
Abstract
Antiretroviral therapy (ART) suppresses HIV but does not eliminate the latent viral reservoir, which persists in programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. Anti-PD-1 therapies have reduced the HIV reservoir in people living with HIV (PLWH) and cancer; however, the individuals who benefit and the mechanisms driving reservoir reduction remain unclear. We performed a prespecified exploratory, longitudinal multiomic profiling of 30 PLWH (29 males and one female) with cancer in the phase 1 CITN-12 clinical trial, in which pembrolizumab was evaluated for safety and preliminary antitumor activity. The therapy was generally well tolerated, with most adverse events graded 1–2 and objective antitumor response observed in five participants (one complete response and four partial responses). Within 24 hours of treatment, we observed an expansion of proliferating HIV-specific effector CD8+ T cells and a decline in plasma TGFβ. Furthermore, among the 14 participants tracked to the end of treatment (ranging from 44 to 315 days after therapy initiation), nine display early induction and sustained expression of interferon-stimulated genes (ISGs), antiviral restriction factors and Toll-like receptor (TLR) signaling and a reduction in the HIV reservoir. Mapping these transcriptomic signatures across more than 1,000 public single-cell RNA sequencing datasets reveals that anti-PD-1-induced programs are present in subsets of across subsets of disease states, indicating that some people already display a heightened antiviral state. Together, these findings define immune pathways that help identify PLWH most likely to experience reservoir decay with anti-PD-1 therapy and suggest that sustained ISG activation may contribute to reservoir reduction and prevention of viral rebound upon ART interruption. ClinicalTrials.gov registration: NCT02595866.
Similar content being viewed by others


Main
ART has improved global mortality rates and outcomes for PLWH1. ART restores CD4+ T cells, normalizes the CD4:CD8 T cell ratio and promotes immune homeostasis1. However, ART is not curative, and interrupting treatment leads to a rapid rebound of plasma viremia2,3. Moreover, HIV persistence drives immune activation and chronic inflammation and increases the risk of non-AIDS comorbidities such as cancer and cardiovascular disease. Studies show that even with long-term ART, latent HIV persists in a small pool of CD4+ T cells, posing a major impediment to a cure4,5. This HIV reservoir is enriched in memory CD4+ T cells that express immune checkpoint proteins (PD-1, CTLA-4, TIGIT and TIM-3)6,7,8,9; the expression of these proteins impairs homeostatic proliferation that supports reservoir persistence10,11,12,13.
Monoclonal antibody-based immunotherapies targeting immune checkpoint proteins have shown clinical efficacy in several types of cancer and have been shown to restore CD8+ T cell effector function14,15,16,17. In PLWH, inadequate immune reconstitution on ART is associated with an increase in exhausted PD-1-expressing CD8+ T cells and impaired innate immune function6,18,19,20,21. It is posited that anti-PD-1 therapy could promote HIV reactivation in CD4+ T cells, rescue antiviral CD8+ T cells and rejuvenate innate immune responses. Indeed, immune checkpoint blockade (ICB) in ex vivo and non-human primate studies has led to a transient increase in viral transcription and reduction in viral DNA, indicative of latency reversal22,23,24,25,26. Clinically, ICB in PLWH and cancer has demonstrated acceptable safety profiles27,28,29 and has been associated with increases in cell-associated and plasma HIV RNA, consistent with reversal of viral latency27,30. However, its long-term effects on cell-associated HIV DNA, a marker of the latent reservoir, remain uncertain due to small cohort sizes, limited follow-up and an incomplete understanding of the underlying mechanisms. Additionally, assessment of the impact of ICB on the HIV reservoir in PLWH without cancer has been constrained by immune-mediated adverse events in early trials31. Nonetheless, PLWH continue to be at increased cancer risk and can benefit from interventions that include ICB.
In this study of a cohort of PLWH with cancer treated with the immune checkpoint blocker anti-PD-1, we provide experimental evidence for the critical role of a cascade integrating myeloid cell-specific interferon (IFN) responses, effector CD8+ T cell activity and reduced TGFβ signaling. This cascade is initiated as early as 1 day after anti-PD-1 treatment initiation and is associated with a reduced viral reservoir during the 2-year follow-up. Insights into the upstream mechanisms that restrict the HIV reservoir in PLWH hold crucial implications for advancing strategies aimed at HIV eradication.
Results
To elucidate the mechanisms triggered by the blockade of PD-1 engagement that contribute to the reduction of the latent HIV reservoir, we analyzed longitudinal samples collected during the Cancer Immunotherapy Trials Network-12 trial (CITN-12; NCT02595866)27,30. Detailed baseline demographic and clinical characteristics of study participants, including age, sex, cancer type and ART regimen, are provided in Supplementary Data 1. This trial was a multicenter phase 1 study in which pembrolizumab, an anti-PD-1 antibody, was administered to PLWH with cancer27,30 (Fig. 1a). The CITN-12 immunologic analyses presented here represent a prespecified exploratory component of the clinical trial, with assays and collection timepoints defined prospectively to capture immune and virologic dynamics after anti-PD-1 administration. A total of 30 PLWH who were virally suppressed on ART—having received at least 4 weeks of continuous ART prior to initiation—were treated with the standard pembrolizumab regimen (200 mg administered intravenously every 3 weeks for up to 2 years), with all participants completing a minimum of two treatment cycles (Fig. 1a,b and Supplementary Data 1, D1). Participants were classified into three cohorts based on their baseline CD4+ T cell counts: cohort 1 (100−199 CD4+ T cells per microliter, n = 6), cohort 2 (200−350 CD4+ T cells per microliter, n = 12) and cohort 3 (>350 CD4+ T cells per microliter, n = 12) (Fig. 1a,b). These participants presented with 11 different cancers, including AIDS-associated cancers such as Kaposi sarcoma and non-Hodgkin lymphoma (Fig. 1a,b and Extended Data Fig. 1a). The distribution of cancer subtypes was not significantly associated with the cohort based on CD4+ T cell numbers (Supplementary Data 1, D1). Notably, complete cancer remission was observed in one participant, and partial remission occurred in four participants (Fig. 1b and Supplementary Data 1, D1). Samples were collected pretreatment (C01D01), at 24 hours (C01D02), at 1 week (C01D08) and at end of treatment (EOT) from participants spanning three CD4+ T cell count cohorts who presented with 11 different cancer types. We applied a comprehensive multiomic approach (Extended Data Fig. 1b) that included profiling plasma cytokines and bulk peripheral blood mononuclear cell (PBMC) transcriptomes to define immune alterations within 24 hours of treatment that were associated with previously reported decline in latent HIV (cell-associated HIV DNA) and transient increase in viral reactivation (HIV RNA)30. Subsequently, we investigated (1) the persistence of these transcriptomic signatures until EOT, (2) the association with HIV outcomes (for example, plasma HIV RNA and cell-associated HIV DNA levels) and (3) their generalizability and mechanistic roles across immune cell subsets using single-cell and in vitro validation approaches.

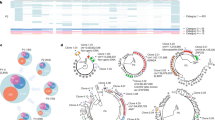
a, Samples were collected from 30 PLWH encompassing 11 different cancer types, stratified into three pre-therapy peripheral CD4+ T cell count-based groups. b, The table lists participant study ID, CD4+ T cell count cohort (cohort 1: <200 CD4+ T cells per µl blood; cohort 2: 200–350 CD4+ T cells per µl blood; cohort 3: >350 CD4+ T cells per µl blood), absolute CD4+ T cell counts per participant, cancer type (NHL, non-Hodgkin lymphoma; KS, Kaposi sarcoma; SCC, squamous cell carcinoma), assessed cancer response to immunotherapy (CR, complete response; DP, disease progression; NA, not applicable; NE, not evaluable; PR, partial response; SD, stable disease) and EOT day, defined as the number of days since the first anti-PD-1 infusion. Anti-PD-1 infusions were administered in 21-day cycles, with the number of cycles varying among individuals. c, SLEA scores of transcriptomic modules significantly modulated between C01D01 and C01D02. In brief, GSEA using the C7 (Immunologic Signature) gene set collection from the MSigDB was performed, and leading-edge genes from significant gene sets (P < 0.05) were collapsed into transcriptomic modules. SLEA scores for seven modules were calculated and displayed in a row (z-score)-normalized heatmap. Modulation of transcription factor (TF) targets (from the ChEA database) between C01D01 and C01D02 was similarly tested by GSEA, and SLEA scores of leading-edge genes are represented in a corresponding heatmap. Each column represents one participant; column headers indicate whether the module score corresponds to C01D01 (black) or C01D02 (green). Samples with higher SLEA scores for a given module are shown in red; blue indicates reduced expression. d, An integrated mixOmics model combining SLEA scores of transcriptomic modules and plasma cytokine levels at C01D01 and C01D02. In the network, nodes represent cytokines (squares) or gene set/transcriptomic modules (circles). Node color indicates upregulation (red) or downregulation (blue) at C01D02 versus C01D01. Edge color represents the Pearsonʼs correlation coefficient between nodes, with red denoting positive correlations and blue denoting negative correlations. e, Column-clustered (correlation distance; complete linkage) heatmap of SLEA scores for transcriptomic modules distinguishing C01D02 from C01D01 for 14 PLWH at EOT, separating two participant groups. Sample numbers for each analysis and timepoint are detailed in Supplementary Data 1, D3. hr, hours; us, unspliced; VL, viral load.
Multiomic analyses reveal rapid induction of innate IFN response within 24 hours of anti-PD-1 therapy
We employed bulk transcriptomic profiling (Supplemental Fig. 1a,b) to identify gene expression modules that were altered between C01D01 and C01D02. To derive these modules, we first performed gene set enrichment analysis (GSEA) using Immunologic Signature gene sets from the Molecular Signatures Database (MSigDB) C7 collection. Pathways significantly altered at C01D02 relative to C01D01 were then collapsed based on overlap in leading-edge genes (Jaccard index ≥ 0.25), and the combined leading-edge genes were used to define each transcriptomic module. In parallel, we used GSEA to assess enrichment of transcription factor target sets curated in the ChIP-X Enrichment Analysis (ChEA) database, which compiles experimentally validated transcription factor–target interactions from multiple chromatin immunoprecipitation followed by sequencing (ChIP−seq) and ChIP-on-chip (ChIP-chip) studies. We next calculated sample-level module and transcription factor target scores to quantify relative expression of these gene sets per participant32 (Fig. 1c). We observed that monocyte function (modules 4 and 5), IFN/viral responses (modules 2 and 3) and CD8 T cell activation (module 1) modules were significantly upregulated at C01D02 (Fig. 1c and Supplemental Data 1). We also observed an increase in transcription factors regulating IFN responses (IRFs, STAT1), monocyte effector function (RELA, STAT4) and T cell activation (STAT5) (Extended Data Fig. 1c,d), whereas immune checkpoint blockers (such as PDCD1, TIGIT, IL10RA) showed reduced expression at C01D02 (Extended Data Fig. 2a). T cell stemness, WNT/β-catenin signaling33 and regulatory T (Treg) modules along with expression of genes regulated by TCF1 and β-catenin and epigenetic regulators such as EGR1 and EZH2 (ref. 34) were reduced at C01D02 (Extended Data Fig. 2b,c and Supplementary Fig. 1c).
Plasma cytokine profiling (Supplementary Fig. 2a,b) identified significant upregulation of innate cytokines, including IFNβ, IL-6 and IFNγ (TH1 response) at C01D02 and C01D08 (Extended Data Fig. 3a and Supplementary Fig. 2c), whereas TGFβ1 levels were significantly reduced at C01D02 (Extended Data Fig. 3b). Notably, levels of TH2 (IL-4) and TH17 (IL-17A) response cytokines only increased by C01D08 (Extended Data Fig. 3c). Next, we generated an integrated transcriptome−cytokine network (Fig. 1d) to demonstrate the positive association between transcriptomic modules—containing gene sets for monocyte function, ISGs and effector CD8+ T cells—and plasma IFNγ, IL-6 and IFNβ levels. Conversely, lower plasma TGFβ levels correlated with reduced expression of WNT signaling and Treg modules. These results show that anti-PD-1 therapy reduces plasma TGFβ levels and promotes a rapid (24-hour) increase in proinflammatory and antiviral innate responses.
Anti-PD-1-induced transcriptomic modules stratify participants by HIV reservoir outcomes at EOT
The anti-PD-1-induced transcriptomic modules described above remained stable across the 14 PLWH tracked through EOT (Supplementary Fig. 2d). However, these modules stratified participants into two distinct groups at EOT (Fig. 1e and Supplementary Fig. 2e). Group 1 (ISGhi) (n = 9) samples expressed ISG/antiviral (modules 2 and 3), monocyte (modules 4 and 5) and CD8 effector genes (module 1). Longitudinally, group 1 participants maintained elevated ISG and expression of antiviral modules through four cycles of therapy (approximately 3–4 months), indicating persistent immune reprogramming (Extended Data Fig. 4a). By contrast, group 2 (ISGlo) PLWH (n = 5), including those who received extended treatment (more than four cycles), exhibited lower ISG module expression at EOT, suggesting that these differences in immune responsiveness were not a consequence of duration of therapy. Group 2 samples also showed increased expression of WNT (module 6) and Treg gene sets (module 7) (Fig. 1e).
Head-to-head comparison of samples from group 1 and group 2 participants at each timepoint (Fig. 2a) revealed that group 1 exhibited significantly higher expression of ISG (modules 2 and 3) (Fig. 2b) and monocyte (modules 4 and 5) (Fig. 2c) at C01D01, whereas group 2 showed elevated expression of WNT signaling (module 6) (Fig. 2d). After PD-1 blockade, both groups showed significant increase in ISG and monocyte modules and decrease in WNT signaling/Treg modules (Fig. 2b–d). By EOT, group 1 retained elevated ISG and monocyte module expression (Fig. 2b,c) and showed a significant increase in the effector CD8+ T cell function module (module 1) (Fig. 2d), which was not elevated at baseline. By contrast, group 2 showed persistently elevated WNT and Treg module expression, with diminished ISG and monocyte responses (Fig. 2b,c). Together, these data reveal that transcriptional differences between group 1 (ISGhi) and group 2 (ISGlo) participants are detectable at baseline and become more pronounced after PD-1 blockade, with a subset of participants exhibiting durable ISG and myeloid antiviral module expression—highlighting sustained immune reprogramming that may underpin therapeutic responsiveness.

a, Row (z-score)-normalized heatmaps show transcriptomic module expression across participants and timepoints (C01D01, C01D02 and EOT). Each row represents a module, and each column represents one participant (n = 13). Cell-associated HIV DNA copies (per 106 PBMCs) are annotated per participant per timepoint. Transcriptomic modules significantly associated with HIV DNA counts (P < 0.05 by GSEA) at EOT clustered participants into two subgroups (group 1 and group 2; headers above the heatmap). Data from participant 29 were unavailable at C01D02 (Supplementary Data 1, D5). b–d, SLEA scores of transcriptomic modules were calculated for each participant and plotted per group (group 1, n = 9; group 2, n = 5) and timepoint (C01D01, C01D02 and EOT). Each point represents one biological replicate (one participant sample). b, IFN response and antiviral module scores were significantly higher in group 1 than group 2 at C01D01 and remained elevated at EOT. c, TLR signaling module scores were higher in group 1 at baseline and remained significantly elevated at EOT. d, Effector T cell module scores did not differ at C01D01 but increased significantly in group 1 at EOT. Group 2 showed higher expression of WNT signaling and Treg cell modules at C01D01 and EOT. Plots show mean ± s.e.m. per group per timepoint. Mann–Whitney U-tests compared group 1 and group 2 at each timepoint. e, Cell-associated HIV DNA counts were compared between groups (n = 14 participants: group 1, n = 9; group 2, n = 5) in a pairwise manner across timepoints. HIV DNA counts were significantly lower in group 1 at C01D01, decreased further by C01D02 and remained low at EOT. Mann–Whitney U-tests compared groups at each timepoint, and Wilcoxon matched-pairs signed-rank tests assessed within-participant changes from baseline. Median values with IQR are shown. f, log2 fold change in inducible HIV-1 RNA (EOT versus C01D01) across participants stratified by group. Each bar represents one participant (group 1, n = 9; group 2, n = 5). Blue bars indicate group 1 (lower HIV DNA), where eight of nine participants showed decreased inducible RNA at EOT; pink bars indicate group 2 (higher HIV DNA), where four of five participants exhibited increased inducible RNA. g, UMAP of PBMCs from 12 participants at C01D01, eight at C01D02 and 10 at C01D08 analyzed by 3′ scRNA-seq. Cells were processed using Seurat and SingleR and annotated according to Monaco et al.62 via the celldex package. h, Frequencies of major annotated PBMC subsets were quantified per participant per timepoint (for the samples in g) as a percentage of total PBMCs. Significant increases in monocytes and decreases in NK and CD8+ T cells were observed over time. Stacked bar plots show mean ± s.e.m. subset frequencies across participants. Mann–Whitney U-tests compared each timepoint to baseline. i, Genes from each bulk transcriptomic module were extracted, and Seurat’s AddModuleScore function was used to compute per-cell module scores across subsets. Dot plots show cell frequencies (dot size) and relative expression (dot color). TLR signaling modules were highly expressed in monocytes but not in other subsets, whereas IFN and antiviral modules were broadly expressed at lower levels. j, To assess generalizability of gene signatures identified in anti-PD-1 therapy responders, scRNA-seq data were analyzed using the SCimilarity platform and compared against approximately 1,000 public datasets. WNT canonical pathway, Treg, effector CD8+ T cell, ISG and TLR signatures showed high similarity scores in subsets of individuals across Healthy, Cancer and Infectious Disease datasets. Unless otherwise indicated, all statistical tests were two-sided, with multiple comparison adjustments applied where specified; otherwise, exact nominal P values are shown in the figures. 24h post-Tx, 24 hours after therapy; ~, approximately; FC, fold change; NK, natural killer; sc, single-cell; TF, transcription factor.
Transcriptome-based stratification identifies ISGhi participants with reduced HIV reservoirs after PD-1 blockade
In line with previous data30, HIV DNA copies per million cells were significantly reduced at C01D02 (P = 0.009, median 1.8× decrease), whereas plasma HIV RNA levels significantly increased at C01D08 (P < 0.05, median 1.67× increase) (Extended Data Fig. 4b) and were associated with high IFNγ and low IL-4 concentrations in the plasma (Extended Data Fig. 5a). Group 1 participants had median HIV DNA levels of 76.18 copies per million CD4+ T cells (interquartile range (IQR): 35.98–214) at C01D01 compared to 287.2 copies per million cells (IQR: 172.8–659.4) in group 2 (P < 0.05). By EOT, group 1 exhibited a significant median 2.39-fold reduction in HIV DNA levels (P < 0.05), whereas group 2 showed no significant change in HIV DNA levels (Fig. 2e and Extended Data Fig. 4b). We also observed that eight of nine individuals in group 1 exhibited a decrease in the frequency of CD4+ T cells with inducible HIV RNA at EOT, consistent with overall reservoir contraction. By contrast, four of five individuals in group 2 showed an increase in inducible HIV-1 RNA at EOT (Fig. 2f). Of note, HIV DNA copies per million cells were positively correlated with the frequency of cells harboring inducible HIV (Extended Data Fig. 5b). Although both parameters also associated with other virologic markers, no significant group differences were observed in plasma HIV RNA, unspliced HIV RNA or HIV RNA/HIV DNA ratio (Extended Data Fig. 5c–e). Notably, the two groups could not be distinguished based on CD4+ T cell counts, cancer type or clinical response to therapy (Extended Data Fig. 5f). These results demonstrate that the subset of PLWH with preexisting myeloid ISG modules, further induced by anti-PD-1, also exhibits a significant decline in HIV DNA levels, reflective of a decrease in the HIV reservoir.
High ISG and low WNT module expression are immune subset specific and conserved across the human population
Next, we profiled immune cell subsets at C01D01, C01D02 and C01D08 using single-cell RNA sequencing (scRNA-seq) (Fig. 2g and Supplementary Data 1). A significant increase in monocyte frequencies—particularly the classical monocyte subset—at C01D02 compared to C01D01 (approximately 15% of PBMCs, P < 0.05) was confirmed by in silico deconvolution (Fig. 2h, Supplementary Fig. 3a and Extended Data Fig. 6a,b). This was concomitant with a decrease in natural killer and CD8 T cell frequencies (Fig. 2h). Quantification of expression scores per subset (Fig. 2i) for the modules (Fig. 1) revealed enrichment of TLR modules in monocytes, IFN modules in both T cells and myeloid cells and Treg/WNT modules in lymphoid subsets (Fig. 2i and Supplementary Fig. 3b,c). ISG and antiviral modules were upregulated in TEMRA/TEM CD8 T cells, TH1/TH17/Treg CD4+ T cells and all monocyte subsets, with IFNG transcripts enriched in effector CD8+ and CD4+ TH1 cells and IFNB and IL6 transcripts highest in monocytes (Supplementary Fig. 4a,b). To identify differences in scRNA-seq-based profiles between group 1 and group 2, we analyzed module expression in monocytes and CD8+ T cells at C01D01, C01D02 and C01D08 in a subset of participants from both groups. Despite limited sample size, group 1 (versus group 2) monocytes showed higher IFN and TLR module expression at baseline and 24 hours after anti-PD-1, whereas CD8+ T cells from group 1 displayed stronger induction of innate and antiviral modules after treatment (Extended Data Fig. 6c). These results suggest that early antiviral programming in monocytes precedes effector T cell responses and could prevail prior to therapy. In parallel, we mapped these modules to publicly available35 whole-blood single-cell datasets from healthy individuals, as well as people with cancer or infections, and observed that a subset of individuals, regardless of disease state, consistently exhibited high expression of ISG modules and low expression of Treg and WNT signaling modules (Fig. 2j). These findings demonstrate that high ISG and low Treg/WNT signaling module expression represent a broader immune activation state in humans, relevant across diseases and interventions.
Increased antiviral gene expression in ISG+ monocytes within 24 hours of anti-PD-1 treatment is linked to HIV DNA decline at EOT
Using scRNA-seq-based profiling, we identified three distinct subclusters of monocytes and quantified the expression of ISG and TLR modules within each (Fig. 3a; classical/intermediate subsets mapped in Extended Data Fig. 6d). We did not observe a significant change in subpopulation frequencies after anti-PD-1 (Extended Data Fig. 6e). However, when contrasting transcriptional profiles against C01D01, we observed that the expression of genes such as IFIT2, B2M, CD86 and CXCL10 was significantly increased at both C01D02 and C01D08 after treatment in monocyte cluster 2 (ISGhi) (Fig. 3b and Extended Data Fig. 6f,g). This ISGhi population also showed reduced expression of TLR module genes (Fig. 3c and Extended Data Fig. 6e). By contrast, the monocyte subsets expressing TLR modules (clusters 0 and 1) did not show significant transcriptional changes after PD-1 therapy (Fig. 3c and Extended Data Fig. 6f,g). We inferred potential ligand−receptor interactions using NicheNet36 to identify the putative ligands upstream of the genes expressed by the ISGhi monocytes (Fig. 3d). In line with heightened plasma IFNβ and IFNγ (Fig. 3c), IFNB1 and IFNG were identified as key upstream cytokines driving elevated expression of genes in ISGhi monocytes (Fig. 3d). These results demonstrate that blocking PD-1 engagement significantly increases monocyte frequencies and antiviral ISG expression in a specific subset of monocytes.

a, The subset of cells labeled ‘Monocytes’ was extracted from scRNA-seq data on PBMCs (Fig. 2g; n = 30; number of samples per timepoint listed in Supplementary Data 1, D3) and analyzed with UMAP and unsupervised clustering in Seurat. The 607 genes most highly expressed after anti-PD-1 treatment were used for clustering, revealing three major clusters. Differential gene expression analyses were performed between C01D02 and C01D01 to identify genes induced after therapy. b, Volcano plots show that, unlike clusters 0 and 1, cluster 2 monocytes exhibited a greater number of genes significantly upregulated at C01D02 versus C01D01, including IFN-inducible genes such as B2M, CD86 and CXCL10. Differential expression was calculated using MAST implemented in Seurat. c, TLR, antiviral and ISG module scores for monocytes were mapped onto the UMAP shown in a. Cluster 2 monocytes displayed reduced TLR module expression and increased ISG/antiviral module expression. In these UMAPs, higher module expression is shown in red, whereas blue indicates lower expression. d, Genes differentially expressed in cluster 2 monocytes (versus all other monocytes) were analyzed using the NicheNet package in R to identify upstream regulators. The analysis was restricted to cytokines measured in plasma. Prioritized ligands—those most strongly correlated with the expression of predicted target genes—are listed to the left. The regulatory potential of these ligands, based on NicheNet’s multistudy reference database, is represented in the heatmap on the right, where deeper purple denotes greater regulatory potential. Details of sample numbers for each analysis and timepoint are provided in Supplementary Data 1, D3. In b, Wilcoxon matched-pairs signed-rank tests were used to assess significant differences from C01D01. Unless otherwise indicated, all statistical tests were two-sided, with multiple comparison adjustments applied where specified; otherwise, exact nominal P values are shown in the figures. FC, fold change; P_val_adj, adjusted P value.
Increased effector ISGhi CD8 T cells within 24 hours of anti-PD-1 treatment is associated with HIV DNA decay at EOT
Four subclusters of CD8+ T cells were identified (Fig. 4a), with clusters 1 and 3 exhibiting high effector gene expression (expressed GZMB, PRF1, IFNG and TNF). Compared to cluster 1, cluster 3 was enriched for ISGs at C01D01, which were further induced at C01D02 (Fig. 4b,c). The frequency of cluster 3 cells significantly increased at C01D02, whereas cluster 1 expanded by C01D08 (Fig. 4d). Cell subset annotation revealed that both clusters included a mix of effector and TEMRA populations (Supplemental Fig. 3c), and flow cytometry confirmed the increase in effector (CD27−CD45RA+) CD8+ T cells by C01D02 (Fig. 4e and Extended Data Fig. 7a,b). Notably, both clusters 1 and 3 exhibited increased ISG expression, consistent with effector cell differentiation (Extended Data Fig. 7c).

a, Cells annotated as ‘CD8+ T cells’ were extracted from scRNA-seq data on PBMCs (Fig. 2g; n = 30; number of samples per timepoint listed in Supplementary Data 1, D3). Four major clusters were identified. b, Genes from each bulk transcriptomic module were extracted, and Seurat’s AddModuleScore function was used to calculate module scores per cluster. Dot plots depict the proportion of cells (dot size) and the average module expression (color intensity). Cluster 0 contained the highest frequency of cells and the greatest expression of WNT signaling, TCF7 and CTNNB1 target genes and was annotated as the naive subset using the SingleR package (Monaco et al.62, celldex database). Clusters 1 and 2 were annotated as central and effector memory T cells (TCM and TEM), respectively, and cluster 3 as the ISG-expressing subset. c, Feature plots of IFNG, TNF, GZMB and PRF1 show that cells expressing these effector genes (red) are enriched in clusters 1 and 3 (TEM and ISG subsets). d, The top panels show relative changes in the frequency of each CD8+ T cell cluster at C01D02 and C01D08 compared to C01D01. The bottom heatmap depicts changes in module scores, represented as the signed log(P) value (sign(fold change) × log(P value)); red indicates increased expression, and blue indicates decreased expression. e, Flow cytometry gating of CD8+ T cells (TEM: CD45RA−CD27−; TEMRA: CD45RA+CD27−) revealed significant decreases in TCM/TTM and increases in TEMRA frequencies after therapy. f, HIV-specific CD8+ T cell proliferation was quantified by IFNγ and Ki-67 co-expression in PBMCs stimulated with an HIV Gag peptide pool. Representative gating is shown (top), and frequencies of IFNγ+Ki-67+ cells (of total CD8+ T cells) are plotted below, showing significant increases at C01D02. g, Genes upregulated in effector and ISG CD8+ T cell clusters (versus other CD8+ clusters) were analyzed using NicheNet to identify upstream regulators, restricted to cytokines measured in plasma. Prioritized ligands (those most correlated with target gene expression) are listed, and their regulatory potential, based on NicheNet’s reference database, is visualized in the heatmap (purple indicates higher potential). h, Frequencies of clusters 1 (TEM) and 3 (ISG) among total CD8+ T cells were inversely correlated with plasma TGFβ2 and TGFβ1 concentrations, respectively (ρ and P values shown; Spearman’s correlation). i, Genes downregulated in effector and ISG CD8+ T cell clusters (versus other CD8+ clusters) were analyzed using NicheNet as in g. Prioritized ligands for downregulated targets are listed, and their regulatory potential is shown (purple indicates higher potential). Details of sample numbers for each analysis and timepoint are provided in Supplementary Data 1, D3. For d–f, Wilcoxon matched-pairs signed-rank tests were used to assess differences from C01D01. In d, *P < 0.05 and **P < 0.01. In h, Spearman’s correlation was used. Unless otherwise indicated, all statistical tests were two-sided, with multiple comparison adjustments applied where specified; otherwise, exact nominal P values are shown in the figures. Avg Exp, average expression; freq., frequency; NS, not significant.
HIV-specific response profiling was performed by stimulating PBMCs with Gag peptide pools in vitro, revealing an increase in the frequency of Ki-67+, IFNγ-producing CD8+ T cells at C01D02 (Fig. 4f and Supplementary Data 1, D7). To further characterize these responses, we performed unsupervised clustering of flow cytometry data, incorporating both HIV-specific (IFNγ+ and/or TNF+) CD8+ T cells and randomly sampled total CD8+ T cells from unstimulated and Gag-stimulated conditions (Extended Data Fig. 8a). Among the resulting clusters, cluster 8—enriched in group 1—exhibited high levels of IFNγ, T-bet, Ki-67, Id2 and Tox and expanded by C01D08 (Extended Data Fig. 8b,c). By contrast, group 2 participants showed predominant enrichment in cluster 1 (reduced Ki-67, T-bet and Tox despite high IFNγ levels) throughout the longitudinal follow-up (Extended Data Fig. 8d,e). At C01D01, 12.87% of Gag-specific IFNγ+CD8+ T cells in group 1 expressed Ki-67 compared to 3.01% in group 2. This proliferative subset expanded more robustly in group 1 at C01D02 (29.9% versus 15.5%) and C01D08 (54.1% versus 36.1%) (Extended Data Fig. 8f). These findings suggest that group 1 PLWH harbor a preexisting pool of poised, functional effector CD8+ T cells responsive to PD-1 blockade.
To identify upstream ligands of effector CD8+ T cell genes, we used NicheNet36 analysis and identified IFNβ1 and IFNγ as top predicted inducers of ISGs and effector genes such as TNF (Fig. 4g). Simultaneously, reduced plasma levels of TGFβ were associated with increased frequencies of effector CD8+ T cells (Fig. 4h and Extended Data Fig. 3a); concomitant downregulation of genes involved in quiescence (ASXL1, BCL9L) and TGFβ signaling (PEMPA1, SKIL) (Fig. 4i) was observed. Together, these findings link antiviral effector responses with reservoir reduction and suggest that PD-1 blockade rapidly induces ISG+ effector CD8+ T cells in group 1 participants.
Persistent CD4+ T cell ISG expression linked to anti-PD-1 response and HIV reservoir decline
Similar to our findings in CD8+ T cells, both flow cytometry and CIBERSORT analysis of bulk PBMC RNA-seq data demonstrated a decline in CD27+CD45RA− (central and transitional memory) CD4+ T cell subsets after anti-PD-1 therapy across the cohort (Extended Data Fig. 9a,b and Supplementary Data 1, D6). Annotation of CD4+ T cells identified eight subsets—including naive, Treg and effector populations—with effector modules (for example, IL15RA and TNF) upregulated and WNT signaling downregulated during the first week of therapy (Extended Data Fig. 9c–e). Unbiased subclustering revealed distinct populations: naive cells (cluster 1), ISGhi effector/memory cells (cluster 3), ISGlo effector/memory cells (cluster 0) and Treg cells (cluster 2) (Fig. 5a). Notably, cluster 3 was maintained during the first week, whereas cluster 0 declined significantly over the same period (Fig. 5b–d) and was associated with elevated plasma HIV RNA levels (Fig. 5e). Functional validation using in vitro stimulation with HIV Gag peptide pools showed increased frequencies of HIV-specific IFNγ+TNF+CD4+ T cells in all but two participants within 24 hours of treatment (Fig. 5f and Supplementary Data 1, D7). Moreover, residual plasma viremia correlated with the frequency of HIV-specific CD4+ T cells, plasma IFNγ levels and expression of both effector and ISG modules (Fig. 5f and Extended Data Fig. 10a,b), suggesting an acute immune response to antigen released from the HIV reservoir.

a, Cells annotated as ‘CD4+ T cells’ were extracted from scRNA-seq data on PBMCs (Fig. 2g; n = 30; number of samples per timepoint listed in Supplementary Data 1, D3). Five major clusters were identified. b, Genes from each bulk transcriptomic module were extracted, and Seurat’s AddModuleScore function was used to calculate module scores per cluster. Dot plots display the proportion of cells (dot size) and average expression (color intensity) for each module. Cluster 1 contained the highest frequency of cells and the strongest expression of WNT signaling, TCF7 and CTNNB1 targets and was annotated as the naive subset using the SingleR package (Monaco et al.62, celldex database). Clusters 0 and 3 were annotated as effector T helper memory (TEM) cells; cluster 3 showed the highest expression of ISG modules. c, Feature plots display module scores for effector T cell, WNT signaling, antiviral and ISG modules across CD4+ T cells. Higher relative module expression per cell is indicated in red; lower expression is shown in blue. d, The top panels show relative changes in the frequency of each CD4+ T cell cluster at C01D02 and C01D08 compared to C01D01. The bottom heatmap depicts module score changes represented as the signed log(P) value (sign(fold change) × log(P value)); red indicates increased expression, and blue indicates decreased expression. e, Correlation analysis showing that increases in plasma HIV RNA across timepoints were associated with decreases in the frequency of cluster 0 (ISG-low effector cells). f, HIV-specific CD4+ T cell responses were measured in PBMCs stimulated with an HIV Gag peptide pool. Flow cytometry plots (top) illustrate gating for IFNγ and TNF co-expression. Frequencies of IFNγ+TNF+ cells (as a proportion of total CD4+ T cells) increased in five of seven participants at C01D02 (right). Lines connect longitudinal samples from each participant; red lines indicate an increase relative to the preceding timepoint; and blue lines indicate a decrease. Across all timepoints, the proportion of IFNγ+TNF+CD4+ T cells was positively correlated with plasma HIV RNA (bottom). In d and f, Wilcoxon matched-pairs signed-rank tests were used to assess changes from C01D01. In d, *P < 0.05 and **P < 0.01. In e and f, Spearman’s correlations were used (ρ and P values shown). Unless otherwise indicated, all statistical tests were two-sided, with multiple comparison adjustments applied where specified; otherwise, exact nominal P values are reported in the figures. freq., frequency; NS, not significant.
Subset-specific ISG profiles and their in vitro induction are associated with reduced HIV DNA levels
To identify molecular signatures and cytokines present at baseline (C01D01) and dynamically induced by C01D02, we generated weighted feature scores using data from both timepoints (Fig. 6a). This model revealed that higher baseline expression of IFN-inducible, antiviral and monocyte TLR modules—combined with elevated IFNγ/IFNβ and reduced TGFβ levels at C01D02—predicted lower HIV DNA at EOT (Fig. 6a). Notably, predicted latent HIV DNA levels derived from these weighted features significantly correlated with HIV DNA levels (Fig. 6b). Given the prominent role of antiviral and immunomodulatory ISG responses, we next deconvoluted cell subset-specific ISG expression profiles across myeloid and T cell populations (Fig. 6c and Supplementary Fig. 5). This analysis revealed distinct ISG-expressing subsets within both compartments. ISG+ monocytes upregulated genes involved in IFN/antiviral signaling (IRF7, IRF9, STAT1, BST2), antigen presentation (B2M) and effector T cell recruitment/differentiation (CXCL10, IL15). Simultaneously, ISG+CD8+ and ISG+CD4+ T cells showed elevated expression of receptors responsive to myeloid-derived signals (IL15RA, TNF) and mediators of T helper polarization (IL12RB1). Notably, these ISG signatures were significantly enriched in group 1 participants—those with lower HIV DNA—at both C01D01 and EOT (Fig. 6c and Supplementary Fig. 5).

a, Ridge regression model weights of features associated with anti-PD-1 therapy at baseline (C01D01) and 24 hours after therapy (C01D02) that correlated with frequencies of HIV DNA-containing cells at EOT. b, Correlation between predicted and actual HIV DNA levels per million CD4+ T cells based on the ridge model (ρ = 0.59, P = 0.035). c, Model illustrating cell-subset-specific ISG expression. The left heatmap shows ISG expression across monocyte, CD4+ and CD8+ T cell subclusters (red, high; gray, low). The middle bar plots display the relative (z-scaled) fold change in gene expression at C01D02 versus C01D01 (values >0 indicate upregulation). The right heatmap shows expression of monocyte-specific and T-cell-specific ISGs at EOT in group 1 versus group 2 participants, normalized per gene (row z-score; red, increased expression; blue, decreased expression). Bar plots above indicate total HIV DNA levels per participant at EOT. d, In vitro stimulation assay. Memory CD4+ T cells and CD8-depleted PBMCs from five donors (n = 5) were cultured with or without innate immune stimuli (IFNα, IFNγ, poly I:C or R848). Mean fluorescence intensity (MFI) of the antiviral proteins APOBEC3G and IFIT1 in memory CD4+ T cells was quantified by flow cytometry. Each point represents a biological replicate (donor); bars denote the mean ± s.e.m. Comparisons between memory CD4+-only cultures and CD8-depleted PBMC co-cultures for each condition were performed using paired t-tests. e,f, In vitro HIV challenge assay. Memory CD4+ T cells and CD8-depleted PBMCs from five donors (n = 5) were infected with HIV in the presence or absence of IFN agonists or signaling inhibitors. e, Representative flow cytometry plots show p24+ cells after 4 days of infection. f, Frequencies of p24+ cells (of total CD3+ T cells) were quantified per donor under each stimulation condition. Treatment with IFNα, IFNγ, poly I:C or R848 reduced infection, whereas blockade of IFN signaling (using a combination of antibodies against the IFNα/β receptor chain (IFNARi) and IFNγ) increased infection frequency. Comparisons among stimulation conditions (control, IFNα, IFNγ, poly I:C and R848) were analyzed by Friedman’s test followed by Dunn’s post hoc test. All bar and scatter plots display mean ± s.e.m. Unless otherwise indicated, all statistical tests were two-sided, with multiple comparison adjustments applied where specified; otherwise, exact nominal P values are reported in the figures. FMO, fluorescence minus one; Unstim, unstimulated.
To evaluate whether innate immune stimulation enhances antiviral responses, we treated memory CD4+ T cells or CD8-depleted PBMCs from healthy donors with type I/II IFNs or TLR agonists (poly I:C and R848). Stimulation led to increased expression of antiviral proteins, that is, IFIT1 (also elevated in group 1 PLWH after anti-PD-1 therapy) and APOBEC3G in CD4+ T cells. This effect was amplified in the presence of innate immune cells, such as monocytes (Fig. 6d). Cultures were then challenged with HIV in vitro, with or without the addition of IFN antagonists. Stimulation with type I/II IFNs or TLR agonists significantly reduced HIV infection (Fig. 6e,f). TLR agonists were particularly effective at suppressing infection when innate immune cells were present. This antiviral control was abrogated by the inclusion of type I and II IFN antagonists, especially in cultures containing innate immune cells. Although both IFN classes provided antiviral protection, type I IFNs more robustly induced antiviral protein expression in CD4+ T cells.
Discussion
We identified a multiomic response signature in PLWH and cancer, characterized by transcriptomic, cellular and cytokine changes within 24 hours of anti-PD-1 infusion. These changes were prominently observed in both lymphoid and myeloid cells from a subset of participants with reduced HIV reservoir (group 1). Notably, innate response modules (TLR and ISGs) from this signature were elevated both prior to and during therapy in group 1. Our data indicate that anti-PD-1 therapy releases checkpoint inhibition between tumor and myeloid cells, or myeloid and T cells, triggering type I/II IFN production that drives HIV reactivation, antiviral ISG upregulation and expansion of HIV-specific CD8+ T cells—responses that, along with reduced TGFβ signaling and Treg activity, promote clearance of HIV-infected cells (Extended Data Fig. 10c).
Recent studies have shown that interventions that target ICBs promote the upregulation of defined immune activation signatures predictive of good clinical response37 that highlight the expression of ISGs38 in T and myeloid cells37. Our findings provide in vivo evidence of PD-1/programmed death ligand 1 (PD-L1) blockade’s rapid (24-hour) effects on innate immune effector differentiation, suggesting that this therapy triggers early epigenetic reprogramming via TLR activation and increases chromatin accessibility at cytokine loci—hallmarks of trained immunity, typically induced through histone modifications and metabolic rewiring by agents such as β-glucan or Bacillus Calmette−Guérin (BCG)39, which have also shown promise in combination with ICB40,41,42,43,44. In parallel, we observed that these TLR-driven innate responses were accompanied by enhanced effector T cell activity and higher expression of IFN/antiviral modules across all immune cell subsets (natural killer cells, B cells and CD4+ and CD8+ T cells). Refractoriness to viral dissemination in ISG-expressing cells was demonstrated by Sandler et al.45 in non-human primates, where IFN-α2a infusion resulted in delayed simian immunodeficiency virus acquisition in rhesus macaques. Similarly, recent work by Wimmers et al.46 showed that chromatin accessibility and the expression of transcription factors regulating antiviral genes—induced by an AS03-adjuvanted H5N1 influenza virus vaccine—enables myeloid cells to resist infection by Zika and Dengue viruses, despite these viruses not being included in the vaccine46. Higher overall survival rates in people with cancer are also observed when both myeloid and effector T cells express ISGs, implying the functional synergy between ISG-expressing immune subsets47. We observed that ISG expression by myeloid cells coincided with high ISG expression in T cells (and validated in vitro; Fig. 6d–f) and increased frequencies of proliferating (Ki-67+) HIV-specific CD8+ T cells, indicating that a poised innate immune response coincides with improved adaptive immune function. The antigen-specific response measured here (that is against HIV Gag protein) captures only a subset of HIV-responsive cells and does not account for diverse tumor-specific responses; future studies in uniform cancer cohorts with tumor antigen reagents are needed to clarify antitumor and antiviral immunity in PLWH receiving PD-1 blockade and will likely reflect findings from lymphocytic choriomeningitis virus (LCMV) models where PD-1 blockade restored TH1 CD4+ T cell help for CD8+ responses48. Although these findings provide mechanistic insight into how PD-1 blockade shapes antiviral immunity in PLWH with cancer, several limitations should be considered. The absence of intact proviral quantification using the Intact Proviral DNA Assay (IPDA) limited availability of viable PBMCs across timepoints, and lack of tissue sampling precluded precise measurement of replication-competent and tissue-resident reservoirs. Although total HIV DNA, cell-associated HIV RNA, inducible HIV-1 RNA and plasma viral load measurements showed consistent trends, denser longitudinal sampling and inclusion of tissue-based assays will be needed to refine the timing and durability of reservoir decay. Clinically, treatment duration, early therapy discontinuation and underlying cancer type—among other variables—may have influenced therapeutic response; however, the study was not powered to stratify outcomes by cancer stage or tumor subtype. Future studies with larger, more homogeneous cohorts and stratified designs will be required to rigorously evaluate these clinical and virologic associations.
Overall, our data support a model in which PD-1 blockade rapidly reprograms immune responses in both T and myeloid cells. Notably, a decline in latent HIV precedes the rise in plasma viremia. In group 1, reservoir reduction may be driven by (1) early antiviral gene expression in myeloid cells at baseline and 24 hours, coupled with reduced TGFβ and increased HIV-specific T cell responses by 1 week; (2) reactivation of HIV transcription in effector CD4+ T cells; and (3) decreased CD4+ T cell susceptibility to infection due to ISG induction, limiting reservoir seeding in tissues with suboptimal drug penetration49. Notably, we identified an ‘IFN-high’ signature present before and after anti-PD-1 therapy that predicts clinical response and could help pre-select participants most likely to benefit. This signature is associated with lower Treg module expression, is generalizable across individuals with or without cancer and overlaps by 36% with the DURVAST study’s clinical benefit signature50, suggesting that enhancing IFN responses may improve cancer outcomes after anti-PD-1 treatment. In PLWH, clinical trials have shown that pegylated IFN-α2a/b can reduce cell-associated HIV DNA and, in some cases, enable transient control of viremia after analytical treatment interruption51,52. However, chronic IFN exposure may also drive immune exhaustion and promote reservoir persistence, underscoring the importance of optimizing the timing, dose and duration of innate immune stimulation in combination strategies. IL-15 superagonists have demonstrated safety in phase 1 HIV53 and cancer54 trials, with ongoing phase 2 studies (for example, QUILT-3.055). In parallel, TGFβ signaling—known to promote a repressive chromatin architecture, dampen IFN responses55,56,57,58,59 and enable latent HIV persistence in CD4+ T cells60,61—has emerged as a critical barrier to HIV cure strategies. The observed antagonism between TGFβ and IFN signaling further supports the rationale for dual targeting, highlighting the potential of integrating type I IFNs and/or TGFβ inhibitors to enhance anti-PD-1 therapies and reduce the HIV reservoir.
Methods
Study design
All study procedures involving human participants were conducted in accordance with all relevant ethical regulations and the principles of the Declaration of Helsinki. The study protocol was approved by the institutional review boards at Fred Hutchinson Cancer Center, the National Cancer Institute, New York University Langone Medical Center, Johns Hopkins University, Yale University, Mount Sinai School of Medicine, the University of California, San Francisco (Zuckerberg and Parnassus campuses), Louisiana State University Health Sciences Center, the University of Alabama at Birmingham, Roswell Park Cancer Center and the University of Maryland. Written informed consent was obtained from all participants prior to enrollment and sample collection. Participant confidentiality was maintained throughout the study by deidentifying all data and specimens. The research at Case Western Reserve University and Emory University was performed in compliance with institutional, national and international guidelines governing research on human subjects and biospecimen use. Participants in this study were part of the CITN-12 trial (NCT02595866), a multicenter, open-label, non-randomized phase 1 study of participants (n = 30) living with HIV and advanced cancer27,30. We analyzed samples from a cohort of 30 virally suppressed individuals with HIV and advanced cancer, encompassing 11 distinct cancer types. These participants, who were stratified by their baseline CD4+ T cell counts, demonstrated clinical benefit from anti-PD-1 therapy, including objective cancer responses and evidence of HIV reservoir modulation. (Supplementary Data 1, D1 and D2). Participant demographics reflected the underlying composition of the CITN-12 parent trial, in which 30 of 34 participants were male and four were female. The present multiomic substudy was performed on the first 30 participants for whom matched biospecimens were available, comprising 29 males and one female. Sex was self-reported for all participants. Gender identity data were not separately collected. The predominance of male participants reflects the demographics of PLWH and cancer enrolled in US-based trials, which largely recruit from populations with higher HIV prevalence among men who have sex with men. No sex-stratified analyses were performed due to the limited number of female participants. Informed consent was obtained for publication of indirect identifiers and full demographic details27,30. Additionally, in the context of HIV outcomes, Uldrick et al. previously showed that a reduction in cells harboring HIV DNA, transcription and elevated plasma viremia was observed using the first week of therapy30. The participants received a pembrolizumab regimen (200 mg administered intravenously every 3 weeks for up to 2 years), with a minimum of two treatment cycles. Participants were stratified into three cohorts based on their baseline CD4+ T cell counts: cohort 1 (100−199 CD4+ T cells per microliter, n = 6), cohort 2 (200−350 CD4+ T cells per microliter, n = 12) and cohort 3 (>350 CD4+ T cells per microliter, n = 12) (Fig. 1a,b). These participants presented with 11 different cancers, including AIDS-associated cancers such as Kaposi sarcoma and non-Hodgkin lymphoma (Fig. 1a,b and Extended Data Fig. 1a). The distribution of cancer subtypes was not significantly associated with the cohort based on CD4+ T cell numbers (Extended Data Fig. 1a). Notably, complete cancer remission was observed in one participant, and partial remission occurred in four participants (Supplementary Data 1, D1). Demographic data, clinical data, cancer profiling data, response to therapy data and all omic data collected are included in Supplementary Data 1, D1.
Sample collection and processing
Peripheral blood was collected longitudinally from study participants at the following timepoints: pretreatment (C01D01), 24 hours after treatment (C01D02), 1 week after treatment (C01D08) and at EOT (Supplementary Data 1, D3). Blood was collected in acid-citrate dextrose (ACD) tubes (BD Biosciences), and PBMCs were isolated using Ficoll-Hypaque-based density centrifugation. The virological and immunological assays run at each timepoint per PLWH (plasma and PBMCs) are summarized in Supplementary Data 1, D3. It should be noted that bulk RNA-seq of PBMCs and plasma cytokine analyses were prioritized during the study, whereas single-cell and flow cytometry-based readouts were assessed on limited samples (depending on sample availability). In brief, fresh blood was diluted in HBSS (without calcium or magnesium), and PBMCs were isolated using Ficoll-Hypaque density gradient centrifugation. PBMCs were resuspended in freezing media (RPMI media with 10% dimethylsulfoxide (DMSO) and 12.5% human serum albumin) and frozen in aliquots of 5–20 million cells per cryovial. Cells were stored in temperature-monitored LN2 until use. Plasma was isolated from peripheral blood using a high-speed centrifugation procedure63. Prior to use, PBMCs were gradually thawed in a 37 °C water bath and resuspended in prewarmed complete RPMI (RPMI + 10% FBS + 1% penicillin−streptomycin) and then centrifuged at 400g for 7 minutes at room temperature. Cells were then resuspended in complete RPMI and benzonase nuclease (1 µl of benzonase/1 ml of media) for 10 minutes at 37 °C to avoid cell clumping. Cells were then counted using trypan blue dye and a Countess automated cell counter (Invitrogen), washed once more and resuspended in complete RPMI media at distinct concentrations based on desired downstream assay.
Quantification of plasma cytokines
Custom MesoScale Discovery Human U-Plex assays were used to assess the following cytokines in participant plasma: IL-17A, IL-1β, IL-2, IL-4, IL-6, IL-8, IL-9, IP-10, VEGF, TNF, TGFβ1, TGFβ2, TGFβ3, IFN-α2a, IFNβ, IL-10, IL-15, IL-16, IL-18, IL-22 and IL-7. Plasma cytokines were assessed at the following timepoints: pretreatment (C01D01), 24 hours after treatment (C01D02) and 1 week after treatment (C01D08). Assays were carried out according to the manufacturer’s instructions using 25 µl of undiluted plasma in duplicate wells. TGFβ1/TGFβ2/TGFβ3 cytokines were assayed on a separate plate, and plasma was pretreated with acid, followed by base neutralization to activate the TGF family proteins. Plates were read on the MESO QuickPlex SQ 120MM (MesoScale Discovery) instrument, and cytokine levels were calculated using a standard curve of known cytokine quantities. Prior to univariate analyses, exploratory principal component analysis (PCA) and hierarchical clustering were performed to confirm the inverse correlation between TGFβ levels and inflammatory cytokines, as shown in Supplementary Fig 2a,b.
Quantification of HIV RNA and DNA
Plasma HIV viral load was quantified in study participants63. In brief, a quantitative real-time polymerase chain reaction (qPCR)-based single-copy assay (HMMCgag) was used to target the noncoding 5′ region of the HIV gag gene. Cell-associated HIV DNA (LTR-gag) was measured as described64,65. Single-copy plasma HIV RNA was measured at multiple timepoints, including baseline (C01D01), 24 hours after treatment (C01D02) and 1 week after treatment (C01D08). In addition, plasma viremia was monitored prior to each of the first four anti-PD-1 cycles and, subsequently, after every three treatment cycles30. Longitudinal measurements of HIV RNA and DNA were used to evaluate virological outcomes and compared between participant groups over the course of therapy.
RNA-seq
Bulk RNA-seq was performed on whole blood collected in PAXgene tubes from participants at the following timepoints: C01D01 (n = 29), C01D02 (n = 28) and EOT (n = 14). These extracted RNA samples were stored at a temperature of −80 °C until they were ready for the preparation of mRNA libraries. The quantity and integrity of the RNA were confirmed through microelectrophoresis using the High Sensitivity RNA ScreenTape Kit (Agilent Technologies) on the 2200 TapeStation system (Agilent Technologies). After this, the construction of mRNA libraries was performed according to the Illumina TruSeq V2 library preparation kit. The validation of these libraries occurred through microelectrophoresis on a 2100 Bioanalyzer system (Agilent Technologies), quantification using Kapa Library Quantification Kits (Roche) and eventual pooling and clustering on Illumina TruSeq V2 flow cells. A paired-end 100-cycle run was performed on the Illumina HiSeq 2500 platform, averaging 43 million paired-end reads per sample.
Single-cell RNA-seq was conducted on PBMCs from participants across distinct timepoints: C01D01 (n = 12), C01D02 (n = 8) and C01D08 (n = 10). The sequencing protocol adhered to the recommended guidelines of the 3′ V3 chemistry kit by 10x Genomics. Initially, single-cell gel beads-in-emulsion (GEMs) were generated by combining 350,000–400,000 PBMCs, barcoded Single Cell 3′ v3.1 beads and partitioning oil. These GEMs underwent processing via a 10x Chromium Controller machine (10x Genomics). Subsequent steps encompassed gel bead elimination, cell lysis and reverse transcription in a C1000 Touch Thermal Cycler (Bio-Rad) to yield barcoded cDNA. To extract oil residues, the GEM mixture underwent purification using Dynabeads followed by cDNA amplification. After amplification, cDNA was purified using SPRIselect reagent. Evaluation of cDNA quality and quantity occurred using a 2100 Bioanalyzer (Agilent Technologies). Further stages entailed fragmentation, end repair, A-tailing and adaptor ligation in accordance with the manufacturer’s protocol. Library quality was verified using the Bioanalyzer. These libraries were subsequently sent to the Beijing Genomics Institute and sequenced on a DNBseq-T7 (MGI Tech) machine, targeting 20,000 reads per cell. Data analysis involved uploading FASTQ files to the 10x Genomics cloud platform using Cell Ranger, without applying depth normalization. The filtered count matrix from the analysis was processed through the Seurat package66 in R for subsequent analysis. Cell annotation was accomplished using SingleR67, a reference expression dataset derived from the MonacoImmuneData atlas62 in the celldex R package67. To enhance analysis quality, the DoubletFinder package68 in R was employed to identify and remove doublet cells. Exploration of differential gene expression was facilitated using the MAST R package69. Cell clusters were visually represented using uniform manifold approximation and projection (UMAP). PCA was performed to assess global differences among the clusters.
Bioinformatic analysis
Multivariate model of plasma cytokines and transcriptomic modules associated with treatment and HIV RNA
Feature selection was performed using least absolute shrinkage and selection operator (LASSO)70, implemented through the glmnet R package. The LASSO technique was used to determine the combination of plasma cytokines that best associated with treatment 24 hours after treatment compared to baseline and also to identify the combination of transcriptomic modules that best predicted the increase in HIV RNA over time. The model was optimized using leave-one-out cross-validation, and the features with the least cross-validated mean square error (MSE) were identified.
Processing of RNA-seq data
An integrated automated pipeline developed in-house was used to preprocess FASTQ files per sample. The raw sequencing reads were trimmed off any adapter sequence contaminants using Trimmomatic71. The trimmed reads were aligned to the Ensembl version of the human genome (GRCh38) using the STAR aligner72. The gene abundance was estimated by counting the number of reads mapping to the exons unique to the transcripts of a gene using HTSeq73. The obtained gene counts were normalized using trimmed mean of M values (TMM) by correction for the library size74. Normalized gene expression data from whole-blood RNA-seq samples collected at pretreatment (C01D01), 24 hours after treatment (C01D02) and EOT were analyzed using PCA. Significant transcriptomic shifts were detected at both C01D02 and EOT relative to baseline (C01D01), as assessed by Wilcoxon test (P < 0.05) (Supplementary Fig. 1a,b).
Differential gene expression analysis
The differences in transcriptomic profiles between the timepoints were determined by fitting a generalized linear model (GLM) to each gene. Gene expression was treated as the dependent or response variable and timepoint as the independent variable. A likelihood ratio test was then performed to test whether the coefficients or fold changes differed from zero. The obtained P values were then corrected for multiple comparisons using the Benjamini−Hochberg method, and P values less than 5% were reported as significant. All these tests were performed as part of the edgeR package in R75.
Pathway enrichment analysis
To test the enrichment of pathways/gene sets among the differentially expressed genes, a GSEA76 pre-ranked with 1,000 permutations was performed using the R package fgsea. The Hallmark gene sets and the Immunologic Signature gene sets (C7)77 from the MSigDB, along with transcription factor targets from the ChEA database78, were used as gene set databases. The differentially expressed genes were pre-ranked by the decreasing order of their −log10(P) times their signed (log fold change) to identify pathways upregulated and downregulated comparing timepoints. The obtained pathway P values were corrected for multiple comparisons using the Benjamini−Hochberg method. Pathways enriched at a P value of less than 5% were considered significant. Enriched pathways were visualized using the R package enrichmentmap. See Supplementary Data 1, D9 for differentially expressed transcription factor targets. Differentially enriched transcription factor targets are listed in Supplementary Data 1, D10. Notably, although pathway modules did not significantly differ across the three pretreatment CD4+ T cell-defined cohorts, they were consistently modulated in all groups at 24 hours after infusion (Supplementary Fig. 1c).
SCimilarity analyses to map the modules to publicly available datasets
To map transcriptional modules from the CITN-12 cohort onto a large, publicly available single-cell atlas, we used the SCimilarity framework. The analysis was conducted using SCimilarity model version 1.1 (cellquery_model_v1.1), which enables efficient cell similarity searches across a reference database of 22.7 million single-cell transcriptomes. The query dataset consisted of scRNA-seq profiles generated from PBMC samples collected in the CITN-12 study at multiple timepoints (C01D01, C01D02 and C01D08). This dataset was formatted for SCimilarity by computing low-dimensional embeddings using the pretrained cellquery model. Transcriptomic modules of interest (as defined in Fig. 1 and Supplementary Data 1, D9 and D10) were scored at the single-cell level, and the resulting module scores were appended to the query scRNA-seq object using the Seurat framework. SCimilarity was used to identify phenotypically similar cells from the precomputed reference database. For each query cell, similarity scores to all database cells were computed. To ensure high-confidence mappings, only the top 1% of most similar cells (that is, 99th percentile matches) were retained for downstream analysis. To focus on biologically relevant contexts, matched cells were filtered to include only those originating from ex vivo PBMC samples derived from healthy individuals, people with cancer or people with infectious diseases. For each module, median similarity scores were calculated per matched sample across the retained database cells. These values were visualized using a row-normalized heatmap to enable comparison of module expression patterns across diverse immune conditions.
Immune cell-type-specific transcriptional signatures module analysis
The significant Immunologic Signature gene sets (of C7 from MSigDB) that were differentially expressed between timepoints were grouped into modules of related gene sets based on a Jaccard coefficient of 0.25—that is, gene sets were grouped into modules if they had a minimum of 25% gene overlap between every pair of gene sets. The minimum module size contained at least three gene sets. The modules were obtained independently for the upregulated and downregulated gene sets (Supplementary Data 1, D9).
SLEA
SLEA32 was used to represent the expression of modules by calculating the z-score of the pathway per sample. The mean expression value of genes enriched in a module was compared to the mean expression of random sets of genes of the same module size for 1,000 permutations for every sample. The difference between the observed and expected mean expression values for each gene set was determined as the SLEA z-score.
Transcriptomic deconvolution
CIBERSORT79 was used to infer immune cell subset frequencies from whole-blood transcriptomic data using a reference expression set of distinct hematopoietic cell states that were purified from umbilical cord blood and peripheral blood (Gene Expression Omnibus (GEO) accession ID: GSE24759). Genes of the reference set were assigned to a cell type if they were expressed at a positive fold change at a P value less than 5% in that cell type compared to all the other cell types. This reference expression matrix of genes by cell type was used with CIBERSORT to infer cell subset frequencies at baseline, at 24 hours after treatment and at C01D08 of treatment. The frequencies derived via CIBERSORT were compared to the flow cytometry-based frequencies obtained at C01D01 (that is, pretreatment) and were significantly correlated. The cell subset frequencies obtained via CIBERSORT were further used on the whole-blood RNA-seq data to obtain immune cell subset-specific gene expression. Deconvolution was performed using least squares fit to model the whole-blood gene expression measures across samples as the function of contributions of immune subset-specific gene expression weighted by the corresponding cell frequencies of those subsets obtained via CIBERSORT. Linear regression was applied separately on baseline and 24 hours after treatment. The regression coefficients were used as surrogates for estimated cell subset-specific average gene expression. The difference in the average cell subset-specific gene expression estimates was used as the level of gene expression change per immune cell subset between 24 hours after treatment compared to baseline.
Integration between transcriptomic modules and plasma cytokines
A projection-based approach using mixOmics80 was used to integrate the change in plasma cytokine expression with the change in transcriptomic module expression. To minimize the technical effect specific to each data type, a sparse least square regression was used to generate a unique scale for each data type and project all data types on the same scale to assess correlation. Pearsonʼs correlation between the features of the two data types and a P value based on the distribution of the correlation coefficients were calculated. Pearsonʼs correlations below a P value cutoff of 0.05 were considered significant.
In vitro HIV infection assay with ISG-inducing compounds
To validate ex vivo findings that activation of TLR3, TLR7/TLR8 and IL-15 pathways induces antiviral responses and restricts HIV infection, we conducted an in vitro infection assay using PBMCs freshly isolated from five healthy human donors. Cells were cultured in AIM-V medium supplemented with 10% serum replacement (Corning, 355500) and 10 mM HEPES. The experimental design consisted of two arms. In arm 1, memory CD4+ T cells were directly isolated from PBMCs using a negative selection enrichment kit (STEMCELL Technologies, 19157). In arm 2, CD8+ T cells were first depleted from the PBMCs, and the remaining cell population was cultured for stimulation. In both arms, cells were subjected to one of the following stimulation conditions for 24 hours: unstimulated control, IFNγ (5 ng ml−1; PeproTech, 300-02), IFNβ (5 ng ml−1; PeproTech, AF-300-02B), poly I:C (10 ng ml−1; InvivoGen, tlrl-pic-5), R848 (10 ng ml−1; InvivoGen, tlrl-r848-1) or IL-15 (100 ng ml−1; PeproTech, 200-15). To evaluate whether the antiviral effects were mediated by IFN signaling, each stimulation condition was also tested in the presence of neutralizing antibodies against IFNAR (10 µg ml−1; PBL Assay Science, 21385) and IFNγ (10 µg ml−1; Miltenyi Biotec, 130-095-743). At 16 hours after stimulation, activation of antiviral signaling pathways—including phosphorylation of STAT1, IRF3 and IRF7 as well as expression of IFIT1 and APOBEC3G—was assessed. At 24 hours, memory CD4+ T cells were isolated from arm 2, and cells from both arms were then infected with the p89.6 dual-tropic HIV-1 strain by spinoculation in the presence of saquinavir to ensure single-round infection. Viral replication was quantified by measuring intracellular p24 levels at day 4 after infection.
Ex vivo HIV-specific response profiling
HIV-specific PBMC responses were assayed using flow cytometry. After the thawing procedure, PBMCs were resuspended at 1.0 × 107 cells per milliliter, and 100 µl (or 1.0 × 106 cells) was added per well of a 96-well U-bottom plate. Cells were unstimulated, stimulated with 2 µg ml−1 HIV-1 PTE Gag peptide or stimulated with 2 µg ml−1 Staphylococcal enterotoxin B (SEB) in the presence of 10 µg ml−1 brefeldin A (BFA). After 6 hours of stimulation, cells were stained with DAPI LIVE/DEAD stain to discriminate between live and dead cells. After a wash with FACS buffer (PBS + 4% human serum), cells were incubated for 20 minutes at room temperature with a surface antibody staining cocktail mix diluted in Brilliant Stain Buffer (BD Biosciences).
Flow cytometry
Flow cytometry staining was performed using surface, intracellular and phospho-specific protocols depending on the experimental design. Surface staining was carried out at 37 °C for 20 minutes in 25 µl of PBS supplemented with 2% FBS (PBS + 2% FBS). After incubation, cells were washed with 150 µl of PBS + 2% FBS buffer. For intracellular staining, cells were washed and then incubated in fixation/permeabilization solution (BD Biosciences, FoxP3 Transcription Factor Kit) for 30 minutes at 4 °C. After fixation, cells were washed with the accompanying fixation/permeabilization buffer and incubated with intracellular antibody cocktails—prepared in the same buffer—for 45 minutes at 4 °C. For phospho-flow staining, cells were fixed in 50 µl of BD Phosflow Fix Buffer I (cat. no. 557870) for 30 minutes at 4 °C, washed with 150 µl of PBS + 2% FBS and permeabilized using 50 µl of cold BD Phosflow Perm Buffer III (cat. no. 558050) on ice for 15 minutes. Cells were washed twice with 150 µl of 1× permeabilization buffer (Invitrogen, 00-8333), and all residual buffer was carefully removed. Intracellular staining for phospho-proteins was performed by adding antibodies diluted in 25 µl of 1× permeabilization buffer (Invitrogen) and gently mixing. Cells were incubated for 60 minutes at 4 °C and subsequently washed with 150 µl of 1× permeabilization buffer. After the final wash, cells were resuspended in 100 µl of PBS for acquisition. All centrifugation steps were carried out at room temperature at 500g for 5 minutes. Data were acquired using a BD FACSymphony A5 flow cytometer operated via BD FACSDiva software, collecting a minimum of 100,000 live cells per sample. Flow cytometry data were analyzed using FlowJo software (version 10.8.1). High-dimensional flow cytometry data were analyzed using unsupervised methods to visualize and identify distinct immune cell populations. After compensation, quality control and gating to exclude debris, doublets and dead cells, events corresponding to viable single cells were exported for downstream analysis. Live CD3+CD4+ T cells or live CD3+CD8+ T cells were selected and exported for clustering analysis. Marker expression values were biexponentially transformed, and data from all samples within each panel were concatenated for joint analysis. Dimensionality reduction was performed using UMAP for two-dimensional visualization of cellular heterogeneity. UMAP projections were used to display marker density and distribution across cell subsets. Unbiased clustering was then performed using the PhenoGraph algorithm (https://github.com/jacoblevine/PhenoGraph) with the number of nearest neighbors (k) set to 60, enabling the identification of phenotypically distinct cellular clusters. Cluster-level marker expression was summarized using median fluorescence intensity (MFI) and visualized via heatmaps in R to compare marker profiles across identified populations. UMAP plots and cluster overlays were generated using FlowJo and R, and representative density plots were used to highlight markers of interest. Antibodies used for HIV-specific profiling are as follows: BUV496 anti-human CD3, BUV563 anti-human CD4, BUV615 anti-human CD45RA, BUV661 anti-human CD27, BUV737 anti-human CD8, BUV805 anti-human CD45RO, BV480 anti-human CD14, BV480 anti-human CD19, BV650 anti-human CD69, BV750 anti-human CCR7, BB515 anti-human CCR5, BB630 anti-human SLAMF6, BB660 anti-human CD101, PE-Cy7 anti-human PD-1, PE-Cy5 anti-human CD95, APC-R700 anti-human CD28, APC-Cy7 anti-human CD39, BUV395 anti-human BCL2, BV421 anti-human TCF7, BV570 anti-human Ki-67, BV605 anti-human IRF4, BV711 anti-human EOMES, BV786 anti-human TBET, BB700 anti-human IFNγ, BB750 anti-human NR4A1, PE anti-human TOX, PE-CF594 anti-human TNF and APC anti-human ID2. Antibodies for in vitro experiments are as follows. The staining panel included: LIVE/DEAD BV510, Life Technologies, L34957; CD3 BUV615, BD Biosciences, 612992, clone UCHT1; CD4 BV605, BioLegend, 317438, clone OKT4; CD45RA BV650, BioLegend, 304136, clone HI100; CD27 AF647, BD Biosciences, 567026, clone O323; CCR7 BUV563, BD Biosciences, 741317, clone 3D12; pIRF3 PerCP, Novus Biologicals, BS-3195R-PERCP, clone polyclonal; pIRF7 BUV496, BD Bioscience, custom, clone K47-671; pSTAT1 (p701) AF488, BD Biosciences, AB_2737715, clone 4a; IFIT1 APC, Novus Biologicals, NBP2-71005APC, clone OTI3G8; APOBEC3G AF700, Novus Biologicals, NBP1-77206AF700, clone polyclonal; and p24 RD1, Beckman Coulter, 6604667, clone KC57. All antibodies were commercially purchased and validated by the vendor, and the details of the clone, fluorophore and dilution used per stain are provided in Supplementary Data 1, D11.
Statistical analyses and reproducibility
Whole-blood, PBMC and plasma samples were collected longitudinally from participants and varied based on availability (see Supplementary Data 1, D3 for sample availability per omic). No replicates were analyzed or added at any timepoint. The samples were collected based on the clinical trial design (see ‘Study design’ section above), and no further statistical tests were done to predetermine the sample size. We were not blinded to the outcomes tested. Samples were run in a single batch for plasma cytokine and single-cell analyses. Whole-blood RNA-seq was done in two batches, and transcript count matrices were corrected using the ComBat package in R. The uncorrected and corrected count matrices will be made available on the GEO upon finalization and before publication of this paper. FlowJo (version 10.10.0), GraphPad Prism (version 10.2.3) and R (version 4.4.0 in RStudio 2024.04.1 Build 748) were used to generate figures and for statistical analyses. The Benjamini−Hochberg test was used to correct for multiple testing during whole-blood RNA-seq data analyses. All figure legends indicate the P values and statistical tests used. The figure panels were assembled using Adobe Illustrator 2024 (version 28.5).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Expression set objects for all bulk RNA-seq data are accessible online as a digital object on Zenodo (https://doi.org/10.5281/zenodo.11200738)81. Cytokine, flow cytometry, transcriptome module scores, viral readouts and clinical readouts are provided in Excel format in the supplementary material. All available bulk RNA-seq and scRNA-seq FASTQ files are available on the GEO under accession numbers GSE298837 and GSE298838, respectively.
Code availability
Code used for sequencing and omic (standalone and integration) analyses was generated using the packages and pipelines cited above. The code is accessible online as a digital object on Zenodo (https://doi.org/10.5281/zenodo.11200738)81. Additional analysis pipelines—including workflows for scRNA-seq and bulk RNA-seq preprocessing, pathway enrichment, multiomic integration and single-cell module scoring—can be made available upon reasonable request to the senior author, A.A.S. (ashish.arunkumar.sharma@emory.edu). All requests will be processed within 2–4 weeks of receipt, and all requested code will be made available online for at least 1 year via Zenodo.
References
Landovitz, R. J., Scott, H. & Deeks, S. G. Prevention, treatment and cure of HIV infection. Nat. Rev. Microbiol. 21, 657–670 (2023).
Chun, T. W., Davey, R. T., Engel, D., Lane, H. C. & Fauci, A. S. Re-emergence of HIV after stopping therapy. Nature 401, 874–875 (1999).
Davey, R. T.Jr et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl Acad. Sci. USA 96, 15109–15114 (1999).
Finzi, D. et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278, 1295–1300 (1997).
Finzi, D. et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 5, 512–517 (1999).
Porichis, F. & Kaufmann, D. E. Role of PD-1 in HIV pathogenesis and as target for therapy. Curr. HIV/AIDS Rep. 9, 81–90 (2012).
Massanella, M., Fromentin, R. & Chomont, N. Residual inflammation and viral reservoirs: alliance against an HIV cure. Curr. Opin. HIV AIDS 11, 234–241 (2016).
Fromentin, R. et al. CD4+ T cells expressing PD-1, TIGIT and LAG-3 contribute to HIV persistence during ART. PLoS Pathog. 12, e1005761 (2016).
Cai, C. W. & Sereti, I. Residual immune dysfunction under antiretroviral therapy. Semin. Immunol. 51, 101471 (2021).
D’Souza, M. et al. Programmed death 1 expression on HIV-specific CD4+ T cells is driven by viral replication and associated with T cell dysfunction. J. Immunol. 179, 1979–1987 (2007).
Chomont, N. et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 15, 893–900 (2009).
Evans, V. A. et al. Programmed cell death-1 contributes to the establishment and maintenance of HIV-1 latency. AIDS 32, 1491–1497 (2018).
Fromentin, R. et al. PD-1 blockade potentiates HIV latency reversal ex vivo in CD4+ T cells from ART-suppressed individuals. Nat. Commun. 10, 814 (2019).
Hiam-Galvez, K. J., Allen, B. M. & Spitzer, M. H. Systemic immunity in cancer. Nat. Rev. Cancer 21, 345–359 (2021).
Ribas, A. & Wolchok, J. D. Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018).
Waldman, A. D., Fritz, J. M. & Lenardo, M. J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol. 20, 651–668 (2020).
Huang, A. C. et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 (2017).
Trautmann, L. et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 12, 1198–1202 (2006).
Day, C. L. et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443, 350–354 (2006).
Petrovas, C. et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med. 203, 2281–2292 (2006).
Macatangay, B. J. C. et al. T cells with high PD-1 expression are associated with lower HIV-specific immune responses despite long-term antiretroviral therapy. AIDS 34, 15–24 (2020).
Velu, V. et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 458, 206–210 (2009).
Dyavar Shetty, R. et al. PD-1 blockade during chronic SIV infection reduces hyperimmune activation and microbial translocation in rhesus macaques. J. Clin. Invest. 122, 1712–1716 (2012).
Mylvaganam, G. H. et al. Combination anti-PD-1 and antiretroviral therapy provides therapeutic benefit against SIV. JCI Insight 3, e122940 (2018).
Rahman, S. A. et al. PD-1 blockade and vaccination provide therapeutic benefit against SIV by inducing broad and functional CD8+ T cells in lymphoid tissue. Science Immunol. 6, eabh3034 (2021).
Velu, V. et al. PD-1 blockade following ART interruption enhances control of pathogenic SIV in rhesus macaques. Proc. Natl Acad. Sci. USA 119, 2202148119 (2022).
Uldrick, T. S. et al. Assessment of the safety of pembrolizumab in patients with HIV and advanced cancer—a phase 1 study. JAMA Oncol. 5, 1332−1339 (2019).
Lurain, K., Ramaswami, R., Yarchoan, R. & Uldrick, T. S. Anti-PD-1 and anti-PD-L1 monoclonal antibodies in people living with HIV and cancer. Curr. HIV/AIDS Rep. 17, 547–556 (2020).
Delyon, J. et al. PD-1 blockade with pembrolizumab in classic or endemic Kaposi’s sarcoma: a multicentre, single-arm, phase 2 study. Lancet. Oncol. 23, 491–500 (2022).
Uldrick, T. S. et al. Pembrolizumab induces HIV latency reversal in people living with HIV and cancer on antiretroviral therapy. Science Transl. Med. 14, eabl3836 (2022).
Gay, C. L. et al. Suspected immune-related adverse events with an anti-PD-1 inhibitor in otherwise healthy people with HIV. J. Acquir. Immune Defic. Syndr. 87, e234–e236 (2021).
Gundem, G. & Lopez-Bigas, N. Sample-level enrichment analysis unravels shared stress phenotypes among multiple cancer types. Genome Med. 4, 28 (2012).
Zhao, X., Shan, Q. & Xue, H. H. TCF1 in T cell immunity: a broadened frontier. Nat. Rev. Immunol. 22, 147–157 (2022).
Gonzalez, M. E. et al. EZH2 expands breast stem cells through activation of NOTCH1 signaling. Proc. Natl Acad. Sci. USA 111, 3098–3103 (2014).
Heimberg, G. et al. A cell atlas foundation model for scalable search of similar human cells. Nature 638, 1085–1094 (2025).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020).
Chen, P. L. et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 6, 827–837 (2016).
Acha-Sagredo, A. et al. A constitutive interferon-high immunophenotype defines response to immunotherapy in colorectal cancer. Cancer Cell 43, 292–307 (2025).
Netea, M. G. et al. Trained immunity: a program of innate immune memory in health and disease. Science 352, aaf1098 (2016).
Geng, Q. et al. β-glucan combined with Envafolimab and Endostar as immune rechallenge for metastatic non-small cell lung cancer. BMC Immunol. 25, 60 (2024).
Hu, X. et al. PD-L1 antibody enhanced beta-glucan antitumor effects via blockade of the immune checkpoints in a melanoma model. Cancer Immunol. Immunother. 72, 719–731 (2023).
Song, Q. et al. A β-1,3/1,6-glucan enhances anti-tumor effects of PD1 antibody by reprogramming tumor microenvironment. Int. J. Biol. Macromol. 279, 134660 (2024).
Wang, M. et al. β-glucan combined with PD-1/PD-L1 checkpoint blockade for immunotherapy in patients with advanced cancer. Front. Pharmacol. 13, 887457 (2022).
Wang, M. et al. β-glucan: a potent adjuvant in immunotherapy for digestive tract tumors. Front. Immunol. 15, 1424261 (2024).
Sandler, N. G. et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature 511, 601–605 (2014).
Wimmers, F. et al. The single-cell epigenomic and transcriptional landscape of immunity to influenza vaccination. Cell 184, 3915–3935 (2021).
Boukhaled, G. M. et al. Pre-encoded responsiveness to type I interferon in the peripheral immune system defines outcome of PD1 blockade therapy. Nat. Immunol. 23, 1273–1283 (2022).
Barber, D. L. et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006).
Fletcher, C. V. et al. Persistent HIV transcription and variable antiretroviral drug penetration in lymph nodes during plasma viral suppression. AIDS 36, 985–990 (2022).
Gonzalez-Cao, M. et al. Assessment of the feasibility and safety of durvalumab for treatment of solid tumors in patients with HIV-1 infection: the phase 2 DURVAST study. JAMA Oncol. 6, 1063–1067 (2020).
Papasavvas, E. et al. Safety, immune, and antiviral effects of pegylated interferon alpha 2b administration in antiretroviral therapy-suppressed individuals: results of pilot clinical trial. AIDS Res. Hum. Retroviruses 37, 433–443 (2021).
Papasavvas, E. et al. Comparable HIV suppression by pegylated-IFN-α2a or pegylated-IFN-α2b during a 4-week analytical treatment interruption. AIDS 35, 2051–2054 (2021).
Miller, J. S. et al. Safety and virologic impact of the IL-15 superagonist N-803 in people living with HIV: a phase 1 trial. Nat. Med. 28, 392–400 (2022).
Wrangle, J. M. et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol. 19, 694–704 (2018).
Sanjabi, S., Oh, S. A. & Li, M. O. Regulation of the immune response by TGF-β: from conception to autoimmunity and infection. Cold Spring Harb. Perspect. Biol. 9, a022236 (2017).
Grunwell, J. R. et al. TGF-β1 suppresses the type I IFN response and induces mitochondrial dysfunction in alveolar macrophages. J. Immunol. 200, 2115–2128 (2018).
Guerin, M. V. et al. TGFβ blocks IFNα/β release and tumor rejection in spontaneous mammary tumors. Nat. Commun. 10, 4131 (2019).
Hamon, P. et al. TGFβ receptor inhibition unleashes interferon-β production by tumor-associated macrophages and enhances radiotherapy efficacy. J. Immunother. Cancer 10, e003519 (2022).
Gauthier, T. & Chen, W. IFN-γ and TGF-β, crucial players in immune responses: a tribute to Howard Young. J. Interferon Cytokine Res. 42, 643–654 (2022).
Yim, L. Y. et al. Transforming growth factor β signaling promotes HIV-1 infection in activated and resting memory CD4+ T cells. J. Virol. 97, e0027023 (2023).
Kulpa, D. A. et al. Differentiation into an effector memory phenotype potentiates HIV-1 latency reversal in CD4+ T cells. J. Virol. 93, e00969-19 (2019).
Monaco, G. et al. RNA-seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 26, 1627–1640 (2019).
Somsouk, M. et al. The immunologic effects of mesalamine in treated HIV-infected individuals with incomplete CD4+ T cell recovery: a randomized crossover trial. PLoS ONE 9, e116306 (2014).
Elliott, J. H. et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 10, e1004473 (2014).
Zerbato, J. M. et al. Multiply spliced HIV RNA is a predictive measure of virus production ex vivo and in vivo following reversal of HIV latency. eBioMedicine 65, 103241 (2021).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019).
McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337 (2019).
Finak, G. et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278 (2015).
Tibshirani, R. Regression shrinkage and selection via the Lasso. J. R. Stat. Soc. Ser. B Methodol. 58, 267–288 (1996).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Lachmann, A. et al. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26, 2438–2444 (2010).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Rohart, F., Gautier, B., Singh, A. & Lê Cao, K. A. mixOmics: an R package for ʼomics feature selection and multiple data integration. PLoS Comput. Biol. 13, e1005752 (2017).
Sharma, A. Code and data for manuscript: blocking PD-1 engagement rapidly upregulates innate antiviral and immune functions that associate with HIV reservoir decay. Zenodo https://doi.org/10.5281/zenodo.11200738 (2024).
Acknowledgements
This study was funded by ZIA BC 011700 Patient Oriented Research in Viral Malignancies and HIV-associated Cancers; grant UM1AI164560 (principal investigator (PI), Deeks), National Institute of Allergy and Infectious Diseases (NIAID), Delaney AIDS Research Enterprise to Cure HIV (DARE); and grant P30AI027757 (PI, Baeten), University of Washington Center for AIDS Research. This study was sponsored by the National Cancer Institute (NCI) Cancer Therapy Evaluation Program. The multiomic assays, analyses and salary support for R.-P.S. and A.A.S. were provided by UM1AI164561 (PI, Sekaly, Chanda, Cannon) and P01AI178376 (PI, Sekaly). This research was also supported, in part, by the Intramural Research Program of the National Institutes of Health (NIH), NCI, through Intramural Project ZIA BC010888. The contributions of the NIH authors were made as part of their official duties as NIH federal employees, are in compliance with agency policy requirements and are considered Works of the United States Government. However, the findings and conclusions presented in this paper are those of the authors and do not necessarily reflect the views of the NIH or the US Department of Health and Human Services. Studies in the laboratory of S.R.L. were funded by National Health and Medical Research Council of Australia APP1138581, APP2026490, APP1052979 and APP1149990 and NIH DARE UM1AI164560, U19 AI096109 and UM1AI126611. DARE UM1AI164560 also funded research in the laboratory of N.C. Research in the laboratory of J.M.-P. was supported by the Spanish Ministry of Science, Innovation and Universities (grants PID2022-139271OB-I00 and CB21/13/ 0006), the NIH/NIAID (1 UM1 AI164561-01 and 1P01AI178376-01) and Research Centers of Catalonia (grant 2021 SGR00452). This work was also supported by 1U01CA154967 and Cancer Center Support Grant P30 CA015704 from the NCI.
Author information
Authors and Affiliations
Contributions
A.T., J.L.L.C.A., A.N.P. and V.S. performed data analysis and curation, developed and executed computational workflows, generated figures and visualizations and contributed to writing, review and editing of the paper. M.B.L. conducted in vitro experimental validation and data analysis. A.B.E. and K.G. reviewed and edited the paper. G.P.S., S.K.B. and S.K. generated and analyzed multiomic data. A.R. and S.R.L. contributed experimental data for reservoir quantification. S.R.L. also assisted in data interpretation and paper review. N.C. performed TILDA and HIV DNA measurements and contributed to paper review. F.M., R.Y., K.L. and R.R. provided clinical oversight and patient care and contributed to data collection and paper editing. R.Y. conceptualized the clinical protocol under which participants were treated and approved the final manuscript. E.S. planned the study and contributed to writing and editing of the paper. B.W.H., L.D., S.P.F. and M.A.C. provided support for investigation, resources and funding acquisition and contributed to paper writing and editing. J.M.-P. and A.B.E. reviewed and edited the paper. T.S.U. and S.G.D. planned the study, collected and analyzed data and contributed to paper writing and editing. A.A.S. performed experimental omic work, analyzed data and led writing and editing of the paper. R.-P.S. planned the study, collected and analyzed data and led writing and editing of the paper.
Corresponding author
Ethics declarations
Competing interests
R.Y., R.R. and K.L. report receiving research support from Celgene (now Bristol Myers Squibb), CTI BioPharma (a Sobi A.B. company), PDS Biotech and Janssen Pharmaceuticals through Cooperative Research and Development Agreements (CRADAs) with the NCI. R.Y., R.R. and K.L. also report receiving drugs for clinical trials from Merck, EMD-Serano and Lilly and preclinical material from Lentigen Technology through CRADAs or Material Transfer Agreements (MTAs) with the NCI. R.Y. is a co-inventor on US Patent 10,001,483 titled ‘Methods for the treatment of Kaposi’s sarcoma or KSHV-induced lymphoma using immunomodulatory compounds and uses of biomarkers’. An immediate family member of R.Y. is a co-inventor on patents or patent applications related to internalization of target receptors, epigenetic analysis and ephrin tyrosine kinase inhibitors. All rights, titles and interests to these patents have been assigned to the US Department of Health and Human Services; the government conveys a portion of the royalties it receives to its employee inventors under the Federal Technology Transfer Act of 1986 (P.L. 99-502). S.R.L. receives honoraria from Gilead Sciences, Merck, AbbVie, Esfam and Immunocore as a member of their scientific advisory boards. J.M.-P. has received research funding, consultancy fees and lecture sponsorships from and has served on advisory boards for various companies (Merck Sharp & Dohme, Gilead Sciences, Grifols and ViiV Healthcare) outside the scope of this article. T.S.U. is a full-time employee of Regeneron Pharmaceuticals. A.T. and A.N.P. are independent consultants and list their consulting entities as primary affiliations that have no financial or commercial interest in the subject matter of this study. S.G.D. receives research support from Gilead. He is also a member of the scientific advisory board for Tendel. S.G.D. has consulted for AbbVie, Hologic and ViiV. The other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Alison Farrell and Lia Parkin, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Transcriptional module activation and transcription-factor targeting 24 h post–PD-1 blockade.
(a) Distribution of cancer types across cohorts, stratified by AIDS-defining versus non–AIDS-defining status. (b) Schematic of sample processing and experimental workflow. Plasma cytokines (23 analytes; Meso Scale Discovery), bulk RNA-seq, plasma viral load, and cell-associated HIV DNA were profiled 24 h post–PD-1 blockade (C01D02). Transcriptomic modules derived from these assays were assessed for persistence through EOT, association with HIV-reservoir dynamics, and validated by single-cell assays (scRNA-seq and flow cytometry). (c) Heatmaps of gene-expression modules upregulated 24 h post-treatment, including interferon-stimulated genes (ISGs), antiviral effectors, TLR/IL-1 signaling, and immune-checkpoint blockade modules. Columns are participants grouped by time point; values are z-scores (row-normalized). Spearman correlations are reported with ρ and P values. (d) Transcription-factor target network highlighting enrichment of upregulated targets 24 h post-treatment. Green edges indicate co-expression or known physical interactions. Major hubs (for example, STAT1, IRF1, RELA) regulate post-treatment changes. Unless otherwise indicated, tests were two-sided; multiple-comparison adjustments were applied where specified; otherwise, exact nominal P values are shown in the figures.
Extended Data Fig. 2 Downregulation of exhaustion-associated genes and TF targets 24 h post–PD-1 blockade.
(a) Expression of selected exhaustion-associated surface markers (PDCD1, CD248, TIGIT, IL10RA, BMPR1A, CD5) decreased at C01D02 (green) versus C01D01 (black). Expression in copies per million; P values from paired two-sided Wilcoxon tests. (b) Heatmaps and overlap networks for three modules significantly altered post-treatment: naïve CD4⁺/CD8⁺ T-cell and WNT-signaling module; effector CD8⁺ T-cell function module; and regulatory T-cell module. Heatmaps display z-scored expression (rows, genes; columns, participants). Network nodes represent gene sets; edges denote ≥25% gene overlap; color indicates normalized enrichment score (NES). (c) TF regulatory network of targets significantly downregulated 24 h post-treatment. Nodes are TF targets colored by log2 fold change; edges indicate co-expression or known interactions. Unless otherwise indicated, tests were two-sided with multiple-comparison adjustments where specified; otherwise, exact nominal P values are shown.
Extended Data Fig. 3 Plasma cytokine dynamics following PD-1 blockade.
(a–b) Log-transformed concentrations of cytokines significantly altered at C01D02 versus C01D01. IFN-γ, IFN-β, and IL-6 increased; TGF-β1 and TGF-β2 decreased. P values from paired two-sided Wilcoxon tests. (c) Longitudinal IL-17A and IL-4 concentrations: all participants (left) and stratified by decreasing versus increasing values at C01D08 (middle, right); percentages denote the proportion per stratum. Unless otherwise indicated, tests were two-sided with multiple-comparison adjustments where specified; otherwise, exact nominal P values are shown.
Extended Data Fig. 4 Longitudinal dynamics of immune transcriptional programs and HIV DNA after PD-1 blockade.
(a) Persistence of module activity across treatment cycles by group (14 participants total; Group 1 n = 9, Group 2 n = 5). Each point is the median SLEA z-score for a participant sample; jitter plots show mean ± s.e.m. Group 1 (blue) shows sustained antiviral/innate modules (Modules 2, 3, 5); Group 2 (pink) shows enrichment of naïve T-cell/WNT (Module 6) and Treg programs (Module 7). Mann–Whitney U tests compare groups at each time point. (b) Longitudinal total HIV DNA in PBMCs (copies per 10⁶ cells) across C01D01, C01D02, C01D08, C02D01, and EOT. Left: all 29 participants (paired reductions at C01D02 and EOT). Middle/right: trajectories for Group 1 (n = 9) and Group 2 (n = 5). Dots are participants; lines connect within-participant samples; medians ± IQR are overlaid. Wilcoxon matched-pairs signed-rank tests assess within-participant change from C01D01. Unless otherwise indicated, tests were two-sided with multiple-comparison adjustments where specified; otherwise, exact nominal P values are shown.
Extended Data Fig. 5 Immune and viral landscape differences by participant group and myeloid-driven, tissue-wide signatures of PD-1 blockade.
(a) Correlations between plasma HIV RNA and cytokines at each time point. IFN-γ correlates positively and IL-4 negatively with plasma HIV RNA (Spearman ρ and P shown). (b) Correlation between HIV DNA and inducible HIV-1 RNA (log-transformed copies per 10⁶ CD4⁺ T cells) across participants (Spearman ρ and P shown). (c) Pairwise correlation heatmap among virologic markers (total HIV DNA, inducible HIV-1 RNA [TILDA], cell-associated HIV RNA, plasma viral load [pVL]). Also shown: plasma HIV RNA over time (C01D01, C01D02, C01D08, EOT), with significant reductions at C01D02 and EOT in Group 1 (paired two-sided Wilcoxon test). (d) Unspliced HIV RNA (us-HIV RNA; copies per 10⁶ PBMCs) measured at C01D01, C01D02, and EOT in 14 participants (Group 1 n = 9; Group 2 n = 5). Points are participant samples; medians ± IQR shown. Group comparisons by Mann–Whitney U test (no significant differences detected). (e) Plasma viral load (copies per mL) by group across time (same participants as in d). Dotted line indicates assay limit of detection. Medians ± IQR per time point; Mann–Whitney U tests compare groups. (f) Summary table of clinical characteristics for Groups 1 and 2 (cohort, baseline CD4 count, cancer type, and treatment response). Unless otherwise indicated, tests were two-sided with multiple-comparison adjustments where specified; otherwise, exact nominal P values are shown.
Extended Data Fig. 6 Monocyte and CD8⁺ T-cell transcriptional responses following PD-1 blockade.
(a) UMAP of PBMCs from scRNA-seq annotated with fine immune-cell labels (SingleR package in R). (b) Frequencies of fine-labeled immune subsets across C01D01, C01D02, and C01D08, showing increased classical monocytes and decreased naïve CD8⁺ T cells at C01D02. (c) SLEA module scores in monocytes (top) and CD8⁺ T cells (bottom), by response group (G1, G2) and time point (C01D01, C01D02, C01D08). Columns are participants; rows are modules. (d) UMAP of monocyte subclusters from scRNA-seq with fine-label annotations. (e) Frequencies of monocyte subclusters and module expression across time (C01D01 n = 12, C01D02 n = 8, C01D08 n = 10). Left: stacked bars show subcluster proportions within total monocytes (mean ± s.e.m.). Right: mean module scores (for example, TLR-signaling and ISG modules) within each subcluster. Subcluster-frequency comparisons across time by Mann–Whitney U tests. (f) Number of differentially expressed genes (DEGs) per monocyte cluster across time points. (g) Heatmap of top DEGs in ISG-high monocyte cluster 2 comparing post-treatment time points to baseline. Unless otherwise indicated, tests were two-sided with multiple-comparison adjustments where specified; otherwise, exact nominal P values are shown.
Extended Data Fig. 7 Flow cytometry gating strategy and CD8⁺ T-cell effector function increases following PD-1 blockade.
(a) Representative gating for CD4⁺/CD8⁺ T cells and memory subsets (naïve; central/transitional memory, TCM/TTM; effector memory, TEM; terminally differentiated effector memory, TEMRA). (b) Intracellular staining of IFN-γ (c), TNF (d), and Ki67 (e) in CD8⁺ T cells under FMO, unstimulated, SEB-stimulated (positive control), and HIV Gag peptide pool–stimulated conditions. (c) Volcano plots of DEGs in effector CD8⁺ T-cell clusters (clusters 1 and 3) at 1 week post-treatment, including increased cytotoxicity-associated genes (for example, GZMB, B2M). Unless otherwise indicated, tests were two-sided with multiple-comparison adjustments done in the panel c.
Extended Data Fig. 8 Functional profiling and activation of HIV-specific CD8⁺ T cells across participant groups.
(a) UMAP of HIV-specific CD8⁺ T cells reveals eight transcriptionally distinct clusters. (b) Cluster composition across groups and time points (C01D01, C01D02, C01D08); pie charts show relative abundances. Cluster 8 is consistently enriched in Group 1. (c) Violin plots of IFN-γ mean fluorescence intensity (MFI) across clusters, with higher IFN-γ in clusters 1 and 8. (d) Heatmap of marker expression (row-normalized MFI) across clusters, conditions (unstimulated vs gag-stimulated), groups, and time points. Equal cell numbers per cluster were randomly sampled. Cluster 8 shows elevated effector markers (for example, CD38, GZMB, T-bet) and is enriched in Group 1; cluster 1 is elevated in Group 2. (e) Frequencies of proliferating HIV-specific CD8⁺ T cells (Ki-67⁺ IFN-γ⁺) following gag stimulation across time; both groups increase post-therapy. (f) Proliferative responses (% Ki-67⁺ among gag-specific IFN-γ⁺ CD8⁺ T cells) are higher in Group 1 across time points, including at baseline (C01D01). Unless otherwise indicated, tests were two-sided with multiple-comparison adjustments where specified; otherwise, exact nominal P values are shown.
Extended Data Fig. 9 Increased CD4⁺ T-cell effector function following PD-1 blockade.
(a) Gating strategy for CD4⁺ T-cell memory subsets (naïve; TCM/TTM; TEM; TEMRA). During week 1, TCM/TTM frequencies decrease significantly. (b) CIBERSORT deconvolution of bulk RNA-seq from 30 participants showing relative frequencies of naïve and central-memory CD4⁺ T cells at C01D01, C01D02, and EOT. Each point is a participant sample; colored lines connect time points per participant indicating decreases (blue) or increases (red) relative to the preceding time point. (c) UMAP of CD4⁺ T cells annotated with fine labels (SingleR package in R) across week-1 samples. (d) Effector-function module scores across CD4⁺ T-cell subsets at C01D01 (n = 12), C01D02 (n = 8), and C01D08 (n = 10). Points are participant samples; comparisons across time by Wilcoxon matched-pairs signed-rank test versus C01D01; mean ± s.e.m. shown. (e) WNT-signaling module scores analyzed as in panel d. Unless otherwise indicated, tests were two-sided with multiple-comparison adjustments where specified; otherwise, exact nominal P values are shown. (TCM/TTM, central or tissue-resident memory; TEM, effector memory; TEMRA, terminally differentiated effector memory).
Extended Data Fig. 10 Integrated Plasma cytokines and transcriptomic modules associated with control of viral outcomes.
(a) Correlation network linking plasma viral load (VL), cytokines, and HIV-specific CD4⁺ T cells. Higher VL correlates positively with IFN-γ, IFN-β, IL-6, and the frequency of HIV-specific CD4⁺ T cells, and negatively with IL-4 (Spearman ρ, P < 0.05). (b) SLEA of transcriptomic modules at C01D01, C01D02, and EOT, ranked by VL. Higher VL associates with increased ISG, TLR1/2-signaling, and effector T-cell signatures, while β-catenin (CTNNB1) target gene expression is inversely associated with viremia. (c) Proposed model of anti–PD-1–mediated HIV-reservoir reduction in ART-treated individuals. Unless otherwise indicated, tests were two-sided with multiple-comparison adjustments where specified; otherwise, exact nominal P values are shown.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–5 with legends.
Supplementary Data 1 (download XLSX )
The Excel file contains 11 sheets (D1–D11) providing source data for figures related to the CITN-12 cohort: (D1) Demographic and clinical outcomes, (D2) Virological outcomes, (D3) Omic IDs and data availability, (D4) Plasma cytokine readouts per timepoint per participant, (D5) Bulk transcriptomic module scores per timepoint per participant, (D6) T cell subset frequencies per timepoint per participant, (D7) Polyfunctional T cell frequencies per timepoint per participant, (D8) Unbiased CD8+ T cell cluster frequencies per timepoint per participant, (D9) List of genes per module altered upon anti-PD-1 treatment, (D10) List of transcription factor targets per module altered upon anti-PD-1 treatment and (D11) Commercial flow cytometry antibody details.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Talla, A., Azevedo, J.L.L.C., Latif, M.B. et al. Innate antiviral and immune functions associated with the HIV reservoir decay after anti-PD-1 therapy. Nat Med 32, 505–517 (2026). https://doi.org/10.1038/s41591-025-04139-y
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41591-025-04139-y
