
Abstract
Despite the remarkable benefits of nusinersen and other disease-modifying therapies in spinal muscular atrophy (SMA), patients may still experience clinical manifestations of the disease. Here we assessed the potential for high-dose nusinersen to rapidly slow neurodegeneration and lead to improved outcomes for patients. The global, three-part, phase 2/3 DEVOTE trial evaluated the efficacy and safety of high-dose nusinersen (50-mg loading dose; 28-mg maintenance dose) in individuals with SMA. In Part B, treatment-naive individuals (n = 75) were randomized 2:1 to 50/28 mg or 12/12 mg nusinersen. In a supportive open-label cohort (Part C), nusinersen-experienced individuals (12/12 mg for more than 1 year) were enrolled. The primary endpoint (Part B infantile-onset participants) was a 6-month change in the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) total score comparing 50/28 mg with matched ENDEAR participants (n = 20) who received sham. DEVOTE met its primary endpoint: at day 183, the CHOP-INTEND total score significantly improved (+15.1 points) in those who received 50/28 mg nusinersen and worsened (−11.1 points) in matched ENDEAR participants who received sham (difference, 26.19 (95% confidence interval = 20.7 to 31.74); statistical testing was performed using the joint-rank test where the difference in ranks was 26.06 (95% confidence interval = 17.9 to 34.2; P < 0.0001). The safety profile of 50/28 mg nusinersen was similar to the 12/12 mg regimen. The data support that high-dose nusinersen provides benefit in patients with SMA, with a generally well-tolerated safety profile. ClinicalTrials.gov registration: https://clinicaltrials.gov/study/NCT04089566. EudraCT no: 2019-002663-10.
Similar content being viewed by others


Main
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disease characterized by alpha-motor neuron degeneration which leads to progressive skeletal muscle weakness and atrophy1. SMA is caused by homozygous loss of function mutations of the SMN1 gene. The paralogous SMN2 gene produces primarily truncated and rapidly degraded survival motor neuron (SMN) protein, but also small amounts of full-length SMN2,3,4,5. SMA disease severity correlates inversely with SMN2 copy number; a lower SMN2 copy number is associated with lower SMN protein levels, earlier symptom onset and a more severe phenotype2,6.
Nusinersen, an intrathecally administered antisense oligonucleotide, modifies SMN2 precursor mRNA splicing to increase expression of full-length SMN protein and compensate for the loss of expression from SMN1. Nusinersen was the first disease-modifying therapy (DMT) approved for individuals with SMA of all ages7,8. The current dosing regimen consists of four 12-mg loading doses followed by 12-mg maintenance doses every 4 months (12/12 mg). The clinical pharmacokinetics, safety and efficacy of nusinersen have been evaluated across a broad range of ages and SMA phenotypes9,10,11,12,13,14. Benefits of nusinersen include improvements in survival, motor function outcomes and reduction in plasma neurofilament levels, a biomarker of neurodegeneration10,15,16.
Despite the remarkable impact that nusinersen and other DMTs have had on the disease, many patients still manifest functional impairment and require more complete and sustained motor neuron protection allowing potential for greater benefit. Long-term follow-up of infants treated early has shown delayed milestones that may represent an unveiling or emergence of weakness not evident when motor functions had not yet developed. This may be due to (1) motor neuron degeneration before treatment (either because of prenatal pathology or delayed diagnosis and treatment)17,18, (2) insufficient motor neuron protection on treatment (for example, because of inadequate SMN2 modulation or incomplete motor neuron transduction) and (3) comorbid features that often accompany SMA, such as joint contractures, scoliosis or pulmonary restrictions. In this study, we assessed the potential for high-dose nusinersen to rapidly slow neurodegeneration and lead to improved outcomes for patients.
The effects of high-dose nusinersen were assessed in the three-part, phase 2/3 DEVOTE trial, informed by the results of studies using lower doses of nusinersen, population pharmacokinetics and exposure–response modeling, and nonhuman primate safety studies10,11,14,19,20,21,22. Previously published results from DEVOTE Part A, an initial safety and tolerability assessment, showed that 28-mg loading and maintenance doses of nusinersen were generally well tolerated and no new safety concerns were identified22.
We report results from Parts B and C of the DEVOTE study that examined the safety and efficacy of high-dose nusinersen (50-mg loading doses and 28-mg maintenance doses (50/28 mg)). The primary component of the study was the Part B treatment-naive infantile-onset SMA cohort, which was powered to assess the primary and key secondary endpoints in the high-dose group compared with a matched subset of the sham arm in the ENDEAR trial. Secondary endpoints were also included to compare high-dose nusinersen with 12/12 mg nusinersen; however, the study was not powered for these comparisons. We also report further evidence of efficacy and safety in the smaller treatment-naive later-onset SMA cohort (Part B), and in participants who transitioned from the 12/12 mg regimen (Part C).
Results
Participant disposition
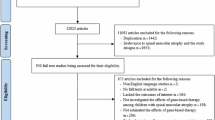
Parts B and C of the DEVOTE trial screened 170 patients and enrolled 139 participants between 12 November 2020 and 8 August 2023. In the Part B cohort with infantile-onset SMA, 50 participants received 50/28 mg nusinersen (35 (70%) completed the trial) and 25 received 12/12 mg (13 (52%) completed). The matched sham arm from ENDEAR included 20 participants. For the cohort with later-onset SMA, 16 received 50/28 mg (all completed the trial) and eight received 12/12 mg (seven (87.5%) completed). Comparator groups for the DEVOTE cohort with later-onset SMA included matched participants from CHERISH who had received sham (16 participants) and 12/12 mg nusinersen (32 participants). In Part C, 40 participants were enrolled (all completed the trial). Screening failures and treatment discontinuations are shown in Fig. 1.

The Part B cohort with infantile-onset SMA was compared with a matched group of 20 participants from ENDEAR who had received sham. The Part B cohort with later-onset SMA was compared with two matched groups of participants from CHERISH who had received sham (n = 16) and 12/12 mg nusinersen (n = 32).
Participants’ characteristics across all DEVOTE cohorts and matched comparator groups are shown in Table 1.
Participants in the cohort with infantile-onset SMA in Part B had a mean age at first dose of 18.4 (s.d. = 9.15) weeks and disease duration of 9.6 (s.d. = 5.26) weeks in the 50/28 mg group compared with 22.0 (s.d. = 7.96) weeks and 11.1 (s.d. = 4.92) weeks, respectively, in the matched sham group. All had two SMN2 copies. Prespecified matching helped minimize imbalance between groups; however, DEVOTE participants were younger at symptom onset with lower Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) scores indicating more severe disease relative to the ENDEAR matched sham group.
In the cohort with later-onset SMA in Part B, nearly all participants (22/24 (92%)) had three SMN2 copies and a median age of 6.1 (Q1, 4.2; Q3, 8.2) years at first dose in the 50/28 mg group compared with 5.7 (Q1, 2.8; Q3, 8.5) in the 12/12 mg group. The demographic characteristics and disease characteristics of the DEVOTE treatment groups and CHERISH comparator groups were generally similar. However, baseline Hammersmith Functional Motor Scale Expanded (HFMSE) scores, Revised Upper Limb Module (RULM) scores and World Health Organization (WHO) motor milestones were lower in the DEVOTE 12/12 mg group compared with the 50/28 mg, CHERISH matched sham and CHERISH matched 12/12 mg groups.
Of the 40 participants enrolled in Part C, two participants (5%) had infantile-onset SMA (both had three SMN2 copies) and 38 (95%) had later-onset SMA. Part C participants had 1–4 SMN2 copies, were aged 4–65 years at the time of first study dose and had received 12/12 mg for a median of 3.9 years before transitioning to 50/28 mg in DEVOTE.
Primary outcomes
For the primary endpoint, assessed in the Part B cohort with infantile-onset SMA, the 50/28 mg group experienced a statistically significant improvement in CHOP-INTEND at day 183 compared with the ENDEAR matched sham group, accounting for death using the joint-rank test (difference in ranks was 26.06 (95% confidence interval (CI) = 17.9 to 34.2); P < 0.0001). The least-squares mean (LSM) change was a 15.1-point (95% CI = 12.4 to 17.8) improvement with 50/28 mg and an 11.1-point (95% CI = −15.9 to −6.2) worsening in the matched sham (difference, 26.19 (95% CI = 20.7 to 31.74)) (Fig. 2a and Table 2).

a, The primary analysis was performed using ranking based on the CHOP-INTEND score at day 183 or day of death and fitting an ANCOVA model adjusting for disease duration and baseline CHOP-INTEND to determine the difference in ranks via the joint-rank test. MI was used to impute missing data. b, Results are from an ANCOVA model with adjustment for participants’ disease duration, baseline log plasma NfL and baseline CHOP-INTEND score. MI was used to impute missing data. In the table, the LSGM ratio from the ANCOVA is shown; the P value was determined using the joint-rank test. c, The P value was determined using the log-rank test stratified according to disease duration (≤12 weeks or >12 weeks). P values were obtained by comparing the two treatment groups indicated per row in each table above each chart. Comparisons between 50/28 mg and 12/12 mg, and between 50/28 mg and matched sham, were performed as separate analyses. In addition to the P value, rankings, changes from baseline in actual scores and ratios are shown. For the EFS and OS, comparisons were similarly made between the two treatment groups. Because of hierarchical testing, none of the time-to-event endpoints were considered statistically significant. Where P < 0.05 but hierarchical testing had stopped, the P value was considered nominally significant and is denoted. aP value was considered nominally significant. Study day 1 was the baseline. All statistical tests were two-sided. LSGM, least-squares geometric mean.
Secondary outcomes
Part B cohort with infantile-onset SMA: motor function
The proportion of Hammersmith Infant Neurological Exam Section 2 (HINE-2) responders at day 183 was significantly higher in the 50/28 mg group compared with the ENDEAR matched sham group (P < 0.0001), as was the magnitude of improvement in mean HINE-2 score at day 183 (P < 0.0001) (Table 2 and Extended Data Fig. 1).
While the study was not powered to detect statistically significant differences between the active treatment groups, secondary endpoints were included in the hierarchy of testing. For CHOP-INTEND, the LSM ranked score from baseline at day 302 was numerically higher in the 50/28 mg group than the 12/12 mg group, indicating greater improvement on CHOP-INTEND when accounting for death; the LSM actual score was numerically greater in the 12/12 mg group (Table 2 and Extended Data Fig. 2). For HINE-2, both the ranked and actual scores trended in favor of the 50/28 mg group.
Part B cohort with infantile-onset SMA: neurodegeneration
Reduction in plasma neurofilament light chain (NfL) levels from baseline at day 183 were significantly greater in the 50/28 mg group (94% reduction) compared with the ENDEAR matched sham group (30% reduction) (P < 0.0001), indicating a substantial slowing of neurodegeneration in response to high-dose nusinersen (Table 2 and Fig. 2b). More rapid reductions in NfL levels were observed in the 50/28 mg group (88%) compared with the 12/12 mg group (77%) at day 64 (nominally significant P = 0.0050; joint-rank test) (Fig. 2b).
Part B cohort with infantile-onset SMA: event-free survival
The 50/28 mg group experienced a lower risk of death or permanent ventilation relative to the ENDEAR matched sham group (hazard ratio (HR) = 0.322; nominal P = 0.0006; log-rank test); results were similar for survival (Table 2 and Fig. 2c). The 50/28 mg group experienced a trend toward a lower risk of death or permanent ventilation relative to the 12/12 mg group (HR = 0.701; P = 0.2775; log-rank test); results were again similar for survival.
Part B cohort with infantile-onset SMA: other key outcomes
The proportion of participants hospitalized and time in hospital attributed to serious adverse events (SAEs), the proportion of participants experiencing serious respiratory events and the proportion of time participants used ventilation support were all numerically lower in the 50/28 mg group compared with the 12/12 mg group (Extended Data Table 1). Of those participants with an opportunity to improve (those with a suck or swallow deficit on HINE-1 at baseline (n = 23 in the 50/28 mg group and n = 11 in the 12/12 mg group), 39% of the 50/28 mg group and 9% of the 12/12 mg group experienced improvement from baseline to day 302. Additionally, a numerically larger proportion of investigators and caregivers of participants in the 50/28 mg group reported a response of ‘much improved’ or ‘very much improved’ from baseline to day 302 on the Clinical Global Impression of Change than was reported for the 12/12 mg group.
Part B cohort with later-onset SMA (treatment-naive participants, n = 24 randomized 2:1): motor function
Participants in the 50/28 mg group experienced numerically greater improvements on both HFMSE and RULM from baseline to day 302 compared with the 12/12 mg group (Fig. 3a,c). However, these trends were variable over time, which is consistent with the limited sample size. More consistent differentiation was observed between the 50/28 mg group and prespecified matched comparator groups from CHERISH (sham and 12/12 mg), both of which provided larger sample sizes (Figs. 3b,d).

Data in the tables represent the mean (s.d.). a,c, Study day 1 was the baseline and MI was used for missing data. b,d, Data were based on MI and ANCOVA with treatment as a fixed effect and adjustment for each participant’s age at first dose, baseline log plasma NfL and baseline HFMSE or RULM score. CHERISH matched 12/12 mg and sham groups indicate prespecified subsets of participants matched to the characteristics of the DEVOTE 50/28 mg group. Analysis days 183 and 279 in DEVOTE correspond with days 169 and 274 in CHERISH, respectively.
Part B cohort with later-onset SMA: neurodegeneration
Greater reductions in plasma NfL levels were observed in the 50/28 mg group compared with the 12/12 mg group at day 64 (nominally significant P = 0.0495) (Extended Data Fig. 4).
Part C previously-treated participants (n = 40 open-label): motor function
After transition to the 50/28 mg regimen, participants experienced a mean improvement on HFMSE of 1.8 points (s.d. = 3.99) from baseline to day 302, with a 2.3-point (s.d. = 3.95) mean improvement in the adult subgroup (n = 24) (Supplementary Fig. 1a). Overall, 53% of participants (n = 20/38) experienced an increase in HFMSE score from baseline to day 302 (Extended Data Fig. 5a).
On RULM, participants experienced a mean improvement of 1.2 points (s.d. = 2.14) from baseline to day 302, with a 0.9-point (s.d. = 1.89) improvement in the adult subgroup (Supplementary Fig. 1b). Of those with an opportunity to improve (a score below the maximum possible at baseline; n = 26), 16 (62%) experienced an increase in RULM score from baseline to day 302 (Extended Data Fig. 5b).
Part C previously-treated participants: neurodegeneration
Given prior treatment with nusinersen, older age and more advanced disease, plasma NfL levels were low at baseline in DEVOTE Part C (5.2 pg ml−1; Table 1) and remained low throughout the study.
Parts B and C: pharmacokinetics
The 50/28 mg regimen in both Part B cohorts with infantile-onset and later-onset SMA achieved higher plasma and cerebrospinal fluid (CSF) trough concentrations than the 12/12 mg regimen (Supplementary Notes (Results, p. 24) and Supplementary Figs. 2 and 3). Day 15 CSF trough concentrations of nusinersen were approximately twofold higher after the first dose of 50 mg compared with the first dose of 12 mg nusinersen (Supplementary Fig. 4). Although the increase in CSF exposures was less than dose-proportional, 50/28 mg achieved higher CSF trough levels more quickly (one dose of 50 mg versus three doses of 12 mg) (Supplementary Fig. 5). Plasma concentrations of nusinersen increased approximately linearly with dose across all measured time points. In Part C, pre-dose CSF concentrations of nusinersen increased rapidly after the 50-mg loading dose on day 1; the higher concentrations were maintained during the maintenance dose period (Supplementary Notes: Results, p. 25). No nusinersen accumulation within plasma was observed after multiple doses in Parts B and C.
Safety
The safety profile of the 50/28 mg regimen was broadly consistent with the known safety profile of the 12/12 mg regimen.
Across Part B, 66 treatment-naive participants (50 infantile-onset, 16 later-onset) received 50/28 mg nusinersen, for a total of 50.06 participant-years; 33 treatment-naive participants (25 infantile-onset, eight later-onset) received 12/12 mg nusinersen for 21.44 participant-years. In Part C, 40 participants received 50/28 mg nusinersen for a total of 34.52 participant-years.
Across all groups treated with 50/28 mg in Parts B and C, most adverse events (AEs) were mild to moderate in severity and did not lead to treatment discontinuation; all AEs that led to study withdrawal were fatal AEs and were assessed as unrelated to treatment by the investigator (Table 3). Many of the commonly reported AEs were consistent with events occurring in the natural history of SMA, common conditions in the general population or events observed in the context of a lumbar puncture procedure. Few AEs were considered related to study treatment across all groups (<7% across all Part B; 20% in both Part C and the ENDEAR matched sham). Across all groups, only one participant in the Part B infantile-onset 12/12 mg group experienced a treatment-related SAE (respiratory failure).
In the Part B cohort with infantile-onset SMA of 50 participants, the most common AEs (≥15% of participants) in the 50/28 mg group were pneumonia (10 (20%)), respiratory failure (10 (20%)), pyrexia (9 (18%)), coronavirus disease 2019 (COVID-19) (8 (16%)) and upper respiratory tract infection (8 (16%)). In the ENDEAR matched sham group, there was a higher incidence of respiratory failure (8 (40%)), pyrexia (9 (45%)) and upper respiratory infection (5 (25%)), and a lower incidence of pneumonia (1 (5%)) and COVID-19 (0 (0%)) compared with the Part B infantile-onset 50/28 mg group. COVID-19 was not relevant at the time of ENDEAR; the higher incidence of pneumonia is discussed below. Overall, the frequency of participants reporting severe AEs in the 50/28 mg (22/50 (44%)) and 12/12 mg groups (14/25 (56%)) was lower compared with the ENDEAR matched sham group (18/20 (90%)). The frequency of participants reporting SAEs was lower in the 50/28 mg group (30/50 (60%)) compared with the 12/12 mg (18/25 (72%)) and ENDEAR matched sham groups (19/20 (95%)) (Table 3). There were no treatment-related SAEs in the 50/28 mg group. SAEs that occurred in ≥10% participants in the 50/28 mg group, which also had a frequency ≥5% in the 50/28 mg group compared with the ENDEAR matched sham, were pneumonia (seven (14%) versus 1 (5%)) and pneumonia aspiration (seven (14%) versus one (5%)). Of seven participants with pneumonia aspiration in the 50/28 mg group, five experienced the event before day 35, with two events occurring on day 1. All events were assessed as unrelated to study treatment by the investigator.
The frequency of fatal AEs was lower in participants treated with nusinersen across all groups compared with matched sham (Table 3). All fatal AEs occurred in the Part B cohort with infantile-onset SMA, and the frequency in the 50/28 mg group (10/50 (20%)) was lower than in the 12/12 mg (6/25 (24%)) or ENDEAR matched sham groups (11/20 (55%)).
Safety profiles in the Part B later-onset and Part C cohorts were similar to that in the Part B cohort with infantile-onset SMA. Across Parts B and C, there were no AEs of meningitis, hydrocephalus, thrombocytopenia or renal/liver failure reported, and no clinically relevant trends related to nusinersen in laboratory evaluations, electrocardiograms or vital signs.
Exploratory outcomes and post hoc analyses
Exploratory outcomes and post hoc analyses are described in the Supplementary Notes: Results, p. 26.
Discussion
The DEVOTE study demonstrated the safety and efficacy of high-dose nusinersen for the treatment of SMA. In treatment-naive infantile-onset participants, the 50/28 mg group experienced significantly greater reductions in neurofilament levels, significantly greater improvement in function (CHOP-INTEND) and milestone attainment (HINE-2), and nominally significantly prolonged event-free survival (EFS) relative to the prespecified, matched natural history comparator group (subgroup of the ENDEAR sham arm). While the study was not powered to detect statistically significant differences between the active treatment groups, the totality of data supports the potential for the novel 50/28 mg regimen to further optimize benefit relative to the 12/12 mg regimen. The 50/28 mg regimen slowed neurodegeneration more rapidly, as evidenced by nominally significantly greater reductions in plasma NfL at day 64. Over time, the 50/28 mg group experienced trends of greater improvement across nearly all measures assessed, including CHOP-INTEND (when accounting for mortality), HINE-2, EFS, overall survival (OS), serious respiratory events and hospitalizations due to SAEs. Data from treatment-naive later-onset participants in Part B further support the benefits of the 50/28 mg regimen. Consistent with the trajectory observed in the infantile-onset population, the 50/28 mg regimen again led to more rapid lowering of plasma NfL relative to the 12/12 mg regimen. The 50/28 mg group experienced trends of greater improvement on HFMSE and RULM but the results were highly variable over time, which is consistent with the small sample size (n = 24, randomized 2:1). Prespecified comparisons to matched subgroups from the CHERISH trial (12/12 mg and sham control groups) enabled contextualization of the improvements seen in the 50/28 mg group relative to larger populations. Part C enrolled a heterogeneous population by design, with a wide range of ages, disease phenotypes, ambulatory status, time on prior commercial nusinersen and baseline HFMSE and RULM scores. Some participants were at the upper end of HFMSE and RULM scoring at baseline; thus, they may have had a limited opportunity for additional improvement in scores. Regardless, observed improvements on HFMSE and RULM were in line with what has been previously reported after initiation of the 12/12 mg regimen in treatment-naive patients23 and exceeded what would be expected in a population that had been on nusinersen for several years (median 3.9 years at study entry)24,25.
Secondary endpoints comparing 50/28 mg with 12/12 mg showed trends in favor of the high-dose regimen, suggesting that high-dose nusinersen may drive greater benefits in individuals with SMA. However, the potential for greater benefit with the 50/28 mg regimen should be considered in the context of the risk profile. Importantly, the safety of the 50/28 mg regimen is broadly consistent with the known safety profile of the 12/12 mg regimen. The most common AEs were generally consistent with (1) the types and severity of AEs reported in people living with SMA, (2) common conditions in the pediatric population or (3) events observed in the context of a lumbar puncture procedure. Of note, in Part B (cohorts with infantile-onset and later-onset SMA), there was a lower incidence of SAEs in the 50/28 mg group compared with both the 12/12 mg and matched sham groups. Continued follow-up in the ongoing ONWARD study (ClinicalTrials.gov registration: NCT04729907) will further inform the longer-term safety profile of the 50/28 mg regimen, which has been well established with over a decade of follow-up for the 12/12 mg regimen.
Before DEVOTE, 12 mg was the highest dose of nusinersen evaluated in people living with SMA, a meaningfully lower dose level than those used for antisense oligonucleotides in other disease areas. Given the safety profile of the 12/12 mg regimen of nusinersen, data from high-dose toxicology studies in nonhuman primates and exposure–response data from humans, there was an opportunity to explore a novel, high-dose regimen to help further optimize benefit for patients.
The DEVOTE trial was powered to compare treatment-naive infantile-onset participants randomized to receive high-dose nusinersen with a matched subset of the ENDEAR sham control group. Participants were matched with the ENDEAR control group because it was infeasible to (1) randomize participants to a placebo/sham cohort in regions with approved therapies and (2) enroll enough participants to formally power the study for comparison of the 50/28 mg and 12/12 mg groups. While use of similar eligibility criteria and the prespecified matching algorithm accounted for differences in key baseline characteristics, the broad geographical scope and inclusion of new sites in the DEVOTE trial introduced additional complexity, including the impact of differences in standard of care. For example, inconsistencies in the availability of cough assist machines or ventilators in some countries can meaningfully affect outcomes for patients and trial results. As discussed above, this supportive equipment was not provisioned to sites with identified gaps until later in the study. In one country, ten of the 13 participants enrolled in the Part B cohort with infantile-onset SMA met the definition for permanent ventilation or died during the study, highlighting the impact of such differences in standard of care. With the evolving treatment landscape, an innovative trial design and inclusion of new countries and sites around the world was important in testing this hypothesis.
Perhaps most notably, changes in neurofilament levels, a marker of axonal injury and neurodegeneration26,27 were evaluated as a key secondary endpoint in the trial. In SMA, neurofilament levels correlate with disease phenotype and age, with the highest levels in the youngest patients with the most severe phenotype. Treatment with nusinersen has been previously shown to drive robust, sustained reductions of neurofilament, and these early reductions preceded demonstrable clinical benefit over time10,15. Recognizing that many of our traditional clinical outcome measures were designed over a decade ago, when trials were assessing an investigational treatment relative to the progressive natural history of SMA, these measures may have more limited utility to assess the relative benefit of different DMTs, dosing regimens or treatment paradigms (for example, monotherapy versus combination or sequential therapy). Future studies should consider incorporation of more sensitive and objective measures, such as neurofilament levels, to help address these important questions for the field. Neurofilament levels can help us understand whether a therapy or combination of therapies are arresting motor neuron degeneration sufficiently and quickly enough, recognizing that the downstream impact of failing to do so may not be clinically evident for months or even years.
Potential limitations that should be considered when interpreting these study results are primarily due to the DEVOTE study not being powered to detect differences between the 50/28 mg and 12/12 mg cohorts. As such, additional factors, such as the limitations of traditional clinical outcome measures that were not created to evaluate the relative benefit of different DMTs, and the impact of differences in standard of care across a diverse study population, may have a larger impact on the reported results than if a larger population were enrolled.
In conclusion, data from DEVOTE Parts B and C support the potential of the high-dose regimen of nusinersen to provide greater benefit compared with the 12/12 mg regimen while maintaining a similar safety profile. DEVOTE also offers important insights for the SMA field around trial design, trial conduct in new countries and sites with evolving standard of care, and the need for objective and sensitive outcome measures to assess new therapeutic approaches and ultimately inform an improved future state for SMA care.
Methods
Study design
DEVOTE was a global, three-part phase 2/3 study. Part A evaluated initial safety and tolerability of 28/28 mg nusinersen22. Part B was a randomized, double-blind evaluation of the efficacy and safety of 50/28 mg nusinersen in treatment-naive participants with infantile-onset SMA (pivotal trial population). Part B also included a supportive, smaller cohort of treatment-naive participants with later-onset SMA. Part C was an open-label cohort that evaluated 50/28 mg nusinersen in participants with SMA who transitioned from the 12/12 mg nusinersen regimen (Extended Data Fig. 6).
Ethics approval and consent
DEVOTE was conducted in accordance with the Good Clinical Practice Guidelines of the International Council for Harmonization and according to the ethical principles outlined in the 2013 Declaration of Helsinki. The protocol was approved by relevant ethics committees and institutional review boards (Supplementary Notes: Methods, p. 22). Safety data, including unblinded data from Part B, were reviewed on an ongoing basis by an Independent Data Monitoring Committee. Signed informed consent from the patient or their parent or guardian in the case of young children was mandatory. DEVOTE was registered with ClinicalTrials.gov (registration: NCT04089566) and EudraCT (no. 2019-002663-10).
While the DEVOTE protocol stipulated a requirement for participants to be offered standard of care in line with SMA consensus guidelines28,29, or provided nutritional and respiratory support per the investigator’s judgment, standard of care varied across regions. Importantly, cough assist machines and ventilators had to be provided, where needed, to sites mid-study, after it was identified that they were not available for supportive care of study participants. Ventilators were provided, when required, on a site or per-patient basis.
Participants
In Part B, individuals eligible for the cohort with infantile-onset SMA were aged between 8 days (>1 week) and 7 months, had two SMN2 copies and had SMA symptom onset at ≤6 months of age (Extended Data Fig. 6). Individuals eligible for the cohort with later-onset SMA were aged from 2 to <10 years, had SMA symptom onset at >6 months of age and could sit independently but had never walked independently. Part C participants had infantile-onset or later-onset SMA, were ambulatory or nonambulatory, and were required to have received 12/12 mg nusinersen for ≥1 year before screening. Full inclusion and exclusion criteria are included in the Supplementary Notes: Methods, p. 3.
Participants were recruited across 44 sites in 18 countries. The full list of investigators, study site personnel and study sites is included in the Supplementary Notes: Methods, p. 15.
Randomization and masking
In both Part B cohorts, treatment-naive individuals were randomized 2:1 to receive 50/28 mg (investigational group) or 12/12 mg nusinersen (control group), respectively. Randomization was stratified according to disease duration for the cohort with infantile-onset SMA (≤12 weeks and more than 12 weeks from age at symptom onset to age at informed consent), and according to age at informed consent for the cohort with later-onset SMA (<6 years and ≥6 years), which is consistent with studies CS3B and CS4.
Procedures
In both Part B cohorts, participants randomized to the control groups received 12-mg loading doses on days 1, 15, 29 and 64, followed by 12-mg maintenance doses on days 183 and 279. Participants randomized to the investigational groups received 50-mg loading doses on days 1 and 15, followed by 28-mg maintenance doses on days 135 and 279 (Extended Data Fig. 6). For the dosing days that were not common between the nusinersen regimen, participants received a sham procedure in lieu of a lumbar puncture and dosing to ensure that the blind was maintained.
All participants in Part C received a single bolus dose of 50 mg nusinersen (administered 4 months (±14 days) after their most recent 12-mg maintenance dose) followed by two 28-mg maintenance doses on days 121 and 241 (Extended Data Fig. 6).
Outcomes
The DEVOTE study was formally powered to evaluate the prespecified primary endpoint for the Part B cohort with infantile-onset SMA; the change in CHOP-INTEND score from baseline at day 183 in the 50/28 mg group versus a matched historical comparator group from the sham arm of the ENDEAR (registration: NCT02193074) trial (‘matched sham group’)11. Key secondary endpoints in the Part B cohort with infantile-onset SMA were tested in a prespecified hierarchical order (Extended Data Table 2). Other secondary endpoints are listed in the Supplementary Notes: Methods, p. 10. For the Part B cohort with later-onset SMA, key secondary efficacy endpoints for the 50/28 mg group were: NfL (change from baseline versus DEVOTE 12/12 mg); HFMSE at day 302 (change from baseline versus DEVOTE 12/12 mg and versus matched sham and 12/12 mg groups from the CHERISH trial (registration: NCT0229253712); RULM at day 302 (same comparisons as HFMSE); and safety.
Safety and tolerability were the primary endpoints in the supportive Part C transition cohort. Key secondary efficacy endpoints included change from baseline at day 302 in HFMSE and RULM.
Exploratory outcomes and post hoc analyses are described in the Supplementary Notes: Methods, p. 10.
Statistical analysis
For the primary endpoint and selected secondary endpoints in the pivotal Part B cohort with infantile-onset SMA, a matched sham comparator group was prespecified from the ENDEAR trial because of the equivocal ethics of a placebo-controlled study given the progressive nature of SMA and the availability of approved treatments. To support this, inclusion criteria for the DEVOTE cohort with infantile-onset SMA were selected to be similar to those from ENDEAR. A prespecified matching algorithm was then used to select sham arm participants from ENDEAR who were comparable at baseline to the 50/28 mg group based on two prognostic factors: disease duration and CHOP-INTEND score (Supplementary Notes: Methods, p. 11).
A prespecified hierarchical testing procedure was used for the primary and key secondary endpoints (Extended Data Table 2). Statistical comparisons for the primary and key secondary endpoints were performed sequentially, with each successive analysis requiring statistical significance of the preceding comparison. Once an endpoint in the hierarchy was found to be not statistically significant, subsequent endpoints with P < 0.05 were reported as nominally significant.
A sample size of approximately 50 participants in the 50/28 mg group was estimated to provide at least 99% power for the primary endpoint to detect an improvement of 24 points on CHOP-INTEND and a 23% survival rate benefit (versus the matched sham group from ENDEAR) at day 183, based on the joint-rank test at a two-sided significance level of 0.05. Part B was not sufficiently powered to detect statistical differences between the 50/28 mg and 12/12 mg groups as this would have required a prohibitively large number of treatment-naive infantile-onset participants; the 12/12 mg group was included to provide contemporaneous supportive evidence (Supplementary Notes: Methods, p. 15).
Multiple imputation (MI) was used to account for missing data. Joint-rank methodology was used to account for death in the analysis of functional endpoints (Supplementary Notes: Methods, p. 11)30. Comparisons of endpoints between treatment groups were based on analysis of covariance (ANCOVA) models for continuous endpoints, including as part of the joint-rank test analysis, logistic regressions or Fisher’s exact test for binary endpoints, and log-rank tests for the survival endpoints (in Part B). EFS was determined in a blinded fashion by a central, independent Endpoint Adjudication Committee.
The sample size (n = 24) for the Part B cohort with later-onset SMA was not based on statistical considerations and was not powered to detect statistically significant differences between the 50/28 mg and 12/12 mg groups. In addition to the DEVOTE 12/12 mg group, the 50/28 mg group was also compared to a subset of matched participants from the CHERISH trial. Specifically, prespecified propensity-score matching was used to select participants from the sham and 12/12 mg arms from CHERISH who were comparable at baseline to the 50/28 mg group on key prognostic factors (age, HFMSE and RULM). Further details on matching are provided in the Supplementary Notes: Methods, p. 14.
The sample size (up to 40 participants) for Part C was not based on statistical considerations nor powered to detect statistically significant improvements; descriptive statistics were performed.
Key prognostic covariates were considered to be disease duration in infantile-onset SMA and age in later-onset SMA, which were stratified for in the randomization, included in matching and adjusted for in the analysis models. In addition, baseline function, assessed using CHOP-INTEND or HFMSE, was also included in any matching and adjusted for in the analysis model. To be parsimonious in covariate selection, given the relatively small sample size, selection was limited to two covariates and sex was not included in the analysis model.
Statistical analyses were performed using the SAS software v.9.4.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Individual participant data collected during the trial may be shared after anonymization and upon approval of the research proposal. Biogen commits to sharing patient-level data, study-level data, case report forms and protocols with qualified scientific researchers who provide a methodologically sound proposal. Biogen reviews all data requests internally based on review criteria and in accordance with our Clinical Trial Transparency and Data Sharing Policy. Deidentified data and documents will be shared under agreements that further protect against participant reidentification. To request access to data, please visit https://vivli.org/. Initial requests are typically acknowledged within 2 weeks, while full data approval may take longer depending on the complexity and nature of the request. Data are controlled to protect participant privacy and comply with ethical and regulatory standards, ensuring secure access and preventing misuse.
References
Russman, B. S. Spinal muscular atrophy: clinical classification and disease heterogeneity. J. Child Neurol. 22, 946–951 (2007).
Farrar, M. A. & Kiernan, M. C. The genetics of spinal muscular atrophy: progress and challenges. Neurotherapeutics 12, 290–302 (2015).
Darras, B. T., Jones, H. R. J., Ryan, M. M. & De Vivo, D. C. in Neuromuscular Disorders of Infancy, Childhood, and Adolescence: a Clinician’s Approach (eds Darras, B. T. et al.) 117–145 (Academic Press, 2015).
Mailman, M. D. et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med. 4, 20–26 (2002).
Monani, U. R. et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183 (1999).
Arnold, W. D., Kassar, D. & Kissel, J. T. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve 51, 157–167 (2015).
Biogen. Prescribing Information: SPINRAZA 12 mg solution for injection. Initial U.S. Approval: 2016 (revised April 2024) www.accessdata.fda.gov/drugsatfda_docs/label/2024/209531s013s014lbl.pdf (2024).
Biogen Netherlands. Summary of Product Characteristics: Spinraza 12 mg solution for injection. First authorisation: 30 May 2021 (latest renewal 21 January 2022) www.ema.europa.eu/en/documents/product-information/spinraza-epar-product-information_en.pdf (2025).
Darras, B. T. et al. Nusinersen in later-onset spinal muscular atrophy: long-term results from the phase 1/2 studies. Neurology 92, e2492–e2506 (2019).
De Vivo, D. C. et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul. Disord. 29, 842–856 (2019).
Finkel, R. S. et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 377, 1723–1732 (2017).
Mercuri, E. et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 378, 625–635 (2018).
Hagenacker, T. et al. Effectiveness of nusinersen in adolescents and adults with spinal muscular atrophy: systematic review and meta-analysis. Neurol. Ther. 13, 1483–1504 (2024).
Crawford, T. O. et al. Continued benefit of nusinersen initiated in the presymptomatic stage of spinal muscular atrophy: 5-year update of the NURTURE study. Muscle Nerve 68, 157–170 (2023).
Darras, B. T. et al. Neurofilament as a potential biomarker for spinal muscular atrophy. Ann. Clin. Transl. Neurol. 6, 932–944 (2019).
Cordts, I. et al. Long-term dynamics of CSF and serum neurofilament light chain in adult patients with 5q spinal muscular atrophy treated with nusinersen. Neurology 104, e213371 (2025).
Kong, L. et al. Boosting neuregulin 1 type-III expression hastens SMA motor axon maturation. Acta Neuropathol. Commun. 11, 53 (2023).
Kong, L. et al. Impaired prenatal motor axon development necessitates early therapeutic intervention in severe SMA. Sci. Transl. Med. 13, eabb6871 (2021).
Finkel, R. S. et al. Scientific rationale for a higher dose of nusinersen. Ann. Clin. Transl. Neurol. 9, 819–829 (2022).
Finkel, R. S. et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388, 3017–3026 (2016).
Acsadi, G. et al. Safety and efficacy of nusinersen in spinal muscular atrophy: the EMBRACE study. Muscle Nerve 63, 668–677 (2021).
Finkel, R. S. et al. DEVOTE study exploring higher dose of nusinersen in spinal muscular atrophy: study design and part A results. J. Neuromuscul. Dis. 10, 813–823 (2023).
Coratti, G. et al. Motor function in type 2 and 3 SMA patients treated with nusinersen: a critical review and meta-analysis. Orphanet J. Rare Dis. 16, 430 (2021).
Günther, R. et al. Long-term efficacy and safety of nusinersen in adults with 5q spinal muscular atrophy: a prospective European multinational observational study. Lancet Reg. Health Eur. 39, 100862 (2024).
Łusakowska, A. et al. Long-term nusinersen treatment across a wide spectrum of spinal muscular atrophy severity: a real-world experience. Orphanet J. Rare Dis. 18, 230 (2023).
Gaetani, L. et al. Neurofilament light chain as a biomarker in neurological disorders. J. Neurol. Neurosurg. Psychiatry 90, 870–881 (2019).
Khalil, M. et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 14, 577–589 (2018).
Mercuri, E. et al. Diagnosis and management of spinal muscular atrophy: Part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 28, 103–115 (2018).
Finkel, R. S. et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul. Disord. 28, 197–207 (2018).
Berry, J. D. et al. The Combined Assessment of Function and Survival (CAFS): a new endpoint for ALS clinical trials. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 162–168 (2013).
Acknowledgements
We thank the participants and their families, investigators, clinical monitors, study coordinators, physical therapists and laboratory technicians. The full list of investigators, study site personnel and study sites is included in the Supplementary Notes: Methods, p. 15. Writing and editorial support for the preparation of this manuscript was provided by D. Gothard, Oxford PharmaGenesis; funding was provided by Biogen. The DEVOTE trial was sponsored by Biogen. Biogen was involved in designing the study, data collection, analysis and interpretation, report writing, and the decision to submit for publication.
Author information
Authors and Affiliations
Contributions
Conceptualization: R.S.F., T.O.C., E.M., J.W.D., J.M., R. Farewell and S.F. Data Curation: R.S.F., T.O.C., E.M., M.d.M.G.R., J.W.D. and J.S. Data Interpretation: R.S.F., T.O.C., C.J.S., J.W.D., P.S., B.T., A.D.P., J.I., R.L., J.S., M.M., R. Foster, R. Farewell and S.F. Statistical Analysis: P.S., B.T., G.G. and R. Foster. Writing – Original Draft Preparation: R.S.F., T.O.C., J.W.D., E.B., R.L., J.S., R. Foster, R. Farewell and S.F. Writing – Review & Editing: all authors. R.S.F., T.O.C. and G.G. directly accessed and verified the underlying data reported in this manuscript.
Corresponding authors
Ethics declarations
Competing interests
R.S.F. reports receiving grant support (institution) from Biogen, Dyne, Genentech, Genethon, Italfarmaco, Novartis, Roche, Sarepta and Scholar Rock; royalties from the Children’s Hospital of Philadelphia and Elsevier; advisory boards of Astellas, Biogen, Dyne, Genentech, Italfarmaco, Novartis, ReveraGen, Roche, Sarepta and Scholar Rock; honoraria and travel expenses from Novartis; patents from Roche; data safety monitoring boards of Ionis and University of Texas Southwestern; a leadership or fiduciary role for Neuromuscular Disorders (editorial board member), Journal of Neuromuscular Diseases (editorial board member) and the Pediatric Neuromuscular Clinical Research Network for SMA (steering committee member). T.O.C. reports funding from AveXis, Biogen, Catalyst, Cure SMA, Cytokinetics, Marathon, Novartis, Roche, Sarepta and the SMA Foundation; and consulting fees, advisory board and data safety monitoring board for Biogen. E.M. reports funding from AveXis, Biogen, Epirium, FamiglieSMA Italy, Ionis, Italian Telethon, NMD, Novartis, Roche and Scholar Rock. C.J.S. reports grant support from Actio Biosciences, Biogen and Roche; royalties from Elsevier; honoraria from Biogen, Genentech, Regeneron and Roche; travel to meetings support from Biogen, Regeneron and Roche; data safety monitoring board for Biogen; leadership or fiduciary role for the Peripheral Nerve Society (President); advisory boards for Cure SMA, the Charcot–Marie–Tooth Association, the Charcot–Marie–Tooth Research Foundation, the Kennedy’s Disease Association, the Muscular Dystrophy Association and the Packard Center and Merkin Center; and receipt of drugs from Ionis and Roche. M.d.M.G.R. reports honoraria, expert testimony, travel expenses and data safety monitoring board support from Biogen. J.W.D. reports grant support (institution) from AMO Pharma, AnnJi Pharmaceutical, Astellas Gene Therapies, Avidity Biosciences, Biogen, CureSMA, Genentech, Ionis, the Muscular Dystrophy Association, the Myotonic Dystrophy Foundation, NMD Pharma, Novartis, Roche, Sanofi, Sarepta, Scholar Rock and the SMA Foundation; royalties (individual and institution) from Athena Diagnostics; and consulting fees from Avidity Biosciences, Biogen, Epirium Bio, Kate Therapeutics, Novartis, PepGen, Roche/Genentech Pharmaceuticals, Sanofi, Sarepta Therapeutics, Scholar Rock, Solid Biosciences and Vertex Pharmaceuticals. J.M. reports grant support (institution) from Genentech and Scholar Rock; honoraria from Roche and Scholar Rock; and data safety monitoring boards for Argenx, Biogen, Roche and Scholar Rock. P.S., B.T., A.D.P., E.B., J.I., R.L., G.G., R. Foster, R. Farewell and S.F. are employees of Biogen and may hold stock. J.S. and M.M. were employees of Biogen at the time of contributing to this work.
Peer review
Peer review information
Nature Medicine thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Jerome Staal, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Part B infantile-onset cohort – HINE-2.
Study day 1 is baseline. All P values were obtained from comparing the two treatment groups indicated per row in each table above each chart. In the treatment columns, rankings and changes from baseline in actual scores are shown; the LSM differences correspond to the differences between the two listed treatments. Results shown are from ANCOVA model with adjustment for participants’ disease duration, baseline HINE-2 score and baseline CHOP-INTEND score. Missing data was handled with multiple imputation. All statistical tests were two sided. ANCOVA, analysis of covariance; CHOP-INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; HINE-2, Hammersmith Infant Neurological Exam section 2; LSM, least-squares mean; MI, multiple imputation; SE, standard error.
Extended Data Fig. 2 Part B infantile-onset cohort – CHOP-INTEND.
aP value considered nominally significant. Study day 1 is baseline. All P values were obtained from comparing the two treatment groups indicated per row in each table above each chart. In the treatment columns, rankings and changes from baseline in actual scores are shown; the LSM differences correspond to the differences between the two listed treatments. Results shown are from an ANCOVA model with adjustment for participants’ disease duration and baseline CHOP-INTEND score. Missing data was handled with multiple imputation. All statistical tests were two sided. ANCOVA, analysis of covariance; CHOP-INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; CI, confidence interval; JRT, joint-rank test; LSM, least-squares mean; MI, multiple imputation; SE, standard error.
Extended Data Fig. 3 Part B infantile-onset – Overall survival.
aIndicates nominal significance; P value is from log-rank test stratified by disease duration ( ≤12 weeks or >12 weeks). All P values were obtained from comparing the two treatment groups indicated per row in each table above each chart. Due to the hierarchical testing, none of the time-to-event endpoints were considered statistically significant. Where a P value was <0.05 but hierarchical testing had stopped, the P value was considered nominally significant and is denoted (a). All statistical tests were two sided. CI, confidence interval; EFS, event-free survival; HR, hazard ratio.
Extended Data Fig. 4 Part B later-onset cohort – Plasma NfL levels.
aIndicates nominal significance. Study day 1 is baseline. The P value is obtained from comparing the two treatment groups indicated. In the treatment columns, the LSGM ratio to baseline is shown; the LSGM ratio corresponds to the difference between the two listed treatments. Results shown are from an ANCOVA model with adjustment for age at screening, baseline log plasma NfL and baseline HFMSE. Missing data was handled with multiple imputation. All statistical tests were two sided. ANCOVA, analysis of covariance; CI, confidence interval; LSGM, least-squares geometric mean; MI, multiple imputation; NfL, neurofilament light chain.
Extended Data Fig. 5 Part C – HFMSE (A) and RULM (B).
Study day 1 is baseline. Observed data and mean change are shown. Two participants did not have HFMSE/RULM scores collected at baseline and are not included in the analysis. HFMSE, Hammersmith Functional Motor Scale Expanded; RULM, Revised Upper Limb Module; SD, standard deviation; SE, standard error.
Extended Data Fig. 6 Parts B and C – Study design.
DEVOTE was powered to assess the efficacy of the 50/28 mg regimen in infantile-onset participants as compared to a prespecified matched sham group from ENDEAR. Inclusion of the 12/12 mg dosing regimen in Part B was intended to provide supportive evidence, but the study was not sufficiently powered to detect statistically significant differences between the 50/28 mg and 12/12 mg groups. aA prespecified hierarchical testing procedure was used for the primary and secondary endpoints for Part B infantile-onset (see Extended Data Table 2). bENDEAR matched sham, CHERISH matched sham and CHERISH matched 12/12 mg indicate prespecified subsets of participants matched to characteristics of the DEVOTE 50/28 mg group (see Extended Data Table 2). CHOP-INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; EFS, event-free survival; HFMSE, Hammersmith Functional Motor Scale Expanded; HINE-2, Hammersmith Infant Neurological Exam section 2; NfL, neurofilament light chain; OS, overall survival; RULM, Revised Upper Limb Module; SMA, spinal muscular atrophy; SMN2, survival motor neuron 2.
Extended Data Fig. 7 Part B infantile-onset cohort – CHOP-INTEND sensitivity analyses.
CHOP-INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; CI, confidence interval; ITT, intent-to-treat; LS, least-squares; MI, multiple imputation; PP, per protocol.
Supplementary information
Supplementary Information (download PDF )
Supplementary Notes (Methods and Results), Figs. 1–6 and References.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Finkel, R.S., Crawford, T.O., Mercuri, E. et al. High-dose nusinersen for spinal muscular atrophy: a phase 3 randomized trial. Nat Med 32, 1095–1104 (2026). https://doi.org/10.1038/s41591-025-04193-6
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41591-025-04193-6
