
Abstract
Large genomic programs have contributed to improving drug development in cancer. To assess the potential benefit of using larger gene panels to guide molecular-based treatments, we conducted a multicenter randomized trial in patients with advanced and/or metastatic solid cancer. Molecular alterations were determined using either a panel of 324 cancer-related genes (Foundation OneCDX (F1CDX)) or a limited panel of 87 single-nucleotide/indel genes and genome-wide copy number variations (CTL) and reviewed by a molecular tumor board to identify molecular-based recommended therapies (MBRTs). Using paired data from both panels for each patient, the primary endpoint was the proportion of patients with an MBRT identified. Main secondary endpoints included the number of patients with at least one actionable alteration leading to MBRT identification, the number of patients with and without MBRTs initiated, progression-free survival, best overall response, duration of response and safety. Among the 741 patients screened, 45.7% had quality-checked tumor samples. MBRTs were identified with F1CDX in 175 (51.6%) patients and with CTL in 125 (36.9%) patients, translating to a significant increase of 14.8 percentage points (P < 0.001) with the more comprehensive gene panel versus the more limited panel, meeting the primary endpoint. However, no differences in clinical outcomes were observed in these patients with advanced and/or metastatic cancer in need of treatment beyond standard genomic alterations. These findings illustrate the potential for larger gene panels to increase the number of molecularly matched therapies. Larger studies are needed to assess the clinical benefit of expanded MBRTs. ClinicalTrials.gov registration: NCT03163732.
Similar content being viewed by others
Main
Genomic profiling efforts from the International Cancer Genomics Consortium, The Cancer Genome Atlas and many other programs have identified actionable molecular alterations in all cancer types1,2,3,4,5,6. However, while substantial acceleration and better accessibility in genetic sequencing have been achieved, large programs to guide cancer treatment still face challenges. The number of patients effectively treated with molecular-based recommended therapies (MBRTs) remains low.
We previously reported the use of next-generation sequencing (NGS) with panels of 59–69 genes and microarray-based comparative genomic hybridization in the ProfiLER-01 study. Molecular profiling in enrolled patients with metastatic cancer allowed proposing MBRTs in 27% of the patients, including 6% in whom an MBRT was initiated7. Similar studies, namely SHIVA4, MPACT8 and MATCH9, also reported that the rates of MBRT initiation have been regularly disappointing and below 9.5%, with a short duration of clinical benefits.
More recently, several large genomic programs aimed to establish standardized high-throughput whole-genome sequencing (WGS) in patients with cancer or rare diseases10,11. For example, the 100,000 Genomes Project study, a UK government initiative conducted within the National Health Service in England, aimed to link genomic and real-world clinical data. Cancer patients showed a high prevalence of genetic variants, and a multidisciplinary molecular tumor board (MTB) intensively discussed the cases to determine clinical actionability and provide MBRTs. The correlation between WGS and patient outcomes allowed confirming prognostic signatures and/or predictive genomic biomarkers11. However, to date, the clinical impact of a large NGS panel (such as WGS) on MBRT initiations (through clinical trial inclusions or off-label prescription of targeted agents) and outcomes remains unclear.
In an attempt to assess this impact, the multicenter prospective randomized study ProfiLER-02 aimed to evaluate the added value of a larger number of screened genomic biomarkers from the gene panel Foundation OneCDX (F1CDX panel, 324 genes) compared to our home-based molecular profiling panel (CTL panel with limited biomarker detection, 87 genes) to identify more MBRTs in patients with advanced cancer.
Results
Description of the cohort
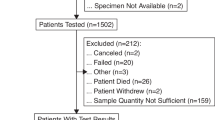
From June 2017 to June 2019, 741 patients with advanced and/or metastatic solid tumors receiving anticancer treatment in the first- or second-line setting were screened, and 339 patients with tumor samples quality-controlled by the referent pathologist were randomized (intention-to-treat (ITT) population) (Figs. 1 and 2 and Extended Data Fig. 1). Among these 339 patients, NGS panels used archived (>3 months) tumor samples in 260 (76.7%) patients and fresh de novo tumor biopsy samples (≤3 months before randomization) in 79 (23.3%) patients.

Patients with tumor sample available (ITT population, n = 339) were allocated to randomization to F1CDX (n = 171) or CTL (n = 168). Molecular profile analysis failed in 13 patients for both panels (F1CDX: n = 5, CTL: n = 8), and 17 patients had a molecular profile achieved from a single panel (F1CDX: n = 10, CTL: n = 7). Molecular profiles achieved with both panels were available for 309 patients (per protocol population). Two gene panels were used: F1CDX (324 genes) and a home-based limited panel (CTL) (87 genes). At patient progression, the MTB reviewed genomic alterations matching with at least one MBRT based on the first panel as defined per randomization. In the case of failure (no MBRT identified with the first panel), the second panel was disclosed. Among the 339 patients, an MBRT was identified in 192 (56.6%) patients and an MBRT was initiated in 51 (15%) patients. Of note, 17 deaths occurred during screening, and tumor samples were not collected from these patients. A total of 58 patients died after randomization and before MTB review. However, their molecular profiles had been considered for MTB review at the date of data cutoff, and MBRTs were recorded (no MBRT: n = 25 (F1CDX: 12, CTL: 13); MBRTs: n = 33 (F1CDX: 19, CTL: 14)). A total of 233 deaths occurred during the study period.

a,b, PFS in patients with subsequent treatment line (n = 163; F1CDX: 79, CTL: 84) (a), and in patients with MBRT initiated after MTB#1 (n = 43; F1CDX: 24, CTL: 19) (b). PFS was estimated from first molecular tumor board (MTB#1). PFS was set to 0 in patients with no MBRT (F1CDX: 30; CTL: 43). KM est., Kaplan–Meier estimate.
Patient, tumor and treatment characteristics are presented in Table 1 (n = 339). In the ITT population, female patients (n = 186) accounted for 54.9% of the population. At inclusion, the median age was 57 (19–85) years and 222 (65.5%) patients had metastasis. At inclusion, 193 (56.9%) patients were receiving treatment in the first-line setting, 107 (31.6%) patients were receiving second-line treatment and 2 patients were receiving third-line treatment (protocol deviation). The most common tumor types were gliomas (n = 81, 23.9%), gynecological cancers (n = 44, 13.0%), sarcomas (n = 41, 12.1%), breast cancers (n = 30, 8.8%) and genitourinary cancers (n = 28, 8.3%).
Primary endpoint
Using paired data issued from both panels for each patient, the proportion of patients for whom an MBRT was identified was 51.6% using the F1CDX panel (n = 175) and 36.9% using the CTL panel (n = 125), corresponding to a significant increase in MBRT identification of 14.8 percentage points using F1CDX versus CTL (McNemar test: P < 0.001) (Table 2a,b). MBRTs were exclusively identified with one panel in 84 (24.8%) patients (F1CDX+/CTL−: 67, 19.8%; F1CDX−/CTL+: 17, 5.0%) (Table 2a,b, Fig. 2 and Extended Data Fig. 1).
Therefore, the study met the primary endpoint, reaching the expected increase in MBRTs of at least 10 percentage points with F1CDX versus CTL considering at least 20% discordant pairs (Table 2a,b).
Prespecified sensitivity analysis of the primary endpoint
A sensitivity analysis was performed including the subgroup of patients with molecular profiles achieved with both panels (n = 309); 170 patients (55%) had MBRTs identified based on the F1CDX panel compared to 123 (39.8%) patients in whom MBRTs were identified with the CTL panel. The rate of discordant pairs (MBRTs identified exclusively with one panel) was 24.9% (n = 77) (F1CDX+/CTL−: 62, 20.1%; F1CDX−/CTL+: 15, 4.9%). The sensitivity analysis, which showed a 15.2 percentage points increase in MBRT identification with F1CDX, confirmed the results regarding the expected increase in MBRTs (McNemar test: P < 0.001) (Table 2c).
Secondary endpoints
Number of patients with at least one alteration and MBRT identified
Among the 339 patients, 192 (56.6%) had at least one actionable alteration leading to the identification of an MBRT regardless of the panel used (both panels: n = 108, exclusively F1CDX: n = 67, exclusively CTL: n = 17) (Table 2b and Extended Data Fig. 2). Patients had MBRTs mainly targeting the PI3K–AKT–mTOR pathway (both panels: n = 45, exclusively F1CDX: n = 22, exclusively CTL: n = 14), homologous recombination deficiency (both panels: n = 21, F1CDX: n = 12, CTL: n = 5), the BRAF pathway (both panels: n = 7, F1CDX: n = 3, CTL: n = 0) or the HER2 pathway (both panels: n = 6, F1CDX: n = 5, CTL: n = 2) (Extended Data Fig. 3a). F1CDX-based MBRTs included those identified according to alterations in tumor mutational burden status (n = 36), gene rearrangements (n = 10) and BAP1 alterations (n = 6) exclusively investigated with the F1CDX panel.
Number of patients with MBRTs initiated
Among the 339 patients, 51 had an MBRT initiated regardless of the panel used. F1CDX led to MBRT initiation in 48 (14.2%) patients, and CTL led to MBRT initiation in 30 (8.8%) patients, that is, an increase of 5.4 percentage points in patients with MBRTs initiated based on F1CDX (McNemar test: P < 0.001). A total of 27 (8.0%) patients had MBRTs initiated based on both panels, 3 (0.9%) additional patients had MBRTs initiated exclusively with CTL and 21 (6.2%) additional patients had MBRTs initiated exclusively with F1CDX, leading to 24 (7.1%) discordant results (Table 2d). Of note, most decisions occurred at the first MTB review (MTB#1: n = 43; F1CDX: n = 24, CTL: n = 19); some decisions were made at MTB#2 (CTL: n = 8).
The initiated MBRTs were more often PI3K–AKT–mTOR inhibitors (everolimus; both panels: n = 7, exclusively F1CDX: n = 3, exclusively CTL: n = 2), PARP inhibitors (olaparib; both panels: n = 7, exclusively F1CDX: n = 2, exclusively CTL: n = 0) or anti-PD-L1 ± anti-CTLA4 therapy (durvalumab, tremelimumab; both panels: n = 0, exclusively F1CDX: n = 6, exclusively CTL: n = 0) (Extended Data Fig. 3b). Among the 51 patients with MBRTs initiated, 34 patients had access to an experimental therapy provided in a clinical trial and 17 patients received off-label or recently approved MBRTs.
Number of patients with no MBRT initiated and reasons for noninitiation
A total of 89 (F1CDX: n = 48, CTL: n = 41) patients had MBRTs identified but not initiated. The main reasons for noninitiation were early death before MTB review (n = 34; F1CDX: n = 19, CTL: n = 15), clinical deterioration or early death within 2 months after MTB review (n = 20; F1CDX: n = 10, CTL: n = 10) or investigator decision (n = 14; F1CDX: n = 8, CTL: n = 6). Few patients had no disease progression documented before MTB review (trial discontinuation: n = 10 (F1CDX: n = 3, CTL: n = 7); investigator mistake: n = 3 (F1CDX: n = 2, CTL: n = 1)).
Progression-free survival
In the ITT population (patients with qualified tumor samples, n = 339), the median time from randomization to MTB#1 was 7.62 (0.8–48.1) months. MTB#1 used, as defined per randomization, either the F1CDX panel (n = 171) or the CTL panel (n = 168) to identify MBRTs (Fig. 1). Patient, tumor and treatment characteristics in the total population and according to the first panel revealed are presented in Table 1.
The median progression-free survival (PFS) (considering PFS = 0 for patients with no MBRT, n = 73; F1CDX: n = 30, CTL: n = 43) from the date of MTB#1 in patients in whom treatment was initiated after MTB#1 (n = 163; F1CDX: n = 79, CTL: n = 84) was 1.4 (95% confidence interval (CI) 0.0–2.1) months in patients allocated to the F1CDX panel first and 0 (95% CI 0–1.4) months in those allocated to the CTL panel first (P = 0.3409) (Fig. 2a).
Best overall response
The best overall response (BOR) was complete response in 1 patient (F1CDX), partial response in 4 patients (F1CDX: n = 1, CTL: n = 3), stable disease in 12 patients (F1CDX: n = 6, CTL: n = 6) and progression in 21 patients; 13 responses were nonevaluable.
Duration of response
The duration of objective response was reported in five patients who received different targeted treatments or immunotherapy alone or in combination (olaparib: 18 months (CTL); encorafenib–binimetinib: 15.6 months (CTL); rucaparib–atezolizumab: 4 months (CTL); erdafitinib: 9 months (early discontinuation due to toxicity) (F1CDX); durvalumab–tremelimumab: 4 months (F1CDX)).
Safety
No adverse events related to the study procedures were reported during the study period.
Exploratory endpoints
Number of MBRTs per patient and type of MBRTs
Among the 293 molecular alterations identified based on F1CDX and 196 molecular alterations identified based on CTL in the 192 (56.6%) patients with MBRTs, a total of 259 MBRTs were provided based on F1CDX versus 164 MBRTs provided based on CTL. Compared to CTL, the F1CDX panel allowed identifying more MBRTs per patient (patients with only one MBRT, F1CDX: n = 106, CTL: n = 96; patients with two MBRTs, F1CDX: n = 58, CTL: n = 21; patients with three or more MBRTs, F1CDX: n = 11, CTL: n = 8). The MBRTs provided were immunotherapies (F1CDX: n = 39, CTL: n = 6) or targeted agents (F1CDX: n = 155, CTL: n = 122). Two patients had two targeted MBRTs (binimetinib, encorafenib), whereas two patients received both targeted and immuno-MBRTs (atezolizumab, rucaparib).
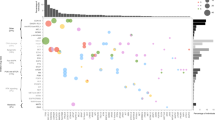
Post hoc analyses according to disease localization reported the number of patients with any MBRT (gynecological cancer, n = 12; glioma, n = 10; sarcoma, n = 7) and the detailed MBRTs initiated (Extended Data Fig. 4 and Extended Data Table 1). Patients with molecular alterations were further classified according to the European Society for Medical Oncology (ESMO) Scale for Clinical Actionability of molecular Targets (ESCAT) and were mainly categorized into ESCAT II or III (ESCAT IIA,B: n = 70, ESCAT IIIA: n = 91) (Extended Data Table 2). The characteristics of patients with or without treatment recommendation or initiation are detailed in Extended Data Table 3.
Survival in patients with MBRTs initiated
In patients with MBRTs initiated after MTB#1 (n = 43; F1CDX: n = 24, CTL: n = 19), the median PFS from MTB#1 was 1.9 (95% CI 1.4–2.9) months with F1CDX and 2.6 (95% CI 1.9–4.0) months with CTL (hazard ratio 0.76, 95% CI 0.41–1.40) (Fig. 2b). Of them, 42 patients were alive at 12 months (F1CDX: n = 22, CTL: n = 20), including 7 patients with no disease progression at 12 months (F1CDX: n = 4, CTL: n = 3) (P = 0.3745) (Fig. 2b).
Discussion
The multicenter prospective ProfiLER-02 trial met its primary endpoint with a more than 10 percentage points increase in treatment recommendations using the commercially available panel F1CDX compared to our homemade control gene panel (CTL). The ProfiLER-02 trial showed that the F1CDX panel increased the total number of MBRTs in this specific population of patients who did not present approved predictive biomarkers to guide MBRTs routinely used in specific cancer types. The trial also showed an increased number of patients in whom MBRTs were effectively initiated using the F1CDX panel compared to our control local gene panel.
In real-world clinical oncology, the effectiveness of routine use of NGS is important. To characterize genomic profiles and select approved molecular-guided therapies for patients with advanced cancer, the US Food and Drug Administration and the ESMO recommend the use of multigene panel testing, especially for advanced non-small cell lung, prostate and ovarian cancers, as well as cholangiocarcinomas12. In early-stage cancers, except for rare cancers, comprehensive genomic profiling remains futile, with no direct impact on the individualized treatment strategy13. For patients with metastatic cancers for whom approved standard-of-care therapies are exhausted, genomic panel testing has been demonstrated to be effective. Some real-world studies showed the positive impact of NGS-based multigene tests on patient outcomes, particularly for early-phase clinical trial screening and rare cancers5,8,13,14,15 or as part of programs at large academic medical centers, such as the MSK-IMPACT at the Memorial Sloan Kettering Cancer Center16. Many reports and ongoing multicenter pan-tumor clinical trials, including TAPISTRY (NCT04589845) or DETERMINE (NCT05722886), underline the potential efficacy of large comprehensive gene profiling, with favorable antitumor response rates16,17 or improved patient survival18,19.
The question of the number of genes to be tested remains controversial20. As the landscape of actionable alterations continues to expand, there is an ongoing need to reconsider the size of the pan-tumor NGS gene panels. Recently, the 100,000 Genomes Project study, gathering WGS data from 10,470 patients, identified 330 cancer driver genes. Besides 55% of the tumors harboring a potentially actionable mutation, many oncogenic mutations resulted in ‘undruggable’ proteins (that is, proteins that cannot be considered targets of MBRTs)21. Furthermore, only a few patients had the opportunity to initiate the recommended therapy, mainly due to the lack of early-phase clinical trials. Although the initiation of off-label MBRTs is not allowed outside a clinical trial in France, 17 patients received recently approved or off-label MBRTs with specific informed consent. This reflects the considerable imbalance between the development of large molecular oncology programs and critical challenges in the clinical setting that still need to be overcome, including (1) administrative limitations to simultaneously opening recruitment in numerous investigation sites as required for these large multicentric programs, (2) inadequate access to genomic DNA- and RNA-sequencing platforms, (3) slow delivery of validated histological and genomic results and (4) scarce access to dedicated clinical trials providing first-in-class compounds in investigational sites. Nevertheless, the ProfiLER-02 study showed that, compared to CTL, F1CDX led to a 14.8 percentage points increase in the proportion of patients with MBRTs, which translated into a slight but significant improvement that may support further use of such large gene panels.
Of note, molecular alterations were mainly qualified as ESCAT II or III, which confirmed the initially targeted study population (without routine predictive biomarkers for approved targeted agents) and emphasized the critical need for MBRTs in this difficult-to-treat patient population. However, the cancer types in this series were not representative of the global incidence of cancers, as attested by the considerable overrepresentation of gliomas and sarcomas and underrepresentation of lung and colorectal cancers. Indeed, the study mainly included patients with no already identified actionable targets or drivers and no standard therapeutic options.
The PI3K–mTOR signaling pathway is known to be dysregulated in a wide spectrum of carcinomas. Mutations in kinase domains or loss of PTEN can contribute to accelerating carcinogenesis and cancer progression. Small-molecule inhibitors of the PI3K–mTOR pathway have exhibited clinical efficacy in various cancers22, but intrinsic and acquired resistance limits their therapeutic efficacy23. Furthermore, correlations between PI3K–mTOR pathway alterations and response to therapy have not been demonstrated in prospective clinical studies. Our study reported only 12 patients treated with everolimus although 81 tumors harbored alterations in the PI3K–AKT–mTOR pathway.
In addition, two patients received a PARP inhibitor combined with a PD-L1 inhibitor based on abnormalities identified in both pathways and one patient received a combination of targeted therapies (binimetinib + encorafenib). No other patient received combined targeted agents. The MTB prioritized the MBRT to treat patients with single-agent therapy.
Performance specifications in sensitivity, reproducibility and accuracy are similar regardless of the panel used for NGS genomic profiling, but differences and discordances in MBRT recommendations mainly rely on the biomarkers and genes investigated for pathognomonic and therapeutic alterations. Immunotherapy has revolutionized cancer management, and the ProfiLER-02 study highlights the need for enhanced and dedicated therapeutic biomarker detection. Recently identified biomarkers, such as homologous recombination deficiency and microsatellite instability status, were not yet investigated when we designed the ProfiLER-02 study.
These results show that wide-ranging studies involving an extended genome size (CTL: 12.5 Mb, F1CDX: 1.2 Mb) or a large number of genes (genome-wide ~15,000 genes; F1CDX: 324 genes) did not allow better identification of patients who may benefit from dedicated treatment but underlined the need for collaborative efforts to significantly increase accuracy in biomarker selection.
We have presented a cost-effectiveness analysis performed in a randomized population of patients with MBRTs who were evaluable for PFS (n = 163; F1CDX: 79, CTL: 84), which showed an incremental cost-effectiveness ratio of below €100,000 per life year gained24. Updated results related to this cost-effectiveness analysis will be published in the near future.
Current data on the use of large panels provide interesting results in terms of genomic identification, with translation into clinical benefits remaining disappointing thus far. As data supporting the validity for further development of liquid biopsy and circulating tumor DNA (ctDNA) testing become available, clinical indications will expand. In turn, increasing use of such noninvasive, quick and comprehensive analyses is expected, and cost decrease and cost-effectiveness studies may soon demonstrate that complete ctDNA NGS would benefit patients without burdening the healthcare system. In addition, rapid advances in overcoming technical barriers, decreasing sequencing costs, the availability of more effective bioinformatic tools and improved access to MBRTs recently showed encouraging developments that should not be neglected. Such approaches open huge opportunities in predictive biomarker assessment, which may soon provide major benefits to this population in need of treatment beyond standard genomic alterations.
Some limitations have to be underlined. These results should be interpreted with caution considering the small number of patients in whom an MBRT was initiated (the main limitation resulting from the critical lack of accessibility of MBRTs) and the aggressiveness of most of the included cancers (gliomas and sarcomas). Furthermore, tight timeframes are required for faster decision-making in line with the already deteriorated health status of these pretreated patients with metastatic disease in critical need of innovative therapeutic options.
In conclusion, the F1CDX panel enabled a 14.8 percentage points increased rate of actionable alterations identified and a 5.4 percentage points increase in treatment initiations compared to the CTL panel. However, no differences in clinical outcomes were observed in this population of patients with advanced and/or metastatic cancer in need of treatment beyond standard genomic alterations. Further developments in DNA- and RNA-sequencing technology, including combined advances in chemistry, bioinformatics and artificial intelligence, associated with other -omics technologies, are required to overcome the current challenges, promote the use of functional genomics and contribute to translating such approaches into efficient routine clinical applications.
Methods
Study design and participants
This multicenter randomized prospective study, ProfiLER-02, was performed in 12 institutions after obtaining approval from the Ethics Committee of Lyon Sud-Est IV on 23 May 2017 and after receiving authorization from the national competent authority on 15 May 2017. This study was conducted in accordance with the Good Clinical Practice guidelines of the International Conference on Harmonization and the Declaration of Helsinki, as well as relevant French and European laws and directives. Several on-study protocol amendments occurred, and the main changes in successive versions of the protocol (protocol v1.0, 10 February 2017; v2.0, 29 January 2018; v3.0, 08 October 2018; v4.0, 05 February 2019; v5.0, 07 April 2019), with the respective approval dates and authorized investigation sites, are detailed at the end of the Methods. All patients provided written informed consent. This trial is registered with ClinicalTrials.gov (NCT03163732).
The study enrolled adult patients with cancer who were receiving first- or second-line chemotherapy for any type of advanced and/or metastatic solid tumors and had available tumor samples (a <3 months archived tumor formalin-fixed and paraffin-embedded (FFPE) block or an at least 10-mm-diameter sample from a de novo tumor biopsy or a <3 years archived sample of a primary or metastatic lesion). The patients had an Eastern Cooperative Oncology Group performance status (ECOG-PS) of 0–1, adequate organ and marrow function and no routine predictive tumor biomarkers for standard therapies. Patients should not progress during the next 45 days after having provided written consent to achieve results from NGS profiles before progression. All inclusion and exclusion criteria are presented at the end of the Methods.
Procedures
The study was proposed to patients during their first or second line of therapy for advanced and/or metastatic solid cancer. FFPE tumor specimens from archival samples of the primary tumor, local relapse or metastasis or from a de novo biopsy were collected and checked for quality (>20–30% tumor cells) and quantity (>5 mm area, 90 µm depth) by a central pathology review (Centre Léon Bérard, Lyon, France). Samples not meeting the quality criteria resulted in patient screening failure. This study was classified as ‘Interventional research involving the human person other than health products’ category 1 (articles L1121-1 and L5311-1 from the French Code of Public Health), and only adverse events and serious adverse events or reactions related to study procedures (including biopsies or punctures and blood sample collection) had to be reported by the investigators.
Genomic molecular profiles were determined concomitantly using NGS and two different gene panels: (1) a panel of 324 cancer-related genes (F1CDX) provided by Foundation Medicine and (2) a panel of 87 cancer-related genes (CTL) routinely used at Centre Léon Bérard (Extended Data Fig. 1 and Supplementary Table 1). F1CDX used targeted high-throughput hybridization-based capture technology for detecting substitutions, insertion and deletion alterations (indels), copy number alterations in 324 genes and selected gene rearrangements, as well as genomic signatures including tumor mutational burden. CTL used genomic DNA extracted from FFPE tissues using the Forma Pure RNA kit (Beckman Coulter, #C19158) following the manufacturer’s instructions. DNA concentrations were determined using a Qubit fluorometer (Invitrogen), absorbance was measured with a NanoDrop spectrophotometer (Thermo Fisher Scientific), and quality profiles were assessed on a TapeStation instrument (Agilent Technologies). A total of 200 ng input DNA per sample was used for SureSelect XT Low Input library preparation (Agilent Technologies). The library preparations were constructed according to the SureSelect XT Low Input Target Enrichment System for Illumina Paired-End Multiplexed Sequencing Library protocol (version C2, July 2019) and sequenced on an Illumina NovaSeq 6000 platform (2 × 151 bp). The quality of sequencing data was checked using FastQC (v0.11.5) and SAMtools (v1.3.1) for DNA and FastQC and RSeQC for RNA. Statistical analyses were performed using R v3.5.3 (R Foundation for Statistical Computing). Mutations were annotated with Annovar (v02-2016) and VEP (release 92). We used an ‘in-house’ pipeline based on the allelic frequencies for the detection of copy number variations on the whole-genome backbone, using two negative controls as the reference level.
Randomization, MTB and treatment decision
Registration and randomization used an interactive web response system (IWRS, Ennov) with a permuted block design. A statistician was in charge of generating the randomization list. No access to the eCRF randomization module was provided during the study. The panel order was assigned by the IWRS. The randomization was not stratified.
The randomization defined (in a 1:1 ratio) the molecular panel to disclose (either F1CDX or CTL) at MTB#1. At randomization request, the IWRS automatically sent an email with the NGS panel assigned at MTB#1 to biologists only. Other study collaborators remained blinded. At documented patient progression, MTB#1 reviewed molecular data from the NGS panel assigned by randomization, and amplifications, deletions and mutations (that is, actionable alterations) were used to identify MBRTs that could be initiated; the second panel remained unrevealed. Molecular data from the other NGS panel were reviewed only in the case of recommendation failure based on the first panel revealed (Extended Data Fig. 1). Genomic alterations were subsequently interpreted according to the classification proposed by Leichsenring et al.25.
If an MBRT was initiated based on the first panel, data from the second panel were revealed at clinical or radiological progression and/or unacceptable toxicity under MBRT treatment and were eventually used to guide another line of MBRT (Extended Data Fig. 1).
An MBRT was defined as a molecular-targeted agent recommended outside its European Medicines Agency approval. Patients with an MBRT initiated were followed according to the related clinical trial protocol they were referred to or according to the routine medical practice for off-label use of MBRTs.
Statistics and reproducibility
Sample size calculation
Sample size calculation was performed considering the patient as their own control; we reported the MBRTs and therapies initiated regardless of the order of panel disclosure defined by randomization. We expected a 10% increased rate of MBRTs with F1CDX versus CTL. A total of 289 patients with results achieved using both panels (pairs) were needed to detect with 98% power a 10% difference in proportions, with 20% discordant pairs in proportions using a McNemar test of equality of paired proportions at a 5% two-sided significance level (Table 2 and Extended Data Fig. 1).
Considering that approximately 10% of the patients would be nonevaluable for the primary endpoint (that is, the second panel is not disclosed at the time of the final analysis due to nonprogressive disease or technical failure with one of the panels), 320 patients had to be randomized.
Considering a rate of randomization of 40%, approximately 600 patients had to be screened to ensure the randomization of 320 patients and a total of 289 patients with molecular profiles achieved with both panels.
General statistical methods
All statistical methods were based on the International Conference on Harmonization E9 (‘Statistical principles for clinical trials’) guidelines.
Qualitative variables were described with frequencies and percentages, and quantitative variables were described using median and range (min–max). Comparisons between groups used the chi-square test for qualitative variables and the Kruskal–Wallis test for quantitative variables.
Patient characteristics and other baseline data (demographics, disease characteristics and clinical and biological data) were summarized per randomization panel order and in total to characterize the study population and ensure the initial comparability.
Statistical analyses were performed using SAS software v9.4 or later.
Analysis population
The global analysis population included all enrolled patients. The ITT population included all randomized patients, that is, patients with at least one molecular profile based on one panel (n = 339), assuming that a nonconclusive analysis with one panel was considered no MBRT. A prespecified sensitivity analysis of the primary endpoint was performed in the population of patients with molecular profiles achieved with both panels (n = 309) (Extended Data Fig. 2). A PFS subgroup analysis included all patients for whom an MBRT was initiated.
Analyses were performed on clinical data from patients enrolled from June 2017 to June 2019, and molecular analyses were completed and examined by the MTB before 11 November 2021.
Study endpoints
Primary endpoint
The primary endpoint was the proportion of patients with an MBRT identified using the large (324 cancer-related genes) F1CDX panel versus the limited control panel (CTL) of 87 cancer-related genes routinely used at the Centre Léon Bérard, considering paired data issued from both panels for each patient.
Secondary endpoints
The main secondary endpoints included the proportion of patients with at least one actionable alteration leading to MBRT identification; the proportion of patients for whom an MBRT was effectively initiated, described using paired data issued from both panels for each patient; the proportion of discriminant cases, that is, patients with an MBRT identified exclusively with one panel; the number of patients with no MBRT initiated with reasons for noninitiation; and the clinical benefit comparing PFS according to Response Evaluation Criteria in Solid Tumors (RECIST 1.1), the BOR according to RECIST 1.1 and the duration of response between the two randomization arms.
Efficacy analysis
Primary endpoint
The primary endpoint analysis was performed on the ITT population using paired data issued from both panels for each patient, regardless of the result of the randomization. Patients for whom no MBRT can be identified from a panel as defined per randomization were considered a screening failure using this panel. For each patient, conclusive analyses were based on molecular profiles achieved using F1CDX (ΠF1CDX) and CTL (ΠCTL), and the number (%) of patients with a conclusive analysis was reported. The ‘+’ symbol indicates that an MBRT was identified based on the displayed panel, whereas the ‘−’ sign means no MBRT was identified. The number (%) of patients with a conclusive analysis was reported. ΠF1CDX and ΠCTL were compared using a McNemar test of equality of paired proportions at a 5% two-sided significance level. The proportions of discriminant cases (ΠF1CDX+/CTL− + ΠF1CDX−/CTL+) were presented.
MBRTs that could be initiated | CTL panel | |||
|---|---|---|---|---|
+ | − | Total | ||
F1CDX panel | + | ΠF1CDX+/CTL+ | ΠF1CDX+/CTL− | ΠF1CDX |
− | ΠF1CDX−/CTL+ | ΠF1CDX−/CTL− | ||
Total | ΠCTL | |||
Patients for whom no MBRT can be identified from a panel as defined per randomization were considered a failure case with this panel.
A sensitivity analysis was performed including patients for whom results were achieved with both panels.
Secondary endpoints
The proportion of patients with at least one actionable alteration leading to the identification of an MBRT and the proportion of patients for whom an MBRT was effectively initiated were described using paired data issued from both panels for each patient. ΠF1CDX and ΠCTL were compared using a McNemar test of equality of paired proportions at a two-sided significance level of 5%. The proportions of discriminant cases (ΠF1CDX+/CTL− + ΠF1CDX−/CTL+) were presented.
The proportion of patients for whom an MBRT was finally not initiated was described per randomization arm, with the corresponding reasons, using paired data issued from both panels for each patient. The clinical endpoints were described in randomized patients and compared between arms according to the first panel as defined per randomization, considering censures at the cutoff date (30 June 2021).
PFS was calculated from the date of the first treatment initiation after MTB#1 to the date of first progression, subsequent treatment line initiation or death, whichever occurred first. Data of patients with no event at the time of the analysis were censored at the cutoff date. In patients with no MBRT following MTB#1, PFS was set to 0. PFS was estimated using the Kaplan–Meier method. The hazard ratio for progression between the two groups was presented with its 95% CI.
The duration of follow-up was estimated using the reverse Kaplan–Meier method and expressed with the Q1–Q3 interval. The BOR according to RECIST 1.1 under MBRT treatment was presented by randomization group.
The duration of response in patients with a complete or partial response as the BOR from the start of the MBRT until the end of treatment was calculated from the time of the first documented response (complete response or partial response) until the first documented progression or death due to the underlying cancer or censored at the date of the last tumoral assessment in the case of no event.
Safety
This study was classified in category 1 of ‘Interventional research involving the human person other than health products’ (articles L1121-1 and L5311-1 from the French Code of Public Health). Only adverse events and serious adverse events or reactions related to the study procedures (including de novo tumor biopsies and blood sample collection) as per the protocol had to be reported by the investigators. No data regarding treatment-related adverse events were collected.
Exploratory endpoints
The exploratory endpoints included the number of MBRTs per patient and the type of MBRTs; PFS subgroup analysis in patients with an MBRT initiated; the number of patients with alterations and MBRTs and the type of MBRTs; the number of patients with at least one MBRT according to pathway; the number of patients with at least one MBRT initiated, according to molecular pathway-based recommendations, using both panels. According to disease localization, the number of patients with any MBRT initiated; the number of patients for whom specific MBRTs were initiated, those with treatment initiated outside of MBRTs or those with no MBRT initiated; and the number of patients according to the ESCAT classification were also analyzed. The characteristics of patients with or without a treatment recommendation and those with or without treatment initiation were also examined.
Inclusion and exclusion criteria
Changes according to protocol amendments are indicated below in bold italics.
Inclusion criteria:
-
1.
Male or female patients aged ≥18 years at the time of signing the informed consent form (ICF).
-
2.
Currently treated by a first or a second line of chemotherapy for advanced cancer (local relapse or metastatic; immunotherapies, endocrine therapies and targeted therapies are not considered a line of chemotherapy).
-
3.
Histologically confirmed diagnosis of advanced (local relapse or metastatic), incurable solid tumors from any histological type (except those listed in exclusion criterion no. 3).
-
4.
Availability of an adequate tumor sample to be sent imperatively to the Centre Léon Bérard within 15 days after ICF signature, either (1) a tumor archival FFPE block not older than 3 months before ICF signature or, if not available, (2) a dedicated biopsy from one accessible lesion visible by medical imaging and accessible to percutaneous sampling with a diameter of at least 10 mm or, if not feasible, (3) an archival tumor sample (primary tumor or metastatic lesion) not older than 3 years at the time of ICF signature. The quality (at least 20–30% tumor cells) and quantity (sample size surface area >5 mm2) of the tumor samples have to be confirmed mandatorily within 7 days by a central pathological review before confirming inclusion.
-
5.
The patient’s disease is not susceptible to progress during the next 45 days following ICF signature.
-
6.
ECOG-PS 0–1.
-
7.
Adequate organ and marrow functions based on medical records (within 21 days before randomization), as defined below.
-
a.
Hemoglobin ≥9.0 g dl−1
-
b.
Absolute neutrophil count ≥1.5 × 109 cells per liter
-
c.
Platelets ≥100 × 109 cells per liter
-
d.
Lymphocytes ≥1 × 109 cells per liter
-
e.
Serum creatinine clearance >50 ml min−1 per 1.73 m2 using the Modification of Diet in Renal Disease or Chronic Kidney Disease Epidemiology Collaboration equation
-
f.
Aspartate aminotransferase and alanine aminotransferase ≤2.5 × the upper limit of normal (ULN) (up to 5 × ULN may be tolerated in case of liver metastases)
-
g.
Serum bilirubin ≤1.5 × ULN (in the absence of Gilbert’s syndrome)
-
a.
-
8.
The patient should understand, sign and date the written voluntary ICF before any protocol-specific procedures are performed. The patient should be able and willing to comply with study visits and procedures as per protocol.
-
9.
The patient must be covered by medical insurance.
Exclusion criteria (patients eligible for this study must not meet any of the following criteria):
-
1.
Inability to take oral medications or significant nausea and vomiting, malabsorption, external biliary shunt or significant bowel resection that would preclude adequate absorption of oral medications.
-
2.
Any clinically significant and/or uncontrolled medical disease that could compromise the patient’s ability to tolerate an anticancer treatment and its procedures (these conditions include, but are not limited to, severely impaired lung function, active gastrointestinal tract ulceration, acute or chronic uncontrolled liver disease or severe renal disease, uncontrolled diabetes, history of HIV infection or active viral infection (hepatitis B or C virus), history of organ allograft or use of immunosuppressive treatment).
-
3.
A patient with the following advanced cancers:
-
Cancer bearing one of the oncogenic driver mutations:
-
∘ Colorectal cancer: KRAS, NRAS, HRAS and BRAF
-
∘ Lung cancer: ALK, EGFR, ROS or MET
-
∘ Breast cancers: RH+ and/or HER2+
-
-
High-grade serous ovarian cancers, platinum-sensitive
-
Liposarcoma
-
Melanoma: BRAF.
-
4.
A patient with a nonassessable tumor sample.
-
5.
A patient already included in this study for one type of cancer (cannot be included a second time for the same cancer or any other type of cancer).
-
6.
A pregnant or breastfeeding woman.
Protocol amendments
The detailed amendments to the protocol, synopsis, information notice/ICF and investigator/site list are listed below.
-
Protocol v1.0, 10 February 2017 (approval received from the health authority, 16 May 2017; favorable opinion from the ethics committee, 23 May 2017)
-
Protocol v2.0, 29 January 2018 (approval received from the health authority, 13 April 2018; favorable opinion from the ethics committee, 06 April 2018)
-
Protocol–synopsis v2.0, 29 January 2018
-
Information notice/ICF v2.0, 15 February 2018
-
Investigator/site list v2, 13 February 2018
-
-
Increase the number of patients to be included from 250 to 300 to reach the target of 236 randomized patients.
-
Modification of three inclusion criteria (inclusion criterion nos. 2, 4 and 7e).
-
Modification of patient follow-up time from 12 to 24 months, with no impact on patient safety.
-
Extension of the recruitment period and modification of the trial duration.
-
Clarification of the study’s secondary objectives, with no impact on patient safety.
-
Correction of two errors in section VIII ‘Safety’, with no impact on patient safety.
-
Update of the investigator list:
Register 18 additional investigators at previously registered research sites:
-
Centre Léon Bérard (LYON): DOUBLET Louis, DUFRESNE Armelle, EBERST Lauriane, MORICEAU Guillaume, REURE Juliette, ROCHEFORT Pauline
-
Institut Bergonié (BORDEAUX): CABART Mathilde
-
Institut Claudius Regaud (TOULOUSE): BETRIAN-LAGARDE Sarah, CHEVREAU Christine, COTTURA Ewa-Anna, DALENC Florence, MOUREY Loïc, POUESSEL Damien, RABEAU Audrey, UNG Mony, VALENTIN Thibaud
-
AP-HM Hôpital Nord (MARSEILLE): BOYER Arnaud, JEANSON Arnaud
Delete one investigator:
-
∘ Institut Bergonié (BORDEAUX): LE MOULEC Sylvestre
Declare seven additional investigation sites:
-
∘ Centre Georges François Leclerc (DIJON): GHIRINGHELLI François (principal investigator (PI)), BENGRINE LEFEVRE Leïla, DESMOULINS Isabelle, FAVIER Laure
-
∘ Centre Jean Perrin (CLERMONT-FERRAND): DURANDO Xavier (PI), BERNADACH Maureen, DUBOIS Sophie, DUBRAY LONGERAS Pascale, MASSON Morgane, MOURET REYNIER Marie-Ange
-
∘ Centre Eugene Marquis (RENNES): DE LA MOTTE ROUGE Thibault (PI), BOUSSEMART Lise, CROUZET Laurence, DIERAS Véronique, DINULESCU Monica, EDELINE Julien, LAGUERRE Brigitte, LARIBLE Claire, LE DU Fanny, LEFEUVRE Claudia, LESIMPLE Thierry, LESOURD Samuel, LIEVRE Astrid, PERRIN Christophe, PRACHT Marc, VAULEON Elodie
-
∘ Institut du Cancer (MONTPELLIER): TOSI Diego (PI), LOPEZ MARTINEZ Ernesto José, VIALA Marie
-
∘ Institut de Cancérologie Lucien Neuwirth (SAINT ETIENNE): COLLARD Olivier (PI), FOURNEL Pierre, GUILLOT Aline, JACQUIN Jean-Philippe, RIVOIRARD Romain, SABAN-ROCHE Léa, VASSAL Cécile
-
∘ Hôpital Tenon (PARIS): CADRANEL Jacques (PI), CANELLAS Anthony, EPAUD Christelle, FALLET Vincent
-
∘ Institut Paoli Calmettes (MARSEILLE): BERTUCCI François (PI), GILABERT Marine, GONCALVES Anthony, GRAVIS Gwenaëlle, MONNEUR Audrey, PROVENSAL Magali
-
Protocol–synopsis v2.0, 29 January 2018
-
Information notice/ICF v2.0, 15 February 2018
-
Investigator/site list v3, 08 June 2018
-
-
Update of the investigator list:
-
Declare 15 investigators in an additional investigation site:
-
∘ ICO site René Gauducheau (ST-HERBLAIN): CAMPONE Mario (PI), BERTON-RIGAUD Dominique, BOMPAS Emmanuelle, BOURBOULOUX Emmanuelle, DU RUSQUEC Pauline, FRENEL Jean-Sébastien, GOURMELON Carole, HIRET Sandrine, RAIMBOURG Judith, RAOUL Jean-Luc, ROBERT Marie, ROLLAND Frédéric, ROLLOT Audrey, SENELLART Helene, VANSTEENE Damien
-
Declare 14 investigators in an additional investigation site:
-
∘ Centre Oscar Lambret (LILLE): PENEL Nicolas Paul (PI), CARNOT Aurélien, ABDEDDAIM Cyril, DANSIN Eric, EL HAJBI Farid, KACZMAREK Emilie, LAESTADIUS Fredrik, LEFEBVRE Gautier, MAILLIEZ Audrey, PANNIER Diane, RYCKEWAERT Thomas, BONNETERRE Jacques, CHEVALIER-PLACE Annick F., STERN Natacha
-
Protocol v3.0, 08 October 2018 (approval received from the health authority, 19 November 2018; favorable opinion from the ethics committee, 19 November 2018).
-
Protocol–synopsis v3.0, 08 October 2018
-
Information notice/ICF v3.0, 08 October 2018
-
Investigator/site list v4, 08 October 2018
-
-
Modification of inclusion criterion no. 2 to allow the inclusion of patients receiving second-line chemotherapy.
-
Update of the number/list of genes in the two panels (information deleted from the study title).
-
Compliance with the recent European Data Protection Regulation.
-
Update of the investigator list.
-
Declare 38 investigators in investigation sites already declared:
-
∘ Institut Claudius Regaud-IUCT (TOULOUSE): GLADIEFF Laurence, LARRIEU-CIRON Delphine
-
∘ Centre Georges François Leclerc-CGFL (DIJON): AYATI Siavoshe, COUDERT Bruno, GUION Jean-Florian, HENNEQUIN Audrey, ISAMBERT Nicolas, LAGRANGE Aurélie, ZANETTA Sylvie, VINCENT Julie, PARADIS Armony, LADOIRE Sylvain, HERVIEU Alice
-
∘ Centre Jean PERRIN (CLERMONT-FERRAND): POIRIER Camille
-
∘ Centre Eugène Marquis (RENNES): LIEVRE Astrid, RIGAULT Eugenie
-
∘ Institut du Cancer Montpellier-ICM (MONTPELLIER): ADENIS Antoine, ALEXANDRE Marie, AZRIA David, BOISSELIER Pierre, CHARISSOUX Marie, CRISTOL-DALSTEIN Laurence, CUPISSOL Didier, DARLIX Amélie, D’HONDT Véronique, FABBRO Michel, FAVIER Catherine, FIRMIN Nelly, GALLET-SUCHET Blandine, GUIU Séverine, JACOT William, MAZARD Thibault, PORTALES Fabienne, POUDEROUX Stéphane, QUANTIN Xavier, RIOU Olivier, SAMALIN Emmanuelle, YCHOU Marc
-
Protocol v4.0, 05 February 2019 (health authority, nonsubstantial modification; favorable opinion from the ethics committee, 08 March 2019)
-
Synopsis v3.0, from 08 October 2018
-
Protocol v4.0, from 05 February 2019
-
Information notice/ICF v4.0, from 05 February 2019
-
Investigator/site list v5, from 05 February 2019
-
-
As requested by ROCHE, the destination of tumor samples was changed from the United States to Germany.
-
Update of the investigator list:
-
Declaration of one investigator in an additional investigation site:
-
∘ Hôpital Privé Jean Mermoz (LYON): DERBEL MILED Olfa
-
Declaration of three investigators and one departure in investigation sites already declared:
-
∘ Centre Léon Bérard (LYON): declaration of VINCENEUX Armelle and departure of DERBEL MILED Olfa
-
∘ Institut Bergonié (BORDEAUX): declaration of BOURCIER Kevin
-
∘ Institut Claudius Regaud (TOULOUSE): declaration of DESLANDRES Marion
-
Protocol v5.0, 07 April 2019 (approval received from the health authority, 22 May 2019; favorable opinion from the ethics committee, 20 June 2019)
-
Synopsis v4.0, 07 April 2019
-
Protocol v5.0, 07 April 2019
-
Information notice/ICF v5.0, 07 April 2019
-
Investigator/site list v5, from 05 February 2019
-
-
Increase the number of patients to be included (from 300 to 600) considering the current randomization rate of around 40% due to abnormalities in the selected tumor sample (insufficient tumor area and/or percentage of tumor cells).
-
Increase the number of patients to be randomized (from 236 to 320) considering the failure rate of molecular analyses and the increase in the number of double-panel patients evaluable for the primary endpoint (from 236 to 289), enabling statistical analysis power to be increased to 98%.
-
Extension of the inclusion period by 6 months (30 months instead of the 24 months initially planned), with modification of the total duration of the study.
-
Synopsis v4.0, 07 April 2019
-
Protocol v5.0, 07 April 2019
-
Information notice/ICF v5.0, 07 April 2019
-
Investigator/site list v6, 03 January 2020
-
-
Update of the investigator list (change of the PI already declared in the investigation site)
-
AP-HM (MARSEILLE): departure in mid-January 2020 of the PI (BARLESI Fabrice), replaced by GREILLIER Laurent already declared as an investigator.
-
List of authorized investigation sites
Centre Léon Bérard, Lyon, France; Institut Claudius Regaud, Toulouse, France; Centre Oscar Lambret, Lille, France; AP-HM-Hôpital Nord, Marseille, France; Centre Georges François Leclerc, Dijon, France; Centre Eugène Marquis, Rennes, France; Institut de Cancérologie ICM, Montpellier, France; Centre Jean Perrin, Clermont-Ferrand, France; Institut de Cancérologie Lucien Neuwirth, Saint-Priest-en-Jarrez, France; Institut de Cancérologie de l’Ouest, site René Gauducheau, Saint Herblain, France; Institut Paoli Calmettes, Marseille, France; Institut Bergonié, Bordeaux, France.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Anonymized individual data, including selected actionable gene alterations, detailed alterations, ESCAT class, pharmaceutical class proposed by the MTB and initiated, and MBRTs initiated, are available via Zenodo at https://doi.org/10.5281/zenodo.14794347 (ref. 26).
References
Hudson, T. J. et al. International network of cancer genome projects. Nature 464, 993–998 (2010).
Hollstein, M., Alexandrov, L. B., Wild, C. P., Ardin, M. & Zavadil, J. Base changes in tumour DNA have the power to reveal the causes and evolution of cancer. Oncogene 36, 158–167 (2017).
Hahn, W. C. et al. An expanded universe of cancer targets. Cell 184, 1142–1155 (2021).
Le Tourneau, C. et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 16, 1324–1334 (2015).
Flaherty, K. T. et al. The Molecular Analysis for Therapy Choice (NCI-MATCH) trial: lessons for genomic trial design. J. Natl. Cancer Inst. 112, 1021–1029 (2020).
Flaherty, K. T. et al. Molecular landscape and actionable alterations in a genomically guided cancer clinical trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). J. Clin. Oncol. 38, 3883–3894 (2020).
Trédan, O. et al. Molecular screening program to select molecular-based recommended therapies for metastatic cancer patients: analysis from the ProfiLER trial. Ann. Oncol. 30, 757–765 (2019).
Chen, A. P. et al. Molecular profiling-based assignment of cancer therapy (NCI-MPACT): a randomized multicenter phase II trial. JCO Precis. Oncol. https://doi.org/10.1200/po.20.00372 (2021).
Murciano-Goroff, Y. R., Drilon, A. & Stadler, Z. K. The NCI-MATCH: a national, collaborative precision oncology trial for diverse tumor histologies. Cancer Cell 39, 22–24 (2021).
Auzanneau, C. et al. Feasibility of high-throughput sequencing in clinical routine cancer care: lessons from the cancer pilot project of the France Genomic Medicine 2025 plan. ESMO Open 5, e000744 (2020).
Sosinsky, A. et al. Insights for precision oncology from the integration of genomic and clinical data of 13,880 tumors from the 100,000 Genomes Cancer Programme. Nat. Med. 30, 279–289 (2024).
Mosele, M. F. et al. Recommendations for the use of next-generation sequencing (NGS) for patients with advanced cancer in 2024: a report from the ESMO Precision Medicine Working Group. Ann. Oncol. 35, 588–606 (2024).
Colomer, R. et al. Usefulness and real-world outcomes of next generation sequencing testing in patients with cancer: an observational study on the impact of selection based on clinical judgement. EClinicalMedicine 60, 102029 (2023).
Massard, C. et al. High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: results of the MOSCATO 01 trial. Cancer Discov. 7, 586–595 (2017).
Tsimberidou, A. M. et al. Precision medicine: preliminary results from the Initiative for Molecular Profiling and Advanced Cancer Therapy 2 (IMPACT2) study. NPJ Precis. Oncol. 5, 21 (2021).
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 (2017).
Trédan, O. et al. Sorafenib in molecularly selected cancer patients: final analysis of the MOST-Plus sorafenib cohort. Cancers (Basel) 15, 3441 (2023).
Gibbs, S. N. et al. Comprehensive review on the clinical impact of next-generation sequencing tests for the management of advanced cancer. JCO Precis. Oncol. 7, e2200715 (2023).
Kato, S. et al. Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat. Commun. 11, 4965 (2020).
Chakravarty, D. et al. Somatic genomic testing in patients with metastatic or advanced cancer: ASCO provisional clinical opinion. J. Clin. Oncol. 40, 1231–1258 (2022).
Kinnersley, B. et al. Analysis of 10,478 cancer genomes identifies candidate driver genes and opportunities for precision oncology. Nat. Genet. 56, 1868–1877 (2024).
Yang, J. et al. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol. Cancer 18, 26 (2019).
Ali, E. S. et al. Recent advances and limitations of mTOR inhibitors in the treatment of cancer. Cancer Cell Int. 22, 284 (2022).
Perrier, L. et al. EE608 Cost effectiveness analysis of a large (Foundation Medicine) versus a home-based medium gene panel for exome sequencing: results of the ProfiLER 02 randomized clinical trial. Value Health 25, S175 (2022).
Leichsenring, J. et al. Variant classification in precision oncology. Int. J. Cancer 145, 2996–3010 (2019).
Trédan, O. PROFILER02. Zenodo https://doi.org/10.5281/zenodo.14794347 (2024).
Acknowledgements
We thank the participating patients and families, the site staff from each of the study sites not listed as coauthors and the MTB members, as well as S. Darnis, an employee of the Centre Léon Bérard, for medical editorial assistance with the manuscript. This work received partial funding from Roche Pharma AG. The F1CDX panel was provided for free by Foundation Medicine. This work was supported by the Integrated Cancer Research Site LYriCAN (INCa-DGOS-Inserm_12563), the Institut National du Cancer (NetSARC, InterSARC), Agence Nationale de la Recherche (ANR) LabEx DEvweCAN (ANR-10-LABX 0061), PIA Institut Convergence Francois Rabelais PLAsCAN (17-CONV-0002), Fondation ARC contre le Cancer, La Ligue contre le Cancer (Canopée) and the European Community (EURACAN, EC 739521). The funders had no role in study design, data collection, data analysis, data interpretation or writing of the report. The Centre Léon Bérard, as the sponsor, was responsible for trial conception and coordination, data analysis and publication writing. All authors were involved in the writing and reviewing of the report and the decision for publication. O.T., S.C. and D. Pérol had full access to all study data and full responsibility to submit the manuscript for publication. The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
O.T., D. Pérol and J.Y.B. contributed to the trial conception and design. All authors contributed to data collection. S.C. did the statistical analysis and contributed together with O.T., D. Pérol and J.Y.B. to data analysis and interpretation. O.T., D. Pérol and J.Y.B. supervised the study. All authors reviewed the report for intellectual content, provided comments and gave final approval for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Alastair Greystoke, Nirupa Murugaesu, Benedikt Westphalen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Anna Maria Ranzoni, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Trial design.
R: randomization; F1CDX panel (Foundation One, Inc); CTL: Control panel; MBRT: Molecular based recommended therapy. MTB: Molecular Tumor Board.
Extended Data Fig. 2 Paired analysis including prespecified sensitivity analysis.
Two gene panels were used F1CDX (324 genes), Home-based limited panel (CTL) (87 genes). ‡ At patient progression, the MTB reviewed genomic alterations matching with at least one MBRT, based on the first panel as defined per randomization. In the case of failure (no MBRT identification with the first panel), the second panel was disclosed.
Extended Data Fig. 3 Patients with molecular based treatment recommendation (MBRT) and MBRT initiated using F1CDX® or CTL panel (N = 339).
a) Patients with ≥1 molecular based treatment recommendation (MBRT) according to pathway (N = 192), b) Patients with ≥1 MBRT initiated, according to molecular pathway-based recommendations (N = 51), using F1CDX® or CTL panel (N = 339).
Extended Data Fig. 4 Number of patients with MBRT initiated (all MBRTs) according to disease localization (N = 51).
Number of treatments initiated by tumor types.
Supplementary information
Supplementary Information (download PDF )
Supplementary Table 1. Changes over time of the ProfiLER-02 CTL panel.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Trédan, O., Pouessel, D., Penel, N. et al. Broad versus limited gene panels to guide treatment in patients with advanced solid tumors: a randomized controlled trial. Nat Med 31, 1502–1508 (2025). https://doi.org/10.1038/s41591-025-03613-x
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41591-025-03613-x
This article is cited by
-
Critical evaluation of the ProfiLER-02 study design and outcomes
Nature Medicine (2025)
-
Signalomics for molecular tumor boards and precision oncology of breast and gynecological cancers
Molecular Systems Biology (2025)
-
Targeting racial disparities in breast cancer: mechanistic insights and therapeutic potential of African medicinal plants
Medical Oncology (2025)
