDCS device and their level of certainty, rating from ‘1’ for ‘very uncertain’ to ‘5’ for to ‘very certain’. A guess of active tDCS was made by 77.6% in the active treatment arm and 59.3% in the sham treatment arm; the difference was significant (P = 0.01). The certainty of having received active tDCS was rated highly by 57.6% (38 out of 66 guesses) in the active arm and 41.7% (20 out of 48 guesses) in the sham arm, as measured by a rating of 4 or 5, while certainty was rated low by 16.7% (11 out of 66 guesses) in the active treatment arm and 18.8% (9 out of 48 guesses) in the sham treatment arm, as measured by a rating of 1 or 2 (Supplementary Tables 36 and 39).
Adverse events and safety
At week 10, reports of skin redness ((active = 54 (63.5%); sham = 15 (18.5%), P < 0.001), skin irritation ((active = 6 (6.9%); sham = 0 (0%), P = 0.03) and trouble concentrating ((active = 12 (14.1%); sham = 3 (3.7%), P = 0.03) were greater in the active treatment arm relative to the sham treatment arm. There were no differences in headache, neck pain, scalp pain, itching, burning sensation, sleepiness or acute mood changes between treatment arms. Two participants in the active group described developing ‘burns’ at the left anode site. When reviewed, they might have been caused by using sponges that had dried out. Neither developed into residual skin lesions or scarring. Participants had not contacted the 24-h contact number; both had informed the research team at their following study visit, which was 1–2 weeks afterward. There were no visible lesions at the study visits. One participant had taken a break from the sessions for 4 days and the burn had fully healed. The second participant was experiencing dry skin at the electrode site and was advised that they could take a break from the sessions until the skin had healed; however, they did not take a break until after the next study visit, 3 weeks later, when they were advised to take a break from the sessions to allow the dry skin and tenderness to heal. The participant then missed the next three stimulations; the skin was no longer tender but it was still dry at the week 10 end-of-study visit. There were no serious adverse events related to the device; no participants developed mania or hypomania (Tables 3 and 4 and Supplementary Tables 24–29).
Discussion
In this international, multisite, sham-controlled, RCT of home-based tDCS treatment for MDD, a 10-week course of active stimulation was associated with significantly greater improvements in depressive symptoms, clinical response and remission rates compared to sham stimulation. Improvements were evident in both clinician-rated depressive symptom ratings (HDRS and MADRS) and in self-reported ratings (MADRS-s). The clinical significance of the outcomes is highlighted by high rates of treatment response and remission that were 2–3 times greater in the active treatment arm compared to the sham treatment arm. Clinical efficacy was demonstrated in a wide range of forms of MDD, from first-episode MDD to individuals having a history of recurrent episodes and participants with treatment-resistant depression.
Meta-analyses of clinic-based tDCS sessions reported that active tDCS is associated with greater improvements in depressive symptoms, clinical response and clinical remission rates compared to sham tDCS, particularly in first-episode and recurrent MDD6,32,33,34. However, in a recent large trial, Burkhardt et al.35 did not observe any significant effects of adjunct tDCS treatment to antidepressant medication in a 6-week trial. In the present trial, we had a comparable inclusion criteria for treatment-resistant depression but a longer 10-week treatment duration. Burkhardt et al.35 included participants with MDD with persistent depressive symptoms of at least moderate severity while taking a selective serotonin reuptake inhibitor for a minimum of 4 weeks. Similarly, our inclusion criteria were participants with MDD with persistent depressive symptoms of at least moderate severity while taking antidepressant medication for a minimum of 6 weeks at the point of screening. Our inclusion criteria met the UK National Institute for Health and Care Excellence definition of treatment-resistant depression24,25; 63% of our sample fulfilled these criteria. However, treatment-resistant depression is negatively correlated with clinical efficacy to tDCS treatment6,32,33,34. This is a clinical definition that can be further delineated by the number and types of failed treatment trials. Our exclusion criteria included having a history of poor treatment response to two or more antidepressant medications, which reflects increased severity of treatment-resistant depression. About 12–17% of participants in the study by Burkhardt et al.35 had such a history of treatment failure, which could have affected their observed lack of clinical effects. The level of depressive symptom severity and mean ages were comparable in Burkhardt et al.35 and in the present trial, while age at onset was younger in the present trial by about 10 years and we did not have an upper age limit.
Furthermore, the clinical effects of tDCS continue to increase for up to 10 weeks20. In the present trial, we found strong clinical efficacy and safety with our 10-week home-based protocol. This is in contrast with recent home-based tDCS trials; all had 6-week treatment durations and two trials had small sample sizes (n = 11 (ref. 18) and n = 58 (ref. 19))17,18,19. A single-blind RCT of tDCS augmentation to antidepressant medication, consisting of hybrid clinic-based and home-based tDCS sessions, reported significant improvements in depressive symptoms in the active group as measured by self-reported symptoms rating but not in clinician-based ratings19. In a large RCT (n = 210), no significant effects were observed between three treatment arms: active tDCS, active tDCS combined with a digital psychological intervention (double-active); and sham tDCS combined with internet browsing (double-sham). In the present trial, clinical treatment effects were evident at 10 weeks. Longer treatment durations may be necessary to observe clinical efficacy36; in their meta-analysis, Nikolin et al.20 reported that effect sizes continued to increase with longer treatment durations.
We found a high level of safety in the present trial. Safety was monitored using real-time assessments by videoconferencing and a dedicated contact number with 24-h access to researchers. A recent trial ended early because of adverse events involving skin lesions, which were the result of an accumulation of electrical burns in five participants in the active tDCS group from a total enrollment of 11 participants with MDD18. Electrical burns can be an unanticipated side effect; they are usually caused by the application of tap water to moisten sponges37, insufficient moistening with conductive saline solution38 or preexisting skin lesions. In the present trial, we had two incidents of reported electrical burns; both participants reported these during the study visit. Both were probably caused by insufficient sponge moistening; neither instance of electrical burn developed into residual skin lesions or scarring, and participants were keen to continue the tDCS sessions after a brief break. There were no serious adverse events related to the device and no incidents of serious suicide risk. However, active stimulation was associated with higher rates of skin redness, irritation and dry skin relative to sham treatment39,40.
During the tDCS sessions, participants were asked to sit or lie down and to avoid engaging in activities that might compromise safety or device functionality. Their activities had not been recorded by the research team. State-dependent effects of tDCS stimulation are possible; an interaction between external stimulation, location and internal state of the region or network has been observed41,42. The type of task activity during stimulation can influence cognitive enhancement in healthy participants43 and treatment response in clinical samples44. Concurrent administration of active tDCS and cognitive control training (CCT) has been associated with sustained improvements in depressive symptoms compared to active tDCS plus sham CCT or sham tDCS plus CCT44. However, a 6-week trial of cognitive behavioral therapy (CBT) with three treatment arms—CBT alone, CBT plus active tDCS and CBT plus sham tDCS—in a sample of 126 participants with MDD, reported no significant effects between groups45.
Blinding is key in RCTs to mitigate potential biases that can impact on the outcome. Procedures involve the establishment and maintenance of blinding, measures to prevent unblinding and assessment of successful blinding46,47. To establish blinding in the present trial, all participants and researchers were blinded to treatment arm allocation; the placebo-sham control intervention was identical in appearance to the active intervention. Furthermore, when using the sham device, there was brief stimulation at the start and at the end of each session to mimic active tDCS sensations to aid in blinding and to balance potential nocebo effects across groups48. To maintain blinding, the treatment protocol and study visits were identical in both treatment arms. All participants maintained their ongoing treatments throughout the trial, and all participants used the active tDCS device in the subsequent open-label phase of the trial to incorporate real-life clinical care while balancing expectations between groups and to limit attrition47. The tDCS treatment arms were described as ‘active’ or ‘inactive’ stimulation by researchers during the trial to maintain comparable phrasing and reduce potential negative connotations associated with the words ‘placebo’ or ‘sham’. Outcome assessors were blinded to group allocation as a second independent researcher was present for the clinical ratings47. Ethicality was assessed a priori and worsening of symptoms was included as a withdrawal criterion. An automatic email report was sent to all research team members when unblinding occurred as notification and to prevent potential concealment of any accidental unblinding. Timing of the blinded assessment questionnaire at the end of the blinded treatment phase, rather than at time points throughout the trial, reduced the influence of potential interjections.
In the blinding assessment, participants were asked to guess if they had been receiving the ‘active’ or ‘sham’ treatment and the certainty of their guess, ranging from ‘very uncertain’ to ‘very certain’ on a five-point scale. Participants who felt ‘very uncertain’ of their guess are comparable to participants guessing ‘don’t know’. More participants in the active treatment arm guessed that they were receiving active tDCS (77.6%) compared to participants in the sham treatment arm (59.3%). However, a moderate proportion were ‘very uncertain’ about their guess in the active (16.7%) and sham (18.8%) treatment arms; endorsement of being ‘very certain’ in the active (57.6%) and sham (41.7%) treatment arms was limited, with no significant differences between treatment arms. It is possible that participants who believed that they were in the active treatment arm were more likely to show a placebo response. However, in their meta-analysis of antidepressant medication RCTs, Lin et al.49 found no association between blinding effects and treatment effect sizes. The 2010 Consolidated Standards of Reporting Trials (CONSORT) guidance recommends specifying how blinding is established but no longer recommends reporting on how the success of blinding is assessed because healthcare providers and participants are likely to know if the primary outcome has been achieved by participants, making interpretation more difficult because responses might reflect accurate assumptions about the efficacy of the intervention rather than a failure of blinding50. Moreover, significant clinical efficacy was maintained for active relative to sham treatment in participants who had made a guess of ‘active’ treatment; the placebo response rate in the sham treatment arm in the present trial (26.9%) was lower than placebo response rates observed in a sham group (36%)19 and double-sham group (38%)17 that had included in-person study visits at the clinical research center17,19 and weekly online visits for 6 weeks19.
Limitations of the present trial include the lack of a ‘don’t know’ option in the blinding assessment. Well-executed blinding to treatment allocation should lead participants to be uncertain of which treatment they are receiving. By including a ‘don’t know’ option, it should be possible to calculate a proposed index of blinding51. Differences in head sizes, individual anatomical features and the positioning of devices among users may lead to unique configurations of electric field density within the brain52,53. Interindividual variations in tDCS can be partially explained due to differences in electric fields54. The tDCS device used in the present study has undergone electric field modeling, indicating that the device targets areas within the prefrontal cortex linked to MDD pathophysiology52. While participants were taught how to use the device and positioning had been observed in real time, variations in positioning could potentially affect electric field intensity and in turn treatment outcomes52. All clinical rating scale assessments were performed using videoconferencing, although no significant differences were found between face-to-face and videoconferencing HDRS ratings conducted within the same day55; we sought to have a second team member to perform clinical ratings to maintain blinding and ensure validity. Video consultation for clinical assessment and mental health treatment has become more common in recent years and is as effective as face-to-face visits for improving clinical outcomes and providing patients with more flexibility56,57. Regarding quality of life, there was no significant difference between groups in a self-report measure. The scores on the quality-of-life measure were relatively high at baseline and both treatment arms reported some improvement in quality of life that was not statistically significant. MDD is more common in women and the present study consisted of a larger proportion of female participants as expected. All participants self-reported their sex. An effect of sex or gender on clinical efficacy was not expected, although this warrants further investigation. Ethnic diversity in the present sample was limited and a history of hospital admissions was an exclusion criterion that may limit the generalizability of the findings.
In summary, a 10-week course of home-based active tDCS was associated with greater improvements in depressive symptoms, clinical response and remission in participants with MDD with at least a moderate severity of depressive symptoms compared to sham tDCS. Efficacy was observed in participants who were taking antidepressant medication indicative of treatment-resistant depression or undergoing psychotherapy, as well as participants who were treatment-free. All participants had real-time remote supervision visits. High acceptability and safety were observed in the present trial. Home-based tDCS could be a potential first-line treatment for MDD as it demonstrates efficacy, acceptability and safety; however, ongoing safety monitoring is required.
Methods
Ethics and study design
The study was a multisite, double-blind, placebo-controlled, randomized, superiority controlled trial of 10-week home-based tDCS treatment for MDD followed by a 10-week open-label treatment. Participants were recruited from throughout England and Wales (UK) and Texas (USA). Recruitment sites were at the University of East London in London, UK and at the University of Texas Health Science Center in Houston, Texas, USA, respectively.
All participants provided written informed consent. Ethical approval was provided by the South Central-Hampshire B Research Ethics Committee (ref. 22/SC/0023) and the WIRB-Copernicus Group International Review Board (ref. 1324775). ClinicalTrials.gov registration: NCT05202119. Research execution included local research assistants who are included as coauthors. The study protocol is available in the Supplementary Information.
Participants
Participants were adults with MDD aged 18 years or older, in a current depressive episode as determined by the DSM-5 (ref. 21) criteria and assessed in a structed clinical interview (Mini-International Neuropsychiatric Interview (MINI) v.7.0.2 (ref. 22)). Inclusion criteria included: having at least moderate severity of depressive symptoms, as measured by score of 16 or greater on the 17-item HDRS23; being treatment-free or taking stable antidepressant medication or undergoing psychotherapy for at least 6 weeks before enrollment and being agreeable to maintaining the same treatment throughout the trial; being under care of general practitioner or psychiatrist. Exclusion criteria included: having treatment-resistant depression, defined as inadequate clinical response to two or more trials of antidepressant medication at an adequate dose and duration; high suicide risk based on the Columbia Suicide Severity Rating Scale (C-SSRS) Triage and Risk Identification Screener60; having a comorbid psychiatric disorder; taking medications that affect cortical excitability (for example, benzodiazepines, epilepsy medication); and contraindications to tDCS. Sex was determined by participant self-report; there was no exclusion of males or females and no upper limit on how many participants of each sex or gender could enroll61. The full inclusion and exclusion criteria are presented in the Supplementary Notes—Inclusion and exclusion criteria.
Procedures
Participants were recruited through the Flow Neuroscience website, email lists and social media posts. Individuals completed an online pre-screening form, hosted by a contract research organization, followed by a telephone call with a contract research organization member. Individuals then provided written informed consent and had an assessment with a research team member using Microsoft Teams videoconferencing. All participants were registered with a primary care physician as an inclusion criterion (Supplementary Notes—Inclusion and exclusion criteria; Supplementary Table 1). Research team members completed training in clinical trial ethics and procedures, namely good clinical practice, MINI interview schedule, C-SSRS and clinical rating scales. The site principal investigators were consultant psychiatrists and reviewed the eligibility of each participant and clinical assessments. Participants were compensated £30 or US$60 for each study visit during the blinded phase of the trial. Participants enrolled in the UK were able to keep the tDCS device after trial completion.
Randomization
Participants were randomly assigned to either sham or active tDCS treatment at a 1:1 ratio, which was performed independently in UK and USA. Block randomization, which is a form of stratified random sampling, was used with permuted block sizes of four and six. This was conducted by the sponsor, Flow Neuroscience, and stored in a dedicated database, which was not accessible to research team members.
Intervention
Active and sham tDCS was administered using the Flow FL-100 device. The device was a headset placed over the forehead with two prepositioned conductive rubber electrodes, each 23 cm2. Electrodes were fixed with approximate placement of the anode over F3 (left DLPFC) and the cathode over F4 (right DLPFC) based on international 10–20 electroencephalography system52.
Active stimulation consisted of 2 mA direct current stimulation for 30 min with gradual ramp up over 120 s at the start and ramp down over 15 s at end of the session. Sham stimulation with the same device and app was used to resemble the active intervention and to receive the treatment schedule. An initial ramp up from 0 to 1 mA over 30 s then ramp down to 0 mA over 15 s was repeated at the end of the session to cause a tingling sensation that mimics active stimulation.
The 10-week RCT consisted of five tDCS sessions per week for 3 weeks followed by three tDCS sessions per week for 7 weeks. The tDCS parameters were based on meta-analyses, which demonstrated that treatment effects are most evident for a 30-min stimulus duration for at least 20 sessions (2-mA current) in MDD32,33,34.
At week 10, participants and researchers were informed of treatment arm allocation. The 10-week open-label phase consisted of active tDCS sessions for all participants. Participants who received active tDCS treatment were offered three sessions per week for 10 weeks; participants in the sham treatment arm were offered the active tDCS stimulation schedule, that is, five sessions per week for three weeks then three sessions per week for 7 weeks.
tDCS stimulation was provided using a study-specific installation of the app that connected to the headset via Bluetooth. Researchers had access to remote monitoring, with real-time data use to monitor compliance. Researchers received training to use the headset and were present by videoconferencing for the initial session to support participants who were at home, with app-guided training to demonstrate electrode placement, consisting of video and augmented reality via the device camera. All remaining tDCS sessions were completed by the participants at home, without the presence of a researcher. Participants were asked to have video and microphone on during the initial session. Participants were advised to sit or lie down during use, not to use the headset outdoors, close to water, while driving, during any activity that could lead to a risk of injury, while intoxicated or incapacitated, or in environments with strong magnetic fields.
Blinding
Participants and research team members were blinded to group allocation. We sought to have the same research team member present for the same participant at each study visit. A second research team member joined the clinical reviews for independent rating and would not be present while adverse events or stimulation was discussed to prevent any potential bias. Ratings were cross-checked and reviewed by the site principal investigators.
At week 10, after completion of all assessments and before unblinding, participants were asked whether they thought they had been using the ‘active’ or ‘sham’ tDCS device and how certain they were, as measured by a rating on a scale from 1 (‘very uncertain’) to 5 (‘very certain’). Once this had been completed, the research team member accessed the online remote monitoring system to unblind allocation and informed the participant of group allocation. At the point of unblinding, an automatic email notification was sent to the principal investigator and research team members that unblinding had occurred.
Outcomes
The primary outcome was the adjusted mean group difference in depressive symptom severity between active and sham treatment arms as measured using the 17-item HDRS23 at week 10 (end of treatment) compared to baseline.
Depressive symptom severity was measured by clinician-rated scales, the HDRS and MADRS26, and self-report scale, the MADRS-s27, suicide ideation and attempts using the C-SSRS60, and manic symptoms using the YMRS29 at baseline and at weeks 1, 4, 7, 10 and 20. Anxiety symptoms were measured using the (HAM-A)30 and quality of life was measured using the EQ-5D-3L58,59, consisting of five dimensions (mobility, self-care, usual activities, pain and discomfort) at baseline and at weeks 10 and 20.
Secondary outcomes were the adjusted mean group difference in depressive symptom severity between active and sham treatment arms as measured using the MADRS and MADRS-s at week 10 compared to baseline; clinical response defined as a minimum of 50% reduction from baseline in HDRS, MADRS and MADRS-s at week 10; clinical remission defined as an HDRS score of 7 or less, MADRS score of 10 or less and MADRS-s score of 12 or less; and quality of life as measured by the EQ-5D-3L at week 10.
Exploratory outcomes included correlation between adherence to stimulation and HDRS, MADRS decrease in active treatment arm at week 10; changes in anxiety symptoms from baseline to week 10; and presence of hypomanic and manic symptoms at week 10.
Exploratory outcomes in neuropsychological functioning were assessed using the RAVLT30 total learning score for memory and verbal learning, and the SDMT31 for psychomotor speed and visuospatial attention, assessed at baseline, and then at weeks 10 and 20. Order and versions were counterbalanced. The written SDMT was chosen to reduce the chance of task interference resulting from a poor internet signal. SDMT was mailed to participants, completed using pen and paper during the session, and recorded using a screenshot.
Treatment acceptability was assessed using our treatment acceptability questionnaire13 at baseline, and then at weeks 10 and 20. The full description of the exploratory outcomes is presented in Supplementary Tables 16, 19, 21, 23–35, 37, 38 and 46–53 and Supplementary Figs. 1–6 and 10–12.
Safety
Adverse events were assessed at each visit; participants were able to contact the research team using a dedicated contact number at any time. The tDCS Adverse Events Questionnaire39 was administered at weeks 10 and 20.
Sample size
Sample size calculation was based on Brunoni et al.36, with a two-sample t-test for the mean difference, with 80% power and one-sided type 1 error (0.025), resulting in a sample size of 176 participants with MDD. To increase power to 87.6%, sample size was increased to 216. Assuming a 20% attrition rate, the total sample size was 270 participants. A prespecified interim analysis was performed when 90 participants with MDD completed week 10, which included both futility assessment and sample size reestimation62. The interim analysis was used to modify the trial in two ways for the primary end point, to declare the trial futile and stop enrollment or to specify the number of participants between 100 and 270 to power the trial based on promising zone methodology63,64.
Statistical analysis
The ITT analysis included all randomized participants classified according to the intended treatment. Participants excluded before randomization were considered screen failures. The modified ITT analysis set included ITT participants who received at least one tDCS session (active or sham) and excluded participants randomized in error. The per-protocol analysis set consisted of participants in the modified ITT analysis set, participants with a device failure within the 10-week randomized trial and participants with deviation from the clinical investigation plan caused by the investigational device or by problems regarding tolerability. It excluded participants who took a new medication or treatment during the trial (listed as exclusion criteria), participants who did not meet the inclusion criteria or fulfilled the exclusion criteria, participants who had performed fewer than ten sessions during the first 3 weeks and participants with major protocol violations that would be expected to confound clinical assessment (Supplementary Information—Statistical Analysis Plan, Section 2).
The primary effectiveness outcome was the estimated mean group difference in HDRS scores in participants randomized to active and sham treatments using a mixed model for repeated measures (MMRM). The model included the HDRS baseline value, antidepressant medication status, psychotherapy treatment, age and sex. Missing data were categorized according to the reason for missingness (missing at random or not) and differentially imputed based on that classification. If P values were less than a one-sided P = 0.025, then the end point would be declared positive (Supplementary Information—Statistical Analysis Plan, Sections 3.1–3.1.4, 4 and 5).
MMRM allows for the inclusion of data from all time points in the model and not only baseline and week 10 end-of-treatment values; it allows for the inclusion of participants with missing week 10 values. The MMRM approach is a direct likelihood approach. The MMRM parameters were estimated using SAS PROC MIXED (SAS Institute) v.9.4 or higher. In a matrix equation, the MMRM can be expressed as Yi = Xiβ + Ziu + ei, where β is the vector of the fixed-effect regression parameters (for the overall mean change, the treatment effect θ, a vector of post-baseline time effects τ, a vector of treatment-by-time interaction effects η and a vector of covariate effects φ that includes baseline HDRS, and, optionally, other covariates selected a priori). X is a design matrix for the fixed effects and Z is a design matrix used to account for other random effects u, if any are included. Key assumptions are about e, the random error vector. The expected value is zero, that is, E(e) = 0. An unstructured covariance is assumed, requiring estimation of variances at each visit and all pairwise covariances, that is, Var(e) = σe2Vunstructured (ref. 65).
If the primary end point is met, the secondary end points can be tested based on a hierarchical approach. As specified in the protocol, the Hochberg66,67 approach was used to control multiplicity (Supplementary Table 11). The Hochberg correction rank-orders the end points based on the size of the P value, ranking them from largest to smallest, and compares those values to a sequentially decreasing alpha level to determine whether the null hypothesis should be rejected. Secondary outcomes were HDRS clinical response and remission, EQ-5D-3L change and change in ratings, response and remission in MADRS and MADRS-s (Supplementary Information—Statistical Analysis Plan, Sections 3.1.5–3.1.9).
Exploratory end points were analyzed through summary statistics as the mean and s.d. or percentages and ORs. The two groups were compared using a Student’s t-test or Fisher’s exact test as appropriate. Spearman correlation was used to assess the association between two continuous variables; 95% CIs were presented. The percentages of participants who correctly guessed the arm that they were in were compared using a Fisher’s exact test. Subgroup analyses of primary and secondary end points were conducted through stratification according to antidepressant use at baseline and site (Supplementary Information—Statistical Analysis Plan, Sections 3.1.10 and 8).
Standard deviations are provided based on Cochran’s68 conversion of s.e. to s.d. weighted by sample size. Type 1 errors were controlled by only testing the three named secondary end points after meeting the primary end point; nominal P values are provided for all other evaluations.
Full description of the statistical analyses and handling of missing data can be found in Supplementary Information.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The deidentified individual participant data and the data dictionary that support the findings of this study are available from the academic researchers or the sponsor beginning 6 months after publication because of legal reasons. However, restrictions apply to the availability of these data; thus, they are not publicly available. The Statistical Analysis Plan is available in the Supplementary Information. A data request and brief analysis plan will be required in accordance with the ethics committee requirements. These will be reviewed by the lead, study steering committee and study sponsor. A data transfer agreement will have to be completed before any data being shared. After completion of the data transfer agreement, data will be shared as password-protected files. Data sharing will abide by the rules and policies defined by the sponsor, relevant institutional review boards, as well as local, state and federal laws and regulations. Rights and privacy of individuals participating in the research will be protected at all times. Approval will not be provided for commercial use of the data. Requests can be made to C.H.Y.F. (cynthia.fu@kcl.ac.uk).
Code availability
The analysis code for the longitudinal model is provided in the Supplementary Information. The full code used for the data analysis will be available from the sponsor beginning 6 months after publication of the trial results.
References
World Health Organization. Depression and Other Common Mental Disorders: Global Health Estimates (2017); https://apps.who.int/iris/bitstream/handle/10665/254610/WHO-MSD-MER-2017.2-eng.pdf
Cuijpers, P. et al. The effects of psychotherapies for major depression in adults on remission, recovery and improvement: a meta-analysis. J. Affect. Disord. 159, 118–126 (2014).
Rush, A. J. et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am. J. Psychiatry 163, 1905–1917 (2006).
Woodham, R., Rimmer, R. M., Mutz, J. & Fu, C. H. Y. Is tDCS a potential first line treatment for major depression? Int. Rev. Psychiatry 33, 250–265 (2021).
Creutzfeldt, O. D., Fromm, G. H. & Kapp, H. Influence of transcortical d-c currents on cortical neuronal activity. Exp. Neurol. 5, 436–452 (1962).
Mutz, J. et al. Comparative efficacy and acceptability of non-surgical brain stimulation for the acute treatment of major depressive episodes in adults: systematic review and network meta-analysis. BMJ 364, 1079 (2019).
Nitsche, M. A. et al. Pharmacological modulation of cortical excitability shifts induced by transcranial direct current stimulation in humans. J. Physiol. 553, 293–301 (2003).
Monte-Silva, K. et al. Induction of late LTP-like plasticity in the human motor cortex by repeated non-invasive brain stimulation. Brain Stimul. 6, 424–432 (2013).
Reato, D., Rahman, A., Bikson, M. & Parra, L. C. Low-intensity electrical stimulation affects network dynamics by modulating population rate and spike timing. J. Neurosci. 30, 15067–15079 (2010).
Keeser, D. et al. Prefrontal transcranial direct current stimulation changes connectivity of resting-state networks during fMRI. J. Neurosci. 31, 15284–15293 (2011).
Ironside, M. et al. Effect of prefrontal cortex stimulation on regulation of amygdala response to threat in individuals with trait anxiety: a randomized clinical trial. JAMA Psychiatry 76, 71–78 (2019).
Moffa, A. H. et al. Efficacy and acceptability of transcranial direct current stimulation (tDCS) for major depressive disorder: an individual patient data meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 99, 109836 (2020).
Woodham, R., Rimmer, R. M., Young, A. H. & Fu, C. H. Y. Adjunctive home-based transcranial direct current stimulation treatment for major depression with real-time remote supervision: an open-label, single-arm feasibility study with long term outcomes. J. Psychiatr. Res. 153, 197–205 (2022).
Alonzo, A. et al. Pilot trial of home-administered transcranial direct current stimulation for the treatment of depression. J. Affect. Disord. 252, 475–483 (2019).
Borrione, L., Klein, I., Razza, L. B., Suen, P. & Brunoni, A. R. Use of app-based psychological interventions in combination with home-use transcranial direct current stimulation for the treatment of major depressive disorder: a case series. J. Affect. Disord. 288, 189–190 (2021).
Rimmer, R. M., Woodham, R. D., Cahill, S. & Fu, C. H. Y. Acceptability of home-based transcranial direct current stimulation (tDCS) in major depression: a qualitative analysis of individual experiences. Ment. Health Rev. J. 29, 79–91 (2024).
Borrione, L. et al. Home-use transcranial direct current stimulation for the treatment of a major depressive episode: a randomized clinical trial. JAMA Psychiatry 81, 329–337 (2024).
Kumpf, U. et al. TDCS at home for depressive disorders: an updated systematic review and lessons learned from a prematurely terminated randomized controlled pilot study. Eur. Arch. Psychiatry Clin. Neurosci. 273, 1403–1420 (2023).
Oh, J., Jang, K.-I., Jeon, S. & Chae, J.-H. Effect of self-administered transcranial direct stimulation in patients with major depressive disorder: a randomized, single-blinded clinical trial. Clin. Psychopharmacol. Neurosci. 20, 87–96 (2022).
Nikolin, S. et al. Time-course of the tDCS antidepressant effect: an individual participant data meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 125, 110752 (2023).
Diagnostic and Statistical Manual of Mental Disorders (DSM-5) (American Psychiatric Association, 2013).
Sheehan, D. V. et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J. Clin. Psychiatry 59, 22–33 (1998).
Hamilton, M. A rating scale for depression. J. Neurol. Neurosurg. Psychiatry 23, 56–62 (1960).
Kessler, D. S. et al. Mirtazapine added to SSRIs or SNRIs for treatment resistant depression in primary care: phase III randomised placebo controlled trial (MIR). BMJ 363, k4218 (2018).
Wiles, N. et al. Clinical and cost-effectiveness of cognitive behavioural therapy as an adjunct to pharmacotherapy for treatment resistant depression in primary care: the CoBalT randomised controlled trial. Health Technol. Assess. 18, 1–167 (2014).
Montgomery, S. A. & Åsberg, M. A new depression scale designed to be sensitive to change. Br. J. Psychiatry 134, 382–389 (1979).
Svanborg, P. & Åsberg, M. A comparison between the Beck Depression Inventory (BDI) and the self-rating version of the Montgomery Åsberg Depression Rating Scale (MADRS). J. Affect. Disord. 64, 203–216 (2001).
Hamilton, M. The assessment of anxiety states by rating. Br. J. Med. Psychol. 32, 50–55 (1959).
Young, R. C., Biggs, J. T., Ziegler, V. E. & Meyer, D. A. A rating scale for mania: reliability, validity and sensitivity. Br. J. Psychiatry 133, 429–435 (1978).
Rey, A. L’Examen Clinique En Psychologie (Presses Universitaires de France, 1964).
Smith, A. Symbol Digit Modalities Test (Western Psychological Services, 1991).
Brunoni, A. R. et al. Transcranial direct current stimulation for acute major depressive episodes: meta-analysis of individual patient data. Br. J. Psychiatry 208, 522–531 (2016).
Meron, D., Hedger, N., Garner, M. & Baldwin, D. S. Transcranial direct current stimulation (tDCS) in the treatment of depression: systematic review and meta-analysis of efficacy and tolerability. Neurosci. Biobehav. Rev. 57, 46–62 (2015).
Mutz, J., Edgcumbe, D. R., Brunoni, A. R. & Fu, C. H. Y. Efficacy and acceptability of non-invasive brain stimulation for the treatment of adult unipolar and bipolar depression: a systematic review and meta-analysis of randomised sham-controlled trials. Neurosci. Biobehav. Rev. 92, 291–303 (2018).
Burkhardt, G. et al. Transcranial direct current stimulation as an additional treatment to selective serotonin reuptake inhibitors in adults with major depressive disorder in Germany (DepressionDC): a triple-blind, randomised, sham-controlled, multicentre trial. Lancet 402, 545–554 (2023).
Brunoni, A. R. et al. Trial of electrical direct-current therapy versus escitalopram for depression. N. Engl. J. Med. 376, 2523–2533 (2017).
Frank, E. et al. Anodal skin lesions after treatment with transcranial direct current stimulation. Brain Stimul. 3, 58–59 (2010).
Kortteenniemi, A., Lehto, S. M. & Javadi, A.-H. Delayed, distant skin lesions after transcranial direct current stimulation. Brain Stimul. 12, 204–206 (2019).
Brunoni, A. R. et al. A systematic review on reporting and assessment of adverse effects associated with transcranial direct current stimulation. Int. J. Neuropsychopharmacol. 14, 1133–1145 (2011).
Moffa, A. H. et al. Safety and acceptability of transcranial direct current stimulation for the acute treatment of major depressive episodes: analysis of individual patient data. J. Affect. Disord. 221, 1–5 (2017).
Sathappan, A. V., Luber, B. M. & Lisanby, S. H. The dynamic duo: combining noninvasive brain stimulation with cognitive interventions. Prog. Neuropsychopharmacol. Biol. Psychiatry 89, 347–360 (2019).
Edgcumbe, D. R., Thoma, V., Rivolta, D., Nitsche, M. A. & Fu, C. H. Y. Anodal transcranial direct current stimulation over the right dorsolateral prefrontal cortex enhances reflective judgment and decision-making. Brain Stimul. 12, 652–658 (2019).
Pisoni, A. et al. Cognitive enhancement induced by anodal tDCS drives circuit-specific cortical plasticity. Cereb. Cortex 28, 1132–1140 (2018).
Segrave, R. A., Arnold, S., Hoy, K. & Fitzgerald, P. B. Concurrent cognitive control training augments the antidepressant efficacy of tDCS: a pilot study. Brain Stimul. 7, 325–331 (2014).
Aust, S. et al. Efficacy of augmentation of cognitive behavioral therapy with transcranial direct current stimulation for depression. JAMA Psychiatry 79, 528–537 (2022).
Kolahi, J., Bang, H. & Park, J. Towards a proposal for assessment of blinding success in clinical trials: up-to-date review. Community Dent. Oral Epidemiol. 37, 477–484 (2009).
Hohenschurz-Schmidt, D. et al. Recommendations for the development, implementation, and reporting of control interventions in efficacy and mechanistic trials of physical, psychological, and self-management therapies: the CoPPS Statement. BMJ 381, e072108 (2023).
Evers, A. W. M. et al. What should clinicians tell patients about placebo and nocebo effects? Practical considerations based on expert consensus. Psychother. Psychosom. 90, 49–56 (2021).
Lin, Y.-H. et al. Assessment of blinding in randomized controlled trials of antidepressants for depressive disorders 2000–2020: a systematic review and meta-analysis. eClinicalMedicine 50, 101505 (2022).
Moher, D. et al. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. BMJ 340, c869 (2010).
Bang, H., Ni, L. & Davis, C. E. Assessment of blinding in clinical trials. Control. Clin. Trials 25, 143–156 (2004).
Borrione, L. et al. The Flow brain stimulation headset for the treatment of depression: overview of its safety, efficacy and portable design. Expert Rev. Med. Devices 17, 867–878 (2020).
Opitz, A., Paulus, W., Will, S., Antunes, A. & Thielscher, A. Determinants of the electric field during transcranial direct current stimulation. Neuroimage 109, 140–150 (2015).
Laakso, I., Mikkonen, M., Koyama, S., Hirata, A. & Tanaka, S. Can electric fields explain inter-individual variability in transcranial direct current stimulation of the motor cortex? Sci. Rep. 9, 626 (2019).
Kobak, K. A. A comparison of face-to-face and videoconference administration of the Hamilton Depression Rating Scale. J. Telemed. Telecare 10, 231–235 (2004).
Schiller, C. E. et al. Efficacy of virtual care for depressive disorders: systematic review and meta-analysis. JMIR Ment. Health 10, e38955 (2023).
Scott, A. M. et al. Telehealth v. face-to-face provision of care to patients with depression: a systematic review and meta-analysis. Psychol. Med. 52, 2852–2860 (2022).
Brooks, R. EuroQol: the current state of play. Health Policy 37, 53–72 (1996).
Rabin, R. & de Charro, F. EQ-5D: a measure of health status from the EuroQol Group. Ann. Med. 33, 337–343 (2001).
Posner, K. et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am. J. Psychiatry 168, 1266–1277 (2011).
Heidari, S., Babor, T. F., De Castro, P., Tort, S. & Curno, M. Sex and Gender Equity in Research: rationale for the SAGER guidelines and recommended use. Res. Integr. Peer Rev. 1, 2 (2016).
Lachin, J. M. A review of methods for futility stopping based on conditional power. Stat. Med. 24, 2747–2764 (2005).
Keenan, B. T. & Maislin, G. The use of ‘Fuzzy Promising Zones’ to mitigate against operational bias in adaptive sample size re-estimation designs. In Proc. JSM 2014 (ed. Opsomer, J. D.) 1493–1505 (American Statistical Association, 2014).
Hsiao, S. T., Liu, L. & Mehta, C. R. Optimal promising zone designs. Biom. J. 61, 1175–1186 (2019).
Verbeke, G. & Molenberghs, G. Linear Mixed Models for Longitudinal Data (Springer, 2000).
Hochberg, Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika 75, 800–802 (1988).
Dmitrienko, A. & D’Agostino, R. Sr Traditional multiplicity adjustment methods in clinical trials. Stat. Med. 32, 5172–5218 (2013).
Higgins, J. et al. (eds) Cochrane Handbook for Systematic Reviews of Interventions (Wiley, 2023).
Acknowledgements
C.H.Y.F. has received research grants from the following: research grant funding on behalf of the University of East London from Flow Neuroscience (no. R102696); research grant funding from the National Institute of Mental Health (NIMH) (no. R01MH134236), the Baszucki Brain Research Fund Milken Institute (no. BD0000009), the Rosetrees Trust (no. CF20212104), the International Psychoanalytic Society (no. 158102845), Medical Research Council (MRC) (no. G0802594), the National Association for Research on Schizophrenia And Depression (NARSAD) and the Wellcome Trust. S.S. acknowledges the following funding: research grant funding on behalf of the University of Texas Health Science Center at Houston from Flow Neuroscience and paid advisory boards for the following companies: Worldwide Clinical Trials and Inversago and Vicore Pharma. S.S. is a full-time employee of Intra-Cellular Therapies. He has received grants and research support from NIMH, United States (no. 1R21MH119441-01A1), NIMH (no. 1R21MH129888-01A1), the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) (no. 1R21HD106779-01A1), the Substance Abuse and Mental Health Services Administration (no. 6H79FG000470-01M003) and the Fizer foundation. A.H.Y.’s independent research is funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at the South London and Maudsley NHS Foundation Trust and King’s College London. He is principal investigator for the following studies: the Restore-Life VNS registry study funded by LivaNova; ESKETINTRD3004: ‘An Open-label, Long-term, Safety and Efficacy Study of Intranasal Esketamine in Treatment-resistant Depression’; The Effects of Psilocybin on Cognitive Function in Healthy Participants; The Safety and Efficacy of Psilocybin in Participants with Treatment-Resistant Depression (P-TRD); A Double-Blind, Randomized, Parallel-Group Study with Quetiapine Extended Release as Comparator to Evaluate the Efficacy and Safety of Seltorexant 20 mg as Adjunctive Therapy to Antidepressants in Adult and Elderly Patients with Major Depressive Disorder with Insomnia Symptoms Who Have Responded Inadequately to Antidepressant Therapy (Janssen); An Open-label, Long-term, Safety and Efficacy Study of Aticaprant as Adjunctive Therapy in Adult and Elderly Participants with Major Depressive Disorder (MDD) (Janssen); A Randomized, Double-blind, Multicentre, Parallel-group, Placebo-controlled Study to Evaluate the Efficacy, Safety, and Tolerability of Aticaprant 10 mg as Adjunctive Therapy in Adult Participants with Major Depressive Disorder (MDD) with Moderate-to-severe Anhedonia and Inadequate Response to Current Antidepressant Therapy; and A Study of Disease Characteristics and Real-life Standard of Care Effectiveness in Patients with Major Depressive Disorder (MDD) With Anhedonia and Inadequate Response to Current Antidepressant Therapy Including an SSRI or SNR (Janssen). He is UK Chief Investigator for the following studies: Novartis MDD study no. MIJ821A12201; Compass; and the COMP006 and COMP007 studies. Grant funding (past and present) includes: the NIMH (USA); Canadian Institutes of Health Research (Canada); NARSAD (USA); Stanley Medical Research Institute (USA); MRC (UK); Wellcome Trust (UK); Royal College of Physicians of Edinburgh; BMA (UK); UBC-VGH Foundation (Canada); WEDC (Canada); CCS Depression Research Fund (Canada); Michael Smith Foundation for Health Research (Canada); NIHR (UK); Janssen (UK) and EU Horizon 2020. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. R.M.-V. is the principal investigator for the following grants: National Institutes of Health (nos. R21HD106779 and R21MH129888) and Milken Institute (no. BD-0000000081). This study was funded by a grant from Flow Neuroscience to the study’s Chief Investigator and UK principal investigator to C.H.Y.F., and to the USA principal investigator, S.S. The funder provided the tDCS devices. The funder had no role in data analysis, data interpretation or writing of the manuscript.
Author information
Authors and Affiliations
Contributions
C.H.Y.F. was the chief investigator of the study. She designed the study, led its conduct, was principal investigator for the UK site, led the interpretation of data and wrote the first draft of the manuscript with R.D.W. S.S. was the principal investigator at the USA site until March 2023. R.M.-V. was the principal investigator at the USA site from March 2023. J.C.S. and A.H.Y. contributed to the study design, conduct and interpretation of the data. R.D.W. was the study coordinator. A.-R.G.-N., G.S., H.H., N.L. and P.J.L. contributed to data acquisition at the UK study site. M.R., P.O. and S.S.K. contributed to data acquisition at the USA study site. D.M. was the lead statistician. He supervised the design of the Statistical Analysis Plan, the statistical analysis and the reporting of the clinical trial results. L.H. provided statistical support. All authors approved submission of the manuscript for publication.
Corresponding author
Ethics declarations
Competing interests
C.H.Y.F. reports the following competing interests: research grant funding on behalf of the University of East London from Flow Neuroscience (no. R102696); research grant funding from NIMH (no. R01MH134236), the Baszucki Brain Research Fund Milken Institute (no. BD0000009), the Rosetrees Trust (no. CF20212104), the International Psychoanalytic Society (no. 158102845), the MRC (no. G0802594), NARSAD and the Wellcome Trust. She is Associate Editor of Psychoradiology and Section Editor of the Brain Research Bulletin. A.H.Y. reports the following competing interests: paid lectures and advisory boards for the following companies with therapies used in affective and related disorders: Flow Neuroscience, Novartis, Roche, Janssen, Takeda, Noema Pharma, Compass, AstraZeneca, Boehringer Ingelheim, Eli Lilly, LivaNova, Lundbeck, Sunovion, Servier, LivaNova, Janssen, Allegan, Bionomics, Sumitomo Dainippon Pharma, Sage, Novartis and Neurocentrx. He is principal investigator for the following studies: the Restore-Life VNS registry study funded by LivaNova; ESKETINTRD3004: ‘An Open-label, Long-term, Safety and Efficacy Study of Intranasal Esketamine in Treatment-resistant Depression’; The Effects of Psilocybin on Cognitive Function in Healthy Participants; The Safety and Efficacy of Psilocybin in Participants with Treatment-Resistant Depression (P-TRD); A Double-Blind, Randomized, Parallel-Group Study with Quetiapine Extended Release as Comparator to Evaluate the Efficacy and Safety of Seltorexant 20 mg as Adjunctive Therapy to Antidepressants in Adult and Elderly Patients with Major Depressive Disorder with Insomnia Symptoms Who Have Responded Inadequately to Antidepressant Therapy (Janssen); An Open-label, Long-term, Safety and Efficacy Study of Aticaprant as Adjunctive Therapy in Adult and Elderly Participants with Major Depressive Disorder (MDD) (Janssen); A Randomized, Double-blind, Multicentre, Parallel-group, Placebo-controlled Study to Evaluate the Efficacy, Safety, and Tolerability of Aticaprant 10 mg as Adjunctive Therapy in Adult Participants with Major Depressive Disorder (MDD) with Moderate-to-severe Anhedonia and Inadequate Response to Current Antidepressant Therapy; A Study of Disease Characteristics and Real-life Standard of Care Effectiveness in Patients with Major Depressive Disorder (MDD) With Anhedonia and Inadequate Response to Current Antidepressant Therapy Including an SSRI or SNR (Janssen). He is UK Chief Investigator for the following studies: Novartis MDD study no. MIJ821A12201; Compass; and the COMP006 and COMP007 studies. Grant funding (past and present) includes: NIMH (USA); CIHR (Canada); NARSAD (USA); the Stanley Medical Research Institute (USA); MRC (UK); the Wellcome Trust (UK); the Royal College of Physicians of Edinburgh; the British Medical Association (UK); the VGH & UBC Foundation (Canada); WEDC (Canada); the CCS Depression Research Fund (Canada); the Michael Smith Foundation for Health Research (Canada); NIHR (UK). Janssen (UK) and EU Horizon 2020. He is the Editor of the Journal of Psychopharmacology and Deputy Editor of BJPsych Open. He has no shareholdings in pharmaceutical companies. S.S. reports the following competing interests: research grant funding on behalf of the University of Texas Health Science Center at Houston from Flow Neuroscience; paid advisory boards for the following companies: Worldwide Clinical Trials and Inversago; and Vicore Pharma. He is a full-time employee of Intra-Cellular Therapies. He has received grants and research support from NIMH (USA) (no. 1R21MH119441-01A1), NIMH (no. 1R21MH129888-01A1), NICHD (no. 1R21HD106779-01A1), SAMHSA (no. 6H79FG000470-01M003) and Fizer foundation. He has received research funding as a principal investigator or study/subinvestigator from or participated as consultant/speaker for Flow Neuroscience, COMPASS Pathways, LivaNova, Janssen, Relmada and the Psychiatry Education Forum. Intra-Cellular Therapies or National Institutes of Health (NIH) or SAMHSA or any other organizations had no role in study design and conduct; the collection, management, analysis and interpretation of the data; the preparation, review or approval of the manuscript; and the decision to submit the manuscript for publication. The study’s content is solely the responsibility of the authors and does not necessarily represent the official views of the Intra-Cellular Therapies or NIH or SAMHSA. R.M-V. has received consulting fees from Eurofarma Pharmaceuticals, Abbott and BioStrategies group; has research contracts with Boerhinger Ingelheim and Janssen Pharmaceuticals; and has received speaker fees from Otsuka, EMS and Cristalia. He is a member of the scientific boards of Symbinas Pharmaceuticals and Allergan. He is also the principal investigator for the following grants: NIH (nos. R21HD106779 and R21MH129888), Milken Institute (no. BD-0000000081). D.M. and L.H. work for Biomedical Statistical Consulting; they provide statistical support to MCRA and received payments from Flow Neuroscience. A.-R.G.-N., G.S., H.H., J.C.S., M.R., N.L., P.J.L., P.O., R.D.W. and S.S.K. declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Ulrich Mansmann, Nolan Williams and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Jerome Staal, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Change in Montgomery-Åsberg Depression Rating Scale (MADRS) ratings over time.
Estimated mean MADRS rating scores from baseline to week 10 in the modified intention-to-treat analysis sample (n = 173) in active tDCS and sham tDCS treatment arms. Error bars represent ± 1 standard error (SE). MADRS scores range from 0 to 60 with higher values indicating more severe depressive symptoms. A significant improvement was observed in the change in MADRS ratings from baseline to week 10 in the active tDCS treatment arm, MADRS change 11.31 ± 8.81(standard deviation (SD)) (mean week 10 MADRS 12.46 ± 1.09 (SE)) as compared to sham tDCS treatment arm, MADRS change 7.74 ± 8.47 (SD) (mean week 10 MADRS 15.30 ± 1.07 (SE)) (95% CI 1.1 to 6.1, p = 0.006). The difference in change scores was also significant at week 4 (95% CI 1.2 to 5.5, p = 0.003) and week 7 (95% CI 1.1 to 5.8, p = 0.005) with a greater score decrease in the active treatment arm. Fully Conditional Specification (FCS) approach was used to produce 20 multiply imputed completed data sets. The FCS approach accommodates nonmonotonicity in the pattern of missing data and requires regression models to be specified for each variable with missing values needing imputation. All models included age, sex, in psychotherapy at baseline, use of any antidepressants at baseline and treatment group. The resulting completed datasets were combined using Rubin’s Rules. ** = p < 0.01.
Extended Data Fig. 2 Change in Montgomery-Åsberg Depression Rating Scale-Self report (MADRS-s) ratings over time.
Estimated mean MADRS-s rating scores from baseline to week 10 in the modified intention-to-treat analysis sample (n = 173) for the active tDCS and sham tDCS treatment arms. Error bars represent ± 1 standard error (SE). MADRS-s scores range from 0 to 60 with higher values indicating more severe depression. A significant improvement was observed in the change in MADRS-s ratings from baseline to week 10 in the active tDCS treatment arm, MADRS-s change 9.90 ± 8.94 (standard deviation (SD)) (mean week 10 MADRS-s 16.60 ± 1.18 (SE)) as compared to sham tDCS treatment arm, MADRS-s change 6.23 ± 9.13 (SD) (mean week 10 MADRS-s 19.55 ± 1.16 (SE)) (95% CI 0.9 to 6.4, p = 0.009). The difference in change scores was also significant at week 4 (95% CI 0.3 to 4.9, p = 0.030) with a greater score decrease in the active treatment arm. Fully Conditional Specification (FCS) approach was used to produce 20 multiply imputed completed data sets. The FCS approach accommodates nonmonotonicity in the pattern of missing data and requires regression models to be specified for each variable with missing values needing imputation. All models included age, sex, in psychotherapy at baseline, use of any antidepressants at baseline and treatment group. The resulting completed datasets were combined using Rubin’s Rules. * = p < 0.05, ** = p < 0.01.
Supplementary information
Supplementary Information (download PDF )
Supplementary Notes, Tables 1–53, Figs. 1–13, Statistical Analysis Plan and Study Protocol.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Woodham, R.D., Selvaraj, S., Lajmi, N. et al. Home-based transcranial direct current stimulation treatment for major depressive disorder: a fully remote phase 2 randomized sham-controlled trial. Nat Med 31, 87–95 (2025). https://doi.org/10.1038/s41591-024-03305-y
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41591-024-03305-y
This article is cited by
-
Home-based transcranial direct current stimulation (tDCS) for bipolar depression: effects on quality of life and functioning—an open-label study
Quality of Life Research (2026)
-
Effect of home-based transcranial direct current stimulation (tDCS) on cognitive functioning in bipolar depression: an open-label, single-arm acceptability and feasibility study
International Journal of Bipolar Disorders (2025)
-
Acceptability of home-based transcranial direct current stimulation (tDCS) in bipolar depression: thematic analysis of individual views
BMC Psychiatry (2025)
-
Effects of transcranial direct current stimulation and mirror therapy on mental health and dopamine level among spastic quadriplegic cerebral palsy children
BMC Pediatrics (2025)
-
Aberrant brain functional network in COPD patients with cognitive impairment: clinical manifestations, mechanisms and therapeutic strategies
Journal of Neuroinflammation (2025)
